
Conditional Ablation of Nonmuscle Myosin II-B Delineates Heart Defects in Adult Mice
16
Liu, Jeffrey A. Towbin, Michael D. Schneider, Robert S. Adelstein and Qize Wei Xuefei Ma, Kazuyo Takeda, Aman Singh, Zu-Xi Yu, Patricia Zerfas, Anthony Blount, Chengyu Conditional Ablation of Nonmuscle Myosin II-B Delineates Heart Defects in Adult Mice Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2009 American Heart Association, Inc. All rights reserved. is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Circulation Research doi: 10.1161/CIRCRESAHA.109.200303 2009;105:1102-1109; originally published online October 8, 2009; Circ Res. http://circres.ahajournals.org/content/105/11/1102 World Wide Web at: The online version of this article, along with updated information and services, is located on the http://circres.ahajournals.org/content/suppl/2009/10/08/CIRCRESAHA.109.200303.DC1.html Data Supplement (unedited) at: http://circres.ahajournals.org//subscriptions/ is online at: Circulation Research Information about subscribing to Subscriptions: http://www.lww.com/reprints Information about reprints can be found online at: Reprints: document. Permissions and Rights Question and Answer about this process is available in the located, click Request Permissions in the middle column of the Web page under Services. Further information Editorial Office. Once the online version of the published article for which permission is being requested is can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Circulation Research in Requests for permissions to reproduce figures, tables, or portions of articles originally published Permissions: by guest on August 3, 2014 http://circres.ahajournals.org/ Downloaded from by guest on August 3, 2014 http://circres.ahajournals.org/ Downloaded from by guest on August 3, 2014 http://circres.ahajournals.org/ Downloaded from by guest on August 3, 2014 http://circres.ahajournals.org/ Downloaded from by guest on August 3, 2014 http://circres.ahajournals.org/ Downloaded from by guest on August 3, 2014 http://circres.ahajournals.org/ Downloaded from by guest on August 3, 2014 http://circres.ahajournals.org/ Downloaded from by guest on August 3, 2014 http://circres.ahajournals.org/ Downloaded from by guest on August 3, 2014 http://circres.ahajournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Conditional Ablation of Nonmuscle Myosin II-B Delineates Heart Defects in Adult Mice

Liu, Jeffrey A. Towbin, Michael D. Schneider, Robert S. Adelstein and Qize WeiXuefei Ma, Kazuyo Takeda, Aman Singh, Zu-Xi Yu, Patricia Zerfas, Anthony Blount, Chengyu
Conditional Ablation of Nonmuscle Myosin II-B Delineates Heart Defects in Adult Mice
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2009 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.109.2003032009;105:1102-1109; originally published online October 8, 2009;Circ Res.
http://circres.ahajournals.org/content/105/11/1102World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2009/10/08/CIRCRESAHA.109.200303.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from

Conditional Ablation of Nonmuscle Myosin II-B DelineatesHeart Defects in Adult Mice
Xuefei Ma, Kazuyo Takeda, Aman Singh, Zu-Xi Yu, Patricia Zerfas, Anthony Blount, Chengyu Liu,Jeffrey A. Towbin, Michael D. Schneider, Robert S. Adelstein, Qize Wei
Rationale: Germline ablation of the cytoskeletal protein nonmuscle myosin II (NMII)-B results in embryoniclethality, with defects in both the brain and heart. Tissue-specific ablation of NMII-B by a Cre recombinasestrategy should prevent embryonic lethality and permit study of the function of NMII-B in adult hearts.
Objective: We sought to understand the function of NMII-B in adult mouse hearts and to see whether the braindefects found in germline-ablated mice influence cardiac development.
Methods and Results: We used a loxP/Cre recombinase strategy to specifically ablate NMII-B in the brains or heartsof mice. Mice ablated for NMII-B in neural tissues die between postnatal day 12 and 22 without showing cardiacdefects. Mice deficient in NMII-B only in cardiac myocytes (B�MHC/B�MHC mice) do not show brain defects.However, B�MHC/B�MHC mice display novel cardiac defects not seen in NMII-B germline-ablated mice. Most ofthe B�MHC/B�MHC mice are born with enlarged cardiac myocytes, some of which are multinucleated, reflectinga defect in cytokinesis. Between 6 to 10 months, they develop a cardiomyopathy that includes interstitial fibrosisand infiltration of the myocardium and pericardium with inflammatory cells. Four of 5 B�MHC/B�MHC heartsdevelop marked widening of intercalated discs.
Conclusions: By avoiding the embryonic lethality found in germline-ablated mice, we were able to study the function ofNMII-B in adult mice and show that absence of NMII-B in cardiac myocytes results in cardiomyopathy inthe adult heart. We also define a role for NMII-B in maintaining the integrity of intercalated discs. (CircRes. 2009;105:1102-1109.)
Key Words: nonmuscle myosin II-B � cardiomyopathy � intercalated discs
Nonmuscle myosin II (NMII) plays an important role inmaintaining the integrity of the actin–myosin cytoskel-
eton, which, in turn, helps to determine cell shape andfunctions in cell migration, cell polarity, and cytokinesis.Like conventional myosin IIs, such as skeletal, cardiac, andsmooth muscle myosins, NMII consists of a pair of heavychains and 2 pairs of light chains. Three isoforms of thenonmuscle myosin heavy chain (NMHC), II-A, II-B, andII-C, which are encoded by 3 genes, Myh9, -10, and -14,respectively, and are located on different chromosomes, havebeen identified in humans and mice.1–3 Although there issome overlap in the localization of these 3 isoforms, growingevidence suggests that they perform distinct functions duringcell migration and embryonic development.4–6 Ablation ofNMII-A in mice results in lethality at embryonic day (E)6.5because of the lack of a normal functioning visceralendoderm that results in a markedly abnormal body pattern.These embryos fail to undergo gastrulation.7 In contrast,
ablation of NMII-B in mice results in embryonic lethalitybetween E14.5 and birth, with defects in the brain andheart,8,9 suggesting that NMII-B is critical for the develop-ment of both. Unfortunately, the embryonic lethality inNMII-B–null mice has impeded further efforts to understandthe physiological roles of NMII-B in adult mice. Hypomor-phic mice expressing low amounts of NMHC II-B cansurvive to adulthood and also display defects in both brainsand hearts; however, severe NMII-B hypomorphs also diebefore adulthood.10 Moreover, because the physiologicalactivities of the heart are continuously regulated by thenervous system, questions arise as to whether any of the heartdefects in NMII-B–ablated or hypomorphic mice are second-ary to the brain defects.
In this study, we ablated NMHC II-B in mice, either in thenervous system alone or in the cardiac myocytes alone, usinga loxP/Cre recombinase strategy. We crossed the NMHC II-Bfloxed mice with a line of mice expressing Cre recombinase
Original received December 8, 2008; resubmission received May 4, 2009; revised resubmission received September 17, 2009; accepted September 24, 2009.From the Laboratory of Molecular Cardiology (X.M., K.T., A.S., A.B., R.S.A., Q.W.), Pathology Core Facility (Z.-X.Y.), and Transgenic Mouse Core
Facility (C.L.), National Heart, Lung, and Blood Institute; and Division of Veterinary Resources (P.Z.), NIH, Bethesda, Md; Cincinnati Children’sHospital Medical Center (J.A.T.), Ohio; and National Heart and Lung Institute (M.D.S.), Imperial College, London, United Kingdom. Present addressfor Q.W.: Department of Biochemistry, Kansas State University, Manhattan.
Correspondence to Robert S. Adelstein, MD, Laboratory of Molecular Cardiology, NHLBI, NIH, Building 10, Room 6C-103, 10 Center Dr, MSC-1583,Bethesda, MD 20892. E-mail [email protected]
© 2009 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.109.200303
1102 by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from

regulated by the neural cell–specific nestin promoter to ablateNMHC II-B in the nervous system.11 In separate experiments,we crossed the NMHC II-B floxed mice with a line of miceexpressing Cre recombinase under control of the �-myosinheavy chain (�MHC) promoter to ablate NMII-B in cardiacmyocytes.12 Below, we present results showing that NMII-Bplays distinct physiological roles in the brain and heart andprovide evidence that absence of NMII-B in the cardiacmyocytes (and not in the nonmyocytes) results in myocyteenlargement and cardiomyopathy. Moreover, we demonstratea role for NMII-B in the intercalated disc (ID) of adult mice.
MethodsAn expanded Methods section is available in the Online DataSupplement at http://circres.ahajournals.org.
AnimalsAll experiments were conducted following animal protocols ap-proved by Animal Care and Use Committee of the National Heart,Lung, and Blood Institute. Nestin-Cre transgenic mice were fromThe Jackson Laboratory (no. 003771).
Histology, Microscopy, and ImmunoblottingHematoxylin/eosin (H&E) and immunofluorescence staining, elec-tron microscopy, and immunoblotting were performed as describedpreviously.8
Measurement of the Cross-Sectional Area of theCardiac MyocytesThe size of cardiac myocytes was measured following wheat germagglutinin staining using a Zeiss measuring tool.
EchocardiographyEchocardiography was performed using an Acuson Sequoia 256cimaging system with the 15L8 multifrequency transducer. Quantitationwas performed using M-mode with Prosolv Software version 3.0.
ElectrocardiographyThree-lead electrocardiograms were recorded with a model MAC1200 (GE Medical Systems).
Data and Statistical AnalysisThe data were expressed as means�SD. Student’s t test was used tocompare data between 2 groups.
ResultsGeneration of Neural- and Cardiac-SpecificNMHC II-B Knockout MiceTo ablate NMHC II-B specifically in the brain or in the heart,we used a loxP/Cre recombinase strategy to delete exon 2, thefirst coding exon of Myh10 (see Figure 1). Using thegene-targeting method, we generated a mouse line designatedBflox/Bflox in which both the neomycin resistance (Neor)expression cassette and exon 2 of Myh10 are flanked by loxPsites (Figure 1c). Bflox/Bflox mice express normal amounts ofNMHC II-B protein and are indistinguishable from wild-typemice. We crossed Bflox/Bflox mice with 2 different transgeniclines expressing Cre recombinase under control of differentpromoters. To ablate NMHC II-B in neural tissue, we used amouse line expressing Cre recombinase with a nestin pro-moter and a nervous system–specific enhancer11 (Figure 1e)to generate B�/Bnest and Bnest/Bnest mice. To ablate NMHCII-B in cardiac myocytes, we used a mouse line expressingCre recombinase driven by the �MHC promoter12 (Figure 1f)to generate B�/B�MHC and B�MHC/B�MHC mice. The trans-genic mouse lines nestin-Cre and �MHC-Cre are well char-acterized and demonstrate that nestin-Cre mice express func-tional Cre recombinase in the nervous system starting atE10.511 and that �MHC-Cre mice express functional Crerecombinase specifically in cardiac myocytes starting at E9.0and at high levels by E11.5.13
Non-standard Abbreviations and Acronyms
Cre Cre recombinase
DORV double outlet of the right ventricle
E embryonic day
H&E hematoxylin/eosin
ID intercalated disc
MHC cardiac myosin heavy chain
NMII nonmuscle myosin II
NMHC nonmuscle myosin heavy chain
VSD ventricular septal defect
Figure 1. Schematic representation of thestrategy used to generate neural- or cardiacmyocyte–specific NMHC II-B knockout mice. a,Portion of the wild-type NMHC II-B gene locusincluding exon 2. b, Targeting construct.Neomycin-resistance cassette (Neor) and exon2 are flanked by loxP sites. c, Bflox allele isgenerated after homologous recombination. d,NMHC II-B–ablated allele (Bnest or B�MHC) lack-ing exon 2 is generated in the presence of Crerecombinase. e, The Cre expression cassetteunder the control of nestin promoter in theneural-specific Cre transgenic mice. f, The Creexpression cassette under the control of the�MHC promoter in the cardiac myocyte–spe-cific Cre transgenic mice. Black boxes in a, b,and c indicate exon 2; arrowheads, lox P sites.
Ma et al Conditional Ablation of Nonmuscle Myosin II-B 1103
by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from

Nestin-Cre–Specific Ablation of NMHC II-B in theMouse BrainImmunoblot analysis of Bnest/Bnest mice on postnatal day(P)18 shows that NMHC II-B protein is markedly reduced inthe cerebellum (Figure 2A, compare lanes 1 and 2), as well asthroughout the entire brain (data not shown) but not in thehearts of Bnest/Bnest mice (Figure 2A, lanes 3 and 4). Immu-nofluorescence staining using an antibody for NMHC II-Bconfirms that NMHC II-B protein is ablated in the cerebellumof these mice at P18 (Figure 2B). As shown in the figure,Purkinje cells in Bflox/Bflox mice express high levels ofNMHC II-B (red; Figure 2B, a). However, the NMHC II-Bprotein level is significantly reduced in the cerebellar Pur-kinje cells of Bnest/Bnest mice (Figure 2B, compare a with e).In contrast, staining for calbindin (Purkinje cell marker) isunaltered in these cells (Figure 2B, compare b with f). Figure2B (d and h) confirms the loss of NMHC II-B (yellow in dand green in h) in the cerebellar Purkinje cells. Both immu-noblot analysis and immunofluorescence staining confirmthat NMHC II-B protein is also significantly reduced in thecerebral cortex of Bnest/Bnest mice (data not shown).
All of the Bnest/Bnest mice die between P12 and P22 as a resultof a severe hydrocephalus, which causes enlargement of thelateral ventricles and a paper-thin cortex, with absence of mostbrain cortical tissue (Figure 2C, compare b and d). Figure 2C (c)also shows an underdeveloped cerebellum (ellipse), which cor-relates with defects in motor activity in these mice and whichwas also seen in hypomorphic mice that have a point mutation inNMHC II-B.14 The arrows in Figure 2C (c) point to deformitiesfollowing the decompression of the lateral ventricles and loss ofcerebral–spinal fluid. Of note is our finding that, similar tohypomorphic mice with a point mutation in NMHC II-B,15 thespinal canal of Bnest/Bnest mice is completely ablated at P7(Figure 2D). This is consistent with a role for NMII-B incell–cell adhesion in the spinal canal.
Sectioning of the hearts confirms that there is none of thecardiac abnormalities in the Bnest/Bnest mice that are found in
B�/B� mice. Specifically, there is no evidence for a ventric-ular septal defect (VSD), double outlet of the right ventricle(DORV), myocyte hypertrophy, decreased myocyte number,or increased cardiac myocyte binucleation, as seen in B�/B�
mice.8,16 This demonstrates that ablation of NMHC II-B inthe nervous system does not affect cardiac development.B�/Bnest mice appear normal in all respects.
Pathological Changes in the Heart Following�MHC-Cre–Specific Ablation of NMHC II-BTo address the role of NMII-B in heart development and in theadult mouse, we crossed the Bflox/Bflox mice with �MHC-Cremice (Figure 1f), so that ablation would occur specifically in thecardiac myocytes starting at midgestation or after approximatelyE11.5 to avoid the early lethality found in germline-ablatedB�/B� mice. Similar to other investigators who have used thisparticular line of mice expressing Cre recombinase,17 we foundno adverse effects of the enzyme on the tissues in which it wasexpressed (see also Figures 4 and 6 and Online Figure I). TheNMHC II-B protein level is significantly reduced in the hearts ofB�MHC/B�MHC mice compared to Bflox/Bflox mice at P0, asdemonstrated in the immunoblot in Figure 3A. However, theNMHC II-B level is not affected in the brain of B�MHC/B�MHC
mice (Figure 3A). The presence of residual NMHC II-B in theheart of B�MHC/B�MHC mice can be attributed to nonmyocytes inthe heart, which continue to express wild-type amounts ofNMHC II-B. Immunofluorescence staining of E13.5 mouseheart sections using antibodies to NMHC II-A, II-B, and II-Chelps to clarify the ablation of NMHC II-B from the myocytesalone. It also demonstrates that ablation of NMHC II-B incardiac myocytes has no effect on NMHC II-A and II-Cexpression. Figure 3B (a and d) shows that NMHC II-A (green)is only present in the nonmyocytes in the heart. This is indicatedby the lack of green signal coincident with desmin (red) in thecardiac myocytes, because NMHC II-A is not present at this agein these cells. In contrast, in Figure 3B (b), NMHC II-B (green)and desmin (red) costain the myocytes (yellow) and stain the
Figure 2. NMHC II-B expression levels and pheno-type changes in Bnest/Bnest mice. A, Immunoblotanalysis of whole tissue lysates from the cerebel-lum and the heart of Bnest/Bnest and Bflox/Bflox miceusing antibodies specific for NMHC II-B and�-tubulin (loading control) as indicated. B, Immu-nofluorescence staining of sections from the cere-bellum of Bflox/Bflox (a through d) and Bnest/Bnest (ethrough h) mice at P18 using antibodies specificfor NMHC II-B (red) and calbindin (green); DAPI isblue. Calbindin is a marker for Purkinje cells andDAPI for nuclei. G indicates the granular layer and Mthe molecular layer in the cerebellum. C (a and c),Photographs of the brain from Bflox/Bflox (a)and Bnest/Bnest (c) mice. Arrows in c indicate thehydrocephalus-induced collapse of the cerebral cor-tex after fixation. Dashed ellipse encloses cerebel-lum. b and d, Coronal sections of the brains fromBflox/Bflox (b) and Bnest/Bnest (d) mice following H&Estaining. D (a and b) shows the spinal canal at P7.The canals of Bflox/Bflox mice (a) are narrow butpatent at this age (boxed areas, enlarged in insets,arrow). In b, the canal of a Bnest/Bnest mouse is com-pletely obliterated.
1104 Circulation Research November 20, 2009
by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from
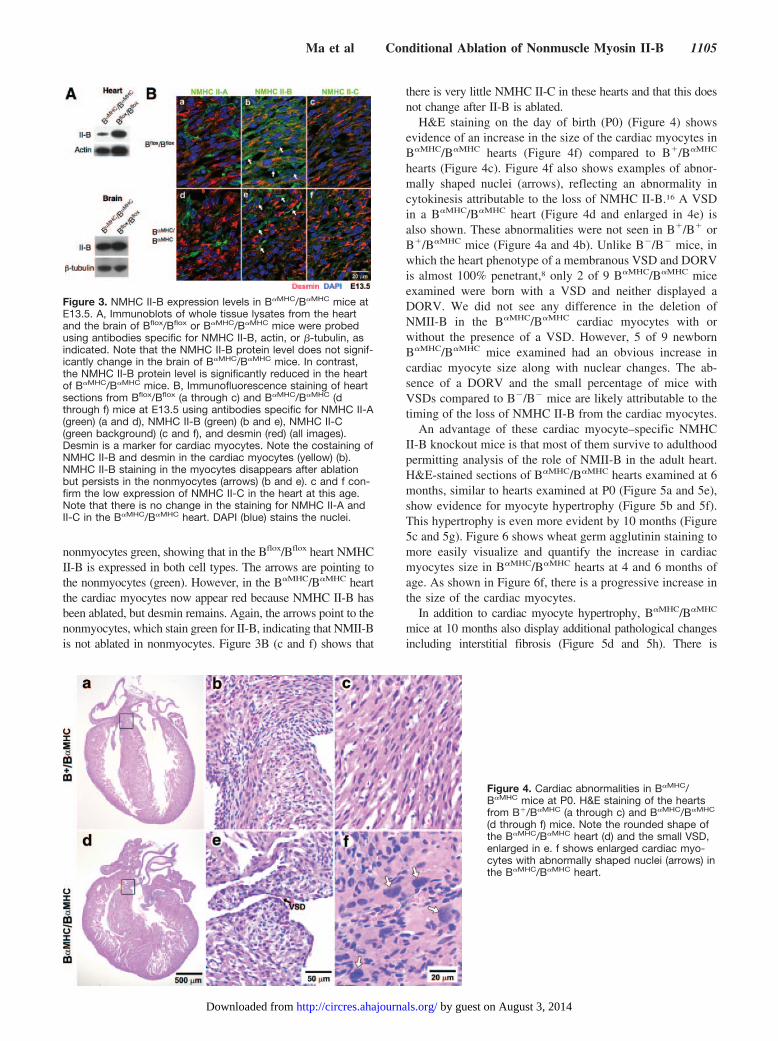
nonmyocytes green, showing that in the Bflox/Bflox heart NMHCII-B is expressed in both cell types. The arrows are pointing tothe nonmyocytes (green). However, in the B�MHC/B�MHC heartthe cardiac myocytes now appear red because NMHC II-B hasbeen ablated, but desmin remains. Again, the arrows point to thenonmyocytes, which stain green for II-B, indicating that NMII-Bis not ablated in nonmyocytes. Figure 3B (c and f) shows that
there is very little NMHC II-C in these hearts and that this doesnot change after II-B is ablated.
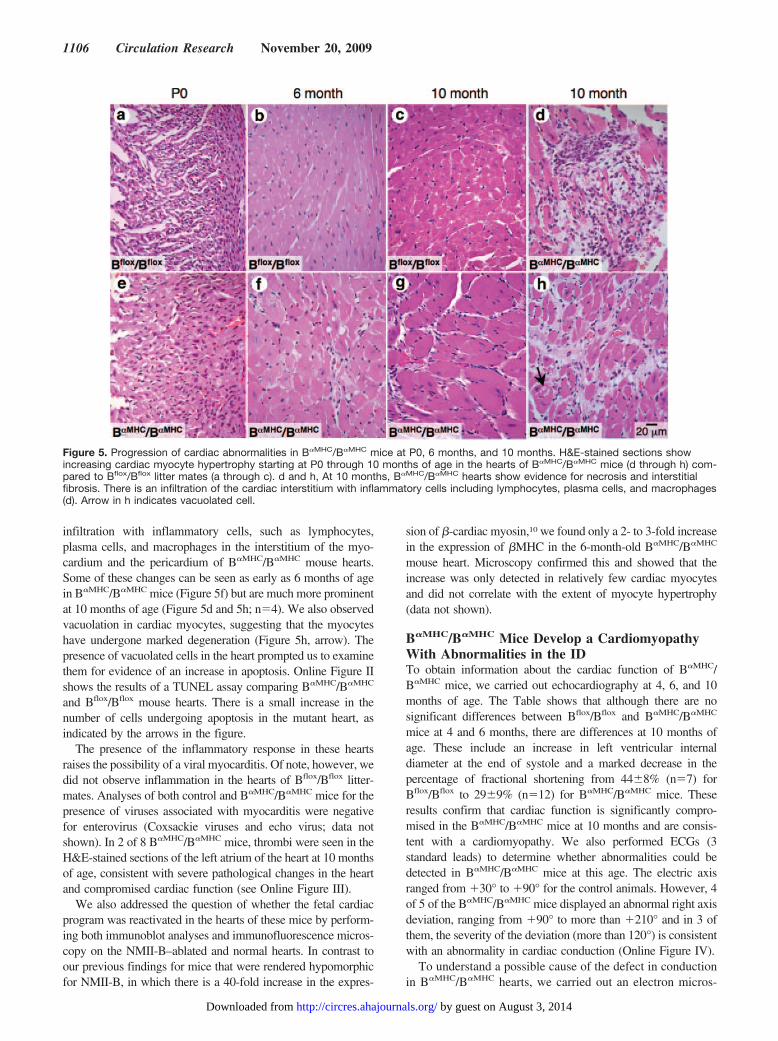
H&E staining on the day of birth (P0) (Figure 4) showsevidence of an increase in the size of the cardiac myocytes inB�MHC/B�MHC hearts (Figure 4f) compared to B�/B�MHC
hearts (Figure 4c). Figure 4f also shows examples of abnor-mally shaped nuclei (arrows), reflecting an abnormality incytokinesis attributable to the loss of NMHC II-B.16 A VSDin a B�MHC/B�MHC heart (Figure 4d and enlarged in 4e) isalso shown. These abnormalities were not seen in B�/B� orB�/B�MHC mice (Figure 4a and 4b). Unlike B�/B� mice, inwhich the heart phenotype of a membranous VSD and DORVis almost 100% penetrant,8 only 2 of 9 B�MHC/B�MHC miceexamined were born with a VSD and neither displayed aDORV. We did not see any difference in the deletion ofNMII-B in the B�MHC/B�MHC cardiac myocytes with orwithout the presence of a VSD. However, 5 of 9 newbornB�MHC/B�MHC mice examined had an obvious increase incardiac myocyte size along with nuclear changes. The ab-sence of a DORV and the small percentage of mice withVSDs compared to B�/B� mice are likely attributable to thetiming of the loss of NMHC II-B from the cardiac myocytes.
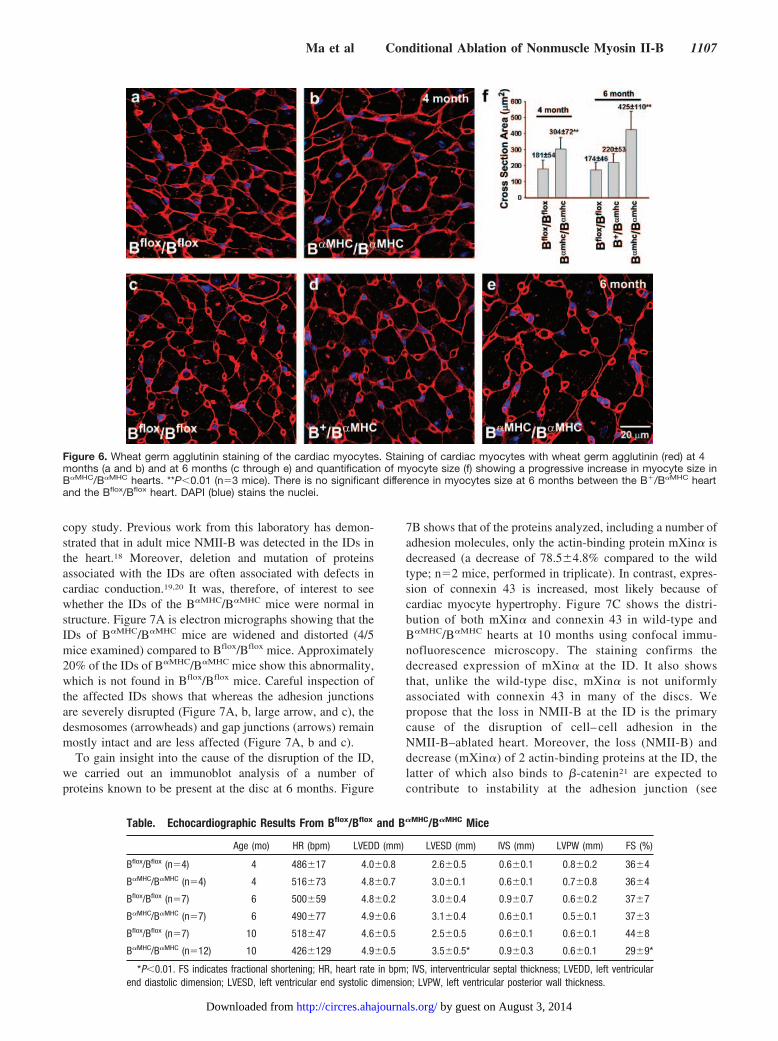
An advantage of these cardiac myocyte–specific NMHCII-B knockout mice is that most of them survive to adulthoodpermitting analysis of the role of NMII-B in the adult heart.H&E-stained sections of B�MHC/B�MHC hearts examined at 6months, similar to hearts examined at P0 (Figure 5a and 5e),show evidence for myocyte hypertrophy (Figure 5b and 5f).This hypertrophy is even more evident by 10 months (Figure5c and 5g). Figure 6 shows wheat germ agglutinin staining tomore easily visualize and quantify the increase in cardiacmyocytes size in B�MHC/B�MHC hearts at 4 and 6 months ofage. As shown in Figure 6f, there is a progressive increase inthe size of the cardiac myocytes.
In addition to cardiac myocyte hypertrophy, B�MHC/B�MHC
mice at 10 months also display additional pathological changesincluding interstitial fibrosis (Figure 5d and 5h). There is
Figure 3. NMHC II-B expression levels in B�MHC/B�MHC mice atE13.5. A, Immunoblots of whole tissue lysates from the heartand the brain of Bflox/Bflox or B�MHC/B�MHC mice were probedusing antibodies specific for NMHC II-B, actin, or �-tubulin, asindicated. Note that the NMHC II-B protein level does not signif-icantly change in the brain of B�MHC/B�MHC mice. In contrast,the NMHC II-B protein level is significantly reduced in the heartof B�MHC/B�MHC mice. B, Immunofluorescence staining of heartsections from Bflox/Bflox (a through c) and B�MHC/B�MHC (dthrough f) mice at E13.5 using antibodies specific for NMHC II-A(green) (a and d), NMHC II-B (green) (b and e), NMHC II-C(green background) (c and f), and desmin (red) (all images).Desmin is a marker for cardiac myocytes. Note the costaining ofNMHC II-B and desmin in the cardiac myocytes (yellow) (b).NMHC II-B staining in the myocytes disappears after ablationbut persists in the nonmyocytes (arrows) (b and e). c and f con-firm the low expression of NMHC II-C in the heart at this age.Note that there is no change in the staining for NMHC II-A andII-C in the B�MHC/B�MHC heart. DAPI (blue) stains the nuclei.
Figure 4. Cardiac abnormalities in B�MHC/B�MHC mice at P0. H&E staining of the heartsfrom B�/B�MHC (a through c) and B�MHC/B�MHC
(d through f) mice. Note the rounded shape ofthe B�MHC/B�MHC heart (d) and the small VSD,enlarged in e. f shows enlarged cardiac myo-cytes with abnormally shaped nuclei (arrows) inthe B�MHC/B�MHC heart.
Ma et al Conditional Ablation of Nonmuscle Myosin II-B 1105
by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from
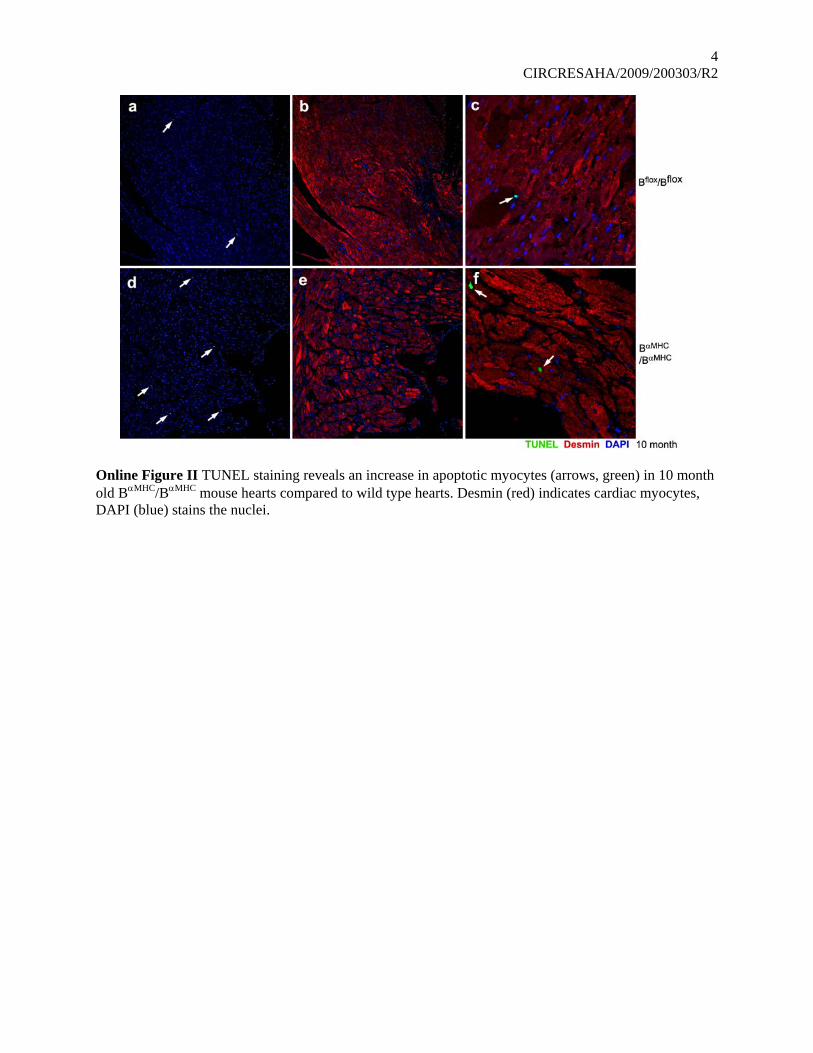
infiltration with inflammatory cells, such as lymphocytes,plasma cells, and macrophages in the interstitium of the myo-cardium and the pericardium of B�MHC/B�MHC mouse hearts.Some of these changes can be seen as early as 6 months of agein B�MHC/B�MHC mice (Figure 5f) but are much more prominentat 10 months of age (Figure 5d and 5h; n�4). We also observedvacuolation in cardiac myocytes, suggesting that the myocyteshave undergone marked degeneration (Figure 5h, arrow). Thepresence of vacuolated cells in the heart prompted us to examinethem for evidence of an increase in apoptosis. Online Figure IIshows the results of a TUNEL assay comparing B�MHC/B�MHC
and Bflox/Bflox mouse hearts. There is a small increase in thenumber of cells undergoing apoptosis in the mutant heart, asindicated by the arrows in the figure.
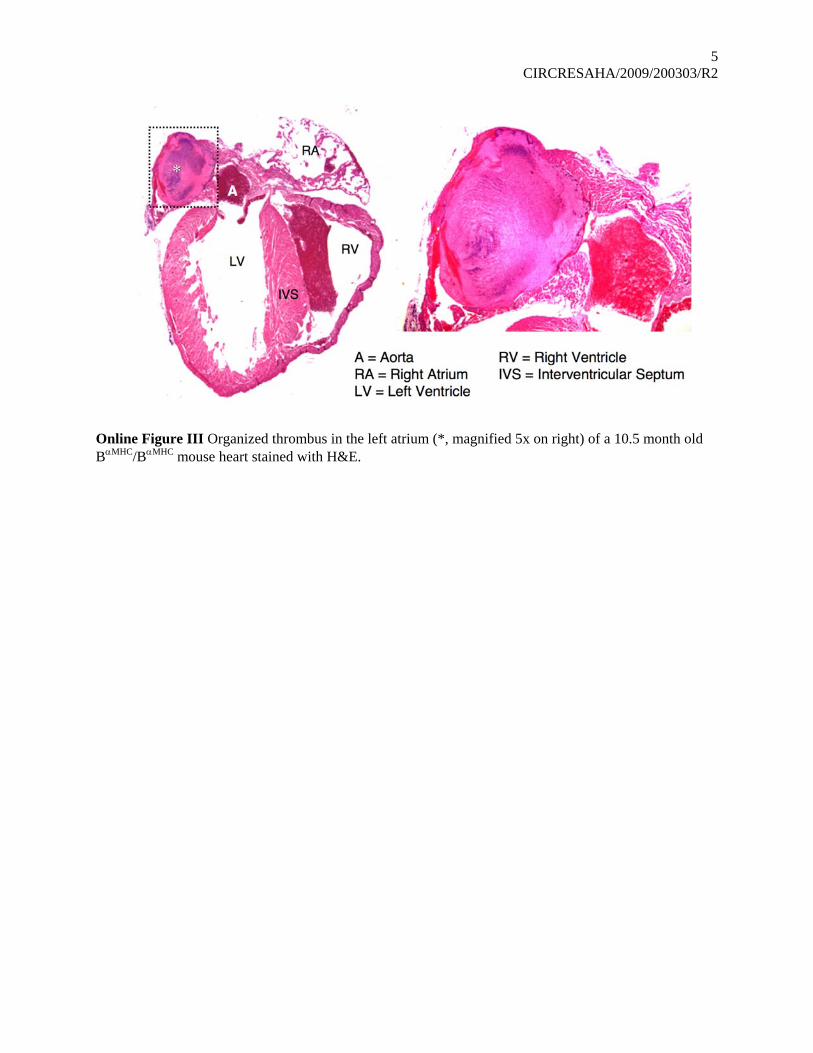
The presence of the inflammatory response in these heartsraises the possibility of a viral myocarditis. Of note, however, wedid not observe inflammation in the hearts of Bflox/Bflox litter-mates. Analyses of both control and B�MHC/B�MHC mice for thepresence of viruses associated with myocarditis were negativefor enterovirus (Coxsackie viruses and echo virus; data notshown). In 2 of 8 B�MHC/B�MHC mice, thrombi were seen in theH&E-stained sections of the left atrium of the heart at 10 monthsof age, consistent with severe pathological changes in the heartand compromised cardiac function (see Online Figure III).
We also addressed the question of whether the fetal cardiacprogram was reactivated in the hearts of these mice by perform-ing both immunoblot analyses and immunofluorescence micros-copy on the NMII-B–ablated and normal hearts. In contrast toour previous findings for mice that were rendered hypomorphicfor NMII-B, in which there is a 40-fold increase in the expres-
sion of �-cardiac myosin,10 we found only a 2- to 3-fold increasein the expression of �MHC in the 6-month-old B�MHC/B�MHC
mouse heart. Microscopy confirmed this and showed that theincrease was only detected in relatively few cardiac myocytesand did not correlate with the extent of myocyte hypertrophy(data not shown).
B�MHC/B�MHC Mice Develop a CardiomyopathyWith Abnormalities in the IDTo obtain information about the cardiac function of B�MHC/B�MHC mice, we carried out echocardiography at 4, 6, and 10months of age. The Table shows that although there are nosignificant differences between Bflox/Bflox and B�MHC/B�MHC
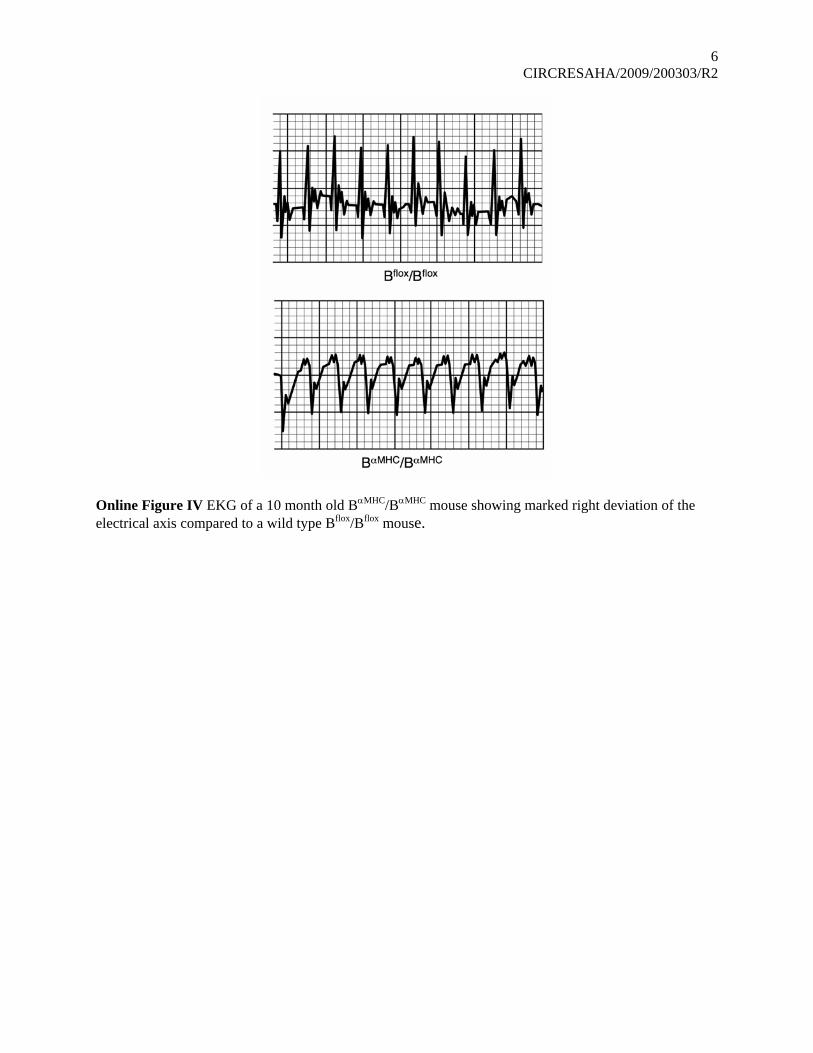
mice at 4 and 6 months, there are differences at 10 months ofage. These include an increase in left ventricular internaldiameter at the end of systole and a marked decrease in thepercentage of fractional shortening from 44�8% (n�7) forBflox/Bflox to 29�9% (n�12) for B�MHC/B�MHC mice. Theseresults confirm that cardiac function is significantly compro-mised in the B�MHC/B�MHC mice at 10 months and are consis-tent with a cardiomyopathy. We also performed ECGs (3standard leads) to determine whether abnormalities could bedetected in B�MHC/B�MHC mice at this age. The electric axisranged from �30° to �90° for the control animals. However, 4of 5 of the B�MHC/B�MHC mice displayed an abnormal right axisdeviation, ranging from �90° to more than �210° and in 3 ofthem, the severity of the deviation (more than 120°) is consistentwith an abnormality in cardiac conduction (Online Figure IV).
To understand a possible cause of the defect in conductionin B�MHC/B�MHC hearts, we carried out an electron micros-
Figure 5. Progression of cardiac abnormalities in B�MHC/B�MHC mice at P0, 6 months, and 10 months. H&E-stained sections showincreasing cardiac myocyte hypertrophy starting at P0 through 10 months of age in the hearts of B�MHC/B�MHC mice (d through h) com-pared to Bflox/Bflox litter mates (a through c). d and h, At 10 months, B�MHC/B�MHC hearts show evidence for necrosis and interstitialfibrosis. There is an infiltration of the cardiac interstitium with inflammatory cells including lymphocytes, plasma cells, and macrophages(d). Arrow in h indicates vacuolated cell.
1106 Circulation Research November 20, 2009
by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from
copy study. Previous work from this laboratory has demon-strated that in adult mice NMII-B was detected in the IDs inthe heart.18 Moreover, deletion and mutation of proteinsassociated with the IDs are often associated with defects incardiac conduction.19,20 It was, therefore, of interest to seewhether the IDs of the B�MHC/B�MHC mice were normal instructure. Figure 7A is electron micrographs showing that theIDs of B�MHC/B�MHC mice are widened and distorted (4/5mice examined) compared to Bflox/Bflox mice. Approximately20% of the IDs of B�MHC/B�MHC mice show this abnormality,which is not found in Bflox/Bflox mice. Careful inspection ofthe affected IDs shows that whereas the adhesion junctionsare severely disrupted (Figure 7A, b, large arrow, and c), thedesmosomes (arrowheads) and gap junctions (arrows) remainmostly intact and are less affected (Figure 7A, b and c).
To gain insight into the cause of the disruption of the ID,we carried out an immunoblot analysis of a number ofproteins known to be present at the disc at 6 months. Figure
7B shows that of the proteins analyzed, including a number ofadhesion molecules, only the actin-binding protein mXin� isdecreased (a decrease of 78.5�4.8% compared to the wildtype; n�2 mice, performed in triplicate). In contrast, expres-sion of connexin 43 is increased, most likely because ofcardiac myocyte hypertrophy. Figure 7C shows the distri-bution of both mXin� and connexin 43 in wild-type andB�MHC/B�MHC hearts at 10 months using confocal immu-nofluorescence microscopy. The staining confirms thedecreased expression of mXin� at the ID. It also showsthat, unlike the wild-type disc, mXin� is not uniformlyassociated with connexin 43 in many of the discs. Wepropose that the loss in NMII-B at the ID is the primarycause of the disruption of cell– cell adhesion in theNMII-B–ablated heart. Moreover, the loss (NMII-B) anddecrease (mXin�) of 2 actin-binding proteins at the ID, thelatter of which also binds to �-catenin21 are expected tocontribute to instability at the adhesion junction (see
Figure 6. Wheat germ agglutinin staining of the cardiac myocytes. Staining of cardiac myocytes with wheat germ agglutinin (red) at 4months (a and b) and at 6 months (c through e) and quantification of myocyte size (f) showing a progressive increase in myocyte size inB�MHC/B�MHC hearts. **P�0.01 (n�3 mice). There is no significant difference in myocytes size at 6 months between the B�/B�MHC heartand the Bflox/Bflox heart. DAPI (blue) stains the nuclei.
Table. Echocardiographic Results From Bflox/Bflox and B�MHC/B�MHC Mice
Age (mo) HR (bpm) LVEDD (mm) LVESD (mm) IVS (mm) LVPW (mm) FS (%)
Bflox/Bflox (n�4) 4 486�17 4.0�0.8 2.6�0.5 0.6�0.1 0.8�0.2 36�4
B�MHC/B�MHC (n�4) 4 516�73 4.8�0.7 3.0�0.1 0.6�0.1 0.7�0.8 36�4
Bflox/Bflox (n�7) 6 500�59 4.8�0.2 3.0�0.4 0.9�0.7 0.6�0.2 37�7
B�MHC/B�MHC (n�7) 6 490�77 4.9�0.6 3.1�0.4 0.6�0.1 0.5�0.1 37�3
Bflox/Bflox (n�7) 10 518�47 4.6�0.5 2.5�0.5 0.6�0.1 0.6�0.1 44�8
B�MHC/B�MHC (n�12) 10 426�129 4.9�0.5 3.5�0.5* 0.9�0.3 0.6�0.1 29�9*
*P�0.01. FS indicates fractional shortening; HR, heart rate in bpm; IVS, interventricular septal thickness; LVEDD, left ventricularend diastolic dimension; LVESD, left ventricular end systolic dimension; LVPW, left ventricular posterior wall thickness.
Ma et al Conditional Ablation of Nonmuscle Myosin II-B 1107
by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from

Discussion). Of note, no defects in the brain or otherorgans were found in B�MHC/B�MHC mice at any age.
DiscussionWe have previously reported that, as early as E11.5, globalablation of NMHC II-B in mice resulted in hydrocephalusassociated with defects in cell–cell adhesion of the neuralepithelial cells lining the spinal canal and cerebral ventri-cles.9,15 In Bnest/Bnest mice, ablation of NMHC II-B wasinitiated at E10.5, controlled by the nestin promoter, which isconsistent with the delayed onset of hydrocephalus. Ofparticular note, despite the death of these mice between 12 to22 days of age, most likely resulting from severe hydroceph-alus, there were no abnormalities found in the heart.
In addition to learning whether the defects we found in theB�/B� mouse hearts were related directly or indirectly tothose found in the nervous system, we also wanted to studythe role of NMII-B in the adult mouse heart. Most B�MHC/B�MHC mice manifested progressive cardiac abnormalities,starting with myocyte hypertrophy, which was apparent asearly as P0 and increased during postnatal development to 6and 10 months of age. At 10 months, there was also evidenceof myocyte vacuolation and cell degeneration, interstitialfibrosis, and an infiltration of the cardiac tissue with inflam-matory cells. We hypothesize that the cardiac phenotype inthe B�MHC/B�MHC mice is initiated by abnormalities specificto the cardiac myocytes, because NMII-B is ablated in thesecells but not in the nonmyocytes in these mice. This loss ofNMII-B (and lack of compensation by NMII-A or II-C)results in a failure in cytokinesis, as manifested bymultinucleation and the abnormal nuclei found in these cells.It most likely contributes to abnormal enlargement of thecardiac myocytes, as well as their decreased numbers at P0.We, therefore, reasoned that the interstitial fibrosis andinfiltration of inflammatory cells are secondary to the primary
abnormality in the cardiac myocytes, which is most likelymyocyte degeneration.
The pathological changes in the hearts of cardiac-specificNMHC II-B knockout mice are in agreement with theechocardiographic and ECG studies. The marked decrease inthe fractional shortening at 10 months is consistent with thecompromised contractility of cardiac muscle. The abnormal-ities noted in ECGs (an abnormal electrical axis) could reflectthe striking defects found in the IDs. Previous work hasshown that in the adult heart NMII-B is localized to the Z-linesand IDs.18 The IDs are composed of adherens junctions, desmo-somes, and gap junctions that form cell–cell boundaries andconnections between cardiac myocytes and allow the myocar-dium to function in synchrony. As noted above, work from anumber of laboratories has shown that NMIIs play an importantrole in cell–cell adhesion7,15,20,22 and that abnormalities in anumber of adhesion proteins result in either loss or structuralchanges in the cardiac IDs.20,23,24 Figure 7A provides evidencethat loss of NMII-B primarily affects the adhesion junctionsrather than the gap junctions or desmosomes. Moreover, B�MHC/B�MHC hearts at 6 months show a milder defect in the adhesionjunction of the IDs and no defects in the desmosomes and gapjunctions (data not shown).
We have analyzed the expression of a number of IDproteins and found a significant decrease in the expression ofmXin� in B�MHC/B�MHC hearts compared to the wild-typehearts. Mice ablated for mXin� also show abnormal IDs.19
Unlike the mXin� knockout hearts, NMII-B–ablated heartsshow no decrease in expression levels of N-cadherin or�-catenin. Moreover, there was no change in the distributionof �-catenin. We, therefore, attribute the primary cause of thedisruption of the IDs to the loss of NMII-B. We speculate thatthe decrease of mXin� is secondary to the loss of NMII-B,and the mechanism of this decrease is of ongoing interest.The decrease in both of these proteins, one of which (mXin�)has been demonstrated to also bind to �-catenin,21 wouldexplain the marked disruption of the IDs.
Figure 7. Abnormalities in theIDs in B�MHC/B�MHC mice at 6and 10 months. A, Electronmicroscopic sections of Bflox/Bflox (a) and B�MHC/B�MHC (band c) left ventricles at 10months. b shows a less-affected ID than c. Both b(large arrow) and c show thatthe adhesion type junctions ofthe B�MHC/B�MHC cardiac myo-cytes are severely distorted,whereas the structures of thedesmosomes (arrowheads)and gap junctions (smallarrows) remain intact. Thestructure between the whitearrows shows a normal adhe-sion junction (a). Similar resultswere found for 4 other wild-type and 3 other B�MHC/B�MHC
mice. B, Immunoblot analysisfor proteins associated withthe ID at 6 months. Samples
are from 2 wild-type and 2 NMII-B–ablated hearts. Immunoblot analysis was repeated 3 times and quantified using an Odyssey InfraredImaging System. C, Confocal immunofluorescence microscope images stained, as indicated for wild-type and NMII-B–ablated mousehearts at 10 months. Arrows indicate IDs where mXin� is decreased.
1108 Circulation Research November 20, 2009
by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from

The finding of a role for NMII-B in the cardiac ID is similarto the findings for NMII-B in the spinal canal. Our hypothesis isthat NMII exerts tension and stabilizes actin filaments, which, inturn, are required for maintenance of adhesion complexesbetween cells or, in this case, between the cardiac myocytes. Theloss of NMII-B from the adhesion complex could, therefore,result in the gradual deterioration in the cardiac adhesioncomplex, including the loss of mXin�, over a period of time, andthis would account for our failure to observe abnormal discs inB�/B� mice, which died before birth. Interestingly, generationof mice in which NMII-A replaced NMII-B did not producedefects in the IDs.25 This is consistent with the hypothesis that incases in which NMIIs are apparently playing a structural ratherthan a motor role, 1 isoform is more likely to substitute for theother in vivo, as well as in cultured cells.6 When myosin isplaying more of a motor role, for example, in neural cellmigration, because of significant differences in the kinetics ofMgATP hydrolysis and actin-binding properties between themyosin isoforms, successful substitution is much less likely, atleast in vivo.15 These findings further support the idea thatdisruption of the IDs in B�MHC/B�MHC mice is attributable to theloss of NMII and is secondary to the development of thecardiomyopathy.
These conditionally ablated mice demonstrate that thedefects we observed in the hearts and brains of the B�/B�
mice are independent of each other. The availability ofNMHC II-B floxed mice will allow conditional ablation ofNMHC II-B in a variety of tissues and cells and thus help tofurther define its role both in situ and in vivo.
AcknowledgmentsWe are grateful to Charles W. Birdsall (National Heart, Lung, andBlood Institute) and to the NIH Mouse Imaging Facility for help withmouse echocardiograms and ECGs. We thank Douglas R. Rosing,(National Heart, Lung, and Blood Institute) for expert advice onECGs. We acknowledge the professional skills and advice ofChristian A. Combs and Daniela Malide of the Light MicroscopyCore Facility (National Heart, Lung, and Blood Institute), regardingmicroscopy-related experiments performed in this study. We thankthe members of the Laboratory of Molecular Cardiology for sugges-tions and criticism, in particular, Mary Anne Conti and SachiyoKawamoto. We thank Stephanie Jackson for editorial assistance.
Sources of FundingThis work was supported by the Division of Intramural Research,National Heart, Lung, and Blood Institute, NIH.
DisclosuresNone.
References1. Bresnick AR. Molecular mechanisms of nonmuscle myosin-II regulation.
Curr Opin Cell Biol. 1999;11:26–33.2. Conti MA, Adelstein RS. Nonmuscle myosin II moves in new directions.
J Cell Sci. 2008;121:11–18.3. Krendel M, Mooseker MS. Myosins: tails (and heads) of functional
diversity. Physiology (Bethesda). 2005;20:239–251.4. Betapudi V, Licate LS, Egelhoff TT. Distinct roles of nonmuscle myosin
II isoforms in the regulation of MDA-MB-231 breast cancer cellspreading and migration. Cancer Res. 2006;66:4725–4733.
5. Sandquist JC, Swenson KI, Demali KA, Burridge K, Means AR. Rhokinase differentially regulates phosphorylation of nonmuscle myosin IIisoforms A and B during cell rounding and migration. J Biol Chem.2006;281:35873–35883.
6. Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF.Regulation of protrusion, adhesion dynamics, and polarity by myosinsIIA and IIB in migrating cells. J Cell Biol. 2007;176:573–580.
7. Conti MA, Even-Ram S, Liu C, Yamada KM, Adelstein RS. Defects incell adhesion and the visceral endoderm following ablation of nonmusclemyosin heavy chain II-A in mice. J Biol Chem. 2004;279:41263–41266.
8. Tullio AN, Accili D, Ferrans VJ, Yu ZX, Takeda K, Grinberg A,Westphal H, Preston YA, Adelstein RS. Nonmuscle myosin II-B isrequired for normal development of the mouse heart. Proc Natl Acad SciU S A. 1997;94:12407–12412.
9. Tullio AN, Bridgman PC, Tresser NJ, Chan CC, Conti MA, Adelstein RS,Hara Y. Structural abnormalities develop in the brain after ablation of thegene encoding nonmuscle myosin II-B heavy chain. J Comp Neurol.2001;433:62–74.
10. Uren D, Hwang HK, Hara Y, Takeda K, Kawamoto S, Tullio AN, Yu ZX,Ferrans VJ, Tresser N, Grinberg A, Preston YA, Adelstein RS. Genedosage affects the cardiac and brain phenotype in nonmuscle myosinII-B-depleted mice. J Clin Invest. 2000;105:663–671.
11. Betz UA, Vosshenrich CA, Rajewsky K, Muller W. Bypass of lethalitywith mosaic mice generated by Cre-loxP-mediated recombination. CurrBiol. 1996;6:1307–1316.
12. Gaussin V, Van de PT, Mishina Y, Hanks MC, Zwijsen A, HuylebroeckD, Behringer RR, Schneider MD. Endocardial cushion and myocardialdefects after cardiac myocyte-specific conditional deletion of the bonemorphogenetic protein receptor ALK3. Proc Natl Acad Sci U S A. 2002;99:2878–2883.
13. McFadden DG, Barbosa AC, Richardson JA, Schneider MD, SrivastavaD, Olson EN. The Hand1 and Hand2 transcription factors regulateexpansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development. 2005;132:189–201.
14. Ma X, Kawamoto S, Hara Y, Adelstein RS. A point mutation in the motordomain of nonmuscle myosin II-B impairs migration of distinct groups ofneurons. Mol Biol Cell. 2004;15:2568–2579.
15. Ma X, Bao J, Adelstein RS. Loss of cell adhesion causes hydrocephalusin nonmuscle myosin II-B-ablated and mutated mice. Mol Biol Cell.2007;18:2305–2312.
16. Takeda K, Kishi H, Ma X, Yu ZX, Adelstein RS. Ablation and mutationof nonmuscle myosin heavy chain II-B results in a defect in cardiacmyocyte cytokinesis. Circ Res. 2003;93:330–337.
17. Lombardi R, Dong J, Rodriguez G, Bell A, Leung TK, Schwartz RJ,Willerson JT, Brugada R, Marian AJ. Genetic fate mapping identifies secondheart field progenitor cells as a source of adipocytes in arrhythmogenic rightventricular cardiomyopathy. Circ Res. 2009;104:1076–1084.
18. Takeda K, Yu ZX, Qian S, Chin TK, Adelstein RS, Ferrans VJ. Non-muscle myosin II localizes to the Z-lines and intercalated discs of cardiacmuscle and to the Z-lines of skeletal muscle. Cell Motil Cytoskeleton.2000;46:59–68.
19. Gustafson-Wagner EA, Sinn HW, Chen YL, Wang DZ, Reiter RS, LinJL, Yang B, Williamson RA, Chen J, Lin CI, Lin JJ. Loss of mXinalpha,an intercalated disk protein, results in cardiac hypertrophy and cardiomy-opathy with conduction defects. Am J Physiol Heart Circ Physiol. 2007;293:H2680–H2692.
20. Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD,Radice GL. Induced deletion of the N-cadherin gene in the heart leads todissolution of the intercalated disc structure. Circ Res. 2005;96:346–354.
21. Choi S, Gustafson-Wagner EA, Wang Q, Harlan SM, Sinn HW, Lin JL,Lin JJ. The intercalated disk protein, mXinalpha, is capable of interactingwith beta-catenin and bundling actin filaments. J Biol Chem. 2007;282:36024–36036.
22. Shewan AM, Maddugoda M, Kraemer A, Stehbens SJ, Verma S, KovacsEM, Yap AS. Myosin 2 is a key Rho kinase target necessary for the localconcentration of E-cadherin at cell-cell contacts. Mol Biol Cell. 2005;16:4531–4542.
23. Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S,Nadvoretskiy VV, DeFreitas G, Carabello B, Brandon LI, Godsel LM,Green KJ, Saffitz JE, Li H, Danieli GA, Calkins H, Marcus F, Towbin JA.Desmosomal dysfunction due to mutations in desmoplakin causesarrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res.2006;99:646–655.
24. Li J, Levin MD, Xiong Y, Petrenko N, Patel VV, Radice GL. N-cadherinhaploinsufficiency affects cardiac gap junctions and arrhythmic suscep-tibility. J Mol Cell Cardiol. 2008;44:597–606.
25. Bao J, Ma X, Liu C, Adelstein RS. Replacement of nonmuscle myosinII-B with II-A rescues brain but not cardiac defects in mice. J Biol Chem.2007;282:22102–22111.
Ma et al Conditional Ablation of Nonmuscle Myosin II-B 1109
by guest on August 3, 2014http://circres.ahajournals.org/Downloaded from

1 CIRCRESAHA/2009/200303/R2
SUPPLEMENT MATERIAL
Detailed Materials and Methods Generation of targeting vector and Bflox/Bflox mice
The targeting vector includes a 6.5-kb genomic DNA fragment containing the first coding exon (exon2) of NMHC II-B. A 1.8-kb PGK-Neomycin resistance (Neor) expression cassette was inserted into a unique BstEII site, 230bp upstream of exon2. Both the PGK-Neor expression cassette and exon2 were flanked by loxP sites (Figure 1b). The linearized targeting vector was electroporated into CMT-1 embryonic stem (ES) cells derived from 129S6SvEv mice. G418-resistant (0.20 mg/ml) ES cell clones were screened for successful homologous recombination by restriction enzyme digestion with BamHI and Southern blot analysis using the BamHI-SpeI fragment (indicated in Figure 1c) as a probe. One positive ES cell clone was microinjected into blastocysts which were transplanted into pseudo-pregnant mice to generate chimeric mice. The chimeras were crossed to C57BL/6J mice to produce germ-line transmitted, heterozygous mice. Mice were genotyped by PCR analysis (primers: 5’GACCGCTACTATTCAGGACTTATC and 5’CAGAGAAACGATGGGAAAGAAAGC). The products are 250bp for wild type and 350bp for the floxed allele.
Histology, immunofluorescence microscopy and immunoblot analysis
Brains and hearts were fixed in 3.7% paraformaldehyde in PBS or frozen in OCT. H&E staining, immunofluorescence staining and immunoblotting were performed according to standard procedures as described previously.1 The antibodies used for immunofluorescence staining were: NMHC II-B (rabbit, 1:1000),2 calbindin (mouse, 1:1000, Sigma), connexin 43 (rabbit, 1:100, Cell Signalling), and desmin (mouse, 1:100, Dako). The antibodies used in immunoblot analysis were: α-actinin (mouse, 1:200, Abcam), connexin 43 (rabbit, 1:1000, Cell Signalling), N-cadherin (mouse, 1:2000, Invitrogen), α-catenin (mouse , 1:500, BD Biosciences), β-catenin (mouse, 1:1000, Zymed), γ-catenin (rabbit, 1:1000, Cell Signalling), mXinα (rabbit, gift from Dr. Jim Lin, Departments of Biological Sciences, University of Iowa, 1:10,000), NMHC II-B (rabbit, 1:50,000), tubulin (rabbit, 1:1000, Sigma) and actin (mouse, 1:5000, Sigma), GAPDH (mouse, 1:5000, Abcam). Wheat germ agglutinin (WGA) staining and measurement of the cross-sectional area of the cardiac myocytes Paraffin embedded heart sections were first dewaxed and rehydrated by standard procedures. Following antigen retrieval in citric acid buffer (10 mM, pH 6.0), the sections were blocked with 1% BSA/5% goat serum in PBS for 1 hour at room temperature and then incubated with Alexa Fluor® 594-WGA (10 µg/ml, Invitrogen) and DAPI in blocking solution for 1 hour at room temperature. The slides were then washed and mounted using Prolong Anti-fade mount media (InVitrogen). Confocal images were captured using a Zeiss LSM 510 Meta Confocal Microscope and the cross sectional area of the cells was measured with a Zeiss measuring tool. We measured all myocytes in the confocal image and calculated their average size. TUNEL assay The TUNEL assay was carried out using the In Situ Cell Death Detection Kit, Fluorescein following the manufacturer’s instructions (Roche Applied Science). After TUNEL staining, the heart sections were stained with antibodies to desmin to identify cardiac myocytes and counter stained with DAPI to visualize nuclei.

2 CIRCRESAHA/2009/200303/R2
Echocardiography
The mice were anesthetized with 1.5% isoflurane and imaged in the supine position. Three standard lead echocardiography was performed using an Acuson Sequoia 256c imaging system with the 15L8 multi-frequency transducer. All images were obtained at a frequency of 15MHz. Multiple 2-D and M-mode images of the left ventricle (LV) were obtained on each subject. Quantitative assessments of LV size, function and wall thickness were performed using M-mode echocardiography. The M-mode measurements were performed off-line on Prosolv Software Version 3.0. Three measurements were performed for each parameter and averaged.
Electrocardiography
The mice were anesthetized with isoflurane and analyzed in a supine position after echocardiography. Electrocardiograms were recorded with a model MAC 1200, G.E. Medical Systems.
Electron Microscopy
Heart tissue was fixed with 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer, postfixed with 1% OsO4 in 0.1 M phosphate buffer and embedded in Polybed 812 (Polysciences). Ultrathin sections were stained with uranyl acetate and lead citrate and examined with a transmission electron microscope (JEOL). Data and Statistical Analysis The data were expressed as mean ± SD. Student’s t-test was used to compare the data between two groups.
3 CIRCRESAHA/2009/200303/R2
Supplement Figures and Figure Legends
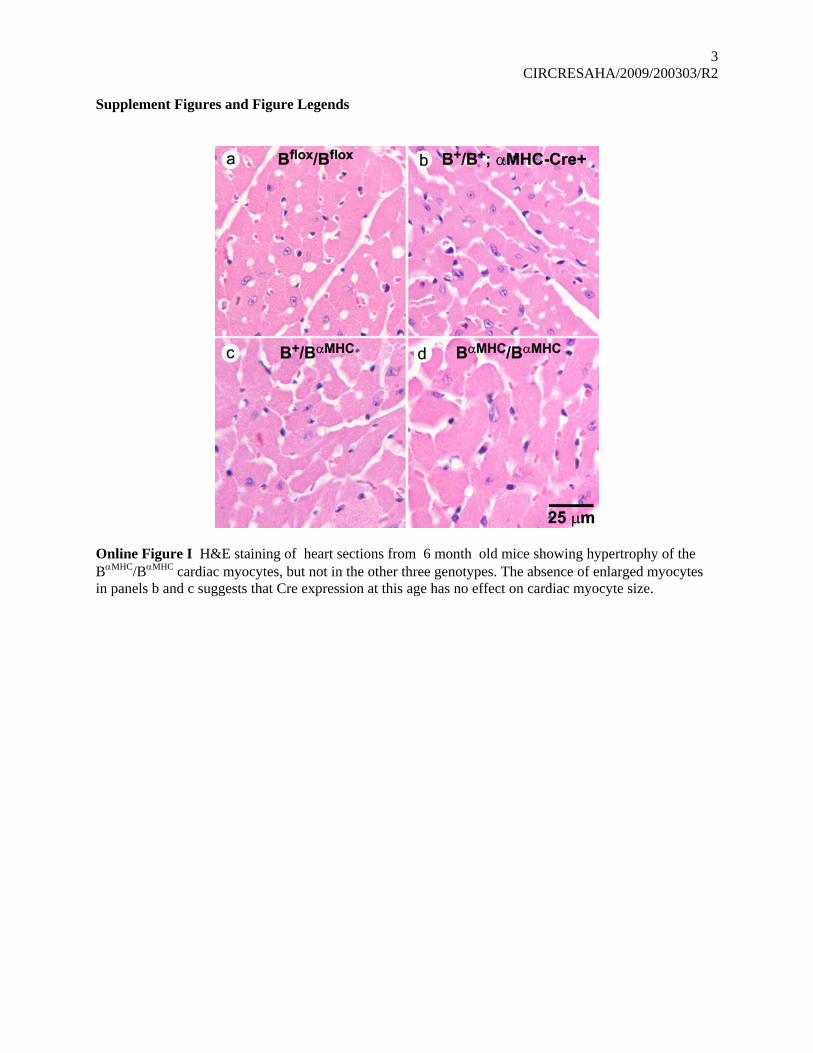
Online Figure I H&E staining of heart sections from 6 month old mice showing hypertrophy of the BαMHC/BαMHC cardiac myocytes, but not in the other three genotypes. The absence of enlarged myocytes in panels b and c suggests that Cre expression at this age has no effect on cardiac myocyte size.
4 CIRCRESAHA/2009/200303/R2
Online Figure II TUNEL staining reveals an increase in apoptotic myocytes (arrows, green) in 10 month old BαMHC/BαMHC mouse hearts compared to wild type hearts. Desmin (red) indicates cardiac myocytes, DAPI (blue) stains the nuclei.
5 CIRCRESAHA/2009/200303/R2
Online Figure III Organized thrombus in the left atrium (*, magnified 5x on right) of a 10.5 month old BαMHC/BαMHC mouse heart stained with H&E.
6 CIRCRESAHA/2009/200303/R2
Online Figure IV EKG of a 10 month old BαMHC/BαMHC mouse showing marked right deviation of the electrical axis compared to a wild type Bflox/Bflox mouse.

7 CIRCRESAHA/2009/200303/R2
References
1. Tullio AN, Accili D, Ferrans VJ, Yu ZX, Takeda K, Grinberg A, Westphal H, Preston YA, Adelstein RS. Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc Natl Acad Sci U S A. 1997;94:12407-12412.
2. Phillips CL, Yamakawa K, Adelstein RS. Cloning of the cDNA encoding human nonmuscle myosin heavy chain-B and analysis of human tissues with isoform-specific antibodies. J Muscle Res Cell Motil. 1995;16:379-389.




















