
Comparative Genomics and Transcriptional Analysis of Prophages Identified in the Genomes of...
18
10.1128/AEM.72.5.3130-3146.2006. 2006, 72(5):3130. DOI: Appl. Environ. Microbiol. Douwe van Sinderen Klaenhammer, Gerald F. Fitzgerald, Paul W. O'Toole and Zink, Erasmo Neviani, Jim Steele, Jeff Broadbent, Todd R. J. Claesson, Yin Li, Sinead Leahy, Carey D. Walker, Ralf Marcus Altermann, Rodolphe Barrangou, Stephen McGrath, Marco Ventura, Carlos Canchaya, Valentina Bernini, Eric casei Lactobacillus , and Lactobacillus salivarius , Lactobacillus gasseri Genomes of Analysis of Prophages Identified in the Comparative Genomics and Transcriptional http://aem.asm.org/content/72/5/3130 Updated information and services can be found at: These include: SUPPLEMENTAL MATERIAL Supplemental material REFERENCES http://aem.asm.org/content/72/5/3130#ref-list-1 at: This article cites 42 articles, 24 of which can be accessed free CONTENT ALERTS more» articles cite this article), Receive: RSS Feeds, eTOCs, free email alerts (when new http://journals.asm.org/site/misc/reprints.xhtml Information about commercial reprint orders: http://journals.asm.org/site/subscriptions/ To subscribe to to another ASM Journal go to: on December 22, 2013 by guest http://aem.asm.org/ Downloaded from on December 22, 2013 by guest http://aem.asm.org/ Downloaded from
Transcript of Comparative Genomics and Transcriptional Analysis of Prophages Identified in the Genomes of...

10.1128/AEM.72.5.3130-3146.2006.
2006, 72(5):3130. DOI:Appl. Environ. Microbiol. Douwe van SinderenKlaenhammer, Gerald F. Fitzgerald, Paul W. O'Toole and Zink, Erasmo Neviani, Jim Steele, Jeff Broadbent, Todd R.J. Claesson, Yin Li, Sinead Leahy, Carey D. Walker, Ralf
MarcusAltermann, Rodolphe Barrangou, Stephen McGrath, Marco Ventura, Carlos Canchaya, Valentina Bernini, Eric casei
Lactobacillus, and Lactobacillus salivarius, Lactobacillus gasseriGenomes of
Analysis of Prophages Identified in the Comparative Genomics and Transcriptional
http://aem.asm.org/content/72/5/3130Updated information and services can be found at:
These include:
SUPPLEMENTAL MATERIAL Supplemental material
REFERENCEShttp://aem.asm.org/content/72/5/3130#ref-list-1at:
This article cites 42 articles, 24 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, May 2006, p. 3130–3146 Vol. 72, No. 50099-2240/06/$08.00�0 doi:10.1128/AEM.72.5.3130–3146.2006Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Comparative Genomics and Transcriptional Analysis of ProphagesIdentified in the Genomes of Lactobacillus gasseri,
Lactobacillus salivarius, and Lactobacillus casei†Marco Ventura,1,2* Carlos Canchaya,1 Valentina Bernini,2 Eric Altermann,3 Rodolphe Barrangou,3
Stephen McGrath,1 Marcus J. Claesson,1 Yin Li,1 Sinead Leahy,1 Carey D. Walker,4Ralf Zink,5 Erasmo Neviani,2 Jim Steele,6 Jeff Broadbent,7 Todd R. Klaenhammer,3
Gerald F. Fitzgerald,1 Paul W. O’Toole,1 and Douwe van Sinderen1
Alimentary Pharmabiotic Centre and Department of Microbiology, Bioscience Institute, National University of Ireland,Western Road, Cork, Ireland1; Department of Genetics, Anthropology and Evolution, University of Parma, Parma, Italy2;
Genomic Sciences Program and Southeast Dairy Foods Research Center, North Carolina State University, Raleigh,North Carolina3; Chr. Hansen, Milwaukee, Wisconsin4; Department of Nutrition and Health, Cognis, Dusseldorf,
Germany5; Department of Food Sciences, University of Wisconsin—Madison, Madison, Wisconsin6;and Department of Nutrition and Food Sciences, Utah State University, Logan, Utah7
Received 21 December 2005/Accepted 16 February 2006
Lactobacillus gasseri ATCC 33323, Lactobacillus salivarius subsp. salivarius UCC 118, and Lactobacillus caseiATCC 334 contain one (LgaI), four (Sal1, Sal2, Sal3, Sal4), and one (Lca1) distinguishable prophage se-quences, respectively. Sequence analysis revealed that LgaI, Lca1, Sal1, and Sal2 prophages belong to thegroup of Sfi11-like pac site and cos site Siphoviridae, respectively. Phylogenetic investigation of these newlydescribed prophage sequences revealed that they have not followed an evolutionary development similar to thatof their bacterial hosts and that they show a high degree of diversity, even within a species. The attachmentsites were determined for all these prophage elements; LgaI as well as Sal1 integrates in tRNA genes, whileprophage Sal2 integrates in a predicted arginino-succinate lyase-encoding gene. In contrast, Lca1 and the Sal3and Sal4 prophage remnants are integrated in noncoding regions in the L. casei ATCC 334 and L. salivariusUCC 118 genomes. Northern analysis showed that large parts of the prophage genomes are transcriptionallysilent and that transcription is limited to genome segments located near the attachment site. Finally, pulsed-field gel electrophoresis followed by Southern blot hybridization with specific prophage probes indicates thatthese prophage sequences are narrowly distributed within lactobacilli.
Sequencing of bacterial genomes has revealed a large num-ber of assumed prophage sequences, which contribute signifi-cantly to bacterial interstrain variability (7). Genome compar-ison of several Streptococcus pyogenes strains associated withdifferent types of streptococcal disease showed that prophagesrepresent up to 16% of the DNA content of the bacterialchromosome (7). Unfortunately, detailed analyses of pro-phages from sequenced nonpathogenic lactic acid bacteria(LAB) are still limited (7). Genome analysis of Lactococcuslactis IL-1403 revealed three cos site prophages and threeprophage remnants (9). In Lactobacillus johnsonii NCC 533DNA microarray analysis indicated that prophage DNA rep-resents almost half of identified strain-specific DNA (39). Re-cently, it has been shown that the Lactobacillus plantarumWCFS1 genome contains four prophage elements that shareclosely related genes encoding structural proteins with eachother (41).
The increasing number of available bacterial genome se-quences has contributed to the understanding of prophage
genome distribution and evolution. The mosaic pattern andlocalized diversity of many different prophage genomes areobvious from comparative analyses of prophage genome con-tent and organization, as well as similarities of orthologousgene products encoded by these elements (26).
Prophage-like elements and prophage remnants have beenidentified in almost all bacterial genomes sequenced so far (7),suggesting that this group of mobile elements is widespread inbacteria and may be considered to represent a useful tool inorder to investigate bacterial evolution. Prophages contribute asubstantial share of the mobile DNA of their bacterial hostsand seem to influence the short-term evolution of pathogenicbacteria. In fact, a large part of bacterial DNA is acquiredhorizontally by transformation, conjugation, or transduction.In this context, phages are the most efficient gene transferparticles developed during evolution and thus may be consid-ered as important vectors for the lateral transfer of DNAbetween bacterial strains (8).
Recently, the identification of prophage-like elements ingenomes of bifidobacteria, which were generally known to beinfected by bacteriophages, indicated that prophage sequencescould be used to investigate genome bacterial evolution (42).
Sequence data from Lactobacillus phages are available fordifferent species of lactobacilli (Lactobacillus gasseri, L. johnso-nii, L. plantarum, Lactobacillus casei, and Lactobacillus del-brueckii). Interestingly, no significant DNA sequence similarity
* Corresponding author. Mailing address: Department of Genetics,Anthropology and Evolution, University of Parma, Parco Area delleScienze 11/a, 43100 Parma, Italy. Phone: 39 521 906236. Fax: 39 521905604. E-mail: [email protected].
† Supplemental material for this article may be found at http://aem.asm.org/.
3130
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

was detected among Lactobacillus phages infecting distinctbacterial species (10), suggesting the presence of a barrierlimiting transfer of phage genes across Lactobacillus species.However, this finding may be biased by the limited amount ofcurrently available Lactobacillus prophage sequences.
Prophages are not only important genetic elements that im-part bacterial genome variability. In fact, prophages may alsoconfer a diverse array of phenotypic traits to their hosts, in-cluding those that govern the course and the pathobiology ofbacterial infections. Prophages in pathogenic strains such as S.pyogenes contribute important virulence factors, which are in-dicated as lysogenic conversion factors to the lysogenic hostand which have been demonstrated to contribute to the eco-logical fitness of the host (6, 7). Based on evolutionary reason-ing, prophages are postulated to contribute genes that increasethe fitness of lysogenic bacteria in their specific ecologicalniche (4) as well as genes encoding immunity factors thatprevent infection from other bacteriophages. This realizationhas changed our understanding of the phage-bacterium inter-action from being a simple parasite-host relationship to a mu-tually beneficial genomic coevolution.
Interestingly, comparative genomics identified putative lyso-genic conversion genes downstream of the lysis cassette andwithin the lysogeny module (7, 40, 43). Moreover, transcriptionstudies in L. johnsonii, L. plantarum, L. lactis, and Streptococ-cus thermophilus prophages demonstrated that these genes,together with those encoding immunity against phage superin-fection and those maintaining the lysogenic state, belong to thesmall number of prophage genes transcribed during the lyso-genic state (5, 38, 41, 43).
In prophages of bacterial pathogens, genes predicted to en-code virulence factors, including a wide range of superantigensand enzymes possibly involved in pathogenicity (DNase, hy-luronidase, and phospholipase [4, 6]) were all located betweenthe lysin and right prophage attachment site.
In the present report, we extend the transcription andgenomic knowledge of lactobacilli prophages through analysisof three additional genome sequences. Members of the genusLactobacillus are common inhabitants of the gastrointestinalenvironments where they reach high levels of colonization.They have also been isolated in decaying plant material andmany fermented food products. L. gasseri, Lactobacillus saliva-rius, and L. casei species are human gut commensal membersor food isolates (14, 17, 18). Sequencing of L. gasseri ATCC33323 as well as L. salivarius subsp. salivarius UCC 188 and L.casei ATCC 334 allowed the identification and the subsequentanalysis of prophage sequences with respect to their gene con-tent, transcription profile, distribution in other Lactobacillusstrains, and comparison to other Lactobacillus prophage se-quences.
MATERIALS AND METHODS
Bacterial strains and culture conditions. Lactobacillus strains were grownaerobically in MRS broth (Difco) and incubated for 16 h at 37°C. The bacterialstrains used and their origins are listed in Table 1.
Sequence analysis. The prophage sequences of LgaI, Sal1, Sal2, Sal3, Sal4, andLca1 were retrieved from the genome sequencing projects in L. gasseri ATCC33323, L. salivarius UCC 118, and L. casei ATCC 334.
Open reading frames (ORFs) were predicted using the ORF Finder (NCBI),taking ATG, GTG, and TTG as possible start codons and requiring a minimumsize of 50 amino acids. Nucleotide and predicted amino acid sequences were
compared with sequences in public sequence databases (GenBank, EMBL, PIR-Protein, SWISS-PROT, and PROSITE) using BLAST (3), PSI-BLAST, andFASTA (23). A scan for tRNA genes was performed using the tRNAscan-SEprogram (24). Motif searches were performed by using the Pfam database.
RNA isolation and Northern blot analysis. Total RNA was isolated by resus-pending bacterial cell pellets in Trizol (GibcoBRL, United Kingdom), addingglass beads (106 �m; Sigma), and disrupting cells with a Mini-Beadbeater-8 celldisruptor (Biospec Products) as described by Ventura et al. (38). Northern blotanalysis of prophages was carried out using 15-�g aliquots of RNA isolated from10 ml of Lactobacillus strains, collected at an optical density at 600 nm of 0.4, 0.8,and 1.2. The RNA was separated in a 1.5% agarose-formaldehyde denaturinggel, transferred to a Zeta-Probe blotting membrane (Bio-Rad, United Kingdom)according to Sambrook and Russell (37), and fixed by UV cross-linking using aStratalinker 1800 (Stratagene). Prehybridization and hybridization were carriedout at 65°C in 0.5 M NaHPO4 (pH 7.2), 1.0 mM EDTA, and 7.0% sodiumdodecyl sulfate (SDS). Following 18 h of hybridization, the membrane was rinsedtwice for 30 min at 65°C in 0.1 M NaHPO4 (pH 7.2), 1.0 mM EDTA, and 1%SDS and twice for 30 min at 65°C in 0.1 mM NaHPO4 (pH 7.2), 1.0 mM EDTA,and 0.1% SDS; the membrane was then exposed to X-OMAT autoradiographyfilm (Eastman Kodak).
Probes for Northern blot hybridization were labeled with �-32P using a ran-dom-primed DNA labeling system (Boehringer Mannheim GmbH) and purifiedwith Nuc-Trap probe purification columns (Stratagene).
DNA amplification of the attB sites. A 520-bp PCR fragment corresponding tothe attB region of prophage LgaI was generated using the primer combination1612 A and 1674, while PCR fragments corresponding to the attB region ofprophages Sal1, Sal2, Sal3, or Sal4 were generated using the primer sets Sal1-attB1 and Sal1-attB2, Sal2-attB1 and Sal2-attB2, Sal3-attB1 and Sal3-attB2, andSal4-attB-uni and Sal4-attB-rev, respectively (Table 2 gives details of the primersequences). The 750 bp encompassing the attB site of Lca1 prophage was gen-erated using the primer set Lca1-attB-uni and Lca1-attB-rev (Table 2). Finally,all PCR fragments corresponding to the attB site of LgaI, Sal1, Sal2, Sal3, Sal4,and Lca1 were verified by sequence analysis on both strands.
Excision of L. gasseri, L. salivarius, and L. casei prophages following additionof mitomycin C or hydrogen peroxide. Lactobacillus cultures were grown untilthey reached the mid-exponential growth phase, at which point mitomycin C(Sigma, United Kingdom) was either added to reach a final concentration of 1,2, 3, or 5 �g/ml or not added (control). Furthermore, possible induction of LgaI,Sal1, Sal2, Sal3, Sal4, or Lca1 prophage was assayed by adding hydrogen perox-ide to a final concentration of 2 mM. Growth was allowed to continue for 12 hat 37°C, after which cells were collected by centrifugation at 8,000 � g for 15 min.DNA was extracted as described previously (38). The occurrence of excision ofLgaI, Sal1, Sal2, Sal3, Sal4, and Lca1 prophages was monitored by PCR with the
TABLE 1. Bacterial strains and their origins
Species Straina Origin
L. salivarius subsp. salivarius UCC 118 HumanL. salivarius subsp. salivarius DSM 20555 SalivaL. salivarius subsp. salivarius DSM 20492 Human salivaL. salivarius subsp. salicinus DSM 20554 SalivaL. salivarius LMG 14477 Parakeet with sepsisL. salivarius LMG 14476 Cat with myocarditisL. casei ATCC 334 Emmental cheeseL. casei NCDO 1202 CheeseL. casei NCDO 1205 CheeseL. casei IMPC 21060 CheeseL. casei NCDO 151 CheeseL. casei NCDO 155 Mouse fecesL. gasseri ATCC 33323 HumanL. gasseri ATCC 4693 UnknownL. gasseri DSM 20077 Human fecesL. gasseri UCC 874 UnknownL. gasseri UCC 2493 UnknownL. gasseri IMPC 4B2 Human fecesL. gasseri UCC 907 Unknown
a ATCC, American Type Culture Collection; DSM, Deutsche Sammlung fuerMikroorganismen; LMG, LMG Bacteria Collection Universiteit Gent; UCC,University College Cork; NCDO, National Collection of Dairy Organisms; JCM:Japanese Collection of Microorganisms; IMPC: Istituto di Microbiologia Uni-versita’ Cattolica, Piacenza, Italy.
VOL. 72, 2006 COMPARATIVE GENOMICS OF LACTOBACILLUS PROPHAGES 3131
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

primer combinations (reverse) LgaI int gene and LgaI-orf1672, Sal1-attP1 primerand (forward) Sal1-attP2 primer, Sal2-attP1 primer and Sal2-attP2 primer, Sal3-attP1 primer and Sal3-attP2 primer, Sal4-attP1 primer and Sal4-attP2 primer,and Lca1-attP1 primer and Lca1-attP2 primer, which are specific for circularizedLgaI, Sal1, Sal2, Sal3, Sal4, or Lca1 phage to generate amplicons of 450, 631, 900,1,600, or 2,100 bp, respectively (Table 2). In the same PCR, positive controls arerepresented by the amplification of ORF 1555, ORF 794, or ORF 64 in prophageLgaI, Sal2, or Lca1, respectively. Amplifications were performed with a Biometrathermocycler with the following temperature profiles: 1 cycle of 95°C for 10 min;35 cycles of 95°C for 30 s, 54°C for 30 s, and 72°C for 1 min.
PFGE and Southern blotting. Agarose-embedded bacterial cells were pre-pared as described by Walker et al. (44). For digestion with restriction endo-nucleases, bacterial cells embedded in agarose blocks were treated with 50 U ofSmaI or XhoI (Roche Molecular, United Kingdom) as described by the manu-facturer. Digestion was stopped by washing the blocks for 20 min in Tris-EDTAbuffer. Pulsed-field gel electrophoresis (PFGE) was performed by a contour-clamped homogeneous electric field mode in a CHEF-DRII apparatus (Bio-Rad). All DNA samples were separated in 1% agarose gels in 0.5� Tris-borate-EDTA buffer, cooled to 14°C, for 20 h at 6 V/cm with a ramping pulse time from1 to 20 s.
Southern blots of agarose gels were performed on Hybond N� membranes(Amersham, United Kingdom) following the protocols of Sambrook and Russell(37). The filters were hybridized with radiolabeled PCR-generated probes de-scribed in the text. Subsequent prehybridization, hybridization, and autoradiog-raphy were carried out according to Sambrook and Russell (37). Each filter washybridized using a set of two probes corresponding to different prophage regions.A specific prophage hybridization signal was identified when both probes gave anidentical hybridization signal in terms of size and intensity, whereas a cross-hybridization signal designation was observed when the probes used gave adifferent hybridization signal in terms of intensity and/or size.
Proteomic tree analysis. The phylogenetic analysis was performed as describedpreviously (35). Every amino acid sequence, including sequences that encodehypothetical proteins, deduced from identified ORFs in the LgaI, Sal1, Sal2,Sal3, Sal4, and Lca1 prophage sequences, was compared to all ORF-derivedproteins deposited in the NCBI phage and prophage genome database, whichcontained annotated protein sequences from 476 bacteriophage and prophagegenomes as described previously (35).
Phylogenetic analysis. Phylogeny calculations were performed using thePHYLIP package (12). The final distance matrix to build the tree was calculatedby the neighbor-joining method as implemented in the neighbor module ofPHYLIP.
Nucleotide sequence accession numbers. The sequence of L. gasseri LgaIprophage has been deposited in the GenBank database under accession numberAAABO02000006.1. The L. salivarius Sal1, Sal2, Sal3, and Sal4 prophage se-quences are contained in the genome sequences of L. salivarius subsp. salivariusUCC 118, accession number CP000233. The L. casei Lca1 prophage sequencehas been deposited under accession number DQ411856.
RESULTS
Prophage content of L. gasseri ATCC 33323, L. salivariussubsp. salivarius UCC 118 and L. casei ATCC 334 genomes. Aspreviously described (7), integrases and/or cI repressors areuseful markers for the identification of mobile DNA elementssuch as prophages in bacterial genomes. L. gasseri ATCC33323 harbors six predicted integrase genes in its 2-Mb ge-nome. BLAST analysis of the surrounding region of ORF 372(int) revealed that this gene belongs to a 40,085-bp-long (LgaI)prophage. The screening for integrase genes and cI repressorsin the sequenced L. salivarius UCC 118 strain revealed thepresence of four apparent genomically complete or partialprophages that are represented by prophage-like sequences48,625 bp long (Sal1), 40,288 bp long (Sal2), 12,529 bp long(Sal3), and 10,315 bp long (Sal4). Finally, the sequenced L.casei ATCC 334 genome is predicted to contain a 46,986-bp-long (Lca1) prophage.
Genome analysis of prophage LgaI. The predicted prophageLgaI extends from ORF 372 (integrase gene) to ORF 1673 onthe genome of L. gasseri ATCC 33323. These ORFs areflanked by a 47-bp repeat, indicating the existence of attL andattR sites. Moreover, PCR primers placed in the adjacentORFs 1612 and 1672 resulted in a 520-bp amplicon withgenomic DNA from L. gasseri ATCC 33323 (Fig. 1b), indicat-ing a low frequency of excision of LgaI, and from other L.gasseri strains that, based on Southern blot hybridization withspecific LgaI probes, appeared to lack prophage LgaI (data notshown). The size of the amplicons corresponds exactly to theATCC 33323 genome minus the LgaI prophage. Sequencing ofthis PCR product identified the presence of a single copy of the47-bp repeat region, suggesting an attB site (Fig. 1a and b).Phage integration complements the 3� end of a tRNAArg gene.
The data thus indicate an integrase-mediated insertion ofprophage LgaI into a tRNAArg gene that is functionally recon-stituted following prophage integration.
Database matches allowed a tentative subdivision of theLgaI prophage genome into functional modules (Fig. 1c). Pro-phage LgaI shares the modular organization of the Sfi11-likepac site Siphoviridae phages (27). The lysogeny module waspredicted to extend from ORF 372 to ORF 1614 and includesthe integrase gene, the genetic switch region, and a likelysuperinfection exclusion gene sie (Table 3). The sie gene en-codes a protein closely related to a predicted surface-exposedlipoprotein encoded by S. thermophilus bacteriophage TP-J34at a corresponding position, where many phages from LABencode phage resistance functions (28). Downstream of thelysogeny module, several ORFs encoded proteins with similar-ities to DNA replication proteins from Lactococcus and Lac-tobacillus phages (29). Database matches included a replicationinitiation protein, a helicase-like protein, and a single-stranded
TABLE 2. Oligonucleotides used in this study
Oligonucleotide Sequence (5�33�)
1612A ..................................GTAATCGCACTTCTTATTTG1674 .....................................GTTTGGCTGATAGTACCAAAGSal1-attB1 ...........................CACTAAATGTAAACCATCAAGSal1-attB2 ...........................GATGAACCGTTCATTTATAATGSal2-attB1 ...........................GAGAGTGGATTATTATATGGTGSal2-attB2 ...........................GACTTGCAAGAAGATAAAGAAGSal3-attB1 ...........................GATATGCAGAGGAGATATTTGSal3-attB2 ...........................GATGACTTCAATCTCTACATTTTCTCSal4-attB-uni.......................GAAAGATATAAGTTTATGGGAGSal4-attB-rev.......................CACCTTTAAGTTTACGTGAGLca-attB-uni........................GAAGAGAATTCATAGTTCACTGLca-attB-rev........................GAGACGTGATTTAATGGCAGLgaI int................................CTTTACGACATTCTCAAGTTGLgaI-orf1672 .......................CTTCAACGAACTCACCTCCSal1-attP1............................CAGCAATATTAACTGGATTTATCSal1-attP2............................CTTCCACAGTCTGCGACATACSal2-attP1............................GAGTTTCAGAAGGTAGTAACSal2-attP2............................GTTATGGTGTAAACGTGTATACSal3-attP1............................CTTAGCTCTAACGTGGCATGSal3-attP2............................CTATTATAGATTTTGCTAATGAACSal4-attP1............................CTTGAGAGCTTAACCAATCSal4-attP2............................CATAGCAATGTACAAACAACACLca1-attP1...........................GATTGCTGCCAACAAACCACLca1-attP2...........................GAGACGTGATTTAATGGCAGORF 1555-uni ....................GTTTGATACACAGTATTAGCATCORF 1555-rev.....................GAAGTGTATAGCAAGTCCGAG394-uni.................................GTCTCTAGGATTAGAAGCTG394-rev.................................CTATAGATATTCCAGTAGTAACORF 64-uni ........................GTACCATGTCGCTTCGTGORF 66-rev.........................GATGGCTGATATTGAAGCTG
3132 VENTURA ET AL. APPL. ENVIRON. MICROBIOL.
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from
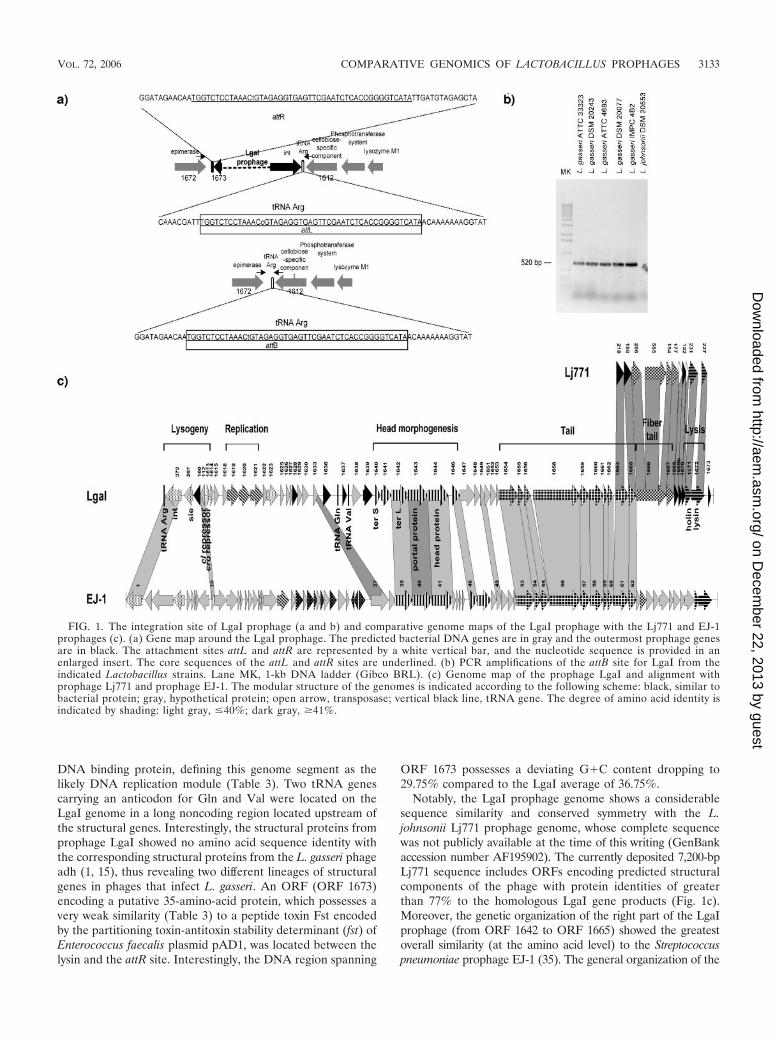
DNA binding protein, defining this genome segment as thelikely DNA replication module (Table 3). Two tRNA genescarrying an anticodon for Gln and Val were located on theLgaI genome in a long noncoding region located upstream ofthe structural genes. Interestingly, the structural proteins fromprophage LgaI showed no amino acid sequence identity withthe corresponding structural proteins from the L. gasseri phageadh (1, 15), thus revealing two different lineages of structuralgenes in phages that infect L. gasseri. An ORF (ORF 1673)encoding a putative 35-amino-acid protein, which possesses avery weak similarity (Table 3) to a peptide toxin Fst encodedby the partitioning toxin-antitoxin stability determinant (fst) ofEnterococcus faecalis plasmid pAD1, was located between thelysin and the attR site. Interestingly, the DNA region spanning
ORF 1673 possesses a deviating G�C content dropping to29.75% compared to the LgaI average of 36.75%.
Notably, the LgaI prophage genome shows a considerablesequence similarity and conserved symmetry with the L.johnsonii Lj771 prophage genome, whose complete sequencewas not publicly available at the time of this writing (GenBankaccession number AF195902). The currently deposited 7,200-bpLj771 sequence includes ORFs encoding predicted structuralcomponents of the phage with protein identities of greaterthan 77% to the homologous LgaI gene products (Fig. 1c).Moreover, the genetic organization of the right part of the LgaIprophage (from ORF 1642 to ORF 1665) showed the greatestoverall similarity (at the amino acid level) to the Streptococcuspneumoniae prophage EJ-1 (35). The general organization of the
FIG. 1. The integration site of LgaI prophage (a and b) and comparative genome maps of the LgaI prophage with the Lj771 and EJ-1prophages (c). (a) Gene map around the LgaI prophage. The predicted bacterial DNA genes are in gray and the outermost prophage genesare in black. The attachment sites attL and attR are represented by a white vertical bar, and the nucleotide sequence is provided in anenlarged insert. The core sequences of the attL and attR sites are underlined. (b) PCR amplifications of the attB site for LgaI from theindicated Lactobacillus strains. Lane MK, 1-kb DNA ladder (Gibco BRL). (c) Genome map of the prophage LgaI and alignment withprophage Lj771 and prophage EJ-1. The modular structure of the genomes is indicated according to the following scheme: black, similar tobacterial protein; gray, hypothetical protein; open arrow, transposase; vertical black line, tRNA gene. The degree of amino acid identity isindicated by shading: light gray, �40%; dark gray, �41%.
VOL. 72, 2006 COMPARATIVE GENOMICS OF LACTOBACILLUS PROPHAGES 3133
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

TABLE 3. Database matches for LgaI, Sal1, Sal2, Sal2, Sal3, Sal4, and Lca1 prophages
ORF Representative similarity to protein in database % Identity E value
LgaI372 Integrase, S. thermophilus 29 �35207 Lipoprotein, S. thermophilus bacteriophage TP-J34 28 �17112 Putative repressor, Streptococcus agalactiae prophage 48 �101613 Cro-like regulatory protein, S. thermophilus bacteriophage Sfi21 37 �41619 Putative DNA recombinase, bacteriophage SPBc2 19 �31620 Single-strand binding protein, bacteriophage LL-H 33 �61621 Replication initiation protein, bacteriophage Tuc2009 46 �241623 Antirepressor, S. thermophilus bacteriophage Sfi21 41 �391640 Terminase, Lactococcus bacteriophage bIL310 50 �151641 Putative HNH homing endonuclease, Lactococcus bacteriophage bIL170 37 �261642 Terminase large subunit, L. johnsonii prophage Lj965 27 �231643 Portal protein, bacteriophage SPP1 25 �311644 Minor head protein, L. johnsonii prophage Lj965 21 �121646 Scaffolding protein, Listeria innocua phage A118 34 �171654 Protein XkdK, phage-like element PBX 31 �441655 Protein XkdM, phage-like element PBX 35 �141656 Protein XkdN, phage-like element PBX 29 �81658 Putative minor tail protein, S. pneumonie phage EJ-1 39 �761659 Protein XkdP, phage-like element PBX 20 �71666 Late gene cluster, ORF585, L. johnsonii prophage Lj771 73 �1121667 Late gene cluster, ORF177, L. johnsonii prophage Lj771 88 �831671 Putative holing, L. johnsonii prophage Lj965 37 �61672 Putative lysin, L. johnsonii prophage Lj928 63 �1051672 Lysin, L. gasseri bacteriophage adh 69 �1271673 Peptide toxin Fst, E. faecalis 42 1.2
Sal1729 Integrase, Lactococcus bacteriophage BK5-T 30 �48733 Transposase, L. johnsonii NCC533 50 �43740 Superinfection immunity protein, L. johnsonii prophage LJ965 40 �18741 Metallo proteinase protein, bacteriophage �105 35 �5742 Repressor protein, S. pyogenes phage 3.15 36 �13752 Helicase, S. pyogenes phage 3.16 35 �16756 NTP binding protein, bacteriophage �AT3 25 �6758 DNA polymerase, Burkholderia cenocepacia phage BcepB1A 25 �22759 Primase, Lactococcus bacteriophage �31 27 �4760 Primase, Lactobacillus plantarum prophage Lp4 33 �30764 Hypothetical protein, Fusibacterium nucleatum subsp. vincentii ATCC 49256 phage 35 �12765 Integrase, Lactococcus bacteriophage BK5-T 28 �32781 Endonuclease family protein, prophage � ba02 45 �22782 Phage terminase small subunit, Enterococcus faecium 33 �8783 Terminase large subunit, Lactococcus bacteriophage BK5-T 27 �28784 Portal protein, Staphylococcus aureus temperate phage phiSLT 45 �79785 ClpP protease, S. thermophilus bacteriophage 7201 37 �19786 Minor head protein, prophage � ba02 45 �81787 DNA packaging protein, S. thermophilus phage Sfi19 32 �5788 Head-tail adaptor, Enterococcus faecium phage 29 �4789 Head-tail joining protein, S. pyogenes phage 315.2 29 �6790 Head-tail joining protein, S. pyogenes MGAS8232 22 �03791 Major tail protein, S. thermophilus phage Sfi19 25 �05794 Tape measure protein, S. thermophilus phage 7201 20 �29796 Phage-related tail protein, Pediococcus pentosaceus ATCC 25745 36 �06797 Endolysin, L. plantarum prophage Lp1 50 �49799 Minor capsid protein, bacteriophage �g1e 50 �44805 Lysin, Lactococcus bacteriophage LC3 48 �100
Sal2236 Integrase, L. plantarum Lp3 35 64240 Superinfection immunity protein, phage �R73 26 �8242 CI repressor, S. pneumonie phage EJ-1 40 �38250 Cro repressor, Lactococcus bacteriophage BK5-T 51 �10251 Antirepressor protein, S. thermophilus phage Sfi21 65 �83256 Replisome organizer protein, Lactococcus bacteriophage BK5-T 40 �22264 Helicase protein, Enterococcus faecium 25 �3271 Endonuclease, enterobacteria phage RB49 27 3.1279 HNH endonuclease family protein, prophage L54a 43 �10280 Terminase small subunit, S. thermophilus 7201 55 �42281 Terminase large subunit, Lactococcus phage BK5-T 68 �99283 Portal protein, S. thermophilus Sfi21 51 �103
Continued on facing page
3134 VENTURA ET AL. APPL. ENVIRON. MICROBIOL.
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

regions spanning ORF 1642 to ORF 1664 and ORF 1654 to ORF1665, respectively, showed a one-to-one correspondence to simi-larly sized genes in the head morphogenesis and tail morphogen-esis clusters of EJ-1 prophage (Fig. 1c).
Genome analysis of L. salivarius Sal1, Sal2, Sal3, and Sal4prophages. Based on the presumptive prophage genomelength, only prophages Sal1 and Sal2 appear to be complete,whereas Sal3 and Sal4 appear to be prophage remnants. Theexact length of all L. salivarius prophages was determined bysequencing the PCR product achieved using primers placed inbacterial genes that flank each of the assumed prophages instrain UCC 118 and/or in other L. salivarius strains.
Genome analysis of prophage Sal1. The predicted prophageSal1 in L. salivarius subsp. salivarius UCC 118 extends fromORF 729 (integrase gene) to ORF 805 (lysin gene) (Fig. 2a).These ORFs are flanked by a 14-bp repeat, indicating theexistence of putative attL and attR sites. Moreover, PCR-prim-ers (Sal1-attB1 and Sal1-attB2) placed in the flanking bacterialORF 808 and ORF 728 genes, encoding an rRNA methylase
protein and a exodeoxyribonuclease V �-chain protein, yieldeda 1.6-kb amplicon with genomic DNA from L. salivarius subsp.salivarius DSM 20555 and L. salivarius subsp. salivarius LMG14447, a size which accords with a chromosome lacking pro-phage sequence at this site. Sequencing of this PCR productidentified the presence of a single copy of a 14-bp repeatregion, suggesting an attB site (Fig. 2a). Phage integrationcomplements the 3� end of a tRNA carrying the anticodonspecific for Ser.
Database matches allowed the subdivision of the Sal1 pro-phage genome into functional modules (Fig. 3 and Table 3). Inthis prophage-like element, the lysogenic module is limited toa region spanning ORF 729 to ORF 746. The next ORFs (ORF752 to ORF 760) constitute the likely DNA replication mod-ule. Downstream of the replication module an endonucleasegene (ORF 781) is notable in Sal1. Related endonucleases areintron-associated in a number of phage systems (13). Identifi-cation of likely structural modules such as the head mor-phogenesis genes (from ORF 782 to ORF 786), DNA pack-
TABLE 3—Continued
ORF Representative similarity to protein in database % Identity E value
284 Clp protease, S. thermophilus Sfi21 52 �54285 Major head protein, Lactococcus phage BK5-T 63 �132294 Tail tape measure protein, S. thermophilus Sfi21 32 �77296 Endolysin, L. plantarum prophage Lp1 48 �40300 Endonuclease, lambda Sa2 bacteriophage 20 �10304 Lysin, Lactococcus bacteriophage LC3 49 �100
Sal31648 Integrase, L. plantarum prophage Lp3 36 �661649 Transcriptional regulator, Bacillus halodurans C-125 39 �71650 Antirepressor protein, Staphylococcus aureus phage phiSLT 30 �111658 Primase, helicase protein, L. plantarum prophage Lp4 25 �13
Sal41189 Integrase, L. plantarum prophage Lp3 36 �671190 Superinfection immunity protein, bacteriophage phi-105 27 �51191 Transcription repressor, Lactococcus phage ps2 46 �141192 Repressor protein, bacteriophage TPW22 47 �71193 Transcriptional regulator, E. faecalis V583 46 �111202 DNA primase protein, Lactobacillus acidophilus NCFM 41 �83
Lca174 Integrase, L. casei bacteriophage A2 27 �971 Chromosome partitioning ATPase, ParA family, E. faecalis V583 40 �4964 Transcriptional repressor, bacteriophage 2638A 33 �1762 Putative transcriptional regulator, Burkholderia pseudomallei K96243 38 �660 Helicase, L. plantarum prophage Lp2 34 �1852 Putative SSB protein, bacteriophage �AT3 29 �0751 Replication protein, Lactococcus prophage BK5-T 36 �0850 Putative DNA replication protein, bacteriophage �AT3 93 �13434 Aspartate/tyrosine/aromatic aminotransferase, L. casei ATCC 334 98 �2231 Putative terminase small subunit, bacteriophage �g1e 46 �3330 Terminase large subunit, L. johnsonii prophage Lj965 64 �15329 Putative portal protein, Lactococcus bacteriophage ul36 39 �7828 Minor head protein, S. thermophilus bacteriophage 2972 30 �2923 Transposase subunit B, Lactobacillus sakei 46 �5922 Probable transposase, Lactobacillus sakei plasmid 51 �1518 Head-tail joining protein, Lactococcus bacteriophage TP901-1 35 �1117 Major structural protein, bacteriophage Tuc2009 30 �1116 Major tail protein, Lactococcus bacteriophage ul36 29 �1113 Tape measure protein, L. plantarum bacteriophage �JL-1 34 �12512 Transposase and inactivated derivatives, IS30 family S. pyogenes M49 591 41 �5611 Tail component, bacteriophage �AT3 30 �6610 Host interaction protein, L. casei bacteriophage A2 57 �1246 Putative holin protein, bacteriophage �AT3 92 �535 Lysin, Lactococcus bacteriophage TP901-1 43 �93
VOL. 72, 2006 COMPARATIVE GENOMICS OF LACTOBACILLUS PROPHAGES 3135
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

aging genes (from ORF 787 to ORF 790), and tail morphogenesisgenes (from ORF 791 to ORF 796) was possible on the basisof observed homology to genes in lactococcal and streptococ-cal phages with low G�C content. Further upstream, a gene(ORF 797) that possesses a glycosyl hydrolase family motif(Pfam 01183) and is similar to the endolysin detected in thegenome of L. plantarum Lp2 prophage was identified; sepa-rated from this gene by four anonymous ORFs, a lysis cassetteformed by only the lysin gene (ORF 805) was also identified(Fig. 3).
Genome analysis of prophage Sal2. Prophage-like elementSal2 is located between a gene (ORF 235) encoding an am-monium transporter on one side and a gene (ORF 305) en-coding an arginino-succinate lyase on the other side, whilebeing flanked by a 24-bp repeat (Fig. 2b). Using primers tar-geting the sequences of the bacterial genes flanking the pro-phage, we amplified a 750-bp-long DNA segment from severalL. salivarius strains (Fig. 2b). Sequencing of this 750-bp am-plicon identified the presence of a single copy of a 24-bp repeat
suggesting an attB site. The 24-bp core sequence overlaps the5� end of the arginino-succinate lyase-encoding gene, whichappeared to be reconstructed upon phage integration (Fig. 2b).The arginino-succinate lyase is an enzyme of the urea cyclewhich splits arginino-succinate to fumarate and arginine (30),so its preservation from gene interruption appeared to behighly important for maintaining correct cell metabolism. Da-tabase matches also allowed a tentative subdivision of the Sal2prophage genome into functional modules. The modular or-ganization was typical for temperate phages from low-GC-content gram-positive bacteria (Fig. 3). The likely extent of thelysogeny module is from ORF 236 to ORF 251. The lysogenymodule of Sal2 prophage contains two genes (ORF 247 andORF 250) whose deduced protein products show a high levelof protein identity to Cro-like proteins and a gene (ORF 242)that resembles the cI repressor gene (Table 3). However, theseputative cI- and cro-like genes were not located adjacent toeach other as is commonly found for other phages. The fol-lowing genes were assigned to the DNA replication module.
FIG. 2. Sal1 (a), Sal2 (b), Sal3 (c), and Sal4 (d) prophage integration and PCR amplification of the attB site of each prophage are indicatedin each panel. The predicted bacterial DNA genes are in shown gray, and the outermost prophage genes are shown in black. The attachment sitesattL and attR are represented by a black vertical bar, and the nucleotide sequence is provided. The core sequences of the attL and attR sites areunderlined.
3136 VENTURA ET AL. APPL. ENVIRON. MICROBIOL.
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

Across the DNA packaging, head, and tail fiber modules,prophage Sal2 shared sequence similarity with S. thermophilusphage 7201 and L. lactis BK5-T phage. Furthermore, severalgenes encoding hypothetical proteins of unknown function arelocated between the tail fiber module and the host lysis clusterin Sal2 prophage. ORF 300 displays a noteworthy homology tothe HNH family of homing endonucleases found in manyphages (Table 3). These endonucleases are often part of bac-teriophage intron systems that give rise to modular enzymes.Sal2 prophage possesses a very similar genetic organization tothe region encompassing ORF 279 to ORF 304 with an aminoacid similarity greater than 40% with the homologous region ofSal1 prophage. Moreover, other regions of similarity wereidentified that encompassed the integrase gene, an endonucle-ase gene, and genes encoding hypothetical proteins of un-known function (Fig. 3).
Genome analysis of Sal3 and Sal4 prophages. Prophage-likeelement Sal3 extends from ORF 1648 to ORF 1665, whereasthe likely extent of prophage Sal4 is from ORF 1189 to ORF
1205. Sal3 and Sal4 prophages are flanked by a 19-bp repeatand a 21-bp repeat, respectively. Most of the L. salivariusstrains from our collection yielded a 300-bp or 700-bp PCRproduct when primers (the pair Sal3-attB1 and Sal3-attB2 andthe pair Sal4-attB1 and Sal4-attB2) were placed in the bacte-rial genes that surround the Sal3 or Sal4 prophage in L. sali-varius UCC 118, respectively. Interestingly, using L. salivariusUCC 118 genomic DNA as a template also generated PCRproducts using these set of primers, thereby indicating that asubfraction of an L. salivarius UCC 118 culture may have beensubject to excision of these phages. These PCR products con-tained the 19-bp or the 21-bp sequences, and the sequences tothe right and left of these deduced attB sites were identical tothose abutting the likely attL and attR sites (Fig. 2c and d).
As shown in Fig. 3 and Table 3, the genetic structures of Sal3and Sal4 appear to have been shaped by multiple DNA dele-tion and rearrangement events. In fact, their limited lengthsand the absence of a region encoding structural phage com-ponents suggest that one or more major DNA deletion events
FIG. 3. Comparative genome maps of the Sal1, Sal2, Sal3, and Sal4 prophages. Genes sharing similarity are linked by shading. Probablefunctions of encoded proteins identified by bioinformatic analysis are noted. The modular structure of the genomes is indicated as described inthe legend of Fig. 1. The degree of amino acid identity is indicated by shading as described in the legend to Fig. 1.
VOL. 72, 2006 COMPARATIVE GENOMICS OF LACTOBACILLUS PROPHAGES 3137
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from
may have occurred. Comparison of the Sal3 and Sal4 prophagegenome sequences showed significant sequence similarity atthe DNA level. Strikingly, the similarity extended to the wholeprophage genome in a patchwork fashion (Fig. 3).
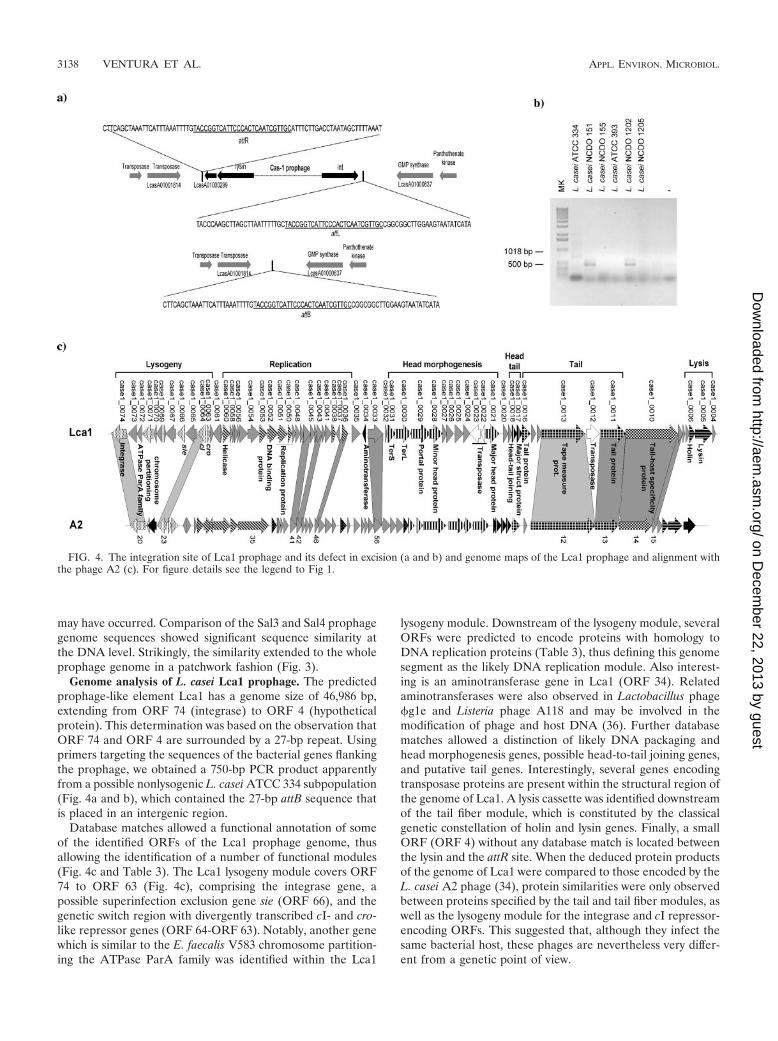
Genome analysis of L. casei Lca1 prophage. The predictedprophage-like element Lca1 has a genome size of 46,986 bp,extending from ORF 74 (integrase) to ORF 4 (hypotheticalprotein). This determination was based on the observation thatORF 74 and ORF 4 are surrounded by a 27-bp repeat. Usingprimers targeting the sequences of the bacterial genes flankingthe prophage, we obtained a 750-bp PCR product apparentlyfrom a possible nonlysogenic L. casei ATCC 334 subpopulation(Fig. 4a and b), which contained the 27-bp attB sequence thatis placed in an intergenic region.
Database matches allowed a functional annotation of someof the identified ORFs of the Lca1 prophage genome, thusallowing the identification of a number of functional modules(Fig. 4c and Table 3). The Lca1 lysogeny module covers ORF74 to ORF 63 (Fig. 4c), comprising the integrase gene, apossible superinfection exclusion gene sie (ORF 66), and thegenetic switch region with divergently transcribed cI- and cro-like repressor genes (ORF 64-ORF 63). Notably, another genewhich is similar to the E. faecalis V583 chromosome partition-ing the ATPase ParA family was identified within the Lca1
lysogeny module. Downstream of the lysogeny module, severalORFs were predicted to encode proteins with homology toDNA replication proteins (Table 3), thus defining this genomesegment as the likely DNA replication module. Also interest-ing is an aminotransferase gene in Lca1 (ORF 34). Relatedaminotransferases were also observed in Lactobacillus phage�g1e and Listeria phage A118 and may be involved in themodification of phage and host DNA (36). Further databasematches allowed a distinction of likely DNA packaging andhead morphogenesis genes, possible head-to-tail joining genes,and putative tail genes. Interestingly, several genes encodingtransposase proteins are present within the structural region ofthe genome of Lca1. A lysis cassette was identified downstreamof the tail fiber module, which is constituted by the classicalgenetic constellation of holin and lysin genes. Finally, a smallORF (ORF 4) without any database match is located betweenthe lysin and the attR site. When the deduced protein productsof the genome of Lca1 were compared to those encoded by theL. casei A2 phage (34), protein similarities were only observedbetween proteins specified by the tail and tail fiber modules, aswell as the lysogeny module for the integrase and cI repressor-encoding ORFs. This suggested that, although they infect thesame bacterial host, these phages are nevertheless very differ-ent from a genetic point of view.
FIG. 4. The integration site of Lca1 prophage and its defect in excision (a and b) and genome maps of the Lca1 prophage and alignment withthe phage A2 (c). For figure details see the legend to Fig 1.
3138 VENTURA ET AL. APPL. ENVIRON. MICROBIOL.
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

Transcription analysis. Analysis of the transcription ofprophage genes was conducted by performing Northern blothybridization using DNA probes that cover a large portion ofthe L. gasseri LgaI and the L. salivarius Sal1, Sal2, Sal3, andSal4 prophage genomes (Fig. 5; see also Fig. S1 in the supple-mental material). From these results it is obvious that largeregions of these prophage genomes are transcriptionally silent.No transcription of the cro-like and the antirepressor geneswas detected, which frequently constitute early or middle lytictranscripts in phages from LAB (43). Also the DNA packagingand head morphogenesis, tail, and tail fiber genes did notappear to be transcribed. In contrast, probes corresponding toparts of the lysogeny module of these prophages revealed ineach case prophage-specific transcripts.
LgaI. Using an LgaI ORF 112 (cI)-specific probe, a tran-script of 1.4 kb was detected. According to its estimated length,this transcript would cover the cI repressor gene, ORF 160(encoding a hypothetical protein) and ORF 207 (encoding aputative superinfection exclusion protein) (Fig. 5a). In fact,when an ORF 207-specific probe was used, the 1.4-kb mRNA
species was also detected. In addition, a 0.7-kb signal withgreater autoradiographic intensity was identified (see Fig. S1ain the supplemental material). A probe corresponding to ORF1673, located between the phage lysin and attR, hybridized witha prominent but small transcript of 0.6 kb (Fig. 5a; see also Fig.S1a in the supplemental material). The length of the transcriptpredicts termination between ORF 1673 and the right attach-ment site. No transcripts were observed using probes coveringseveral structural genes and the lysin gene.
Sal1. The lysogeny module of Sal1 gave rise to a smallnumber of transcripts (Fig. 5b; see Fig. S1b in the supplemen-tal material). A probe corresponding to the cI repressor (ORF742) yielded a 1.5-kb signal, which was also revealed with aprobe covering the putative superinfection exclusion gene(ORF 740). The 1.5-kb mRNA thus spanned ORFs 742 and740. The likely constellation of the transcripts from the lysog-eny module of Sal1 is depicted in Fig. 5b. Furthermore, a seriesof mRNAs with sizes ranging from 3.5, 2.9, and 2.0 to 1.5 kbwere detected in the lysogeny module when an ORF 733-specific probe was employed (Fig. 5b; see Fig. S1b in the
FIG. 5. Schematic representation of the transcription pattern of prophage LgaI (a), Sal1 (b), Sal2 (c), and Sal4 (d). The relative position andextent of the probes used for Northern blot analysis are provided as horizontal lines just below each of the schematically displayed phage genomes.The thin arrows just below the probe positions indicate the deduced position and direction of the transcripts; the numbers above the thin arrowsrefer to the estimated length of the transcripts.
VOL. 72, 2006 COMPARATIVE GENOMICS OF LACTOBACILLUS PROPHAGES 3139
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from
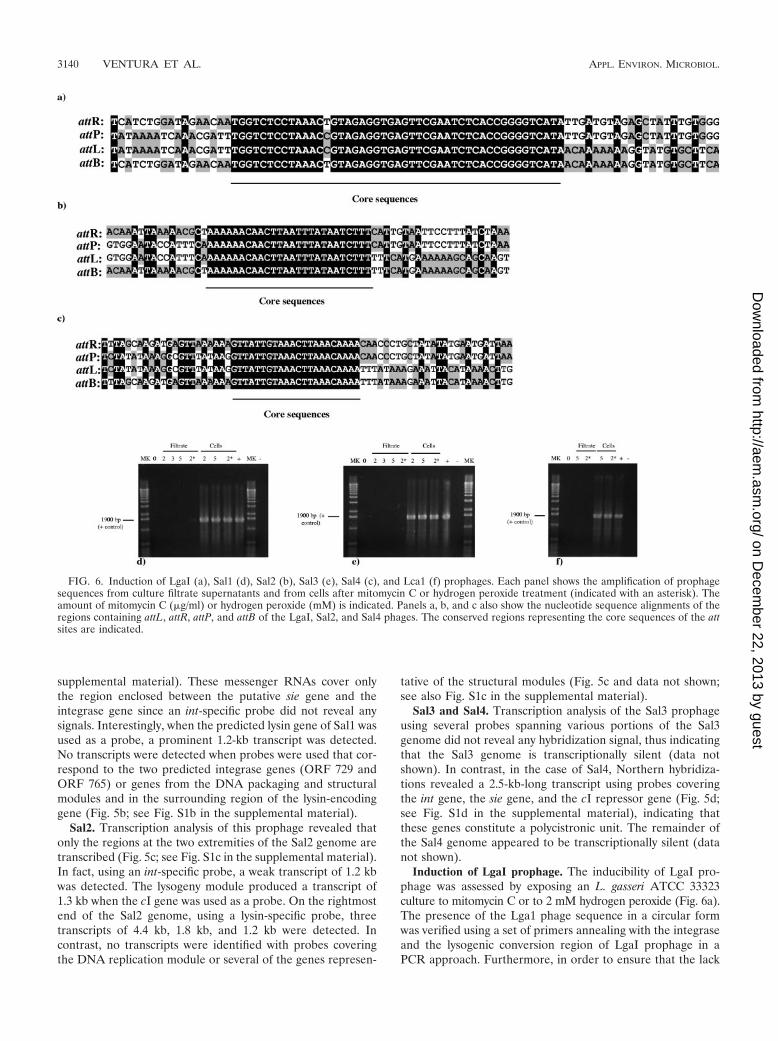
supplemental material). These messenger RNAs cover onlythe region enclosed between the putative sie gene and theintegrase gene since an int-specific probe did not reveal anysignals. Interestingly, when the predicted lysin gene of Sal1 wasused as a probe, a prominent 1.2-kb transcript was detected.No transcripts were detected when probes were used that cor-respond to the two predicted integrase genes (ORF 729 andORF 765) or genes from the DNA packaging and structuralmodules and in the surrounding region of the lysin-encodinggene (Fig. 5b; see Fig. S1b in the supplemental material).
Sal2. Transcription analysis of this prophage revealed thatonly the regions at the two extremities of the Sal2 genome aretranscribed (Fig. 5c; see Fig. S1c in the supplemental material).In fact, using an int-specific probe, a weak transcript of 1.2 kbwas detected. The lysogeny module produced a transcript of1.3 kb when the cI gene was used as a probe. On the rightmostend of the Sal2 genome, using a lysin-specific probe, threetranscripts of 4.4 kb, 1.8 kb, and 1.2 kb were detected. Incontrast, no transcripts were identified with probes coveringthe DNA replication module or several of the genes represen-
tative of the structural modules (Fig. 5c and data not shown;see also Fig. S1c in the supplemental material).
Sal3 and Sal4. Transcription analysis of the Sal3 prophageusing several probes spanning various portions of the Sal3genome did not reveal any hybridization signal, thus indicatingthat the Sal3 genome is transcriptionally silent (data notshown). In contrast, in the case of Sal4, Northern hybridiza-tions revealed a 2.5-kb-long transcript using probes coveringthe int gene, the sie gene, and the cI repressor gene (Fig. 5d;see Fig. S1d in the supplemental material), indicating thatthese genes constitute a polycistronic unit. The remainder ofthe Sal4 genome appeared to be transcriptionally silent (datanot shown).
Induction of LgaI prophage. The inducibility of LgaI pro-phage was assessed by exposing an L. gasseri ATCC 33323culture to mitomycin C or to 2 mM hydrogen peroxide (Fig. 6a).The presence of the Lga1 phage sequence in a circular formwas verified using a set of primers annealing with the integraseand the lysogenic conversion region of LgaI prophage in aPCR approach. Furthermore, in order to ensure that the lack
FIG. 6. Induction of LgaI (a), Sal1 (d), Sal2 (b), Sal3 (e), Sal4 (c), and Lca1 (f) prophages. Each panel shows the amplification of prophagesequences from culture filtrate supernatants and from cells after mitomycin C or hydrogen peroxide treatment (indicated with an asterisk). Theamount of mitomycin C (�g/ml) or hydrogen peroxide (mM) is indicated. Panels a, b, and c also show the nucleotide sequence alignments of theregions containing attL, attR, attP, and attB of the LgaI, Sal2, and Sal4 phages. The conserved regions representing the core sequences of the attsites are indicated.
3140 VENTURA ET AL. APPL. ENVIRON. MICROBIOL.
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

of any PCR product was attributable to the absence of circu-larized phage DNA target rather than a failure of the ampli-fication reaction, a second pair of primers (1555-uni and 1555-rev) targeting a 1,600-bp region within the prophage genomewas used as a positive control. Specific 450-bp amplicons withprimers running outward from the ends of the linear prophagegenomes were achieved with DNA isolated from cells follow-ing the addition of mitomycin C or hydrogen peroxide (datanot shown), indicating that free circularized phage genomicDNA is present. Spontaneous excision of LgaI was also ob-served (data not shown). When the 450-bp amplicon, whichcorresponds to the LgaI attP site, was sequenced and alignedwith the LgaI attL, attR, and attB sites, a common 47 bp witha single variable nucleotide was found (Fig. 6a). In this com-mon core, DNA strand exchange is expected to occur duringphage genome integration into the bacterial chromosome.
L. salivarius Sal1, Sal2, Sal3, and Sal4 prophage induction.In order to detect excised and circularized Sal1, Sal2, Sal3, orSal4 phage genome following treatment of L. salivarius UCC118 cultures with mitomycin C or to 2 mM hydrogen peroxide,a PCR product of 631 bp, 900 bp, 1,600 bp, or 2,100 bp,respectively, should be obtained with primers placed in theleft- and rightmost part of each prophage genome and runningoutward from each of these four prophages. The PCR primerpair 394-1 and 394-2 and the pair 394-1 and 392-2, which aretargeted to amplify internal Sal1 prophage genes and provideamplicons of 1,900 bp or 2,100 bp, respectively, were used asPCR positive controls.
Amplicons of 900 bp and 2,100 bp were obtained with prim-ers placed at the periphery of the Sal2 and Sal4 prophages,respectively, which indicates that circularized bacteriophageswere obtained after either mitomycin C or hydrogen peroxidetreatment (data not shown). In contrast, no such PCR productswere obtained when DNA extracted from a noninduced UCC118 culture was used (data not shown). When the 900-bp am-plicon, which corresponds to the Sal2 attP site, was sequencedand aligned with the Sal2 attL, attR, and attB sites, a common24-bp core region was found (Fig. 6b). In this common core,DNA strand exchange is expected to occur during phagegenome integration into the bacterial chromosome. Simi-larly, when the 2,100-bp PCR product, which corresponds tothe attP site of Sal4, was sequenced and aligned with theSal4 attL, attR, and attB sites, a common 21-bp core regionwas found (Fig. 6c).
In contrast, no specific amplicons were obtained using prim-ers running out of the Sal1 or Sal3 prophage genome, indicat-ing that free circularized Sal1 and Sal3 phage genomes are notpresent following mitomycin C or hydrogen peroxide treat-ment (Fig. 6d and e).
L. casei Lca1 prophage induction. The inducibility of Lca1prophage was assessed using an identical procedure as outlinedfor the L. salivarius prophages. A PCR primer pair (Lca1-attP-uni and Lca1-attP-rev) was designed at each border of the Lca1prophage, and each was directed outward from the prophagesequences. Moreover, another PCR primer pair (64-1 and 66-2)which targets ORF 64 was used as a PCR positive control. DNAisolated from cells upon the addition of different concentrationsof mitomycin C or hydrogen peroxide yielded amplicons only withprimers placed within prophage sequences (positive control),whereas no specific amplicons were achieved using primers run-
ning out of the prophage genome, indicating that no free circu-larized phage genomes are present (Fig. 6f). Furthermore, noPCR products suggestive of prophage excision were obtained withprimers running out of the prophage genome in the culture su-pernatants after filtration (Fig. 6f).
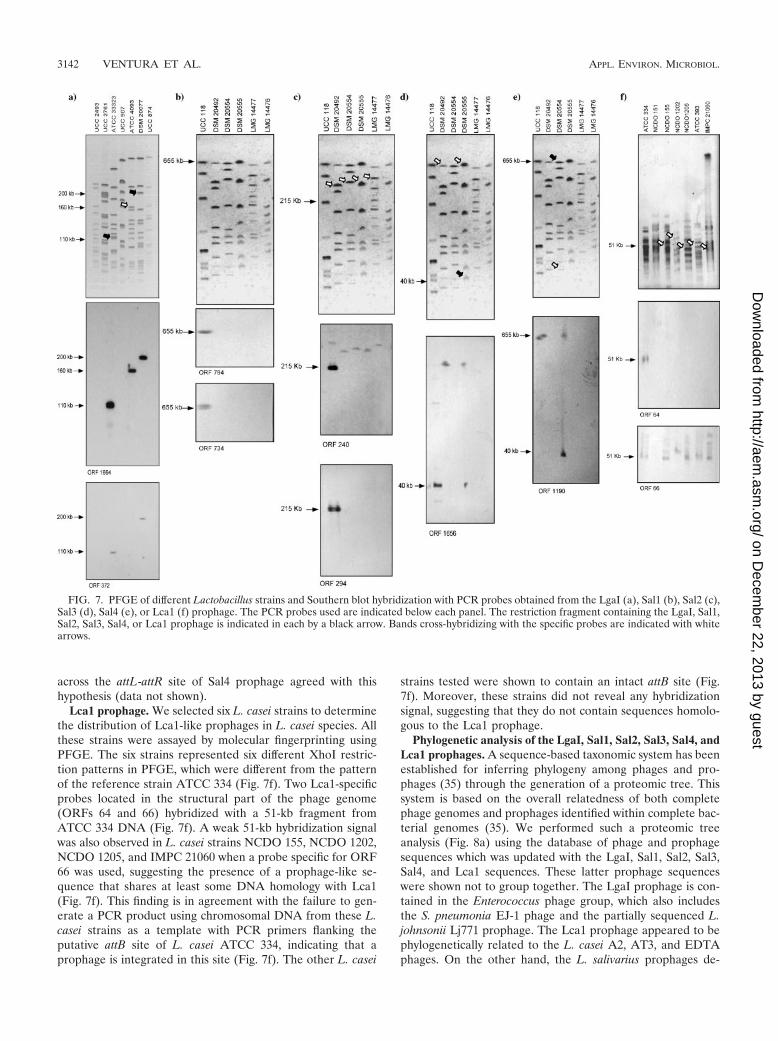
Distribution of L. gasseri, L. salivarius, and L. casei pro-phages. To analyze the distribution of prophage sequences indifferent lactobacillus species, we performed a Southern hy-bridization of PFGE-separated chromosomal digests using aset of probes specific for prophages LgaI, Sal1, Sal2, Sal3, Sal4,and Lca1 (Fig. 7).
LgaI prophage. Seven L. gasseri strains were employed toanalyze the distribution of LgaI prophages (Fig. 7a). The sevenstrains represented seven different SmaI restriction patterns inPFGE, and all were distinct from the pattern of the referencestrain ATCC 33323. In two different hybridization experimentsusing an ORF 1664 (encoding a hypothetical protein)-specificprobe or an ORF 372 (int)-specific probe, hybridization signalswere obtained to a 110-kb fragment from L. gasseri ATCC33323. The probes were selected in a manner that representsthe two borders of the LgaI prophage genome. With the ORF1664-specific probe, a 200-kb hybridization signal in strainDSM 20077 and a 160-kb hybridization signal in strain ATCC4693 were obtained, whereas the ORF 372-specific probeyielded a hybridization signal of 200 kb in strain DSM 20077.Although the LgaI prophage may be restricted to the ATCC33323 strain, it appears that homologs of individual genes arefound in distinctly different L. gasseri strains. This could indi-cate the presence of phages that belong to a lineage related toLgaI (or phage remnants) in L. gasseri species.
Sal1, Sal2, Sal3, and Sal4 prophages. We used all L. saliva-rius strains deposited in several international bacteria collec-tions (i.e., ATCC, DSM, LMG, and JCM) to analyze the dis-tribution of the Sal1, Sal2, Sal3, and Sal4 prophages. All fivestrains used represented five different SmaI restriction patternsin PFGE analysis. Two Sal1-specific probes, located in thelysogeny module (ORF 734) and in the structural encodingregion (ORF 794), hybridized exclusively with a 655-kb frag-ment from UCC 118 DNA (Fig. 7b), suggesting the absence ofSal1-like prophages in the tested strains or at least the absenceof homologous regions of the probes used. A Sal2-specificprobe covering the ORF 294 hybridized exclusively with a215-kb fragment in the UCC 118 strain. In contrast, a probecorresponding to the sie gene (ORF 240) produced hybridiza-tion signals with all four L. salivarius strains (Fig. 7c), suggest-ing the presence of prophages containing sie-like genes in thesestrains.
A Sal3-specific probe covering the ORF 1656 hybridizedwith a fragment of about 40 kb from L. salivarius UCC 118 andL. salivarius DSM 20555 and also produced hybridization sig-nals with a fragment of 655 kb in L. salivarius DSM 20492 andL. salivarius DSM 20555 (Fig. 7d). This would therefore indi-cate that Sal3 prophage-like elements are also present in otherL. salivarius strains. Similarly, a Sal4-specific probe (ORF1190) hybridized with a 655-kb SmaI fragment in L. salivariusUCC 118 as well as in L. salivarius DSM 20554 (Fig. 7e). Thelatter results therefore suggest that this small prophage is notspecific to the UCC 118 strain. Since the prophages in the twostrains are found on SmaI fragments of the same size, theymight be located at corresponding sites in these strains. PCR
VOL. 72, 2006 COMPARATIVE GENOMICS OF LACTOBACILLUS PROPHAGES 3141
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from
across the attL-attR site of Sal4 prophage agreed with thishypothesis (data not shown).
Lca1 prophage. We selected six L. casei strains to determinethe distribution of Lca1-like prophages in L. casei species. Allthese strains were assayed by molecular fingerprinting usingPFGE. The six strains represented six different XhoI restric-tion patterns in PFGE, which were different from the patternof the reference strain ATCC 334 (Fig. 7f). Two Lca1-specificprobes located in the structural part of the phage genome(ORFs 64 and 66) hybridized with a 51-kb fragment fromATCC 334 DNA (Fig. 7f). A weak 51-kb hybridization signalwas also observed in L. casei strains NCDO 155, NCDO 1202,NCDO 1205, and IMPC 21060 when a probe specific for ORF66 was used, suggesting the presence of a prophage-like se-quence that shares at least some DNA homology with Lca1(Fig. 7f). This finding is in agreement with the failure to gen-erate a PCR product using chromosomal DNA from these L.casei strains as a template with PCR primers flanking theputative attB site of L. casei ATCC 334, indicating that aprophage is integrated in this site (Fig. 7f). The other L. casei
strains tested were shown to contain an intact attB site (Fig.7f). Moreover, these strains did not reveal any hybridizationsignal, suggesting that they do not contain sequences homolo-gous to the Lca1 prophage.
Phylogenetic analysis of the LgaI, Sal1, Sal2, Sal3, Sal4, andLca1 prophages. A sequence-based taxonomic system has beenestablished for inferring phylogeny among phages and pro-phages (35) through the generation of a proteomic tree. Thissystem is based on the overall relatedness of both completephage genomes and prophages identified within complete bac-terial genomes (35). We performed such a proteomic treeanalysis (Fig. 8a) using the database of phage and prophagesequences which was updated with the LgaI, Sal1, Sal2, Sal3,Sal4, and Lca1 sequences. These latter prophage sequenceswere shown not to group together. The LgaI prophage is con-tained in the Enterococcus phage group, which also includesthe S. pneumonia EJ-1 phage and the partially sequenced L.johnsonii Lj771 prophage. The Lca1 prophage appeared to bephylogenetically related to the L. casei A2, AT3, and EDTAphages. On the other hand, the L. salivarius prophages de-
FIG. 7. PFGE of different Lactobacillus strains and Southern blot hybridization with PCR probes obtained from the LgaI (a), Sal1 (b), Sal2 (c),Sal3 (d), Sal4 (e), or Lca1 (f) prophage. The PCR probes used are indicated below each panel. The restriction fragment containing the LgaI, Sal1,Sal2, Sal3, Sal4, or Lca1 prophage is indicated in each by a black arrow. Bands cross-hybridizing with the specific probes are indicated with whitearrows.
3142 VENTURA ET AL. APPL. ENVIRON. MICROBIOL.
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

FIG. 8. (a) Phage proteomic tree illustrating the relationship between LgaI, Sal1, Sal2, Sal3, Sal4, and Lca1 prophages and other sequencedphages and prophages. (b) Phylogenetic tree based on the 16S rRNA gene from various Lactobacillus strains. The tree is based on the more closelyrelated phage and prophage sequences of LgaI, Sal1, Sal2, Sal3, Sal4, and Lca1 deduced on the basis of a previous proteomic tree based on 476sequenced phage genomes and prophages (data not shown). In panel b, bootstrap values are reported for a total of 1,000 replicates.
3143
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

scribed here are not located within the same phylogeneticgroup. In fact, the two largest L. salivarius prophages, i.e., Sal1and Sal2, are closely related to the Bacillus licheniformisphages, whereas the two prophage remnants Sal3 and Sal4cluster with a number of Bacillus subtilis and E. faecalis phages(Fig. 8a).
Furthermore, we compared the evolutionary development ofthese prophage sequences and those of their bacterial hosts. Aphylogenetic analysis using a classical molecular marker suchas the 16S rRNA gene sequences revealed significant discrep-ancies with the evolutionary development of the phage se-quences (Fig. 8b). The phylogenetic tree of the bacterial hostsclearly showed a branching of close bacterial taxa such as L.gasseri and L. johnsonii, whereas in the proteomic tree, L.gasseri phages, such as LgaI and adh, do not cluster with L.johnsonii phages, such as Lj928 and Lj965.
DISCUSSION
Many bacterial genome sequences deposited in public data-bases contain integrated phage DNA, which often constitutes asizable part of the total bacterial DNA. Among the availableLactobacillus genomes, which include bacteria of relevant in-dustrial and ecological interest, all sequenced species except L.delbrueckii subsp. bulgaricus contain prophage sequences. Pro-phages can be present in many different forms ranging frominducible prophages to prophages showing apparent (small)deletions, insertions, and rearrangements, to prophage rem-nants that appear to have lost most of their genome. Theamount of prophage sequence in Lactobacillus genomes isvariable, ranging from polylysogenic strains, e.g., L. plantarumWCFS1 (20) and L. johnsonii NCC 533 (33), to strains con-taining prophage remnants, e.g., Lactobacillus acidophilusNCFM genome (2). In contrast, some strains that have beensubjected to extensive industrial processing such as Lactoba-cillus helveticus DPC4571 do not contain any prophage-likesequence (M. Callanan, personal communication). The host-parasite interaction constitutes a highly dynamic equilibrium.Prophages will kill the lysogenic cell upon their induction.
The genome sequencing of L. gasseri ATCC 33323, L. sali-varius UCC 118, and L. casei ATCC 334 contributed six newprophage sequences to the already existing repertoire of Lac-tobacillus prophage sequences (40–41). This extended databasewill not only substantially increase the volume of the currentphage database but also provide a greater phylogenetic breadth tothe present database. Interestingly, phylogenetic relationships ofcurrently available Lactobacillus prophage sequences revealed avery different phylogenetic image from that of their bacterialhosts. This indicates that prophage sequences and bacterial hostshave followed a different evolutionary development. These find-ings are in contrast to the phage-host coevolution hypothesis ofRohwer et al. (35), perhaps as a consequence of the enlargedLactobacillus phage database. These observations indicate thatLactobacillus prophage sequences are an example of genetic mo-saicism apparently arising from nonhomologous recombinationbetween ancestral sequences following a web-like, rather than atree-like, phylogeny.
Notably, with respect to overall genome organization, theLgaI, Sal1, Sal2, Sal3, Sal4, and Lca1 prophages were shown tobelong to the group of Sfi11-like pac site Siphoviridae (34),
which also contain L. plantarum prophages Lp1 and Lp2 (41),L. johnsonii prophages Lj928 and Lj965 (40), �g1e (21), and L.delbrueckii phage LL-H (31). However, very limited sequencesimilarity was observed between the presumptive structuralphage proteins of these new prophage sequences. The struc-tural gene cluster of LgaI and Lca1 prophage sequences be-longed to a widely distributed lineage of Sfi11-like phagesdetected in LAB genera, e.g., S. pyogenes and L. lactis. Sincesequence-related phages were isolated from different bacterialgenera, it is unlikely that these new Sfi11-like phages haveevolved within the confines of one of these bacterial genera. Infact, Sfi11-like phages share part of the genome organizationand even distant sequence relatedness with lambdoid phagesinfecting gram-negative bacteria, suggesting descent from astructural phage module of a common but rather distant an-cestor (34).
Interestingly, phages isolated from closely related taxa thatoccupy very similar ecological niches could be used as a testcase in order to investigate lateral gene transfer between Lac-tobacillus phages. In this context, L. gasseri and L. johnsonii aretwo closely related species belonging to the L. acidophiluscluster B that share the same ecological niches (gastrointesti-nal tract of human and animals). The genome similarity shownby the LgaI prophage and by the available partial Lj771 pro-phage sequence reflects the overall similarity of the genomesequences of their hosts (unpublished results). Comparativephage genomics has suggested that phages may have evolvedthrough exchange of functional modules, individual genes, orgene segments via various genetic recombination events (26).Since L. gasseri and L. johnsonii share the same ecologicalniche, it is possible that horizontal gene transfer and recombi-nation events may have occurred between some phages of L.gasseri and L. johnsonii origin. Alternatively, a common ances-tor phage infecting one species may have acquired the capacity,not necessarily via horizontal gene transfer and/or recombina-tion events, to infect the other bacterial species. The high DNAsimilarity shown by LgaI and Lj771 (10) and EJ-1 (36) maysuggest a capacity for interspecies cross-infection similar tothat reported for L. plantarum and L. brevis phages (25). Fur-thermore, the predicted existence of DNA uptake systems invarious sequenced lactobacilli (2, 20, 33) may also have al-lowed modular exchanges between bacteriophages that infectdifferent species.
Previous phage transcription studies in Streptococcus, Lac-tococcus, and Lactobacillus have shown that large parts of theprophage genome were transcriptionally silent during the ly-sogenic state, while genes near either attachment site werehighly transcribed (38, 40, 41, 43). A similar pattern was iden-tified for L. gasseri LgaI and L. salivarius Sal1, Sal2, Sal3, andSal4. Transcription analysis revealed the presence of mRNAencompassing the presumed phage repressor and superinfec-tion exclusion genes and the lack of expression of the cro-likegene. Furthermore, all prophage genome regions correspond-ing to the structural part of the phage and DNA replicationappeared to be transcriptionally silent. Interestingly, the lysin-encoding genes in Sal1 and Sal2 prophages were shown to betranscribed, although the significance of this finding is stillunknown in terms of culture lysis. In fact, either the transcriptmay not be translated in significant amounts or the corre-sponding enzyme is not functional. Another hypothesis is that
3144 VENTURA ET AL. APPL. ENVIRON. MICROBIOL.
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

it acts as a hok/sok-like portioning system; however, no homol-ogy to these systems has been identified. The experimentalobservations accord with the theoretical expectations in thatthe genes encoding immunity functions (cI and sie) should beexpressed during the lysogenic state. Notably, the genes lo-cated between the lysin and the attR (presumed lysogenic con-version region) were highly transcribed in the LgaI prophage.A previous study identified an alternative candidate for thelysogenic conversion region within the lysogeny module (41).Of note, in Sal1 the DNA region encompassing ORFs 448 to442, which encodes a putative transposase and hypotheticalprotein, was transcribed during the lysogenic state. In phagesinfecting bacterial pathogens, this region carries genes thatmay contribute to a selective advantage to the lysogens (11,19). In many dairy prophages it has been reported that thetranscription of genes located in the lysogenic conversion re-gion is more prominent than that of the phage repressor (38,43). These observations suggest a physiological function forthese prophage genes in the lysogen. So far, the lack of func-tional characterization to support the bioinformatic predic-tions for these phage genes makes it difficult to speculate aboutpossible lysogenic conversion phenotypes. Conversely, theLgaI genome contains a small gene within the lysogenic con-version region, which showed a very limited similarity at theamino acid level with peptide toxin Fst encoded by the partoxin-antitoxin stability determinant. These systems have beendetected in different bacteria, including in the pau-LA III rem-nant of L. acidophilus NCFM (2) and in E. coli phage P1 (16,32). These par systems act in general as a maintenance killersystem of prophages or plasmids. However, the identity be-tween the LgaI gene and fst is very low, and therefore nodefinitive role can be attributed to these genes in this L. gasseriprophage.
Paradoxically, all prophage sequences that have been iden-tified in Lactobacillus genomes appear to be very stable sinceattempts to induce their excision have failed so far (40, 41).However, LgaI and Sal2 appeared to be complete phages thatcan be excised from their bacterial host. Interestingly, althoughthe Sal4 prophage-like element appears to represent a defi-cient bacteriophage, it was shown to be inducible and perhapsconstitutes a functional satellite phage that can become mobilein a manner similar to that described for the cryptic myco-phages Rv1 and Rv2 (22).
The mobility of Lactobacillus prophage DNA was assessedby PFGE hybridization. As demonstrated in this study, DNAfrom the majority of a representative set of strains for each ofthe species tested indicates a narrow range of distribution in L.gasseri, L. salivarius, and L. casei species. This observationconfirms a previous study where two L. johnsonii NCC 533prophages were found to constitute a substantial part of thestrain-specific DNA (39). Similarly, prophages of L. plantarumWCFS1 seem to be narrowly distributed within their own bac-terial species (41).
A number of additional Lactobacillus genomes are currentlybeing sequenced, making it likely that other prophage se-quences will be identified. This increased resource of (Lacto-bacillus) phage sequences will expand our ability to provideanswers by comparative genomics to questions such as thoseconcerning horizontal versus vertical DNA transfer within dif-ferent species of Lactobacillus.
ACKNOWLEDGMENTS
This work was financially supported by the Department of Agricul-ture and Food FIRM Program under the National Development Plan2000–2006, by the Higher Education Authority Programme for Re-search in Third Level Institutions (PRTLI3), by the Science Founda-tion Ireland Alimentary Pharmabiotic Centre located at UniversityCollege Cork, by a Marie Curie Development Host Fellowship(HPMD-2000-00027) to M.V., by an IRCSET Embark postdoctoralfellowship scheme 2005 to C.C., and by Dairy Management Inc. Effortsat North Carolina State University were supported by the North Caro-lina Dairy Foundation, Southeast Dairy Foods Research Center, andDanisco USA, Inc. (Madison, WI).
Genome sequencing of L. gasseri was carried out by the JointGenome Institute of the U.S. Department of Energy and FidelitySystems, Inc., in conjunction with the Lactic Acid Bacteria GenomeConsortium.
Finally, we thank Paul Ross, Mike Callanan, and Tom Beresford forsharing with us their unpublished results on L. helveticus DPC4571genome sequences.
REFERENCES
1. Altermann, E., J. R. Klein, and B. Henrich. 1999. Primary structure andfeatures of the genome of the Lactobacillus gasseri temperate bacteriophage�adh. Gene 236:333–346.
2. Altermann, E., W. M. Russell, M. A. Azcarate-Peril, R. Barrangou, B. L.Buck, O. McAuliffe, N. Souther, A. Dobson, T. Duong, M. Callanan, S. Lick,A. Hamrick, R. Cano, and T. R. Klaenhammer. 2005. Complete genomesequence of the probiotic lactic acid bacterium Lactobacillus acidophilusNCFM. Proc. Natl. Acad. Sci. USA 102:3906–3912.
3. Altschul, S. F., T. L. Madden, A. A. Shaffer, J. Zhang, Z. Zhang, W. Miller,and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generationof protein database search programs. Nucleic Acids Res. 25:3389–3402.
4. Beres, S. B., G. L. Sylva, K. D. Barbian, B. Lei, J. S. Hoff, N. D. Mammarella,M. Y. Liu, J. C. Smoot, S. F. Porcella, L. D. Parkins, D. S. Campbell, T. M.Smith, J. K. McCormick, D. Y. Leung, P. M. Shlievert, and J. M. Musser.2002. Genome sequence of a serotype M3 strain of group A Streptococcus:phage-encoded toxins, the high-virulence phenotype and clone emergence.Proc. Natl. Acad. Sci. USA 99:10078–10083.
5. Boyce, J. D., B. E. Davidson, and A. J. Hiller. 1995. Identification of pro-phage genes expressed in lysogens of the Lactococcus lactis bacteriophageBK5-T. Appl. Environ. Microbiol. 61:4099–4104.
6. Boyd, E. F., and H. Brussow. 2002. Common themes among bacteriophage-encoded virulence factors and diversity among the bacteriophages involved.Trends Microbiol. 10:521–529.
7. Canchaya, C., C. Proux, A. Bruttin, and H. Brussow. 2003. Prophage genom-ics. Microbiol. Mol. Biol. Rev. 67:238–276.
8. Casjens, S., G. Hatfull, and R. Hendrix. 1992. Evolution of dsDNA tailed-bacteriophage genomes. Semin. Virol. 3:383–397.
9. Chopin, A., A. Bolotin, A. Sorokin, S. D. Ehrlich, and M. Chopin. 2001.Analysis of six prophages in Lactococcus lactis IL1403: different geneticstructure of temperate and virulent phage populations. Nucleic Acids Res.29:644–651.
10. Desiere, F., and H. Brussow. 2000. Comparative genomics of the late genecluster from Lactobacillus phages. Virology 275:294–305.
11. Desiere, F., W. M. McShan, D. van Sinderen, J. J. Ferretti, and H. Brussow.2001. Comparative genomics reveals close genetic relationships betweenphages from dairy bacteria and pathogenic streptococci: evolutionary impli-cations for prophage-host interactions. Virology 288:325–341.
12. Felsenstein, J. 1993. PHYLIP (phylogeny inference package) version 3.5c.Department of Genetics, University of Washington, Seattle, Wash.
13. Foley, S., A. Bruttin, and H. Brussow. 2000. Widespread distribution of agroup I intron and its three deletion derivatives in the lysin gene of Strep-tococcus thermophilus bacteriophages. J. Virol. 74:611–618.
14. Fujishawa, T., Y. Benno, T. Yaeshima, and T. Mitsuoka. 1992. Taxonomicstudy of the Lactobacillus acidophilus group, with recognition of Lactobacil-lus gallinarum sp. nov. and Lactobacillus johnsonii sp. nov. and synonym ofLactobacillus acidophilus group A3 (Johnson et al. 1980) with type-strain ofLactobacillus amylovorus (Nakamura 1981). Int. J. Syst. Bacteriol. 42:487–491.
15. Hendrich, B., B. Binishofer, and U. Blasi. 1995. Primary structure andfunctional analysis of the lysis genes of Lactobacillus gasseri bacteriophagephi adh. J. Bacteriol. 177:723–732.
16. Jansen, R., J. D. van Embden, W. Gaastra, and L. M. Schouls. 2002. Iden-tification of a novel family of sequence repeats among prokaryotes. OMICS6:23–33.
17. Johnson, J. L., C. F. Phelps, C. S. Cummins, J. London, and F. Gasser. 1980.Taxonomy of the Lactobacillus acidophilus group. Int. J. Syst. Bacteriol.30:53–68.
18. Kandler, O., and N. Weiss. 1986. Regular, nonsporing gram-positive rods, p.
VOL. 72, 2006 COMPARATIVE GENOMICS OF LACTOBACILLUS PROPHAGES 3145
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from

1208–1260. In P. H. A. Sneath, N. S. Mair, M. E. Sharpe, and J. G. Holt (ed.),Bergey’s manual of systematic bacteriology, vol. 2. William & Wilkins, Bal-timore, Md.
19. Kaneko, J., T. Kimura, S. Narita, T. Tomita, and Y. Kamio. 1998. Completenucleotide sequence and molecular characterization of the temperate staph-ylococcal bacteriophage PVL carrying Panton-Valentine leukocidin genes.Gene 215:57–67.
20. Kleerebezem, M., J. Boekhorst, R. van Kranenburg, D. Molenaar, O. P.Kuipers, R. Leer, R. Tarchini, S. A. Peters, H. M. Sandbrink, M. W. Fiers,W. Stiekema, R. M. Lankhorst, P. A. Bron, S. M. Hoffer, M. N. Groot, R.Kerkhoven, M. de Vries, B. Ursing, W. M. de Vos, and R. J. Siezen. 2003.Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl.Acad. Sci. USA 100:1990–1995.
21. Kodaira, K. I., M. Kakikawa, N. Watanabe, M. Hirakawa, K. Yamada, andA. Taketo. 1997. Genome structure of the Lactobacillus temperate phage�g1e: the whole genome sequence and the putative promoter/repressorsystem. Gene 187:45–53.
22. Lindqvist, B. H., G. Deho, and R. Calendar. 1993. Mechanisms of genomepropagation and helper exploitation by satellite phage P4. Microbiol. Rev.57:683–702.
23. Lipman, D. J., and W. R. Pearson. 1985. Rapid and sensitive protein simi-larity searches. Science 227:1435–1441.
24. Lowe, T. M., and S. R. Eddy. 1997. tRNAscan-SE: a program for improveddetection of transfer RNA genes in genomic sequence. Nucleic Acids Res.25:955–964.
25. Lu, Z., F. Breidt, Jr., H. P. Fleming, E. Altermann, and T. R. Klaenhammer.2003. Isolation and characterization of a Lactobacillus plantarum bacterio-phage, �JL-1, from a cucumber fermentation. Int. J. Food Microbiol. 84:225–235.
26. Lucchini, S., F. Desiere, and H. Brussow. 1999. Comparative genomics ofStreptococcus thermophilus phage species supports a modular evolution the-ory. J. Virol. 73:8647–8656.
27. Lucchini, S., F. Desiere, and H. Brussow. 1999. Similarly organized lysogenymodules in temperate Siphoviridae from low GC content gram-positive bac-teria. Virology 263:427–435.
28. McGrath, S., G. F. Fitzgerald, and D. van Sinderen. 2002. Identification andcharacterization of phage-resistance genes in temperate lactococcal bacte-riophages. Mol. Microbiol. 43:509–520.
29. McGrath, S., J. Seegers, G. F. Fitzgerald, and D. van Sinderen. 1999. Mo-lecular characterization of a phage-encoded resistance system in Lactococcuslactis. Appl. Environ. Microbiol. 65:1891–1899.
30. Mendz, G. L., and S. L. Hazell. 1996. The urea cycle of Helicobacter pylori.Microbiology 142:2959–2967.
31. Mikkonen, M., L. Dupont, T. Alatossava, and P. Ritzenthaler. 1996. Defec-tive site-specific integration elements are present in the genome of virulent
bacteriophage LL-H of Lactobacillus delbrueckii. Appl. Environ. Microbiol.62:1847–1851.
32. Pecota, D. C., C. S. Kim, K. Wu, K. Gerdes, and T. K. Wood. 1997. Com-bining the hok/sok, parDE, and pnd postsegregational killer loci to enhanceplasmid stability. Appl. Environ. Microbiol. 63:1917–1924.
33. Pridmore, R. D., B. Berger, F. Desiere, D. Vilanova, C. Barretto, A. C. Pittet,M. C. Zwahlen, M. Rouvet, E. Altermann, R. Barrangou, B. Mollet, A.Mercenier, T. Klaenhammer, F. Arigoni, and M. A. Schell. 2004. The ge-nome sequence of the probiotic intestinal bacterium Lactobacillus johnsoniiNCC 533. Proc. Natl. Acad. Sci. USA 101:2512–2517.
34. Proux, C., D. van Sinderen, J. Suarez, P. Garcia, V. Ladero, G. F. Fitzgerald,F. Desiere, and H. Brussow. 2002. The dilemma of phage taxonomy illus-trated by comparative genomics of Sfi21-like Siphoviridae in lactic acid bac-teria. J. Bacteriol. 184:6026–6036.
35. Rohwer, F., and R. Edwards. 2002. The phage proteomic tree: a genomebased taxonomy for phage. J. Bacteriol. 184:4529–4535.
36. Romero, P., R. Lopez, and E. Garcia. 2004. Genomic organization andmolecular analysis of the inducible prophage EJ-1, a mosaic myovirus froman atypical pneumococcus. Virology 322:239–252.
37. Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratorymanual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor,N.Y.
38. Ventura, M., A. Bruttin, C. Canchaya, and H. Bruessow. 2002. Transcriptionanalysis of Streptococcus thermophilus phages in the lysogenic state. Virology302:21–32.
39. Ventura, M., C. Canchaya, and H. Brussow. 2003. Integration and distribu-tion of Lactobacillus johnsonii prophages. J. Bacteriol. 185:4603–4608.
40. Ventura, M., C. Canchaya, and H. Brussow. 2004. The prophages of Lacto-bacillus johnsonii NCC 533: comparative genomics and transcription analysis.Virology 320:229–242.
41. Ventura, M., C. Canchaya, M. Kleerebezem, W. M. de Vos, R. Siezen, and H.Brussow. 2003. The prophages sequences of Lactobacillus plantarum strainWCFS1. Virology 316:245–255.
42. Ventura, M., J. H. Lee, C. Canchaya, R. Zink, S. Leahy, J. A. Moreno-Munoz, M. O’Connell-Motherway, D. Higgins, G. F. Fitzgerald, D. J.O’Sullivan, and D. van Sinderen. 2005. Prophage-like elements in bifidobac-teria: insights from genomics, transcription, integration, distribution andphylogenetic analysis. Appl. Environ. Microbiol. 71:8692–8705.
43. Ventura, M., S. Foley, A. Bruttin, C. Canchaya, and H. Brussow. 2003.Transcription mapping as a tool in phage genomics: the case of temperateStreptococcus thermophilus phage Sfi21. Virology 296:62–76.
44. Walker, D. C., H. S. Girgis, and T. R. Klaenhammer. 1999. The groESLchaperone operon of Lactobacillus johnsonii. Appl. Env. Microbiol. 65:3033–3041.
3146 VENTURA ET AL. APPL. ENVIRON. MICROBIOL.
on Decem
ber 22, 2013 by guesthttp://aem
.asm.org/
Dow
nloaded from




















