
Role of Distant Al Atoms in Alkaline Earth Zeolites for Stabilization of Hydroxyl Groups
12
Published: December 22, 2011 r2011 American Chemical Society 2399 dx.doi.org/10.1021/jp205028c | J. Phys. Chem. C 2012, 116, 2399–2410 ARTICLE pubs.acs.org/JPCC Role of Distant Al Atoms in Alkaline Earth Zeolites for Stabilization of Hydroxyl Groups Alexander V. Larin,* ,† Andrey A. Rybakov, † and Georgy M. Zhidomirov †,‡ † Lomonosov MSU, Moscow, Russia ‡ Boreskov Institute of Catalysis, Novosibirsk, Russia b S Supporting Information 1. INTRODUCTION Multivalent cationic form zeolites are an important class of catalysts possessing acid character. The crucial role of water in the chemical activity of cationic form zeolites is often explained by its possible dissociation and formation of Brønsted groups. 110 Breck discussed the interpretation of water cleavage over divalent cations in detail in his monography 11 after numerous works. 18 The explanation that the high electric field produced by divalent cations is the main reason for the formation of structural hydroxyls has remained popular until nowadays. For example, for the alkaline earth (AE) cations the following reaction is suggested 2,3,5 Me 2þ ðH 2 OÞþ½Si O Al f MeOH þ þ Si OðHÞ Al ð1Þ The appearance of IR peaks of Me(OH) + and of the hydroxyls close to that of Brønsted groups was recorded 111 and served to approve the water cleavage (1) as due to the high electric field near the divalent Me 2+ cation. Such a step (1) is involved in the tentative mechanisms suggested for alcohol dehydration, 12 oxo-cluster formation, 13 alkane oxidations, 14 alkene protonation, 15 and alkene oxida- tions 1517 and alkene isomerizations. 15 (We cite here only the articles whose authors directly addressed the reaction 1 in their publications, but this dissociation is implicitly suggested in a much wider series of papers. D.W. Breck 11 cited this reaction for a broad series of catalytic implementations.) The types of hydroxyl groups are of fundamental importance for zeolite activity, but it was not yet discussed theoretically for AE zeolites to our best knowledge. The dissociative adsorption of a water molecule on divalent metal cations in zeolites was considered only in a few papers as compared to the molecular adsorption. Particularly, Zn ion-exchange zeolites attracted more attention due to their activity as catalysts of the dehydrogenation reaction of small alkanes. 1820 So, the calculations of the interaction of Zn 2+ , localized in the six-membered cycle of Y zeolite with a water molecule, showed that the energy of molecular adsorption is quite significant, being equal to 29 kcal/mol. 18 The authors of ref 18 were unable to find any structure corresponding to the heterolytic dissociation of water (eq 1) over the same 6R window of the Y type. All optimizations resulted in molecular adsorption regardless of the starting point. In contrast to the Y type fragment, the structures corresponding to water heterolytic dissociation on Zn 2+ in various five-membered (5R) zeolite cycles were found. The process (1) was calculated as enough exothermic (21.628.1 kcal/mol). However, in this work 18 there was no comparison between the energies of the molecular and dissociative adsorption over the 5R cycles. It was provided in refs 19 and 20 for Zn 2+ in 5R and in the α-position of ZSM-5, and it was found that molecular adsorption of Zn(H 2 O) 2+ is more stable than the dissociative one (eq 1) on 11.94 kcal/mol for these cluster models. Hence, the theoretical justification of reaction 1 for any zeolite remains uncertain. The experimental spectra of the products of water cleavage deserve much more attention since they are unique evidence of reaction 1. Resuming this analysis, only the band at 3740 3750 cm 1 was definitely assigned (silanol groups). The main resume regarding experimental spectra for MeMOR is the sim- ilarity between the spectra for all AE cations containing two strong bands at 3745 and 3616 cm 1 . (Slightly shifted 3620 cm 1 was measured 10 with an accuracy of 5 cm 1 so that Received: May 30, 2011 Revised: December 21, 2011 ABSTRACT: The traditional point of view about water cleavage at divalent cations in alkaline earth (AE) mordenite (MOR) and faujasite (FAU) zeolites was not supported by energy analysis at both the isolated cluster level and the computations with periodic conditions if the cation is located in the vicinity of both Al atoms. A new hypothesis about the kinetic (and not thermodynamic) stabilization of hydroxyl groups near one noncompensated distant Al atom is verified theoretically to explain the acidic nature of the AE cationic MOR and FAU forms. This model allows the development of qualitative interpretation of the temperature varia- tions in the IR spectra upon hydration, dehydration, and rehydration.
-
Upload
moscowstate -
Category
Documents
-
view
1 -
download
0
Transcript of Role of Distant Al Atoms in Alkaline Earth Zeolites for Stabilization of Hydroxyl Groups

Published: December 22, 2011
r 2011 American Chemical Society 2399 dx.doi.org/10.1021/jp205028c | J. Phys. Chem. C 2012, 116, 2399–2410
ARTICLE
pubs.acs.org/JPCC
Role of Distant Al Atoms in Alkaline Earth Zeolites for Stabilization ofHydroxyl GroupsAlexander V. Larin,*,† Andrey A. Rybakov,† and Georgy M. Zhidomirov†,‡
†Lomonosov MSU, Moscow, Russia‡Boreskov Institute of Catalysis, Novosibirsk, Russia
bS Supporting Information
1. INTRODUCTION
Multivalent cationic form zeolites are an important class ofcatalysts possessing acid character. The crucial role of water inthe chemical activity of cationic form zeolites is often explainedby its possible dissociation and formation of Brønsted groups.1�10
Breck discussed the interpretation of water cleavage over divalentcations in detail in his monography11 after numerous works.1�8
The explanation that the high electric field produced by divalentcations is the main reason for the formation of structuralhydroxyls has remained popular until nowadays. For example,for the alkaline earth (AE) cations the following reaction issuggested2,3,5
Me2þðH2OÞ þ ½Si�O� Al�� f MeOHþ
þ Si�OðHÞ � Al ð1Þ
The appearance of IR peaks of Me(OH)+ and of the hydroxylsclose to that of Brønsted groups was recorded1�11 and served toapprove the water cleavage (1) as due to the high electric fieldnear the divalent Me2+ cation.
Such a step (1) is involved in the tentative mechanismssuggested for alcohol dehydration,12 oxo-cluster formation,13
alkane oxidations,14 alkene protonation,15 and alkene oxida-tions15�17 and alkene isomerizations.15 (We cite here only thearticles whose authors directly addressed the reaction 1 in theirpublications, but this dissociation is implicitly suggested in amuch wider series of papers. D.W. Breck11 cited this reaction fora broad series of catalytic implementations.) The types ofhydroxyl groups are of fundamental importance for zeoliteactivity, but it was not yet discussed theoretically for AE zeolitesto our best knowledge. The dissociative adsorption of a watermolecule on divalent metal cations in zeolites was consideredonly in a few papers as compared to the molecular adsorption.
Particularly, Zn ion-exchange zeolites attracted more attentiondue to their activity as catalysts of the dehydrogenation reactionof small alkanes.18�20 So, the calculations of the interaction ofZn2+, localized in the six-membered cycle of Y zeolite with awater molecule, showed that the energy of molecular adsorptionis quite significant, being equal to 29 kcal/mol.18 The authorsof ref 18 were unable to find any structure corresponding tothe heterolytic dissociation of water (eq 1) over the same 6Rwindow of the Y type. All optimizations resulted in molecularadsorption regardless of the starting point. In contrast to the Ytype fragment, the structures corresponding to water heterolyticdissociation on Zn2+ in various five-membered (5R) zeolitecycles were found. The process (1) was calculated as enoughexothermic (21.6�28.1 kcal/mol). However, in this work18 therewas no comparison between the energies of the molecular anddissociative adsorption over the 5R cycles. It was provided inrefs 19 and 20 for Zn2+ in 5R and in theα-position of ZSM-5, andit was found that molecular adsorption of Zn(H2O)
2+ is morestable than the dissociative one (eq 1) on 11.94 kcal/mol forthese cluster models. Hence, the theoretical justification ofreaction 1 for any zeolite remains uncertain.
The experimental spectra of the products of water cleavagedeserve much more attention since they are unique evidence ofreaction 1. Resuming this analysis, only the band at 3740�3750 cm�1 was definitely assigned (silanol groups). The mainresume regarding experimental spectra for MeMOR is the sim-ilarity between the spectra for all AE cations containing twostrong bands at 3745 and 3616 cm�1. (Slightly shifted3620 cm�1 was measured10 with an accuracy of 5 cm�1 so that
Received: May 30, 2011Revised: December 21, 2011
ABSTRACT: The traditional point of view about water cleavage atdivalent cations in alkaline earth (AE) mordenite (MOR) and faujasite(FAU) zeolites was not supported by energy analysis at both the isolatedcluster level and the computations with periodic conditions if the cation islocated in the vicinity of both Al atoms. A new hypothesis about the kinetic(and not thermodynamic) stabilization of hydroxyl groups near onenoncompensated distant Al atom is verified theoretically to explain theacidic nature of the AE cationic MOR and FAU forms. This model allowsthe development of qualitative interpretation of the temperature varia-tions in the IR spectra upon hydration, dehydration, and rehydration.
2400 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
we adopted the later value of 3616 cm�1 9 for all MeMOR.Finally, the peak at 3745 cm�1 also varies within 5 cm�1 indifferent works.2,5,9,10.) A more complex structure is observed inthe MeY spectra (Table 1). All of them contain the same silanolgroup at 3740 or 3745 cm�1 and the series of three peaks at 3685,3645, and 3540 cm�1. The positions of the lower frequenciesslightly vary with cation, while the CaY spectra do not contain theline 3540 cm�1 at 350 �C but involve 3590 and 3520 cm�1 in anearlier work of Ward.2 However, this band at 3540 cm�1 isobservable in the CaY spectra at other temperatures.2 Thefrequencies lower than 3600 cm�1 have not probably been mea-sured or presented for all zeolites in the spectra obtained byKustov et al.10 Above 3600 cm�1, the spectra of ref 10 do notdeviate essentially versus ref 2 with the exception of the line3600 cm�1 measured for MgY (3595 cm�1 for CaY).
The problem of the stabilization of dissociative form wasearlier considered by one of us (A.V.L.) while optimizing thegeometry of water molecules in LiABW, NaNAT, and BaEDIzeolites.21�23 Starting from distorted XRD geometries such asthe water molecule in the LiABW zeolite,24 i.e., one of the O�Hbond lengths takes 1.09 Å, the optimization restores it up to aconventional distance of 1.01 Å using the CRYSTAL code.25
Moreover, within the AIM approach,26 we have analyzed elec-tron densities at the bond critical points (BCP) of O�H valenceand O 3 3 3H hydrogen bonds. It was calculated that the waterdistortion in cationic form zeolites is pretty close to the one in thepure water clusters for which the dissociation was not observed.22
Hence, we have questioned at which conditions the proposedmechanism (1) of water dissociation takes place.
In this work, we have provided a systematic study of thereaction 1 for faujasite and mordenite zeolites. Below we shalladdress the model with distant Al atoms in the framework anddivalent cations located near one of them.27 Such a type ofstabilization of divalent cations was proved by experimentaldata28 and was confirmed by whole agreement of the theoreticalcalculations of H2,
29 CH4,30 and C2H6
31 of the molecular andthermal dissociative adsorption with experimental data. Asproposed in our work, the location of a proton near other Alatom whose charge is not compensated by Me2+ can explain the“kinetic” type of the stabilization of hydroxyl groups in the AEzeolites. We shall compare the energies of water dissociation onclosely located Al atoms at the isolated cluster level and in themordenite (MOR) framework with varied |Al 3 3 3Al| distancesusing the periodic approach.
Suggesting the kinetic model for proton stabilization, weshould characterize the main features of such a process. (1)
The energy barriers between the proton sites determine the siteoccupation; (2) consequent heating and cooling steps do notrestore the same site occupation (and respective IR spectra). Thefirst property suggests that the system can partially lose its acidicproperties at higher temperatures when some protons cansteeply accumulate enough energy to pass the barriers and toform water. It cannot happen under thermodynamic controlwhen the dissociated hydroxyl group remains more stable thanwater. Inverse formation of acid OH groups from water isforbidden by the upper temperature limit of catalytic cracking(673K), above which no new acidic sites are formed in the zeoliteunder water influence.32 Then the acidity will be determined byhydroxyls already formed at lower temperatures.
2. COMPUTATIONAL DETAILS
The isolated cluster approach was performed usingGAUSSIAN0333 at the B3LYP and MP2 levels using the6-31G*(Si, Al, O, H, Ca, Mg)/LANL2DZ(Sr, Ba) basis set.Two types of clusters have been used. The first is the eight-membered ring (8R) of MOR, and the second is the Y zeolitefragment that includes two 6R and 4Rwindows (6R + 4R) havingone common Si�O�Si moiety. More technical details about thecluster models are presented in refs 34�36. Different cappinggroups (H, OH) near T-atoms of the 8R ring were tested. Whileadding the OH groups instead of capping H-atoms, a specialprocedure to start from the GULP-optimized O-positions nearthe 8R of the MOR was applied.34�36 Deprotonation energieswere calculated with the cluster approach allowing the opti-mization of positions of all atoms in an anion with the excep-tion of capping hydrogens after deletion of the proton understudy using redundant coordinates (see Supporting Informa-tion, part S1). To determine the reaction barrier for H2Odissociation, we applied the QST3 algorithm as implemented withGAUSSIAN03.33
At the periodic level, we optimized reactant and productgeometries for the reaction between H2O and Me cations,Me =Mg, Ca, Sr, and Ba, in theMOR zeolite with different distancesbetween Al atoms using the VASP.37 The projected-augmentedwave (PAW) method38 and the PW91 functional39 were used.The energy cutoff was set to 500 eV. For calculating the mini-mum energy path between reagents and products, we used theclimbing image nudged elastic band (NEB) method.40,41 Vibra-tional frequencies were calculated using the finite differencemethod as implemented in VASP. Small displacements (0.015 Å)of the atoms from the MeH2O(MOR) species were used toestimate the numerical Hessian matrix. The rest of the zeoliteatoms were kept fixed at their equilibrium positions.
The CRYSTAL06 code42 was applied to calculate the heatof water adsorption in MgPHI within the framework of crystal-line orbitals as a linear combination of the atomic orbitals(CO LCAO) method. The 88-511G*(Mg, Al)/88-31G*(Si)/8-411G*(O)/511G*(H) basis set has been used with B3LYP(30% ofHartree�Fock exchange) to totally optimize theMgPHIcell with and without a water molecule. The sp/d-exponentsat Mg, Al, Si, and O atoms are (0.688, 0.28)/0.54, (0.59, 0.35)/0.51, 0.193/0.161, and (0.5, 0.191)/0.47 au�2.42 The s/p-exponents at H atoms are (0.5, 0.13)/0.3 au�2.42 The tolerancesof 5, 5, 5, 7, and 10 were used. Visualization at both cluster andperiodic levels was realized with the MOLDRAW code.43
Table 1. Experimental Spectra (cm�1) of AE Form Y ZeolitesMeasured at Room Temperature after Calcinations at 350 or500�C
350 �C2,a 500 �C10
type Mg Ca Sr Mg Ca Sr
silanol 3740 3740 3740 3745 3745 3745
Me�OH2 3688 3690 3700 3690 3685 3685
Me�OH2 3642 3640 3639 3645 3645 3645
Si�OH�Al � 3590 3570 3600 3595 �Si�OH�Al 3540 3520 � � � �
aThe spectra after other calcination temperatures (110, 230, and 480 �C)have also been measured.

2401 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
3. RESULTS
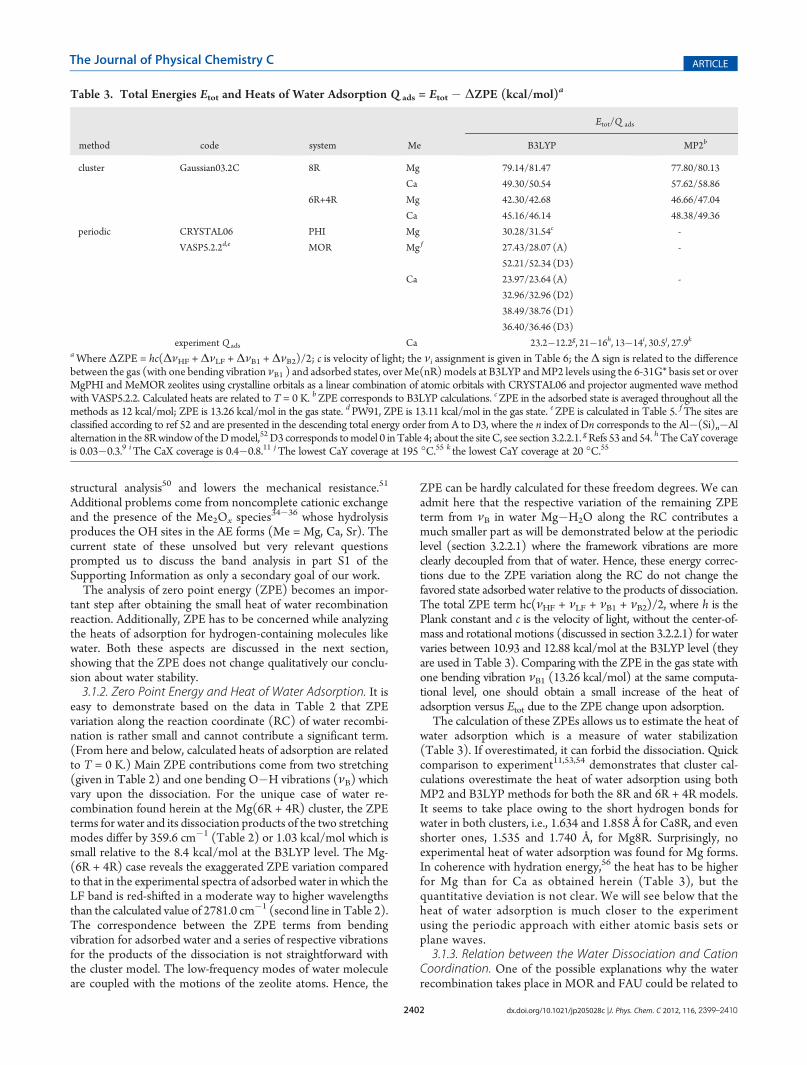
3.1. Cluster Models with Short Al 3 3 3Al Distances. A seriesof optimizations were performed considering the interactionbetweenH2O andMe cations with both 8R (|Al 3 3 3Al| = 8.27 Å)and 6R + 4R (|Al 3 3 3Al| = 8.55 Å) cluster models for Me = Mg,Ca, Sr, and Ba. The geometries of such systems converge to wateradsorption starting from either adsorbed water or from thedissociated form over all AE cations (Table 2).3.1.1. Water Dissociation with the Cluster Model. The H�O
rupture is easier in the 6R + 4R cluster selected herein due to theupper oxygen in the 4R position that is closely located toward theproton of the water molecule above the cation coordinated to the6R window (Figure 1). However, only for Mg in the 6R + 4Rcluster we observed a local minimum between the reagent, i.e.,MgOH+, and the proton attached to the Si�O�Al species(Figure 1a) and the product, i.e., Mg(H2O)
2+ (Figure 1c),separated by a minor activation barrier of the inverse reaction1. The Mg(H2O)
2+ is more stable by 8.4 and 9.9 kcal/mol at theB3LYP andMP2 levels, respectively, with the barriers of 1.44 and2.51 kcal/mol (imaginary frequency of 867.8i cm�1 withB3LYP). Hence, it is difficult to believe that Mg can initiatewater cleavage in the case of the 6R + 4R fragment. According tothe calculations from amore accurate electron correlation (MP4)level, the barrier for proton jump from O(1) to O(4) type inHCHA resulted in a 13% increase of the MP2 barrier.44 Wesuppose that such a barrier increase from 2.51 to 2.84 kcal/moldoes not forbid water dissociation over the Mg(6R + 4R) cluster.The highest intensity ILF/IHF ratio of 121.57 (Table 2) was
calculated namely for the OH vibration in the Si�OH�Alspecies obtained due to water dissociation over Mg(6R + 4R)(Figure 1) through all the cluster models. This agrees withthe domination of the acid OH band of the Si�OH�Al groupin the most acid MeMOR zeolites as compared with MeY.9
(Below (see section 3.2.2.3) we will explain the closenessbetween water adsorption at the cluster Me(6R + 4R) andperiodic MeMOR models in opposition to a difference between
the adsorption at the cluster Me8R and periodic MeMORmodels.) As a result, the fine IR structure below silanol bandswas suppressed in the most acid MeMOR forms,9,10 while theweak bands at the domain of 3700�3500 cm�1 were observedfor less acidic MeY forms, i.e., with a lower intensity of the OHband at the Si�OH�Al position (Table 2). This coincidencebetween the highest intensity calculated for even less probablewater dissociation over Mg(6R + 4R) and the experimentalintensity ILF/IHF ratio demonstrates that the proposed mechan-ism of water dissociation does not contradict the experimentalspectra. However, the full band assignment presents an extremelyhard task. The main problem of the assignment appears due tothe variation of the cationic site occupationswithwater content.45�47
Water is not unique in this respect because pyridine48 and CO2
(or respective carbonate)49 can act in the same style. Togetherwith redistribution of hydrated cations between other sites, thefraction of the nonlocalized Me+q(H2O)n cations due to theirhighmobility grows for both q = 1 and 2.45 This effect hinders theassignment, but most probably it cannot increase the probabi-lity of water dissociation. While varying water content andthus increasing the cation coordination, one shields its electricfield and partly decreases the cation charge. This decreases thepossibility of water dissociation. If water could dissociate over adehydrated sample, this possibility could disappear over the samehydrated form. Regarding the presence of CO2, the positionsof cations can vary due to coordination by carbonates formedin zeolites and following drift together49 that complicates the
Table 2. Intensities (I, KM/mol) and Band Positions(ν, cm�1) of High-Frequency (HF) and Low-Frequency (LF)O�H Stretching Vibrations of Water Adsorbed at Me(8R) orMe(6R+4R) Calculated at the B3LYP/6-31G* Level
HF LF
Me cluster νHF IHF νLF ILF ILF/IHF νHF � νLF
Mg 6R + 4R 3802.0 141.0 3491.9 522.0 3.70 310.1
6R + 4Ra 3793.8 12.9 2781.0 1568.2 121.57 1012.8
8R 3347.8 1058.7 2565.9 2475.1 2.34 781.9
8Rb 3208.0 1090.7 2423.9 143.6 0.132 784.1
Ca 6R + 4R 3800.9 89.2 3035.9 1285.0 14.40 766.1
8R 3569.9 519.2 3085.6 1465.1 2.82 484.3
8Rb 3433.4 532.4 2896.8 1562.1 2.93 536.6
Sr 6R + 4R 3801.1 84.5 2967.2 1357.7 16.07 833.9
8R 3661.7 299.2 3279.4 966.5 3.23 382.3
8Rb 3537.5 303.7 3103.4 1045.6 3.44 434.1
Ba 6R + 4R 3802.8 76.9 3006.6 1278.6 16.62 796.2
8R 3723.0 171.2 3404.1 681.4 3.98 318.9
8Rb 3600.8 169.0 3230.8 752.1 4.45 370.0a Local minimum for dissociated form, not observed for other cations atthe cluster level. b PW91.
Figure 1. Reaction complex (a), transition state (b), and product (c) ofthe proton transfer from the Si�O�Al site to theMg�OH group in theMg(6R + 4R) cluster. Relative energies are given in kcal/mol. Imaginaryfrequency is 869.2i cm�1 at the B3LYP/6-31G* level. The color code is:O in red, Si in yellow, Al in violet, Mg in dark cyan, and H in gray.
2402 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
structural analysis50 and lowers the mechanical resistance.51
Additional problems come from noncomplete cationic exchangeand the presence of the Me2Ox species
34�36 whose hydrolysisproduces the OH sites in the AE forms (Me = Mg, Ca, Sr). Thecurrent state of these unsolved but very relevant questionsprompted us to discuss the band analysis in part S1 of theSupporting Information as only a secondary goal of our work.The analysis of zero point energy (ZPE) becomes an impor-
tant step after obtaining the small heat of water recombinationreaction. Additionally, ZPE has to be concerned while analyzingthe heats of adsorption for hydrogen-containing molecules likewater. Both these aspects are discussed in the next section,showing that the ZPE does not change qualitatively our conclu-sion about water stability.3.1.2. Zero Point Energy and Heat of Water Adsorption. It is
easy to demonstrate based on the data in Table 2 that ZPEvariation along the reaction coordinate (RC) of water recombi-nation is rather small and cannot contribute a significant term.(From here and below, calculated heats of adsorption are relatedto T = 0 K.) Main ZPE contributions come from two stretching(given in Table 2) and one bending O�H vibrations (νB) whichvary upon the dissociation. For the unique case of water re-combination found herein at the Mg(6R + 4R) cluster, the ZPEterms for water and its dissociation products of the two stretchingmodes differ by 359.6 cm�1 (Table 2) or 1.03 kcal/mol which issmall relative to the 8.4 kcal/mol at the B3LYP level. The Mg-(6R + 4R) case reveals the exaggerated ZPE variation comparedto that in the experimental spectra of adsorbed water in which theLF band is red-shifted in a moderate way to higher wavelengthsthan the calculated value of 2781.0 cm�1 (second line in Table 2).The correspondence between the ZPE terms from bendingvibration for adsorbed water and a series of respective vibrationsfor the products of the dissociation is not straightforward withthe cluster model. The low-frequency modes of water moleculeare coupled with the motions of the zeolite atoms. Hence, the
ZPE can be hardly calculated for these freedom degrees. We canadmit here that the respective variation of the remaining ZPEterm from νB in water Mg�H2O along the RC contributes amuch smaller part as will be demonstrated below at the periodiclevel (section 3.2.2.1) where the framework vibrations are moreclearly decoupled from that of water. Hence, these energy correc-tions due to the ZPE variation along the RC do not change thefavored state adsorbed water relative to the products of dissociation.The total ZPE term hc(νHF + νLF + νB1 + νB2)/2, where h is thePlank constant and c is the velocity of light, without the center-of-mass and rotational motions (discussed in section 3.2.2.1) for watervaries between 10.93 and 12.88 kcal/mol at the B3LYP level (theyare used in Table 3). Comparing with the ZPE in the gas state withone bending vibration νB1 (13.26 kcal/mol) at the same computa-tional level, one should obtain a small increase of the heat ofadsorption versus Etot due to the ZPE change upon adsorption.The calculation of these ZPEs allows us to estimate the heat of
water adsorption which is a measure of water stabilization(Table 3). If overestimated, it can forbid the dissociation. Quickcomparison to experiment11,53,54 demonstrates that cluster cal-culations overestimate the heat of water adsorption using bothMP2 and B3LYP methods for both the 8R and 6R + 4R models.It seems to take place owing to the short hydrogen bonds forwater in both clusters, i.e., 1.634 and 1.858 Å for Ca8R, and evenshorter ones, 1.535 and 1.740 Å, for Mg8R. Surprisingly, noexperimental heat of water adsorption was found for Mg forms.In coherence with hydration energy,56 the heat has to be higherfor Mg than for Ca as obtained herein (Table 3), but thequantitative deviation is not clear. We will see below that theheat of water adsorption is much closer to the experimentusing the periodic approach with either atomic basis sets orplane waves.3.1.3. Relation between the Water Dissociation and Cation
Coordination. One of the possible explanations why the waterrecombination takes place in MOR and FAU could be related to
Table 3. Total Energies Etot and Heats of Water Adsorption Q ads = Etot � ΔZPE (kcal/mol)a
Etot/Q ads
method code system Me B3LYP MP2b
cluster Gaussian03.2C 8R Mg 79.14/81.47 77.80/80.13
Ca 49.30/50.54 57.62/58.86
6R+4R Mg 42.30/42.68 46.66/47.04
Ca 45.16/46.14 48.38/49.36
periodic CRYSTAL06 PHI Mg 30.28/31.54c -
VASP5.2.2d,e MOR Mg f 27.43/28.07 (A) -
52.21/52.34 (D3)
Ca 23.97/23.64 (A) -
32.96/32.96 (D2)
38.49/38.76 (D1)
36.40/36.46 (D3)
experiment Q ads Ca 23.2�12.2g, 21�16h, 13�14i, 30.5j, 27.9k
aWhereΔZPE = hc(ΔνHF + ΔνLF + ΔνB1 + ΔνB2)/2; c is velocity of light; the νi assignment is given in Table 6; theΔ sign is related to the differencebetween the gas (with one bending vibration νB1 ) and adsorbed states, overMe(nR)models at B3LYP andMP2 levels using the 6-31G* basis set or overMgPHI and MeMOR zeolites using crystalline orbitals as a linear combination of atomic orbitals with CRYSTAL06 and projector augmented wave methodwith VASP5.2.2. Calculated heats are related to T = 0 K. bZPE corresponds to B3LYP calculations. cZPE in the adsorbed state is averaged throughout all themethods as 12 kcal/mol; ZPE is 13.26 kcal/mol in the gas state. dPW91, ZPE is 13.11 kcal/mol in the gas state. eZPE is calculated in Table 5. fThe sites areclassified according to ref 52 and are presented in the descending total energy order from A to D3, where the n index of Dn corresponds to the Al�(Si)n�Alalternation in the 8Rwindowof theDmodel,52D3 corresponds tomodel 0 inTable 4; about the siteC, see section 3.2.2.1. gRefs 53 and 54. hTheCaY coverageis 0.03�0.3.9 iThe CaX coverage is 0.4�0.8.11 jThe lowest CaY coverage at 195 �C.55 k the lowest CaY coverage at 20 �C.55

2403 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
the stronger stabilization of the Me2+ cation in the 8R or 6R + 4Rclusters considered herein. One can suppose that the cationbecomes less active owing to its tight binding to the framework sothat water does not dissociate. In ZnZSM-5, the stable geometryof the products of water dissociation was optimized in three 5Rwindowswithout comparisonwith the energy of adsorbedwater.18
We compared the energies of the Zn bonding in the work18 withour results at the cluster level and obtained the same trends. Moreprecisely, the authors found a correlation between the heat ofheterolytic ethane dissociation over Zn2+ and the cation bindingenergies to 4R (16.3 kcal/mol), 5R (35.7�47.2 kcal/mol), and6R (59.7�72.9 kcal/mol) windows. The Zn2+ cation bindingin the most accurate extended 8R model results in the energyof 11.4 kcal/mol, which is even smaller than for the 4R one.18
Such a small energy correlates with the existence of the dis-sociated form in the Zn8R case which is less stable than water by7.5 and 8.8 kcal/mol for the Al�(Si)2�Al and Al�(Si)3�Alalternation in the rings, respectively, at the B3LYP/6-31G* level.In parallel, we tested if the Zn�O distances can serve for such
analysis of cation activity, but no confirmation was obtained. Wecompared the Zn�O distances in the 5R rings and the extended8R model (both without water coordinated to Zn) used in refs34�36 (with OH terminal groups instead of protons). The3-coordinated Zn position optimized with B3LYP/6-31G* (ourbasis set differs compared to the D95(Al, Si, H)/6-311G**(O)/6-31G*(Me) one used in ref 18) led to the Zn�O distances of1.955, 1.970, and 2.190 Å which are pretty close to that in the 5Rwindows with 4-coordinated Zn, i.e., 1.962, 2.029, 2.061, and2.102 Å for one of them. The Zn position in the 6R ring(calculated heat of water adsorption is 29 kcal/mol18) is alsocharacterized by similar or slightly larger Zn�O distances of2.033, 2.038, and 2.126 Å (also in the absence of water). Suchcloseness between the Zn�O bonds shows that only the binding
energy of a lower coordinated cation can serve for accurateevaluation of its activity for water dissociation as proposed in ref18. One should note that the Mg cation has nearly the same3-coordinated position with Mg�O distances of 1.962, 1.966,and 2.234 Å in the extended 8R cluster. The Mg�O distancesoptimized at the periodic level with an accurate atomic basis set42
are only slightly larger than these values (section 3.2.1).To check a specific Zn impact on water dissociation compared
to that of Mg which possesses a similar ionic or covalent radius,analogous calculation for the water recombination has beenperformed at the Zn(6R + 4R) cluster where the dissociationproducts have been already optimized for Mg. It also resulted inthe lower stability of the products of water dissociation asobtained for Mg above with the exothermic effect for therecombination, i.e., 5.38 and 6.20 kcal/mol at the B3LYP andMP2 levels, respectively, using 6-31G*. Going forward, we wouldnote that these exothermic heats of recombination for Mg(6R +4R) (9.9 kcal/mol) and Zn(6R + 4R) (6.20 kcal/mol) at theMP2 level are much smaller than those obtained for the periodicMeMOR models 0 for Mg (19.4 kcal/mol, Figure 2b) and Ca(23.4 kcal/mol, Figure 2d) at the PW91 level with close locationof two Al atoms. For the models 1�3 with distant Al atoms, theheat is closer to the cluster results, i.e., 10.76�12.22 kcal/mol(Table 4).Finally, modeling of H2O cleavage in the 8R cluster with two
closely located Al atoms results in barrierless recombination ofwater. Neither larger capping groups of the 8R ring (OH in theextended 8R model instead of H) near T-atoms attached to Siand Al atoms nor theory level (B3LYP and MP2) shows avaluable influence on the results. Hence, according to our clustercalculations, none of the AE cations split water at both theB3LYP and MP2 levels at the close distance between the Alatoms in both 6R + 4R and 8R models, i.e., |Al 3 3 3Al| < 8 Å. Thesingle case of the recombination over Mg(6R + 4R) is character-ized by too small a barrier (1.44 kcal/mol) to explain a reliablemechanism of hydroxyl formation in zeolites. As a logicalcontinuation, we shall test the stabilization of hydroxyl groupsin the periodic MOR models varying |Al 3 3 3Al| distance.3.2. Periodic PHI and MOR Models. As the first step, we
performed the geometry optimization of the zeolites with two(MgPHI) or one (MeMOR)water molecule(s) per cell using theperiodic boundary conditions with atomic basis sets (section 3.2.1)or plane waves (section 3.2.2), respectively. Then, the samestructures have been optimized without water as well to evaluate
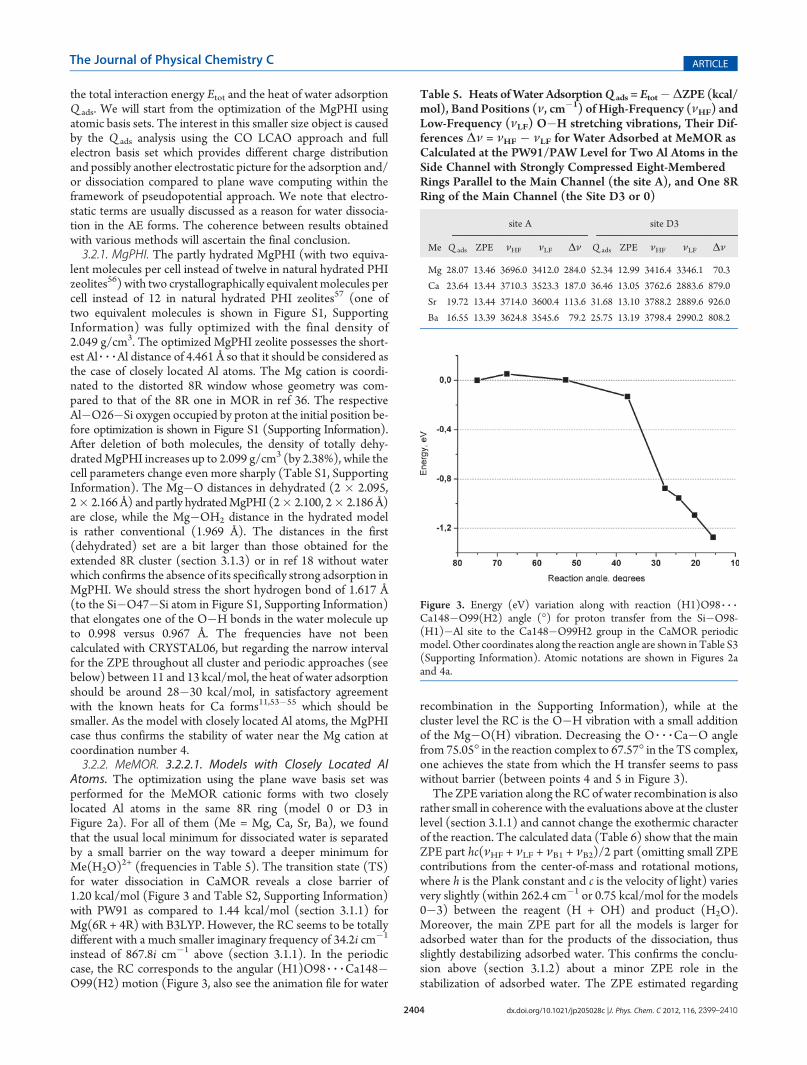
Figure 2. Geometries for reagent (a, c) and product (b, d) of wateradsorption over Ca (a, b) and Mg (c, d) cations in the MeMOR type(model 0 for both cases). Relative energies for (a, b) or (c, d) pairs aregiven in kcal/mol. Products are obtained during geometry optimizationusing VASP without barriers. The dpar and dper in (b) relate to paralleland perpendicular O 3 3 3O diameters of the 8R window (see text). Thecolor code is as in Figure 1 with Ca in dark cyan.
Table 4. Energies U(H + OH) for the Dissociated Form orU(H2O) for Adsorbed Water (Both in eV, 1 eV = 23.05 kcal/mol), Energy Difference ΔU = U(H + OH) � U(H2O) (inkcal/mol) in CaMOR at the Cell Volumes (Å3), and |Al 3 3 3Al|Distances (Å) Optimized at the PAW/PW91 Levela
model
parameter 0b 1 2 3
U(H2O) �1163.09 �1162.58 �1162.64 �1162.60
U(H + OH) �1162.09 �1162.12 �1162.11 �1162.08
ΔU 23.05 10.60 12.22 11.76
|Al 3 3 3Al| 8.31 10.90 11.78 9.80
volume 2813.50 2837.80 2828.66 2830.24aBoth Al atoms are in one 8R window only in the case |Al 3 3 3Al| = 8.31 Å(model 0). bThe model 0 corresponds to D3 in Table 3
2404 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
the total interaction energy Etot and the heat of water adsorptionQ ads. We will start from the optimization of the MgPHI usingatomic basis sets. The interest in this smaller size object is causedby the Q ads analysis using the CO LCAO approach and fullelectron basis set which provides different charge distributionand possibly another electrostatic picture for the adsorption and/or dissociation compared to plane wave computing within theframework of pseudopotential approach. We note that electro-static terms are usually discussed as a reason for water dissocia-tion in the AE forms. The coherence between results obtainedwith various methods will ascertain the final conclusion.3.2.1. MgPHI. The partly hydrated MgPHI (with two equiva-
lent molecules per cell instead of twelve in natural hydrated PHIzeolites56) with two crystallographically equivalentmolecules percell instead of 12 in natural hydrated PHI zeolites57 (one oftwo equivalent molecules is shown in Figure S1, SupportingInformation) was fully optimized with the final density of2.049 g/cm3. The optimized MgPHI zeolite possesses the short-est Al 3 3 3Al distance of 4.461 Å so that it should be considered asthe case of closely located Al atoms. The Mg cation is coordi-nated to the distorted 8R window whose geometry was com-pared to that of the 8R one in MOR in ref 36. The respectiveAl�O26�Si oxygen occupied by proton at the initial position be-fore optimization is shown in Figure S1 (Supporting Information).After deletion of both molecules, the density of totally dehy-dratedMgPHI increases up to 2.099 g/cm3 (by 2.38%), while thecell parameters change even more sharply (Table S1, SupportingInformation). The Mg�O distances in dehydrated (2 � 2.095,2� 2.166 Å) and partly hydratedMgPHI (2� 2.100, 2� 2.186 Å)are close, while the Mg�OH2 distance in the hydrated modelis rather conventional (1.969 Å). The distances in the first(dehydrated) set are a bit larger than those obtained for theextended 8R cluster (section 3.1.3) or in ref 18 without waterwhich confirms the absence of its specifically strong adsorption inMgPHI. We should stress the short hydrogen bond of 1.617 Å(to the Si�O47�Si atom in Figure S1, Supporting Information)that elongates one of the O�H bonds in the water molecule upto 0.998 versus 0.967 Å. The frequencies have not beencalculated with CRYSTAL06, but regarding the narrow intervalfor the ZPE throughout all cluster and periodic approaches (seebelow) between 11 and 13 kcal/mol, the heat of water adsorptionshould be around 28�30 kcal/mol, in satisfactory agreementwith the known heats for Ca forms11,53�55 which should besmaller. As the model with closely located Al atoms, the MgPHIcase thus confirms the stability of water near the Mg cation atcoordination number 4.3.2.2. MeMOR. 3.2.2.1. Models with Closely Located Al
Atoms. The optimization using the plane wave basis set wasperformed for the MeMOR cationic forms with two closelylocated Al atoms in the same 8R ring (model 0 or D3 inFigure 2a). For all of them (Me = Mg, Ca, Sr, Ba), we foundthat the usual local minimum for dissociated water is separatedby a small barrier on the way toward a deeper minimum forMe(H2O)
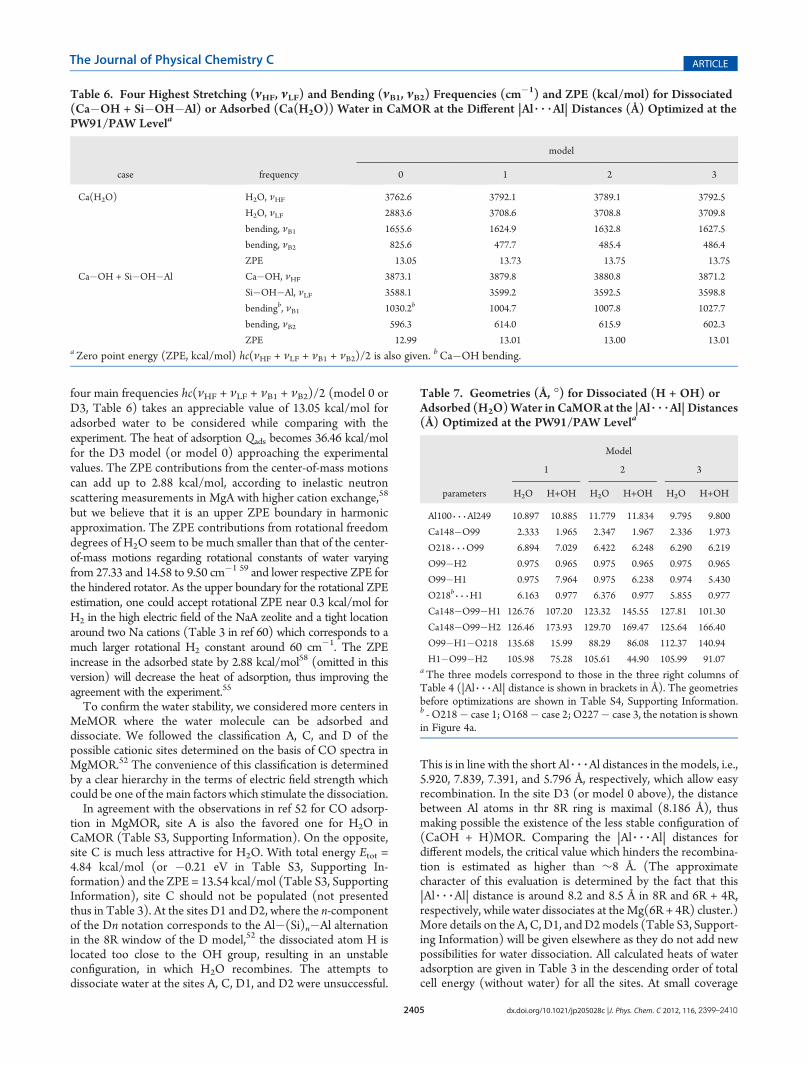
2+ (frequencies in Table 5). The transition state (TS)for water dissociation in CaMOR reveals a close barrier of1.20 kcal/mol (Figure 3 and Table S2, Supporting Information)with PW91 as compared to 1.44 kcal/mol (section 3.1.1) forMg(6R + 4R) with B3LYP. However, the RC seems to be totallydifferent with a much smaller imaginary frequency of 34.2i cm�1
instead of 867.8i cm�1 above (section 3.1.1). In the periodiccase, the RC corresponds to the angular (H1)O98 3 3 3Ca148�O99(H2) motion (Figure 3, also see the animation file for water
recombination in the Supporting Information), while at thecluster level the RC is the O�H vibration with a small additionof the Mg�O(H) vibration. Decreasing the O 3 3 3Ca�O anglefrom 75.05� in the reaction complex to 67.57� in the TS complex,one achieves the state from which the H transfer seems to passwithout barrier (between points 4 and 5 in Figure 3).The ZPE variation along the RC of water recombination is also
rather small in coherence with the evaluations above at the clusterlevel (section 3.1.1) and cannot change the exothermic characterof the reaction. The calculated data (Table 6) show that the mainZPE part hc(νHF + νLF + νB1 + νB2)/2 part (omitting small ZPEcontributions from the center-of-mass and rotational motions,where h is the Plank constant and c is the velocity of light) variesvery slightly (within 262.4 cm�1 or 0.75 kcal/mol for the models0�3) between the reagent (H + OH) and product (H2O).Moreover, the main ZPE part for all the models is larger foradsorbed water than for the products of the dissociation, thusslightly destabilizing adsorbed water. This confirms the conclu-sion above (section 3.1.2) about a minor ZPE role in thestabilization of adsorbed water. The ZPE estimated regarding
Table 5. Heats ofWater AdsorptionQ ads = Etot�ΔZPE (kcal/mol), Band Positions (ν, cm�1) of High-Frequency (νHF) andLow-Frequency (νLF) O�H stretching vibrations, Their Dif-ferences Δν = νHF � νLF for Water Adsorbed at MeMOR asCalculated at the PW91/PAW Level for Two Al Atoms in theSide Channel with Strongly Compressed Eight-MemberedRings Parallel to the Main Channel (the site A), and One 8RRing of the Main Channel (the Site D3 or 0)
site A site D3
Me Q ads ZPE νHF νLF Δν Q ads ZPE νHF νLF Δν
Mg 28.07 13.46 3696.0 3412.0 284.0 52.34 12.99 3416.4 3346.1 70.3
Ca 23.64 13.44 3710.3 3523.3 187.0 36.46 13.05 3762.6 2883.6 879.0
Sr 19.72 13.44 3714.0 3600.4 113.6 31.68 13.10 3788.2 2889.6 926.0
Ba 16.55 13.39 3624.8 3545.6 79.2 25.75 13.19 3798.4 2990.2 808.2
Figure 3. Energy (eV) variation along with reaction (H1)O98 3 3 3Ca148�O99(H2) angle (�) for proton transfer from the Si�O98-(H1)�Al site to the Ca148�O99H2 group in the CaMOR periodicmodel. Other coordinates along the reaction angle are shown in Table S3(Supporting Information). Atomic notations are shown in Figures 2aand 4a.
2405 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
four main frequencies hc(νHF + νLF + νB1 + νB2)/2 (model 0 orD3, Table 6) takes an appreciable value of 13.05 kcal/mol foradsorbed water to be considered while comparing with theexperiment. The heat of adsorption Qads becomes 36.46 kcal/molfor the D3 model (or model 0) approaching the experimentalvalues. The ZPE contributions from the center-of-mass motionscan add up to 2.88 kcal/mol, according to inelastic neutronscattering measurements in MgA with higher cation exchange,58
but we believe that it is an upper ZPE boundary in harmonicapproximation. The ZPE contributions from rotational freedomdegrees of H2O seem to be much smaller than that of the center-of-mass motions regarding rotational constants of water varyingfrom 27.33 and 14.58 to 9.50 cm�1 59 and lower respective ZPE forthe hindered rotator. As the upper boundary for the rotational ZPEestimation, one could accept rotational ZPE near 0.3 kcal/mol forH2 in the high electric field of the NaA zeolite and a tight locationaround two Na cations (Table 3 in ref 60) which corresponds to amuch larger rotational H2 constant around 60 cm�1. The ZPEincrease in the adsorbed state by 2.88 kcal/mol58 (omitted in thisversion) will decrease the heat of adsorption, thus improving theagreement with the experiment.55
To confirm the water stability, we considered more centers inMeMOR where the water molecule can be adsorbed anddissociate. We followed the classification A, C, and D of thepossible cationic sites determined on the basis of CO spectra inMgMOR.52 The convenience of this classification is determinedby a clear hierarchy in the terms of electric field strength whichcould be one of the main factors which stimulate the dissociation.In agreement with the observations in ref 52 for CO adsorp-
tion in MgMOR, site A is also the favored one for H2O inCaMOR (Table S3, Supporting Information). On the opposite,site C is much less attractive for H2O. With total energy Etot =4.84 kcal/mol (or �0.21 eV in Table S3, Supporting In-formation) and the ZPE = 13.54 kcal/mol (Table S3, SupportingInformation), site C should not be populated (not presentedthus in Table 3). At the sites D1 and D2, where the n-componentof the Dn notation corresponds to the Al�(Si)n�Al alternationin the 8R window of the D model,52 the dissociated atom H islocated too close to the OH group, resulting in an unstableconfiguration, in which H2O recombines. The attempts todissociate water at the sites A, C, D1, and D2 were unsuccessful.
This is in line with the short Al 3 3 3Al distances in the models, i.e.,5.920, 7.839, 7.391, and 5.796 Å, respectively, which allow easyrecombination. In the site D3 (or model 0 above), the distancebetween Al atoms in thr 8R ring is maximal (8.186 Å), thusmaking possible the existence of the less stable configuration of(CaOH + H)MOR. Comparing the |Al 3 3 3Al| distances fordifferent models, the critical value which hinders the recombina-tion is estimated as higher than ∼8 Å. (The approximatecharacter of this evaluation is determined by the fact that this|Al 3 3 3Al| distance is around 8.2 and 8.5 Å in 8R and 6R + 4R,respectively, while water dissociates at theMg(6R + 4R) cluster.)More details on the A, C, D1, andD2models (Table S3, Support-ing Information) will be given elsewhere as they do not add newpossibilities for water dissociation. All calculated heats of wateradsorption are given in Table 3 in the descending order of totalcell energy (without water) for all the sites. At small coverage
Table 6. Four Highest Stretching (νHF, νLF) and Bending (νB1, νB2) Frequencies (cm�1) and ZPE (kcal/mol) for Dissociated
(Ca�OH + Si�OH�Al) or Adsorbed (Ca(H2O)) Water in CaMOR at the Different |Al 3 3 3Al| Distances (Å) Optimized at thePW91/PAW Levela
model
case frequency 0 1 2 3
Ca(H2O) H2O, νHF 3762.6 3792.1 3789.1 3792.5
H2O, νLF 2883.6 3708.6 3708.8 3709.8
bending, νB1 1655.6 1624.9 1632.8 1627.5
bending, νB2 825.6 477.7 485.4 486.4
ZPE 13.05 13.73 13.75 13.75
Ca�OH + Si�OH�Al Ca�OH, νHF 3873.1 3879.8 3880.8 3871.2
Si�OH�Al, νLF 3588.1 3599.2 3592.5 3598.8
bendingb, νB1 1030.2b 1004.7 1007.8 1027.7
bending, νB2 596.3 614.0 615.9 602.3
ZPE 12.99 13.01 13.00 13.01aZero point energy (ZPE, kcal/mol) hc(νHF + νLF + νB1 + νB2)/2 is also given.
bCa�OH bending.
Table 7. Geometries (Å, �) for Dissociated (H + OH) orAdsorbed (H2O)Water in CaMOR at the |Al 3 3 3Al| Distances(Å) Optimized at the PW91/PAW Levela
Model
1 2 3
parameters H2O H+OH H2O H+OH H2O H+OH
Al100 3 3 3Al249 10.897 10.885 11.779 11.834 9.795 9.800
Ca148�O99 2.333 1.965 2.347 1.967 2.336 1.973
O218 3 3 3O99 6.894 7.029 6.422 6.248 6.290 6.219
O99�H2 0.975 0.965 0.975 0.965 0.975 0.965
O99�H1 0.975 7.964 0.975 6.238 0.974 5.430
O218b 3 3 3H1 6.163 0.977 6.376 0.977 5.855 0.977
Ca148�O99�H1 126.76 107.20 123.32 145.55 127.81 101.30
Ca148�O99�H2 126.46 173.93 129.70 169.47 125.64 166.40
O99�H1�O218 135.68 15.99 88.29 86.08 112.37 140.94
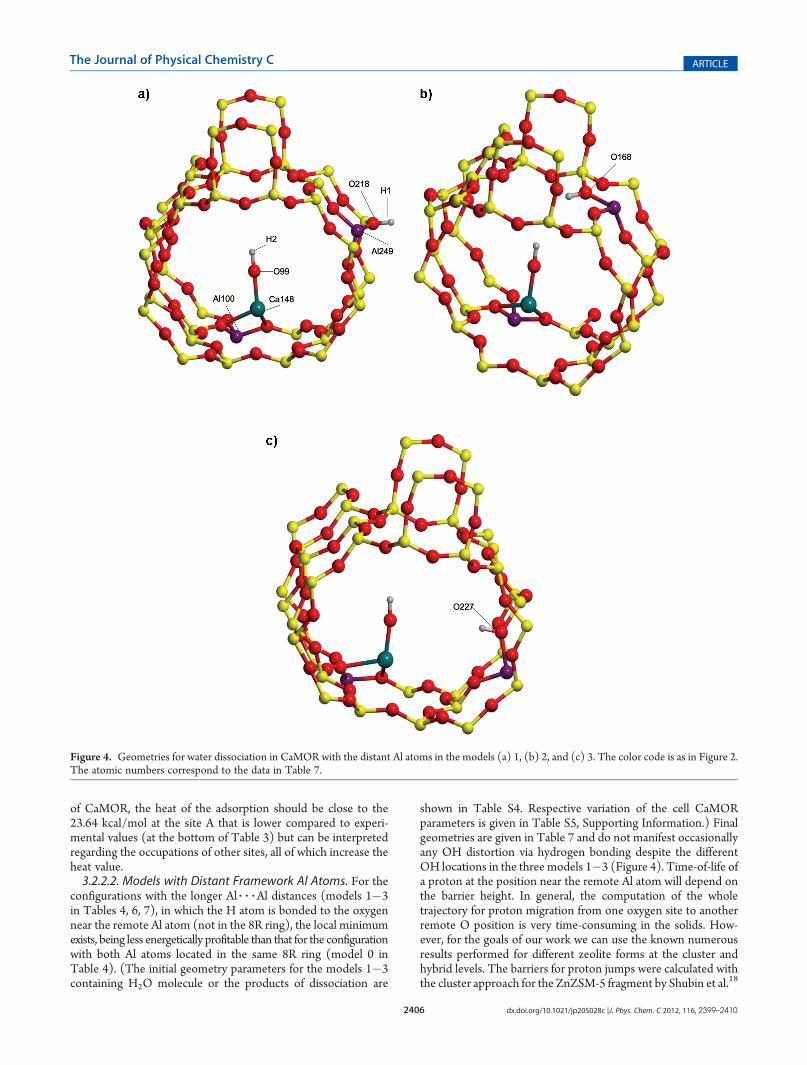
H1�O99�H2 105.98 75.28 105.61 44.90 105.99 91.07aThe three models correspond to those in the three right columns ofTable 4 (|Al 3 3 3Al| distance is shown in brackets in Å). The geometriesbefore optimizations are shown in Table S4, Supporting Information.b - O218� case 1; O168� case 2; O227� case 3, the notation is shownin Figure 4a.
2406 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
of CaMOR, the heat of the adsorption should be close to the23.64 kcal/mol at the site A that is lower compared to experi-mental values (at the bottom of Table 3) but can be interpretedregarding the occupations of other sites, all of which increase theheat value.3.2.2.2. Models with Distant Framework Al Atoms. For the
configurations with the longer Al 3 3 3Al distances (models 1�3in Tables 4, 6, 7), in which the H atom is bonded to the oxygennear the remote Al atom (not in the 8R ring), the local minimumexists, being less energetically profitable than that for the configurationwith both Al atoms located in the same 8R ring (model 0 inTable 4). (The initial geometry parameters for the models 1�3containing H2O molecule or the products of dissociation are
shown in Table S4. Respective variation of the cell CaMORparameters is given in Table S5, Supporting Information.) Finalgeometries are given in Table 7 and do not manifest occasionallyany OH distortion via hydrogen bonding despite the differentOH locations in the three models 1�3 (Figure 4). Time-of-life ofa proton at the position near the remote Al atom will depend onthe barrier height. In general, the computation of the wholetrajectory for proton migration from one oxygen site to anotherremote O position is very time-consuming in the solids. How-ever, for the goals of our work we can use the known numerousresults performed for different zeolite forms at the cluster andhybrid levels. The barriers for proton jumps were calculated withthe cluster approach for the ZnZSM-5 fragment by Shubin et al.18
Figure 4. Geometries for water dissociation in CaMOR with the distant Al atoms in the models (a) 1, (b) 2, and (c) 3. The color code is as in Figure 2.The atomic numbers correspond to the data in Table 7.

2407 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
and the hybrid Qm-Pot scheme by Sierka and Sauer for a series ofH-zeolites.61 In the first work, the barriers were evaluated alongthe proton trajectory from the O atom near one (compensatedby Zn cation) Al atom to the remote one with various barriersranging between 2.4 and 14.2 kcal/mol.18 The periodic correc-tions to the barrier jump values vary from�6.4 to 7.1 kcal/mol forthe intersite jumps.62 Together with these corrections, the cal-culated values are pretty close to the experimental barriers ob-tained fromNMR 1H spectra, i.e., from 2.6 to 10.8 kcal/mol.63�65
Respective calculated values are slightly overestimated with thehybrid Qm-Pot scheme, which considers correlation correctionsto all the barrier values up to the CCSD calculated for one AlO4
tetrahedra.61 More precisely, the lowest activation barriers forproton jump were calculated as 16.3, 16.8, and 12.4 kcal/mol,in HY, HCHA, and HZSM-5 types, respectively.61 Thesevalues approach the activation barrier of electric conductivity(14.1 kcal/mol) in the dehydrated NaA zeolite66 that determinesNa mobility. The Na cation is usually considered as immobile inNaA at room temperature. These high barriers61 can hence resultin the trapping of protons at the Si�O�Al moieties near remoteAl atoms.3.2.2.3. Different Flexibility of the 8R Cluster and MOR
Framework. The higher flexibility of the periodic frameworkresults in qualitatively different water coordination in the MORzeolite relative to that in the 8R cluster for all AE cations with theexception of Mg (Figure 2d). The larger cations are also locatednearly in the 8R plane of MOR (like that for Ca in Figure 2b) sothat the water molecule cannot occupy the same ring due tospatial limits. As a result, H2O is located above the 8R windowand is coordinated via only one H atom in the periodic model(Figure 2b) like in the 6R + 4R cluster (Figure 1c). A similaritybetween νHF or νLF in the Me(6R + 4R) and MeMORmodels isobserved in Figure 5. A relatively large gap is only between νHFvalues forMg. It is also reflected inmuch closer HF frequencies inthe periodicMe(H2O)MORmodel throughoutMe =Ca, Sr, andBa cations (Table 5) and O�H distances, i.e., 0.973, 0.971, and0.970 Å, respectively, as compared to that in the 8R cluster(Table 8). We assign this distinction between water coordinationin the 8R cluster and in MOR to a less flexible 8R cluster model
which cannot allow for the AE cation to be completely coordi-nated by the oxygen atoms of the 8R tightly fixed by cappinghydrogens.Different relaxation of the 8R ring in the Mg8R cluster and
MgMOR periodic model explains the drastic νLF difference(Figure 5). In MgMOR, the O 3 3 3O distance between oppositeoxygens in the perpendicular direction versus the axis of the mainchannel is increased up to 6.722 Å (dper in Figure 4b for Ca case)relative to 6.219 Å in 8R. This parameter defines the differencebetween the hydrogen bonding of the water molecule with the Oatoms in MgMOR or 8R, even if the second 8R “parallel”diameter dpar, i.e., O 3 3 3O distance between opposite oxygensin the direction parallel to the main channel, also varies(Figure 4b). It alternates between the occupied (6.866 Å) andfree (8.248 Å) 8R windows at the lower side (“floor” in Figures 2and 4) of the main MgMOR channels. However, the dpar of the8R window does not vary (6.808 Å) and does not influence thewater coordination. The dpar diameters alternate on both theupper nonoccupied and lower occupied parts of the MgMORmain channels. However, this O 3 3 3O difference is only 0.1 Åbetween neighboring nonoccupied 8R rings at the upper side ofthe channel (“ceiling” in Figures 2 and 4).3.3. Kinetic Stabilization and the Independence of the
O�H Band in the Si�O(H)�Al Moieties of MeMOR on theCation. So, on the basis of the cluster and periodic modeling (seepart S1, Supporting Information), the possible consequence ofthe OH peaks in CaY can be proposed. First, either the higherOH frequency branch of Ca�OHhas a minor intensity as shownwith the cluster computations (by analogy with the dissociationform overMg(6R + 4R) only, Table 2) above, or it is masked by theband of the silanol group at around 3740 cm�1. Second, the peakfrom the Ca(H2O) group falls to the interval 3690�3640 cm�1.
Figure 5. Low-frequency (LF, closed symbols) and high-frequency(HF, open symbols) O�H vibrations in the Me(H2O) calculated in the8R cluster at the B3LYP/6-31G* (triangles down) and PW91/6-31G*(triangles up) levels and in the (6R + 4R) cluster at the B3LYP/6-31G*level (rhomboherda) and in the MeMOR framework calculated at thePW91/PAW level (circles), Me = Mg, Ca, Sr, Ba.
Table 8. O�H Bond Lengths (Å), H�O�H Angle (�), andHydrogen Bond Lengths O 3 3 3H in Water Adsorbed at Me-(6R + 4R) orMe(8R) Clusters Calculated Using the B3LYP orPW91a DFT Functionals and 6-31G* Basis Set or MeMORZeolite Using PW91
Me cluster O�H O 3 3 3H H�O�H
Mg 6R + 4R 0.988 0.970 2.355 - 106.71
8R 1.040 0.994 1.535 1.740 104.23
8Ra 1.060 1.004 1.493 1.726 103.95
MORb 0.996 0.992 2.021 2.050 102.80
Ca 6R + 4R 1.012 0.970 1.722 - 103.87
8R 1.009 0.983 1.635 1.858 101.92
8Ra 1.023 0.993 1.608 1.846 101.52
MORb 1.019 0.973 1.697 - 103.44
Sr 6R + 4R 1.016 0.969 1.652 - 103.44
8R 0.998 0.978 1.715 1.999 100.83
8Ra 1.011 0.987 1.685 1.986 100.39
MORb 1.020 0.971 1.640 - 104.63
Ba 6R + 4R 1.013 0.969 1.635 - 103.31
8R 0.992 0.975 1.787 2.181 100.48
8Ra 1.004 0.984 1.751 2.166 99.93
MORb 1.012 0.970 1.671 - 106.47aAt the PW91 level with Gaussian03.2C; in the gas state B3LYP orPW91 functionals result in the bond length and bond angle of 0.969 Åand 103.87� or 0.976 Å and 103.01�, respectively, in a water moleculeusing the 6-31G* basis set. b PW91with VASP.

2408 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
Third, the OH vibration of the acidic Si�O(H)�Al group with anonbonded proton probably results in the peak between 3540 and3500 cm�1, if one uses the same scaling factor around 0.973 asevaluated from two experimental bands 3640/3740 (see part S1,Supporting Information).On the basis of the peak assignment at 3640�3690 cm�1 to
the Me(H2O) group (3680�3690 cm�1 according to ref 10)with the cluster modeling (see part S1, Supporting Information),we can tentatively assign the OH peaks in Ca�OH and Si�O(H)�Al moieties obtained using the periodic models to theexperimental bands. We note that the Me�OH and Si�O(H)�Al species were not found with the cluster approach(with the exception of Mg(6R + 4R)) because adsorbed watercannot dissociate in the 8R ring (model 0) if the latter containstwo Al atoms. The respective energy difference for the model 0,ΔU = U(H + OH) � U(H2O), is at least as much as two timesgreater than for other models (Table 4). Both the Ca�OH andCa(H2O) groups are less perturbed by changing the position ofthe second Al between the models 1�3 (Table 4). The narrowinterval for the O�H vibrations in the Si�O(H)�Al moieties(Table 6), i.e., between 3588.1 and 3599.5 cm�1, is in a goodagreement with the constant position of the experimental peaknear 36169 or 3620 cm�1 10 for the acidOH group in allMeMOR(Me =Mg, Ca, Sr) forms. The absolute calculated value, i.e., thatrescaled from 3592.5 and 3599.5 cm�1, is slightly overestimatedrelative to the experimental one (3616 cm�1 9), but nearly thesame peak position of the most intense acidic band (Table 6)for the three distinct O�H locations in the CaMOR models(Figure 4) is more important for comparison with experiment. Itis a logic consequence of the remote OH location relative to theAE cation so that the acid Si�O(H)�Al group is not influencedby the cation. These 36169 or 3620 cm�1 10 values for MeMOR(Me = Mg, Ca, Sr) are only slightly larger than experimentalvalues in pure (without cations) H-forms, i.e., 3609 cm�1 forHZSM-5 and 3606 cm�1 for HMOR.28 The small differenceof 10 cm�1 between 3616 cm�1 (CaMOR)9 and 3606 cm�1
(HMOR)28 also supports a weak dependence of the OH peakson the cation.The superior intensity of the peaks of acid OH groups has not
been calculated with the periodic models, but one could appeal tothe intensity ratio ILF/IHF in the 6R + 4R clusters (Table 2) aswe did in the section 3.1.1. Water orientation relative to theMe(6R + 4R) zeolite fragment is pretty similar to that inMeMOR (Table 8, see also section 3.2.2.3). The high ILF/IHFvalues, i.e., between 14.40 and 16.62 from Ca to Ba, are coherentwith the suppressed OH band structure between silanol (3740�3745 cm�1) and Si�OH�Al (3616�3620 cm�1) peaks inCaMOR spectra.9,10 The intensity of the OH band in the Mg�OH group (12.9 KM/mol) is even smaller than that in theMg�OH2 (141.0 KM/mol), so that both of them are notobservable relative to the intensive LF peaks.
4. DISCUSSION
The above proposed assignment of the OH bands does notcontradict with the experimental data. On this basis, we couldpropose a new interpretation of the behavior of the OH groups inAE mordenite and faujasite zeolites. For this, we will first sum-marize the known data. (1) The exact values of the O�Hvibrations depend on temperature of the zeolite activation.2,4
(2) The IR spectra of hydroxyl groups in the AE zeolites dependon thermal or flash activation.4 (3) A nonreversible picture of
hydration and rehydration processes over MeY, Me =Mg, Ca, Sr,Ba, was observed in a wide temperature interval.2,4 The peaks ofthe OH vibrational bands do not keep the same positions afterrehydration at the same temperature. (4) The changes in IRspectra upon rehydration are more or less coherent for all theMeY with the exception of Me = Ba and minor changes in exactO�H frequency values for each AE zeolite.
These variations in the hydroxyl spectra suit well for ametastable system in which acidic protons are captured at theAl�O�Si type oxygens near remote Al atoms whose charges arenot compensated by cations or protons. Their distribution iscontrolled by the jump barrier value shown above and not by adeep potential minimum whose difference can be smaller than thebarrier heights. The jump energies, which proton gains after jump,are within 0.12�4.37, 0.62�3.08, and 0.52�4.94 kcal/mol for HY,HCHA, and HZSM-5 types, respectively. The higher barriers ofthe proton jumps over SiO4 tetrahedra, i.e., 7�14.2 kcal/mol,relative to that over the AlO4 ones, i.e., 2�6.2 kcal/mol, wereobtained in ref 18. This ratio between the barriers over SiO4
and AlO4 tetrahedra permits an explanation of the higheracidity of the AE zeolites with higher Si/Al modulus. Waterrecombines easier in the zeolites through the lower barriersover the AlO4, which occur more frequently at a higher Al con-centration or at a smaller Si/Al ratio. Hence, the OH concen-tration decreases.
It seems that the kinetic control of the hydroxyl distribution isusually masked by the large absolute values of deprotonationenergy (DE) that are considered as a measure of acidity. Thevariation of the large DE values is nevertheless limited by theinterval of one or two tens of kcal/mol for each system (part S1,Supporting Information). Within a specific class of acid protons,the shifts of DE values can be even narrower than the range forchanges of the barrier for proton jumps.18,60 We did not performthe cluster calculations of the DE values which have to operatewith rather large zeolite clusters to take into account all possiblefirst and second neighbors around the Si atom (in the Si�O�Alspecies) in the zeolite under study to be comparable with theexperimental DE values.67,68 The attempts to evaluate DE withthe plane wave basis set seemed to turn out not accurate as only afraction of the DE value was calculated.69
This metastable model allows explaining the O�H peakredistributions between neighboring peaks while heating thesamples, for example, of CaY up to 225 �C: the 3685 cm�1 bandweakens, while that at 3640 cm�1 grows.2 This state possessingthe band at 3640 cm�1 dominates in the initial CaY beforerehydration. Within the framework of this work, the redistribu-tion 3685f 3640 cm�1 corresponds to a partial transformationof adsorbed water at the Me cation to a new acidic state which ispossible to be reached at 225 �C. Such sites at 3640 cm�1 arequickly saturated at 225 �C in the course of rehydration so thattheir intensity becomes comparable to that of the remainingadsorbed water (the peak at 3685 cm�1). We suppose that a partof new OH sites is formed owing to the water interaction withMe2Ox. These interactions are exothermic (at least with Me2Ofor Me = Mg, Ca, Sr, Ba) and barrierless as well (with theexception of Mg36).
The “kinetic” stabilization of hydroxyl groups in the AEzeolites can also explain complex differences between thermaland flash activation.4 In the last case, all the near-Al sites areattainable for protons if they are separated by the barriers whoseheight is less than the energy obtained from the photon by thesystem and transferred to the protons. It has to produce another

2409 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
distribution of hydroxyl groups versus the thermal activation. Theidea about the metastability should be supported by additionalefforts to describe the rich structure of the OH bands involvingboth acidic and nonacidic protons. Such work is in progress.
5. CONCLUSIONS
We analyzed the products of water interaction with the singlealkaline earth (AE) cations in the cationic form of mordenite(MOR) and faujasite (FAU) zeolites. Within the relatively small6R + 4R and 8R zeolite fragments with closely located Al atoms(|Al 3 3 3Al| < 8 Å), no stable minimum was found at the isolated(B3LYP, MP2) cluster level for the dissociation products over allAE cations with the exception of Mg. Our results support theassignment by Angell and Schaffer4 and Bertsch and Habgood70
that the line at 3690 cm�1 corresponds to the water stronglyadsorbed at the AE cation in MeY zeolites and whose secondproton is hydrogen bonded. Finally, we have proposed the se-quence of the OH frequencies combining the cluster and periodiccomputations. For example, in CaY,2 the following order wastentatively proposed: silanols (3750�3740 cm�1) g Ca(OH)+ >Ca(H2O)
2+ (3690�3640 cm�1) > Si�O(H)�Al (3600�3400 cm�1) > Ca(H2O)
2+ with hydrogen bonds (red shift bysome hundreds cm�1 relative to silanol signal). For the other twoAE cationic forms (Mg and Sr), the order of the OH frequenciesshould coincide. The Ba form presents a more complicated case,being very similar to other AE forms at the theoretical level butnot at the experimental one.
At the periodic level, a very small barrier (1.20 kcal/mol withPW91) has been calculated for the water recombination usingthe mordenite (CaMOR) model with two Al atoms in one 8Rring as compared to 1.44/2.51 kcal/mol with B3LYP/MP2 at thecluster level for Mg(6R + 4R). At higher |Al 3 3 3Al| distances, theproducts of the dissociation are less stable than adsorbed water,but relatively high barriers for proton jumps18,44,61 can result inthe “kinetic” capture of protons at the Si�O�Al moieties nearremote Al atoms. The |Al 3 3 3Al| distance of more than 8�9 Å canthus stabilize the products of the dissociation. This model explainsthe independence of the experimentalmost intense acidicOHbandon the AE cation type inMeMOR (Me =Mg, Ca, Sr).10 It is a logicconsequence of its remote location toward theAE cation so that theacidic Si�O(H)�Al group is not influenced by the cation. In theMeY forms with lower Si/Al ratio and higher cation concentration,such a slight dependence on the cation type was observed.2,10 Thismetastable model allows the development of qualitative interpreta-tion of the temperature variations in the IR spectra upon hydration,dehydration, and rehydration.2,4,5 The practical importance of thehydroxyl formation in AE zeolites is related to their activity incatalytic processes like cracking, alkylation, etc. which require appli-cation of solid acids.11Upon rising temperature, the proton transferbetween active sites and respective change of their acidities isdetermined by the barriers for proton jumps and not by deprotona-tion energies as in the thermodynamic model.
’ASSOCIATED CONTENT
bS Supporting Information. Additional details on the bandassignment, deprotonation energies, relation between binding fre-quency of water molecules, and hydrogen bonding, Tables S1�S6,and Figure S1.Movie of the water recombination. Thismaterial isavailable free of charge via the Internet at http://pubs.acs.org.
’AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
’ACKNOWLEDGMENT
The authors deeply acknowledge Prof. D.P. Vercauteren forpermanent help. The authors thank the FUNDP, the FNRS-FRFC, and the Loterie Nationale (convention 2.4578.02) for theuse of the Namur iSCF Centre. AVL thanks RFFI for the grant11-01-00280-a. The authors are extremely grateful to ComputerComplex SKIF of Lomonosov Moscow State University00Chebyshev00 for computational time and to the manager ofthe Complex Dr. S.A. Zhumatii for the help. The authors alsoacknowledge the FNRS and WBI for financial support (WBIwithin the framework of the,Programme de coop�eration 2008-2010 entre la Communaut�e franc-aise de Belgique, la R�egionwallonne et la F�ed�eration de Russie. (convention 2008/18342).
’REFERENCES
(1) Hirschler, A. E. J. Catal. 1963, 2, 428.(2) Ward, J. W. J. Phys. Chem. 1968, 72, 4211.(3) Jacobs, P. A.; Van Cauwelaert, F.; Vansant, E.; et al. J. Chem. Soc.,
Faraday Trans. I 1973, 69, 1056.(4) Angell, C. L.; Schaffer, P. C. J. Phys. Chem. 1965, 69, 3463.(5) Christner, L. G.; Liengme, B. V.; Hall, W. K. Trans. Faraday Soc.
1968, 64, 1679.(6) Ward, J. W. J. Catal. 1968, 10, 34.(7) Uytterhoeven, J. B.; Schoonheydt, R. A.; Liengme, B. V.; Hall,
W. K. J. Catal. 1969, 13, 425.(8) Rees, L. V. C.; Williams, C. J. Trans. Faraday Soc. 1965, 61, 481.(9) Xu, J.; Mojet, B. L.; Lefferts, L. Microporous Mesoporous Mater.
2006, 91, 187.(10) Kustov, L. M.; Borovkov, V. Yu.; Kazansky, V. B. J. Catal. 1981,
72, 149.(11) Breck, D.W., Zeolite molecular sieves - structure, chemistry and use;
J. Wiley & Sons, Inc.: New York, 1974.(12) Ishinaga, Y.; Otsuka, K.; Morikawa, A. Bull. Chem. Soc. Jpn.
1979, 52, 933.(13) Jacobs, P. A.; Van Cauwelaert, F.; Vansant, E.; et al. J. Chem.
Soc., Faraday Trans. I 1973, 69, 1056.(14) Xu, J.; Mojet, B. L.; van Ommen, J. G.; Lefferts, L. Phys. Chem.
Chem. Phys. 2003, 5, 4407.(15) Kao, H.-M.; Grey, C. P.; Pitchumani, K.; Lakshminarasimhan,
P. H.; Ramamurthy, V. J. Phys. Chem. A 1998, 102, 5627.(16) Panov, A. G.; Larsen, R. G.; Totah, N. I.; Larsen, S. C.; Grassian,
V. H. J. Phys. Chem. B 2000, 104, 5706.(17) Xiang, Y.; Larsen, S. C.; Grassian, V. H. J. Am. Chem. Soc. 1999,
121, 5063.(18) Shubin, A. A.; Zhidomirov, G. M.; Yakovlev, A. L.; van Santen,
R. A. J. Phys. Chem. B 2001, 105, 4928.(19) Aleksandrov, H. A.; Vayssilov, G. N.; R€osch, N. J. Mol. Catal. A
2006, 256, 149.(20) Aleksandrov, H. A.; Vayssilov, G. N.; R€osch, N. Stud. Surf. Sci.
Catal. A 2005, 158, 593.(21) Larin, A. V.; Vercauteren, D. P. Surf. Stud. Sci. Cat. 2001,
135, 263.(22) Larin, A. V.; Trubnikov, D. N.; Vercauteren, D. P. Int. J.
Quantum Chem. 2003, 92, 71.(23) Larin, A. V.; Trubnikov, D. N.; Vercauteren, D. P. Int. J.
Quantum Chem. 2005, 102, 971.(24) Krogh Andersen, E.; Ploug-Sorensen, G. Z. Kristallogr. 1986,
176, 67.(25) Saunders, V.R.; Dovesi, R.; Roetti, C.; M. Caus�a, Harrison, N.
M.; Orlando, R.; Zicovich-Wilson, C. M. CRYSTAL98 1.0, User’sManual; Torino, 1999.

2410 dx.doi.org/10.1021/jp205028c |J. Phys. Chem. C 2012, 116, 2399–2410
The Journal of Physical Chemistry C ARTICLE
(26) Bader, R.F.W. Atoms in Molecules: A Quantum Theory; TheInternational Series of Monographs in Chemistry; Clarendon Press:Oxford, UK, 1995.(27) Yakovlev, A. L.; Shubin, A. A.; Zhidomirov, G. M.; van Santen,
R. A. Catal. Lett. 2000, 70, 175.(28) Kazansky, V. B. J. Catal. 2003, 216, 192.(29) Shubin, A. A.; Zhidomirov, G. M.; Kazansky, V. B.; van Santen.,
R. A. Catal. Lett. 2003, 90, 137.(30) Zhidomirov, G. M.; Shubin, A. A.; Kazansky, V. B.; van Santen.,
R. A. Int. J. Quantum Chem. 2004, 100, 489.(31) Zhidomirov, G. M.; Shubin, A. A.; Kazansky, V. B.; van Santen,
R. A. Theor. Chem. Acc. 2005, 114, 90.(32) Corma, A.; Marie, O.; Ortega, F. J. J. Catal. 2004, 222, 338.(33) Frisch, M. J., et al. Gaussian 03, revision C.02; Gaussian, Inc.:
Wallingford, CT, 2004.(34) Zhidomirov, G.M.; Larin, A. V.; Trubnikov, D. N.; Vercauteren,
D. P. J. Phys. Chem. C 2009, 113, 8258.(35) Larin, A. V.; Zhidomirov, G.M.; Trubnikov, D. N.; Vercauteren,
D. P. J. Comput. Chem. 2010, 31, 421.(36) Rybakov, A. A.; Larin, A. V.; Zhidomirov, G. M.; Trubnikov,
D. N.; Vercauteren, D. P. Comput. Theory Chem. 2011, 964, 108.(37) Kresse, G.; Hafner, J. Phys. Rev. B 1993, 47, 558. Kresse, G.;
Furthm€uller, J. ibid. 1996, 54, 11169.(38) Kresse, G.; Joubert, J. Phys. Rev. B 1999, 59, 1758.(39) Perdew, J. P.; Chevary, J. A.; Vosko, S. H.; Jackson, K. A.; et al.
Phys. Rev. B 1992, 46, 6671.(40) Henkelman, G.; Uberuaga, B. P.; et al. J. Chem. Phys. 2000,
113, 9901.(41) Sheppard, D.; Terrell, R.; et al. ibid. 2008, 128, 134106.(42) Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; C. M.
Zicovich-Wilson, Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.;Bush, I.J.; D’Arco, Ph.; Llunell, M.CRYSTAL06 User’s Manual; Universityof Torino: Torino, 2006.(43) Ugliengo, P.; Viterbo, D.; Chiari, G. Z. Kristallogr. 1993, 207, 9.(44) Fermann, J. T.; Blanco, C.; Auerbach, S. J. Chem. Phys. 2000,
112, 6779.(45) Jeffroy, M.; Borissenko, E.; Boutin, A.; Di Lella, A.; Porcher, F.;
Souhassou, M.; Lecomte, C.; Fuchs, A. H. Microporous MesoporousMater. 2011, 138, 45.(46) Barry, T. I.; Lay, L. A. J. Phys. Chem. Solids 1966, 27, 1821.(47) Barry, T. I.; Lay, L. A. J. Phys. Chem. Solids 1968, 29, 1395.(48) Gallezot, P.; Taarit, Y.; Imelik, B. C. R. Acad. Sci., Ser. C 1971,
272, 261.(49) Larin, A. V.; Rybakov, A. A.; Zhidomirov, G. M.; Mace, A.;
Laaksonen, A.; Vercauteren, D. P. J. Catal. 2011, 281, 212.(50) Pulin, A. L.; Fomkin, A. A.; Sinitsyn, V. A.; Pribylov, A. A. Russ.
Chem. Bull. 2001, 50, 60.(51) Baimpos, Th.; Giannakopoulos, I. G.; Nikolakis, V.; Kouzoudis,
D. Chem. Mater. 2008, 20, 1470.(52) Sun, K.; Su, W.; Fan, F.; Feng, Z.; Jansen, T. A. P. J.; van Santen,
R. A.; Li, C. J. Phys. Chem. A 2008, 112, 1352.(53) Keleman, G.; Sch€on, G. J. Mater. Sci. 1992, 27, 6036.(54) Kalogeras, I. M.; Vassilikou-Dova, A. Cryst. Res. Technol. 1996,
31, 693.(55) Romanovsky, B. V.; Topchieva, K. V.; Stolyarova, L. V.; Alekseev,
A. M. Kinet. Katal. 1970, 11, 1525.(56) Ockwig, N. W.; Cygan, R. T.; Criscentia, L. J.; Nenoff, T. M.
Phys. Chem. Chem. Phys. 2008, 10, 800.(57) Rinaldi, R.; Pluth, J. J.; Smith, J. V. Acta Crystallogr. B 1974,
30, 2426.(58) Corsaro, C.; Crupi, V.; Majolino, D.; Parker, S. F.; Venuti, V.;
Wanderlingh, U. J. Phys. Chem. A 2006, 110, 1190.(59) Herzberg, G.,Molecular Spectra and Molecular Structure, Part II;
Van Nostrand Reinhold: New York, 1945; p 488.(60) Larin, A. V.; Cohen de Lara, E. Mol. Phys. 1996, 88, 1399.(61) Sierka, M.; Sauer, J. J. Phys. Chem. B 2001, 105, 1603.(62) Franke, M. F.; Sierka, M.; Simon, U.; Sauer, J. Phys. Chem.
Chem. Phys. 2002, 4, 5207.
(63) Sarv, P.; Tuherm, T.; Lippmaa, E.; Keskinen, K.; Root, A.J. Phys. Chem. 1995, 99, 13763.
(64) Baba, T.; Inoue, Y.; Shoji, H.; Uematsu, T.; Ono, Y.MicroporousMater. 1995, 3, 647.
(65) Baba, T.; Komatsy, N.; Ono, Y.; Sugisawa, H. J. Phys. Chem. B1998, 102, 804.
(66) Fedorov, V. M.; Glazun, B. A.; et al. Izv. Akad. Nauk SSSR, Otd.Chim. Nauk 1964, 11, 1930.
(67) Datka, J.; Broczawik, E.; Gill, B. J. Phys. Chem. 1993, 98, 5622.(68) Datka, J.; Broczawik, E.; Gill, B. Langmuir 1993, 9, 2496.(69) Tielens, F.; Dzwigaj, S. Catal. Today 2010, 152, 66.(70) Bertsch, L.; Habgood, H. W. J. Phys. Chem. 1963, 67, 1621.




















