
Complete suboptimal folding of RNA and the stability of secondary structures
21
Complete Suboptimal Folding of RNA and the Stability of Secondary Structures Stefan Wuchty 1 Walter Fontana 1,2 Ivo L. Hofacker 1 Peter Schuster 1,2 1 Institut fu ¨ r Theoretische Chemie, Universita ¨ t Wien, Wa ¨ hringerstrasse 17, A-1090 Wien, Austria 2 Santa Fe Institute, 1399 Hyde Park Road, Santa Fe, NM 87501 USA Received 13 May 1998; accepted 6 August 1998 Abstract: An algorithm is presented for generating rigorously all suboptimal secondary structures between the minimum free energy and an arbitrary upper limit. The algorithm is particularly fast in the vicinity of the minimum free energy. This enables the efficient approximation of statistical quantities, such as the partition function or measures for structural diversity. The density of states at low energies and its associated structures are crucial in assessing from a thermodynamic point of view how well-defined the ground state is. We demonstrate this by exploring the role of base modification in tRNA secondary structures, both at the level of individual sequences from Esche- richia coli and by comparing artificially generated ensembles of modified and unmodified sequences with the same tRNA structure. The two major conclusions are that (1) base modification consider- ably sharpens the definition of the ground state structure by constraining energetically adjacent structures to be similar to the ground state, and (2) sequences whose ground state structure is thermodynamically well defined show a significant tendency to buffer single point mutations. This can have evolutionary implications, since selection pressure to improve the definition of ground states with biological function may result in increased neutrality. © 1999 John Wiley & Sons, Inc. Biopoly 49: 145–165, 1999 Keywords: RNA secondary structure; suboptimal folding; density of states; tRNA; modified bases; thermodynamic stability of structure; mutational buffering; neutrality; dynamic programming INTRODUCTION The structure of RNA molecules can be discussed at an empirically well established level of resolution known as secondary structure. It refers to a topology of binary contacts arising from specific base pairing, rather than a geometry cast in terms of coordinates and distances (see Figure 1). The driving force behind Correspondence to: Walter Fontana; email: [email protected] Contract grant sponsor: Austrian Fond zur Fo ¨rderung der Wis- senschaftlichen Forschung (FWF) and Santa Fe Institute Contract grant number: 11065-CHE (FWF) Biopolymers, Vol. 49, 145–165 (1999) © 1999 John Wiley & Sons, Inc. CCC 0006-3525/99/020145-21 145
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Complete suboptimal folding of RNA and the stability of secondary structures

Complete Suboptimal Foldingof RNA and the Stability ofSecondary Structures
Stefan Wuchty1
Walter Fontana1,2
Ivo L. Hofacker1
Peter Schuster1,2
1 Institut fur TheoretischeChemie,
Universitat Wien,Wahringerstrasse 17,A-1090 Wien, Austria
2 Santa Fe Institute,1399 Hyde Park Road,
Santa Fe, NM 87501 USA
Received 13 May 1998;accepted 6 August 1998
Abstract: An algorithm is presented for generating rigorously all suboptimal secondary structuresbetween the minimum free energy and an arbitrary upper limit. The algorithm is particularly fastin the vicinity of the minimum free energy. This enables the efficient approximation of statisticalquantities, such as the partition function or measures for structural diversity. The density of statesat low energies and its associated structures are crucial in assessing from a thermodynamic pointof view how well-defined the ground state is. We demonstrate this by exploring the role of basemodification in tRNA secondary structures, both at the level of individual sequences fromEsche-richia coliand by comparing artificially generated ensembles of modified and unmodified sequenceswith the same tRNA structure. The two major conclusions are that (1) base modification consider-ably sharpens the definition of the ground state structure by constraining energetically adjacentstructures to be similar to the ground state, and (2) sequences whose ground state structure isthermodynamically well defined show a significant tendency to buffer single point mutations. Thiscan have evolutionary implications, since selection pressure to improve the definition of groundstates with biological function may result in increased neutrality.© 1999 John Wiley & Sons, Inc.Biopoly 49: 145–165, 1999
Keywords: RNA secondary structure; suboptimal folding; density of states; tRNA; modified bases;thermodynamic stability of structure; mutational buffering; neutrality; dynamic programming
INTRODUCTION
The structure of RNA molecules can be discussed atan empirically well established level of resolution
known as secondary structure. It refers to a topologyof binary contacts arising from specific base pairing,rather than a geometry cast in terms of coordinatesand distances (see Figure 1). The driving force behind
Correspondence to:Walter Fontana; email: [email protected] grant sponsor: Austrian Fond zur Fo¨rderung der Wis-
senschaftlichen Forschung (FWF) and Santa Fe InstituteContract grant number: 11065-CHE (FWF)
Biopolymers, Vol. 49, 145–165 (1999)© 1999 John Wiley & Sons, Inc. CCC 0006-3525/99/020145-21
145

secondary structure formation is the stacking of basepairs. The formation of an energetically favorablehelical region, however, also implies the formation ofan energetically unfavorable loop region. This “frus-trated” energetics leads to a vast combinatorics ofhelix and loop arrangements spanning the structuralrepertoire of an individual RNA sequence.
The secondary structure provides both geometri-cally and thermodynamically a scaffold for the ter-tiary structure. Its free energy accounts for a largeshare of the overall free energy of the full structure.This linkage puts the secondary structure in corre-spondence with functional properties of the tertiarystructure. Consequently, selection pressures becomemanifest at the secondary structure level as evolution-ary conserved base pairs.
A secondary structure can be conveniently dis-cretized as a graph representing a pattern of base paircontacts (Figure 1). This yields a formally well-de-fined combinatorial object that can be subject to math-ematical treatment. Of particular interest are second-ary structures possessing some extremal property withrespect to a given sequence, such as having the largestnumber of admissible base pairs or minimizing thefree energy. The theoretical importance of RNA as amodel system for sequence–structure relations inbiopolymers lies in the fact that structures of this kindcan be computed by dynamic programming.1–5 Thismethod produces a single structure with the desiredextremal property (even in the case of degeneracy). Ithas been stressed,6 however, that this may not ade-quately describe a real situation for two major rea-sons. First, the energy parameters on which the fold-ing algorithm relies are inevitably imprecise. Hence,the true minimum free energy (mfe) structure mightbe one that is suboptimal with respect to the param-eters used. The same might hold because of unknownbiological constraints that may change relative ener-
gies, turning an otherwise suboptimal structure intothe most favorable one. Second, under physiologicalconditions RNA sequences may exist in alternativestates whose energy difference is small. Aside fromtheir possible biological significance, the density andaccessibility of such low lying states may determinehow well defined an mfe structure actually is.
Issues like these have prompted several approachesto generating suboptimal structures.6–8 While repre-senting an improvement, these approaches share oneproblem: they do not computeall suboptimal struc-tures within a given energy range from the mfe. Forexample, a widely used algorithm is Zuker’s exten-sion6 of his own dynamic programming procedure.4 Itgenerates for each admissible base pair in a givensequence the energetically best structure containingthat base pair. Hence, for a sequence of lengthn atmostn(n 2 1)/ 2 suboptimal structures are produced.Furthermore, each base pair present in the mfe struc-ture regenerates by definition the mfe structure as thebest structure containing it. It follows that no struc-tures are generated that differ from the mfe by theabsence of one or more base pairs. In addition, if anmfe structure consists of two substructures A-B con-nected by a stretch of external bases, no suboptimalalternatives will be produced that are suboptimal inboth modules. As a calibration for the number ofstructures missed, consider theEscherichia colise-quences tRNAhis (RH1660) and tRNAser (RS1661)from the EMBL Heidelberg tRNA Database (see Ap-pendix C), which have 5 and 73 structures, respec-tively, within 10% of their minimum free energy. Ofthese, 2 (tRNAhis) and 17 (tRNAser) structures wouldshow up under Zuker’s scheme.
Many of the missing structures may well be clas-sified as “uninteresting” by some account, yet thiscannot be said with certainty for all of them and notfor all accounts. They clearly are relevant in thecalculation of measures for structural well defined-ness, in approximating statistical quantities such asthe partition function, or in calculating the density ofstates at low energies. Among the major benefits of acomplete suboptimal folding procedure is the possi-bility of rigorously analyzing the low energy sectionof the energy landscape on which the actual kineticfolding process occurs.
In this paper we describe a fairly simple algorithmthat generates all suboptimal folds of a sequencewithin a desired energy range from the mfe. The ideaunderlying the algorithm is straightforward, and wetook it literally from Waterman and Byers,9 whodeveloped it in the context of suboptimal solutions tothe shortest path problem in networks. Waterman andByers also applied their scheme to obtain near-opti-
FIGURE 1 An RNA secondary structure graph. Unpairedpositions not enclosed by base pairs, such as free ends orlinks between independent structure modules, are called“external.” Here they are marked by ticks.
146 Wuchty et al.

mal sequence alignments.9 Yet, to our knowledge,their idea has not been exploited to produce subopti-mal solutions to RNA folding, which is somewhatpuzzling, since the energy minimization of secondarystructures is handled by the same technique employedin the shortest path problem or in sequence alignment.We first illustrate the Waterman–Byers scheme for thecase of base pair maximization. While being of the-oretical interest only, the case serves as a pedagogicalexposition of the logic underlying the algorithm. Wethen briefly discuss the more involved case of energyminimization, while relegating excruciating details toappendices A and B. We proceed by applying thealgorithm to study the degree to which a minimumfree energy structure is thermodynamically sharplydefined. We are specifically interested in the role ofbase modification in tRNA sequences to that effect.
MAXIMUM MATCHING AND THEWATERMAN–BYERS SCHEME
The usual formalization5,10 views a secondary struc-ture as a graph whose nodes represent nucleotides atpositionsi 5 1, . . . ,n of an RNA sequence of lengthn. The set of edges connecting the nodes consists oftwo disjoint subsets. One is common to all secondarystructure graphs, while the other is specific to eachsequence. The common set represents the covalentbackbone connecting nodei with node i 1 1, i5 1, . . . ,n 2 1. The sequence-specific part consistsof a set3 of edgesi z j , 3 5 { i z j ui Þ j and j Þ i1 1}, representing admissible hydrogen bonds be-tween the bases at positionsi andj , such that (i) everyedge in3 connects a node to at most one other node,and (ii) the pseudoknot constraint is met. The latterstates that if bothi z j andk z l are in3, then i , k, j implies that i , l , j . Failure to meet thisconstraint results in interactions that are considered tobe tertiary (pseudoknots), or perhaps more to thepoint, computationally and thermodynamically un-wieldy at present. The set of admissible base pairs thatwe shall consider consists of the Watson–Crick pairs{ AU, UA, GC, CG} and {GU, UG}.
The problem of finding the largest possible set3of admissible base pairs compatible with the abovedefinition of a secondary structure is known as “max-imum matching.” Amatchingin an undirected graphG is a set of edges, no two of which have a vertex incommon. Evidently, any set3 of base pairs compliantwith the definition of secondary structure is a match-ing. A matchingM is called a maximum matching, ifno matching contains more edges thanM.
When maximizing base pairing, the basic structuralbuilding block is an individual base pair. This is incontrast to energy minimization, where the buildingblocks to which an energy can be assigned are largerchunks of context known as “loops” (or faces of thesecondary structure graph). It is this property thatmakes maximum matching considerably simpler thanfree energy folding.
The dynamic programming procedure to computethe maximum number of admissible base pairs isstraightforward.1,10 Let Pi , j, i , j , denote the maxi-mum number of base pairs on the sequence segment[ i , j ]. Pi , j can be defined recursively:
Pi, j 5 max$Pi, j21, maxi#l#j22
$~Pi,l21 1 1
1 Pl11,j21!r~al, aj!%%
~1!
whereai { { A, U, G, C} denotes the base at positioni , andr( z , z ) is an indicator function of biophysicallylegal base pairs:
r~ai, aj! 5 H1, if ai andaj can pair;0, otherwise
The recursion in Eq. (1) works by filling theP arrayin such a way that all smaller fragments needed in thecomputation ofPi , j have already been computed. (Forexample, let indexi run from n down to 1, whileindex j sweeps fromi 1 2 to n.) Adding basessequentially at the 39-end, procedure (1) checkswhether a pairing between the added base and someposition downstream improves the total number ofpairs on the segment, as compared to leaving theadded base unpaired. When all is done, the maximumnumber of base pairs isPmax 5 P1,n. A structure withPmax pairs is obtained by tracing back through theParray. Although the backtrack is simple, we shallexplain it in some detail, since it is the key procedurein understanding the Waterman–Byers extension.
Let us define apartial structure6 to be a pair6 5(s; 3) consisting of a stacks of sequence segments{[ i1, i2] z [ i3, i4] z . . .}, and a set3 of base pairs. Acomplete structureis a partial structure whose stack isempty,6 5 (A; 3).
The backtrack starts with the partial structure([1,n]; A), pops the segment from the stack, and follow-ing Eq. (1), checks whether thenth position shallremain unpaired, i.e., whetherP1,n 5 P1,n21. In thatcase, [1,n 2 1] is pushed on the stacks, and theprocedure repeats similarly with6 5 ([1, n 2 1];A). If P1,n Þ P1,n21, the procedure follows thesecond term of Eq. (1), looping overl { [1, n 2 2]
Suboptimal Folding of RNA 147

attempting to find a pairl z n that is consistent withthe value ofP1,n. Such a pair splits the sequence intotwo disjoint substructures on the segments [1,l 2 1]and [l 1 1, n 2 1]. Both segments are pushed on thestack, and the pairl z n is added to3, yielding 6 5([1, l 2 1] z [ l 1 1, n 2 1]; { l z n}). The procedurenow repeats in a similar fashion by popping the nextsegment from the stack. The process bottoms out assegments become too small for holding a base pair.Such segments are popped from the stack withoutcausing other segments to be pushed. Finally, whenthe stack is empty, a complete structure6 5 (A; 3)with Pmax 5 u3u base pairs has been reconstructed.The algorithm is sketched in Table I.
In suboptimal folding we wish to findall structuresthat meet a given suboptimality criterion. In the max-imum matching case the criterion is to haveat leastPmax 2 D base pairs, with 0# D # Pmax. It is herethat the notion of a partial structure becomes useful.65 (s; 3) actually represents asetof structures, all ofwhich have the base pairs3 in common. We shall calla partial structure61 5 (s1; 31) a refinementof apartial structure62 5 (s2; 32), written 61 , 62, if32 # 31 and@ [a, b] { s1 ? [c, d] { s2, such that[a, b] # [c, d], with strict inequality holding at leastonce (otherwise61 5 62).
As before, backtracking consists in the iteratedrefinement of the set of all structures6 5 ([1, n];A). The difference now is that at each stageallrefinements69 are kept that represent sets of (com-plete) structures with at leastPmax 2 D base pairs. Todecide whether an69 qualifies, consider a refinementgenerated from6 5 ([ i , j ] z s; 3) by splittingsegment [i , j ] with a base pairl z j for somel , i # l# j 2 2. This yields the partial structure69 5 ([ i ,l 2 1] z [ l 1 1, j 2 1] z s; 3 ø { l z j }). The
maximum numberP69 of base pairs that any structurein the set represented by69 can have is given by
P69 5 u3u 1 1 1 Pi,l21 1 Pl11,j21 1 O@a,b#{s
Pa,b (2)
where the entries of theP array have been computedin a previous optimization pass. IfP69 is less than therequired minimum,Pmax2 D, the set69 can safely bepruned from further consideration. If, on the otherhand,
P69 $ Pmax 2 D (3)
the partial structure69 is kept for further refinements.Previously we backtracked (1) by refining a par-
ticular partial structure all the way down to a singlecomplete structure. Now, in contrast, we push eachrefinement satisfying the suboptimality criterion (3)on a stackR of partial structures for further iterativerefinement. The algorithm is summarized in Table II.
When choosingD 5 0, the algorithm produces alldegenerate optimal solutions to the maximum match-ing problem. IfD 5 Pmax, the algorithm degeneratesto a systematic construction of all admissible struc-tures on the given sequence. The latter can take a longtime, since the number of structures scales exponen-tially with the sequence length. We performed sanitychecks of our implementation by comparing the num-ber of admissible structures generated by it with out-puts from independent structure counting proceduresbased on the partition function algorithm11,12 and adensity of states algorithm.13
For example, the maximum matching solution forthe E. coli tRNAhis sequence RH1660 has 26 base
Table I Pseudocode for the Backtrack Process in theMaximum Matching Problema
6 5 { s 5 {[1, n]}; 3 5 A}Repeat:If (s 5 A)
Terminate with6 5 { A; 3}[a, b] d sIf ( Pa,b 5 Pa,b21)
[a, b 2 1] f s and repeatIf ( l z b with l { [a, b 2 2] andPa,b
5 Pa,l21 1 Pl11,b21 1 1)3 4 3 ø { l z b}, [ a, l 2 1] f s,
[ l 1 1, b 2 1] f s and repeat
a Thexd s denotes the popping of the first element from thestacks. That element is assigned tox, and is deleted froms. Thex f s denotes the pushing ofx on the stacks.
148 Wuchty et al.

pairs (we require that a hairpin turn must have at least3 unpaired positions). ChoosingD 5 0 we find149,126 structures, all having the maximum of 26base pairs. Two instances are shown in Figure 2. To
find all “ground states” required 125 s CPU time on aSUN Ultra 2 (256 Mb memory) with our prototypeimplementation not tuned for efficiency. There are9,889,659 solutions with 25 or more base pairs (2.2h), and 318,369,772 structures with 24 or more basepairs (68.6 h).
SUBOPTIMAL FREE ENERGY FOLDING
From the thermodynamic point of view, the buildingblocks of secondary structures are loops (Figure 3)—stacked base pairs, internal loops, bulges, and mul-tiloops (i.e., structural elements delimited by morethan one base pair)—rather than individual base pairs.As a consequence, the minimum free energy foldingalgorithm requires a number of distinct arrays (seebelow). This complicates the backtrack procedure.
Like in the previous section, the suboptimal freeenergy backtrack must refine partial structures byexactly reversing the optimization procedure used tosystematically generate structures from smaller seg-ments. If we were not to proceed in this way, theenergy arrays filled during the optimization pass couldnot be used in the pruning criterion. A problem arises,however, when reversal of the optimization procedureyields more than one way of generating thesamestructure. It is in particular the construction of mul-tiloops that needs attention in this regard. Reversingthe usual Zuker–Stiegler procedure4 yields vastamounts of structure repetitions (with the same en-ergy) due to the nonuniqueness of their multiloop
Table II Pseudocode for Generating SuboptimalSolutions to the Maximum Matching Problema
6 5 { s 5 {[1, n]}; 3 5 A}, R 5 A
6 3 RRepeat:if ( R 5 A)
Done(s; 3) d RIf (s 5 A)
Output suboptimal solution (A; 3) and repeat[a, b] d sLet 69 5 ([a, b 2 1] z s; 3)If ( P69 $ Pmax 2 D)
6 6 f RFor eachl z b with l { [a, b 2 2]{
Let 69 5 ([a, l 2 1] z [ l 1 1, b 2 1] z s;3 ø { l z b})
If ( P69 $ Pmax 2 D)69 f R
}If (nothing has been pushed onR since the last repeat)
(s; 3): f RRepeat
a The meaning ofd andf is explained in Table I. Recall that[a, b] d s deletes [a, b] from s. It is understood that in the caseof illegal intervals ([a, b] with a . b), all statements referring tothat interval are skipped.
FIGURE 2 Two solutions maximizing base pairing inE. coli tRNAhis (RH1660). Certainmodified bases in the sequences were replaced by a nonbonding nucleotide N, see Appendix C.
Suboptimal Folding of RNA 149

decomposition. This is obviously irrelevant whentracing back for the optimal structure, but of little usein the systematic generation of suboptimal structures.Our solution to this consists in modifying the Zuker–Stiegler procedure by decomposing multiloops in aunique way (see Appendix A).
A further problem arises from energy contributionsdue to so-called dangling ends. Unpaired bases adja-cent to a helix may lower the energy of a structure bystacking onto their neighboring base pairs. These con-tributions are taken into account for external bases (abase not enclosed in any loop, see Figure 1) adjacentto the 59- and 39-end of a helical region. The sameholds for unpaired nucleotides inside a multiloop ad-jacent to helical regions (Figure 3). Normally, a basemay not simultaneously participate in both interac-tions a 59 dangling end with one helixand a 39dangling end with another. The problem for the sub-optimal backtrack is that, when decomposing a mul-tiloop, we do not know yet whether a base adjacent toa helix is available for a dangling end interaction, thatis, whether that base is unpaired and not alreadyinvolved in another dangling end interaction. We han-dle this situation simply by always adding a danglingend contribution without checking whether the baseinvolved qualifies. This leads to additional dangling
end contributions for helices directly adjacent to oneanother. Incidentally, such helices often do engage instabilizing interactions through coaxial stacking.14
Our treatment of dangling ends thus can actuallyimprove predictions in some cases. Alternatively, thecontributions from dangling ends can be switched offaltogether. They can, however, be substantial. Com-pilations of energy parameters used in our implemen-tation are given in Refs. 15–18.
With the algorithm described in Appendix A theWaterman–Byers scheme can be used to find all sub-optimal structures within a given energy range abovethe minimum free energyEmin. Exactly like in themaximum matching case, we proceed by refining par-tial structures, and checking whether the refinementssurvive a pruning criterion analogous to Eqs. (2 and3). The technical definition of a partial structure needstwo slight amendments. First, in addition to the set ofbase pairs3 and the stack of segmentss, we keeptrack of the total free energyEL6
, of all loopsL6 thatconstitute a given partial structure6. Second, eachsegment on the stacks requires an additional label,indicating in which one of the arraysF5, C, FM, andFM1 (see Appendix A) the best energy attainable onthat segment should be looked up. These labels arealso needed in switching the backtrack between theappropriate arrays. The labels are assigned accordingto how a segment is generated through refinementfrom another segment (see Appendix B).
Let us denote the best possible energy attainable onsegment [i , j ]E with Ei , j, whereE { { F5, C, FM,FM1}. Suppose now that we are refining a partialstructure6 5 ([ i , j ]E z s; 3; EL6
) by reversing theminimization procedure outlined in Table IV of Ap-pendix A. Depending on the equation used, this willyield subintervals of [i , j ]E whose total best possibleenergy we denote here withh to avoid distractingdetails. All (complete) structures represented by theattempted refinement of6 have an energy not lowerthanE6, where
E6 5 h 1 EL61 O
@k,l #E{s
Ek,l (4)
In analogy to Eq. (3), we will accept any refinementfor which
E6 # Emin 1 d (5)
with some desiredd . 0. The strategy for tracingback all suboptimal structures in the energy rangebetweenEmin andEmin 1 d is detailed in Appendix B.
FIGURE 3 Secondary structure elements.
150 Wuchty et al.
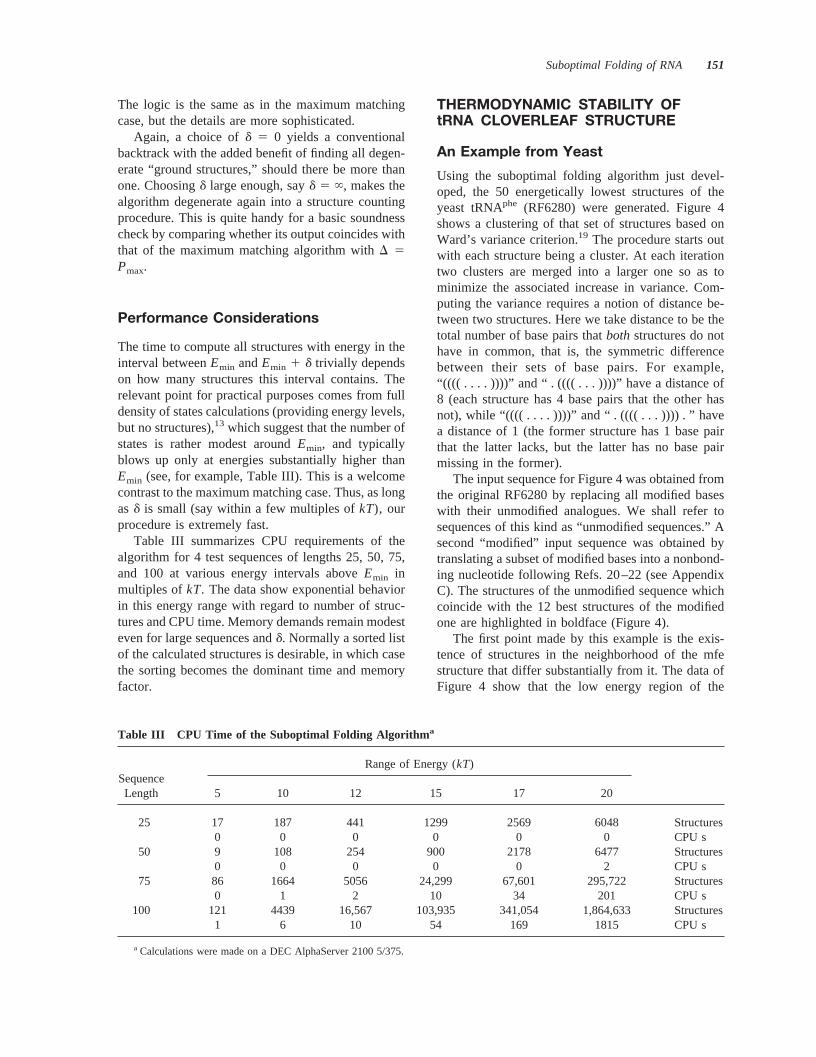
The logic is the same as in the maximum matchingcase, but the details are more sophisticated.
Again, a choice ofd 5 0 yields a conventionalbacktrack with the added benefit of finding all degen-erate “ground structures,” should there be more thanone. Choosingd large enough, sayd 5 `, makes thealgorithm degenerate again into a structure countingprocedure. This is quite handy for a basic soundnesscheck by comparing whether its output coincides withthat of the maximum matching algorithm withD 5Pmax.
Performance Considerations
The time to compute all structures with energy in theinterval betweenEmin andEmin 1 d trivially dependson how many structures this interval contains. Therelevant point for practical purposes comes from fulldensity of states calculations (providing energy levels,but no structures),13 which suggest that the number ofstates is rather modest aroundEmin, and typicallyblows up only at energies substantially higher thanEmin (see, for example, Table III). This is a welcomecontrast to the maximum matching case. Thus, as longasd is small (say within a few multiples ofkT), ourprocedure is extremely fast.
Table III summarizes CPU requirements of thealgorithm for 4 test sequences of lengths 25, 50, 75,and 100 at various energy intervals aboveEmin inmultiples ofkT. The data show exponential behaviorin this energy range with regard to number of struc-tures and CPU time. Memory demands remain modesteven for large sequences andd. Normally a sorted listof the calculated structures is desirable, in which casethe sorting becomes the dominant time and memoryfactor.
THERMODYNAMIC STABILITY OFtRNA CLOVERLEAF STRUCTURE
An Example from Yeast
Using the suboptimal folding algorithm just devel-oped, the 50 energetically lowest structures of theyeast tRNAphe (RF6280) were generated. Figure 4shows a clustering of that set of structures based onWard’s variance criterion.19 The procedure starts outwith each structure being a cluster. At each iterationtwo clusters are merged into a larger one so as tominimize the associated increase in variance. Com-puting the variance requires a notion of distance be-tween two structures. Here we take distance to be thetotal number of base pairs thatboth structures do nothave in common, that is, the symmetric differencebetween their sets of base pairs. For example,“((( ( . . . . ))))” and “ . (((( . . . ))))” have a distance of8 (each structure has 4 base pairs that the other hasnot), while “(((( . . . . ))))” and “ . (((( . . . )))) . ” havea distance of 1 (the former structure has 1 base pairthat the latter lacks, but the latter has no base pairmissing in the former).
The input sequence for Figure 4 was obtained fromthe original RF6280 by replacing all modified baseswith their unmodified analogues. We shall refer tosequences of this kind as “unmodified sequences.” Asecond “modified” input sequence was obtained bytranslating a subset of modified bases into a nonbond-ing nucleotide following Refs. 20–22 (see AppendixC). The structures of the unmodified sequence whichcoincide with the 12 best structures of the modifiedone are highlighted in boldface (Figure 4).
The first point made by this example is the exis-tence of structures in the neighborhood of the mfestructure that differ substantially from it. The data ofFigure 4 show that the low energy region of the
Table III CPU Time of the Suboptimal Folding Algorithm a
SequenceLength
Range of Energy (kT)
5 10 12 15 17 20
25 17 187 441 1299 2569 6048 Structures0 0 0 0 0 0 CPU s
50 9 108 254 900 2178 6477 Structures0 0 0 0 0 2 CPU s
75 86 1664 5056 24,299 67,601 295,722 Structures0 1 2 10 34 201 CPU s
100 121 4439 16,567 103,935 341,054 1,864,633 Structures1 6 10 54 169 1815 CPU s
a Calculations were made on a DEC AlphaServer 2100 5/375.
Suboptimal Folding of RNA 151

unmodified tRNAphe comprises at least two majorclasses of structures. In particular, the mfe structure(219.26 kcal/mol) is in one class, while a structure asclose to it as no. 3 (218.83 kcal/mol) belongs to adifferent class. These classes are split into furtherclusters, and Figure 5 gives an indication of theirstructural diversity. This is a static picture, and noth-ing is said about the barrier between no. 1 and no. 3.By systematically generating the complete configura-tion space around the mfe, our procedure can assist inobtaining either the barrier itself or a lower bound to
it. However, we shall not be concerned with kineticsin this communication.
Both modified and unmodified sequences fold intothe same mfe structure, and the 12 structures withlowest energy of the modified sequence are among the50 lowest structures of the unmodified variant. How-ever, all 12 structures of the modified sequence groupinto the same cluster. This raises the issue about theeffect of tRNA base modification on the density anddiversity of states around the mfe. In cases where theunmodified sequence folds into the correct cloverleaf
FIGURE 4 Similarity clustering of suboptimal structures of yeast tRNAphe. The 50 energeticallylowest structures, ranging from219.26 kcal/mol to217.28 kcal/mol, of yeast tRNAphe (EMBLaccession RF6280) are clustered using Ward’s variance method19 with the symmetric differencedistance as a metric on the structures. Numbers at the leaves of the tree indicate the energy rank ofa suboptimal structure (mfe structure is no. 1). The structures belong to the sequence obtained fromRF6280 by translating modified bases into their corresponding unmodified analogues. Numbers inboldface flag structures that coincide with the 12 lowest suboptimal structures of the sequenceobtained from RF6280 by replacing certain modified bases by a nonbonding nucleotide. Arrowsindicate structures satisfying the definition of Zuker’s suboptimal folding scheme.6
152 Wuchty et al.

structure, modifications that prevent base pairing donot alter the mfe structure. They seem, however, toconstrain structures at low energies to be similar to theground state. This suggests that base modificationimproves the “definition” of the mfe structure. Weshall return to this point in greater detail.
Figure 4 also indicates that Zuker’s subset of sub-optimal structures6 (those that are optimal with re-spect to the choice of a base pair) does indeed con-stitute a representative sample of the structural vari-ability in the vicinity of the mfe (arrows in Figure 4).
Diversity of States in Modified andUnmodified Artificial tRNAs
The previous example suggests a role for modifiedbases in altering the structural states in the vicinity ofthe mfe. Yet, conclusions that rest on the details (inparticular the ordering) of these states for a singlesequence remain susceptible to the same imprecisionsin the available energy parameters as pure mfe fold-ing. One way around this problem is to turn awayfrom the structure prediction and analysis of a singlesequence to a statistical approach23 in which we iden-tify and compare robust properties of specific (naturalor artificial) sequence ensembles. This approach canbe expected to yield conclusions that are robust tovariations in the energy parameters.
FIGURE 6 tRNA secondary structure shared by six sequences fromE. coli. The figure shows thesecondary structure (obtained with the thermodynamic folding algorithm) shared by the six listedtRNA sequences. (The second letter in the accession number identifies the amino acid.) Thestructure has the tRNAasp sequence (RD1660) superimposed, indicating the positions at which anonbonding baseN was placed. The shaded areas in the list of sequences indicate the positions atwhich Ns were placed in inverse folded sequences of the modified sample.
FIGURE 5 Structural diversity near the mfe of yeasttRNAphe. The structures shown correspond to the clusternucleators no. 1, no. 3, no. 11, no. 12, and no. 13 (from leftto right, top to bottom) in Figure 4.
Suboptimal Folding of RNA 153

Using an inverse folding procedure,24,25we gener-ated a pool of 2000 sequences whose mfe structurecoincides with that of the six natural sequences listedin Figure 6. This constitutes an ensemble of unmod-ified sequences, or “unmodified sample” for short.Similarly, we generated a pool of 2000 sequenceswith a nonbonding nucleotide at every position indi-cated in Figure 6. These sequences were chosen so asto have the same mfe structure as the unmodifiedpool. We shall refer to this ensemble as the “modifiedsample.”
For each sequence in both samples we computedthe energy gap between the mfe structure and thesecond best structure. The distribution of these firstgap energies is shown in Figure 7. An immediateobservation is that natural tRNA sequences have largefirst gap energies, located far out in the tail of thedistribution. A more subtle feature, however, is thatthe modified sample exhibits a set of spikes risingdistinctively above a generally flatter background ascompared to the unmodified sample. An analysis ofthe structures associated with the gap energies at thesespikes reveals that the extent to which all major spikesrise above the background is due precisely to thosestructures resulting from the ground state by removingone base pair at either end of a helical region. For
example, 74, 70, 80, 92, and 88% of the structures atgap energies 0.09, 0.17, 0.29, 0.69, and 0.97 kcal/molabove the mfe, respectively, lack either one base pairat the acceptor end of the multiloop or at the loop endof one of the hairpin turns. This “quantized” super-structure in the gap distribution of the modified sam-ple shows that nonbonding bases constrain the secondbest structure to be as similar as possible to the groundstate. This is in marked contrast to the unmodifiedsample, where larger refolds at the first energy levelare considerably more likely. Consider that the energydifference between the ground structure and the sec-ond best structure cannot be larger than the largeststacking energy (2DGGC z CG, which is about 3 kcal/mol at 37°C). To see this, assume thatS is the secondbest structure. IfS were to differ more than the statedamount from the mfe structure, we could construct abetter structure by simply removing a base pair ateither end of some stack in the mfe structure, thuscontradicting the assumption thatS is the second beststructure. Hence, the next structure above the groundstate will be a similar structure with just a base pairremoved from a helix end only if there exists norefolded configuration with a lower energy. Figure 7shows that properly placed nonbonding bases makethe latter possibility distinctively less likely.
FIGURE 7 Distribution of first gap energies. The upper and lower half show the distribution ofgap energies in the unmodified and modified sample, respectively. The dotted vertical lines indicatethe gap energies of the six natural sequences in Figure 6.
154 Wuchty et al.

Structural Stability of SecondaryStructures
In view of the previous analysis we ask whether thedensity of states at low energies and their associatedstructures can be used to quantify the degree to whichan mfe structure is “well defined.” Intuitively, andfrom a static viewpoint, a structure is well defined ifthere are no “substantially different” structures in itsthermodynamic neighborhood. Even in the absence ofa kinetic assessment, criteria of static well definitioncan be useful in identifying parts of an RNA structurewith biological significance.
Extant measures of well definition use McCaskill’spartition function algorithm.11 For example, one mayquantify the most likely state—paired or un-paired—of a positionk in a sequence by the proba-bility of the most probable base pair involvingk,or the probability thatk is unpaired, whichever islarger.26 These base pair probabilities are obtainedfrom the partition functionZ,11 which can also beapproximated with the suboptimal folding procedureby summing over the density of states at low ener-gies.27
At the other extreme one might consider a simpleglobal measure as given by the fraction of the mfestructure in the Boltzmann ensemble:
fmfe 5e2DGmfe/kT
Z5
1
1 1 Oi e2Dgi/kT (6)
whereDgi is the i th gap energyDGi 2 DGmfe. Thiscan also be expressed askT ln fmfe 5 F 2 DGmfe,whereF is the free energy of the Boltzmann ensem-ble. Another such measure is the mean gap energy^Dg&:
^Dg& 5 Oi
~DGi 2 DGmfe!e2DGi/kT
Z(7)
Figure 8 shows the distribution offmfe in the modifiedand unmodified samples, together with the values forthe natural sequences of Figure 6. Again, the lattershow a remarkably highfmfe as compared to bothsamples with the same mfe structure. The comparisonbetween the two samples evidences the role of mod-ified bases in shiftingfmfe to higher values. The badnews, however, is that a highfmfe does not imply alarge separation to the energetically adjacent struc-ture, although the reverse is true (Figure 9).
Neither the quantified pairing state of an individualposition norfmfe provide sufficient information aboutstructuralstability. The former is too local a measureto say much about structural diversity in the vicinityof the mfe, and a lowfmfe can be caused by a numberof similar structures that are energetically nearby themfe. Yet, in the latter case, we would still consider thebasic architecture of the mfe to be well defined.
A simple measure for the structural diversitypresent in the secondary structure configuration space
FIGURE 8 Fraction of mfe structure in the Boltzmann ensemble. The upper and lower half showthe distribution offmfe in the unmodified and modified sample, respectively. The dotted vertical linesindicate thefmfe of the six natural sequences of Figure 6.
Suboptimal Folding of RNA 155

of a sequence is the Boltzmann weighted sum over thestructure distances between thei th configuration andthe ground state. As a structure distance we use theso-called base pair distance, defined as follows: eachposition in structure A that is not paired to the sameposition as in structure B increases the distance by onecount. In this metric one-strand shifts of helical re-gions give large distances. For example, “(((( . . . . ))))”and “ . (((( . . . .))))” have a base pair distance of 9 (all8 paired positions differ), while “(((( . . . . ))))” and“ . (((( . . . )))) . ” have a base pair distance of 2. (Basepair distance is similar but not identical to the sym-metric difference distance used earlier in this section.)
^dbp& 5 Oi
dbp~0, i !e2DGi/kT
Z(8)
where dbp(0, i) denotes the base pair distance be-tween the mfe structure (0) and theith structureabove it.
In Figure 10 we plot for three classes of tRNAsequences the mean gap energy against the mean basepair distance, which we approximated by consideringall structures within 10kT of the mfe. The threeclasses were derived from Steegborn’s compilation28
of E. coli tRNA sequences. The first class (solidcircles in Figure 10) consisted of the natural tRNA
sequences whose modified bases were replaced bynonbonding bases in their original positions (accord-ing to the translations of Appendix C), the secondclass (open circles) had the same amount of nonbond-ing bases, this time in random positions, but so as toyield the same mfe structure as the originals. The thirdclass (crosses) had the modified bases replaced bytheir corresponding unmodified ones. Not all of thelatter had the same mfe structure as their native (i.e.,modified) counterpart. (The algorithm missed the clo-verleaf also for a few modified sequences.) In suchcases we took the lowest lying cloverleaf structure asthe reference (0). As a consequence, the mean gapenergy can become negative. For better readability ofthe plot, we assigned the mean base pair distance thesame sign as the mean gap energy (but it obviouslymeans a positive value).
Figure 10 shows that the natural modified se-quences (solid circles) have by and large very small^dbp& values indicating that most structures close tothe mfe structure are also similar to it. At the sametime the mean gap energy has a wide spread. Thisshows that^dbp& is a better predictor of structuralstability than^Dg&. The same trend is confirmed byplots similar to Figure 10 for the modified and un-modified samples (not shown). Furthermore, se-quences with nonbonding bases at random positions
FIGURE 9 Relation betweenfmfe and the first gap energy. The figure shows a scatter plot wherethe first gap energy of each sequence in the modified sample is plotted against itsfmfe. Recall thatan upper bound forDg1 is about 3 kcal/mol. The figure shows that a highDg1 implies a high mfefraction in the Boltzmann ensemble, but that the reverse is not true. A similar picture holds for theunmodified ensemble.
156 Wuchty et al.

(open circles) have a better defined mfe structure thanunmodified sequences, but not as well defined as theoriginals. Thus, the positioning of the nonbondingbases is important, even when it does not affect themfe structure itself. It is tempting to interpret thesedata as natural tRNA sequences having their non-bonding bases positioned so as to also maximize thedefinition of the ground state structure.
A further assessment of structural well definition isobtained by counting the number of different structure“architectures” as energy increases from the groundstate. By “architecture” we mean a coarse-grainedsecondary structure obtained by disregarding the sizeof loops and helices.29 Such a coarse-grained structureconstitutes an equivalence class of conventional sec-ondary structures with respect to the topological ar-rangement of loops and helices. The upper plot ofFigure 11 shows the density of states at low energies,that is, the number of states existing at any givenenergy up to 15kT from the ground state for theunmodifiedE. coli tRNAlys sequence (RK1660), andup to 30kT for the modified sequence. The lower halfof the plot displays the cumulative count of differentcoarse grained structures encountered since the mfestructure. The difference in the rates by which struc-tural diversity increases is quite impressive, as is thedifference in energy from the ground state at whichdiversity starts rising fast. This indicates once morethat, from a thermodynamic point of view, the mod-
ified sequence is structurally much more stable. Wefound similar observations to hold for the otherE. colitRNA sequences as well.
Finally, Figure 12 shows an intriguing relationshipbetween the thermodynamic stability of a structureand the fraction of neutral mutants accessible by asingle point mutation. Neutral here means that refer-ence sequence and the mutant have the same mfestructure. Each part of Figure 12 plots for each se-quence in the modified and unmodified sample thelogarithms offmfe, ^dbp&, and ^Dg& against the frac-tion of neutral mutants of that sequence. For bothsamples there is a clear correlation between welldefinition of the ground state and the degree to whicha sequence can buffer mutations against altering thatground state. Average neutrality is higher for modi-fied sequences, since their mfe structure is on averagethermodynamically better defined than for unmodifiedsequences. The best predictor of mutational stabilityis again the mean base pair distance, while the meangap energy is virtually insensitive. Small mean basepair distance (high thermodynamic structural stabil-ity) implies high neutrality, but the reverse, while trueto some degree for unmodified sequences, does nothold for modified ones. Given that properly modifiedsequences have intrinsically a better defined groundstate, a high degree of neutrality does no longer dis-criminate between different degrees of well definitionwithin that sample.
FIGURE 10 Structural diversity vs mean gap energy. The figure plots a 10kT approximation tothe mean structure distance, as defined in Eq. (8), and the mean gap energy, as defined in Eq. (7),for E. coli tRNA sequences from the Steegborn compilation.28 Solid circles: natural modifiedsequences. Open circles: sequences with nonbonding nucleotides at random positions but preservingthe mfe structure of their natural modified counterparts. Crosses: unmodified sequences.
Suboptimal Folding of RNA 157

CONCLUDING REMARKS
Following an idea of Waterman and Byers,9 we havedevised and implemented an algorithm that rigorouslygenerates all energetically suboptimal secondarystructures of an RNA sequence within a desired en-ergy range above the minimum free energy. The logicof the algorithm was discussed for the simple case ofbase pair maximization. To implement a suboptimalfolding procedure based on the free energy of struc-tures, we had to modify the Zuker–Stiegler strategyfor free energy minimization. Minimization and sub-optimal backtrack are detailed in the appendices.
Depending on the choice of energy range, thealgorithm has two limiting behaviors. If the intervalabove the minimum free energy is set to zero, alldegenerate ground states are obtained, while a suffi-ciently high energy range yields a systematic structurecounting procedure. Since the density of states isrelatively sparse in the10–15kT vicinity of the min-imum free energy, the algorithm is fast and practicaleven for long sequences. Our implementation and allalgorithms used in this paper are freely available foracademic research,30 and will be integrated in the nextrelease of the Vienna RNA Package.24
A suboptimal folding algorithm that generates rig-orously all suboptimal configurations between theminimum free energy and some chosen upper limit is
important for a meaningful approximation of statisti-cal quantities. Because of this property our algorithmhas the pleasant feature that energy minimization,suboptimal structures, the (truncated) density ofstates, the (truncated) partition function (and otherstatistical quantities derived from it) are unified in asingle procedure and obtainable in the same optimi-zation plus backtracking pass.
In the second part of this contribution we used ourprocedure to compute indicators for the thermody-namic stability of the minimum free energy structureof an RNA sequence. We defined three simple indi-cators capturing in different ways the degree of welldefinition or well determination of the ground statestructure: (a) mean gap energy, that is, the averageenergy separation of configurations in the vicinity ofthe minimum free energy; (b) Boltzmann weightedmean structure distance (here implemented as meanbase pair distance), that is, the average distance be-tween the minimum free energy structure and theconfigurations in its energy neighborhood; and (c)topological diversity, that is, the number of differentcoarse-grained structures in an energy interval aroundthe ground state. These quantities were used to assessthe influence of base modification on the thermody-namic robustness of the ground state structure intRNA sequences. To this end we compared the sta-tistics of these indicators in large samples of modified
FIGURE 11 Diversity of coarse grained structures. The upper half shows the density of states forthe unmodified sample (up to 15kT) on the left, and for the modified sample (up to 30kT) on theright. The lower half is a plot of the cumulative number of different coarse grained architecturesencountered with increasing energy; left curve: unmodified sample, right curve: modified sample.
158 Wuchty et al.

and unmodified artificial sequences whose minimumfree energy structure is identical to that of naturallyoccurring tRNA sequences fromE. coli. The latterwere also studied individually. Base modification wasconsidered here only in its quality of preventing par-ticular positions in the linear sequence from contrib-uting base pairs to the secondary structure.
Our study shows from several perspectives thatbase modification considerably sharpens the definitionof the ground state structure by constraining energet-ically adjacent structures to be similar to the groundstate. Base pair distance turned out to be the bestindicator for how well the ground state is determined.Artificial sequences with nonbonding nucleotides atrandom positions, yet with the natural tRNA clover-leaf pattern as ground state, determine the cloverleafbetter than unmodified sequences, but not as well asnatural sequences with the same secondary structure.
This indicates that certain positions when locked intoa nonbonding state are more effective than others insharpening the thermodynamic definition of the min-imum free energy structure.
There is a noteworthy correlation between the ther-modynamic stability of the minimum free energystructure of a given sequence and its capacity to buffermutations. The better the ground state is defined, themore one-error mutants preserve the minimum freeenergy structure. This may have evolutionary conse-quences at the molecular level. If well definition of asecondary structure is important for biological func-tion, then evolving a sequence that improves the ther-modynamic definition of that structure has as a likelyside effect an increased stability toward point muta-tions–that is, neutrality.
The importance of a rigorous suboptimal foldingalgorithm rests not only with computing criteria for
FIGURE 12 Relationship between thermodynamic stability and neutrality. Each graph (a), (b),and (c) plots one measure of thermodynamic well definition of the ground state vs the fraction ofneutral mutants accessible by one point mutation for each sequence in the unmodified sample (upperplot in each part) and the modified sample (lower plot in each part).
Suboptimal Folding of RNA 159

discerning biologically relevant structures held underselection pressure, or for detecting relevant alternativestates to the ground state. A key issue will be tounravel the kinetic aspects of RNA folding, and tounderstand what makes a sequence fold well. Byproviding access to the complete configuration spaceat low energies, we expect a rigorous suboptimalfolding algorithm to be a valuable tool towards thatgoal.
APPENDICES
A. Optimization with Unique MultiloopDecomposition
In this appendix we explain the modified optimizationprocedure on which we base the suboptimal backtrackdetailed in Appendix B.
We recall here for later reference the usual treat-ment for multiloop energies},5
} 5 }C 1 }I z no. interior pairs
1 }B z no. unpaired bases(9)
where }C denotes the stabilizing energy derivingfrom the multiloop closing pair,}I denotes the sta-bilizing energy for each base pair interior to the mul-tiloop, and}B the destabilizing energy for each un-paired base in the loop.17
As in the maximum matching case, energy arraysare filled in a recursive fashion. LetCi , j be the min-imum free energy on the segment [i , j ], provided thati and j pair with one another. As is well known,5 byvirtue of the additivity of loop energies, the bestenergy attainable on the segment [i , j ], with i z j , isgiven by the energy of the particular loopL closed offby i z j plus the energy of any substructures endingwith a base pairp z q in that loop.
Ci, j 5 minloopsL
closed byi zj
$E~L! 1 Ointerior pairs
pzq{L
Cp,q% (10)
FIGURE 12 (Continued from the previous page.)
160 Wuchty et al.

with Ci ,i 5 `. The minimization in Eq. (10) runs overthree major classes of structures, consisting of variousloop types closed off byi z j (see Figure 13 for aschematic representation).
Ci,j 5 min$
(11)*~i, j!,
minp{@i11, j2m22#q{@p1m11, j21#
$Cp,q 1 (~i, j, p, q!%
mink{@i11,j2m22#
$Fi11,k21M 1 Fk, j21
M1 1 di, j, j215 1 di, j,i11
3 1 }C%
}
The first term,*(i, j), denotes the tabulated free energyof a hairpin loop closed byi z j. The second termconsiders all cases wherei z j closes an interior loop (or
a bulge) whose interior delimiting base pair isp z q. Theloop has a tabulated energy((i, j, p, q), the structure“behind”p z qhas energyCp,q, and the minimum is takenover all admissible pairsp z q. The third term refers tomultiloop structures closed byi z j. A multiloop isconstructed from two pieces with energyFi11,k21
M andFk,j21
M1 (to be explained shortly; see also Figure 13), andthe multiloop closing pairi z j with energy}C [see Eq.(9)]. We also take into account the stabilizing energyfrom dangling ends on the 59- and the 39-side of the pairi z j. Thedi,j,j21
5 denotes the energy contribution of thebase at positionj 2 1 stacking from the 59 direction ontothe pairi z j. Similarly for d3.
As indicated in Figure 13(c),Fi , jM1 denotes the
minimum free energy of the last stem (closed, say, byi z l ) toward the 39-end of the multiloop being con-sidered, including an arbitrary number of unpairedbases at the 39-end. Its energy is the sum of the en-ergy Ci ,l of the structure closed byi z l , the energy}B( j 2 l ) of j 2 l unpaired multiloop bases, the
FIGURE 12 (Continued from the previous page.)
Suboptimal Folding of RNA 161

multiloop energy contribution}I deriving from theinterior pair i z l , and the dangling ends:
Fi, jM1 5 min
l{@i1m11, j#
$Ci,l 1 }B~ j 2 l !
1 di,l,i215 1 di,l,l11
3 1 }I%(12)
The remaining 59 piece of the multiloop structure isfurther split recursively into a 39-stem plus a remain-ing 59 section. The recursion bottoms out when no39-stem is possible. In terms of energies:
Fi, jM 5 min$ min
k{@i1m11, j2m21#
$Fi,k21M 1 Fk, j
M1%, (13)
mink{@i, j2m21#
$Fk, jM1 1 }@~k 2 i !%% (14)
This procedure ensures that there is only one decom-position of a multiloop into substructures, thus en-abling a meaningful suboptimal backtrack.
Finally, all we need is the best free energy on asegment [1, j ], denoted by Fj
5, irrespective ofwhether positionj is paired.Fj
5 is constructed recur-sively, as illustrated in Figure 14,
Fj5 5 min$Fj21
5 , minl{@i, j2m21#
$Fl215 1 Cl, j
1 dl, j,l215 1 dl, j, j11
3 %%(15)
The first term represents the case wherej is leftunpaired. The second term considers all possible po-sitions l that might be paired toj . The free energyEmin of the best structure on the entire sequence isthen given byEmin 5 Fn
5.
FIGURE 13 Schematic representation of the terms in Eq. (11). First term (a): base pairi z j closesa hairpin loop of a certain size. The minimal loop size is 3. Second term (b): base pairi z j closesan interior loop, whose inner base pair isp z q. All possible pairsp z q must be considered. Thirdterm (c): base pairi z j closes a multiloop with a certain number of interior base pairs (solid circles,such ask z l ). Multiloops are divided recursively into substructures, containing the last stem at the39-end (energyFk, j21
M1 ) and the remaining 59 structure (energyFi1l ,k21M ). The remaining structure
is again split in the same way, see Eq. (13). Dangling end contributions are not shown.
FIGURE 14 Schematic representation of Eq. (15). The left scheme and right scheme represent thefirst and second term, respectively, in the minimization of Eq. (15). Dangling end contributions arenot indicated.
162 Wuchty et al.

Table IV summarizes the algorithm for computingthe minimum free energy on a given RNA sequence.It has complexityO(n3), and its implementation isvery fast. A structure corresponding to the minimumfree energy is again obtained by backtracking throughthe various arrays.
In Appendix B we detail the trace back yielding allsuboptimal structures with energies betweenEmin andEmin 1 d, with d . 0 chosen by the user.
B. Suboptimal Backtrack
We label segments [i , j ] with subscriptsF, C, M, andM1, referring to the arraysF5, C, FM, and FM1,respectively. As usual, the backtrack starts with6 5([1, n]F; A; 0). We outline the procedure involved inrefining the partial structure6 5 ([ i , j ]E z s; 3; EL6
)which has just been popped from the partial structurestack R. The segment [i , j ]E is popped from thepartial structure’s segment stack, and refined accord-ing to the markerE.
‹ CaseE 5 F ~backtrack inF5!
The i and j are external bases, and the possible re-finements follow Eq. (15). Leaving the 39 end un-paired, leads to the acceptance condition
Fj215 1 EL6
1 O@k,l #{s
Ek,l # Emin 1 d (16)
If (16) is fulfilled, we push the new partial structure69 5 ([ i , j 2 1]F z s; 3; EL6
) on the stackR forlater refinement.
Next we scan for all possible outermost pairsl z j .If for a particularl z j the criterion
Fl215 1 Cl, j 1 dl, j,l21
5 1 dl, j, j113 1 EL6
1 O@k,l #{s
Ek,l # Emin 1 d(17)
is fulfilled, we push the refinement69 5 ([ l , j ]C z [ i ,l 2 1]F z s; 3; EL6
1 dl , j ,l215 1 dl , j , j11
3 ) on thestack R. Note that we do not to add the base pairl z j here, but we shall do so when refining the interval[ l , j ]C closed by it.
‹ CaseE 5 C ~backtrack inC!
Positioni pairs with j in the popped segment [i , j ]C.We first take it to be a hairpin. If
*~i , j ! 1 EL61 O
@k,l #{s
Ek,l # Emin 1 d (18)
we obtain a refinement of6, 69 5 (s; 3 ø { i z j };EL6
1 *(i , j )), which is pushed on the structurestackR. Next, we construct stacks, interior loops, andbulges by scanning for all admissible pairsp z q, andchecking the condition
Ci, j 1 (~i, j, p, q! 1 EL61 O
@k,l #{s
Ek,l # Emin 1 d (19)
Each time inequality (19) is fulfilled, we obtain arefinement of6, 69 5 ([ p, q]C z s; 3 ø { i z j ,p z q}; EL6
1 ((i , j , p, q)), which is stacked onR.We proceed to construct multiloops in correspon-dence with the third term of Eq. (11). To this end weloop overk, monitoring condition
Fi11,kM 1 Fk11, j21
M1 1 di, j,i115 1 di, j, j21
3 1 }C 1 EL6
1 O@k,l #{s
Ek,l # Emin 1 d(20)
yielding more 6 refinements,69 5 ([k 1 1,j 2 1]M1 z [ i 1 1, k]M z s; 3 ø { i z j }; EL6
1di , j ,i11
5 1 di , j , j213 1 }C), to be pushed on the partial
structure stack.
‹ CaseE 5 M1 ~multiloop backtrack inFM1!
Table IV Recursive Calculation on the MinimumFree Energya
Ci,j 5 min$*~i, j!, mink{@i11, j2m22#l{@k1m11, j21#
$Fk,lB 1 (~i,j,k,l!%, min
k{@i11, j2m22#
$Fi 1 1,k 2 1M 1 Fk, j 2 1
M1 1 }C%%
Fi, jM1 5 min
l{@i1m11, j#
$Ci,l 1 di,l,i 2 15 1 di,l,l 1 1
3 1 }B~j 2 1! 1 }(%
Fi, jM 5 min$ min
k{@i1m11, j2m21#
$Fi,k 2 1M 1 Fk, j
M1%,
mink{@i , j2m21#
$Fk, jM1 1 }B~k 2 i!%%
Fj5 5 min
l{@1, j2m21#
$Fj 2 15 ,Fl 2 1
5 1 Cl, j 1 dl, j,l 2 15 1 dl, j, j 1 1
3 %
a Calligraphic symbols denote tabulated energy parameters fordifferent loop types. Hairpin loops:*(i , j ); interior loops; bulges,and stacks:((i , j , k, l ); the multiloop energy is modeled by thelinear ansatz of Eq. (9). The particular recursion on the multilooparraysFM and FM1 yields a unique decomposition. The overallcalculation proceeds from smaller segments to larger ones. Theminimum free energy on the segment [1,j ] is stored inFj
5. Uponcompletion the minimum free energy is inFn
5.
Suboptimal Folding of RNA 163

Equation (12) is effectively traced back by nibblingaway at the 39-end, and checking for a base pair thatinitiates the stem of theFM1 segment under consid-eration. We first eat way at the 39-end:
Fi, j21M1 1 }B 1 EL6
1 O@k,l #{s
Ek,l # Emin 1 d (21)
If (21) holds, we push69 5 ([ i , j 2 1]M1 z s; 3;EL6
1 }B). When 69 is popped again, the 39 nib-bling will continue.
We next check whetheri andj can pair. If they can,we must consider
Ci, j 1 di, j,i215 1 di, j, j11
3 1 }I 1 EL6
1 O@k,l#{s
Ek,l # Emin 1 d(22)
which leads us to push69 5 [( i , j ]C z s; 3; EL61
di , j ,i215 1 di , j , j11
3 1 }I).
‹ CaseE 5 M ~multiloop backtrack inFM!
To trace back equation (13), we insert the definition ofFM1, (12), into (13). As in theFM1 case, we startnibbling away at the 39-end, and also consider aninterior base pair. This takes partially care of theFM1
term in Eq. (13). The procedure here follows exactlythe E 5 M1 case, except that the nibbled segment[ i , j 2 1], to be pushed, is now markedM.
To complete Eq. (13) we only need to loop overk,considering pairsk 1 1 z j , which fulfill
Fi,kM 1 Ck11, j 1 dk11, j,k
5 1 dk11, j, j113 1 }I 1 EL6
1 O@k,l #{s
Ek,l # Emin 1 d(23)
The corresponding refinements69 5 ([k 1 1, j ]C z[ i , k]M z s; 3; EL6
1 dk11,j ,k5 1 dk11,j , j11
3 1 }I)are pushed onR.
To cover the case in which the multiloop decom-position segment [i , j ]M contains exactly one interiorbase pair, we complete the backtrack of Eq. (14) bylooping overk, searching for pairsk 1 1 z j such that
Ck11, j 1 dk11, j,k5 1 dk11, j, j11
3 1 }I 1 ~k2 i 2 1! z }B
1 EL61 O
@k,l #{s
Ek,l # Emin 1 d(24)
and pushing69 5 ([k 1 1, j ]C z s; 3; EL61
dk11,j ,k5 1 dk11,j , j11
3 1 }I 1 }B) z (k 2 i 1 1).
‹ If 6 5 ~@i , j #E z s; 3; EL6! caused no refinement to
be pushed onR, then push~s; 3; EL6!.
C. Translation of Modified Nucleotides
All tRNA sequences ofE. coli are from the compila-tion of Steegborn,28 which can be obtained via anon-ymous ftp from EMBL Heidelberg, ftp.embl-heidel-berg.de, in directory /pub/databases/trna.
Bases are translated as suggested by Higgs.22 Mostmodified bases occur only in loop regions and aretherefore classified as nonbonding. Only the follow-ing bases are often found in paired regions and aretranslated to their canonical equivalents:
H (?A) Unknown modified adenosineˆ [(Ar(p)] 29-O-ribosyladenosine (phosphat)
, (?C) Unknown modified cytidineB (Cm) 29-O-methylcytidineM (aC4C) N4-acetylcytidine? (m5C) 5-Methylcytidine
; (G) Unknown modified guanosineL (m2G) N2-methylguanosine# (Gm) 29-O-methylguanosineR (m22G) N2,N2-dimethylguanosine
N (?U) Unknown modified uridineJ (Um) 29-O-methyluridineP (psi) Pseudouridine] (m1psi) 1-MethylpseudouridineZ (psi m) 29-O-methylpseudouridine
Several drawings were kindly provided by Jan Cupal. Thiswork was supported by the Austrian Fond zur Fr¨derung derwissenschaftlichen Forschung, project no. 11065-CHE, andby the integrative core research of the Santa Fe Institute.
REFERENCES
1. Nussinov, R.; Piecznik, G.; Griggs, J. R.; Kleitman,D. J. SIAM J Appl Math 1978, 35, 68–82.
2. Waterman, M. S.; Smith, T. F. Math Biosci 1978, 42,257–266.
3. Nussinov, R.; Jacobson, A. B. Proc Natl Acad Sci USA1980, 77, 6309–6313.
4. Zuker, M.; Stiegler, P. Nucleic Acids Res 1981, 9,133–148.
5. Zuker, M.; Sankoff, D. Bull Math Biol 1984, 46, 591–621.
6. Zuker, M. Science 1989, 244, 48–52.
164 Wuchty et al.

7. Nakaya, A.; Yamamoto, K.; Yonezawa, A. ComputApplic Biosci 1995, 11, 685–692.
8. Nakaya, A.; Yonezawa, A.; Yamamoto, K. J Theor Biol1996, 183, 105–117.
9. Waterman, M. S.; Byers, T. Math Biosci 1985, 77,179–188.
10. Waterman, M. S. Introduction to Computational Biol-ogy: Sequences, Maps and Genomes; Chapman & Hall:London, 1995.
11. McCaskill, J. S. Biopolymers 1990, 29, 1105–1119.12. Hofacker, I. L.; Fontana, W.; Stadler, P. F.; Schuster, P.
Free software, http://www.tbi.univie.ac.at/˜ivo/RNA/,1994–1998.
13. Cupal, J.; Hofacker, I. L.; Stadler, P. F. In ComputerScience and Biology 96, Proceedings of the GermanConference on Bioinformatics; Hofsta¨dt, R., Lengauer,T., Loffler, M., Schomburg, D., Ed.; Univerista¨tLeipzig, Leipzig, Germany, 1996; pp 184–186.
14. Walter, A. E.; Turner, D. H.; Kim, J.; Lyttle, M. H.;Muller, P.; Mathews, D. H.; Zuker, M. Proc Natl AcadSci 1994, 91, 9218–9222.
15. Freier, S. M.; Kierzek, R.; Jaeger, J. A.; Sugimoto, N.;Caruthers, M. H.; Neilson, T.; Turner, D. H. Proc NatlAcad Sci USA 1986, 83, 9373–9377.
16. Turner, D. H.; Sugimoto, N.; Freier, S. (1988) Ann RevBiophys Biophys Chem 1988, 17, 167–192.
17. Jaeger, J. A.; Turner, D. H.; Zuker, M. Proc Natl AcadSci USA 1989, 86, 7706–7710.
18. He, L.; Kierzek, R.; SantaLucia, J.; Walter, A.; Turner,D. Biochemistry 1991, 30, 11124.
19. Ward, J. H. J Am Stat Assoc 1963, 58, 236–244.20. Ninio, J. Biochimie 1979, 61, 1133.21. Higgs, P. G. J Phys I (France) 1993, 3, 43.22. Higgs, P. G. J Chem Soc Faraday Trans 1995, 91,
2531–2540.23. Fontana, W.; Konings, D. A. M.; Stadler, P. F.; Schus-
ter, P. Biopolymers 1993, 33, 1389–1404.24. Hofacker, I. L.; Fontana, W.; Stadler, P. F.; Bonhoeffer,
S.; Tacker, M.; Schuster, P. Monatsh Chem 1994, 125,167–188.
25. Schuster, P.; Fontana, W.; Stadler, P. F.; Hofacker, I.Proc Roy Soc (London) B 1994, 255, 279–284.
26. Huynen, M.; Perelson, A.; Vieira, W.; Stadler, P. Com-put Biol 1996, 3(2), 253–274.
27. Wuchty, S. master’s thesis, University of Vienna1998.
28. Steegborn, C.; Steinberg, S.; Huebel, F.; Sprinzl, M.Nucleic Acids Res 1995, 24(1).
29. Shapiro, B. A. CABIOS 1988, 4, 387–393.30. Wuchty, S.; Fontana, W.; Hofacker, I. L. (1998) free
software, http://www.tbi.univie.ac.at/˜ivo/RNA/.
Suboptimal Folding of RNA 165




















