
Theoretical investigation of CO adsorption on Pd(111) and Pd(111) --- Zn systems
Upload
independentCategory
view
1download
0
Surface Science 191 (1987) 203-224 203 North-Holland, Amsterdam
C O L L I S I O N S O F N O ( X 2I I ) W I T H A A g ( l l l ) S U R F A C E :
V A L I D I T Y O F T H E E N E R G Y S U D D E N A P P R O X I M A T I O N
Gregory C. C O R E Y *'* *, Jane E. S M E D L E Y , Mi l la rd H. A L E X A N D E R
Department of Chemistry, University of Maryland, College Park, Maryland 20742, USA
a n d
W i n g - K i L I U *
Guelph- Waterloo Program for Graduate Work in Physics and Department of Physics, University of Waterloo, Waterloo, Ontario, Canada N2L 3GI
Received 16 March 1987; accepted for publication 1 May 1987
Energy sudden calculations of the rotationally inelastic scattering of ground-state NO(X 2II) from an uncorrugated, rigid Ag(111) surface have been carried out. In this approximation, the inelastic transition probabilities are obtained by solution of a fixed-orientation, single-channel Schr~Sdinger equation, which must be solved for each of the two adiabatic potential energy surfaces which arise when a molecule in a 21-I electronic state interacts with a surface. These two potential surfaces were based on a description of the NO-Ag(l l l ) potential used by Muhlhausen, Williams and Tully [J. Chem. Phys. 83 (1985) 2594]. Comparison of the calculated transition probabilities with the results of our previous close-coupling calculations at a collision energy of 6700 cm-1 shows that for this potential, which possesses a deep, strongly anisotropic, and fairly long-ranged attractive region, the sudden approximation predicts significant flux into high rotational levels which are neither energetically nor dynamically accessible. The dependence of the collisional transition probabilities on the symmetry of the electronic wavefunction of the NO A-doublet states, with respect to reflection in the plane of rotation of the molecule, is described reasonably well within the sudden approximation. The sudden results also adequately predict the relative flux into rotational levels associated with the two (~2 =1 /2 and $2 = 3/2) spin-orbit manifolds of NO.
1. I n t r o d u c t i o n
There has been cons iderab le theoret ical in teres t [1_16] in the sca t te r ing of
N O in its X 2yI e lect ronic g r o u n d state f rom A g ( l l l ) surfaces, mo t iva t ed by
the exper imenta l molecu la r b e a m studies of this sys tem [1 -3 ,17 -27] . Mos t
* NSERC of Canada University Research Fellow. * * Present address: Drpartement de Chimie, Universit6 de Montrral, C.P. 6128, Succursale A,
Montrral, Qurbec, Canada H3C 3J7.
0 0 3 9 - 6 0 2 8 / 8 7 / $ 0 3 . 5 0 �9 Elsevier Science Publ ishers B.V.
( N o r t h - H o l l a n d Physics Publ i sh ing Divis ion)

204 (3.C. Core), et al. / Collisions of NO(X 217) with Ag(l l 1)
previous theoretical investigations have ignored the complexities arising from the nonzero spin and orbital electronic angular momentum in the electronic ground state of NO, treating the molecule as if it were in a closed-shell ~Y. state. Recently, however, we have presented several papers [11,12,16,28-32], both formal and computational, in which the open-shell character of NO was specifically included within a fully quantum description of the scattering dynamics. It is only by doing so that one can understand experiments [17,21,23,25] in which the spin-orbit and /o r A-doublet levels [33,34] of the scattered NO molecules are resolved.
Unfortunately, converged quantum close-coupling calculations [16] are expensive in this case, since the number of rotational levels included in the channel basis must be multiplied by a factor of four over that which would be necessary if the molecule were in a ly. electronic state. It is thus desirable to look for simpler ways of treating the scattering dynamics which would, however, still describe accurately the essential quantum features of the scatter- ing of an open-shell molecule. In principle, this would allow one to include the effect of corrugations and surface motion on the scattering dynamics. At present this is virtually impossible to do within a full close-coupling treatment.
Much of our formal analysis [11,12,16] of NO-Ag(111) collisions, as well as earlier studies [6,8] of collisions of NO (treated as a 1Z molecule) and other molecules with surfaces, was based on the energy sudden approximation [13,35-41]. One remaining question is whether this method, which greatly simplifies the scattering calculations, can be used to obtain accurate transition probabilities for inelastic collisions of NO(X 21-I) with Ag(111) surfaces, and, in particular, whether it can describe those features which reflect the open-shell character of the molecule. It is this question which will be addressed in this article.
We shall use the results of our recent close-coupling study [16], which was based on a description of the NO-Ag(111) interaction due to Muhlhausen, Williams and Tully (MWT) [14], to ascertain the validity of the energy sudden approximation for NO-Ag(111) scattering. In the next section we shall review briefly the theory of the scattering dynamics, concentrating on the application of the energy sudden approximation. Then in section 3, we shall describe the interaction potential used in our calculations. The scattering calculations and results will be discussed in sections 4 and 5. We end with a brief conclusion.
2. Collision dynamics: energy sudden limit
Molecules in 21-I electronic states possess either singly- or triply-filled ~r molecular orbitals. Because of the directional properties of the unfilled r orbital, a description of the electrostatic interaction of a / I - s ta te molecule with a surface requires knowledge of two electronic wavefunctions. The degeneracy

G. C Corey et al. / Collisions of NO(X 2fI) with Ag(l 11) 205
of these two electronic wavefunctions of the isolated molecule will be lifted as the molecule approaches a surface. Consequently, as discussed in our earlier papers [11,16], the interaction of a 21-1 molecule with a surface must be described in terms of two electrostatic potential energy surfaces. For an uncorrugated, rigid surface these potentials, which we designate V+(fl, Z ) and V_(f l , Z), will be functions only of r , the angle between the molecular axis and the surface normal, and Z, the distance of the center-of-mass of the molecule above the surface.
In the present application to N O - A g scattering, we assume that fl = 0 corresponds to the N-end of the molecule down and fl = ~r to the O-end down. The two electrostatic potentials are degenerate when the molecule is oriented perpendicular to the surface (fl = 0 and fl = ~r), but differ for all other values of/3. I n particular, when fl = 7r/2 the V+ potential describes the interaction when the electronic wavefunction of the molecule is oriented so that it is symmetric with respect to reflection in a plane parallel to the plane of the surface, whereas the V_ potential describes the interaction when the electronic wavefunction is antisymrnetric with respect to reflection in this plane. When an NO molecule (with a . . .5a227r 1 electron occupancy) lies parallel to the surface, the V+ potential corresponds to the singly-filled 2~r orbital also lying parallel to the surface, while the V_ potential corresponds to the singly-filled 2 ~r orbital pointing down to the surface.
In a pure Hund's case (a) basis [11,33,34] the eigenstates of a molecule in a 2II electronic state can be characterized by the total angular momentum J, its projections M and I2 along space- and body-fixed z-axes, and a symmetry index c (e = + 1). The magnitude of the projection quantum number g2 can be 1 / 2 or 3/2. The symmetry index c designates the two A-doublet states, which are labelled e (with e-- + 1) and f (with e = - 1 ) . The pure Hund's case (a) limit is reached only when BJ << I AI; B and A designate, respectively, the rotational and spin-orbit constants of the molecule. The spin-rotat ion term in the molecular Hamiltonian is diagonal in J, M, and c, but mixes the ~2 = 1 / 2 and 12 -- 3 /2 spin-orbit manifolds. The resultant intermediate coupling wave- functions can be expressed in a pure case (a) basis as [11]:
I J M F l e / f ) =- I JMFtc)
= cos OjIJM, $2 = 1 /2 , e) + sin Oj[JM, ~2 -- 3 /2 , c) (1)
and
[ JMF2e / f ) - I JMF2~)
= - sin 0 s I JM , ~ = 1 /2 , e) + cos Oj [ JM, f2 = 3 /2 , , ) . (2)
Here [JM~2~) is a case (a) wavefunction, the labels F~ and F z designate the lower and upper spin-orbit manifolds, and the mixing angle Oj can be obtained by diagonalization of a 2 x 2 molecular Hamiltonian. In the pure case (a) limit the mixing angle Os equals 0 for a regular 21-I state (A > 0, as, for

206 G.C Core), et al. / Collisions of NO(X2II) with Ag(111)
example, NO) or ~r/2 for an inverted 2II state (A < 0, as, for example, OH(X 2/-/)). At high J the Hund's case (b) limit is approached, with 6j = r
In the application of the energy sudden limit to rotationally inelastic collisions of a molecule in a 1E electronic state with an uncorrugated surface, fl, the angle between the molecular axis and the surface normal, is assumed fixed during the collision. The scattering at fixed orientation is governed by a single-channel SchriSdinger equation. As we have discussed before [32], in the extension of the sudden approximation to collisions of a molecule in a 2II electronic state it is necessary to solve the following single-channel Schrbdinger equation for each of the two adiabatic potentials: (d2/dZ2+k2)C+(fl, Z )= (2m/h2)V+(fl, Z)C+(fl, Z), (3)
where the wavevector k = (2mE)I/2/h, with m and E designating, respec- tively, the mass of the molecule and the total energy. Here V• Z) denotes the symmetric and antisymmetric potential energy surfaces described earlier in this section. These two single-channel Schrrdinger equations are then solved subject to the boundary conditions
lim C+(fl, Z) = 0 (4) Z ----~ - o o
and
lim C• Z) = e - i k z + S• e ikz. (5) Z---* oo
Because the V+ and V_ potentials differ, expect for/3 = 0 and 13 = 7r, the two sudden S functions, S+(/3) and S_(/3), will be equal, in general, only for /3=0 and/3 = ~r.
For rotationally inelastic collisions the state-to-state transition probability is defined by
PJge,,-.J'g'e,', ' = I S:'M'F,','.JMF,, I 2 (6)
As in the case of collisions of molecules in ~E electronic states, the sudden approximation to the S-matrix elements can be evaluated by an expansion in terms of Legendre polynomials. By analogy with the matrix elements of the electrostatic interaction potential [11,16,31,32], for H-state molecules the matrix elements of the sudden S functions can be evaluated most conveniently by expanding the average and difference of S+(/3) and S_(/3), rather than S+(/3) and S_(/3) separately. The average is expanded in ordinary Legendre polynomials, namely
s0(/3) - [ s+( /3) + s _ ( / 3 ) ] / 2 = E S, oe,(cos /3 ). (7) /=0
The expansion coefficients can be obtained by integrating over/3 and using the orthogonality properties of the Legendre polynomials. We find
St0= 2 l + 1 s 4 Pt(cos/3)[S+(/3) + S_(/3)]sin/3 d/3. (8)

G. C. Corey et al. / Collisions of NO(X21-1) with A g( l l l ) 207
The difference between S+(fl) and S_(fl) must, however, be expanded in terms of associated Legendre polynomials of order 2, as follows:
S2(fl) - [S+(/3) - S _ ( B ) ] / 2 = (l + 2)! Sap,2(cos/3). (9)
This equation can be inverted to give
S'2= 4 ( l + 2 ) ! PI2(cos/3)[S+(/3)-S_(fl)] sin/3 d/3. (10)
Note that the $2(/3) function is defined as the difference between the S function for scattering on the V+ potential minus that for scattering on the V_ potential. This is opposite to the definition which is appropriate for gas-phase collisions involving U-state molecules [32]. This difference arises because in the case of gas-phase collisions the symmetry of the electronic wavefunctions, and the labelling of the electrostatic potentials, is defined with respect to reflection in the body-frame xz-plane [31,32]. However, for collisions with surfaces, the symmetry of the electronic wavefunctions is defined with respect to the body-frame yz-plane, which is coincident with the plane of the surface [11]. The symmetry properties of the U-state electronic wavefunctions are not the same for reflection in these two planes [42].
Since the sudden S functions are expanded in terms of the same set of Legendre polynomials used to expand the interaction potential, within the sudden approximation the S-matrix elements in eq, (6) are formally equivalent to the matrix elements of the interaction potential, which we have given earlier [11,16]. The S-matrix element for a transition between the initial state I JMF, e) and the final state I J'M'F,.'d) is given by
( J'M'F/e' I S I JMF,.e) n ,+ , '+ , l : ' s l
=aMM,,--1MLJJ'I/= g ( r 1 v M - M 0 /
)(, , ,)] + B l ( J ' F i ' c ' , JFie 3 3 0
t-~Sl2[Cl(J'fi"" JFir J' JT' -21)
+D,(J'F/E', JF~c) �89 ~ -2 "
In eq. (11) ( : : : ) is a 3j symbol, $/o and S/2 are the expansion coefficients

208 G. C. Core), et al. / Collisions of N O( X 21I) with A g(111)
Table 1 Mixing coefficients in the sudden approximat ion to the S-matr ix elements in an in termediate coupling basis a)
AA J'F,.'d, JF~c) FI' - c o s 0 s, cos Oa cos 0 s, sin Oj Fz' sin 0 s, cos 0 s - s i n Oj, sin 0 s
B,(J'F/g, J~c) Fx' sin Oj, sin 0j sin 0s, cos 0j Fz' cos O.t, sin O s cos Og, sin Og
G ( J ' ~ ' r JF/r F ( sin Oj, cos Oj - s i n Oj, sin Og F2' cos Os, cos Oj - c o s Os, sin Og
Dt( J 'Fi 'c ' , JF,.c ) F ( - c o s Oj, sin Oj - c o s 0 s, cos Og F2' sin 0 s, sin 0 s sin 0 s, cos 0 s
a) See eq. (11) of text.
defined by eqs. (8) and (10), and [JJ'] = (2J + 1)(2J ' + 1) denotes a product of rotational degeneracy factors. The At, Bt, Ct, and D / coefficients, which are products of the sin O., and cos O: factors [eqs. (1) and (2)] for the initial and final states, are given in table 1.
It will be useful in the ensuing discussion to determine simplified expres- sions for the sudden S-matrix elements in eq. (11) which are valid when the initial rotational level at low J can be described in the pure case (a) limit with Og = 0, and the final level at high J ' can be described in the pure case (b) limit with 8j, = r We find, for transitions out of an F1 level:
( J'M'F,.'c'I S I JMFac)
=Sgg, ( - -1)g+~[JJ '] l /2E i 1)S+g'+q t 2 - 5 ) 5 1 1 - ' " ( - ( ~ - M J 0)l
x +st0 _�89 �89 - 2 '
where the plus sign refers to F / = F 1 and the minus sign to F,.'= Fz. The negative sign in front of the cSt2 term in eq. (12) is corrected from eq. (37) of reL [11], which was in error.
From eq. (12) one can show that in the energy sudden limit the probabili- ties for a transition from a low-J, F 1 level into a high J ' level will satisfy the symmetry relations:
PJMFle / f --* J ' M ' F l e / f = P J u & f / e --* J ' M ' F 2 f / e , (13)

G. C. Corey et al. / Collisions of N O( X 2 FI ) with A g( l l l ) 209
and
PJMFIe/f ~ J'M'FI f / e = PJMFI f / e ~ J'M'F2e/f" (14)
Thus if one sums over the initial and final A-doublet levels one predicts that transitions to the F 1 and F 2 levels at high J ' will be equally probable. Also, if we take eqs. (13) and (14) and sum only over the initial A-doublet levels, we find
PJMF 1 -- J 'M'Fte/ f = PJMF, -- J'M'Fzf/e" ( 1 5 )
This implies that of the four possible final levels, for a given J ' , the pair Fie and F 2 f will be populated equally, as will be the pair F2e and F a f-
For a 211 molecule at large J, the wavefunctions of the Fie and Fzf A-doublet states are symmetric [34] with respect to reflection in the plane of rotation of the molecule, and will be probed by P- and R-branch lines of a 2y+_z11 transition (as, for example the A ~ X transition in NO). The wavefunctions of the Fze and Flf levels are antisymrnetrie with respect to reflection in the plane of rotation of the molecule, and will be probed by Q-branch lines of a 2E+-ZlI transition. Thus, within the energy sudden limit, we predict that the final A-doublet levels will be populated in two unequal pairs, with a propensity toward either the two levels which are symmetric with respect to reflections in the plane of rotation or toward the two levels which are antisymmetric.
3. Choice of potential
In our previous close-coupled study [16] we based our description of the NO-Ag(111) potential on the electrostatic interaction potential used by Muhlhausen, Williams and Tully (MWT) [14]. Since this potential was devel- oped for a classical trajectory study of the molecule-surface scattering, in which the spin and electronic angular momenta of the NO molecule were ignored, we equated the MWT potential with the average of the V+ and V_ potentials corresponding to the two different orientations of the singly-filled 2~r orbital of NO. The MWT potential, which takes into account the weak corrugation of the Ag(111) surface, is given as a sum of terms. Some of these involve pair potentials between the N or the O atom in the molecule and the individual Ag atoms in the surface, while other terms depend only on the coordinates Z and ft.
To adapt the MWT potential to our assumptiorr of a rigid, uncorrugated surface, the atom-atom terms were averaged over a unit cell and over all possible azimuthal orientations of the NO axis with respect to the surface normal. This averaged potential was then expanded in a Legendre series
Vavg(/3, Z ) - [V+(/3, Z ) + V ( / 3 , Z ) ] / 2 = ~ Vt0(Z)Pt(cos/3). (16) / = 0

210 G . C Corey et al. / Collisions o f N O ( X 2H) with A g ( l l l )
Table 2 Parameters for the ~o ( Z ) and ~2 ( Z ) coefficients [eqs. (17)-(20),(23)] in the angular expansion of the Vavg(fl, Z ) and Vdlf(fl, Z ) potential energy surfaces; distance is in bohr and energy in hartree (numbers in parentheses denote powers of ten)
C 9.8628 (1) D 2.7401 ( - 1 ) E 1.8481 (-2) Z o 1.8898
I = 0 l = 1 / ~ - 2 1 = 3
A t - 1.65315 - 1.39243 B t - 1.96240 - 1.87826 C t - 3.9665 ( - 1) - 6.034 ( - 2) D t 1.39303 ( - 3 ) - 2.7753 ( - 3 ) - fit 7.57809 ( - 1 ) - 6.9228 ( - 1 ) - z7 5.78 - 5.31 -
The expansion was truncated at the l = 3 term, and the Vt0(Z) coefficients were fit to the following analytic form
V00(Z) = VM(D0, 8o, Z~, Z) + C( Z - Z0) -9 - ( D + E /3 ) ( Z - Zo) -s, (17)
V10(Z) = - C 1 e x p [ - a , z 2 / ( 1 + B ,Z)] , (18)
//20(Z) = VM(D2, 82, Z~, Z) - (2E/S) ( Z - Z0) -3, (19)
Vs0(Z) = - C s e x p [ - A s Z 2 / ( 1 + BsZ)] , (20)
where VM(D I, 8it, ZT, Z) designates a Morse potential, namely
VM(D,, ill, Z~, Z ) = D, e x p [ - f t ( Z - Z~)] ( e x p [ - f t ( Z - Z~)] - 2}. (21)
Values for all the parameters in eqs. (17)-(20) are given in table 2. Knowing Vavg [eq. (16)], one can fully Characterize the V+ and V_ poten-
rials if one also has information on the difference between V+ and V . As discussed briefly in section 2, it is appropriate to expand the difference between V+ and V in terms of associated Legendre polynomials of order 2, namely
,~ 1/2
(/~2)!(l- 2)! ) Vt2pt2 (cos fl). (22) v if(8, z)= [v+(8)- v_(8)]/2= 1=2
Unfortunately, no ab initio calculations of the interaction of NO with Ag( l l l ) have been done, so that no information is available about this difference potential. In our recent close-coupled study [16] of collisions of NO with
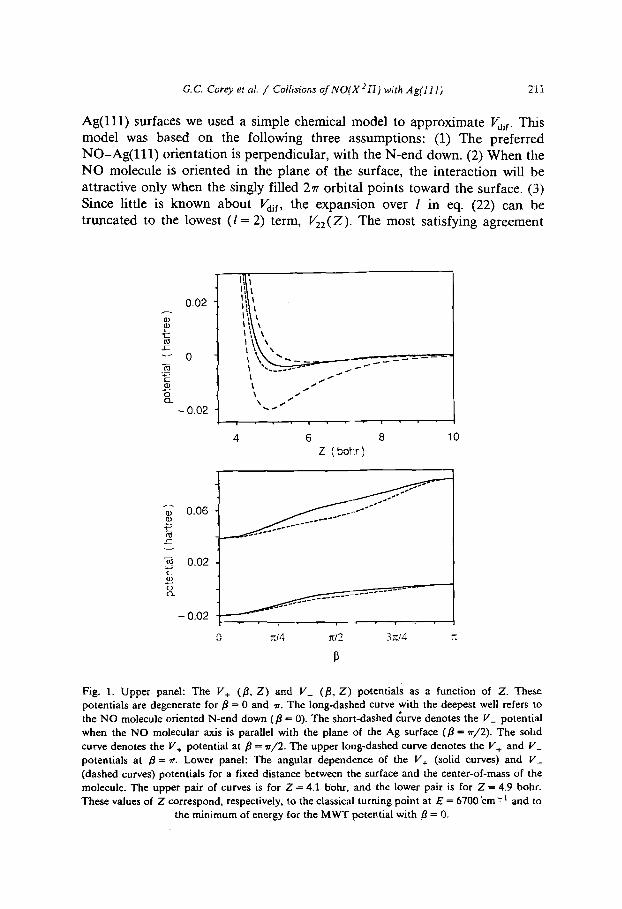
G. C. Corey et aL / Collisions of N O( X 2 IT ) with A g( i t l ) 21~
Ag( l l l ) surfaces we used a simple chemical model to approximate Vai r. This model was based on the following three assumptions: (1) The preferred NO-Ag(111) orientation is perpendicular, with the N-end down. (2) When the NO molecule is oriented in the plane of the surface, the interaction will be attractive only when the singly filled 27r orbital points toward the surface. (3) Since little is known about Vail, the expansion over / in eq. (22) can be truncated to the lowest ( / = 2) term, V22(Z ). The most satisfying agreement
0.02 ~g
~ 0 N
o
- 0.02
,, - - _ _
, �9 , i �9 , �9 L �9 �9 ,
6 8 10 Z (bohr)
"~ 0.06
-~ 0.02
0 o _
- 0.02
Fig. 1. Upper panel: The V+ (/~, Z ) and V_ (,8, Z) potentials as a function of Z. These potentials are degenerate for ,8 = 0 and rr. The long-dashed curve with the deepest well refers to the NO molecule oriented N-end down (,8 = 0). The short-dashed curve denotes the V_ potential when the NO molecular axis is parallel with the plane of the Ag surface (,8 = rr/2). The sohd curve denotes the V , potential at ,8 = 7r/2. The upper long-dashed curve denotes the V+ and V_ potentials at 13 = ~r. Lower panel: The angular dependence of the V+ (solid curves) and V_ (dashed curves) potentials for a fixed distance between the surface and the center-of-mass of the molecule. The upper pair of curves is for Z = 4.1 bohr, and the lower pair is for Z ~ 4.9 bohr. These values of Z correspond, respectively, to the classical turning point at E = 6700 "cm': 1 and to
the minimum of energy for the MWT potential with ~ = 0.

212 G. C Corey et al. / Collisions of N O( X 2 H ) with A g( l l l )
with the experimental data available on NO-Ag(111) scattering was obtained with the following approximation to V=(Z):
V=(Z) = V z o ( Z ) / 2 - [V10(Z)+ V s o ( Z ) ] / 3 , (23)
with Vlo(Z ), Vzo(Z), and Vso(Z ) given by eqs. (18)-(20). The potential defined by eqs. (16)-(23) is designated Potential II in ref. [16]. The corre- sponding V+(fl, Z ) and V ( f l , Z ) potentials are displayed in the upper panel of fig. 1, as a function of Z for fl = 0, rr/2, and rr. For fl = 0 and ~r, the NO molecule is oriented perpendicular to the surface, so that the V+(fl, Z) and V ( f l , Z) potentials are degenerate. Similarly, the lower panel of fig. 1 displays the dependence of the V+(fl, Z) and V_(f l , Z ) potentials on the angle fl for two fixed values of Z, Z = 4.1 bohr and Z = 4.9 bohr, which correspond, respectively, to the classical turning point at E = 6700 cm-1 and the minimum of energy for the MWT potential with fl = 0.
In our previous article we used three different modifications of eq. (23) to explore the dependence of the calculated transition probabilities on the particular choice of the difference between the V+ and V potentials. Since the emphasis of the present article is the assessment of the validity of the energy sudden approximation rather than the dependence of the NO-Ag(111) transition probabilities o.n the details of the potential, we restrict ourselves to only one choice of V+ and V .
4. Scattering calculations
As described in section 2, the two sudden S functions for U-state molecules can be obtained by solving two single-channel fixed-orientation Schr~Sdinger equations for scattering on the V+ and V_ adiabatic potential energy surfaces. The sudden S functions S § and S _ ( f l ) then can be expressed in terms of phase shifts in the usual manner, namely
S • = exp[2i~7 • (24)
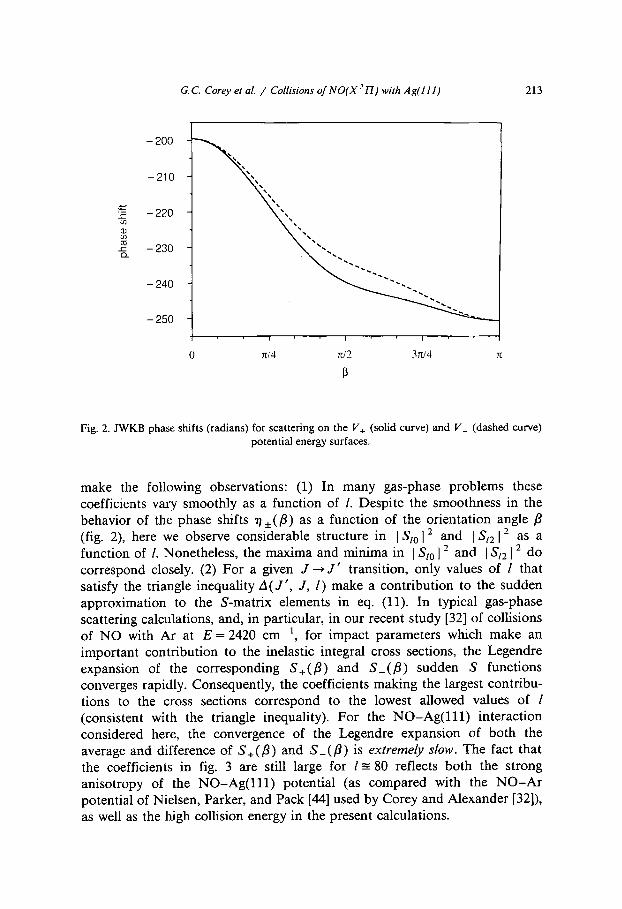
The angle-dependent phase shifts were calculated within the JWKB approxi- mation through a Gauss-Mehler quadrature, as suggested by Pack [43] for molecules in ly, electronic states. These phase shifts, which are displayed in fig. 2, are large and negative but smoothly varying.
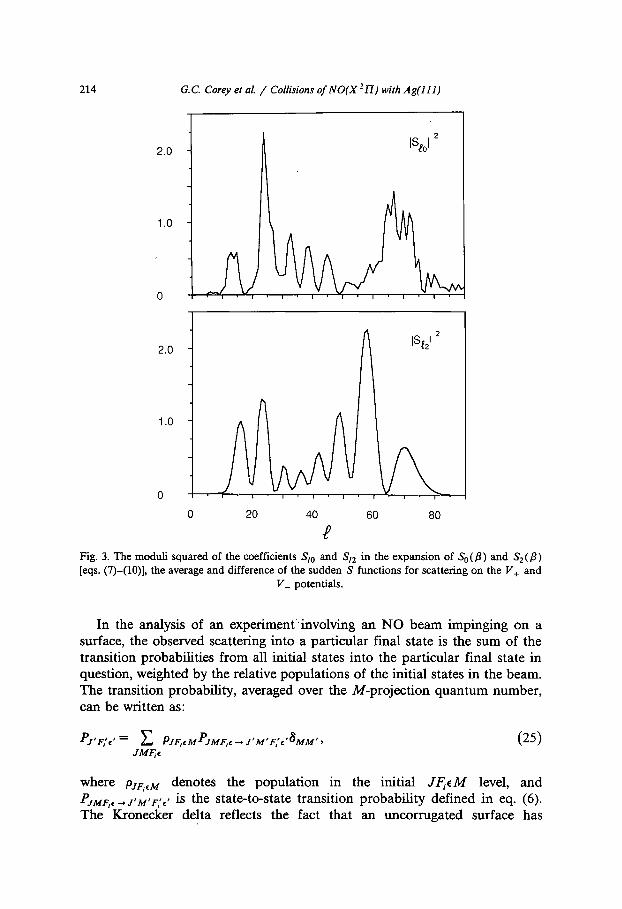
The crucial dynamical quantities in the sudden description of the scattering of a molecule in a zII electronic state with an uncorrugated surface are not the phase shifts ~ +(fl), but rather the coefficients Sto and St2 in the expansion of So(f l) and S2(fl) [eqs. (7)-(10)], the average and difference of the sudden S functions for scattering on the V+ and V_ potentials. The square magnitudes of the St0 and S~2 coefficients are plotted in fig. 3 as a function of l. We can
G.C. Core), et al. / Collisions o f N O( X 2 H ) with A g( l l l ) 213
_c
t - O . .
- 2 0 0
- 2 1 0
- 2 2 0
- 2 3 0
- 2 4 0
- 2 5 0
i I I
0 r d 4 rt/2 3rt/4
Fig, 2. JWKB phase shifts (radians) for scattering on the V+ (solid curve) and V_ (dashed curve) potential energy surfaces.
make the following observations: (1) In many gas-phase problems these coefficients vary smoothly as a function of l. Despite the smoothness in the behavior of the phase shifts 71 • as a function of the orientation angle/3 (fig. 2), here we observe considerable structure in IS~012 and I Snl = as a function of l. Nonetheless, the maxima and minima in Iaz012 and I Snl 2 do correspond closely. (2) For a given J--* J ' transition, only values of l that satisfy the triangle inequality A(J ' , J, I) make a contribution to the sudden approximation to the S-matrix elements in eq. (11). In typical gas-phase scattering calculations, and, in particular, in our recent study [32] of collisions of NO with Ar at E = 2420 cm -1, for impact parameters which make an important contribution to the inelastic integral cross sections, the Legendre expansion of the corresponding S+(/3) and S_(/3) sudden S functions converges rapidly. Consequently, the coefficients making the largest contribu- tions to the cross sections correspond to the lowest allowed values of / (consistent with the triangle inequality). For the N O - A g ( l l l ) interaction considered here, the convergence of the Legendre expansion of both the average and difference of S+(/3) and S_ (/3) is extremely slow. The fact that the coefficients in fig. 3 are still large for l-= 80 reflects both the strong anisotropy of the N O - A g ( l l l ) potential (as compared with the N O - A r potential of Nielsen, Parker, and Pack [44] used by Corey and Alexander [32]), as well as the high collision energy in the present calculations.
214 G.C. Corey et al. / Collisions of NO(X 2 I-I) with A g(111 )
1.0
2.0
2.0 lse012
1.0
I I I i I
2 ISt21
0 20 40 60 80
Fig. 3. The moduli squared of the coefficients Sto and St2 in the expansion of So(fl) and S2(fl) [eqs. (7)-(10)], the average and difference of the sudden S functions for scattering on the V+ and
V_ potentials.
In the analysis of an experiment involving an NO beam impinging on a surface, the observed scattering into a particular final state is the sum of the transition probabilities from all initial states into the particular final state in question, weighted by the relative populations of the initial states in the beam. The transition probability, averaged over the M-projection quantum number, can be written as:
ej,F,,,,= E (25) JMF~.c
where PJF, cM denotes the population in the initial JF~cM level, and PJMF,,-.J'M'F/,' is the state-to-state transition probability defined in eq. (6). The Kronecker delta reflects the fact that an uncorrugated surface has

G.C. Core), et al. / Collisions of NO(X 'H) with A g(l 11) 215
azimuthal symmetry, so that collisions cannot change the magnetic quantum number [11,12].
We assume an unpolarized incident beam, so that PJF,,M is independent of M. We further assume that both A-doublet levels are populated equally, since the A-doublet splitting is too small for significant cooling to take place in the supersonic expansion. If, finally, the distribution of spin-orbit and rotational levels in the incident beam can be characterized by a single temperature T, then the initial populations can be written as
DJF,r = exp ( - [ Ej + A ( i - 1 ) ] / k T }/QrotOso, (26)
where A is the NO spin-orbit constant (A = 123 cm-l) . The quantities Qrot and Q~o designate, respectively, the rotational and spin-orbit partition func- tions. These are defined by
Ofot= 2 ~ (2J + 1) exp( - E j / k T ) (27) J
and
Q~o = 1 + e x p ( - A / k T ) . (28)
The factor 2 in eq. (27) reflects the presence of the degenerate A-doublet levels.
5. Discussion of results
5.1. Transition probabilities summed and averaged over fine-structure states
If we sum the transition probabilities given by eq. (25) over the spin-orbit and A-doublet levels, the resulting probabilities, which we denote P ( J ' ) , can be compared with the results of most of the previous calculations as well as experiments on N O - A g ( l l l ) scattering, in which neither the final A-doublet nor spin-orbit states were resolved. These transition probabilities are dis- played in the right-hand panel of fig. 4 as a function of the final rotational quantum number J ' . The spin-orbit and rotational temperature of the inci- dent beam [eqs. (26)-(28))] was taken to be T = 10 K.
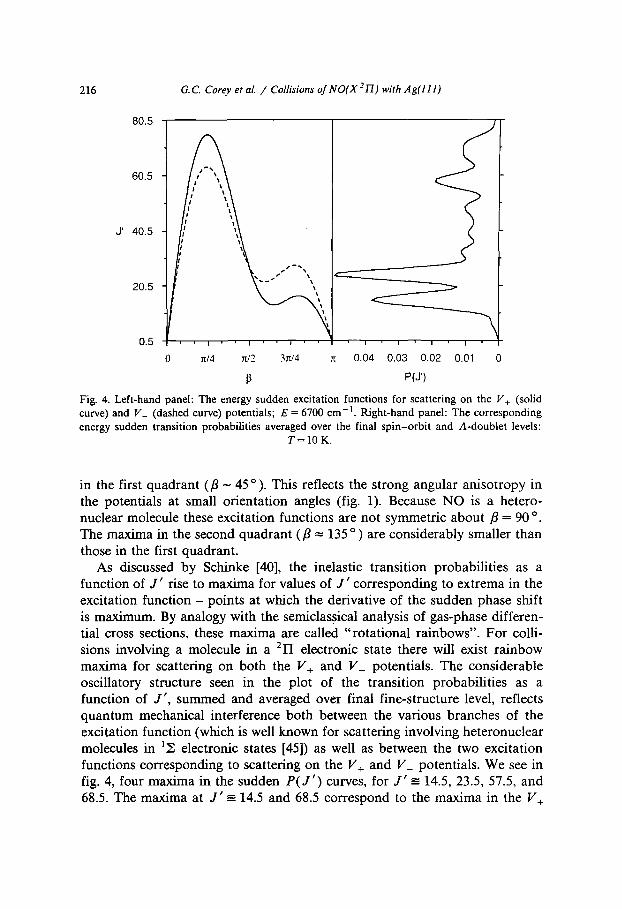
Within a simple impulsive model, in the energy sudden limit the magnitude of the final rotational quantum number J ' as a function of the initial orientation fl of the incident molecule with respect to the surface normal is proportional to the derivative of the phase shift ~ • [eq. (24)]. In the case of the scattering of a molecule in a 2YI electronic state, there will exist two excitation functions ( I J ' I as a function of/3), for scattering on the V+ and V_ potentials. These two excitations functions are plotted in the left-hand panel of fig. 4. The excitation function for both potentials rises to a high maximum
80.5
60.5
J' 40.5
20.5 ' l
\
0.5
216 G.C Corey et al. / Collisions of NO(X 21"I) with Ag(111)
I I I
0 rU4 rt/2 31t/4 ;t
P(J')
~ J
I I I I
0.04 0.03 0.02 0.01 0
Fig. 4. Left-hand panel: The energy sudden excitation functions for scattering on the V§ (solid curve) and V_ (dashed curve) potentials; E = 6700 cm -1. Right-hand panel: The corresponding energy sudden transition probabilities averaged over the final spin-orbi t and A-doublet levels:
T = 1 0 K.
in the first quadrant (fl - 45 o ). This reflects the strong angular anisotropy in the potentials at small orientation angles (fig. 1). Because NO is a hetero- nuclear molecule these excitation functions are not symmetric about fl = 90 o. The maxima in the second quadrant (fl = 135 o) are considerably smaller than those in the first quadrant.
As discussed by Schinke [40], the inelastic transition probabilities as a function of J ' rise to maxima for values of J ' corresponding to extrema in the excitation function - points at which the derivative of the sudden phase shift is maximum. By analogy with the semiclassical analysis of gas-phase differen- tial cross sections, these maxima are called "rotational rainbows". For colli- sions involving a molecule in a 2H electronic state there will exist rainbow maxima for scattering on both the V+ and V_ potentials. The considerable oscillatory structure seen in the plot of the transition probabilities as a function of J ' , summed and averaged over final fine-structure level, reflects quantum mechanical interference both between the various branches of the excitation function (which is well known for scattering involving heteronuclear molecules in ly~ electronic states [45]) as well as between the two excitation functions corresponding to scattering on the V+ and V_ potentials. We see in fig. 4, four maxima in the sudden P(J') curves, for J ' ---- 14.5, 23.5, 57.5, and 68.5. The maxima at J ' -- 14.5 and 68.5 correspond to the maxima in the V+
G. C. Corey et al. / Collisions of NO(X217) with A g( l l l )
0.10 217
13._
0.08
0.06
0.04 ~ . % I " I I
# ~, l I I I l
\, / }, 0.02 / , / . .
" - - ' ' ' ' I ~ i ' I I
0.5 20.5 40.5 60.5 j'
80.5
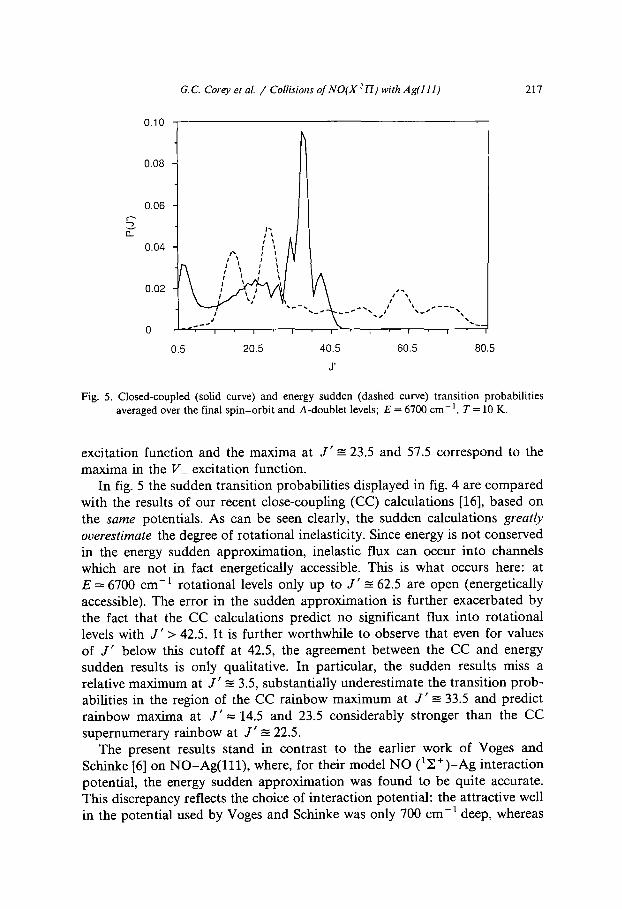
Fig. 5. Closed-coupled (solid curve) and energy sudden (dashed curve) transition probabilities averaged over the final spin-orbit and A-doublet levels; E = 6700 cm -1, T = 10 K.
excitation function and the maxima at J ' ---- 23.5 and 57.5 correspond to the maxima in the V_ excitation function.
In fig. 5 the sudden transition probabilities displayed in fig. 4 are compared with the results of our recent close-coupling (CC) calculations [16], based on the same potentials. As can be seen clearly, the sudden calculations greatly overestimate the degree of rotational inelasticity. Since energy is not conserved in the energy sudden approximation, inelastic flux can occur into channels which are not in fact energetically accessible. This is what occurs here: at E---6700 cm -1 rotational levels only up to J'-=-62.5 are open (energetically accessible). The error in the sudden approximation is further exacerbated by the fact that the CC calculations predict no significant flux into rotational levels with J ' > 42.5. It is further worthwhile to observe that even for values of J ' below this cutoff at 42.5, the agreement between the CC and energy sudden results is only qualitative. In particular, the sudden results miss a relative maximum at J ' ~ 3.5, substantially underestimate the transition prob- abilities in the region of the CC rainbow maximum at J ' -= 33.5 and predict rainbow maxima at J ' = 14.5 and 23.5 considerably stronger than the CC supernumerary rainbow at J ' - 22.5.
The present results stand in contrast to the earlier work of Voges and Schinke [6] on NO-Ag(111), where, for their model NO (1 ~. +)_Ag interaction potential, the energy sudden approximation was found to be quite accurate. This discrepancy reflects the choice of interaction potential: the attractive well in the potential used by Voges and Schinke was only 700 cm- l deep, whereas

218 G. C Corey et al. / Collisions of N O( X 21-l ) with A g( l l l )
the well in the averaged MWT potential is 4300 c m - k As in gas-phase scattering, the energy sudden approximation is expected to be accurate for scattering governed by repulsive potentials. The presence of a deep and anisotropic well (cf. fig. 1) implies that, even at a collision energy of 6700 cm -1, the molecule can be substantially reoriented prior to reaching the repulsive wall of the potential. Thus the assumption of fixed orientaticn scattering will not be physically reasonable.
5.2. Relatioe scattering into the two spin-orbit manifolds
In the experiments of Auerbach [21] and Zare [25] and their co-workers the internal (spin-orbit and rotational) temperature of the incident NO beam is much less than the splitting between the two spin-orbit manifolds (A = 123 cm-l) , so that the impinging molecules are primarily in the lower (F1) spin-orbit manifold. In the case (a) limit the F1 and F 2 manifolds can be characterized by definite values of 12 (~2 = 1 /2 for F~ and ~2 = 3 / 2 for F2). As J increases the 12 = 1 /2 and 12 = 3 /2 wavefunctions become increasingly mixed [eqs. (1) and (2)], although for a given value of J, the splitting between the F 1 and F 2 levels remains roughly constant, and equal to 123 cm -1 in the case of NO.
Since it is possible to resolve spectroscopically the F~ label of the final rotational level, one can compare the scattering into one or the other of the spin-orbit manifolds, as a function of J. As discussed in section 2, within the energy sudden limit, one can formally show that if one sums over the final state A-doublet levels, the transition probability for scattering from a low-J [pure case (a)] level into a high-J F~ [pure case (b)] level will, in fact, be equal to the transition probability for scattering into a high-J F 2 level.
It is obviously worthwhile to determine if this formal prediction is borne out by the N O - A g ( l l l ) transition probabilities calculated here within the energy sudden limit. To do so it is sufficient to examine the following ratio of transition probabilities:
P(F,) + = , ( 2 9 )
P ( F z ) PJ ' , , : I ,F2+ PJ' , ,=-I ,F,
where the quantities PJ','F; are defined in eq. (25). This ratio is displayed in fig. 6.
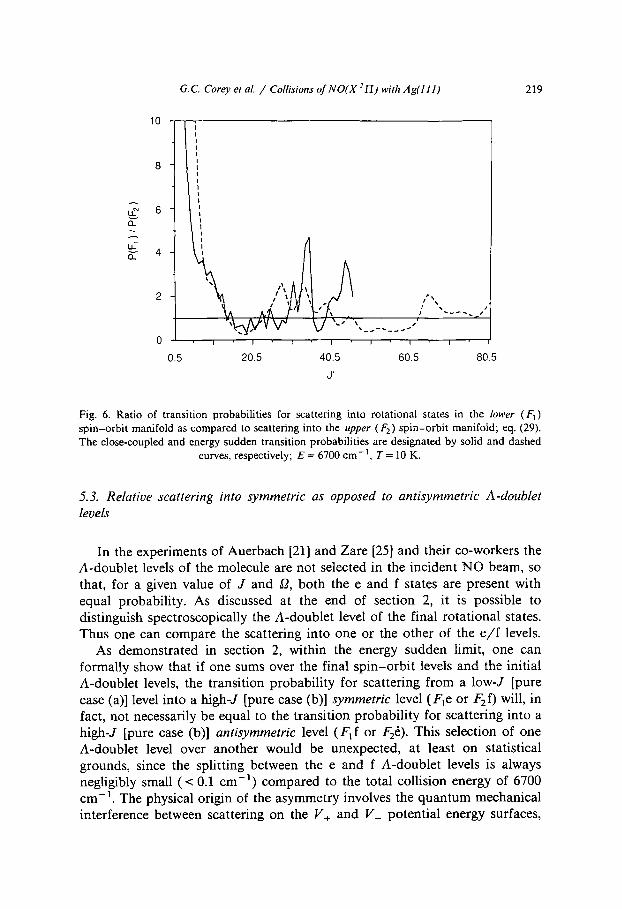
In contrast to the analytic prediction based on eq. (12) the F~ : F 2 ratio oscillates around unity at high J ' . For values of J ' less than the cut-off in the CC transition probabilities ( J ' = 42.5, see fig. 5), the energy sudden and CC results agree reasonably well. In particular, both calculations predict F 1 : F 2 ratios with considerable oscillatory structure that are substantially greater than unity at low J ' , a tendency for preferential population of the higher F2 manifold at intermediate values of J ' and again an F~ : F z ratio greater than unity at higher values of J ' .
G. C. Corey et a L / Collisions of N O( X 217 ) with A g( l l l ) 219
lO
uY 6 0,.
u: 4
0.5 20.5 40.5 60.5 80.5 j ,
Fig. 6. Ratio of transition probabilities for scattering into rotational states in the lower (F 1) spin-orbit manifold as compared to scattering into the upper (F2) spin-orbit manifold; eq. (29). The close-coupled and energy sudden transition probabilities are designated by solid and dashed
curves, respectively; E = 6700 cm-1, T = 10 K.
5.3. Relat ive scattering into symmetr i c as opposed to an t i symmetr ic A-double t
levels
In the experiments of Auerbach [21] and Zare [25] and their co-workers the A-doublet levels of the molecule are not selected in the incident N O beam, so that, for a given value of J and I2, both the e and f states are present with equal probability. As discussed at the end of section 2, it is possible to distinguish spectroscopically the A-doublet level of the final rotational states. Thus one can compare the scattering into one or the other of the e / f levels.
As demonstrated in section 2, within the energy sudden limit, one can formally show that if one sums over the final sp in-orb i t levels and the initial A-doublet levels, the transition probabili ty for scattering from a low-J [pure case (a)] level into a high-J [pure case (b)] symmetr i c level (Fie or F2f) will, in fact, not necessarily be equal to the transition probabihty for scattering into a high-J [pure case (b)] ant isymmetr ic level ( F 1 f or F2~). This selection of one A-doublet level over another would be unexpected, at least on statistical grounds, since the splitting between the e and f A-doublet levels is always negligibly small ( < 0.1 cm -1) compared to the total collision energy of 6700 cm-a. The physical origin of the asymmetry involves the quantum mechanical interference between scattering on the V§ and V_ potential energy surfaces,
220 G.C. Core), et al. / Collisions of NO(X 21-1) with Ag(111)
aY
aY
3
i It ! 2 / ,
I
:, /,, /
I I ! I l ! I I I
0
0.5 20.5 40.5 60.5 80.5 j,
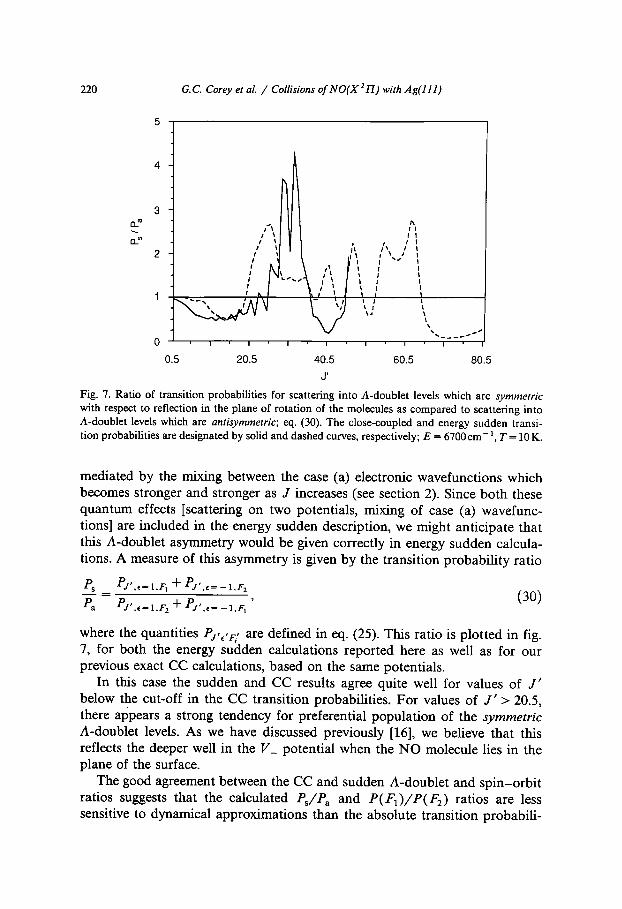
Fig. 7. Ratio of transition probabilities for scattering into A-doublet levels which are symmetric with respect to reflection in the plane of rotation of the molecules as compared to scattering into A-doublet levels which are antisymmetric; eq. (30). The close-coupled and energy sudden transi- tion probabilities are designated by solid and dashed curves, respectively; E = 6700cm-~, T = 10 K.
mediated by the mixing between the case (a) electronic wavefunctions which becomes stronger and stronger as J increases (see section 2). Since both these quantum effects [scattering on two potentials, mixing of case (a) wavefunc- tions] are included in the energy sudden description, we might anticipate that this A-doublet asymmetry would be given correctly in energy sudden calcula- tions. A measure of this asymmetry is given by the transition probability ratio
Ps + P+',,:-I,F2 = + P + ' . , : (30)
where the quantities PJ'c'F/ are defined in eq. (25). This ratio is plotted in fig. 7, for both the energy sudden calculations reported here as well as for our previous exact CC calculations, based on the same potentials.
In this case the sudden and CC results agree quite well for values of J ' below the cut-off in the CC transition probabilities. For values of J ' > 20.5, there appears a strong tendency for preferential population of the symmetric A-doublet levels. As we have discussed previously [16], we believe that this reflects the deeper well in the V_ potential when the NO molecule lies in the plane of the surface.
The good agreement between the CC and sudden A-doublet and spin-orbit ratios suggests that the calculated PJPa and P(FO/P(F2) ratios are less sensitive to dynamical approximations than the absolute transition probabili-

G.C. Corer et al. / Collisions of NO( X : H) with A g( I I I) 221
ties. Our previous CC calculations [16] show that all three quantities are extremely sensitive to the exact form of the V+ and V_ potential energy surfaces used in the calculations. The good agreement in figs. 6 and 7 may then indicate that the energy sudden method can be useful in assessing the dependence of radical-surface bonding on the orientation of the unfilled ~r orbitals in the molecule.
This will be true especially if our model for the difference potential significantly overestimates the depth and the anisotropy in the V. and V potentials wells. The quality of the sudden approximation would improve if the attractive interaction were in fact significantly weaker than the potentials used here. Little is known about the true N O - A g ( l l l ) interaction potential, and some recent work [15] does suggest that the average well depth may be only ~ 2000 cm -~, considerably less than in the potential used here (fig. 1).
6. Conclusion
In this paper we have reviewed the application of the energy sudden approximation to collisions of a molecule in a 21I electronic state with an uncorrugated, rigid surface. Determination of inelastic transition probabilities proceeds by solution of a fixed-orientation, single channel scattering calcula- tion, as in the case of the scattering of a molecule in a ~Y. electronic state. Here, however, this fixed-orientation calculation must be performed twice, once for each of the two potential energy surfaces which describe the interac- tion of a 2H molecule with a surface [32]. These potentials are degenerate when the molecule is oriented perpendicular to the surface but differ when the molecule is oriented in the plane of the surface, where the singly filled ~r-orbital can be oriented either in the plane of the surface or perpendicular to the surface. The desired inelastic transition probabilities can be obtained most conveniently be expanding the sum and the difference of the two sudden S functions in terms of, respectively, Legendre and associated Legendre poly- nomials.
Here we have applied this formalism to the calculation of inelastic transi- tion probabilities for collisions of NO(X 2YI) with Ag( l l l ) surfaces at an energy of 6700 cm-~. The potential energy surfaces used were taken from our previous close-coupling (CC) study of this system [16], and were based on the N O - A g ( l l l ) potential developed by Muhlhausen, Williams and Tully [14].
In contrast to many previous energy sudden studies of gas-phase collisions, the Legendre expansion of the sudden S functions required many (> 80) terms for convergence. This reflects the deep well (4300 cm- ~) and high degree of anisotropy in the potential. Comparison with our previous CC calculations [16] based on the same potentials was disappointing, in light of the many previous applications of the energy sudden approximation to molecule-surface

222 G. C Corey et al. / Collisions of NO(X -'17) with Ag(l 11)
scattering [6,8,13,35-41]. In principle one would have expected this approxi- mation to work well at a collision energy of 6700 cm-~. The sudden approxi- mation predicts significant flux into final rotational levels which are not only energetically but also dynamically forbidden. The rainbow maxima in the transition probabilities as a function of final rotational l eve l - a quantity often used to compare theory with experiment - are not predicted well by the sudden calculations, even for final levels which lie below the CC dynamical threshoM.
We paid particular attention to those features of the scattering which are sensitive to the open-shell character of the NO molecule, namely the depen- dence of the transition probabilities on the final spin-orbit and A-doublet state. For a given final rotational quantum number the ratio of the transition probabilities into the two A-doublet levels of the lower ( F 1) versus upper ( F 2) spin-orbit manifolds and the ratio of the transition probabilities into the symmetric (Fie and F2f)versus antisymmetric (Flf and F2e ) A-doublet levels were predicted satisfactorily.
The disappointing accuracy of the energy sudden calculations reported here in predicting the absolute transition probabilities stands in contrast to many previous studies in which this approximation was found to reproduce accu- rately the results of close-coupling calculations for molecule-surface scatter- ing, in particular the work of Voges and Schinke [6] on N O - A g ( l l l ) scatter- ing. We believe that this discrepancy is a function of the interaction potential used: energy sudden calculations will fail when the potential possesses a deep, strongly anisotropic, and fairly long-ranged attractive region. In this case considerable reorientation of the molecule can occur prior to contact with the repulsive wall. Thus a fixed-orientation treatment of the dynamics will break down.
It is unfortunate that the energy sudden approximation, which has proven so useful in allowing insights into molecule-surface collision dynamic [11-13,37,40] to be drawn merely by examination of the formal expressions for the transition probabilities, is not capable of consistently furnishing accurate values of these transition probabilities. However, little is known about the true NO-Ag interaction potential. If, as discussed at the end of section 5, the attractive anisotropy for this system is substantially weaker than in the potentials used here, the energy sudden approximation may provide a more accurate method for calculating relative transition probabilities.
Quantum scattering calculations are time-consuming due to the large num- ber of channels which must be retained. This problem is particularly acute for collisions involving open-shell molecules, where there exists more than one A-doublet and spin-orbit state associated with each value of the rotational quantum number. Further, at this level of complexity it would be virtually impossible to extend full quantum close-coupling calculations to include the effect of surface corrugations or surface motion. Unfortunately, it is not yet

G. C. Corey et al. / Collisions of N O( X 2 H) with A g(111) 223
clear how to extend classical trajectory calculations to describe successfully scattering involving two potential surfaces which coalesce asymptotically and at certain geometries (NO perpendicular to the surface). The present results suggest that energy sudden calculations, although computationally fast, may not be universally useful for quantitative studies of these systems, or, at least the N O - A g ( l l l ) system. This conclusion is in accord with earlier model studies by Fitz et al. [38] of H 2 and CI 2 colliding with a flat surface. In those studies, based on a purely repulsive potential, the energy sudden approxima- tion also predicted significant flux into closed rotational channels. Hopefully, other approaches, such as the semiclassical techniques recently proposed by Richard and DePristo [46], Sawada and Metiu [47] and Jackson and Metiu [48], or the quantal close-coupling wave packet method of Mowrey and Kouri [49] may provide the means for studying the scattering of NO off corrugated surfaces at nonzero temperatures.
Acknowledgements
The work described here was supported in part by a grant to M.H.A. from the National Science Foundat ion (Grant CHE85-06592), and by a grant to W.K.L. from the National Science and Engineering Research Council of Canada. The authors wish to thank John Tully for his help in using the N O - A g ( l l l ) potential described in ref. [14].
References
[1] D.J. Auerbach, Proceedings of the Fourth International Workshop on Inelastic Ion-Surface Collisions, Phys. Scripta T6 (1983) 122.
[2] J.A. Barker and D.J. Auerbach, Surface Sci. Rept. 4 (1985) 1. [3] M.C. Lin and G. Ertl, Ann. Rev. Phys. Chem. 37 (1986) 587. [4] J.E. Hurst, G.D. Kubiak and R.N. Zare, Chem. Phys. Letters 93 (1982) 235. [5] J.A. Barker, A.W. Kleyn and D.J. Auerbach, Chem. Phys. Letters 97 (1983) 9. [6] H. Voges and R. Schinke, Chem. Phys. Letters 95 (1983) 221; 100 (1983) 245. [7] J.G. Lauderdale, J.F. McNutt and C.W. McCurdy, Chem. Phys. Letters 107 (1984) 43. [8] S. Tanaka and S. Sugano, Surface Sci. 136 (1984) 488. [9] S. Tanaka and S. Sugano, Surface Sci. 143 (1984) L371.
[10] E. Zamir and R.D. Levine, Chem. Phys. Letters 104 (1984) 143. [11] M.H. Alexander, J. Chem. Phys. 80 (1984) 3485. [12] G.C. Corey and W.-K. Liu, Surface Sci. 148 (1984) 675. [13] R. Schinke and R.B. Gerber, J. Chem. Phys. 82 (1985) 1567. [14] C.W. Muhlhausen, L.R. Williams and J.C. Tully, J. Chem. Phys. 83 (1985) 2594. [15] H. Kasai, W. Brenig and H. Mailer, Z. Phys. B (Condensed Matter) 60 (1985) 489. [16] J.E. Smedley, G.C. Corey and M.H. Alexander, J. Chem. Phys., in press. [17] G.M. McClelland, G.D. Kubiak, H.G. Rennagel and R.N. Zare, Phys. Rev. Letters 46 (1981)
831.

224 G.C. Corey et al. / Collisions of NO(X2II) with Ag(111)
[18] A.W. Kleyn, A.C. Luntz and D.J. Auerbach, Phys. Rev. Letters 47 (1981) 1169. [19] A.W. Kleyn, A.C. Luntz and D.J. Auerbach, Surface Sci. 117 (1982) 33. [20] A.C. Luntz, A.W. Kleyn and D.J. Auerbach, Phys. Rev. B25 (1982) 4273. [21] A.C. Luntz, A.W. Kleyn and D.J. Auerbach, J. Chem. Phys. 76 (1982) 737. [22] D.J. Auerbaeh, J. Electron Spectrosc. Related Phenomena 30 (1983) 87. [23] A.C. Luntz, A.W. Kleyn and D.J. Auerbach, Vacuum 33 (1983) 781. [24] A.W. Kleyn, A.C. Luntz and D.J. Auerbach, Surface Sci. 152 (1985) 99. [25] G.D. Kubiak, J.E. Hurst, H.G. Rennagel, G.M. McCleUand and R.N. Zare, J. Chem. Phys.
79 (1983) 5163. [26] C.T. Rettner, F. Fabre, J. Kimman and D.J. Auerbach, Phys. Rev. Letters 55 (1985) 1904. [27] J. Misewich, P.A. Roland and M.M.T. Loy, Surface Sci. 171 (1986) 483. [28] M.H. Alexander, J. Chem. Phys. 76 (1982) 5974. [29] T. Orlikowski and M.H. Alexander, J. Chem. Phys. 79 (1983) 6006; 80 (1984) 4133. [30] M.H. Alexander and T. Orlikowski, J. Chem. Phys. 80 (1984) 1506. [31] M.H. Alexander, Chem. Phys. 92 (1985) 337. [32] G.C. Corey and M.H. Alexander, J. Chem. Phys 85 (1986) 5652. [33] G. Herzberg, Molecular Spectra and Molecular Structure, Vol. 1, Spectra of Diatomic
Molecules, 2nd ed. (Van Nostrand, Princeton, 1950). [34] M.H. Alexander and P.J. Dagdigian, J. Chem. Phys. 80 (1984) 4325. [35] J.E. Adams, Surface Sci. 97 (1980) 43. [36] R.B. Gerber, A.T. Yinnon, Y. Shimoni and D.J. Kouri, J. Chem. Phys. 73 (1980) 4397. [37] R.B. Gerber, L.H. Beard and D.J. Kouri, J. Chem. Phys. 74 (1981) 4709. [38] D.E. Fitz, A.O. Bawagan, L.H. Beard, D.J. Kouri and R.B. Gerber, Chem. Phys. Letters 80
(1981) 537; D.E. Fitz, L.H. Beard and D.J. Kouri, Chem. Phys. 59 (1981) 257.
[39] D.J. Kouri and R.B. Gerber, Israel J. Chem. 22 (1982) 321. [40] R. Schinke, J. Chem. Phys. 76 (1982) 2352. [41] W. Brenig, H. Kasai and H. Miiller, Springer Ser. Solid State Sci. 59 (1984) 2. [42] M. Larsson, Phys. Scripta 23 (1981) 835. [43] R.T Pack, J. Chem. Phys. 60 (1974) 633. [44] G.C. Nielson, G.A. Parker and R.T Pack, J. Chem. Phys. 66 (1977) 1396. [45] R. Schinke and J.M. Bowman, in: Molecular Collision Dynamics, Ed. J.M. Bowman
(Springer, Berlin, 1983) oh. 4. [46] A.M. Richard and A.E. DePristo, Surface Sci. 134 (1983) 338. [47] S. Sawada and H. Metiu, J. Chem. Phys. 84 (1986) 227. [48] B. Jackson and H. Metiu, J. Chem. Phys. 84 (1986) 3535. [49] R.C. Mowrey and D.J. Kouri, Chem. Phys. Letters 119 (1985) 285; J. Chem. Phys. 84 (1986)
6466; R.C. Mowrey, H.F. Bowen and D.J. Kouri, J. Chem. Phys. 86 (1987) 2441.
Copyright © 2022 FDOKUMEN




















