
Coffee component hydroxyl hydroquinone (HHQ) as a putative ligand for PPAR gamma and implications in...
16
RESEARCH Open Access Coffee component hydroxyl hydroquinone (HHQ) as a putative ligand for PPAR gamma and implications in breast cancer Babita Shashni 1 , Karun Sharma 1 , Rumani Singh 2 , Kishore R Sakharkar 3 , Sarinder K Dhillon 4 , Yukio Nagasaki 5,6,7 , Meena K Sakharkar 1,8,9* From Asia Pacific Bioinformatics Network (APBioNet) Twelfth International Conference on Bioinformatics (InCoB2013) Taicang, China. 20-22 September 2013 Abstract Background: Coffee contains several compounds that have the potential to influence breast cancer risk and survival. However, epidemiologic data on the relation between coffee compounds and breast cancer survival are sparse and inconsistent. Results: We show that coffee component HHQ has significant apoptotic effect on MDA-MB-231 and MCF-7 cells in vitro, and that ROS generation, change in mitochondrial membrane permeability, upregulation of Bax and Caspase-8 as well as down regulation of PGK1 and PKM2 expression may be important apoptosis-inducing mechanisms. The results suggest that PPARg ligands may serve as potential therapeutic agents for breast cancer therapy. HHQ was also validated as a ligand for PPARg by docking procedure. Conclusion: This is the first report on the anti-breast cancer (in vitro) activity of HHQ. Background Breast cancer is the fifth most common cancer globally and accounts for the highest morbidity and mortality. It is the second highest occurring cancer in women and one of the leading causes of death [1]. Although anti-estrogens have provided an effective endocrine therapy, a significant proportion of patients have acquired resistance to these drugs. Hence, there is a requirement for alternative thera- peutics to treat breast cancer. Since, cancer cells modify several pathways to achieve continuous progression and survival, and undergo metabolic alterations, it is important that multiple target strategies are used to achieve effective treatment. Several drugs that inhibit metabolism of cancer cells by targeting a variety of molecules (including enzymes) directly or indirectly are currently under clinical trials, hence it is important to screen drugs with a potential to target critical molecules involved in meta- bolic transformation [2]. PPARg receptor is a member of the nuclear receptor superfamily which upon ligand activation undergoes heterodimerization with retinoic acid-like receptor (RXR) and is translocated to the nucleus where it recognizes a specific sequence - the peroxisome proliferator response element (PPRE) located within promoters of target genes, and acts as a transcription regulator for genes involved in proliferation, cell differentiation, apoptosis, angiogenesis, inflammation, organogenesis, and lipid and carbohydrate metabolism and energy homeostasis [3-5]. Two isoforms of PPARg have been identified (PPARg 1 and PPARg 2), with a wide tissue distribution among various animal species [6]. PPARg are expressed in a variety of tumor cells and PPARg agonists e.g Thiazolidinediones (TZDs), and tyrosine based agonists show cytostatic and cytotoxic activity against tumor cells in vitro and in vivo brought about by regulating proteins involved in growth regulatory pathways and cell cycle [7]. TZDs are also reported to * Correspondence: [email protected] 1 Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki, Japan Full list of author information is available at the end of the article Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6 http://www.biomedcentral.com/1471-2164/14/S5/S6 © 2013 Shashni et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Transcript of Coffee component hydroxyl hydroquinone (HHQ) as a putative ligand for PPAR gamma and implications in...

RESEARCH Open Access
Coffee component hydroxyl hydroquinone (HHQ)as a putative ligand for PPAR gamma andimplications in breast cancerBabita Shashni1, Karun Sharma1, Rumani Singh2, Kishore R Sakharkar3, Sarinder K Dhillon4, Yukio Nagasaki5,6,7,Meena K Sakharkar1,8,9*
From Asia Pacific Bioinformatics Network (APBioNet) Twelfth International Conference on Bioinformatics(InCoB2013)Taicang, China. 20-22 September 2013
Abstract
Background: Coffee contains several compounds that have the potential to influence breast cancer risk and survival.However, epidemiologic data on the relation between coffee compounds and breast cancer survival are sparse andinconsistent.
Results: We show that coffee component HHQ has significant apoptotic effect on MDA-MB-231 and MCF-7 cellsin vitro, and that ROS generation, change in mitochondrial membrane permeability, upregulation of Bax andCaspase-8 as well as down regulation of PGK1 and PKM2 expression may be important apoptosis-inducingmechanisms. The results suggest that PPARg ligands may serve as potential therapeutic agents for breast cancertherapy. HHQ was also validated as a ligand for PPARg by docking procedure.
Conclusion: This is the first report on the anti-breast cancer (in vitro) activity of HHQ.
BackgroundBreast cancer is the fifth most common cancer globallyand accounts for the highest morbidity and mortality. It isthe second highest occurring cancer in women and one ofthe leading causes of death [1]. Although anti-estrogenshave provided an effective endocrine therapy, a significantproportion of patients have acquired resistance to thesedrugs. Hence, there is a requirement for alternative thera-peutics to treat breast cancer. Since, cancer cells modifyseveral pathways to achieve continuous progression andsurvival, and undergo metabolic alterations, it is importantthat multiple target strategies are used to achieve effectivetreatment. Several drugs that inhibit metabolism of cancercells by targeting a variety of molecules (includingenzymes) directly or indirectly are currently underclinical trials, hence it is important to screen drugs with
a potential to target critical molecules involved in meta-bolic transformation [2].PPARg receptor is a member of the nuclear receptor
superfamily which upon ligand activation undergoesheterodimerization with retinoic acid-like receptor (RXR)and is translocated to the nucleus where it recognizes aspecific sequence - the peroxisome proliferator responseelement (PPRE) located within promoters of target genes,and acts as a transcription regulator for genes involved inproliferation, cell differentiation, apoptosis, angiogenesis,inflammation, organogenesis, and lipid and carbohydratemetabolism and energy homeostasis [3-5]. Two isoformsof PPARg have been identified (PPARg 1 and PPARg 2),with a wide tissue distribution among various animalspecies [6]. PPARg are expressed in a variety of tumorcells and PPARg agonists e.g Thiazolidinediones (TZDs),and tyrosine based agonists show cytostatic and cytotoxicactivity against tumor cells in vitro and in vivo broughtabout by regulating proteins involved in growth regulatorypathways and cell cycle [7]. TZDs are also reported to
* Correspondence: [email protected] School of Life and Environmental Sciences, University of Tsukuba,Tsukuba, Ibaraki, JapanFull list of author information is available at the end of the article
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
© 2013 Shashni et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative CommonsAttribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction inany medium, provided the original work is properly cited.

induce G0/G1 arrest and apoptosis of malignant cells byupregulation of the tumor suppressor p53, and control ofDNA repair systems and apoptosis [8]. However, the exactmechanism of action and the genes regulated by PPARgand biological functions of this transcription factor are notknown and need elucidation.Also, due to high levels of toxicity associated with TZDs
(e.g. - troglitazone (Rezulin), rosiglitazone (Avandia), andpioglitazone (Actos)), and their recent withdrawal inseveral countries, there is a need to search for newerPPAR drugs that exhibit better efficacy but lesser toxicity.Phytochemicals in dietary components are increasinglybeing used as nutritional supplements in treatment ofdiseases. Due to the plant origin of these supplements theyare considered safe for human consumption [9]. Presentdata reveal that healthy dietary molecules have apleiotropic role and are able to change cell metabolismfrom anabolism to catabolism, modulate energy homeosta-sis and down regulate inflammation by interacting withenzymes, nuclear receptors and transcriptional factors [10].Towards this end developing and positioning knownphytochemicals that bind and activate PPARg with moreefficacy and safety, while promoting health benefits hasbecome an absolute necessity. Also, it is important toidentify the dietary molecules able to influence the courseof the disease, their targets in the cell, and the molecularmechanisms involved.Coffee is one of the most widely consumed beverages in
the world. The health-promoting properties of coffee areoften attributed to its rich phytochemistry, includingcaffeine, chlorogenic acid, caffeic acid, hydroxyl hydroqui-none (HHQ), etc. More recently, coffee consumption hasbeen associated with reductions in the risk of severalchronic diseases, including type 2 diabetes mellitus,Parkinson’s disease and hepatocellular disease [11-13].The association between coffee intake and breast cancerrisk is biologically plausible because of its complexmake-up of chemicals, e.g., caffeine and polyphenoliccompounds such as flavonoids and lignans [14-16].Among them, the relationship between coffee drinkingand breast cancer risk holds great interest. Recent meta-analyses demonstrate inverse associations between coffeeintake and the risk of colon, liver, breast, and endometrialcancer [17-20]. Also, a high daily intake of coffee hasrecently been reported to be associated with a statisticallysignificant decrease in ER-negative breast (ER - EstrogenResponsive) cancer among postmenopausal women [21].A number of previous epidemiologic studies have esti-mated the association between coffee consumption andbreast cancer risk. However, the results are inconsistent[22]. Nevertheless, several reports in literature suggest thatcoffee consumption reduces the risk of cancer, but themolecular mechanisms of its chemopreventive effectsremain unknown. Moreover, the interpretation of
these data has often been limited to the role that caffeineplays [23,24].HHQ is a natural constituent of coffee accounting for
main dry matter constituent in roasted beans. Studiesexploring the effects of this bioactive compound onmammalian cells are limited. HHQ was observed to dockand form hydrogen bonds with PDB ID - 2PRG (PDB 3-Dcrystal structure of the Ligand-binding domain of thehuman peroxisome proliferator activated receptor gammasolved in complexation with Rosiglitazone, the PPARgamma agonist/ligand). The initial purpose of our investi-gation is to determine whether HHQ alters the cellviability in estrogen dependent human breast cancer(MCF-7) and estrogen independent (MDA-MB-231) cellsas a model system. HHQ was observed to decrease cellviability and colony formation in a dose-responsivemanner and ROS was found to significantly increase inHHQ treated cells in a dose-dependent manner inboth the cell lines. We examined the involvement ofROS signaling components (ROS levels and membranepotential) and demonstrated that the selective killing ofcancer cells is mediated by induction of oxidative stressthat leads to apoptosis. These findings were complemen-ted by the finding that caspase-8 is upregulated. Sinceglucose utilization provides a constant energy supply,as well as precursors for de novo macromolecularbiosynthesis, including DNA, RNA, fatty acids and aminoacids that are essential for cell growth and proliferation,we further investigated the effect of HHQ on two keyglycolytic genes PGK1 and PKM2 (at the ATP generationstep) and observed that both the genes are repressed in adose-dependent manner. These data suggest on theapoptotic and anti-cancer properties of HHQ via PPARg.Further investigations on these could possibly help us inunderstanding the molecular mechanisms by whichPPARg regulates disease targets specifically in breastcancer and the use of HHQ in breast cancer therapeutics.
Results and discussionUnderstanding the molecular pathways that link tumorbiology to the staggering array of pathologies and genes isof paramount scientific and medical importance. Though,the complexity of the underlying biochemical and molecu-lar mechanisms of breast cancer make metabolic repro-gramming and transformation in breast cancer unclear,many dietary compounds have been identified as potentialchemopreventive agents. PPARg is an interesting target forcancer therapy as its expression is elevated in tumors andalso because PPARg activation is reported to result indecreased cell proliferation, decreased G0/G1 to S phaseprogression, apoptosis and increased terminal differentia-tion [25-27]. Also, imbalances in expression of targetgenes forms the core of metabolic syndrome and cancerregulation through atherogenic metabolic triad/lipid triad
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 2 of 16

metabolism modulation by PPARs [28]. Concurrently,increased levels of glycolytic proteins are observed inplasmas of women with breast cancer as a result ofupregulation of glycolysis pathway [29].In this paper, we for the first time report on the use of
coffee component HHQ as a potential ligand for PPARgand its role in induction of apoptosis in breast cancer cellsby delineating the glycolytic pathway gene regulation byPPARg activation.
DockingThe three-dimensional protein structures can be used tounderstand ligand binding and to rationally design novelligands as prospective drug compounds. PPAR agoniststypically possess a small polar region and a hydrophobicregion that form hydrogen bonds and hydrophobicinteractions, respectively, within the ligand bindingdomain. Several crystal structures of PPARg in com-plexation with their ligands are available in the proteindata bank (PDB). Here, we used the PDB crystal struc-ture 2PRG (PPARg with ligand Rosiglitazone) for struc-ture-based identification of HHQ as a potential ligandfor PPARg in breast cancer therapeutics. We also com-pared the ligand binding properties of the two com-pounds Rosiglitazone (the known ligand) and HHQ (theproposed novel ligand). The rerank scores for the Rosi-glitazone and HHQ were found to be -122.433 and-33.3562, respectively. The hydrogen bond energy valueswere -5.487 kcal/mol, and -9.460 kcal/mol for Rosiglita-zone and HHQ. Rosiglitazone (the ligand in the crystalstructure 2PRG) forms 3 hydrogen bonds with the2PRG (Gln286, His449, Ser289) and HHQ forms5 hydrogen bonds with 2PRG (Gln286, His449, Tyr473,Ser289, His323). Hydrogen bonding of the ligand toTyr473 is reported to be the key to the stabilization ofthe AF-2 region and it has also been shown that [30]agonistic activity of ligands disappears when Tyr473 ismutated. The importance of His323 and His449 has alsobeen reported [31,32]. Our results show that both theligands have hydrogen bond interactions with key resi-dues - Tyr473 and His 449, thereby providing specificityof interaction that is a fundamental aspect of molecularrecognition. The active site residues that interact withHHQ are shown in Table 1 and the hydrogen bondsbetween each compound and the 2PRG active site areshown in Figure 1.
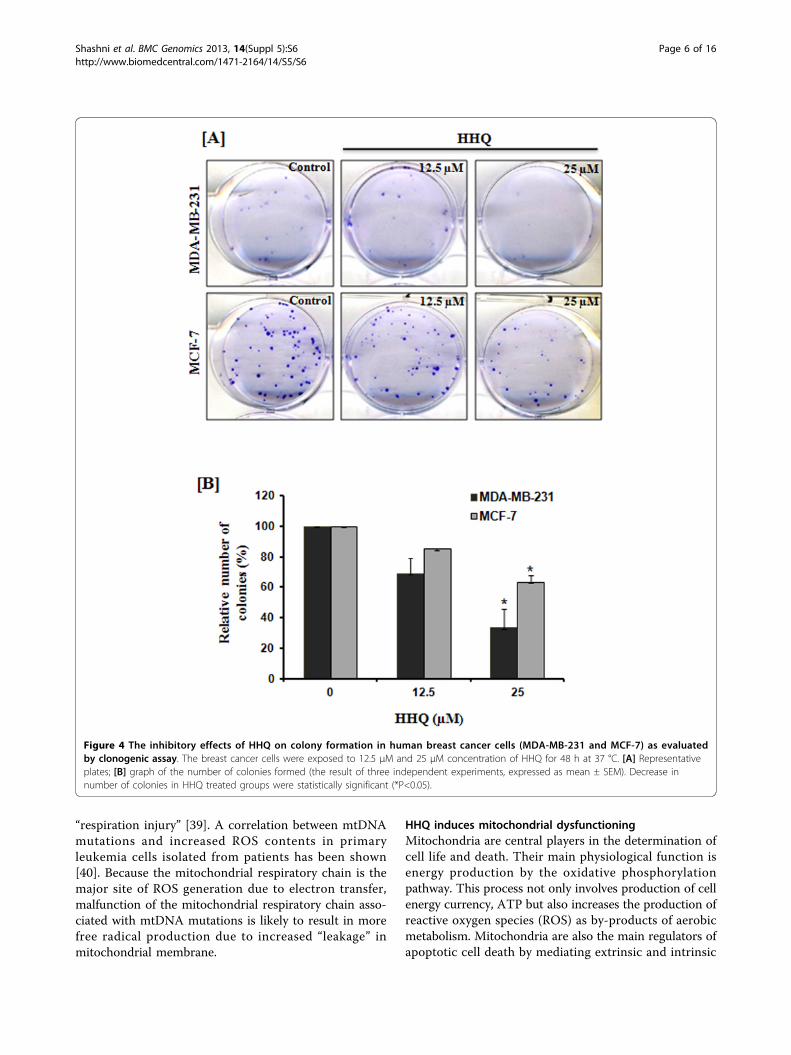
HHQ inhibits cell proliferation and clonogenic survivalTo investigate the potential cell growth inhibition of HHQ,we first examined the effect of HHQ on cell proliferationand clonogenic survival in human breast adenocarcino-mas. For this we chose two breast adenocarcinoma celllines, MDA-MB-231 (estrogen independent) and MCF-7(estrogen dependent). MDA-MB-231 and MCF-7 werecultured in the control medium for 24 h, followed by 48 htreatment with different concentrations of HHQ for cyto-toxic and anti-proliferative studies. HHQ led to decreasein relative density of viable cells as detected by MTT dyereduction assay. A dose-effect relationship was observed asshown in Figure 2. The IC-50 value for MDA-MB-231 andMCF-7 were found to be 25 μM and 50 μM, respectively.We proceeded with 12.5 μM and 25 μM of HHQ to inves-tigate the comparable effects of same concentration onboth the cell lines. At 12.5 μM the relative viabilityin MDA-MB-231 and MCF-7 was 58.2% and 76.3%,respectively, whereas at 25 μM the relative viability inMDA-MB-231 and MCF-7 was 50% and 60.7%, respec-tively. As morphological analyses depict the health ofa cell, we next examined the cells for any altered morphol-ogy on treatment with HHQ. Cells cultured in thepresence of HHQ (12.5 μM and 25 μM) show significantmorphological changes and were found to be reduced incell mass as well as number as compared to control. Also,HHQ treatment induced detachment and rounding inbreast cancer cells as shown by phase contrast images inboth the cell lines (Figure 3 (12.5 μM and 25 μM)).Furthermore, to examine the antitumor activity of HHQon colony forming potential in breast cancer cells weperformed clonogenic assay. This assay is an in vitro assaybased on the ability of a single cell to proliferate anddifferentiate into colonies in response to various insults.HHQ severely affected the colony forming potential ofhuman breast cancer cells - MDA-MB-231 and MCF-7.As shown in Figure 4A and 4B clonogenicity of both thebreast cancer lines were found to be significantly reducedin a concentration-dependent manner after exposureto HHQ.
HHQ induces intracellular ROS generation and cytotoxicityReactive oxygen species (ROS) is a universal entity med-iating apoptosis [33]. ROS act as a secondary messengerin cell signaling and are essential for various biologicalprocesses in normal cells. Disturbances in redox balance
Table 1 Docking results for HHQ and Rosiglitazone.
Drugs Hydrogen bond score (kcal.mol-1)
Number of hydrogenbonds
Residues of PPARg binding site interacting with theligands
Rerankscore
HHQ -9.46033 5 Gln286, His449, Tyr473, Ser289, His323 -33.3562
Rosiglitazone -5.48771 3 Gln286, His449, Ser289 -122.433
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 3 of 16

relate to human pathogenesis including cancers. ROSare constantly generated and eliminated in the biologicalsystem, and play important roles in a variety of normalbiochemical functions and abnormal pathologicalprocesses. Growing evidence suggests that cancer cellsexhibit increased intrinsic ROS stress, due in part tooncogenic stimulation, increased metabolic activity, andmitochondrial malfunction. Since ROS are chemicallyactive and can inflict severe cellular damage, the veryfact that cancer cells are under increased intrinsic ROSstress may also provide a unique opportunity to kill themalignant cells based on their vulnerability to furtherROS insults caused by exogenous agents [34]. HHQ isearlier reported to generate reactive oxygen species(ROS) by autoxidation [35]. Therefore, to get furtherinsights to HHQ induced cytotoxicity in breast cancercells; we examined the intracellular ROS generation. Asshown in Figure 5, intracellular ROS formation was
found to be significantly increased in HHQ treated cellsas compared to control cells in a dose-dependent man-ner. The effective enhancement of ROS production byHHQ correlates to its cytotoxicity nature. Since themitochondrial respiratory chain (electron transportchain) is a major source of ROS generation in the cells,the vulnerability of the mitochondrial DNA to ROSmediated damage has been suggested to be a mechan-ism to amplify ROS stress in cancer cells [36]. Onemajor effect is to generate increased intracellular ROScausing loss of outer mitochondrial membrane perme-ability and induction of apoptosis [37,38]. Another pos-sible mechanism by which cancer cells generateincreased amounts of ROS may involve malfunction ofthe mitochondrial respiratory chain. The fact that cancercells exhibit an increased dependency on glycolysis tomeet their ATP need (the Warburg effect) may reflectan inefficient ATP generation in mitochondria, or
Figure 1 HHQ docked in the ligand binding domain of PPARg protein crystal structure for PDB solved in conjunction withRosiglitazone (PDBID: 2PRG). [A] Represents hydrogen bonds (in black dotted line) observed for Rosiglitazone (in red) with the active siteresidues in the ligand binding domain of PPARg (2PRG). [B] Represents hydrogen bonds (in black dotted line) observed for HHQ (in yellow) withthe active site residues in the ligand binding domain of 2PRG. [C] Represents superposition of the best conformation of Rosiglitazone (in red)and HHQ (in yellow) in the ligand binding domain of 2PRG.
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 4 of 16
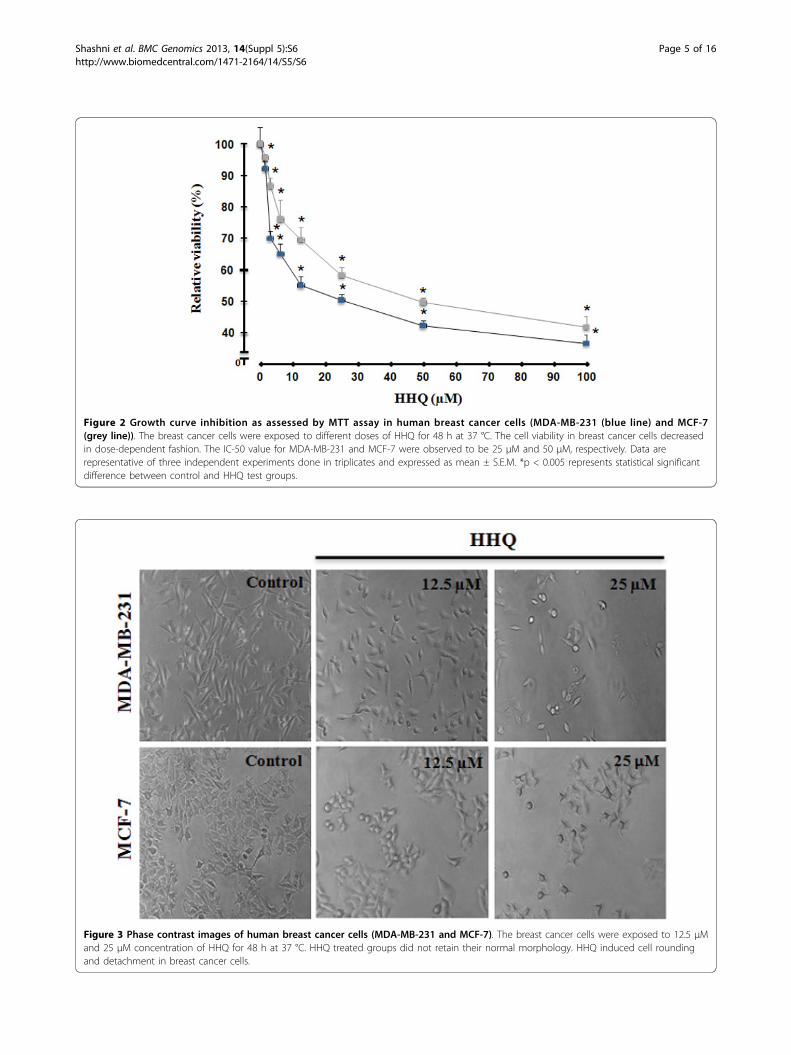
Figure 2 Growth curve inhibition as assessed by MTT assay in human breast cancer cells (MDA-MB-231 (blue line) and MCF-7(grey line)). The breast cancer cells were exposed to different doses of HHQ for 48 h at 37 °C. The cell viability in breast cancer cells decreasedin dose-dependent fashion. The IC-50 value for MDA-MB-231 and MCF-7 were observed to be 25 μM and 50 μM, respectively. Data arerepresentative of three independent experiments done in triplicates and expressed as mean ± S.E.M. *p < 0.005 represents statistical significantdifference between control and HHQ test groups.
Figure 3 Phase contrast images of human breast cancer cells (MDA-MB-231 and MCF-7). The breast cancer cells were exposed to 12.5 µMand 25 µM concentration of HHQ for 48 h at 37 °C. HHQ treated groups did not retain their normal morphology. HHQ induced cell roundingand detachment in breast cancer cells.
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 5 of 16
“respiration injury” [39]. A correlation between mtDNAmutations and increased ROS contents in primaryleukemia cells isolated from patients has been shown[40]. Because the mitochondrial respiratory chain is themajor site of ROS generation due to electron transfer,malfunction of the mitochondrial respiratory chain asso-ciated with mtDNA mutations is likely to result in morefree radical production due to increased “leakage” inmitochondrial membrane.
HHQ induces mitochondrial dysfunctioningMitochondria are central players in the determination ofcell life and death. Their main physiological function isenergy production by the oxidative phosphorylationpathway. This process not only involves production of cellenergy currency, ATP but also increases the production ofreactive oxygen species (ROS) as by-products of aerobicmetabolism. Mitochondria are also the main regulators ofapoptotic cell death by mediating extrinsic and intrinsic
Figure 4 The inhibitory effects of HHQ on colony formation in human breast cancer cells (MDA-MB-231 and MCF-7) as evaluatedby clonogenic assay. The breast cancer cells were exposed to 12.5 µM and 25 µM concentration of HHQ for 48 h at 37 °C. [A] Representativeplates; [B] graph of the number of colonies formed (the result of three independent experiments, expressed as mean ± SEM). Decrease innumber of colonies in HHQ treated groups were statistically significant (*P<0.05).
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 6 of 16
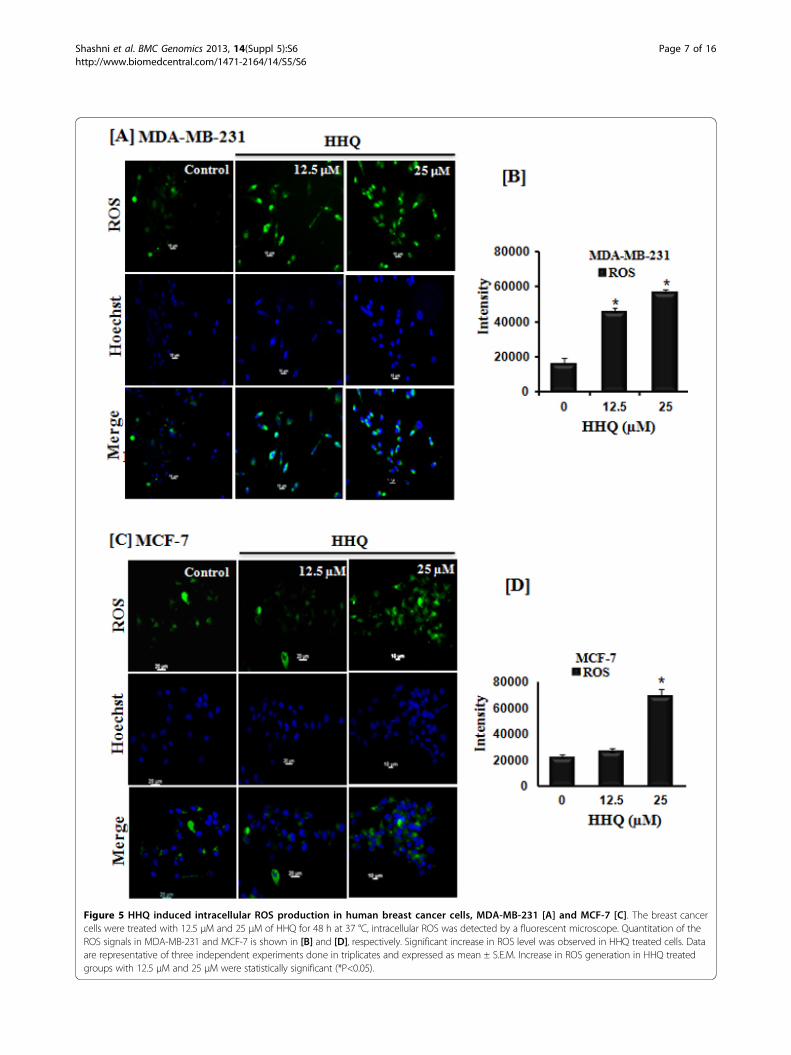
Figure 5 HHQ induced intracellular ROS production in human breast cancer cells, MDA-MB-231 [A] and MCF-7 [C]. The breast cancercells were treated with 12.5 μM and 25 μM of HHQ for 48 h at 37 °C, intracellular ROS was detected by a fluorescent microscope. Quantitation of theROS signals in MDA-MB-231 and MCF-7 is shown in [B] and [D], respectively. Significant increase in ROS level was observed in HHQ treated cells. Dataare representative of three independent experiments done in triplicates and expressed as mean ± S.E.M. Increase in ROS generation in HHQ treatedgroups with 12.5 μM and 25 μM were statistically significant (*P<0.05).
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 7 of 16
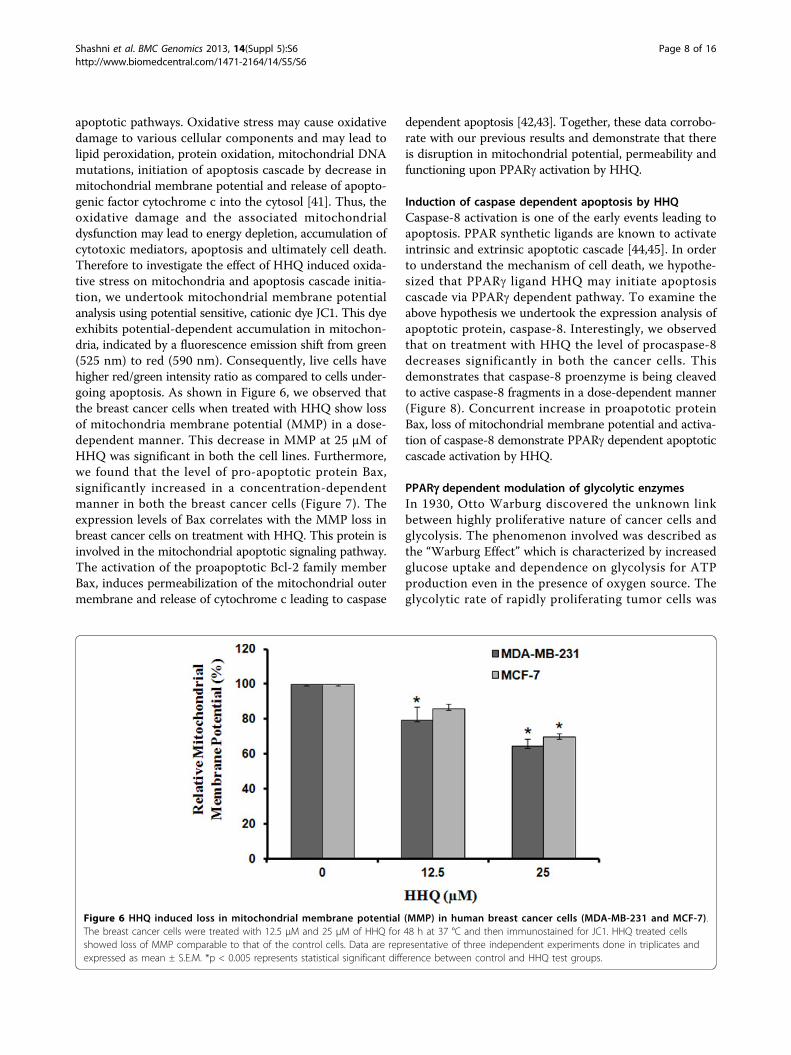
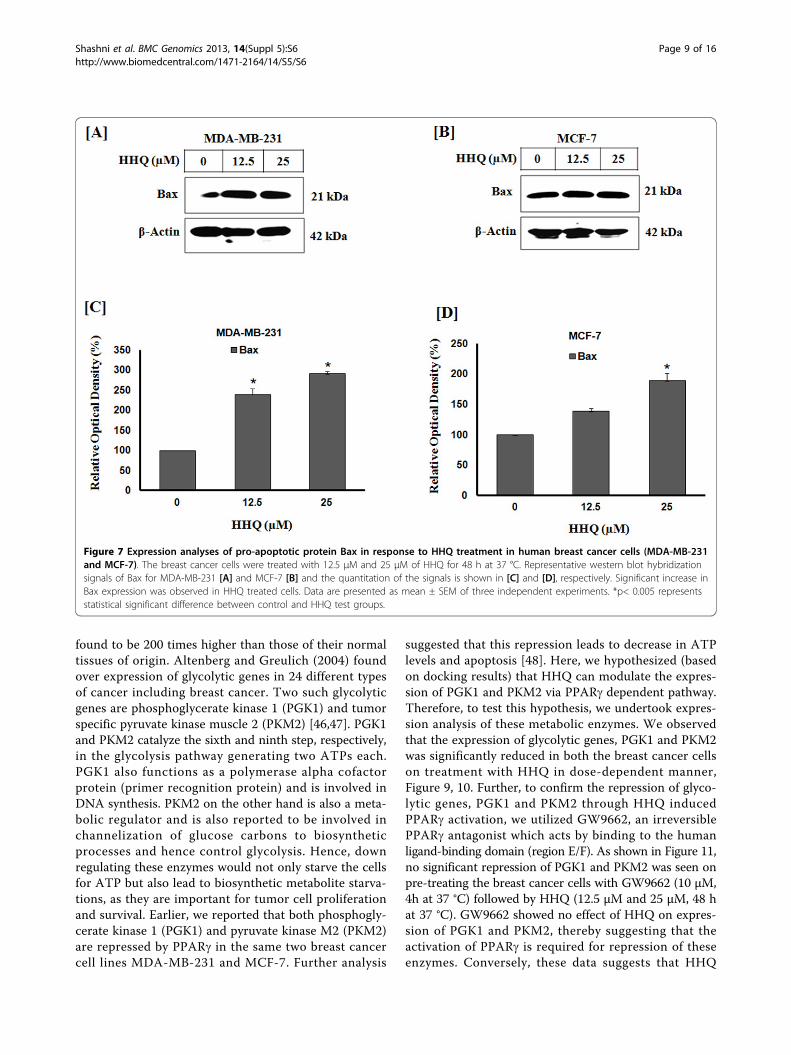
apoptotic pathways. Oxidative stress may cause oxidativedamage to various cellular components and may lead tolipid peroxidation, protein oxidation, mitochondrial DNAmutations, initiation of apoptosis cascade by decrease inmitochondrial membrane potential and release of apopto-genic factor cytochrome c into the cytosol [41]. Thus, theoxidative damage and the associated mitochondrialdysfunction may lead to energy depletion, accumulation ofcytotoxic mediators, apoptosis and ultimately cell death.Therefore to investigate the effect of HHQ induced oxida-tive stress on mitochondria and apoptosis cascade initia-tion, we undertook mitochondrial membrane potentialanalysis using potential sensitive, cationic dye JC1. This dyeexhibits potential-dependent accumulation in mitochon-dria, indicated by a fluorescence emission shift from green(525 nm) to red (590 nm). Consequently, live cells havehigher red/green intensity ratio as compared to cells under-going apoptosis. As shown in Figure 6, we observed thatthe breast cancer cells when treated with HHQ show lossof mitochondria membrane potential (MMP) in a dose-dependent manner. This decrease in MMP at 25 μM ofHHQ was significant in both the cell lines. Furthermore,we found that the level of pro-apoptotic protein Bax,significantly increased in a concentration-dependentmanner in both the breast cancer cells (Figure 7). Theexpression levels of Bax correlates with the MMP loss inbreast cancer cells on treatment with HHQ. This protein isinvolved in the mitochondrial apoptotic signaling pathway.The activation of the proapoptotic Bcl-2 family memberBax, induces permeabilization of the mitochondrial outermembrane and release of cytochrome c leading to caspase
dependent apoptosis [42,43]. Together, these data corrobo-rate with our previous results and demonstrate that thereis disruption in mitochondrial potential, permeability andfunctioning upon PPARg activation by HHQ.
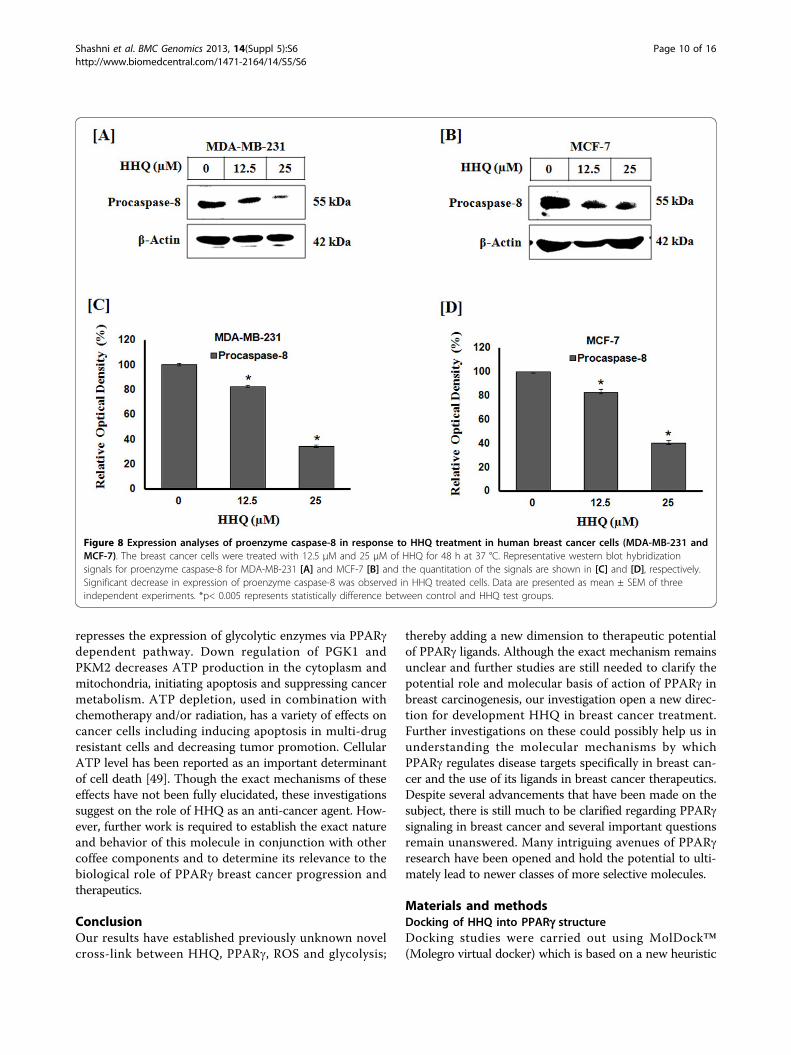
Induction of caspase dependent apoptosis by HHQCaspase-8 activation is one of the early events leading toapoptosis. PPAR synthetic ligands are known to activateintrinsic and extrinsic apoptotic cascade [44,45]. In orderto understand the mechanism of cell death, we hypothe-sized that PPARg ligand HHQ may initiate apoptosiscascade via PPARg dependent pathway. To examine theabove hypothesis we undertook the expression analysis ofapoptotic protein, caspase-8. Interestingly, we observedthat on treatment with HHQ the level of procaspase-8decreases significantly in both the cancer cells. Thisdemonstrates that caspase-8 proenzyme is being cleavedto active caspase-8 fragments in a dose-dependent manner(Figure 8). Concurrent increase in proapototic proteinBax, loss of mitochondrial membrane potential and activa-tion of caspase-8 demonstrate PPARg dependent apoptoticcascade activation by HHQ.
PPARg dependent modulation of glycolytic enzymesIn 1930, Otto Warburg discovered the unknown linkbetween highly proliferative nature of cancer cells andglycolysis. The phenomenon involved was described asthe “Warburg Effect” which is characterized by increasedglucose uptake and dependence on glycolysis for ATPproduction even in the presence of oxygen source. Theglycolytic rate of rapidly proliferating tumor cells was
Figure 6 HHQ induced loss in mitochondrial membrane potential (MMP) in human breast cancer cells (MDA-MB-231 and MCF-7).The breast cancer cells were treated with 12.5 μM and 25 μM of HHQ for 48 h at 37 °C and then immunostained for JC1. HHQ treated cellsshowed loss of MMP comparable to that of the control cells. Data are representative of three independent experiments done in triplicates andexpressed as mean ± S.E.M. *p < 0.005 represents statistical significant difference between control and HHQ test groups.
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 8 of 16
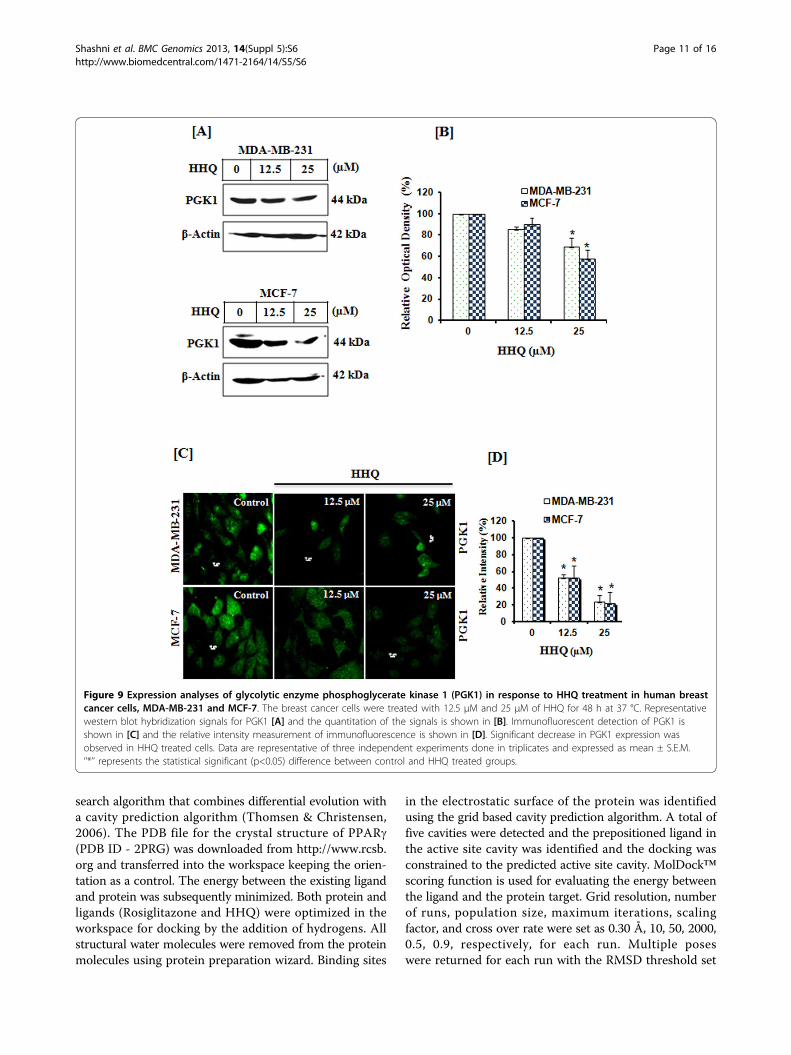
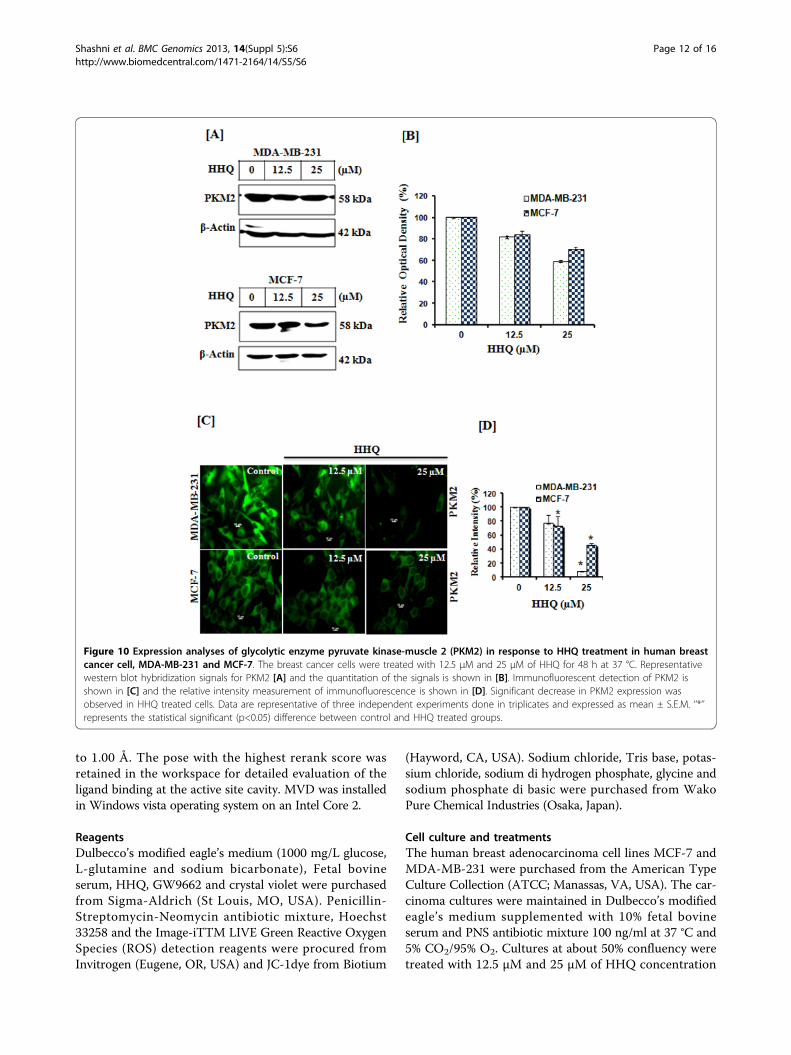
found to be 200 times higher than those of their normaltissues of origin. Altenberg and Greulich (2004) foundover expression of glycolytic genes in 24 different typesof cancer including breast cancer. Two such glycolyticgenes are phosphoglycerate kinase 1 (PGK1) and tumorspecific pyruvate kinase muscle 2 (PKM2) [46,47]. PGK1and PKM2 catalyze the sixth and ninth step, respectively,in the glycolysis pathway generating two ATPs each.PGK1 also functions as a polymerase alpha cofactorprotein (primer recognition protein) and is involved inDNA synthesis. PKM2 on the other hand is also a meta-bolic regulator and is also reported to be involved inchannelization of glucose carbons to biosyntheticprocesses and hence control glycolysis. Hence, downregulating these enzymes would not only starve the cellsfor ATP but also lead to biosynthetic metabolite starva-tions, as they are important for tumor cell proliferationand survival. Earlier, we reported that both phosphogly-cerate kinase 1 (PGK1) and pyruvate kinase M2 (PKM2)are repressed by PPARg in the same two breast cancercell lines MDA-MB-231 and MCF-7. Further analysis
suggested that this repression leads to decrease in ATPlevels and apoptosis [48]. Here, we hypothesized (basedon docking results) that HHQ can modulate the expres-sion of PGK1 and PKM2 via PPARg dependent pathway.Therefore, to test this hypothesis, we undertook expres-sion analysis of these metabolic enzymes. We observedthat the expression of glycolytic genes, PGK1 and PKM2was significantly reduced in both the breast cancer cellson treatment with HHQ in dose-dependent manner,Figure 9, 10. Further, to confirm the repression of glyco-lytic genes, PGK1 and PKM2 through HHQ inducedPPARg activation, we utilized GW9662, an irreversiblePPARg antagonist which acts by binding to the humanligand-binding domain (region E/F). As shown in Figure 11,no significant repression of PGK1 and PKM2 was seen onpre-treating the breast cancer cells with GW9662 (10 μM,4h at 37 °C) followed by HHQ (12.5 μM and 25 μM, 48 hat 37 °C). GW9662 showed no effect of HHQ on expres-sion of PGK1 and PKM2, thereby suggesting that theactivation of PPARg is required for repression of theseenzymes. Conversely, these data suggests that HHQ
Figure 7 Expression analyses of pro-apoptotic protein Bax in response to HHQ treatment in human breast cancer cells (MDA-MB-231and MCF-7). The breast cancer cells were treated with 12.5 μM and 25 μM of HHQ for 48 h at 37 °C. Representative western blot hybridizationsignals of Bax for MDA-MB-231 [A] and MCF-7 [B] and the quantitation of the signals is shown in [C] and [D], respectively. Significant increase inBax expression was observed in HHQ treated cells. Data are presented as mean ± SEM of three independent experiments. *p< 0.005 representsstatistical significant difference between control and HHQ test groups.
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 9 of 16
represses the expression of glycolytic enzymes via PPARgdependent pathway. Down regulation of PGK1 andPKM2 decreases ATP production in the cytoplasm andmitochondria, initiating apoptosis and suppressing cancermetabolism. ATP depletion, used in combination withchemotherapy and/or radiation, has a variety of effects oncancer cells including inducing apoptosis in multi-drugresistant cells and decreasing tumor promotion. CellularATP level has been reported as an important determinantof cell death [49]. Though the exact mechanisms of theseeffects have not been fully elucidated, these investigationssuggest on the role of HHQ as an anti-cancer agent. How-ever, further work is required to establish the exact natureand behavior of this molecule in conjunction with othercoffee components and to determine its relevance to thebiological role of PPARg breast cancer progression andtherapeutics.
ConclusionOur results have established previously unknown novelcross-link between HHQ, PPARg, ROS and glycolysis;
thereby adding a new dimension to therapeutic potentialof PPARg ligands. Although the exact mechanism remainsunclear and further studies are still needed to clarify thepotential role and molecular basis of action of PPARg inbreast carcinogenesis, our investigation open a new direc-tion for development HHQ in breast cancer treatment.Further investigations on these could possibly help us inunderstanding the molecular mechanisms by whichPPARg regulates disease targets specifically in breast can-cer and the use of its ligands in breast cancer therapeutics.Despite several advancements that have been made on thesubject, there is still much to be clarified regarding PPARgsignaling in breast cancer and several important questionsremain unanswered. Many intriguing avenues of PPARgresearch have been opened and hold the potential to ulti-mately lead to newer classes of more selective molecules.
Materials and methodsDocking of HHQ into PPARg structureDocking studies were carried out using MolDock™(Molegro virtual docker) which is based on a new heuristic
Figure 8 Expression analyses of proenzyme caspase-8 in response to HHQ treatment in human breast cancer cells (MDA-MB-231 andMCF-7). The breast cancer cells were treated with 12.5 μM and 25 μM of HHQ for 48 h at 37 °C. Representative western blot hybridizationsignals for proenzyme caspase-8 for MDA-MB-231 [A] and MCF-7 [B] and the quantitation of the signals are shown in [C] and [D], respectively.Significant decrease in expression of proenzyme caspase-8 was observed in HHQ treated cells. Data are presented as mean ± SEM of threeindependent experiments. *p< 0.005 represents statistically difference between control and HHQ test groups.
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 10 of 16
search algorithm that combines differential evolution witha cavity prediction algorithm (Thomsen & Christensen,2006). The PDB file for the crystal structure of PPARg(PDB ID - 2PRG) was downloaded from http://www.rcsb.org and transferred into the workspace keeping the orien-tation as a control. The energy between the existing ligandand protein was subsequently minimized. Both protein andligands (Rosiglitazone and HHQ) were optimized in theworkspace for docking by the addition of hydrogens. Allstructural water molecules were removed from the proteinmolecules using protein preparation wizard. Binding sites
in the electrostatic surface of the protein was identifiedusing the grid based cavity prediction algorithm. A total offive cavities were detected and the prepositioned ligand inthe active site cavity was identified and the docking wasconstrained to the predicted active site cavity. MolDock™scoring function is used for evaluating the energy betweenthe ligand and the protein target. Grid resolution, numberof runs, population size, maximum iterations, scalingfactor, and cross over rate were set as 0.30 Å, 10, 50, 2000,0.5, 0.9, respectively, for each run. Multiple poseswere returned for each run with the RMSD threshold set
Figure 9 Expression analyses of glycolytic enzyme phosphoglycerate kinase 1 (PGK1) in response to HHQ treatment in human breastcancer cells, MDA-MB-231 and MCF-7. The breast cancer cells were treated with 12.5 μM and 25 μM of HHQ for 48 h at 37 °C. Representativewestern blot hybridization signals for PGK1 [A] and the quantitation of the signals is shown in [B]. Immunofluorescent detection of PGK1 isshown in [C] and the relative intensity measurement of immunofluorescence is shown in [D]. Significant decrease in PGK1 expression wasobserved in HHQ treated cells. Data are representative of three independent experiments done in triplicates and expressed as mean ± S.E.M.‘’*’’ represents the statistical significant (p<0.05) difference between control and HHQ treated groups.
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 11 of 16
to 1.00 Å. The pose with the highest rerank score wasretained in the workspace for detailed evaluation of theligand binding at the active site cavity. MVD was installedin Windows vista operating system on an Intel Core 2.
ReagentsDulbecco’s modified eagle’s medium (1000 mg/L glucose,L-glutamine and sodium bicarbonate), Fetal bovineserum, HHQ, GW9662 and crystal violet were purchasedfrom Sigma-Aldrich (St Louis, MO, USA). Penicillin-Streptomycin-Neomycin antibiotic mixture, Hoechst33258 and the Image-iTTM LIVE Green Reactive OxygenSpecies (ROS) detection reagents were procured fromInvitrogen (Eugene, OR, USA) and JC-1dye from Biotium
(Hayword, CA, USA). Sodium chloride, Tris base, potas-sium chloride, sodium di hydrogen phosphate, glycine andsodium phosphate di basic were purchased from WakoPure Chemical Industries (Osaka, Japan).
Cell culture and treatmentsThe human breast adenocarcinoma cell lines MCF-7 andMDA-MB-231 were purchased from the American TypeCulture Collection (ATCC; Manassas, VA, USA). The car-cinoma cultures were maintained in Dulbecco’s modifiedeagle’s medium supplemented with 10% fetal bovineserum and PNS antibiotic mixture 100 ng/ml at 37 °C and5% CO2/95% O2. Cultures at about 50% confluency weretreated with 12.5 µM and 25 µM of HHQ concentration
Figure 10 Expression analyses of glycolytic enzyme pyruvate kinase-muscle 2 (PKM2) in response to HHQ treatment in human breastcancer cell, MDA-MB-231 and MCF-7. The breast cancer cells were treated with 12.5 μM and 25 μM of HHQ for 48 h at 37 °C. Representativewestern blot hybridization signals for PKM2 [A] and the quantitation of the signals is shown in [B]. Immunofluorescent detection of PKM2 isshown in [C] and the relative intensity measurement of immunofluorescence is shown in [D]. Significant decrease in PKM2 expression wasobserved in HHQ treated cells. Data are representative of three independent experiments done in triplicates and expressed as mean ± S.E.M. ‘’*’’represents the statistical significant (p<0.05) difference between control and HHQ treated groups.
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 12 of 16
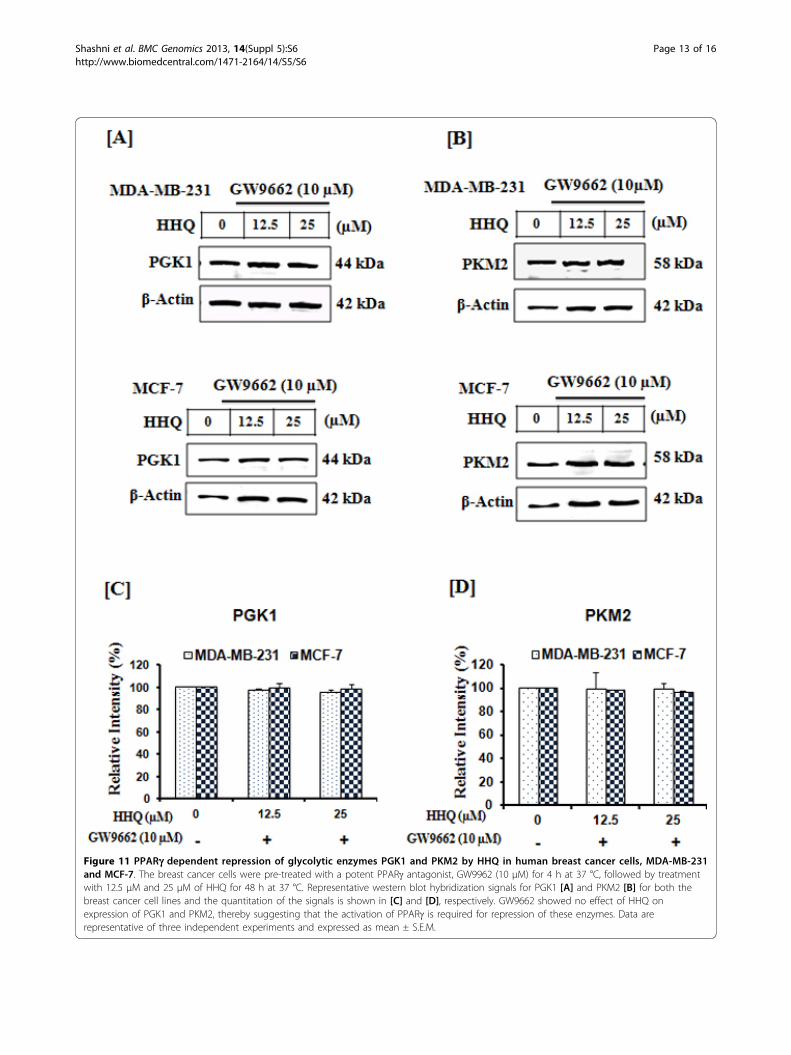
Figure 11 PPARg dependent repression of glycolytic enzymes PGK1 and PKM2 by HHQ in human breast cancer cells, MDA-MB-231and MCF-7. The breast cancer cells were pre-treated with a potent PPARg antagonist, GW9962 (10 μM) for 4 h at 37 °C, followed by treatmentwith 12.5 μM and 25 μM of HHQ for 48 h at 37 °C. Representative western blot hybridization signals for PGK1 [A] and PKM2 [B] for both thebreast cancer cell lines and the quantitation of the signals is shown in [C] and [D], respectively. GW9662 showed no effect of HHQ onexpression of PGK1 and PKM2, thereby suggesting that the activation of PPARg is required for repression of these enzymes. Data arerepresentative of three independent experiments and expressed as mean ± S.E.M.
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 13 of 16

for 48 h at 37 °C and for PPARg activity inhibition studies,the breast cancer cells were pre-treated with GW9662(10 μM, 4h at 37 °C) followed by HHQ (12.5 μM and25 μM, 48 h at 37 °C) and harvested for further use.
Cell proliferation assay and morphological studyHHQ was tested for its anti-proliferative activity on MDA-MB-231 and MCF-7 cells using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromides (MTT) test(Roche, Mannheim, Germany) following manufacturer’sinstructions. The MTT metabolic activity is a colorimetriccell proliferation assay that identifies living cells, and isbased on the cellular conversion of a tetrazolium salt intoinsoluble formazan, which can be quantified by spectropho-tometry. 5 × 103 cells/ well were plated in 96-well platesand grown for 24 h. The cells were then exposed to varyingconcentrations of HHQ for 48 h to find the half maximalinhibitory concentration (IC50) values. The intensity of thereduced dye that corresponds to the viable cells was mea-sured at reference wavelength of 570 nm by Thermo Scien-tific Varioskan Flash Multimode Reader. Morphologicalchanges in breast cancer cells treated with 12.5 µM and25 µM of HHQ concentration for 48 h were examinedby phase contrast microscopy. All experiments wereperformed in triplicates, and the relative cell viability (%)was expressed as a percentage relative to the untreatedcontrol cells.
Clonogenic assayColony formation potential of human breast cancer cellson exposure to HHQ was assessed by clonogenic assay.Clonogenic assay is a cell survival assay based on the abil-ity of a single cell to grow into a colony. To determinelong-term effects, breast cancer cells were treated with12.5 µM and 25 µM of HHQ concentration for 48 h. Atthe end of treatment the breast cancer cells were thenplated at a concentration of 100 cells/well in a new 6-wellplate. The cells were allowed to grow for 14 days to formcolonies. Fresh medium was replaced every third day. Thecolonies were washed with PBS and fixed in pre-chilledmethanol: acetone mixture (1:1) for 10 min at roomtemperature. The colonies were then stained with crystalviolet dye (0.5% in water) at room temperature for over-night. Cells were washed with water and plates werephotographed with image scanner (EPSON GT-1500).Quantitative analysis of the total number of colonies wasperformed with Image J software (National Institutes ofHealth).
Detection of reactive oxygen speciesThe reactive oxygen species were detected by fluorescentstaining using the Image-iTTM LIVE Green Reactive Oxy-gen Species (ROS) Detection Kit (Molecular Probes Inc,USA). The assay is based on a nonfluorescent and cell
permeable 5-(and-6)-carboxy-2’,7’-dichlorodihydrofluores-cein diacetate (carboxy-H2DCFDA). The carboxy-H2DCFDA permeates the live cells and is deacetylated byintracellular esterases. The reduced fluorescein compoundis oxidized by the cellular ROS and emits bright greenfluorescence with excitation/ emission maxima of 495/529nm. The cells were grown on glass cover slips placed in12-well plate and were treated with 12.5 µM and 25 µMconcentration of HHQ for 48 h. Subsequently, cells werefixed and then stained for ROS by following manufac-turer’s instructions. The images were analyzed with Axio-Vision software (version 4.4, Carl Zeiss).
Assessment of mitochondrial membrane potential (ΔΨ)Mitochondrial transmembrane potential was investigatedusing a fluorochrome, JC-1 dye (Biotium). JC-1 (5,5’,6,6’-tetrachloro-1,1’,3,3’ tetra ethyl benzimidazolyl carbocyanine iodide) dye exhibits potential-dependent accumu-lation of red fluorescent J-aggregates in energized mito-chondria. JC-1 exists as a green fluorescent (535 nm)monomer and also accumulates as J-aggregates (595 nm)in the active mitochondria, which stain red. Consequently,healthy cells will exhibit high red/green fluorescenceintensity ratio. In apoptotic cells, mitochondrial depolari-zation is indicated by a decrease in the red/green fluores-cence intensity ratio. Therefore this fluorescence emissionshift from green (~529 nm) to red (~590 nm) is indicativeof mitochondrial depolarization occurring during apopto-sis. After 48 h of 12.5 µM and 25 µM of HHQ treatment,the cells were incubated with 1X JC-1 dye at 37 °C for15 min followed by washes with assay buffer. The redfluorescence (excitation 550 nm, emission 600 nm) andgreen fluorescence (excitation 485 nm, emission 535 nm)were measured using fluorescence Thermo ScientificVarioskan Flash Multimode plate reader. The ratio of redfluorescence to green fluorescence was determined in JC-1stained cells. The relative mitochondrial membranepotential (%) was expressed as a percentage relative to theuntreated control cells.
ImmunocytostainingIn order to find out the modulation of test proteins byHHQ through PPARg, the test protein were immunos-tained. For immunostaining, cells were plated on glasscoverslips in a 12-well plate (104 cells/coverslip). The cellswere allowed to attach for 24 h and then exposed to12.5 µM and 25 µM of HHQ for 48 h. At the end of treat-ment cells were washed with cold PBS three times andthen fixed with pre-chilled methanol: acetone mixture(1:1) for 10 min at room temperature. Fixed cells werewashed twice with PBS, permeabilized with 0.32% TritonX-100 in PBS (PBST) for 10 min and blocked with 2%bovine serum albumin (BSA) in PBS for 30 min. The cellswere then probed with anti-PGK1 (1:250; Abcam,
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 14 of 16

Cambridge, UK), anti-PKM2 (1:250; Abcam, Cambridge,UK), antibodies for overnight at 4°C. After primary anti-body incubation the cells were washed thrice with 0.1%PBST, and then incubated with secondary antibodies(Alexa-594-conjugated goat anti-rabbit and Alexa 488-conjugated goat anti-mouse, (Molecular Probes, Eugene,OR) for 1 h at room temperature followed by 3 timeswashing with 0.1% PBST and then once with PBS. Thecoverslips were then mounted with ProLong+® Gold anti-fade reagent (Molecular Probes, Inc.) and observed undera microscope (Axiovert 200M; Carl Zeiss, Thornwood,NY). The experiment was carried out in duplicate inthree independent experiments. The images were ana-lyzed with Axio Vision software (version 4.4, Carl Zeiss).
Western blot analysisExpression level of indicated proteins was examined byWestern blotting. The control and treated cells were lysedin RIPA lysis buffer (25 mM Tris-HCl (pH 7.6), 1% NP-40,1% sodium deoxycholate, 150 mM NaCl, 0.1% SDS) com-plemented with complete protease inhibitors (Roche) for15 min on ice followed by centrifugation at 14,000 g for15 min at 4 °C. The protein concentration was determinedusing BCA reagents (Pierce, Rockford, IL, USA). Proteinlysate (20 µg) was resolved in 10% in SDS-polyacrylamidegel under standard denaturing conditions according toLaemmli’s method (1970) followed by transfer onto polyvi-nylidene difluoride membranes using a Trans-Blot SD(Bio-Rad, Lewes, E. Sussex, UK.) semi-dry electroblotterfor 30 min at 20 V. Subsequently, the membranes wereblocked for 45 min at room temperature with 2% bovineserum albumin in 0.1% PBST. The membranes were thenprobed with anti-PGK1 (1:1000; Abcam, Cambridge, UK),anti-PKM2 (1:500, Abcam), anti-caspase-8 (1:1000; CellSignaling, Boston, MA, USA), Anti-Bax (1:1000; CellSignaling) and anti-b-actin (1:20,000; Abcam) at 4°Covernight followed by three washes for 5 min each with0.1% PBST. The membranes were then incubated withhorseradish peroxidase conjugated secondary antibody(1:20,000; Abcam) for 45 min at RT and washed thrice for5 min each with 0.1% PBST followed by chemiluminescentdetection using Luminescent Image Analyzer equippedwith charge-coupled device camera (LAS-4000 Ver. 2.1;Fuji Film, Tokyo, Japan).
Data analysisThe captured images for ROS assay and immunostainingwere analyzed using AxioVision software (version 4.4; CarlZeiss). The analysis determined the overall density of ROS,PGK1 and PKM2 immunoreactivity in 5-8 randomlyselected fields in each slide. The mean intensity of ROS,PGK1, and PKM2 immunoreactivity in control andtreated cells were evaluated and presented as a histo-gram. Similarly the Relative Optical Density (%) for
immunoreactive bands in western blotting for PGK1,PKM2 and Caspase-8 were analyzed using Image J software(National Institutes of Health).
Statistical analysisThe quantitative data are representative of three indepen-dent experiments done in triplicates and expressed asmean ± SEM. Statistical analysis was performed using ana-lysis of variance (one way analysis of variance) followed byBonferroni’s on test to determine differences in mean andp < 0.05 was considered as statistically significant.
Competing interestsThe authors declare that they have no conflict of interest.
Authors’ contributionsMKS, YN designed the project. SKD and KRS carried out computationalanalysis. BS, KS and RS performed the experiments. MKS, KRS and BSanalyzed the results and wrote the manuscript. All authors read andapproved the final manuscript.
DeclarationsPublication of this article was funded by University of Tsukuba, Japan.This article has been published as part of BMC Genomics Volume 14Supplement 5, 2013: Twelfth International Conference on Bioinformatics(InCoB2013): Computational biology. The full contents of the supplement areavailable online at http://www.biomedcentral.com/bmcgenomics/supplements/14/S5.
Authors’ details1Graduate School of Life and Environmental Sciences, University of Tsukuba,Tsukuba, Ibaraki, Japan. 2AIST, Tsukuba, Japan. 3OmicsVista, Singapore.4Institute of Biological Sciences, Faculty of Science, University Malaya, KualaLumpur, Malaysia. 5Department of Materials Sciences, Graduate School ofPure and Applied Sciences, University of Tsukuba, Ibaraki, Japan. 6Master’sSchool of Medical Sciences, Graduate School of Comprehensive HumanSciences, University of Tsukuba, Ibaraki, Japan. 7Satellite Laboratory,International Center for Materials Nanoarchitectonics (WPI-MANA), NationalInstitute for Materials Science (NIMS), University of Tsukuba, Ibaraki, Japan.8Department of Biotechnology, University of Pune, Pune, India. 9DrugDiscovery and Development Research Group, College of Pharmacy andNutrition, University of Saskatchewan, Saskatoon, Canada.
Published: 16 October 2013
References1. Mukhopadhyay S, Das SK, Mukherjee S: Expression of Mn-Superoxide
Dismutase Gene in Nontumorigenic and Tumorigenic Human MammaryEpithelial Cells. J Biomed Biotechnol 2004, 4:195-202.
2. Tennant DA, Duran RV, Gottlieb E: Targeting metabolic transformation forcancer therapy. Nat Rev Cancer 2011, 10:267-277.
3. Desvergne B, Wahli W: Peroxisome proliferator-activated receptors:nuclear control of metabolism. Endocr Rev 1999, 20(5):649-88.
4. Theocharis S, Margeli A, Vielh P, Kouraklis G: Peroxisome proliferator-activated receptor-gamma ligands as cell-cycle modulators. Cancer TreatRev 2004, 30(6):545-54.
5. Shashni B, Sakharkar KR, Nagasaki Y, Sakharkar MK: Glycolytic enzymesPGK1 and PKM2 as novel transcriptional targets of PPARγ in breastcancer pathophysiology. J Drug Target 2013, 21(2):161-74.
6. Vidal-Puig AJ, Considine RV, Jimenez-Liñan M, Werman A, Pories WJ,Caro JF, Flier JS: Peroxisome proliferator activated receptor geneexpression in human tissues. Effects of obesity, weight loss, andregulation by insulin and glucocorticoids. J Clin Invest 1997,99(10):2416-22.
7. Vignati S, Albertini V, Rinaldi A, Kwee I, Riva C, Oldrini R, Capella C,Bertoni F, Carbone GM, Catapano CV: Cellular and molecular
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 15 of 16

consequences of peroxisome proliferator-activated receptor-gammaactivation in ovarian cancer cells. Neoplasia 2006, 8:851-861.
8. Liu Y, Meng Y, Liu H, Li J, Fu J, Chen X: Growth inhibition anddifferentiation induced by peroxisome proliferator activated receptorgamma ligand rosiglitazone in human melanoma cell line a375. MedOncol 2006, 23:393-402.
9. Penumetcha M, Santanam N: Nutraceuticals as Ligands of PPARγ. PPARRes 2012, 2012:858352, doi: 10.1155/2012/858352.
10. Riccio P: The molecular basis of nutritional intervention in multiplesclerosis: a narrative review. Complement Ther Med 2011, 19(4):228-37.
11. Higdon JV, Frei B: Coffee and health: a review of recent human research.Crit Rev Food Sci Nutr 2006, 46(2):101-23.
12. Leung WW, Ho SC, Chan HL, Wong V, Yeo W, Mok TS: Moderate coffeeconsumption reduces the risk of hepatocellular carcinoma in hepatitis Bchronic carriers: a case-control study. J Epidemiol Community Health 2011,65(6):556-558.
13. Chou TM, Benowitz NL: Caffeine and coffee: effects on health andcardiovascular disease. Comp Biochem Physiol C Pharmacol ToxicolEndocrinol 1994, 109(2):173-89.
14. Scalbert A, Williamson G: Dietary intake and bioavailability ofpolyphenols. J Nutr 2000, 130:2073-2085.
15. Allred KF, Yackley KM, Vanamala J, Allred CD: Trigonelline is a novelphytoestrogen in coffee beans. J Nutr 2009, 139:1833-1838.
16. Welsch CW, Scieszka KM, Senn ER, Dehoog JV: Caffeine (1, 3, 7trimethylxanthine), a temperate promoter of DMBA-induced ratmammary gland carcinogenesis. Int J Cancer 1983, 32(4):479-484.
17. Larsson SC, Wolk A: Coffee consumption and risk of liver cancer: a meta-analysis. Gastroenterology 2007, 132(5):1740-1745.
18. Bravi F, Bosetti C, Tavani A, et al: Coffee drinking and hepatocellularcarcinoma risk: a meta-analysis. Hepatology 2007, 46(2):430-435.
19. Je Y, Liu W, Giovannucci E: Coffee consumption and risk of colorectalcancer: a systematic review and meta-analysis of prospective cohortstudies. International Journal of Cancer 2009, 124(7):1662-1668.
20. Tang N, Zhou B, Wang B, Yu R: Coffee consumption and risk of breastcancer: a metaanalysis. American Journal of Obstetrics and Gynecology 2009,200(3):290.e1-290.e9.
21. Li J, Seibold P, Chang-Claude J, Flesch-Janys D, Liu J, Czene K,Humphreys K, Hall P: Coffee consumption modifies risk of estrogen-receptor negative breast cancer. Breast Cancer Res 2011, 13(3):R49.
22. Harris HR, Bergkvist L, Wolk A: Coffee and black tea consumption andbreast cancer mortality in a cohort of Swedish women. Br J Cancer 2012,107(5):874-8.
23. Lowcock EC, Cotterchio M, Anderson LN, Boucher BA, El-Sohemy A: Highcoffee intake, but not caffeine, is associated with reduced estrogenreceptor negative and postmenopausal breast cancer risk with no effectmodification by CYP1A2 genotype. Nutr Cancer 2013, 65(3):398-409.
24. Simonsson M, Soderlind V, Henningson M, Hjertberg M, Rose C, Ingvar C,Jernstrom H: Coffee prevents early events in tamoxifen-treated breastcancer patients and modulates hormone receptor status. Cancer CausesControl 2013, 24(5):929-40.
25. Guan YF, Zhang YH, Breyer RM, Davis L, Breyer MD: Expression ofperoxisome proliferator-activated receptor gamma (PPARgamma) inhuman transitional bladder cancer and its role in inducing cell death.Neoplasia 1999, 1:330-339.
26. Suzuki T, Nakagawa T, Endo H, Mitsudomi T, Masuda A, et al: Thesensitivity of lung cancer cell lines to the EGFR-selective tyrosine kinaseinhibitor ZD1839 (’Iressa’) is not related to the expression of EGFR orHER-2 or to K-ras gene status. Lung Cancer 2003, 42:35-41.
27. Burgermeister E, Tencer L, Liscovitch M: Peroxisome proliferator-activatedreceptor-gamma upregulates caveolin-1 and caveolin-2 expression inhuman carcinoma cells. Oncogene 2003, 22:3888-3900.
28. Smith AG, Muscat GE: Orphan nuclear receptors: therapeuticopportunities in skeletal muscle. American Journal of Physiology 2006,291(2):C203-C217.
29. Amon LM, Pitteri SJ, Li CI, et al: Concordant release of glycolysis proteinsinto the plasma preceding a diagnosis of ER+ breast cancer. CancerResearch 2012, 72(8):1935-1942.
30. Nolte RT, Wisely GB, Westin S, et al: Ligand binding and co-activatorassembly of the peroxisome proliferator-activated receptor-γ. Nature1998, 395(6698):137-143.
31. Bruning JB, Chalmers MJ, Prasad S, et al: Partial agonists activate PPARγusing a helix 12 independent mechanism. Structure 2007,15(10):1258-1271.
32. Tsukahara T, Tsukahara R, Yasuda S, Makarova N, Valentine WJ, Allison P,Yuan H, Baker DL, Li Z, Bittman R, Parrill A, Tigyi G: Different residuesmediate recognition of 1-O-oleyl-lysophosphatidic acid and rosiglitazonein the ligand binding domain of PPAR1. J Biol Chem 2006,281(6):3398-407.
33. Haddad JJ: Redox and oxidant mediated regulation of apoptosissignaling pathways: immuno-pharmaco-redox conception of oxidativesiege versus cell death commitment. Int Immunopharmacol 2004,4(4):475-93.
34. Barrera G: Oxidative stress and lipid peroxidation products in cancerprogression and therapy. ISRN Oncol 2012, 2012:137-289.
35. Nishikawa T, Miyahara E, Horiuchi M, Izumo K, Okamoto Y, Kawai Y,Kawano Y, Takeuchi T: Benzene metabolite 1,2,4 benzenetriol induceshalogenated DNA and Tyrosines representing halogenative stress inHL-60 human myeloid cell line. Environ Health Perspect 2012, 120(1):62-7.
36. Pelicano H, Carney D, Huang P: ROS stress in cancer cells and therapeuticimplications. Drug Resist Updat 2004, 7(2):97-110.
37. Qanungo S, Das M, Haldar S, Basu A: Epigallocatechin-3-gallate inducesmitochondrial membrane depolarization and caspase-dependentapoptosis in pancreatic cancer cells. Carcinogenesis 2005, 26:958-967.
38. Cao XH, Wang AH, Wang CL, Mao DZ, Lu MF, Cui YQ, Jiao RZ: Surfactininduces apoptosis in human breast cancer MCF-7 cells through a ROS/JNK-mediated mitochondrial/caspase pathway. Chem Biol Interact 2010,183:357-362.
39. Warburg O: On respiratory impairment in cancer cells. Science 1956,124:269-270.
40. Carew JS, Zhou Y, Albitar M, Carew JD, Keating MJ, Huang P: MitochondrialDNA mutations in primary leukemia cells after chemotherapy: clinicalsignificance and therapeutic implications. Leukemia 2003, 17:1437-1447.
41. Mammucari C, Rizzuto R: Signaling pathways in mitochondrialdysfunction and aging. Mech Ageing Dev 2010, 131(7-8):536-43.
42. Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G: Mitochondrial control ofcellular life, stress, and death. Circ Res 2012, 111(9):1198-207.
43. Sakharkar MK, Shashni B, Sharma K, Kaur S, Ranjekar P, Sakharkar KR:Therapeutic Implications of Targeting Energy Metabolism in BreastCancer. PPAR Res 2013, 2013:109285.
44. Kroemer G, Galluzzi L, Brenner C: Mitochondrial membranepermeabilization in cell death. Physiol Rev 2007, 87(1):99-163.
45. Bonofiglio D, Cione E, Qi H, Pingitore A, Perri M, Catalano S, Vizza D,Panno ML, Genchi G, Fuqua SA, Andò S: Combined low doses of PPARgamma and RXR ligands trigger an intrinsic apoptotic pathway inhuman breast cancer cells. Am J Pathol 2009, 175(3):1270-80.
46. Li S, Zhou Q, He H, Zhao Y, Liu Z: Peroxisome proliferator-activatedreceptor γ agonists induce cell cycle arrest through transcriptionalregulation of Kruppel-like factor 4 (KLF4). J Biol Chem 2013,288(6):4076-84.
47. Altenberg B, Greulich KO: Genes of glycolysis are ubiquitouslyoverexpressed in 24 cancer classes. Genomics 2004, 84:1014-20.
48. Jiang K, He B, Lai L, Chen Q, Liu Y, Guo Q, Wang Q: CyclosporineA inhibits breast cancer cell growth by downregulating the expressionof pyruvate kinase subtype M2. Int J Mol Med 2012, 30:302-8.
49. Martin DS, Bertino JR, Koutcher JA: ATP depletion + pyrimidine depletioncan markedly enhance cancer therapy: fresh insight for a new approach.Cancer Res 2000, 60:6776-6783.
doi:10.1186/1471-2164-14-S5-S6Cite this article as: Shashni et al.: Coffee component hydroxylhydroquinone (HHQ) as a putative ligand for PPAR gamma andimplications in breast cancer. BMC Genomics 2013 14(Suppl 5):S6.
Shashni et al. BMC Genomics 2013, 14(Suppl 5):S6http://www.biomedcentral.com/1471-2164/14/S5/S6
Page 16 of 16




















