
Complex I Dysfunction and Tolerance to Nitroglycerin: An Approach Based on Mitochondrial-Targeted...
10
Complex I Dysfunction and Tolerance to Nitroglycerin An Approach Based on Mitochondrial-Targeted Antioxidants Juan V. Esplugues, Milagros Rocha, Cristina Nun ˜ez, Irene Bosca, Sales Ibiza, Jose R. Herance, Angel Ortega, Juan M. Serrador, Pilar D’Ocon, Victor M. Victor Abstract—Nitroglycerin (GTN) tolerance was induced in vivo (rats) and in vitro (rat and human vessels). Electrochemical detection revealed that the incubation dose of GTN (510 6 mol/L) did not release NO or modify O 2 consumption when administered acutely. However, development of tolerance produced a decrease in both mitochondrial O 2 consumption and the K m for O 2 in animal and human vessels and endothelial cells in a noncompetitive action. GTN tolerance has been associated with impairment of GTN biotransformation through inhibition of aldehyde dehydrogenase (ALDH)-2, and with uncoupling of mitochondrial respiration. Feeding rats with mitochondrial-targeted antioxidants (mitoquinone [MQ]) and in vitro coincubation with MQ (10 6 mol/L) or glutathione (GSH) ester (10 4 mol/L) prevented tolerance and the effects of GTN on mitochondrial respiration and ALDH-2 activity. Biotransformation of GTN requires functionally active mitochondria and induces reactive oxygen species production and oxidative stress within this organelle, as it is inhibited by mitochondrial-targeted antioxidants and is absent in HUVEC 0 cells. Experiments analyzing complex I– dependent respiration demonstrate that its inhibition by GTN is prevented by mitochondrial-targeted antioxidants. Furthermore, in presence of succinate (1010 3 mol/L), a complex II electron donor added to bypass complex I– dependent respiration, GTN-treated cells exhibited O 2 consumption rates similar to those of controls, thus suggesting that complex I was affected by GTN. We propose that, following prolonged treatment with GTN in addition to ALDH-2, complex I is a target for mitochondrially generated reactive oxygen species. Our data also suggest a role for mitochondrial-targeted antioxidants as therapeutic tools in the control of the tolerance that accompanies chronic nitrate use. (Circ Res. 2006;99:1067-1075.) Key Words: nitroglycerin endothelium oxidative stress mitochondria antioxidant T he vasodilatory actions of nitroglycerin (glyceryl trini- trate [GTN]) have generally been attributed to its bio- conversion into the relaxant agent nitric oxide (NO), which acts on the enzyme soluble guanylate cyclase (sGC). 1–3 However, most studies that support the existence of such a pathway have demonstrated increases of NO only when GTN concentrations considerably exceeded the plasma levels reached during clinical dosing. 4 Moreover, the involvement of other NO-related species in the actions of GTN when used at clinically relevant concentrations is also under debate. 5,6 Different enzymes have been implicated in the bioconversion of GTN, in particular, glutathione S-transferases, 7 the cyto- chrome p450 system, 8 and xanthine oxidoreductase, 9 al- though the most recent evidence suggests a central role for mitochondrial aldehyde dehydrogenase (ALDH)-2. 10 –13 The medical use of GTN is limited by the development of tolerance, which occurs following prolonged administration or the application of high doses. This phenomenon has been related to various mechanisms, in particular, desensitization of sGC, 14 and, mainly, impairment of GTN biotransformation by inhibition of ALDH-2. 10 –12 These actions, like others associated with GTN, have been linked to an increase in the production of reactive oxygen species (ROS), 15 as well as mitochondrial dysfunction. 11 The present study confirms the theory that tolerance to GTN (both in vivo and in vitro) is related to the oxidative stress that leads to an inhibition of ALDH-2. Furthermore, through the use of mitochondrial antioxidants such as mitoquinone (MQ), 16,17 or glutathione (GSH) ester, 18 we have confirmed the mitochondria to be the main location for the development of tolerance. Finally, we have identified mitochondrial complex I as a target at which the initial oxidative stress responsible for GTN tolerance takes place. Materials and Methods Umbilical cords obtained from the Department of Gynaecology (Faculty of Medicine of Valencia) were cut into rings (5 mm) and placed in Krebs solution ([in 10 3 mol/L] NaCl 118, KCl 4.75, Original received May 30, 2006; resubmission received September 18, 2006; revised resubmission received October 5, 2006; accepted October 6, 2006. From the Departamento de Farmacologia (J.V.E., M.R., P.D.), Facultad de Medicina, Universitat de Valencia; Unidad Mixta Centro Nacional de Investigaciones Cardiovasculares (CNIC)-Universitat de Valencia (C.N., I.B., S.I., J.M.S., V.M.V.); Instituto de Alta Tecnologia (J.R.H.), Parque de Investigaciones Biomedicas, Barcelona; and Unidad Central de Investigacion (A.O.), Universitat de Valencia, Spain. Correspondence to Dr Victor M. Victor, Unidad Mixta CNIC-Valencia, Departamento de Farmacologia, Avda Blasco Iban ˜ez 15-17, Valencia, Va 46010, Spain. E-mail [email protected] © 2006 American Heart Association, Inc. Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/01.RES.0000250430.62775.99 1067 by guest on February 19, 2016 http://circres.ahajournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Complex I Dysfunction and Tolerance to Nitroglycerin: An Approach Based on Mitochondrial-Targeted...

Complex I Dysfunction and Tolerance to NitroglycerinAn Approach Based on Mitochondrial-Targeted Antioxidants
Juan V. Esplugues, Milagros Rocha, Cristina Nunez, Irene Bosca, Sales Ibiza, Jose R. Herance,Angel Ortega, Juan M. Serrador, Pilar D’Ocon, Victor M. Victor
Abstract—Nitroglycerin (GTN) tolerance was induced in vivo (rats) and in vitro (rat and human vessels). Electrochemicaldetection revealed that the incubation dose of GTN (5�10�6 mol/L) did not release NO or modify O2 consumption whenadministered acutely. However, development of tolerance produced a decrease in both mitochondrial O2 consumptionand the Km for O2 in animal and human vessels and endothelial cells in a noncompetitive action. GTN tolerance has beenassociated with impairment of GTN biotransformation through inhibition of aldehyde dehydrogenase (ALDH)-2, andwith uncoupling of mitochondrial respiration. Feeding rats with mitochondrial-targeted antioxidants (mitoquinone[MQ]) and in vitro coincubation with MQ (10�6 mol/L) or glutathione (GSH) ester (10�4 mol/L) prevented tolerance andthe effects of GTN on mitochondrial respiration and ALDH-2 activity. Biotransformation of GTN requires functionallyactive mitochondria and induces reactive oxygen species production and oxidative stress within this organelle, as it isinhibited by mitochondrial-targeted antioxidants and is absent in HUVEC�0 cells. Experiments analyzing complexI–dependent respiration demonstrate that its inhibition by GTN is prevented by mitochondrial-targeted antioxidants.Furthermore, in presence of succinate (10�10�3 mol/L), a complex II electron donor added to bypass complexI–dependent respiration, GTN-treated cells exhibited O2 consumption rates similar to those of controls, thus suggestingthat complex I was affected by GTN. We propose that, following prolonged treatment with GTN in addition to ALDH-2,complex I is a target for mitochondrially generated reactive oxygen species. Our data also suggest a role formitochondrial-targeted antioxidants as therapeutic tools in the control of the tolerance that accompanies chronicnitrate use. (Circ Res. 2006;99:1067-1075.)
Key Words: nitroglycerin � endothelium � oxidative stress � mitochondria � antioxidant
The vasodilatory actions of nitroglycerin (glyceryl trini-trate [GTN]) have generally been attributed to its bio-
conversion into the relaxant agent nitric oxide (NO), whichacts on the enzyme soluble guanylate cyclase (sGC).1–3
However, most studies that support the existence of such apathway have demonstrated increases of NO only when GTNconcentrations considerably exceeded the plasma levelsreached during clinical dosing.4 Moreover, the involvementof other NO-related species in the actions of GTN when usedat clinically relevant concentrations is also under debate.5,6
Different enzymes have been implicated in the bioconversionof GTN, in particular, glutathione S-transferases,7 the cyto-chrome p450 system,8 and xanthine oxidoreductase,9 al-though the most recent evidence suggests a central role formitochondrial aldehyde dehydrogenase (ALDH)-2.10–13
The medical use of GTN is limited by the development oftolerance, which occurs following prolonged administrationor the application of high doses. This phenomenon has beenrelated to various mechanisms, in particular, desensitization
of sGC,14 and, mainly, impairment of GTN biotransformationby inhibition of ALDH-2.10–12 These actions, like othersassociated with GTN, have been linked to an increase in theproduction of reactive oxygen species (ROS),15 as well asmitochondrial dysfunction.11 The present study confirms thetheory that tolerance to GTN (both in vivo and in vitro) isrelated to the oxidative stress that leads to an inhibition ofALDH-2. Furthermore, through the use of mitochondrialantioxidants such as mitoquinone (MQ),16,17 or glutathione(GSH) ester,18 we have confirmed the mitochondria to be themain location for the development of tolerance. Finally, wehave identified mitochondrial complex I as a target at whichthe initial oxidative stress responsible for GTN tolerancetakes place.
Materials and MethodsUmbilical cords obtained from the Department of Gynaecology(Faculty of Medicine of Valencia) were cut into rings (5 mm) andplaced in Krebs solution ([in �10�3 mol/L] NaCl 118, KCl 4.75,
Original received May 30, 2006; resubmission received September 18, 2006; revised resubmission received October 5, 2006; accepted October 6, 2006.From the Departamento de Farmacologia (J.V.E., M.R., P.D.), Facultad de Medicina, Universitat de Valencia; Unidad Mixta Centro Nacional de
Investigaciones Cardiovasculares (CNIC)-Universitat de Valencia (C.N., I.B., S.I., J.M.S., V.M.V.); Instituto de Alta Tecnologia (J.R.H.), Parque deInvestigaciones Biomedicas, Barcelona; and Unidad Central de Investigacion (A.O.), Universitat de Valencia, Spain.
Correspondence to Dr Victor M. Victor, Unidad Mixta CNIC-Valencia, Departamento de Farmacologia, Avda Blasco Ibanez 15-17, Valencia, Va46010, Spain. E-mail [email protected]
© 2006 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/01.RES.0000250430.62775.99
1067 by guest on February 19, 2016http://circres.ahajournals.org/Downloaded from
CaCl2 1.9, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, and glucose 10.1).Human umbilical vein cells (HUVECs) and human umbilical arterialendothelial cells (HUAECs) were cultured in medium 199 (Cam-brex, Walkersville, Md) as previously described.19 Male Sprague–Dawley rats (200 to 250 g; Harlan, Barcelona, Spain) were decapi-tated and 5 mm rings obtained from their thoracic aortas.
To generate HUVEC�0 cells, 50 ng/mL ethidium bromide wasadded to the medium for 7 days to inhibit mitochondrial genetranscription and activity.11 Pyruvate (110 mg/mL) and uridine (50�g/mL) were used as alternative energy and nucleotide sources.Lack of mitochondrial gene expression was evaluated by Westernblot analysis for cytochrome c oxidase subunit I (Molecular Probes,Leiden, The Netherlands), and the absence of respiratory functionwas measured by analyzing mitochondrial oxygen (O2) consumption.
All protocols complied with European Community guidelines forthe use of experimental animals and were approved by the EthicsCommittee of the University of Valencia.
Vascular Contractility StudiesRings were suspended in an organ bath containing Krebs solution(37°C), as described previously.6 GTN tolerance was induced invitro by incubating rings for 3 hours with GTN (5�10�6 mol/L).Rings from the in vivo nitrate-tolerance experiments were obtainedfrom rats that had been allowed free access for 14 days to tap waterwith or without MQ or triphenylphosphonium (TPP) (5�10�4
mol/L)20 and that had been treated with a GTN patch for the last 3days.15 All of these parameters were selected from preliminaryexperiments and were similar to those used by other authors.15,21,22
Relaxation-response curves were obtained by adding cumulativeconcentrations of GTN (10�10 to 10�4 mol/L) or DETA-NO (10�8 to10�4 mol/L). Some experiments were performed in aortic denudatedrings, and, when necessary, either MQ (10�6 mol/L) or GSH ester(10�4 mol/L) was added 1 hour before the 3-hour GTN incubationperiod and maintained thereafter. In preliminary experiments thelipophilic cation linker TPP (5�10�6 mol/L), responsible for target-ing MQ to mitochondria, showed no effect on vascular responses.When necessary, experiments were performed in the presence ofnon–mitochondrial-targeted antioxidants such as uric acid (UA)(10�4 mol/L, scavenger of ONOO�), ebselen (Eb) (10�4 mol/L;scavenger of ONOO� and H2O2), and tempol (TP) (10�3 mol/L;scavenger of O2
�). The adequate concentration (�log [M]) forproducing 50% relaxation (pEC50) was obtained from a nonlinearregression analysis with GraphPad software.
ALDH ActivityThe activity of ALDH-2 in homogenates of aortic rings wasdetermined by measuring the conversion of propionaldehyde topropionic acid11 and was expressed as the mean rate of absorbance(0.0125 A340 was equivalent to 1 nmol/mg per minute).
Measurements of O2 Consumption andNO ProductionThe aorta from 2 animals (100�1.04 mg wet weight) or humanumbilical arteries or veins (196�2.3 or 199�3.6 mg wet weight,respectively) were cut into rings and resuspended in Krebs solution.
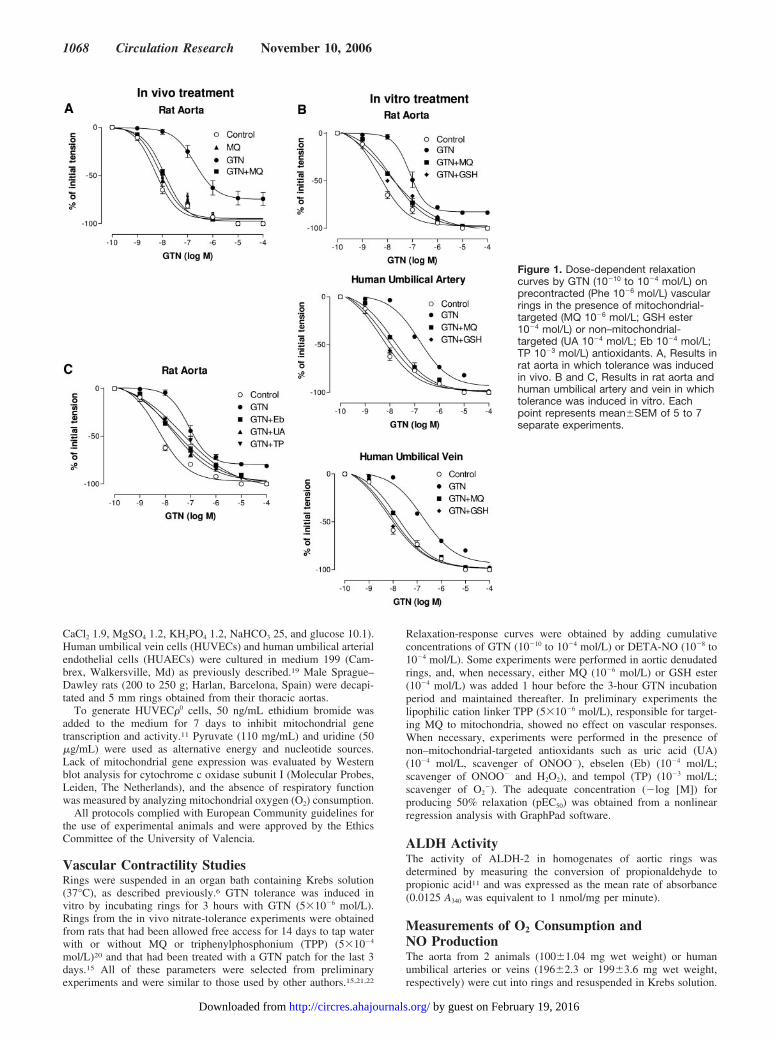
Figure 1. Dose-dependent relaxationcurves by GTN (10�10 to 10�4 mol/L) onprecontracted (Phe 10�6 mol/L) vascularrings in the presence of mitochondrial-targeted (MQ 10�6 mol/L; GSH ester10�4 mol/L) or non–mitochondrial-targeted (UA 10�4 mol/L; Eb 10�4 mol/L;TP 10�3 mol/L) antioxidants. A, Results inrat aorta in which tolerance was inducedin vivo. B and C, Results in rat aorta andhuman umbilical artery and vein in whichtolerance was induced in vitro. Eachpoint represents mean�SEM of 5 to 7separate experiments.
1068 Circulation Research November 10, 2006
by guest on February 19, 2016http://circres.ahajournals.org/Downloaded from

HUVECs, HUVEC�0 cells, and HUAECs were resuspended (5�106
cells/mL) in Krebs supplemented with L-arginine (3�10�4 mol/L)and HEPES (25�10�3 mol/L). Induction of GTN tolerance (in vivoor in vitro) and the use of mitochondrial and non–mitochondrial-targeted antioxidants were similar to those described in the vascularreactivity studies. When administered acutely, the dose of GTN used(5�10�6 mol/L) had no effect on mitochondrial O2 consumption,despite producing maximal relaxations of vascular rings. The afore-mentioned tissues were then placed in a gas-tight chamber, and O2
consumption was measured with a Clark-type O2 electrode (RankBrothers, Bottisham, UK).6 Sodium cyanide (10�3 mol/L) confirmedthat O2 consumption was mainly mitochondrial. Measurements werecollected using the data-acquisition device Duo.18 (WPI, Stevenage,UK). A hyperbolic function was used to describe the relationshipbetween O2 concentration and the rate of O2 consumption (VO2). Themaximal rate of O2 consumption (VO2max) and the apparent O2 affinityMichaelis–Menten constant (Km) (10�6 mol/L) were calculated ac-cording to their analogy with the Michaelis–Menten constant.Trypan blue exclusion test revealed no changes in cell viability.
NO concentration in the cell medium was monitored throughoutthe 3-hour incubation period with GTN by means of an NO electrode(ISO-NOP; WPI, Stevenage, UK) as described previously.23 The NOdonor DETA-NO (10�5 mol/L) was added at the end of eachexperiment as an internal control. Changes in intracellular NO werealso evaluated by incubating cells with the fluorescent probe diami-nodifluorofluorescein diacetate (DAF-FM DA) (1�10�6 mol/L).Thereafter, the medium was changed to HBSS, supplemented withglucose (20�10�3 mol/L), L-arginine (3�10�4 mol/L), and DAF-FM, incubated for 30 minutes and measured using a Fluoroskan platereader (TL, Franklin, Mass). cGMP levels were measured with theBiomol assay kit (Plymouth Meeting, Pa).
Measurement of ROS Production, GSH Content,and Complex I ActivityTwo methods were used to evaluate ROS. Total ROS production wasassessed following incubation (30 minutes) with the fluorescentprobe 2�,7�-dichlorodihydrofluorescein diacetate (DCFH-DA)(5�10�6 mol/L), as described elsewhere.24 Quantitative assessmentof hydrogen peroxide (H2O2) was performed with the Amplex RedH2O2/Peroxidase Assay kit (Molecular Probes, Eugene, Ore).25,26
GSH content was assessed following incubation (30 minutes) withthe fluorescent probe monochlorobimane (MCB) (40�10�6 mol/L).GSH levels were also measured by confocal microscopy (Leica,Heidelberg, Germany), following incubation (30 minutes) with5-chloromethylfluorescein diacetate (CMFDA) (1�10�6 mol/L). Theratio oxidized glutathione (GSSG)/GSH was calculated as previouslydescribed.27 Proteins were determined using the BCA protein assaykit (Pierce, Rockford, Ill).
The activity of mitochondrial complex I was assessed by calcu-lating the NADH oxidation rate.28 Briefly, a cellular homogenate (20�L, 0.3 mg) was added to 1 mL of potassium phosphate buffer (10�2
mol/L) containing NADH (10�4 mol/L) at 37°C. Basal absorbance(340 nm) was recorded for 1 minute, and 5 �L of ubiquinone (10�2
mol/L) was then added, and the rate of NADH oxidation (taken ascomplex I activity) over 2 minutes was measured. The NADHoxidation rate was calculated from the time-dependent decrease inthe slope of absorbance using an extinction coefficient for NADH of6.81�10�3 mol/L cm�1 at 340 nm. Isolated complex I–dependentrespiration was evaluated in digitonin permeabilized cells in thepresence of the complex I substrates malate (0.4�10�3 mol/L) andglutamate (3�10�2 mol/L), the complex II substrate succinate (10�2
mol/L), or the complex I inhibitor rotenone (6�10�6 mol/L).29
Drugs and SolutionsPhenylephrine (Phe), acetycholine (ACh), 1H-[1,2,4]-oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), sodium cyanide, KCl,ubiquinone, succinate, rotenone, glutamate, malate, digitonin, argi-nine, HEPES, uridine, TPP, glucose, 5,5�-dithio-bis(2-nitrobenzoicacid) (DTNB), trypan blue, GSH reductase (GR), NADH, TP, UA,Eb, methylpyrazole, H2O2, and hemoglobin were obtained from
Sigma-Aldrich (St Louis, Mo). Propionaldehyde was from Fluka(Milano, Italy). The GTN used is a clinically used preparation(Solinitrina, Allmirall, Barcelona, Spain). Patches of GTN were fromSchering-Plough (Madrid, Spain). Na-pyruvate was from GIBCO-BRL (Gaithersburg, Md). Ethidium bromide was from SERVA(Heidelberg, Germany). HBSS and medium 199 were from Cambrex(Verviers, Belgium). DETA-NO was from Alexis (San Diego, Calif).DAF-FM, and DCFH-DA were from Calbiochem (San Diego, Calif).MCB and CMFDA were from Molecular Probes (Eugene, Ore). MQwas synthesized according to the published method.30 GSH ester wasprepared as previously described.31
Data AnalysisUnless stated otherwise all values are mean�SEM of at least 5experiments. Statistical analysis was performed with 1-way ANOVAwith post hoc corrections, followed by the Student t test for unpairedsamples (GraphPad Software). Significance was defined as P�0.05.
ResultsTolerance to GTN VasorelaxationFigure 1 shows dose-dependent relaxation curves by GTN(10�10 to 10�4 mol/L) in precontracted (Phe 10�6 mol/L) rings.Figure 1A represents results from rat aorta in which tolerancewas induced in vivo, and Figure 1B and 1C show results fromrat aorta and human umbilical artery and vein in whichtolerance was induced in vitro. Table 1 shows pEC50 values.Continuous treatment with GTN (in vivo or in vitro) causeda significant rightward shift in the GTN concentration-response curves, indicating the development of GTN-inducedvascular tolerance. In GTN-tolerant rat aortas, the maximalGTN relaxation values were significantly diminished(82.2�0.9% of controls, P�0.05). Removal of the endothe-lium enhanced the maximal relaxations values caused by
TABLE 1. pEC50 of the Dose-Dependent Relaxation Curves byGTN in Rat Aortic Rings and Artery and Vein Rings from HumanUmbilical Cord
In Vitro In Vivo
Rat Aorta
HumanUmbilical
Artery
HumanUmbilical
Vein Rat Aorta
Control 8.30�0.10 8.30�0.14 8.21�0.14 8.28�0.06
MQ 8.06�0.11 8.19�0.18 8.09�0.13 8.09�0.07
GSH 8.05�0.09 8.12�0.06 8.10�0.09
Eb 8.28�0.06
UA 8.25�0.07
TP 8.11�0.05
GTN 7.11�0.06* 6.81�0.07* 6.75�0.08* 6.70�0.13*
MQ 7.82�0.07‡ 7.83�0.16‡ 7.80�0.05‡ 7.91�0.04‡
GSH 8.03�0.12‡ 8.12�0.12‡ 8.09�0.11‡
Eb 7.63�0.08†
UA 7.67�0.07†
TP 7.32�0.09†
Concentration range of GTN was 10�10 to 10�4 mol/L). Rat aortic rings andartery and vein rings from human umbilical cord were precontracted with Phe(10�6 mol/L), in the presence of MQ (10�6 mol/L), GSH ester (10�4 mol/L), Eb(10�4 mol/L), UA (10�4 mol/L), or TP (10�3 mol/L). GTN tolerance was inducedin vitro or in vivo. Data are the mean�SEM of 5–7 independent experiments.*P�0.001 vs control, †P�0.05 and ‡P�0.001 vs GTN.
Esplugues et al Mitochondrial Antioxidants and Nitrate Tolerance 1069
by guest on February 19, 2016http://circres.ahajournals.org/Downloaded from

GTN, although they continued to differ from those of controls(91.6�2% of controls, P�0.05 versus both).
Treatment with MQ (in vivo or in vitro) or GSH ester (invitro) during the induction of tolerance restored the acutevasorelaxant effects of GTN, suggesting that mitochondrialROS were involved in the development of GTN tolerance.Addition of non–mitochondrial-targeted antioxidants (UA,Eb, and TP) following the in vitro induction of tolerance onlypartially restored the acute effects of GTN (Figure 1C). pEC50
values with DETA-NO (10�8 to 10�4 mol/L) were similar incontrols (6.06�0.07) and GTN-tolerant (5.96�0.05) aortas.Incubation with DETA-NO (5�10�6 mol/L; 3 hours) did notinduce tolerance to the effects of the GTN (7.98�0.07)administered later.
GTN BiotransformationALDH activity was significantly (P�0.001) diminished inGTN-tolerant aortas when compared with that of controls(1.1�0.1 vs 2.8�0.2 nmol/mg per minute, respectively).
Presence of MQ or GSH ester during the in vitro induction ofGTN tolerance prevented such a decrease (2.7�0.2 and2.5�0.2 nmol/mg per minute, respectively).
Mitochondrial O2 ConsumptionFigure 2 and Table 2 show that the rate of O2 consumption,the apparent Km for O2, and VO2max decreased in vessels inwhich GTN tolerance was induced in vivo (Figure 2A) or invitro (Figure 2B). Treatment with MQ (in vivo or in vitro) orGSH ester (in vitro) during the induction of tolerance pre-vented these inhibitory effects of GTN. Following the in vitroinduction of tolerance, addition of non–mitochondrial-targeted antioxidants partially (P�0.05) restored the inhibi-tory effects of GTN (Table 2). Presence of the NO scavengerhemoglobin (10�5 mol/L) or the sGC inhibitor ODQ (5�10�6
mol/L) did not modify the effects of GTN on O2 consumption(data not shown).
Figure 2C shows representative traces of the electrochem-ical detection of NO in a medium containing HUVECs.
TABLE 2. Modulation of the Apparent Km for O2 and VO2max in Rat Aortic Rings and Artery andVein Rings From Human Umbilical Cord and HUVECs and HUAECs
In Vitro In Vivo
Rat aortaHuman Umbilical
ArteryHuman Umbilical
Vein HUVECs HUAECs Rat Aorta
Km (10�6 mol/L)
Control 29.3�2 30.6�4 28.4�3 27.4�3 28.4�5 28.2�3
MQ 28.3�2 29.2�2 27.8�3 26.4�3 28.2�3 27.7�2
GSH 27.9�2 30.2�2 28.8�3 28.4�3 27.2�3
Eb 28.6�3
UA 29.1�2
TP 28.4�2
GTN 12.2�2* 11.9�3* 11.2�3* 11.4�3* 10.9�3* 11.6�1*
MQ 28.3�3‡ 29.5�3‡ 26.7�3‡ 28.2�4‡ 26.8�4‡ 29.1�2‡
GSH 26.7�4‡ 28.4�3‡ 27.5�3‡ 27.3�4‡ 27.5�5‡
Eb 22.4�3†
UA 24.6�3†
TP 20.8�3†
VO2max (mol O2/minper 10�6 g of protein)
Control 34.1�1 35.3�2 31.8�3 30.4�2 32.2�1 32.4�2
MQ 33.6�1 33.8�3 30.6�3 31.4�3 31.1�1 33.9�4
GSH 32.9�3 33.7�3 31.6�3 31.4�3 32.1�3
Eb 32.2�2
UA 33.4�3
TP 34.1�2
GTN 20.5�2* 22.1�3* 19.3�3* 20.2�3* 18.9�3* 19.4�1*
MQ 35.1�3‡ 34.2�3‡ 33.4�3‡ 32.4�4‡ 33.4�4‡ 33.6�2‡
GSH 33.1�2‡ 32.1�2‡ 34.5�3‡ 33.2�4‡ 34.1�5‡
Eb 25.3�3†
UA 28.2�3†
TP 23.9�2†
Samples were in the presence of MQ (10�6 mol/L), GSH ester (10�4 mol/L), Eb (10�4 mol/L), UA (10�4 mol/L), orTP (10�3 mol/L). GTN tolerance was induced in vitro or in vivo. Data are the mean�SEM of 5–7 independentexperiments. *P�0.001 vs control, †P�0.05 and ‡P�0.001 vs GTN.
1070 Circulation Research November 10, 2006
by guest on February 19, 2016http://circres.ahajournals.org/Downloaded from

Addition of GTN (5�10�6 mol/L) did not induce any NOsignal, whereas a robust increase was noted followingDETA-NO (10�5 mol/L). Similar results were obtained withthe NO-sensitive fluorescent probe DAF-FM (data notshown).
Role of Mitochondria in GTN-InducedcGMP ProductionTo confirm the role of functioning mitochondria in GTNbioactivity, we created HUVEC�0 cells and considered theabsence of cytochrome c oxidase subunit I as an index (Figure3A). These cells were associated with a 99% reduction in O2
Figure 2. Representative traces showing the rate of O2 con-sumption in a closed respiration chamber. A, Traces in the aortaof rats pretreated with MQ (14 days) and in which GTN toler-ance was induced in vivo. B, Traces following preincubationwith GTN (5�10�6 mol/L) and MQ (10�6 mol/L) in rat aorta andhuman umbilical artery and vein in which GTN tolerance wasinduced in vitro (3 hours). C, Electrochemical detection of NOproduction in HUVECs after addition of GTN (5�10�6 mol/L) andDETA-NO (10�5 mol/L).
Figure 3. (Figure 3A) Western blot analysis for cytochrome c oxi-dase in HUVECs and HUVEC�0 cells (in which the expression wasabsent). Tubulin was used as a control charge. B, Representativetraces showing the rate of O2 consumption in HUVEC�0 cells andin HUVECs following in vitro preincubation with GTN (5�10�6
mol/L) or GTN�MQ (10�6 mol/L). C, cGMP levels in HUVECs andHUVEC�0 cells. In some cases, cells were preincubated with GTN(5�10�6 mol/L). Coincubation with MQ (10�6 mol/L) or GSH ester(10�4 mol/L) prevented the effect on cGMP levels of GTN tolerancein HUVECs. Addition of DETA-NO (10�4 mol/L) increased cGMPlevels in both HUVECs and HUVEC�0 cells. Data are mean�SEMof 3 to 5 separate experiments. *P�0.001 vs control, †P�0.001 vscells with mitochondria.
Esplugues et al Mitochondrial Antioxidants and Nitrate Tolerance 1071
by guest on February 19, 2016http://circres.ahajournals.org/Downloaded from

consumption (Figure 3B). In HUVECs (Figure 3B), preincu-bation with MQ during the induction of tolerance preventedthe inhibitory effects of GTN on mitochondrial O2 consump-tion. The addition of GTN increased cGMP levels inHUVECs, but such a response was absent when cells havebeen preincubated with GTN and in HUVEC�0 cells (Figure3C). Coincubation with either MQ or GSH ester preventedthe effects induced by the incubation with GTN in HUVECs.Both HUVECs and HUVEC�0 cells exhibited an increasedcGMP production following addition of DETA-NO (10�4
mol/L). Preincubation of HUVECs with DETA-NO (5�10�6
mol/L; 3 hours) did not modify the increase in cGMP thatfollows acute addition of GTN (data not shown).
ROS Production and GSH LevelsPreincubation of HUVECs with GTN significantly increasedthe fluorescence of DCFH-DA, indicating an augmentedproduction of ROS (Figure 4A). A similar increase wasobserved when the effects of GTN preincubation were eval-uated by confocal microscopy (Figure 4B). Incubation withGTN also increased H2O2 (Figure 4C). In all cases, coincu-bation with MQ and GSH ester reversed the effects of GTN.Similar results were obtained in HUAECs (data not shown).Neither incubation of HUVEC�0 cells with GTN nor acuteadministration of GTN in both HUVECs and HUVEC�0 cellsproduced any increase in ROS-related fluorescence. Pretreat-ment with GTN produced increases in H2O2 in aortic ringswith and without endothelium (Figure 4D), and coincubationwith MQ or GSH ester prevented such an increase (data notshown). Although the removal of the endothelium reducedthe level of fluorescence, the production of ROS continued tobe substantial, thus suggesting that both muscle and endothe-lial cells are capable of generating ROS following incubationwith GTN.
Oxidative stress is related to both an increase in ROSproduction and a decrease in antioxidant content. As shown in
Figure 5A, preincubation of HUVECs with GTN significantlydecreased the fluorescence of MCB, indicating a reduction inGSH levels. A similar diminution in CMFDA fluorescencewas observed when the effects of GTN were evaluated byconfocal microscopy (Figure 5B). Treatment with MQ re-versed these effects, and, as expected, GSH ester boosted thelevels of the fluorescence signal. Figure 5C shows howincubation with GTN increased the GSSG:GSH ratio, anindex of oxidative stress, whereas MQ and GSH esterprevented this effect.
GTN Impairs Mitochondrial O2 Consumption byInhibiting Complex I: Reversal byMitochondrial-Targeted AntioxidantsFigure 6A shows the inhibitory effects of preincubation withGTN on mitochondrial complex I activity, as calculated from therate of NADH oxidation in HUVECs. Coincubation with eitherMQ or GSH ester reversed this effect. The specificity of theaction of GTN was further characterized through an alternativemethod involving cells permeabilized with digitonin and mea-surement of isolated complex I–dependent respiration. Figure6B shows that HUVECs respiring on the complex I substratesmalate (0.4�10�3 mol/L) and glutamate (30�10�3 mol/L) wereinhibited by nearly 90% with the complex I inhibitor rotenone(6�10�6 mol/L). Cells incubated with GTN respired very poorlywith malate and glutamate, whereas rotenone-sensitive respira-tion did not differ to that observed in the absence of the inhibitor.When succinate (10�10�3 mol/L), a complex II electron donor,was added to bypass complex I–dependent respiration, GTN-treated cells exhibited O2 consumption rates similar to those ofcontrols, suggesting that complex I was the main target of GTNpreincubation. Coincubation with MQ or GSH ester preventedthe effects of GTN preincubation on the levels of complexI–dependent respiration.
Figure 4. Effects of preincubation with GTN (5�10�6 mol/L; 3 hours), GTN�MQ (10�6 mol/L),or GTN�GSH ester (10�4 mol/L) on total ROS. A through C, Changes in fluorescence of (A),representative images of (B), and H2O2 production in (C) HUVECs and HUVEC�0 cells. D,H2O2 in aortic rings with or without endothelium. *P�0.001 vs control.
1072 Circulation Research November 10, 2006
by guest on February 19, 2016http://circres.ahajournals.org/Downloaded from

DiscussionThe present study demonstrated how prolonged in vivoadministration or in vitro preincubation with GTN result in areduced vascular relaxation when the drug is administeredacutely, thus resembling the clinical induction of GTNtolerance.11,21,22 The dose of GTN used for incubation doesnot release NO and following the induction of tolerance, itreduced mitochondrial O2 consumption in a noncompetitivemanner in vascular tissue and endothelial cells, which iscontrary to that observed when administered acutely.6 It hasbeen suggested that biotransformation of GTN requires afunctionally active mitochondria and leads to an increasedproduction of ROS and oxidative stress that coincides with animpairment of mitochondrial ALDH-2 activity.10,11 We havedelved further into these processes by using HUVEC�0 cellsand mitochondrial-targeted antioxidants to show that MQ andGSH ester prevent the induction of GTN tolerance and blockany increase in the production of ROS and any reduction inALDH-2 activity within the mitochondria. Finally, we pro-pose that complex I of the electron transport chain is a targetof the ROS that is generated during the induction of GTNtolerance and can cause a decrease in mitochondrial O2
consumption. Collectively, these findings extent the conceptthat mitochondrial dysfunction plays a key role in theappearance of nitrate tolerance.
MQ is the result of covalently linking ubiquinone to a TPPcation, causing a several-hundred-fold accumulation withinthe mitochondria.30 The active antioxidant form of MQ is thereduced ubiquinol form, which is regenerated by the electrontransport chain, and selectively blocks mitochondrial oxida-tive damage by detoxifying ROS.16,17 This and othermitochondrial-targeted antioxidants have been shown to pro-tect this organelle from oxidative stress in isolated cells32 andliving tissues20 in hypoxia,26 apoptosis,29 and cancer.18 In ourexperiments, continuous pretreatment with GTN produced asubstantial reduction in its vasorelaxant effects of GTN whenadministered acutely, but not in those of acute DETA-NO.These experiments argue against desensitization of sGC as a
possible mechanism for GTN tolerance. Administration invivo of MQ prevented the reduction in the relaxant effects ofGTN. This effect was equally reproduced in vitro by MQ andGSH ester, another antioxidant that increases GSH levels inmitochondria.18 Furthermore, as previously shown,11,33 non–mitochondrial-targeted antioxidants such as UA, Eb, and TPpartially reproduced the effects of MQ and GSH ester,probably as a result of their access to the mitochondria.
In further experiments, vascular tissues and cells in whichGTN tolerance had been induced in vivo or in vitro exhibiteda reduction of O2 consumption and the Km for O2 in a way thatindicated of a noncompetitive action. Administration in vivoof MQ or in vitro of either MQ or GSH ester prevented theseeffects. Once more, UA, Eb, and TP partially reproduced theeffects of MQ and GSH ester. In this way, taking into accountthe specificity of these non–mitochondrial-targeted antioxi-dants,33 ONOO�, H2O2, and O2
� would be involved in theimpairment of mitochondrial respiration.
The dose of GTN used in vitro did not result in anelectrochemical detection of NO release, thus confirming ourprevious hypothesis that biotransformation of these clinicallyrelevant doses of GTN does not release free NO.6 InHUVECs, the acute addition of GTN increased cGMP levelsand ALDH-2 activity, whereas this response was absentfollowing preincubation with GTN. Coincubation with eitherMQ or GSH ester prevented the effects induced by incubationwith GTN. In HUVEC�0 cells, GTN-stimulated increases incGMP were abolished, thus confirming the necessity for GTNto be metabolized by functional mitochondria. This corrobo-rates previous results obtained in mouse macrophages andporcine endothelial cells.10,11 Collectively, this evidence givesweight to the idea that ALDH-2 contributes to GTN biocon-version, and the subsequent increase in cGMP levels, whereasinhibition of this enzyme is implicated in GTN tolerance.
It has been widely postulated that continuous exposure toGTN would lead to an increase in the production ofROS,15,34–39 which is related to the development of toleranceand cross-tolerance.11 Our results with varying fluorescence
Figure 5. Effects of preincubation with GTN (5�10�6 mol/L; 3 hours), GTN�MQ(10�6 mol/L), or GTN�GSH ester (10�4 mol/L) on GSH levels in HUVECs, as mea-sured by fluorimetry (mean�SEM of 5 to 7 experiments) (A) or confocal micros-copy (representative images) (B). The GSSG:GSH ratio was measured spectropho-tometrically (C). *P�0.001 vs control.
Esplugues et al Mitochondrial Antioxidants and Nitrate Tolerance 1073
by guest on February 19, 2016http://circres.ahajournals.org/Downloaded from

methods confirm this hypothesis, because incubation withGTN increased the release of ROS in vascular cells andtissues. The mitochondria seems the origin of said ROSproduction, as they were absent in HUVEC�0 cells, whereastreatment with MQ and GSH ester reversed the increase inboth ROS and oxidative stress that followed incubation withGTN. It is important to highlight the important contributionof vascular muscle cells to this genesis of ROS, as demon-strated by the maintenance of a substantial production inendothelium-denudated vessels.
The subsequent oxidative stress and complex I impairmentfollowing long-term treatment with GTN could inactivateALDH-2 or inhibit the ALDH-2 repair system, both of whichresult in the impairment of GTN bioactivation. In addition, apotential interaction with superoxide may decrease the bioavail-ability of the vasodilator released following GTN bioactivation.ROS are highly toxic to various sites of mitochondrial respira-tory chain, and inhibition of complex I would seem to be themost likely consequence.22 We demonstrate that GTN toleranceis accompanied by a marked reduction in NADH oxidation,indicative of a reduction in complex I activity and prevented bymitochondrial-targeted antioxidants. Further experiments ana-lyzing isolated complex I–dependent respiration in permeabil-ized HUVECs demonstrated that in the presence of succinate, acomplex II electron donor added to bypass complex I–depen-
dent respiration, GTN-treated cells exhibited O2 consumptionrates similar to those of controls, thus confirming that continuousexposure to GTN mainly affects complex I. Again, MQ andGSH ester prevented the effects of GTN, highlighting ROS-mediated damage in complex I as the likely cause of therespiration deficiency.
In conclusion, the present study confirms that prolongedexposure to GTN induces oxidative stress and highlights themitochondria as both the source and target of ROS. Inaddition, we propose that the activity of mitochondrialcomplex I is diminished following continuous incubationwith GTN. Our data provide fresh insight into the mecha-nisms responsible for nitrate tolerance, suggesting a potentialrole for mitochondrial-targeted antioxidants as a therapeutictool in the prevention or control of the tolerance thataccompanies the chronic use of GTN in patients.
AcknowledgmentsWe thank B. Normanly for editorial assistance and F. Rodriguez andC. Amezcua for providing help.
Sources of FundingThis study was financed by Generalitat Valenciana grant 2006-341 andContrato-Investigador Fondo de Investigacion Sanitaria (FIS) (CP03/00024) (both to V.M.V.) and Ministerio de Educacion y Ciencia grantSAF2005-01366 (to J.V.E.). V.M.V. is the recipient of a Contrato (FIS).
DisclosuresNone.
References1. Parker JD, Parker JO. Nitrate therapy for stable angina pectoris. N Engl
J Med. 1998;338:520–531.2. Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate
tolerance. Circ Res. 2005;97:618–628.3. Mulsch A, Bara A, Mordvintcev P, Vanin A, Busse R. Specificity of
different organic nitrates to elicit NO formation in rabbit vascular tissuesand organs in vivo. Br J Pharmacol. 1995;116:2743–2749.
4. Artz JD, Toader V, Zavorin SI, Bennet BM, Thatcher GR. In vitroactivation of soluble guanylyl cyclase and nitric oxide release: a com-parison of NO donors and NO mimetics. Biochemistry. 2001;40:9256–9264.
5. Kleschyov AL, Oelze M, Daiber A, Huang Y, Mollnau H, Schulz E, SydowK, Fichtlscherer B, Mulsch A, Munzel T. Does nitric oxide mediate thevasodilator activity of nitroglycerin? Circ Res. 2003;93:104–112.
6. Nunez C, Victor VM, Tur R, Alvarez-Barrientos A, Moncada S,Esplugues JV, D’Ocon P. Discrepancies between nitroglycerin andNO-releasing drugs on mitochondrial oxygen consumption, vasoactivity,and the release of NO. Circ Res. 2005;97:1063–1069.
7. Lau DT, Chan EK, Benet LZ. Glutathione S-transferase-mediated metab-olism of glyceryl trinitrate in subcellular fractions of bovine coronaryarteries. Pharm Res. 1992;9:1460–1464.
8. McDonald BJ, Bennett BM. Biotransformation of glyceryl trinitrate by rataortic cytochrome P450. Biochem Pharmacol. 1993;45:268–270.
9. O’Byrne S, Shirodaria C, Millar T, Stevens C, Blake D, Benjamin N.Inhibition of platelet aggregation with glyceryl trinitrate and xanthineoxidoreductase. J Pharmacol Exp Ther. 2000;292:326–330.
10. Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanismof nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2002;99:8306–8311.
11. Sydow K, Daiber A, Oelze M, Chen Z, August M, Wendt M, Ullrich V,Mulsch A, Schulz E, Keaney JF, Stamler JS, Munzel T. Central role ofmitochondrial aldehyde dehydrogenase and reactive oxygen species in nitro-glycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–489.
12. Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T,Kitagawa K, Nakayama KI, Hess DT, Stamler JS. An essential role formitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation.Proc Natl Acad Sci U S A. 2005;102:12159–12164.
Figure 6. Effects of preincubation with GTN (5�10�6 mol/L; 3hours), GTN�MQ (10�6 mol/L), or GTN�GSH ester (10�4 mol/L)on the activity of complex I in HUVECs, as measured by calcu-lating the NADH oxidation rate (A) or isolated complex I–depen-dent respiration in digitonin-permeabilized cells in the presenceof the complex I substrates malate (0.4�10�3 mol/L) and gluta-mate (3�10�2 mol/L), the complex II substrate succinate(10�2 mol/L), or the complex I inhibitor rotenone (6�10�6 mol/L)(B). Mean�SEM of 5 to 7 experiments. *P�0.001 vs control.
1074 Circulation Research November 10, 2006
by guest on February 19, 2016http://circres.ahajournals.org/Downloaded from

13. Li Y, Zhang D, Jin W, Shao C, Yan P, Xu C, Sheng H, Liu Y, Yu J, XieY, Zhao Y, Lu D, Nebert DW, Harrison DC, Huang W, Jin L. Mito-chondrial aldehyde dehydrogenase-2 (ALDH-2) Glu504Lys poly-morphism contributes to the variation in efficacy of sublingual nitroglyc-erin. J Clin Invest. 2006;116:506–511.
14. Artz JD, Schmidt B, McCracken JL, Marletta MA. Effects of nitroglyc-erin on soluble guanylate cyclase: implications for nitrate tolerance. J BiolChem. 2002;277:18253–18256.
15. Munzel T, Sayegh H, Freeman BA, Tarpey MM, Harrison DG. Evidence forenhanced vascular superoxide anion production in nitrate tolerance. A novelmechanism underlying tolerance and cross-tolerance. J Clin Invest. 1995;95:187–194.
16. Schafer M, Schafer C, Ewald N, Piper HM, Noll T. Role of redoxsignaling in the autonomous proliferative response of endothelial cells tohypoxia. Circ Res. 2003;92:1010–1015.
17. Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targetedantioxidants protect Friedreich Ataxia fibroblasts from endogenous oxi-dative stress more effectively than untargeted antioxidants. FASEB J.2003;17:1972–1974.
18. Ortega AL, Carretero J, Obrador E, Gambini J, Asensi M, Rodilla V,Estrela JM. Tumor toxicity by endothelial cells. Impairment of the mito-chondrial system for glutathione uptake in mouse B16 melanoma cellsthat survive after in vitro interaction with the hepatic sinusoidal endothe-lium. J Biol Chem. 2003;278:13888–13897.
19. Dejana E, Colella S, Languino LR, Balconi G, Corbascio GC, Marchisio PC.Fibrinogen induces adhesion, spreading, and microfilament organization ofhuman endothelial cells in vitro. J Cell Biol. 1987;104:1403–1411.
20. Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, MurphyMP, Sammut IA. Targeting an antioxidant to mitochondria decreasescardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095.
21. Mihm MJ, Coyle CM, Jing L, Bauer JA. Vascular peroxynitrite formation duringorganic nitrate tolerance. J Pharmacol Exp Ther. 1999;291:194–198.
22. Hanspal IS, Magid KS, Webb DJ, Megson IL. The effect of oxidativestress on endothelium-dependent and nitric oxide donor-induced relax-ation: implications for nitrate tolerance. Nitric Oxide. 2002;6:263–270.
23. Ibiza S, Victor VM, Bosca I, Ortega A, Urzainqui A, O’Connor JE,Sanchez-Madrid F, Esplugues JV, Serrador JM. Endothelial nitric oxidesynthase regulates T cell receptor signaling at the immunological synapse.Immunity. 2006;24:753–765.
24. Frost MT, Wang Q, Moncada S, Singer M. Hypoxia accelerates nitricoxide-dependent inhibition of mitochondrial complex I in activated mac-rophages. Am J Physiol Regul Integr Comp Physiol. 2005;288:394–400.
25. Michelakis ED, Hampl V, Nsair A, Wu X, Harry G, Haromy A, Gurtu R,Archer SL. Diversity of mitochondrial function explains differences invascular oxygen sensing. Circ Res. 2002;90:1307–1315.
26. Sanjuan-Pla A, Cervera AM, Apostolova N, Garcia-Bou R, Victor VM,Murphy MP, McCreath KJ. A targeted antioxidant reveals the importanceof mitochondrial reactive oxygen species in the hypoxic signaling ofHIF-1 alpha. FEBS Lett. 2005;579:2669–2674.
27. Victor VM, De la Fuente M. Immune redox state from mice withendotoxin-induced oxidative stress. Involvement of NF-kappaB. FreeRadic Res. 2003;37:19–27.
28. Ragan CI, Wilson MT, Darley-Usmar VM, Lowe PN. In: edited byDarley-Usmar VM, Rickwood D, Wilson MT, eds. Mitochondria: APractical Approach. Oxford, UK: IRL; 1997:79–112.
29. Apostolova N, Cervera AM, Victor VM, Cadenas S, Sanjuan-Pla A,Alvarez-Barrientos A, Esplugues JV, McCreath KJ. Loss of apoptosis-inducing factor leads to an increase in reactive oxygen species, and animpairment of respiration that can be reversed by antioxidants. Cell DeathDiffer. 2006;13:354–357.
30. Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK,Ledgerwood EC, Smith RA, Murphy MP. Selective targeting of aredox-active ubiquinone to mitochondria within cells: antioxidant andantiapoptotic properties. J Biol Chem. 2001;276:4588 – 4596.
31. Martensson J, Meister A. Mitochondria damage in muscle occurs aftermarked depletion of glutathione and is prevented by giving glutathionemonoester. Proc Natl Acad Sci U S A. 1989;86:471–475.
32. Dhanasekaran A, Kotamraju S, Kalivendi SV, Matsunaga T, Shang T,Keszler A, Joseph J, Kalyanaraman B. Supplementation of endothelialcells with mitochondria-targeted antioxidants inhibits peroxide-inducedmitochondrial iron uptake, oxidative damage, and apoptosis. J Biol Chem.2004;279:37575–37587.
33. Hink U, Oelze M, Kolb P, Bachschmid M, Zou MH, Daiber A, MollnauH, August M, Baldus S, Tsilimingas N, Walter U, Ullrich V, Munzel T.Role of peroxynitrite in the inhibition of prostacycline synthase in nitratetolerance. J Am Coll Cardiol. 2003;42:1826–1834.
34. Dikalov S, Fink B, Skatchkov M, Stalleicken D, Bassenge E. Formationof reactive oxygen species by pentaerithrityltetranitrate and glyceryltrinitrate in vitro and development of nitrate tolerance. J Pharmacol ExpTher. 1998;286:938–944.
35. Mulsch A, Oelze M, Kloss S, Mollnau H, Topfer A, Smolenski A, Walter U,Stasch JP, Warnholtz A, Hink U, Meinertz T, Munzel T. Effects of in vivonitroglycerin treatment on activity and expression of the guanylyl cyclase andcGMP-dependent protein kinase and their downstream target vasodilator-stimulated phosphoprotein in aorta. Circulation. 2001;103:2188–2194.
36. Gori T, Parker JD. The puzzle of nitrate tolerance: pieces smaller than wethought? Circulation. 2002;106:2404–2408.
37. Warnholtz A, Mollnau H, Heitzer T, Kontush A, Moller-Bertram T, Lavall D,Giaid A, Beisiegel U, Marklund SL, Walter U, Meinertz T, Munzel T. Adverseeffects of nitroglycerin treatment on endothelial function, vascular nitrotyrosinelevels and cGMP-dependent protein kinase activity in hyperlipidemic Watanaberabbits. J Am Coll Cardiol. 2002;40:1356–1363.
38. Parker JD. Nitrate tolerance, oxidative stress, and mitochondrial function:another worrisome chapter on the effects of organic nitrates. J Clin Invest.2004;113:352–354.
39. Daiber A, Mulsch A, Hink U, Mollnau H, Warnholtz A, Oelze M, MunzelT. The oxidative stress concept of nitrate tolerance and the antioxidantproperties of hydralazine. Am J Cardiol. 2005;96:25–36.
Esplugues et al Mitochondrial Antioxidants and Nitrate Tolerance 1075
by guest on February 19, 2016http://circres.ahajournals.org/Downloaded from

Angel Ortega, Juan M. Serrador, Pilar D'Ocon and Victor M. VictorJuan V. Esplugues, Milagros Rocha, Cristina Nuñez, Irene Bosca, Sales Ibiza, Jose R. Herance,
Mitochondrial-Targeted AntioxidantsComplex I Dysfunction and Tolerance to Nitroglycerin: An Approach Based on
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2006 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/01.RES.0000250430.62775.992006;99:1067-1075; originally published online October 19, 2006;Circ Res.
http://circres.ahajournals.org/content/99/10/1067World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on February 19, 2016http://circres.ahajournals.org/Downloaded from




















