
FT-IR evidence of the interaction of benzothiophene with the hydroxyl groups of H-MFI and H-MOR...
6
FT-IR evidence of the interaction of benzothiophene with the hydroxyl groups of H-MFI and H-MOR zeolites Aı ´da Gutie ´rrez-Alejandre a,b, * , Maria Angeles Larrubia a,c , Jorge Ramirez b,d , Guido Busca a, * a Laboratorio di Catalisi, Dipartimento di Ingegneria Chimica e di Processo, Universita `, Genova, P.le J.F. Kennedy, I-16129 Genova, Italy b UNICAT, Depto. de Ingenierı ´a Quı ´mica, Facultad de Quı ´mica, UNAM, Cd. Universitaria, 04510 Me ´xico D.F., Mexico c Departamento di Ingenierı ´a Quı ´mica, Facultad de Quı ´mica, Universidad de Malaga, 29071 Malaga, Spain d Instituto Mexicano del Petro ´leo, Eje Central La ´zaro Ca ´rdenas No. 152, 07730 Me ´xico D.F., Mexico Received 10 October 2005; received in revised form 9 December 2005; accepted 21 December 2005 Available online 2 February 2006 Abstract The interaction of benzothiophene (BT) with H-MFI and H-MOR has been investigated by FT-IR spectroscopy. The results from BTadsorption on acid H-MFI and H-MOR coupled with the study of adsorbed naphthalene show that two types of interactions occur between BT and the strongly acidic protonic sites of the zeolites: one interaction through the sulfur lone electron pair and another one, similar to that observed with naphthalene, involving the p-type electron cloud. With the external silanols, only a weak hydrogen bonding type interaction with the p-type electron cloud of the aromatic ring of BT was clearly observed. # 2006 Elsevier B.V. All rights reserved. Keywords: Benzothiophene; FT-IR; H-MFI; H-MOR zeolites IR spectroscopy; H-bonding; Adsorption 1. Introduction The development of new catalysts [1,2] and of new adsorbants [3] is of great importance for the elimination of thiophenic sulfur compounds from transport fuels, in order to fulfill the strict sulfur content regulations expected for the near future (15 ppm or less). Materials containing protonic zeolites are among candidates in both fields. Several studies have been reported in recent years concerning the interaction of thiophene with protonic zeolites. Garcia and Lercher [4,5], Geobaldo et al. [6] and Yu et al. [7] reported spectroscopic studies of thiophene interaction with H- MFI and H-Y. Soscun et al. [8], Saintigny et al. [9] and Rozanska et al. [10] reported theoretical studies on the adsorption and reactions of thiophene over protonic zeolites. Garcia and Lercher [4,5] concludes that on H-MFI thiophene is weakly hydrogen bonded to the SiOH groups and that initially, thiophene hydrogen bonds to the Si–OH–Al groups. This interaction is followed by ring opening and oligomerization reactions promoted by the strong acidity of the H-MFI zeolite. These results are consistent with those reported in [6]. According to Yu et al. [11] and Li et al. [12] thiophene converts into benzothiophene and likely higher heteroaromatics plus H 2 S on H-MFI zeolite and this reaction is greatly enhanced by the presence of hydrogen or alkanes. The same group found that thiophene oligomers form, during adsorption and that their size, depends on spatial constraints within zeolite channels. The data reflect specific interactions of thiophene with Brønsted acid sites [13,14]. Much less is known about the interaction of benzothio- phene (BT) with solids. To our knowledge, the only spectroscopic study of benzothiophene adsorbed on solids has been performed by our group and concerns about the interactions with Al, Mg and Zr oxides [15]. The molecular adsorption of benzothiophene on protonic zeolites has been the object of few recent studies [16,17]. A theoretical study has been performed on the isomerization of alkyl–benzothio- phenes on protonic mordenite [18]. Clearly, better knowledge of the interactions of benzothiophenes and aromatics with www.elsevier.com/locate/vibspec Vibrational Spectroscopy 41 (2006) 42–47 * Corresponding author. Tel.: +39 010 3536024; fax: +39 010 3536028. E-mail addresses: [email protected] (A. Gutie ´rrez-Alejandre), Gui- [email protected] (G. Busca). 0924-2031/$ – see front matter # 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.vibspec.2005.12.008
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of FT-IR evidence of the interaction of benzothiophene with the hydroxyl groups of H-MFI and H-MOR...

FT-IR evidence of the interaction of benzothiophene with
the hydroxyl groups of H-MFI and H-MOR zeolites
Aıda Gutierrez-Alejandre a,b,*, Maria Angeles Larrubia a,c,Jorge Ramirez b,d, Guido Busca a,*
a Laboratorio di Catalisi, Dipartimento di Ingegneria Chimica e di Processo, Universita, Genova, P.le J.F. Kennedy, I-16129 Genova, Italyb UNICAT, Depto. de Ingenierıa Quımica, Facultad de Quımica, UNAM, Cd. Universitaria, 04510 Mexico D.F., Mexico
c Departamento di Ingenierıa Quımica, Facultad de Quımica, Universidad de Malaga, 29071 Malaga, Spaind Instituto Mexicano del Petroleo, Eje Central Lazaro Cardenas No. 152, 07730 Mexico D.F., Mexico
Received 10 October 2005; received in revised form 9 December 2005; accepted 21 December 2005
Available online 2 February 2006
Abstract
The interaction of benzothiophene (BT) with H-MFI and H-MOR has been investigated by FT-IR spectroscopy. The results from BT adsorption
on acid H-MFI and H-MOR coupled with the study of adsorbed naphthalene show that two types of interactions occur between BT and the strongly
acidic protonic sites of the zeolites: one interaction through the sulfur lone electron pair and another one, similar to that observed with naphthalene,
involving the p-type electron cloud. With the external silanols, only a weak hydrogen bonding type interaction with the p-type electron cloud of the
aromatic ring of BT was clearly observed.
# 2006 Elsevier B.V. All rights reserved.
Keywords: Benzothiophene; FT-IR; H-MFI; H-MOR zeolites IR spectroscopy; H-bonding; Adsorption
www.elsevier.com/locate/vibspec
Vibrational Spectroscopy 41 (2006) 42–47
1. Introduction
The development of new catalysts [1,2] and of new
adsorbants [3] is of great importance for the elimination of
thiophenic sulfur compounds from transport fuels, in order to
fulfill the strict sulfur content regulations expected for the near
future (15 ppm or less). Materials containing protonic zeolites
are among candidates in both fields.
Several studies have been reported in recent years
concerning the interaction of thiophene with protonic zeolites.
Garcia and Lercher [4,5], Geobaldo et al. [6] and Yu et al. [7]
reported spectroscopic studies of thiophene interaction with H-
MFI and H-Y. Soscun et al. [8], Saintigny et al. [9] and
Rozanska et al. [10] reported theoretical studies on the
adsorption and reactions of thiophene over protonic zeolites.
Garcia and Lercher [4,5] concludes that on H-MFI thiophene is
weakly hydrogen bonded to the SiOH groups and that initially,
* Corresponding author. Tel.: +39 010 3536024; fax: +39 010 3536028.
E-mail addresses: [email protected] (A. Gutierrez-Alejandre), Gui-
[email protected] (G. Busca).
0924-2031/$ – see front matter # 2006 Elsevier B.V. All rights reserved.
doi:10.1016/j.vibspec.2005.12.008
thiophene hydrogen bonds to the Si–OH–Al groups. This
interaction is followed by ring opening and oligomerization
reactions promoted by the strong acidity of the H-MFI zeolite.
These results are consistent with those reported in [6].
According to Yu et al. [11] and Li et al. [12] thiophene
converts into benzothiophene and likely higher heteroaromatics
plus H2S on H-MFI zeolite and this reaction is greatly enhanced
by the presence of hydrogen or alkanes. The same group found
that thiophene oligomers form, during adsorption and that their
size, depends on spatial constraints within zeolite channels. The
data reflect specific interactions of thiophene with Brønsted
acid sites [13,14].
Much less is known about the interaction of benzothio-
phene (BT) with solids. To our knowledge, the only
spectroscopic study of benzothiophene adsorbed on solids
has been performed by our group and concerns about the
interactions with Al, Mg and Zr oxides [15]. The molecular
adsorption of benzothiophene on protonic zeolites has been
the object of few recent studies [16,17]. A theoretical study
has been performed on the isomerization of alkyl–benzothio-
phenes on protonic mordenite [18]. Clearly, better knowledge
of the interactions of benzothiophenes and aromatics with
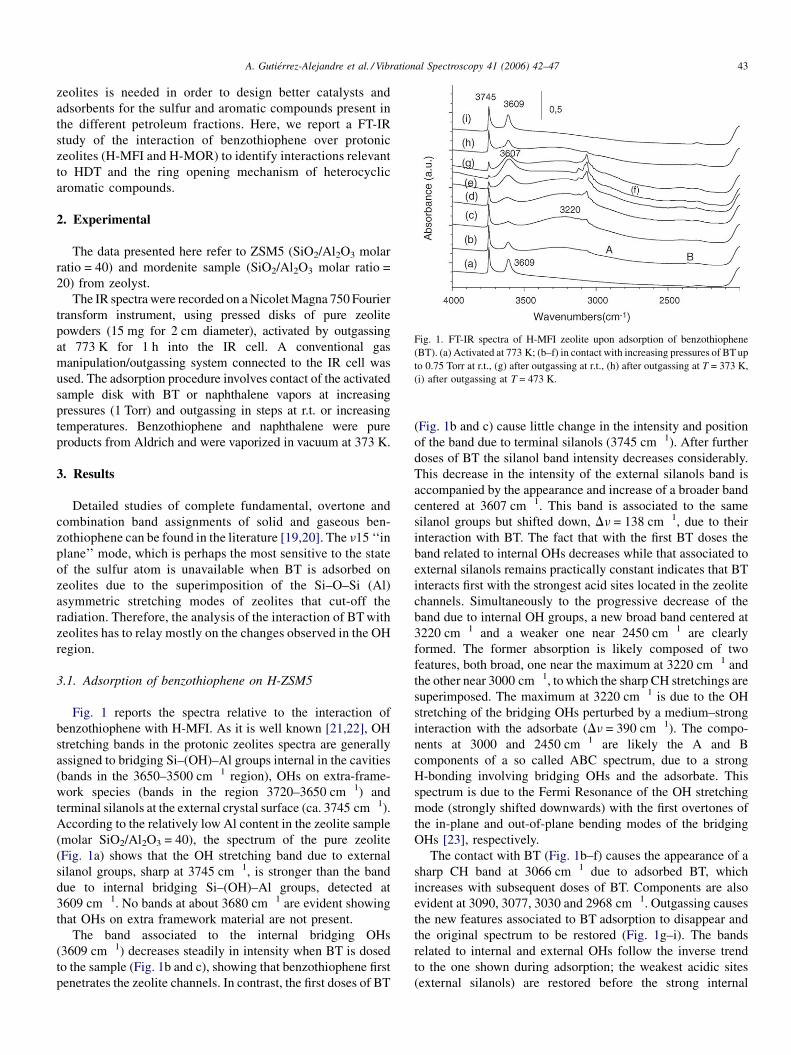
A. Gutierrez-Alejandre et al. / Vibrational Spectroscopy 41 (2006) 42–47 43
Fig. 1. FT-IR spectra of H-MFI zeolite upon adsorption of benzothiophene
(BT). (a) Activated at 773 K; (b–f) in contact with increasing pressures of BT up
to 0.75 Torr at r.t., (g) after outgassing at r.t., (h) after outgassing at T = 373 K,
(i) after outgassing at T = 473 K.
zeolites is needed in order to design better catalysts and
adsorbents for the sulfur and aromatic compounds present in
the different petroleum fractions. Here, we report a FT-IR
study of the interaction of benzothiophene over protonic
zeolites (H-MFI and H-MOR) to identify interactions relevant
to HDT and the ring opening mechanism of heterocyclic
aromatic compounds.
2. Experimental
The data presented here refer to ZSM5 (SiO2/Al2O3 molar
ratio = 40) and mordenite sample (SiO2/Al2O3 molar ratio =
20) from zeolyst.
The IR spectra were recorded on a Nicolet Magna 750 Fourier
transform instrument, using pressed disks of pure zeolite
powders (15 mg for 2 cm diameter), activated by outgassing
at 773 K for 1 h into the IR cell. A conventional gas
manipulation/outgassing system connected to the IR cell was
used. The adsorption procedure involves contact of the activated
sample disk with BT or naphthalene vapors at increasing
pressures (1 Torr) and outgassing in steps at r.t. or increasing
temperatures. Benzothiophene and naphthalene were pure
products from Aldrich and were vaporized in vacuum at 373 K.
3. Results
Detailed studies of complete fundamental, overtone and
combination band assignments of solid and gaseous ben-
zothiophene can be found in the literature [19,20]. The n15 ‘‘in
plane’’ mode, which is perhaps the most sensitive to the state
of the sulfur atom is unavailable when BT is adsorbed on
zeolites due to the superimposition of the Si–O–Si (Al)
asymmetric stretching modes of zeolites that cut-off the
radiation. Therefore, the analysis of the interaction of BT with
zeolites has to relay mostly on the changes observed in the OH
region.
3.1. Adsorption of benzothiophene on H-ZSM5
Fig. 1 reports the spectra relative to the interaction of
benzothiophene with H-MFI. As it is well known [21,22], OH
stretching bands in the protonic zeolites spectra are generally
assigned to bridging Si–(OH)–Al groups internal in the cavities
(bands in the 3650–3500 cm�1 region), OHs on extra-frame-
work species (bands in the region 3720–3650 cm�1) and
terminal silanols at the external crystal surface (ca. 3745 cm�1).
According to the relatively low Al content in the zeolite sample
(molar SiO2/Al2O3 = 40), the spectrum of the pure zeolite
(Fig. 1a) shows that the OH stretching band due to external
silanol groups, sharp at 3745 cm�1, is stronger than the band
due to internal bridging Si–(OH)–Al groups, detected at
3609 cm�1. No bands at about 3680 cm�1 are evident showing
that OHs on extra framework material are not present.
The band associated to the internal bridging OHs
(3609 cm�1) decreases steadily in intensity when BT is dosed
to the sample (Fig. 1b and c), showing that benzothiophene first
penetrates the zeolite channels. In contrast, the first doses of BT
(Fig. 1b and c) cause little change in the intensity and position
of the band due to terminal silanols (3745 cm�1). After further
doses of BT the silanol band intensity decreases considerably.
This decrease in the intensity of the external silanols band is
accompanied by the appearance and increase of a broader band
centered at 3607 cm�1. This band is associated to the same
silanol groups but shifted down, Dn = 138 cm�1, due to their
interaction with BT. The fact that with the first BT doses the
band related to internal OHs decreases while that associated to
external silanols remains practically constant indicates that BT
interacts first with the strongest acid sites located in the zeolite
channels. Simultaneously to the progressive decrease of the
band due to internal OH groups, a new broad band centered at
3220 cm�1 and a weaker one near 2450 cm�1 are clearly
formed. The former absorption is likely composed of two
features, both broad, one near the maximum at 3220 cm�1 and
the other near 3000 cm�1, to which the sharp CH stretchings are
superimposed. The maximum at 3220 cm�1 is due to the OH
stretching of the bridging OHs perturbed by a medium–strong
interaction with the adsorbate (Dn = 390 cm�1). The compo-
nents at 3000 and 2450 cm�1 are likely the A and B
components of a so called ABC spectrum, due to a strong
H-bonding involving bridging OHs and the adsorbate. This
spectrum is due to the Fermi Resonance of the OH stretching
mode (strongly shifted downwards) with the first overtones of
the in-plane and out-of-plane bending modes of the bridging
OHs [23], respectively.
The contact with BT (Fig. 1b–f) causes the appearance of a
sharp CH band at 3066 cm�1 due to adsorbed BT, which
increases with subsequent doses of BT. Components are also
evident at 3090, 3077, 3030 and 2968 cm�1. Outgassing causes
the new features associated to BT adsorption to disappear and
the original spectrum to be restored (Fig. 1g–i). The bands
related to internal and external OHs follow the inverse trend
to the one shown during adsorption; the weakest acidic sites
(external silanols) are restored before the strong internal

A. Gutierrez-Alejandre et al. / Vibrational Spectroscopy 41 (2006) 42–4744
Fig. 2. FT-IR spectra of BT adsorbed on H-MFI. In-plane and out-of-plane
vibrational modes region. From bottom to top: BT liquid, the adsorbed species
in contact with increasing pressures of BT up to 0.75 Torr at r.t., after outgassing
at r.t., after outgassing at T = 373 K, after outgassing at T = 673 K.
Fig. 3. FT-IR spectra of H-MFI zeolite upon adsorption of naphthalene. (a)
Activated at 773 K, (b) naphtalene at r.t., (c) outgassed at r.t., (d) outgassed at
373 K and (e) outgassed at 423 K.
Scheme 1. H-bonding interactions of the naphthalene aromatic electron cloud
with (a) the silanol groups and (b) interaction of naphthalene with the bridging
OHs.
bridging OHs. Almost complete BT desorption is only obtained
by outgassing at 473 K (Fig. 1i).
The spectra in the lower frequency region (Fig. 2) show,
after adsorption and outgassing at r.t., the main BT bands, all
sharp, at 1498 (weak), 1458, 1423 (both very strong), 1346
and 1323 cm�1. In the windows between the zeolite’s
skeletal vibrations (not shown), bands of adsorbed BT at
866, 851, 762, 734 and 688 cm�1 were evident. The bands in
the region above 1300 cm�1 correspond to in-plane vibra-
tional modes of BT. The position of the bands, with respect
to the values reported for liquid BT are shifted few cm�1
except for n13, which is involved in Fermi resonances. The
lower frequency detected bands, in particular those at 851,
734 and 688 cm�1, which correspond to out-of-plane
vibrations (n31, n33 and n34), are also shifted few cm�1.
Upon outgassing from room temperature up to 473 K no
evidence of the formation of reaction products different from
BT itself was found.
The interaction of BT with the zeolite OH groups can take
place in two different modes: through the p electron cloud of
the aromatic ring, or through the sulfur lone electron pair. Each
of these interactions will present a characteristic shift of the
different OH bands perturbed by BT adsorption. To analyze if
the OH band shifts observed upon BT adsorption are purely due
to interactions with the p electron cloud of the aromatic ring,
experiments were made with naphthalene, a molecule with
similar steric hindrance, molecular shape, and aromatic
electronic structure than BT.
3.2. Adsorption of naphthalene on H-MFI
Naphthalene adsorption over H-MFI (Fig. 3) is associated to
the formation of two broad features: one with two unresolved
components near 3620 and 3550 cm�1 and another one which
seems also split with components near 3250 and 3150 cm�1.
According to our previous data concerning IR studies on the
adsorption of hydrocarbons on H-MFI [24], the former feature
is associated to H-bonding interactions of the naphthalene
aromatic electron cloud with the silanol groups, while the latter
is due to the interaction of naphthalene with the bridging OHs.
The different observed shifts, Dn OH 125–200 cm�1 for the
silanol, and 360–460 cm�1 for bridging OHs, agree with the
different acidities of the two OH groups. The splitting for both
bands can be either due to the existence of two families of
silanol groups and two families of bridging OHs with a
remarkably different acidity, or a different steric hindrance, or
(more likely) to the possibility of formation of two types of
adsorption complexes with naphthalene for both kinds of OHs
(e.g. an interaction with fully delocalized p orbitals and an
interaction localized on one C–C bond). In any case, the
interaction of aromatic hydrocarbons with OH groups is
interpreted as evidence of a medium–strong hydrogen bonding
involving the p electron cloud (see Scheme 1) and both
bridging and terminal OHs.
The magnitude of the OH band shifts observed upon
naphthalene adsorption on bridging OHs (Dn OH 360–
460 cm�1) is in the same range to that observed for BT
adsorption (Dn OH 390 cm�1), suggesting that interactions
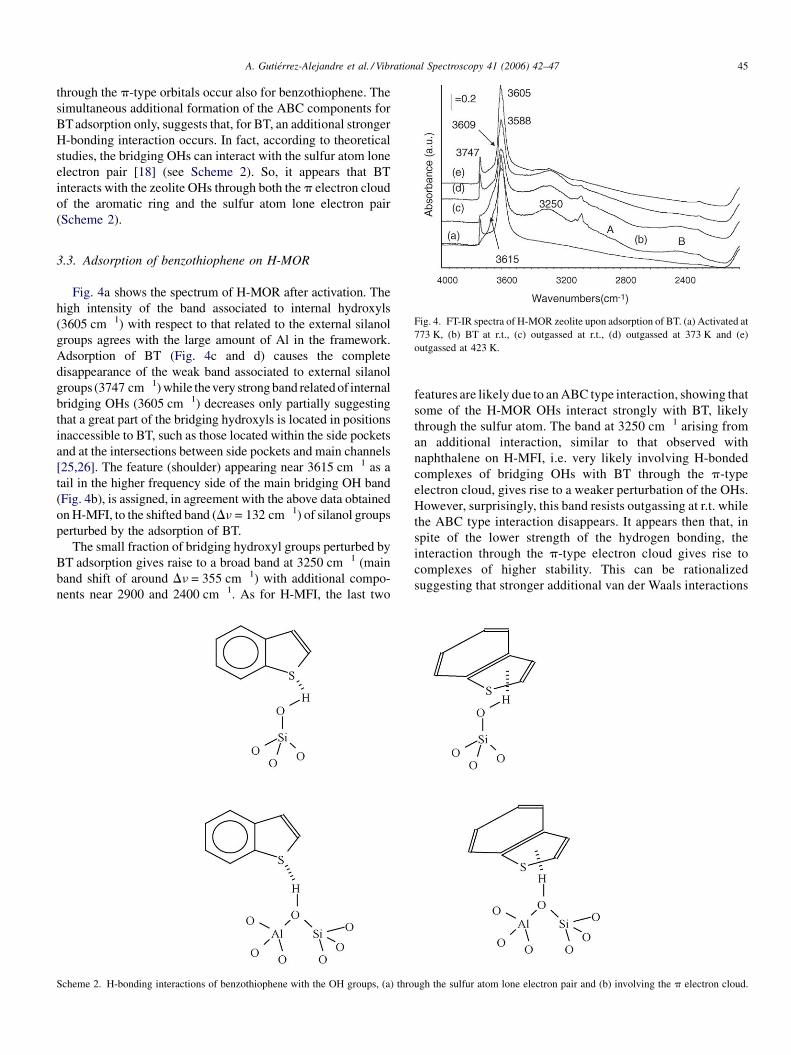
A. Gutierrez-Alejandre et al. / Vibrational Spectroscopy 41 (2006) 42–47 45
Fig. 4. FT-IR spectra of H-MOR zeolite upon adsorption of BT. (a) Activated at
773 K, (b) BT at r.t., (c) outgassed at r.t., (d) outgassed at 373 K and (e)
outgassed at 423 K.
through the p-type orbitals occur also for benzothiophene. The
simultaneous additional formation of the ABC components for
BTadsorption only, suggests that, for BT, an additional stronger
H-bonding interaction occurs. In fact, according to theoretical
studies, the bridging OHs can interact with the sulfur atom lone
electron pair [18] (see Scheme 2). So, it appears that BT
interacts with the zeolite OHs through both the p electron cloud
of the aromatic ring and the sulfur atom lone electron pair
(Scheme 2).
3.3. Adsorption of benzothiophene on H-MOR
Fig. 4a shows the spectrum of H-MOR after activation. The
high intensity of the band associated to internal hydroxyls
(3605 cm�1) with respect to that related to the external silanol
groups agrees with the large amount of Al in the framework.
Adsorption of BT (Fig. 4c and d) causes the complete
disappearance of the weak band associated to external silanol
groups (3747 cm�1) while thevery strong band related of internal
bridging OHs (3605 cm�1) decreases only partially suggesting
that a great part of the bridging hydroxyls is located in positions
inaccessible to BT, such as those located within the side pockets
and at the intersections between side pockets and main channels
[25,26]. The feature (shoulder) appearing near 3615 cm�1 as a
tail in the higher frequency side of the main bridging OH band
(Fig. 4b), is assigned, in agreement with the above data obtained
on H-MFI, to the shifted band (Dn = 132 cm�1) of silanol groups
perturbed by the adsorption of BT.
The small fraction of bridging hydroxyl groups perturbed by
BT adsorption gives raise to a broad band at 3250 cm�1 (main
band shift of around Dn = 355 cm�1) with additional compo-
nents near 2900 and 2400 cm�1. As for H-MFI, the last two
Scheme 2. H-bonding interactions of benzothiophene with the OH groups, (a) thro
features are likely due to an ABC type interaction, showing that
some of the H-MOR OHs interact strongly with BT, likely
through the sulfur atom. The band at 3250 cm�1 arising from
an additional interaction, similar to that observed with
naphthalene on H-MFI, i.e. very likely involving H-bonded
complexes of bridging OHs with BT through the p-type
electron cloud, gives rise to a weaker perturbation of the OHs.
However, surprisingly, this band resists outgassing at r.t. while
the ABC type interaction disappears. It appears then that, in
spite of the lower strength of the hydrogen bonding, the
interaction through the p-type electron cloud gives rise to
complexes of higher stability. This can be rationalized
suggesting that stronger additional van der Waals interactions
ugh the sulfur atom lone electron pair and (b) involving the p electron cloud.

A. Gutierrez-Alejandre et al. / Vibrational Spectroscopy 41 (2006) 42–4746
occur in this case and stabilize these weaker hydrogen bonding
complexes.
Previous IR studies using hindered nitriles as probe
molecules [25,26] allowed distinguishing three families of
bridging OHs in H-MOR. The hydroxyl groups absorbing at
3605 cm�1 are thought to stand near the center of the main
channels. Those responsible for the shoulder at 3588 cm�1 are
likely located inside the side-pockets. Others, absorbing at
3609 cm�1, are likely at the intersection between side pockets
and main channels. The spectra relative to BT adsorbed on H-
MOR indicate that only the external silanols and the bridging
OHs located well in the middle of the H-MOR’s main channels
are actually able to interact with BT. The bridging OHs located
not only in the side pockets (3588 cm�1) but also those at the
intersection of the main channels with the side pockets
(unresolved in the main OH band, near 3609 cm�1), do not
appear to be perturbed. This has been already shown in a
previous study adsorbing highly hindered and rigid molecules
such as pivalonitrile and orthotoluonitrile [25,26]. From these
data, it is evident that only the OHs standing near the middle of
the main channels are able to interact with such large
molecules. This occurs also with BT whose critical radius is
near 6 A and allows its entrance in the main channels of
mordenite even better than in the channels of HMFI.
Upon outgassing at 423 K, increasing BT desorption occurs
and the above discussed OH stretching bands disappear.
However, inspection of the 1600–1300 cm�1 region shows that
at the same time, new bands appear (Fig. 5) at 1590, 1490 and
1365 cm�1. Fig. 5 shows that the spectra of adsorbed and solid
BT are similar with small shifts and intensity modifications.
Therefore, the significant differences observed in the spectrum
of adsorbed BT when the outgassing temperature is raised from
r.t. up to 423 K, are interpreted as evidence of the occurrence of
a BT chemical transformation. The resilient persistence of the
CH stretching band near 3075 cm�1 suggests that the resulting
species is still aromatic. The spectrum of this new species is
similar to that observed after interaction with some metal
oxides and is likely due to a species where the thiophenic ring
Fig. 5. FT-IR spectra of BT adsorbed on H-MOR. In-plane and out-of-plane
vibrational modes region. From bottom to top: BT solid, BT at r.t., outgassed at
r.t., outgassed at 373 K, outgassed at 423 K.
is opened while the aromatic ring is still intact, as suggested
previously [15].
The above results show that under our experimental
conditions (r.t., exposure times of less than 30 min), H-MOR
is active towards BT transformation while H-MFI is not.
4. Discussion
Comparison of the data arising from BT adsorption on the
acid H-MFI and H-MOR zeolites coupled with the study of
adsorbed naphthalene allow concluding that with the external
silanols, only a weak hydrogen bonding type interaction is
observed whose extent is similar to that occurring through the
p-type electron cloud of the aromatic ring of naphthalene. On
the contrary, two types of interactions occur between BT and
the strongly acidic protonic sites of the zeolites studied here.
The interaction of BT with the acidic OHs through the sulfur
lone electron pair is likely responsible for the formation of the
ABC type spectrum typical of strong H-bonding (Figs. 1 and 4).
An additional interaction similar to that observed with naph-
thalene, i.e. involving the p-type electron cloud, also observed,
gives rise to a weaker perturbation of the OHs. This latter
interaction leads to complexes of greater stability than the
former one, in spite of the different strength of the hydrogen
bonding, possibly because in this case additional stronger van
der Waals interactions occur and tend to stabilize the weaker
hydrogen bonding complexes formed with the aromatic ring.
The formation of species different from BT after its
adsorption at r.t. and outgassing at higher temperatures was
observed on H-MOR while on H-MFI was not. It seems likely
that these species are formed by reaction of BT with the zeolite
acid sites, hence, the difference between H-MOR and H-MFI
could be due to differences in the acid strength, in agreement
with the acidity scale for the zeolites studied here, reported
to be H-MOR > H-MFI [27,28]. A transition state shape
selectivity effect (i.e. to produce the transition state needed to
perform this reaction a little large cavity is needed) or
diffusional limitations due to the difference in channel size
between the two zeolites could also play a role in the absence of
reaction products over H-MFI. For H-MOR, the identification
of the resulting product adsorbed species cannot be ascertained
with our experimental data only, nevertheless, the presence of
the typical C–H stretching band at 3075 cm�1 and the aromatic
ring stretching bands at 1600 and 1490 cm�1, after BT
adsorption and outgassing at 423 K, suggests the formation
of an aromatic compound. So, opening of the BT sulfur-
containing ring very likely occurred [15]. Further studies are in
progress to ascertain this suggestion.
In any case, the interaction of acidic protons with the aromatic
electron cloud is considered in basic organic chemistry to be
precursor for electrophilic aromatic substitution. This kind of
reactivity would lead to different reaction products than those
expected from activation of the molecule at the sulfur atom.
These two different reaction paths may give rise to carbon sulfur
bond breaking or, alternatively, to carbon–carbon bond breaking,
making them interesting to hydrodesulfurization and aromatic
ring-opening reactions.

A. Gutierrez-Alejandre et al. / Vibrational Spectroscopy 41 (2006) 42–47 47
Acknowledgments
Authors acknowledge Ministero degli Esteri (Italy) and
CONACyT-SRE (Mexico), PAPIIT IN112802–DGAPA and
IMP-FIES programs for financial support.
References
[1] D.D. Whitehurst, T. Isoda, I. Mochida, Adv. Catal. 42 (1998) 345.
[2] H. Topsøe, B.S. Clausen, in: J.R. Anderson, M. Boudart (Eds.), Catalysis
Science and Technology, vol. 11, Springer, 1996, p. 1.
[3] A.J. Hernandez-Maldonado, R.T. Yang, J. Am. Chem. Soc. 126 (2004)
992.
[4] C.L. Garcia, J.A. Lercher, J. Phys. Chem. 96 (1992) 2669.
[5] C.L. Garcia, J.A. Lercher, J. Mol. Struct. 293 (1993) 235.
[6] F. Geobaldo, G. Turnes Palomino, S. Bordiga, A. Zecchina, C. Otero
Arean, Phys. Chem. Chem. Phys. 1 (1999) 561.
[7] S.Y. Yu, J. Garcia Martinez, W. Li, G.D. Meitzner, E. Iglesia, Phys. Chem.
Chem. Phys. 4 (2002) 1241.
[8] H. Soscun, O. Castellano, J. Hernandez, J. Mol. Struct. 531 (2000) 315.
[9] X. Saintigny, R.A. van Santen, S. Clemendot, F. Hutschka, J. Catal. 183
(1999) 107.
[10] X. Rozanska, R.A. van Santen, F. Hutschka, J. Catal. 200 (2001) 79.
[11] S.Y. Yu, W. Li, E. Iglesia, J. Catal. 187 (1999) 257.
[12] W. Li, S.Y. Yu, E. Iglesia, J. Catal. 203 (2001) 175.
[13] A. Chica, K.G. Strohmaier, E. Iglesia, Langmuir 20 (2004) 10982.
[14] A. Chica, K.G. Strohmaier, E. Iglesia, Appl. Catal. B: Environ. 60 (2005)
223.
[15] M.A. Larrubia, A. Gutierrez-Alejandre, J. Ramırez, G. Busca, Appl. Catal.
A: Gen. 224 (2002) 167.
[16] E. Furuya, K. Sato, T. Kataoka, T. Horiguchi, Y. Otake, Sep. Purif.
Technol. 39 (2004) 73.
[17] F.T.T. Ng, A. Rahman, T. Ohasi, M. Jiang, Appl. Catal. B: Environ. 56
(2005) 127.
[18] X. Rozanska, R.A. van Santen, F. Hutschka, J. Hafner, J. Catal. 205 (2002)
388.
[19] T.D. Klots, W.B. Collier, Spectrochim. Acta A 51 (1995) 1273.
[20] A.A. El-Azhary, Spectrochim. Acta A 55 (1999) 2437.
[21] G. Sastre, V. Fornes, A. Corma, J. Phys. Chem. B 106 (2002) 701.
[22] G. Busca, in: J.L.G. Fierro (Ed.), Metal Oxides: Chemistry and Applica-
tions, CRC Press, Boca Raton, FL, USA, 2005, pp. 247–318.
[23] A.G. Pelmenschikov, R.A. van Santen, J. Phys. Chem. 97 (1993) 10678.
[24] T. Armaroli, M. Bevilacqua, M. Trombetta, A. Gutierrez Alejandre, J.
Ramirez, G. Busca, Appl. Catal. A: Gen. 220 (2001) 181.
[25] M. Bevilacqua, A. Gutierrez Alejandre, C. Resini, M. Casagrande, J.
Ramirez, G. Busca, Phys. Chem. Chem. Phys. 4 (2002) 4575.
[26] M. Bevilacqua, G. Busca, Catal. Commun. 3 (2002) 497.
[27] A. Auroux, Top. Catal. 19 (2002) 205.
[28] Y. Miyamoto, N. Takada, M. Niwa, Micropor. Mesopor. Mater. 40 (2000)
271.




















