
Vasomotor dysfunction after cardiac surgery
13
Review Vasomotor dysfunction after cardiac surgery Marc Ruel a,b,1 , Tanveer A. Khan a,1 , Pierre Voisine a,1 , Cesario Bianchi a,1 , Frank W. Sellke a, * ,1 a Division of Cardiothoracic Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02215, USA b Division of Cardiac Surgery, University of Ottawa, Ottawa, Ontario, Canada Received 24 February 2004; received in revised form 28 June 2004; accepted 23 July 2004; Available online 1 September 2004 Summary Cardiopulmonary bypass and cardioplegic arrest, which allow for support of the circulation and stabilization of the heart during cardiac procedures, are still used for the vast majority of cardiac operations worldwide. However, in addition to a well-recognized systemic inflammatory response, cardiopulmonary bypass and cardioplegic arrest elicit complex, multifactorial vasomotor disturbances that vary according to the affected organ bed, with reduced vascular resistances in the skeletal muscle and peripheral circulation, and increased propensity to spasm in the cardiac, pulmonary, mesenteric and cerebral vascular beds. This article outlines the nature, mechanistic basis, and clinical correlates of the vasomotor alterations encountered in patients undergoing cardiac surgery using cardiopulmonary bypass and cardioplegic arrest. q 2004 Elsevier B.V. All rights reserved. Keywords: Cardioplegia; Cardiopulmonary bypass; Endothelial function; Vascular tone 1. Introduction Continued improvements in cardiopulmonary bypass (CPB) and myocardial protection techniques over the last four decades have made cardiac surgical procedures feasible and safe for the vast majority of patients. Nevertheless, modern CPB and cardioplegic arrest techniques remain associated with an acute transcriptional response of hundreds of genes that primarily affects the largest organ of the body, the endothelium, as well as other constituents of blood vessels [1]. The resultant vasomotor disturbances may clinically manifest as reduced peripheral vascular resist- ances in the skeletal muscle bed and, conversely, increased propensity to spasm in the coronary, pulmonary, mesenteric, and cerebral circulations. Abnormal vascular permeability and secondary tissue edema may in turn contribute to malperfusion and dysfunction of the heart, lungs, brain, kidneys, gastrointestinal tract, and other organs [2–5]. These phenomena are most striking after cardiac operations for patients in cardiogenic shock [6,7] or in patients for whom CPB support of more than 80 min is needed [8–10]. Cardioplegia, whether blood or crystalloid, is also intrinsically associated with functional changes in the coronary vasculature that add to the effects of CPB. This may be observed after even routine cardiac cases, with up to 8% of patients having coronary artery spasm manifested by temporary ST segment elevations on ECG after surgery [11–13], and an even greater proportion exhibiting myo- cardial contractile dysfunction that usually peaks 4–6 h postoperatively [14]. These phenomena may be seen in virtually any patient regardless of age, and irrespective of the presence of atherosclerotic coronary artery disease or other risk factors for endothelial dysfunction [15]. Mechanisms that mediate microvascular alterations after CPB and cardioplegic arrest include activation of the complement system, leukocyte-mediated cytokine release, as well as increases in oxidative stress and disturbances in calcium homeostasis that result from ischemia-reperfusion [3] (Fig. 1). These mechanisms lead to an increased local concentration of nitric oxide from upregulation of inducible nitric oxide synthase, and to the release of inflammatory substances such as thromboxane A 2 and inducible cycloox- ygenase from various types of cells, which result in alterations of vasomotor regulation, endothelial integrity, European Journal of Cardio-thoracic Surgery 26 (2004) 1002–1014 www.elsevier.com/locate/ejcts 1010-7940/$ - see front matter q 2004 Elsevier B.V. All rights reserved. doi:10.1016/j.ejcts.2004.07.040 * Corresponding author. Tel.: C1-617-632-8385; fax: C1-617-632- 8287. E-mail address: [email protected] (F.W. Sellke). 1 The authors have no relationship to disclose pertaining to this research.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Vasomotor dysfunction after cardiac surgery

Review
Vasomotor dysfunction after cardiac surgery
Marc Ruela,b,1, Tanveer A. Khana,1, Pierre Voisinea,1, Cesario Bianchia,1, Frank W. Sellkea,*,1
aDivision of Cardiothoracic Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02215, USAbDivision of Cardiac Surgery, University of Ottawa, Ottawa, Ontario, Canada
Received 24 February 2004; received in revised form 28 June 2004; accepted 23 July 2004; Available online 1 September 2004
Summary
Cardiopulmonary bypass and cardioplegic arrest, which allow for support of the circulation and stabilization of the heart during cardiac
procedures, are still used for the vast majority of cardiac operations worldwide. However, in addition to a well-recognized systemic
inflammatory response, cardiopulmonary bypass and cardioplegic arrest elicit complex, multifactorial vasomotor disturbances that vary
according to the affected organ bed, with reduced vascular resistances in the skeletal muscle and peripheral circulation, and increased
propensity to spasm in the cardiac, pulmonary, mesenteric and cerebral vascular beds. This article outlines the nature, mechanistic basis, and
clinical correlates of the vasomotor alterations encountered in patients undergoing cardiac surgery using cardiopulmonary bypass and
cardioplegic arrest.
q 2004 Elsevier B.V. All rights reserved.
Keywords: Cardioplegia; Cardiopulmonary bypass; Endothelial function; Vascular tone
1. Introduction
Continued improvements in cardiopulmonary bypass
(CPB) and myocardial protection techniques over the last
four decades have made cardiac surgical procedures feasible
and safe for the vast majority of patients. Nevertheless,
modern CPB and cardioplegic arrest techniques remain
associated with an acute transcriptional response of
hundreds of genes that primarily affects the largest organ
of the body, the endothelium, as well as other constituents of
blood vessels [1]. The resultant vasomotor disturbances may
clinically manifest as reduced peripheral vascular resist-
ances in the skeletal muscle bed and, conversely, increased
propensity to spasm in the coronary, pulmonary, mesenteric,
and cerebral circulations. Abnormal vascular permeability
and secondary tissue edema may in turn contribute to
malperfusion and dysfunction of the heart, lungs, brain,
kidneys, gastrointestinal tract, and other organs [2–5]. These
phenomena are most striking after cardiac operations for
1010-7940/$ - see front matter q 2004 Elsevier B.V. All rights reserved.
doi:10.1016/j.ejcts.2004.07.040
* Corresponding author. Tel.: C1-617-632-8385; fax: C1-617-632-
8287.
E-mail address: [email protected] (F.W. Sellke).1The authors have no relationship to disclose pertaining to this research.
patients in cardiogenic shock [6,7] or in patients for whom
CPB support of more than 80 min is needed [8–10].
Cardioplegia, whether blood or crystalloid, is
also intrinsically associated with functional changes in
the coronary vasculature that add to the effects of CPB. This
may be observed after even routine cardiac cases, with up to
8% of patients having coronary artery spasm manifested by
temporary ST segment elevations on ECG after surgery
[11–13], and an even greater proportion exhibiting myo-
cardial contractile dysfunction that usually peaks 4–6 h
postoperatively [14]. These phenomena may be seen in
virtually any patient regardless of age, and irrespective of
the presence of atherosclerotic coronary artery disease or
other risk factors for endothelial dysfunction [15].
Mechanisms that mediate microvascular alterations after
CPB and cardioplegic arrest include activation of the
complement system, leukocyte-mediated cytokine release,
as well as increases in oxidative stress and disturbances in
calcium homeostasis that result from ischemia-reperfusion
[3] (Fig. 1). These mechanisms lead to an increased local
concentration of nitric oxide from upregulation of inducible
nitric oxide synthase, and to the release of inflammatory
substances such as thromboxane A2 and inducible cycloox-
ygenase from various types of cells, which result in
alterations of vasomotor regulation, endothelial integrity,
European Journal of Cardio-thoracic Surgery 26 (2004) 1002–1014
www.elsevier.com/locate/ejcts
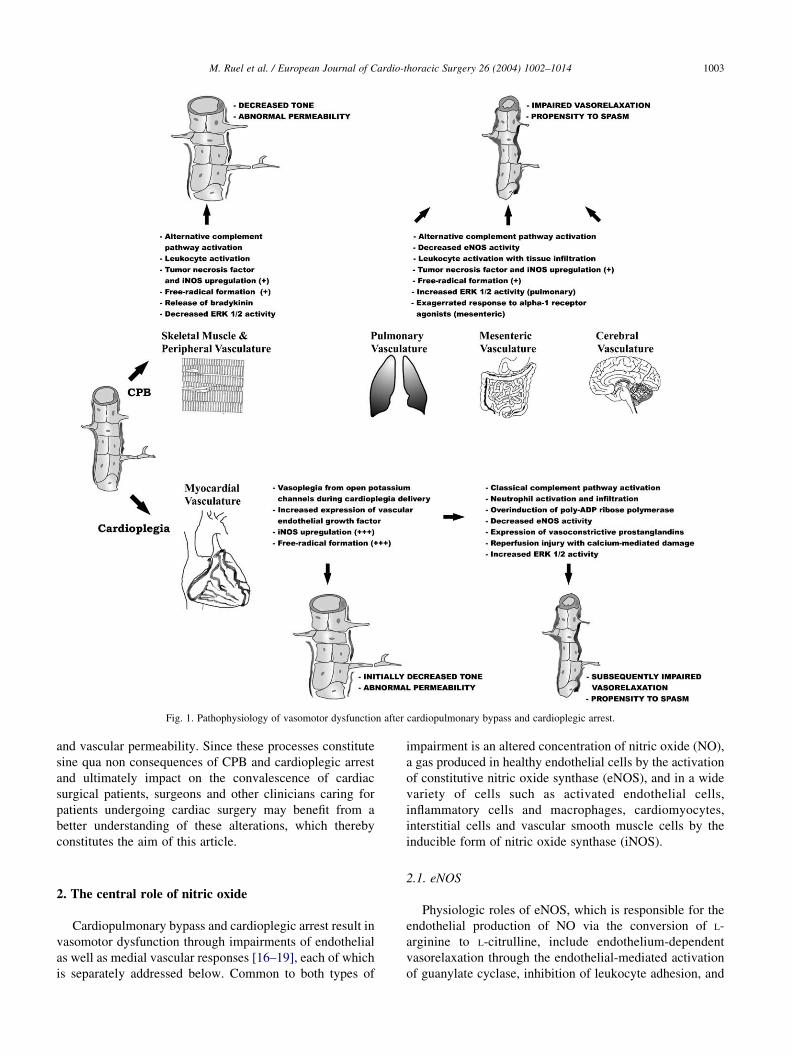
Fig. 1. Pathophysiology of vasomotor dysfunction after cardiopulmonary bypass and cardioplegic arrest.
M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–1014 1003
and vascular permeability. Since these processes constitute
sine qua non consequences of CPB and cardioplegic arrest
and ultimately impact on the convalescence of cardiac
surgical patients, surgeons and other clinicians caring for
patients undergoing cardiac surgery may benefit from a
better understanding of these alterations, which thereby
constitutes the aim of this article.
2. The central role of nitric oxide
Cardiopulmonary bypass and cardioplegic arrest result in
vasomotor dysfunction through impairments of endothelial
as well as medial vascular responses [16–19], each of which
is separately addressed below. Common to both types of
impairment is an altered concentration of nitric oxide (NO),
a gas produced in healthy endothelial cells by the activation
of constitutive nitric oxide synthase (eNOS), and in a wide
variety of cells such as activated endothelial cells,
inflammatory cells and macrophages, cardiomyocytes,
interstitial cells and vascular smooth muscle cells by the
inducible form of nitric oxide synthase (iNOS).
2.1. eNOS
Physiologic roles of eNOS, which is responsible for the
endothelial production of NO via the conversion of L-
arginine to L-citrulline, include endothelium-dependent
vasorelaxation through the endothelial-mediated activation
of guanylate cyclase, inhibition of leukocyte adhesion, and

M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–10141004
attenuation of platelet activation. In addition to these
endothelial effects, eNOS also regulates tone in the vascular
smooth muscle, to which the endothelium signals, and thus
affects medial vasodilatory responses [20].
eNOS activity is decreased after CPB and cardioplegic
arrest as a result of changes in cell membrane potential
[21–23], substrate and cofactor depletion [5,24], alterations
in the concentration or compartmentalization of
intracellular calcium [25,26], and injury to cell membranes,
associated regulatory enzymes, or ion pumps [19].
Following reperfusion after cardioplegic arrest, increased
breakdown of bioavailable NO occurs from increased
oxidative stress secondary to the generation of oxygen-
derived free-radicals [27], and production is further
impaired by exposure of the endothelium to fragments of
activated complement [28,29], activated neutrophils, and
macrophages [3].
2.2. iNOS
On the other hand the inducible form of NOS, iNOS, is
found in increased quantities in the myocardium after
cardioplegic arrest [30,31], and to a lesser extent in other
organs after CPB [32–36]. In contrast to the low
concentrations of NO produced by eNOS which inhibit
adhesion molecule expression, cytokine synthesis, and
leukocyte adhesion, the large amounts of NO generated by
iNOS under stressed local conditions can be toxic and pro-
inflammatory [37], since the excess NO reacts spon-
taneously with the reactive oxygen radicals released under
inflammatory or postischemic conditions by stressed
endothelial cells and activated leukocytes in order to form
peroxynitrite, which may cause cell apoptosis, cell necrosis,
and circulatory shock [38].
3. Endothelial responses
3.1. Complement activation
Activation of the alternative complement pathway occurs
during CPB as soon as blood gets in contact with
components of the extracorporeal circuit [39]. In addition,
cardioplegic arrest activates the classical complement
pathway, which may cause direct endothelial and cardio-
myocyte injury [40]. The anaphylotoxins C3a, C4a and C5a
are released, which promote neutrophil chemotaxis and
adherence, augment the myocardial inflammatory response,
and cause tissue edema [39,41]. Complement fragments like
C5b-9, known as ‘terminal membrane attack complexes’,
further impair endothelial function by causing direct cell
membrane injury and promoting neutrophil chemotaxis
[28]. Complement activation also causes upregulation of
adhesion molecules and increased generation of oxygen-
derived free radicals [42]. Experimentally, exposure of
isolated myocardial microvessels to zymosan-induced
complement-activated serum reduces NO-mediated endo-
thelium-dependent relaxation [28,29,43], suggesting that
activated complement intrinsically causes endothelial
injury, i.e. in tissues isolated from other blood constituents.
In the clinical setting, complement activation may be
partially inhibited by the administration of heparin [44,45],
by the use of heparin-coated circuits [46,47], by systemic
cooling on CPB [44,48], and by the use of complement
inhibitors such as pexelizumab [49] (Table 1).
3.2. Leukocytes and cytokines
3.2.1. Leukocyte activation
Activated leukocytes, through the release of oxygen-
derived free radicals, proteolytic enzymes, and inflamma-
tory cytokines, have been implicated in myocardial and
endothelial damage after ischemia [50], cardioplegia
[31,51], and CPB alone [41]. Focal leukocyte-endothelial
adherence has been identified on electron microscopy
following cardioplegic arrest and reperfusion [17], and
improved myocardial perfusion and recovery of function
have been observed if leukocyte-depleted blood is used to
reperfuse hearts after cardioplegic arrest in animals [51].
Similarly, improved endothelial and vascular smooth
muscle function have been demonstrated when monoclonal
antibodies to adhesion molecules are administered prior to
reperfusion [31,52].
Leukocytes also mediate detrimental myocardial and
systemic effects in response to CPB alone, regardless of the
occurrence of the cardiac-specific ischemia/reperfusion
injury attributed to cardioplegic arrest. For instance, the
expression of selectins is increased after the start of CPB [1,
53], and this process initiates neutrophil rolling, adherence
to the endothelium, and transmigration across the vascular
wall [54]. Granulocyte apoptosis is also decreased after
CPB, effectively prolonging the functional lifespan of
neutrophils [55]. Neutrophil and macrophage infiltration
has been documented in myocardial [56], pulmonary [57],
and mesenteric [41] tissues after CPB, and may clinically
correlate with the degree of post-CPB renal impairment [58].
3.2.2. Cytokine release
Increased circulating levels of tumor necrosis factor-a(TNF-a), interleukin (IL)-6, IL-8, and other cytokines
liberated during CPB have been shown to directly increase
the permeability of blood vessels, mediate inflammation,
and increase the expression of iNOS [30,59–63]. The
expression of vascular endothelial growth factor (VEGF), a
potent vasodilator and inducer of vascular permeability, and
that of its receptor VEGFR-1 are also upregulated after
cardioplegic arrest and reperfusion, resulting in increased
coronary microvascular relaxation responses [64,65]. On
the other hand, responses to another endothelium-dependent
vasodilator, ADP, are unchanged after cardioplegic arrest,
suggesting that the upregulation of VEGF receptors on the
coronary endothelium is selective and may play a role in

Table 1
Overview of vasomotor disturbances due to cardiopulmonary bypass and cardioplegia
Mechanism Site affected Effect Interventions and selected clinical trials
Alternative and
classical complement
activation
Systemic and
myocardial
endothelial cells
Neutrophil chemotaxis
and adherence,
free- radical production
Complement inhibitors. Intravenous Pexelizumab (a C5 complement
inhibitor)!24 h during and after CABG was effective in reducing the
incidence of the composite end-point of death or perioperative
myocardial infarction in 3088 patients [49]
Full systemic heparinization and heparin-coated circuits. Decrease
serum complement complex levels, CK-MB release and systemic
edema in patients after CPB [44–47]
Systemic cooling. Decreases C3a/C5a and elastase release [44,134],
with however no significant difference between 28 and 34 8C [48]
Leukocyte activation
and cytokine release
Systemic and
myocardial endothelial
and medial smooth
muscle cells
Free-radicals,
proteolytic enzymes,
and inflammatory
cytokines production
Leukocyte-depleted cardioplegic solutions. Were effective in
decreasing troponin T release and the incidence of postoperative low
cardiac output in a trial of 160 patients [135]
Antibodies to adhesion molecules. No clinical data. Anti-P-selectin and
anti-CD18 monoclonal antibodies appeared efficacious in reducing lung
and myocardial injury in animals [52,136–138]
Modified ultrafiltration. Decreases systemic cytokine and adhesion
molecules concentrations after CPB, with however no clinically
detectable effect [139]
Free radical formation Myocardial endothelial
cells
Endothelial cell
membrane injury
Blood cardioplegia. Limits superoxide production [140], warm being
potentially better than cold [141]
Free radical scavengers. N-acetylcysteine added to crystalloid
cardioplegia decreases myeloperoxidase and leukocyte activation, but
with no clinical impact [70]. Similarly, the addition of deferoxamine, an
iron chelator, to cardioplegia reduces release of superoxide and lipid
peroxidation products only [142]. Mannitol was ineffective in a small
study [143]
Prostaglandin
production
Systemic, pulmonary,
and myocardial
endothelial cells
Vasoconstriction,
increased permeability
Aspirin. Preoperative use may protect from protamine-induced
pulmonary vasoconstriction [144]. Data sparse with respect to possible
edema reduction
Steroids. Dextamethasone 1 mg/kg IV at induction may limit
extravascular fluid accumulation, inhibit pulmonary vasoconstriction
[145], and 1.5 g IV methylprednisolone over 24 h improves pulmonary
(controversial [146]) and myocardial function, and decreases CK-MB
release [130]
Intracellular calcium
accumulation
Systemic and
myocardial endothelial
and medial smooth
muscle cells
Increased basal
vascular tone and
propensity to spasm
Low calcium blood cardioplegia. Clinical data sparse. Animal data
suggest optimal calcium concentration of crystalloid solution is between
0.4 and 1.2 mmol/l [89,90]. Optimal concentration in warm blood is
undetermined [91]. In human neonates, the addition of nicardipine, a
calcium-channel blocker, to crystalloid cardioplegia may reduce
myocardial injury [147], although nifedipine was not effective
in adults [148]
Magnesium supplementation. 5-10 mmol/l added to cold blood
cardioplegia may be associated with less ATP depletion [149], less
troponin release and possibly improved performance in humans [150].
Magnesium cardioplegia may also decrease the incidence of atrial
fibrillation by increasing serum magnesium levels [97]
Increased iNOS, ERK,
and MAPK expressions
Systemic and
myocardial endothelial
and medial smooth
muscle cells
Increased permeability,
vasodilatation,
maldistribution
of perfusion
Improvements in CPB circuit biocompatibility. No clinical trial has yet
examined the possible impact of HCC or SMA circuits on iNOS, ERK
and MAPK
Increased peroxynitirite
production, with
resultant cell necrosis
and apoptosis
Peroxynitrite (iNOS degradation product) scavenging. The addition of
glutatione to crystalloid cardioplegia decreased neutrophil adhesion and
improved cardiac function a dog model [151]. Clinical data lacking
Minimization of CPB duration. Animal or observational clinical data
only (clinical RCT setting may be unethical)
Avoidance of alpha-adrenergic agonists. Animal or observational
clinical data only (clinical RCT setting may be unethical)
HSP70 induction with IV glutamine. Small animal data only [103]
CABG, coronary artery bypass grafting; CK-MB, creatine kinase MB isoenzyme; CPB, cardiopulmonary bypass; HCC, heparin-coated circuits; HSP, heat
shock protein; IV, intravenous; RCT, randomized controlled trial; SMA, surface modifying additive.
M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–1014 1005

M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–10141006
mediating perioperative increases in myocardial vascular
permeability.
3.3. Ischemia-reperfusion
3.3.1. Free-radicals
Oxidative stresses resulting from ischemia and reperfu-
sion damage not only myocardial endothelial cells after
anoxia and cardioplegia, but also affect systemic, non-
cardiac cells during CPB, largely due to the synthesis of
superoxide anions and hydroxyl radicals, and the interaction
between superoxide anions and NO leading to peroxynitrite
free-radicals [66–68]. In the myocardium, strategies to
decrease oxidative stress associated with cardioplegic arrest
have also been examined; for instance, the addition to
cardioplegic solutions of manganese superoxide dismutase
or deferoxamine, which respectively neutralize superoxide
and hydroxyl free-radicals, has been shown to better
preserve coronary endothelium-dependent relaxation after
cardioplegic arrest in swine [18]. The antioxidant N-
acetylcysteine has been evaluated as a cardioplegia additive
and has resulted in impressive effects in dogs [69] but no
clinically demonstrable benefit a small series of patients
[70]. The well-recognized free-radical scavenging proper-
ties of blood, particularly if warm or tepid, constitute some
of the reasons for its widespread preference over crystalloid
solutions in the composition of cardioplegia, with however
little clinically demonstrable benefit in elective, stable
patients [71–74].
The myocardium can also be protected from ischemia-
reperfusion by inhibiting ‘futile’ cell cycles such as the
poly(ADP-ribose) polymerase nuclear enzyme pathway,
which is overinduced by ischemia and cardioplegic arrest,
consumes ATP, increases the oxygen debt, results in cellular
dysfunction, and yet has little known homeostatic role. In a
recent study in pigs, Khan et al. showed that an intravenous
poly(ADP-ribose) polymerase inhibitor improved myocar-
dial perfusion, reduced the extent of infarction, and
improved cardiac function after regional ischemia and
cardioplegia-CPB [75]. However, no report on the use of
such a strategy in humans is yet available.
Few studies have examined the role of interventions
destined at decreasing systemic oxidative stress during
clinical CPB; in this regard, it is possible that angiotensin-
converting enzyme inhibitors, angiotensin-1 blockers, and
statin drugs could play a role in improving systemic and
myocardial outcomes [76–79], although clinical studies
involving angiotensin-converting enzyme inhibitors have so
far been disappointing [80,81].
3.3.2. Cyclooxygenase
Cardioplegia-reperfusion enhances the myocardial
expression of the inducible isoform of cyclooxygenase
(COX-2), an enzyme involved in prostaglandin synthesis
from the conversion of arachidonic acid. Cyclooxygenase is
implicated in the inflammatory response of diseases such as
rheumatoid arthritis and cancer. In the cardiac and systemic
vasculatures, prostaglandins have either vasodilatory or
vasoconstrictive actions, and their effects on the regulation
of vascular permeability changes are comparable and
synergistic to that of NO. CPB and cardioplegia both result
in the increased expression of COX-2 as well as other
predominantly constrictive prostanglandins, whose overall
effect is vasoconstriction of atrial and ventricular micro-
vessels [82–84]. However, no report is yet available of the
possibly beneficial effects of specific COX-2 inhibitors on
the myocardial and systemic vasculature of patients
operated using cardiopulmonary bypass and cardioplegic
arrest.
3.3.3. Calcium homeostasis
Cardioplegic arrest is associated with cellular depolariz-
ation, calcium (Ca2C) influx through voltage-dependent
Ca2C channels, release of Ca2C from intracellular Ca2C
stores, and increases in intracellular Ca2C concentration, all
of which mediate reperfusion injury. Transsarcolemmal
Ca2C influx via NaC–Ca2C exchange also plays an
important role in ischemia/reperfusion-mediated intracellu-
lar Ca2C accumulation [23,85]. These processes may be
exacerbated by the use of crystalloid over blood cardiople-
gic solutions, as crystalloid-based approaches may increase
myocardial hypoxia during cardioplegic arrest [86,87] and
cause higher accumulations of intracellular Ca2C.
Elevated intracellular Ca2C concentrations also alter the
Ca2C sensitivity of the vascular smooth muscle contractile
apparatus, which is regulated by a myosin light chain kinase
whose activity is in turn governed by Ca2C-calmodulin-
mediated phosphorylation [88]. Consequently, Ca2C over-
loading during cardioplegia constitutes a trigger for the
enhancement of basal vascular tone and agonist-induced
microvascular contraction or spasm after reperfusion,
providing an additional rationale for the use of low Ca2C
concentrations in cardioplegic solutions at a concentration
between 0.4 and 1.2 mmol/l [89,90], with the possible
exception of continuous warm blood cardioplegic tech-
niques [91]. Complete calcium depletion of the cardioplegia
solution should be avoided, as it may paradoxically lead to
increased myocardial injury and necrosis [89,92].
3.3.4. Magnesium
Following crystalloid cardioplegia and reperfusion,
contractile myocardial microvascular responses are
enhanced, whereas intrinsic myogenic contractile responses
are diminished [19]. Supplementation of cardioplegic
solutions with magnesium (Mg2C) at a concentration of
at least 5.0 mM preserves these agonist-induced and
myogenic responses [93]. While the exact causes of this
phenomenon remain unclear, higher local Mg2C concen-
trations may confer protection against endothelial dysfunc-
tion [94], or against alterations of the contractile properties
of vascular smooth muscle by displacing Ca2C from
calcium channels binding sites and hyperpolarizing

M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–1014 1007
sarcolemmal membranes, thus inhibiting Ca2C entry into
cells [95]. Extracellular Mg2C may also act by raising
intracellular Mg2C concentration, thereby reducing the
release of Ca2C from the sarcoplasmic reticulum [12,51], or
by diminishing the depletion of ATP stores and preserving
the intracellular homeostasis of smooth muscle cells. Since
Mg2C supplementation may translate into endothelial
protection and reduce the incidence of coronary vascular
spasm after cardioplegia, its addition to cardioplegic
solutions at a concentration of 5.0 mEq/l or more appears
desirable [90,96]. This was recently shown in a clinical trial
to decrease requirements for internal defibrillation and
temporary epicardial pacing intraoperatively, as well as
decrease the incidence of new postoperative atrial fibrilla-
tion in urgent CABG patients operated using warm blood
cardioplegia [97].
3.4. Apoptosis and heat-shock proteins
Cardioplegic arrest and cardiopulmonary bypass are
intrinsically associated with apoptosis of endothelial and
other vascular cells in the myocardial and systemic vascular
beds of animals and humans [1,98,99]. This is mediated
by a variety of transcriptional genes such as c-FOS and
JUN-b, which are significantly upregulated in peripheral
and myocardial tissues during cardiopulmonary bypass
and cardioplegic arrest [1]. Protection against apoptosis,
necrosis, and detrimental iNOS/peroxynitrite activity may
be afforded by the spontaneously increased production of
cytoplasmic heat shock proteins (HSP) 70, 72, and 73 in
endothelial cells and cardiomyocytes [100–102]. Upregula-
tion of these heat shock proteins could theoretically afford
protection against apoptosis and necrosis. In rats exposed to
CPB, the systemic administration of glutamine, an inducer
of HSP70, resulted in lower plasma concentrations of
interleukin-6 and interleukin-8, preserved eNOS activity,
and attenuated iNOS activity [103]. No such data is yet
available in humans.
Fig. 2. Skeletal muscle expression of activated ERK 1/2 before and after
cardiopulmonary bypass. Immunohistochemistry shows dark staining
localized to peripheral arterioles (black arrows) with decreased expression
after cardiopulmonary bypass (CPB) of activated ERK 1/2, a major
mediator of vascular tone and of the response to alpha-adrenergic agents.
(Adapted from Khan et al. [98]; with permission).
4. Medial responses
Blood flow is regulated in vascular medial smooth
muscle by metabolic, myogenic, and autonomic mechan-
isms, the latter of which is separately discussed below.
Metabolic regulation of vascular medial tone after CPB and
cardioplegic arrest is altered in the peripheral vasculature by
the systemic release of vasoactive substances during CPB,
and in the myocardial circulation by the abnormally
elevated proportion of open potassium channels during
cardioplegia [104]. Myogenic regulation, i.e. the intrinsic
property of medial smooth muscle to regulate vascular
resistance in response to changes in transmural pressure, is
actually preserved in coronary microvessels after CPB and
cardioplegia, but its pressure-diameter relationship is
shifted upward and normalized only in the presence of a
NO antagonist, thus consistent with an increased release of
NO from iNOS [105].
4.1. Autonomic responses
Autonomic medial regulation of peripheral and myocar-
dial arterioles is on the other hand markedly impaired by
CPB. This mechanism, in conjunction with the increased
circulating levels of vasodilator substances and cytokines,
explains much of the decrease in peripheral vascular
resistance frequently observed as patients are being
separated from CPB and thereafter [30,106,107], the
magnitude of which appears to correlate with the duration
of CPB [10]. As previously mentioned, the systemic
induction of iNOS as well as other vasoactive substances
such as bradykinin and VEGF also contributes to this
vasoplegic syndrome [31,64] and to the reductions in
peripheral vascular resistances and increases in vascular
permeability that contribute to the marked generalized
edema frequently observed after cardiac surgery, all with
the potential of culminating in organ malperfusion [85].
Changes in the tissue activity and expression of

M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–10141008
regulatory enzymes such as MAP kinases (MAPK) are now
also being recognized as major mediators of the complex
autonomic vascular responses observed after CPB. In the
coronary and pulmonary circulations, CPB leads to
enhanced vasoconstriction partly as a result of the rapid
induction of ERK1/2 [16,108,109], a subset of MAPK
involved in a variety of signal transduction mechanisms
triggered by a broad range of effectors including TNF-a,
which itself is elevated during CPB and returns to basal
levels after 24 h [110]. ERK1/2 and MAPK in turn induce
interleukin-6 (IL-6) production [111], leading to a cascade
of inflammatory events that include leukocytosis, thrombo-
sis and lymphocyte activation [112]. On the other hand,
recent evidence indicates that ERK1/2 activity is actually
decreased in the non-cardiac, systemic circulation after
CPB, and that this is associated with reduced contractile
responses of peripheral arterioles (Fig. 2) [113]. These
findings implicate MAPK as a major mediator of the site-
specific vasomotor dysfunction observed after CPB, with a
decreased ERK1/2 activity in the peripheral vasculature
associated with a decreased vascular tone, and an increased
ERK1/2 activity in the myocardial circulation associated
with an increased propensity to coronary spasm.
5. Clinical implications
The most frequent clinical scenario resulting from the
adverse endothelial and medial responses to CPB and
cardioplegic arrest described above can be summarized as
follows: (1) decreased basal tone and decreased alpha-
adrenergic microvascular responses in the peripheral/skele-
tal muscle vascular beds [31,64,113], with propensity
to distributive organ malperfusion, (2) initially decreased
coronary vascular tone during cardioplegia [104] followed
by decreased endothelial-mediated relaxation and
increased propensity to spasm [83,84], modifiable in part
by the composition of the cardioplegic solution [18,90,94],
(3) increased vasocontractile responses in the pulmonary
and mesenteric microcirculations [29,32,41] predisposing to
the development of pulmonary shunt and mesenteric
ischemia, respectively, particularly when vasoactive drugs
are administered to regulate blood pressure after CPB [114],
and (4) impaired but as of this writing still incompletely
characterized endothelium-mediated relaxation responses in
the cerebral microvasculature [115] (Fig. 1).
Several approaches introduced over the years to increase
the safety of CPB may have their clinical benefit explained
at least in part by their propensity to limit vasomotor
disturbances after cardiac surgery. For instance, the
increased expression of COX-2 and enhanced contractile
response of coronary arterioles to serotonin after CPB and
cardioplegia [83,84] suggest that the improvements in
coronary bypass graft patency obtained from the periopera-
tive administration of acetylsalicylic acid may not only
derive from its effects on prevention of platelet aggregation
and thrombus formation [116–118], but also from
the prevention of coronary spasm and preservation of
microvascular flow as a result of COX-2 inhibition.
The advantages of blood over crystalloid cardioplegia in
emergency or high-risk cases may result from the inhibitory
effects of blood on oxygen-derived free radical generation,
improved coronary endothelial oxygenation, enhanced
buffering capacity from histidine and other blood proteins,
and better preservation of the morphology of coronary
endothelial cells [3,82]. Antiproteases such as aprotinin, in
addition to inhibiting fibrinolysis and decreasing blood loss
after cardiopulmonary bypass, are also potent inhibitors of
the bradykinin and kallikrein systems [81,119] and may
confer additional clinical benefit by non-specifically redu-
cing the systemic inflammatory response to CPB [120,121],
although the magnitude of this effect remains controversial.
Finally, the inhalational administration of low concen-
trations of NO at 20 ppm for 8 h was recently associated
with decreased myocardial troponin release [122], but it is
reasonable to speculate that high-dose systemic NO
administration, in parallel with the effects of excessive
local production of NO and peroxynitrite from increased
iNOS activity during CPB, might ultimately prove
detrimental.
Other strategies are also available to help prevent
systemic and coronary vasomotor dysfunction after CPB
and cardioplegic arrest (Table 1). Mild systemic cooling and
the administration of heparin, both of which decrease
complement activation [44,45], the utilization of blood-
based cardioplegic solutions [82,123,124], magnesium
supplementation [93] and moderate calcium depletion [15]
of cardioplegic solutions all have beneficial effects on
endothelium-dependent relaxation, myogenic contraction,
responses to adrenergic agonists, and other indices of
myocardial vascular health after CPB and ischemia-
reperfusion. It should be remembered, however, that several
of these strategies may prove beneficial for emergency or
high-risk patients only, as improved outcomes related to
their use during routine operations has not been demon-
strated [125,126].
One standard of practice in need of further research and
foreseeable change, in light of the knowledge gained in
understanding the pathophysiology of vasomotor dysfunc-
tion after CPB, is the routine use of alpha-adrenergic
agonists for the treatment of hemodynamic instability
associated with post-CPB distributive shock. Since periph-
eral arterioles show decreased alpha-adrenergic responsive-
ness after CPB and that these responses are conversely
increased in the mesenteric and coronary circulations, the
widespread use of alpha-adrenergic agonists may constitute
a suboptimal approach for cardiac surgical patients with
vasoplegic syndromes, as these agents can worsen splanch-
nic and cerebral malperfusion, increase pulmonary shunt,
and favor the development of coronary spasm. More
specifically targeted to the underlying cause of vasoplegia
are iNOS/guanylate cyclase inhibitors such as methylene

M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–1014 1009
blue, with which preliminary clinical experience in 54
patients treated a 2 mg/kg intravenous infusion adminis-
tered over 20 min demonstrated relative safety and potential
effectiveness [127]. Other promising approaches in post-
CPB vasoplegic syndromes include ‘protecting’ the mesen-
teric circulation against its exacerbated contractile
responses to alpha-agonists agents with an intravenous
infusion of the eNOS/nitric oxide substrate L-arginine [5].
Several interventions have not delivered on the promises
that experimental data suggested with respect to their
clinical use. This may be related to the falsely exaggerated
effects of CPB in animal models compared to humans (due,
for instance, to less advanced circuits, larger priming to
circulatory volume ratios, lack of a dedicated perfusionist,
non-sterile conditions, larger amounts of embolic material,
etc.), to the rarity and lack of sensitivity of adverse clinical
outcomes, to the large potential for confounding biological
noises such as baseline endothelial function and immuno-
logic response in patients, and to the difficulty in a priori
obtaining a proper treatment effect estimate in order to
perform an adequate sample size calculation prior to
launching these small trials. The clinical use of glucocorti-
costeroids on CPB constitutes one example of disappointing
clinical results, since these agents, despite their theoretical
role in blocking the effects of inflammatory cytokines and
the expression of iNOS and COX-2, have repeatedly not
resulted in appreciable clinical benefit [128,129], with the
possible exception of decreased creatine kinase release and
a reduction in the incidence of postoperative atrial
fibrillation in two recent double-blind randomized con-
trolled trials [130] (Rubens FD et al.; in press). Also
controversial has been the role of inhibiting neutrophil
infiltration with a monoclonal antibody to C5a, which
experimentally results in improved endothelial-dependent
relaxation but no demonstrable benefit on myocardial,
pulmonary, or mesenteric functional recovery [31,131,132],
until one clinical trial demonstrated a dose-dependent
inhibition of the generation of complement byproducts, a
reduction in leukocyte activation, a 40% reduction in
creatine kinase-MB release, a 80% reduction in new
cognitive deficits, and a significant reduction in post-
operative blood loss [133]. More research is needed to
further examine the clinical impact of these and other
mechanistically based interventions on the clinical out-
comes of emergency, high-risk, and perhaps even routine
cardiac surgical patients operated using CPB and cardio-
plegic arrest.
6. Conclusion
Cardiopulmonary bypass and cardioplegia are indispen-
sable tools of the cardiac surgical armamentarium that are
still used for the majority of cardiac operations and will
remain selectively needed for decades to come. However,
these modalities constitute a double-edged sword that lead
to formidable multi-systemic microvascular derangements.
Although most of the pathophysiologic contributors to these
disturbances have been identified and their clinical impact
reasonably well elucidated, therapeutic options to circum-
vent them remain limited. Consequently, the development
and evaluation of new therapeutic modalities oriented at
limiting or eliminating the consequences of vasomotor
dysfunction after cardiac surgery should remain a main
focus of applied cardiac surgical research, as we surgeons
aim at continuously improving the results and safety of the
surgical treatment of heart disease for our patients.
References
[1] Ruel M, Bianchi C, Khan TA, Xu S, Liddicoat JR, Voisine P,
Araujo E, Lyon H, Kohane IS, Libermann TA, Sellke FW. Gene
expression profile after cardiopulmonary bypass and cardioplegic
arrest. J Thorac Cardiovasc Surg 2003;126:1521–30.
[2] Long C, Hu X, Zhang J, Xiu R, Guan Y. Changes of microvascular
vasomotion and oxygen metabolism during cooling and rewarming
period of cardiopulmonary bypass. J Extra Corpor Technol 2003;35:
13–16.
[3] Sellke FW, Boyle Jr EM, Verrier ED. Endothelial cell injury in
cardiovascular surgery: the pathophysiology of vasomotor dysfunc-
tion. Ann Thorac Surg 1996;62:1222–8.
[4] Edmunds Jr LH. Inflammatory response to cardiopulmonary bypass.
Ann Thorac Surg 1998;66:S12–S16 discussion S25–8.
[5] Andrasi TB, Soos P, Bakos G, Stumpf N, Blazovics A, Hagl S,
Szabo G. L-arginine protects the mesenteric vascular circulation
against cardiopulmonary bypass-induced vascular dysfunction.
Surgery 2003;134:72–9.
[6] Locker C, Mohr R, Paz Y, Kramer A, Lev-Ran O, Pevni D, Shapira I.
Myocardial revascularization for acute myocardial infarction:
benefits and drawbacks of avoiding cardiopulmonary bypass. Ann
Thorac Surg 2003;76:771–6 discussion 776–7.
[7] Sergeant P, Meyns B, Wouters P, Demeyere R, Lauwers P. Long-
term outcome after coronary artery bypass grafting in cardiogenic
shock or cardiopulmonary resuscitation. J Thorac Cardiovasc Surg
2003;126:1279–86.
[8] Boldt J, Brenner T, Lehmann A, Suttner SW, Kumle B, Isgro F. Is
kidney function altered by the duration of cardiopulmonary bypass?
Ann Thorac Surg 2003;75:906–12.
[9] Kumle B, Boldt J, Suttner SW, Piper SN, Lehmann A, Blome M.
Influence of prolonged cardiopulmonary bypass times on splanchnic
perfusion and markers of splanchnic organ function. Ann Thorac
Surg 2003;75:1558–64.
[10] Carrel T, Englberger L, Mohacsi P, Neidhart P, Schmidli J. Low
systemic vascular resistance after cardiopulmonary bypass: inci-
dence, etiology, and clinical importance. J Card Surg 2000;15:
347–53.
[11] Buxton AE, Hirshfeld Jr JW, Untereker WJ, Goldberg S,
Harken AH, Stephenson LW, Edie RN. Perioperative coronary
arterial spasm: long-term follow-up. Am J Cardiol 1982;50:444–51.
[12] Lockerman ZS, Rose DM, Cunningham Jr JN, Lichstein E.
Postoperative ST-segment elevation in coronary artery bypass
surgery. Chest 1986;89:647–51.
[13] Skarvan K, Graedel E, Hasse J, Stulz P, Pfisterer M. Coronary artery
spasms after coronary artery bypass surgery. Anesthesiology 1984;
61:323–7.
[14] Royster RL. Myocardial dysfunction following cardiopulmonary
bypass: recovery patterns, predictors of inotropic need, theoretical
concepts of inotropic administration. J Cardiothorac Vasc Anesth
1993;7:19–25.

M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–10141010
[15] Wernovsky G, Wypij D, Jonas RA, Mayer Jr JE, Hanley FL,
Hickey PR, Walsh AZ, Chang AC, Castaneda AR, Newburger JW,
Wessel DL. Postoperative course and hemodynamic profile after
the arterial switch operation in neonates and infants. A comparison
of low-flow cardiopulmonary bypass and circulatory arrest. Circula-
tion 1995;92:2226–35.
[16] Khan TA, Bianchi C, Ruel M, Voisine P, Li J, Liddicoat JR,
Sellke FW. Mitogen-activated protein kinase inhibition and
cardioplegia-cardiopulmonary bypass reduce coronary myogenic
tone. Circulation 2003;108(Suppl 1):II348–II353.
[17] Sellke FW, Shafique T, Schoen FJ, Weintraub RM. Impaired
endothelium-dependent coronary microvascular relaxation after cold
potassium cardioplegia and reperfusion. J Thorac Cardiovasc Surg
1993;105:52–8.
[18] Sellke FW, Shafique T, Ely DL, Weintraub RM. Coronary
endothelial injury after cardiopulmonary bypass and ischemic
cardioplegia is mediated by oxygen-derived free radicals. Circula-
tion 1993;88:II395–II400.
[19] Sellke FW, Friedman M, Dai HB, Shafique T, Schoen FJ,
Weintraub RM, Johnson RG. Mechanisms causing coronary
microvascular dysfunction following crystalloid cardioplegia and
reperfusion. Cardiovasc Res 1993;27:1925–32.
[20] Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA.
Apparent hydroxyl radical production by peroxynitrite: implications
for endothelial injury from nitric oxide and superoxide. Proc Natl
Acad Sci USA 1990;87:1620–4.
[21] Engelman DT, Watanabe M, Engelman RM, Rousou JA, Flack
3rd JE, Deaton DW, Das DK. Constitutive nitric oxide release is
impaired after ischemia and reperfusion. J Thorac Cardiovasc Surg
1995;110:1047–53.
[22] Busse R, Luckhoff A, Pohl U. Role of membrane potential in the
synthesis and release of EDRF. In: Rubanyi GM, editor. Endothelial
regulation of vasomotor tone. New York: Marcel Dekker; 1992. p.
105–20.
[23] He GW, Yang CQ. Hyperkalemia alters endothelium-dependent
relaxation through non-nitric oxide and noncyclooxygenase path-
way: a mechanism for coronary dysfunction due to cardioplegia. Ann
Thorac Surg 1996;61:1394–9.
[24] Cakir O, Oruc A, Eren S, Buyukbayram H, Erdinc L, Eren N. Does
sodium nitroprusside reduce lung injury under cardiopulmonary
bypass? Eur J Cardiothorac Surg 2003;23:1040–5.
[25] Cavallo MJ, Dorman BH, Spinale FG, Roy RC. Myocyte contractile
responsiveness after hypothermic, hyperkalemic cardioplegic arrest.
Disparity between exogenous calcium and beta- adrenergic stimu-
lation. Anesthesiology 1995;82:926–39.
[26] Meldrum DR, Cleveland Jr JC, Sheridan BC, Rowland RT,
Banerjee A, Harken AH. Cardiac surgical implications of calcium
dyshomeostasis in the heart. Ann Thorac Surg 1996;61:1273–80.
[27] Kukreja RC, Hess ML. The oxygen free radical system: from
equations through membrane-protein interactions to cardiovascular
injury and protection. Cardiovasc Res 1992;26:641–55.
[28] Stahl GL, Reenstra WR, Frendl G. Complement-mediated loss
of endothelium-dependent relaxation of porcine coronary arteries.
Role of the terminal membrane attack complex. Circ Res 1995;76:
575–83.
[29] Friedman M, Wang SY, Stahl GL, Johnson RG, Sellke FW. Altered
beta-adrenergic and cholinergic pulmonary vascular responses after
total cardiopulmonary bypass. J Appl Physiol 1995;79:1998–2006.
[30] de Vera ME, Shapiro RA, Nussler AK, Mudgett JS, Simmons RL,
Morris Jr SM, Billiar TR, Geller DA. Transcriptional regulation of
human inducible nitric oxide synthase (NOS2) gene by cytokines:
initial analysis of the human NOS2 promoter. Proc Natl Acad Sci
USA 1996;93:1054–9.
[31] Tofukuji M, Stahl GL, Agah A, Metais C, Simons M, Sellke FW.
Anti-C5a monoclonal antibody reduces cardiopulmonary bypass and
cardioplegia-induced coronary endothelial dysfunction. J Thorac
Cardiovasc Surg 1998;116:1060–8.
[32] Sato K, Li J, Metais C, Bianchi C, Sellke F. Increased pulmonary
vascular contraction to serotonin after cardiopulmonary bypass: role
of cyclooxygenase. J Surg Res 2000;90:138–43.
[33] Tofukuji M, Stahl GL, Metais C, Tomita M, Agah A, Bianchi C,
Fink MP, Sellke FW. Mesenteric dysfunction after cardiopulmonary
bypass: role of complement C5a. Ann Thorac Surg 2000;69:
799–807.
[34] Mayers I, Hurst T, Radomski A, Johnson D, Fricker S, Bridger G,
Cameron B, Darkes M, Radomski MW. Increased matrix metallo-
proteinase activity after canine cardiopulmonary bypass is sup-
pressed by a nitric oxide scavenger. J Thorac Cardiovasc Surg 2003;
125:661–8.
[35] Hayashi Y, Sawa Y, Fukuyama N, Nakazawa H, Matsuda H.
Inducible nitric oxide production is an adaptation to cardiopulmon-
ary bypass-induced inflammatory response. Ann Thorac Surg 2001;
72:149–55.
[36] Hill GE, Springall DR, Robbins RA. Aprotinin is associated with a
decrease in nitric oxide production during cardiopulmonary bypass.
Surgery 1997;121:449–55.
[37] Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide
in inflammation and immune regulation. J Physiol Pharmacol 2003;
54:469–87.
[38] Becker BF, Kupatt C, Massoudy P, Zahler S. Reactive oxygen
species and nitric oxide in myocardial ischemia and reperfusion. Z
Kardiol 2000;89(Suppl 9):IX/88–IX/91.
[39] Chenoweth DE, Cooper SW, Hugli TE, Stewart RW, Blackstone EH,
Kirklin JW. Complement activation during cardiopulmonary bypass:
evidence for generation of C3a and C5a anaphylatoxins. N Engl
J Med 1981;304:497–503.
[40] Pinckard RN, Olson MS, Giclas PC, Terry R, Boyer JT,
O’Rourke RA. Consumption of classical complement components
by heart subcellular membranes in vitro and in patients after acute
myocardial infarction. J Clin Invest 1975;56:740–50.
[41] Sellke FW, Tofukuji M, Stahl GL, Tomita M, Fink MP. Effect of C5a
inhibition on mesenteric dysfunction after cardiopulmonary bypass
(abstract). Shock 1998;9.
[42] Adler S, Baker PJ, Johnson RJ, Ochi RF, Pritzl P, Couser WG.
Complement membrane attack complex stimulates production of
reactive oxygen metabolites by cultured rat mesangial cells. J Clin
Invest 1986;77:762–7.
[43] Stamler A, Wang SY, Li J, Thurer RL, Schoen FJ, Sellke FW.
Moderate hypothermia reduces cardiopulmonary bypass-induced
impairment of cerebrovascular responses to platelet products. Ann
Thorac Surg 1996;62:191–8.
[44] Moore Jr FD, Warner KG, Assousa S, Valeri CR, Khuri SF. The
effects of complement activation during cardiopulmonary bypass.
Attenuation by hypothermia, heparin, and hemodilution. Ann Surg
1988;208:95–103.
[45] Edens RE, Linhardt RJ, Bell CS, Weiler JM. Heparin and derivatized
heparin inhibit zymosan and cobra venom factor activation of
complement in serum. Immunopharmacology 1994;27:145–53.
[46] Belboul A, Lofgren C, Storm C, Jungbeck M. Heparin-coated
circuits reduce occult myocardial damage during CPB: a random-
ized, single blind clinical trial. Eur J Cardiothorac Surg 2000;17:
580–6.
[47] Jansen PG, te Velthuis H, Huybregts RA, Paulus R, Bulder ER, van
der Spoel HI, Bezemer PD, Slaats EH, Eijsman L, Wildevuur CR.
Reduced complement activation and improved postoperative
performance after cardiopulmonary bypass with heparin-coated
circuits. J Thorac Cardiovasc Surg 1995;110:829–34.
[48] Lindholm L, Bengtsson A, Hansdottir V, Lundqvist M, Rosengren L,
Jeppsson A. Regional oxygenation and systemic inflammatory
response during cardiopulmonary bypass: influence of temperature
and blood flow variations. J Cardiothorac Vasc Anesth 2003;17:
182–7.
[49] Verrier ED, Shernan SK, Taylor KM, Van de Werf F, Newman MF,
Chen JC, Carrier M, Haverich A, Malloy KJ, Adams PX, Todaro TG,

M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–1014 1011
Mojcik CF, Rollins SA, Levy JH. Terminal complement blockade
with pexelizumab during coronary artery bypass graft surgery
requiring cardiopulmonary bypass: a randomized trial. J Am Med
Assoc 2004;291:2319–27.
[50] Dreyer WJ, Michael LH, West MS, Smith CW, Rothlein R,
Rossen RD, Anderson DC, Entman ML. Neutrophil accumulation
in ischemic canine myocardium. Insights into time course,
distribution, and mechanism of localization during early reperfusion.
Circulation 1991;84:400–11.
[51] Kawata H, Sawatari K, Mayer Jr JE. Evidence for the role of
neutrophils in reperfusion injury after cold cardioplegic ischemia in
neonatal lambs. J Thorac Cardiovasc Surg 1992;103:908–17
discussion 917–8.
[52] Kawata H, Aoki M, Hickey PR, Mayer Jr JE. Effect of antibody
to leukocyte adhesion molecule CD18 on recovery of neonatal
lamb hearts after 2 h of cold ischemia. Circulation 1992;86:
II364–II370.
[53] Chen YF, Tsai WC, Lin CC, Lee CS, Huang CH, Pan PC, Chen ML,
Huang YS. Leukocyte depletion attenuates expression of neutrophil
adhesion molecules during cardiopulmonary bypass in human
beings. J Thorac Cardiovasc Surg 2002;123:218–24.
[54] Mulligan MS, Polley MJ, Bayer RJ, Nunn MF, Paulson JC,
Ward PA. Neutrophil-dependent acute lung injury. Requirement
for P-selectin (GMP-140). J Clin Invest 1992;90:1600–7.
[55] Chello M, Mastroroberto P, Quirino A, Cuda G, Perticone F,
Cirillo F, Covino E. Inhibition of neutrophil apoptosis after coronary
bypass operation with cardiopulmonary bypass. Ann Thorac Surg
2002;73:123–9 discussion 129–30.
[56] Vakeva AP, Agah A, Rollins SA, Matis LA, Li L, Stahl GL.
Myocardial infarction and apoptosis after myocardial ischemia and
reperfusion: role of the terminal complement components and
inhibition by anti-C5 therapy. Circulation 1998;97:2259–67.
[57] Friedman M, Wang SY, Sellke FW, Cohn WE, Weintraub RM,
Johnson RG. Neutrophil adhesion blockade with NPC 15669
decreases pulmonary injury after total cardiopulmonary bypass.
J Thorac Cardiovasc Surg 1996;111:460–8.
[58] Rinder CS, Fontes M, Mathew JP, Rinder HM, Smith BR. Neutrophil
CD11b upregulation during cardiopulmonary bypass is associated
with postoperative renal injury. Ann Thorac Surg 2003;75:899–905.
[59] Butler J, Chong GL, Baigrie RJ, Pillai R, Westaby S, Rocker GM.
Cytokine responses to cardiopulmonary bypass with membrane and
bubble oxygenation. Ann Thorac Surg 1992;53:833–8.
[60] Dauber IM, Parsons PE, Welsh CH, Giclas PC, Whitman GJ,
Wheeler GS, Horwitz LD, Weil JV. Peripheral bypass-induced
pulmonary and coronary vascular injury. Association with increased
levels of tumor necrosis factor. Circulation 1993;88:726–35.
[61] Downing SW, Edmunds Jr LH. Release of vasoactive substances
during cardiopulmonary bypass. Ann Thorac Surg 1992;54:1236–43.
[62] Wan S, LeClerc JL, Vincent JL. Cytokine responses to cardiopul-
monary bypass: lessons learned from cardiac transplantation. Ann
Thorac Surg 1997;63:269–76.
[63] Borgermann J, Friedrich I, Flohe S, Spillner J, Majetschak M,
Kuss O, Sablotzki A, Feldt T, Reidemeister JC, Schade FU.
Tumor necrosis factor-alpha production in whole blood after
cardiopulmonary bypass: downregulation caused by circulating
cytokine-inhibitory activities. J Thorac Cardiovasc Surg 2002;
124:608–17.
[64] Tofukuji M, Metais C, Li J, Franklin A, Simons M, Sellke FW.
Myocardial VEGF expression after cardiopulmonary bypass
and cardioplegia. Circulation 1998;98:II242–II246 discussion
II247–8.
[65] Sellke FW, Wang SY, Stamler A, Lopez JJ, Li J, Simons M.
Enhanced microvascular relaxations to VEGF and bFGF in
chronically ischemic porcine myocardium. Am J Physiol 1996;
271:H713–H720.
[66] Clermont G, Vergely C, Jazayeri S, Lahet JJ, Goudeau JJ, Lecour S,
David M, Rochette L, Girard C. Systemic free radical activation is
a major event involved in myocardial oxidative stress related to
cardiopulmonary bypass. Anesthesiology 2002;96:80–7.
[67] Ochoa JJ, Vilchez MJ, Ibanez S, Huertas JR, Palacio MA, Munoz-
Hoyos A. Oxidative stress is evident in erythrocytes as well as
plasma in patients undergoing heart surgery involving cardiopul-
monary bypass. Free Radic Res 2003;37:11–17.
[68] Ulus AT, Aksoyek A, Ozkan M, Katircioglu SF, Basu S.
Cardiopulmonary bypass as a cause of free radical-induced oxidative
stress and enhanced blood-borne isoprostanes in humans. Free Radic
Biol Med 2003;34:911–7.
[69] Fischer UM, Cox Jr CS, Allen SJ, Stewart RH, Mehlhorn U,
Laine GA. The antioxidant N-acetylcysteine preserves myocardial
function and diminishes oxidative stress after cardioplegic arrest.
J Thorac Cardiovasc Surg 2003;126:1483–8.
[70] Vento A, Nemlander A, Aittomaki J, Salo J, Karhunen J, Ramo OJ.
N-Acetylcysteine as an additive to crystalloid cardioplegia increased
oxidative stress capacity in CABG patients. Scand Cardiovasc J
2003;37:349–55.
[71] Biagioli B, Borrelli E, Maccherini M, Bellomo G, Lisi G,
Giomarelli P, Sani G, Toscano M. Reduction of oxidative stress
does not affect recovery of myocardial function: warm continuous
versus cold intermittent blood cardioplegia. Heart 1997;77:465–73.
[72] Young JN, Choy IO, Silva NK, Obayashi DY, Barkan HE.
Antegrade cold blood cardioplegia is not demonstrably advan-
tageous over cold crystalloid cardioplegia in surgery for congenital
heart disease. J Thorac Cardiovasc Surg 1997;114:1002–8 discus-
sion 1008–9.
[73] Elwatidy AM, Fadalah MA, Bukhari EA, Aljubair KA, Syed A,
Ashmeg AK, Alfagih MR. Antegrade crystalloid cardioplegia vs
antegrade/retrograde cold and tepid blood cardioplegia in CABG.
Ann Thorac Surg 1999;68:447–53.
[74] Jacquet LM, Noirhomme PH, Van Dyck MJ, El Khoury GA,
Matta AJ, Goenen MJ, Dion RA. Randomized trial of intermittent
antegrade warm blood versus cold crystalloid cardioplegia. Ann
Thorac Surg 1999;67:471–7.
[75] Khan TA, Ruel M, Bianchi C, Voisine P, Komjati K, Szabo C,
Sellke FW. Poly(ADP-ribose) polymerase inhibition improves
postischemic myocardial function after cardioplegia-cardiopulmon-
ary bypass. J Am Coll Surg 2003;197:270–7.
[76] Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M,
Laufs U. Oxygen free radical release in human failing myocardium is
associated with increased activity of rac1-GTPase and represents a
target for statin treatment. Circulation 2003;108:1567–74.
[77] Hamilton CA, Miller WH, Al-Benna S, Brosnan MJ, Drummond RS,
McBride MW, Dominiczak AF. Strategies to reduce oxididative
stress in cardiovascular disease. Clin Sci (Lond) 2004.
[78] Chello M, Mastroroberto P, Patti G, D’Ambrosio A, Morichetti MC,
Di Sciascio G, Covino E. Simvastatin attenuates leucocyte–
endothelial interactions after coronary revascularisation with
cardiopulmonary bypass. Heart 2003;89:538–43.
[79] Lazar HL, Bao Y, Zhang Y, Bernard SA. Pretreatment with statins
enhances myocardial protection during coronary revascularization.
J Thorac Cardiovasc Surg 2003;125:1037–42.
[80] Walter T, Helber U, Bail D, Heller W, Hoffmeister HM. Influence of
ACE inhibition on myocardial damage, the Kallikrein–Kinin system
and hemostasis during cardiopulmonary bypass surgery. Thorac
Cardiovasc Surg 2002;50:150–4.
[81] Campbell DJ, Dixon B, Kladis A, Kemme M, Santamaria JD.
Activation of the kallikrein–kinin system by cardiopulmonary
bypass in humans. Am J Physiol Regul Integr Comp Physiol 2001;
281:R1059–R1070.
[82] Sellke FW, Shafique T, Johnson RG, Dai HB, Banitt PF, Schoen FJ,
Weintraub RM. Blood and albumin cardioplegia preserve endo-
thelium-dependent microvascular responses. Ann Thorac Surg 1993;
55:977–85.

M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–10141012
[83] Metais C, Li J, Simons M, Sellke FW. Serotonin-induced coronary
contraction increases after blood cardioplegia-reperfusion: role of
COX-2 expression. Circulation 1999;100:II328–II334.
[84] Metais C, Bianchi C, Li J, Simons M, Sellke FW. Serotonin-induced
human coronary microvascular contraction during acute myocardial
ischemia is blocked by COX-2 inhibition. Basic Res Cardiol 2001;
96:59–67.
[85] Yuan Y, Granger HJ, Zawieja DC, DeFily DV, Chilian WM.
Histamine increases venular permeability via a phospholipase C-NO
synthase-guanylate cyclase cascade. Am J Physiol 1993;264:
H1734–H1739.
[86] Ibrahim MF, Venn GE, Young CP, Chambers DJ. A clinical
comparative study between crystalloid and blood-based St Thomas’
hospital cardioplegic solution. Eur J Cardiothorac Surg 1999;15:
75–83.
[87] Quinlan GJ, Westerman ST, Mumby S, Pepper JR, Gutteridge JM.
Plasma hypoxanthine levels during crystalloid and blood cardiople-
gias: warm blood cardioplegia increases hypoxanthine levels with a
greater risk of oxidative stress. J Cardiovasc Surg (Torino) 1999;40:
65–9.
[88] Pfitzer G. Invited review: regulation of myosin phosphorylation in
smooth muscle. J Appl Physiol 2001;91:497–503.
[89] Yamamoto F, Braimbridge MV, Hearse DJ. Calcium and cardiople-
gia. The optimal calcium content for the St Thomas’ Hospital
cardioplegic solution. J Thorac Cardiovasc Surg 1984;87:908–12.
[90] Kronon MT, Allen BS, Hernan J, Halldorsson AO, Rahman S,
Buckberg GD, Wang T, Ilbawi MN. Superiority of magnesium
cardioplegia in neonatal myocardial protection. Ann Thorac Surg
1999;68:2285–91 discussion 2291–2.
[91] Nakamura Y, Kuroda H, Takemoto N, Ohgi S, Mori T. Risk of low
calcium and high magnesium in continuous warm hyperkalemic
cardioplegia. Ann Thorac Surg 1999;68:1295–301.
[92] Rebeyka IM, Axford-Gatley RA, Bush BG, del Nido PJ, Mickle DA,
Romaschin AD, Wilson GJ. Calcium paradox in an in vivo model of
multidose cardioplegia and moderate hypothermia. Prevention with
diltiazem or trace calcium levels. J Thorac Cardiovasc Surg 1990;99:
475–83.
[93] Matsuda N, Tofukuji M, Morgan KG, Sellke FW. Coronary
microvascular protection with mg2C: effects on intracellular
calcium regulation and vascular function. Am J Physiol 1999;276:
H1124–H1130.
[94] Tofukuji M, Stamler A, Li J, Franklin A, Wang SY, Hariawala MD,
Sellke FW. Effects of magnesium cardioplegia on regulation of the
porcine coronary circulation. J Surg Res 1997;69:233–9.
[95] Cartier R, Pellerin M, Hollmann C, Pelletier LC. Effects of pressure
and duration of hyperkalemic infusions on endothelial function. Ann
Thorac Surg 1993;55:700–5.
[96] Tsukube T, McCully JD, Faulk EA, Federman M, LoCicero 3rd J,
Krukenkamp IB, Levitsky S. Magnesium cardioplegia reduces
cytosolic and nuclear calcium and DNA fragmentation in the
senescent myocardium. Ann Thorac Surg 1994;58:1005–11.
[97] Yeatman M, Caputo M, Narayan P, Lotto AA, Ascione R, Bryan AJ,
Angelini GD. Magnesium-supplemented warm blood cardioplegia in
patients undergoing coronary artery revascularization. Ann Thorac
Surg 2002;73:112–8.
[98] Aebert H, Kirchner S, Keyser A, Birnbaum DE, Holler E,
Andreesen R, Eissner G. Endothelial apoptosis is induced by
serum of patients after cardiopulmonary bypass. Eur J Cardiothorac
Surg 2000;18:589–93.
[99] Yeh CH, Wang YC, Wu YC, Chu JJ, Lin PJ. Continuous tepid
blood cardioplegia can preserve coronary endothelium and amelio-
rate the occurrence of cardiomyocyte apoptosis. Chest 2003;123:
1647–54.
[100] Aebert H, Cornelius T, Birnbaum DE, Siegel AV, Riegger GA,
Schunkert H. Induction of early immediate genes and programmed
cell death following cardioplegic arrest in human hearts. Eur
J Cardiothorac Surg 1997;12:261–7.
[101] Qing M, Vazquez-Jimenez JF, Schumacher K, Bhardwaj RS,
Klosterhalfen B, Minkenberg R, Messmer BJ, von Bernuth G,
Seghaye MC. Moderate hypothermia during cardiopulmonary
bypass increases intramyocardial synthesis of heat shock protein
72. J Thorac Cardiovasc Surg 2002;124:724–31.
[102] Demidov ON, Tyrenko VV, Svistov AS, Komarova YY,
Karpishenko AI, Margulis BA, Shevchenko YL. Heat shock proteins
in cardiosurgery patients. Eur J Cardiothorac Surg 1999;16:444–9.
[103] Hayashi Y, Sawa Y, Fukuyama N, Nakazawa H, Matsuda H.
Preoperative glutamine administration induces heat-shock protein 70
expression and attenuates cardiopulmonary bypass-induced inflam-
matory response by regulating nitric oxide synthase activity.
Circulation 2002;106:2601–7.
[104] Wang SY, Friedman M, Johnson RG, Zeind AJ, Sellke FW.
Adenosine triphosphate-sensitive KC channels mediate postcardio-
plegia coronary hyperemia. J Thorac Cardiovasc Surg 1995;110:
1073–82.
[105] Wang SY, Friedman M, Franklin A, Sellke FW. Myogenic reactivity
of coronary resistance arteries after cardiopulmonary bypass and
hyperkalemic cardioplegia. Circulation 1995;92:1590–6.
[106] Wang SY, Stamler A, Li J, Johnson RG, Sellke FW. Decreased
myogenic reactivity in skeletal muscle arterioles after hypothermic
cardiopulmonary bypass. J Surg Res 1997;69:40–4.
[107] Wang SY, Friedman M, Johnson RG, Weintraub RM, Sellke FW.
Adrenergic regulation of coronary microcirculation after extracor-
poreal circulation and crystalloid cardioplegia. Am J Physiol 1994;
267:H2462–H2470.
[108] Araujo EG, Bianchi C, Sato K, Faro R, Li XA, Sellke FW.
Inactivation of the MEK/ERK pathway in the myocardium during
cardiopulmonary bypass. J Thorac Cardiovasc Surg 2001;121:
773–81.
[109] Khan TA, Bianchi C, Araujo EG, Ruel M, Voisine P, Sellke FW.
Activation of pulmonary mitogen-activated protein kinases during
cardiopulmonary bypass. J Surg Res 2003;115:56–62.
[110] Marano CW, Garulacan LA, Laughlin KV, Igidbashian L, Trace C,
Goldman SM, Sutter FP, Reichard Jr GA, Mullin JM. Plasma
concentrations of soluble tumor necrosis factor receptor I and tumor
necrosis factor during cardiopulmonary bypass. Ann Thorac Surg
2000;70:1313–8.
[111] Hayashi Y, Sawa Y, Nishimura M, Tojo SJ, Fukuyama N,
Nakazawa H, Matsuda H. P-selectin participates in cardiopulmonary
bypass-induced inflammatory response in association with nitric
oxide and peroxynitrite production. J Thorac Cardiovasc Surg 2000;
120:558–65.
[112] Van den Berghe G. Novel insights into the neuroendocrinology of
critical illness. Eur J Endocrinol 2000;143:1–13.
[113] Khan TA, Bianchi C, Araujo EG, Ruel M, Voisine P, Li J,
Liddicoat JR, Sellke FW. Cardiopulmonary bypass reduces periph-
eral microvascular contractile function by inhibition of mitogen-
activated protein kinase activity. Surgery 2003;134:247–54.
[114] Allen KB, Salam AA, Lumsden AB. Acute mesenteric ischemia after
cardiopulmonary bypass. J Vasc Surg 1992;16:391–5 discussion
395–6.
[115] Cooper WA, Duarte IG, Thourani VH, Nakamura M, Wang NP,
Brown 3rd WM, Gott JP, Vinten-Johansen J, Guyton RA. Hypother-
mic circulatory arrest causes multisystem vascular endothelial
dysfunction and apoptosis. Ann Thorac Surg 2000;69:696–702
discussion 703.
[116] Chesebro JH, Clements IP, Fuster V, Elveback LR, Smith HC,
Bardsley WT, Frye RL, Holmes Jr DR, Vlietstra RE, Pluth JR,
Wallace RB, Puga FJ, Orszulak TA, Piehler JM, Schaff HV,
Danielson GK. A platelet-inhibitor-drug trial in coronary-artery
bypass operations: benefit of perioperative dipyridamole and aspirin
therapy on early postoperative vein-graft patency. N Engl J Med
1982;307:73–8.
[117] Gavaghan TP, Gebski V, Baron DW. Immediate postoperative
aspirin improves vein graft patency early and late after coronary

M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–1014 1013
artery bypass graft surgery. A placebo-controlled, randomized study.
Circulation 1991;83:1526–33.
[118] Mangano DT. Aspirin and mortality from coronary bypass surgery.
N Engl J Med 2002;347:1309–17.
[119] Mojcik CF, Levy JH. Aprotinin and the systemic inflammatory
response after cardiopulmonary bypass. Ann Thorac Surg 2001;71:
745–54.
[120] Hill GE, Alonso A, Spurzem JR, Stammers AH, Robbins RA.
Aprotinin and methylprednisolone equally blunt cardiopulmonary
bypass-induced inflammation in humans. J Thorac Cardiovasc Surg
1995;110:1658–62.
[121] Schmartz D, Tabardel Y, Preiser JC, Barvais L, d’Hollander A,
Duchateau J, Vincent JL. Does aprotinin influence the inflammatory
response to cardiopulmonary bypass in patients? J Thorac Cardio-
vasc Surg 2003;125:184–90.
[122] Gianetti J, Del Sarto P, Bevilacqua S, Vassalle C, De Filippis R,
Kacila M, Farneti PA, Clerico A, Glauber M, Biagini A. Sup-
plemental nitric oxide and its effect on myocardial injury and
function in patients undergoing cardiac surgery with extracorporeal
circulation. J Thorac Cardiovasc Surg 2004;127:44–50.
[123] Wang SY, Stamler A, Tofukuji M, Deuson TE, Sellke FW. Effects of
blood and crystalloid cardioplegia on adrenergic and myogenic
vascular mechanisms. Ann Thorac Surg 1997;63:41–9.
[124] Murphy CO, Pan C, Gott JP, Guyton RA. Microvascular reactivity
after crystalloid, cold blood, and warm blood cardioplegic arrest.
Ann Thorac Surg 1995;60:1021–7.
[125] Shapira N, Kirsh M, Jochim K, Behrendt DM. Comparison of
the effect of blood cardioplegia to crystalloid cardioplegia on
myocardial contractility in man. J Thorac Cardiovasc Surg 1980;80:
647–55.
[126] Shanewise JS, Kosinski AS, Coto JA, Jones EL. Prospective,
randomized trial comparing blood and oxygenated crystalloid
cardioplegia in reoperative coronary artery bypass grafting.
J Thorac Cardiovasc Surg 1998;115:1166–71.
[127] Leyh RG, Kofidis T, Struber M, Fischer S, Knobloch K,
Wachsmann B, Hagl C, Simon AR, Haverich A. Methylene blue:
the drug of choice for catecholamine-refractory vasoplegia after
cardiopulmonary bypass? J Thorac Cardiovasc Surg 2003;125:
1426–31.
[128] Boscoe MJ, Yewdall VM, Thompson MA, Cameron JS. Comp-
lement activation during cardiopulmonary bypass: quantitative study
of effects of methylprednisolone and pulsatile flow. Br Med J (Clin
Res Ed) 1983;287:1747–50.
[129] Andersen LW, Baek L, Thomsen BS, Rasmussen JP. Effect of
methylprednisolone on endotoxemia and complement activation
during cardiac surgery. J Cardiothorac Anesth 1989;3:544–9.
[130] Giomarelli P, Scolletta S, Borrelli E, Biagioli B. Myocardial and
lung injury after cardiopulmonary bypass: role of interleukin (IL)-
10. Ann Thorac Surg 2003;76:117–23.
[131] Park KW, Tofukuji M, Metais C, Comunale ME, Dai HB, Simons M,
Stahl GL, Agah A, Sellke FW. Attenuation of endothelium-
dependent dilation of pig pulmonary arterioles after cardiopulmon-
ary bypass is prevented by monoclonal antibody to complement C5a.
Anesth Analg 1999;89:42–8.
[132] Tofukuji M, Stahl GL, Metais C, Tomita M, Agah A, Bianchi C,
Fink MP, Sellke FW. Mesenteric dysfunction after cardiopulmonary
bypass: role of complement C5a. Ann Thorac Surg 2000;69:
799–807.
[133] Fitch JC, Rollins S, Matis L, Alford B, Aranki S, Collard CD,
Dewar M, Elefteriades J, Hines R, Kopf G, Kraker P, Li L, O’Hara R,
Rinder C, Rinder H, Shaw R, Smith B, Stahl G, Shernan SK.
Pharmacology and biological efficacy of a recombinant, humanized,
single-chain antibody C5 complement inhibitor in patients under-
going coronary artery bypass graft surgery with cardiopulmonary
bypass. Circulation 1999;100:2499–506.
[134] Menasche P, Peynet J, Haeffner-Cavaillon N, Carreno MP, de
Chaumaray T, Dillisse V, Faris B, Piwnica A, Bloch G, Tedgui A.
Influence of temperature on neutrophil trafficking during clinical
cardiopulmonary bypass. Circulation 1995;92:II334–II340.
[135] Palatianos GM, Balentine G, Papadakis EG, Triantafillou CD,
Vassili MI, Lidoriki A, Dinopoulos A, Astras GM. Neutrophil
depletion reduces myocardial reperfusion morbidity. Ann Thorac
Surg 2004;77:956–61.
[136] Hayashi Y, Sawa Y, Nishimura M, Tojo SJ, Ichikawa H, Satoh H,
Yamaguchi T, Suhara H, Ohtake S, Matsuda H. P-selectin
monoclonal antibody may attenuate the whole body inflammatory
response induced by cardiopulmonary bypass. Asaio J 2000;46:
334–7.
[137] Aoki M, Jonas RA, Nomura F, Kawata H, Hickey PR. Anti-CD18
attenuates deleterious effects of cardiopulmonary bypass and
hypothermic circulatory arrest in piglets. J Card Surg 1995;10:
407–17.
[138] Mayers I, Hurst T, Johnson D, Cujec B, Ang LC, Thomson D,
Fox JA, Blank GS, Saxena A, Richardson JS. Anti-CD18 antibodies
improve cardiac function following cardiopulmonary bypass in dogs.
J Crit Care 1996;11:189–96.
[139] Grunenfelder J, Zund G, Schoeberlein A, Maly FE, Schurr U,
Guntli S, Fischer K, Turina M. Modified ultrafiltration lowers
adhesion molecule and cytokine levels after cardiopulmonary bypass
without clinical relevance in adults. Eur J Cardiothorac Surg 2000;
17:77–83.
[140] Kalawski R, Balinski M, Bugajski P, Wysocki H, Olszewski R,
Siminiak T. Stimulation of neutrophil activation during coronary
artery bypass grafting: comparison of crystalloid and blood
cardioplegia. Ann Thorac Surg 2001;71:827–31.
[141] Mezzetti A, Calafiore AM, Lapenna D, Deslauriers R, Tian G,
Salerno TA, Verna AM, Bosco G, Pierdomenico SD, Caccurullo F.
Intermittent antegrade warm cardioplegia reduces oxidative stress
and improves metabolism of the ischemic-reperfused human
myocardium. J Thorac Cardiovasc Surg 1995;109:787–95.
[142] Drossos G, Lazou A, Panagopoulos P, Westaby S. Deferoxamine
cardioplegia reduces superoxide radical production in human
myocardium. Ann Thorac Surg 1995;59:169–72.
[143] Larsen M, Webb G, Kennington S, Kelleher N, Sheppard J, Kuo J,
Unsworth-White J. Mannitol in cardioplegia as an oxygen free
radical scavenger measured by malondialdehyde. Perfusion 2002;17:
51–5.
[144] Comunale ME, Maslow A, Robertson LK, Haering JM,
Mashikian JS, Lowenstein E. Effect of site of venous protamine
administration, previously alleged risk factors, and preoperative use
of aspirin on acute protamine-induced pulmonary vasoconstriction.
J Cardiothorac Vasc Anesth 2003;17:309–13.
[145] von Spiegel T, Giannaris S, Wietasch GJ, Schroeder S, Buhre W,
Schorn B, Hoeft A. Effects of dexamethasone on intravascular and
extravascular fluid balance in patients undergoing coronary bypass
surgery with cardiopulmonary bypass. Anesthesiology 2002;96:
827–34.
[146] Chaney MA, Nikolov MP, Blakeman B, Bakhos M, Slogoff S.
Pulmonary effects of methylprednisolone in patients undergoing
coronary artery bypass grafting and early tracheal extubation. Anesth
Analg 1998;87:27–33.
[147] Mori F, Miyamoto M, Tsuboi H, Noda H, Esato K. Clinical trial of
nicardipine cardioplegia in pediatric cardiac surgery. Ann Thorac
Surg 1990;49:413–7 discussion 417-8.
[148] Flameng W, De Meyere R, Daenen W, Sergeant P, Ngalikpima V,
Geboers J, Suy R, Stalpaert G. Nifedipine as an adjunct to St Thomas’
Hospital cardioplegia. A double-blind, placebo-controlled, random-
ized clinical trial. J Thorac Cardiovasc Surg 1986;91:723–31.
[149] Caputo M, Bryan AJ, Calafiore AM, Suleiman MS, Angelini GD.
Intermittent antegrade hyperkalaemic warm blood cardioplegia

M. Ruel et al. / European Journal of Cardio-thoracic Surgery 26 (2004) 1002–10141014
supplemented with magnesium prevents myocardial substrate
derangement in patients undergoing coronary artery bypass surgery.
Eur J Cardiothorac Surg 1998;14:596–601.
[150] Ji B, Feng Z, Liu J, Long C. Myocardial protection related to
magnesium content of cold blood hyperkalemic cardioplegic
solutions in CABG. J Extra Corpor Technol 2002;34:107–10.
[151] Nakamura M, Thourani VH, Ronson RS, Velez DA, Ma XL,
Katzmark S, Robinson J, Schmarkey LS, Zhao ZQ, Wang NP,
Guyton RA, Vinten-Johansen J. Glutathione reverses endothelial
damage from peroxynitrite, the byproduct of nitric oxide degra-
dation, in crystalloid cardioplegia. Circulation 2000;102:
III332–III338.




















