
A Uniform Geometrical Theory of Diffraction Model of Very ...
Upload
independentCategory
view
0download
0
Mass spectral study of geometrical (€)- and (2)-isomers
M. Vairamani* and M. Saraswathi lndian Institute of Chemicat Technology, Hyderabad 5UU UU7, lndin
I. INTRODUCTION
Stereochemistry controls the physical and chemical properties of molecules to a great extent. Various physical methods like UV, IR, and NMR spectroscopy and x-ray diffraction give valuable information regarding the orientation of the different atoms in the molecule (1,2). Mass spectrometry provides information about the molecular weight, molecular formula, and some structural features at the expense of picograms of the sample. Despite the excess energy deposited during the ionization of the molecule in the gas phase, it has been shown repeatedly that the original geometry of the molecule is retained in the ionized molecule also. This is often reflected in the characteristic fragmentation of stereoisomers. Review articles which appeared in 1968, 1976, and 1977 covered the importance of this aspect (3-5). Professor Mandelbaum, in 1983, very elegantly summarized various stereochemical effects observed in mass spectral studies (6). Geometrical isomers across the C=C bond are often encountered in various organic reactions and in natural products. Although NMR offers an easy way of differentiating these molecules, mass spectrometry has the advantage of analyzing mixtures by combining separation techniques like GC and HPLC with it. The utility of mass spectral techniques in differentiating geometrical isomers was realized by Natalis in 1965 (7). Some interesting early examples are quoted in Mandelbaum’s article also (6). In this review we have summarized the mass spectral differentiation of ( E ) - and (Z)-isomers across the C=C bond using electron ionization, chemical ionization, desorption, metastable, and collisionally activated dissociation techniques covering the literature between 1966 and 1990.
11. ELECTRON IONIZATION
Isomeric 2-butene-1,4-dicarboxylic acids and their homologues and deriva- tives are the compounds most widely studied by mass spectrometry using various techniques. Although these compounds are expected to show large differences in their mass spectra due to the two functional groups, each study has resulted in new findings. Bowie et al. studied the mass spectral fragmenta- tion of various dialkyl fumarates and maleates (1-16) with the help of high- resolution mass spectrometry (8 ) . The mass spectra of compounds 1-16 show
*Corresponding author.
Mass Spectrometry Reviews, 1992, 10, 491-517 0 1992 by John Wiley & Sons, Inc. CCC 0277-7037/92/060491-27$04.00

492 VAIRAMANI AND SARASWATHI
0
E z
n-C3H7 5 6
i - C3H7 7 8
n - CkHg 9 10
i - CkHg 11 12
S - C t H g 13 14
CH2 = CH - CH2 15 16
c6 H5 17 18
H 21 22
differences in the relative abundance of the ion at m l z 99 between ( E ) - and (Z)-isomers. This ion was assigned a cyclic structure a, and it is more abundant in the (Z)-isomers than in the (E)-isomers. The low abundance of ion a in the (E)-isomers was explained in terms of the requirement of additional energy for the isomerization about the double bond leading to the (Z)-isomers before fragmentation. The ion at m l z 99 has, however, a different elemental composi- tion in some esters (3,4). In addition to that, the ratio of R' to (R- H)' is always higher for the maleates when R = C,H,, C,H,, and this difference was attributed to the stability of the departing radical b, which is cyclic.
Meyerson et al . also explored the mass spectra of dimethyl fumarate (1) and maleate (2) in detail and found that the facile loss of methoxy radical is observed from the molecular ion of the (Z)-isomer (2) (9). This was also explained in terms of stability of the cyclic ion c in 2. In the (E)-isomer (1) the loss of methoxy radical from the molecular ion is followed by the facile elimination of CO. The mass spectra of diphenyl fumarate (17) and maleate (18)

MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 493
0 II
k C / C
II '0
RO' '0' /
H"\C
b
were studied by Harrison and Kallury, and the fragmentation patterns of 17 and 18 are similar to the mass spectral fragmentation of dimethyl fumarate and maleate (9,lO).
Large and Saunders reported the mass spectra of bis(trimethylsily1) fumarate (19) and maleate (20) (11). It was shown that 19 can be differentiated from 20 by the relative abundance of the ion at m l z 147, which is more abundant in the (Z)-isomer (20). This was explained in terms of the formation of a cyclic ion d for the (M - CHJ+ ion, which is the precursor for the ion at m l z 147. A different structure was proposed for the ion at m l z 245 formed from the (E)-isomer (19).
0 I I
Benoit et al. studiec the mass spectra fragmentation of fumaric acid (21), maleic acid (22), mesaconic acid (23), and citraconic acid (24) with the help of D-labeled compounds (12). It was found that the loss of CO, from the (Z)- isomers (22,24) is a dominant process, and that this process is preceded by a hydrogen transfer from one of the carboxyl groups to the other (Scheme I). On the contrary, loss of H,O from the molecular ion is observed as a facile process in compounds 21 and 23.
Gil-Av et al. reported the mass spectra of isomeric (E,E)- (25), (Z,Z)- (26), and ( E , Z ) - (27) 3,4-diethylmuconic acid dimethyl esters (13). The mass spectra of (E,E)- (25) and (Z,Z)- (26) diethylmuconic acid dimethyl esters show the

494 VAIRAMANI AND SARASWATHI
II C
HO-C’ ‘H II 0
23 -
H I
,0-H
\ I c=c I \
\ I
/ \ H H
COOH o=c + to2 H OOC - t - c=c
H H
l-co*
H I +
/o\ \ c= c.
o=c
I \ H H
m l z 72
Scheme I
t I C
H’ ‘C-OH II 0
2 4 -
H ..
‘c02 \ /OH o=c + \ I
1-c.’
\ * c=c I \
H H
m l z 72
difference in the abundance of the ion at m / z 194, which is formed by the loss of methanol from the molecular ion. The proximity of an allylic hydrogen to the methoxyl group was shown to be a suitable geometry for the facile loss of methanol from 25 (Scheme 11). In the (Z,Z)-isomer (26) the ion at m / z 194 is insignificant. The (M - CH,)’ and (M - COOCH,)’ ions are more abundant in the spectra of 26 and 27. The facile formation of the (M - COOCH,)’ ion in 26 was explained in terms of formation of a cyclic ion e (Scheme 111). This process is not prominent in 25 because the compound has to undergo isomerization in order to form the cyclic ion. Methyl 3,4-di-n-propylmuconates (28-30) also give similar results (14).
The mass spectral fragmentation of many tiglates (31) and angelates (32) has been reported (15). The geometrically allowed 1,5-hydrogen migration involv- ing the methyl hydrogens leads to the loss of ROH, resulting in the ion at m/z 82. This process is significant in angelates where the methyl and the ester (COOR) groups are on the same side of the double bond. In the absence of such geometry tiglates show the loss of the alkoxy radical (OR) from the molecular ion as the dominant process and this leads to an ion at m / z 83. Differences are

MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 495
R R R R R R
Me02C
c02Me Mc02C COple M
MeO2C CO2Me
- e 25 - ( M - C H ~ O H i'
mlz 194
- e 26 - ( M - C H 3 0 ) +
m l z 195 Scheme I1
also noticed in the abundances of ions at m l z 101 and 100, which arise by the loss of alkene and alkenyl radicals when the R is other than methyl.
Liebich et al. studied the mass spectra dimethyl and diethyl mesaconates (33,35) and citraconates (34,36) (16). The (Z)-isomer 34 gives a molecular ion under electron ionization, whereas the (E)-isomer 33 does not. The ( M - OCH,)+ and (M - OCH, - CO)' ions are more significant in the (Z)-isomers (34,36), whereas ions formed by the loss of methanol followed by CO loss from the M" are more abundant in 33 and 35 (E)-isomers. The extensive loss of an alkoxy radical from 34 and 36 occurs because of the formation of the stable cyclic ion c in these compounds, as in the case of dimethyl maleate (9). Preferential loss of ROH from the (E)-isomers (33,35) was studied in detail with the help of mixed esters and deuterium-labeled compounds by Bornstein et al. (17). These experiments showed the participation of both of the ester groups in the elimination of ROH (Scheme IV). Transfer of one of the methyl
- e 26 - -
H3 CO e
m l z 167
Scheme I11
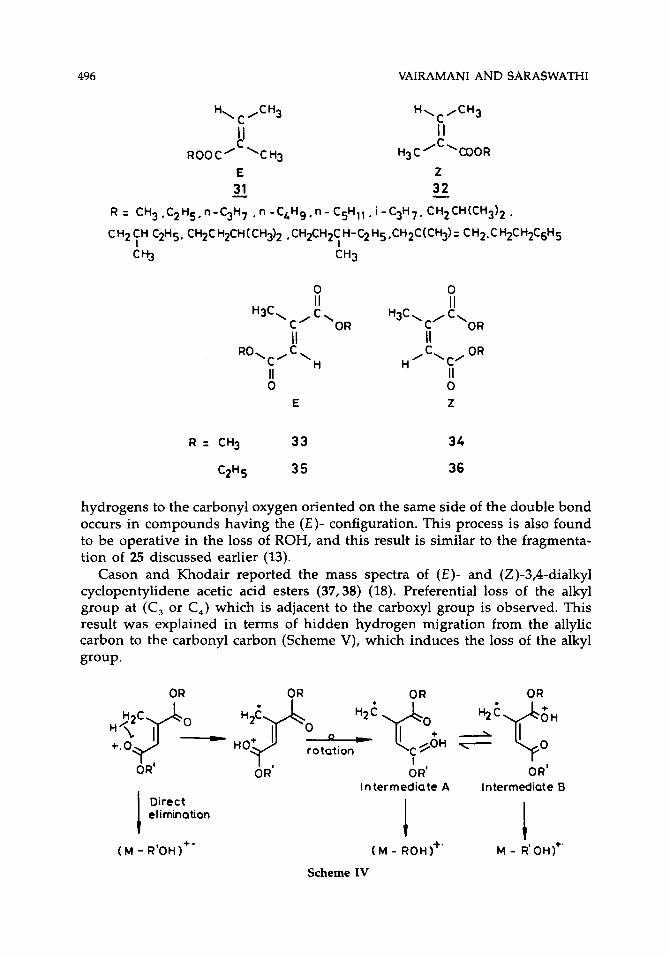
496 VAIRAMANI AND SARASWATHI
0 0
E z
R = CH3 33
C2H5 35
34
36
hydrogens to the carbonyl oxygen oriented on the same side of the double bond occurs in compounds having the ( E ) - configuration. This process is also found to be operative in the loss of ROH, and this result is similar to the fragmenta- tion of 25 discussed earlier (13).
Cason and Khodair reported the mass spectra of (E)- and (Z)-3,4-dialkyl cyclopentylidene acetic acid esters (37,38) (18). Preferential loss of the alkyl group at (C, or C,) which is adjacent to the carboxyl group is observed. This result was explained in terms of hidden hydrogen migration from the allylic carbon to the carbonyl carbon (Scheme V), which induces the loss of the alkyl group. H?p - H2$ 0, ' 2 ' q 0 + 2 % ' f H 0
+.O H O+ rotation @H 7
I 0 R' OR' OR' OR'
Intermediate A Intermediate B
M - R'oH)+'
Direct elimination
( M - ROH)+'
Scheme IV
( M - R'OH I+ '

MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 497
C,-z .OH
H3c3g - CH3
c=o
OCZHS C2H5 H3c3! I; + I
E - 37
+
0C2H5 O C ~ H5
38 - Scheme V
Merksammer and Mandelbaum also studied the mass spectra of ( E ) - and (Z)-3,5-dialkyl cyclohexylidene acetic acid esters (39-44) and ( E ) - and (2)-3- alkyl-2-alkenoates (45-50) (19). The stereospecific loss of the alkyl group which is homoallylic and adjacent to the carboxymethyl group is also observed in 39-50 (Scheme VI).
Natalis and Franklin reported differences in the mass spectra of (E)- and (Z)-isomers of di- tert-butylethylenes (51,52) in 1966 (20). Although the ioniza-
Me02C 'ITR2 TR2 z C02 Me
44
E Z

498 VAIRAMANI AND SARASWATHI
OMe I
Scheme VI
tion potentials are found to be the same for both isomers, the appearance po- tentials for the major ions are smaller for the (Z)-isomer (52), and this differ- ence results in the lower abundance of the molecular ion in the (Z)-isomer.
There are hardly any differences between the EI mass spectra of ( E ) - and (Z)-2-butenes (21). Sunner studied charge-transfer mass spectra of ( E ) - and (Z)-2-butenes by using slow positive ions for ionization in a tandem mass spectrometer (22). It was shown that the isomers can be differentiated by charge-transfer mass spectrometry utilizing incident COS', Kr+, N i , and F' ions. The variation of charge-transfer cross sections with energy was assumed to be responsible for the observed differences. In a later study, Selim calculated the appearance energy values for the C3Hl ion generated from ( E ) - and (Z)-2-butenes separately (23). The heat of formation of the C3H: ion from the (Z)-2-butene was found to be 279.4 kcal - mol-', while a value of 296.6 kcale mol-' was obtained for the formation of the C H' ion from (E)-2-butene. The large difference in the appearance energy of C3H3 ions produced from the two isomers was explained by proposing propargyl ion and allenyl ion as the C3Hl ion from the (Z)- and (E)-isomers, respectively.
The mass spectra of the (E)- and (Z)-isomers of 2-octenes, 3-octenes, and 4-octenes do not show significant differences (24). The EI mass spectra of fifteen pairs of ( E ) - and (Z)-2-methoxymethoxy 4-substituted stilbenes have been reported (25). Although marginal differences in the abundances of different ions are noticed, a detailed investigation has not been done.
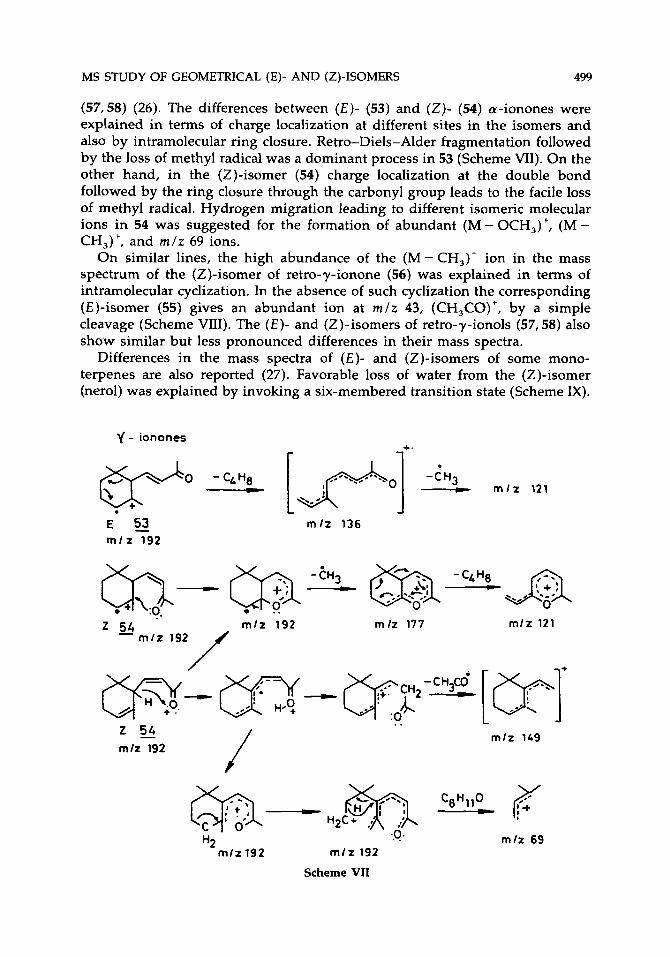
Significant differences are reported between the mass spectra of (E)- and (Z)-isomers of a-ionones (53,54), retro-y-ionones (55,56), and retro- y-ionols
3 + 3
MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 499
(57,58) (26). The differences between (€)- (53) and (Z)- (54) a-ionones were explained in terms of charge localization at different sites in the isomers and also by intramolecular ring closure. Retro-Diels-Alder fragmentation followed by the loss of methyl radical was a dominant process in 53 (Scheme VII). On the other hand, in the (Z)-isomer (54) charge localization at the double bond followed by the ring closure through the carbonyl group leads to the facile loss of methyl radical. Hydrogen migration leading to different isomeric molecular ions in 54 was suggested for the formation of abundant (M - OCH,)+, (M - CH,)+, and r n l z 69 ions.
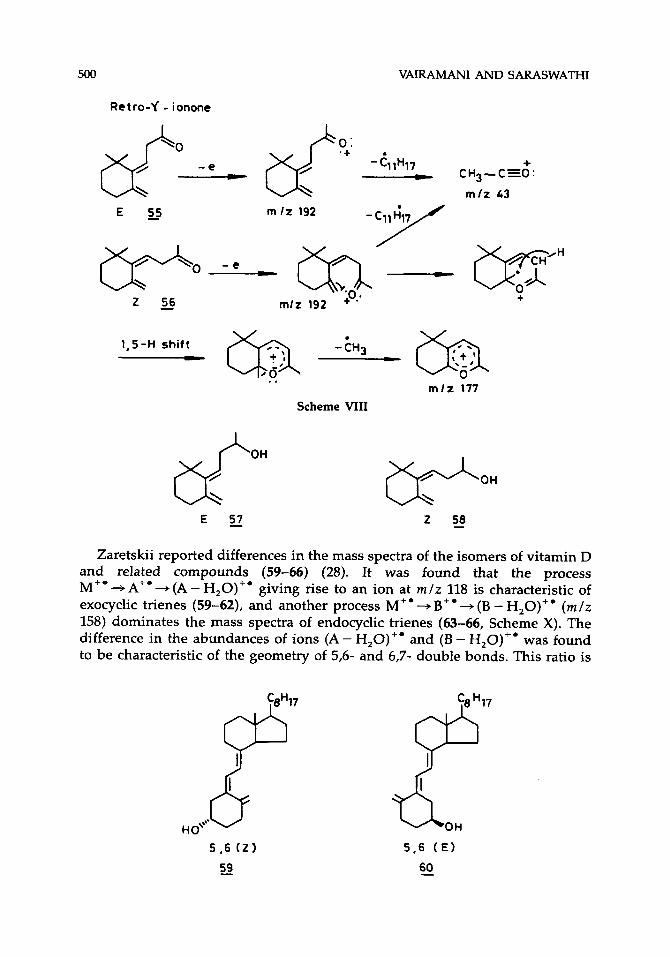
On similar lines, the high abundance of the (M - CH,)+ ion in the mass spectrum of the (Z)-isomer of retro-y-ionone (56) was explained in terms of intramolecular cyclization. In the absence of such cyclization the corresponding (E)-isomer (55) gives an abundant ion at r n l z 43, (CH,CO)', by a simple cleavage (Scheme VIII). The ( E ) - and (2)-isomers of retro-7-ionols (57,58) also show similar but less pronounced differences in their mass spectra.
Differences in the mass spectra of ( E ) - and (Z)-isomers of some mono- terpenes are also reported (27). Favorable loss of water from the (Z)-isomer (nerol) was explained by invoking a six-membered transition state (Scheme IX).
m l t 192 m h 177 5 4 - m l z 192 f' m l z 121
/ (p-(y%wpy(yr / + : ..* H . 2 -- :o ..
@J-- H2c.sjo\ ce.llo_
m l z 169 / 2 % m l t 192
m l z 69 ..o *
m l t 192 rnlz 192 H2
Scheme VII
500 VAIRAMANI AND SARASWATHI
Retro-f - ionone
1,s-H shift - -tH3 (yJ c, ..
m l z 177
Scheme VIII
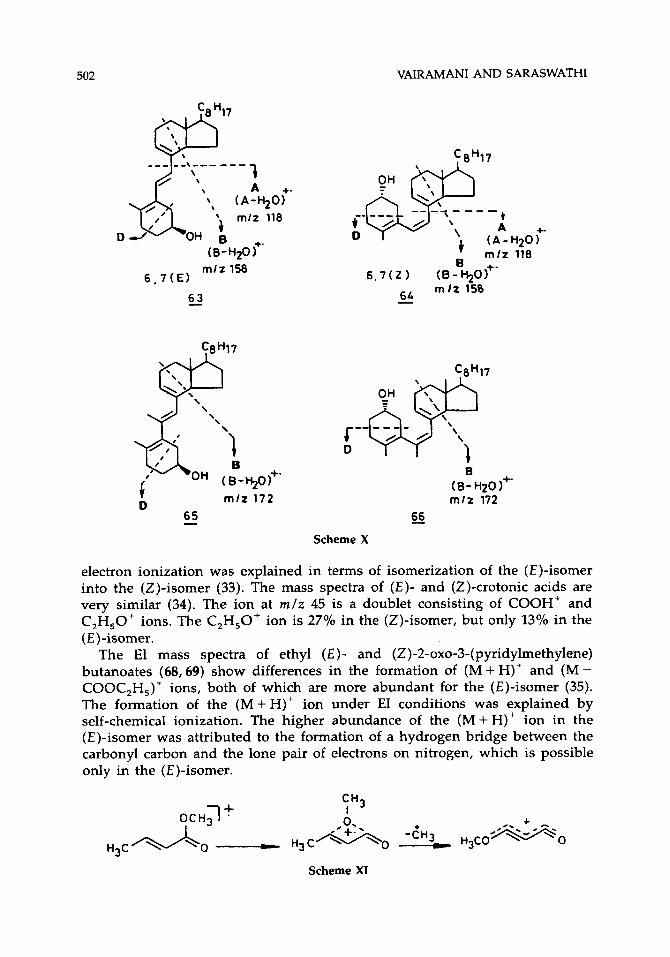
Zaretskii reported differences in the mass spectra of the isomers of vitamin D and related compounds (59-66) (28). It was found that the process M+' + A+' + (A - H,O)+' giving rise to an ion at m /z 118 is characteristic of exocyclic trienes (59-62), and another process M" 3 B" + (B - H,O)+' (m /z 158) dominates the mass spectra of endocyclic trienes (63-66, Scheme X). The difference in the abundances of ions (A - H,O)+' and (B - H20)+' was found to be characteristic of the geometry of 5,6- and 6,7- double bonds. This ratio is
'eH17
o"' 5 . 6 2 ti!)
ss

MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 501
greater for vitamin D, (59) and epi-vitamin D, (61), which have (2)-5,6- geometry as compared to the corresponding (E)-5,6-isomers (60-62). This difference was explained in terms of isomerization of 59 and 61 into the respective previtamin D, forms [(Z)-5,6], which show decreased abundance of (A - H,O)''. This kind of isomerization is less favored in (E)-5,6-isomers (60-62) because of their greater stability. Along similar lines, the observed difference in the intensity ratio of (B - H,O)+/(A - H,O)+' between tachysterol [(E)-6,7] (63) and previtamin D, [(Z)-6,7)] (64) was explained on the basis of isomerization of 64 into vitamin D, (59), which results in higher abundance of (A - H,O)+' in 64. 6-Methyl tachysterol (65) and 6-methyl previtamin D, (66), however, do not show differences in their mass spectra, because they are known to exist in equilibrium. The p-nitrobenzoyl derivatives of 59-62 show larger differences than the underivatized compounds (29).
The mass spectra of dimethyl (E )- and (Z)-octa-2,6-dienoic acids are reported (30). The difference between the geometrical isomers is not significant. Only the ratio of (M - CH,)+/M+' is higher for the (E)-isomer. The mass spectral study of ( E ) - and (Z)-a,@-difluorocinnamic acid is reported (31). The low abundance of fragment ions in the (E)-isomer as compared to the (2)-isomer was attributed to the higher stability of the (E)-isomer. The EI mass spectra of methyl ( E ) - and (Z)-2-butenoates showed differences in the relative abund- ances of M+' and (M-OCH,)' ions (32). The unusual formation of the (M - CH,)+ ion from the (E)-isomer (67) was explained by a concerted [1,3] rearrangement of the methoxy group to the p-carbon atom (Scheme XI). The loss of ROH (R=CH,, C,H,) from (E)-methyl and ethyl crotonates under
I -C-
I -C-
1
Scheme IX
- c - I - c - I
502 VAIRAMANI AND SARASWATHI
mlz 158 6 , 7 ( E l
6_3
t A
( A - D
+ m / z B
6 , 7 ( Z ) (8-%o)+' 6L m l t 150 -
C8H17
+. H20 1 118
65 - 66 Scheme X
electron ionization was explained in terms of isomerization of the ( E ) - * isomer into the (Z)-isomer (33). The mass spectra of ( E ) - and (Z)-crotonic acids are very similar (34). The ion at m / z 45 is a doublet consisting of COOH' and C,H,O+ ions. The C,H,O+ ion is 27% in the (Z)-isomer, but only 13% in the (0- isomer.
The El mass spectra of ethyl (E)- and (Z)-2-0~0-3-(pyridylmethylene) butanoates (68,69) show differences in the formation of (M + H)+ and (M - COOC,H,)' ions, both of which are more abundant for the (E)-isomer (35). The formation of the (M+H)' ion under EI conditions was explained by self-chemical ionization. The higher abundance of the (M + H)+ ion in the (E)-isomer was attributed to the formation of a hydrogen bridge between the carbonyl carbon and the lone pair of electrons on nitrogen, which is possible only in the (E)-isomer.
CH,
Scheme XI

MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 503
E 68 -
CH3
Z 69 -
Korhonen studied the mass spectra of (E)- and (2)-isomers of ethyl 2,3- dichloro, bromochloro and dibromopropionates (36). Preferential loss of the bulky halogen atom at the C, carbon from the (Z)-isomers was reported uniformly for all the compounds. The abundances of the corresponding ions from the (E)-isomers are relatively less. Differences in the abundances of ions (M-OCH,)' and (M-X)+ in the mass spectra of methyl esters of (E)- and (Z)-isomers of 2-butenoic acid containing bromine and/or chlorine atoms are also reported (37). The mass spectra of methyl esters of 2-alkenoic acids show differences in the abundances of ions at rnlz 113 and 87 among (E)- and (Z)-isomers (38). Both nonbranched and branched (Z)-isomers show a greater abundance of the ion at rnlz 113 than the (E)-isomers.
111. CHEMICAL IONIZATION
The chemical ionization mass spectra (CIMS) of diethyl fumarate (3) and maleate (4) have been reported (39). The mass spectra contain only the (M + H)' ion when recorded at low source temperature (130 "C). When the spectra were recorded at a higher temperature (280 "C), some differences in the fragmentation of these isomers were observed. The (Z)-isomer (4) shows a facile loss of ethanol leading to an ion at m / z 127. The cyclic structure c was proposed for this ion. The ions that cannot acquire cyclic structure c for stabilization show consecutive loss of ethylene, and thus facile loss of ethylene is observed in the (E)-isomer (3). Similar behavior is observed in diethyl phthalates and isophthalates. Proximity of the two carboxyethyl groups makes the formation of the cyclic ion more favorable.
The hydrogen and methane CIMS of (E)- and (Z)-isomeric dicarboxylic acids (21-24) show significant differences among the isomers (10). The abundance of the (M + H)+ ion is always less for the maleic (22) and citraconic (24) acids and the (MH-H,O)+ ion is the most abundant ion. On the contrary, fumaric (21) and mesaconic (23) acids show highly abundant (M+H)' ions. Also, the (E)-isomers (21,23) show higher abundance of ions due to the loss of CO from the (MH - H,O)+ ion, especially under hydrogen CI conditions. This was explained in terms of higher exothermicity of protonation under hydrogen CI conditions. Some minor differences in the abundances of ions formed by the loss of CO and CO, from (M+H)+ are also reported. A similar trend in the abundances of the (M + H)+ and (MH - C,H,OH)+ ions was reported for the diethyl fumarate (3) and maleate (4). In addition, loss of C,H, from the (MH - C,H,OH)+ ion leads to an abundant ion for the (Z)-isomer, and this

504 VAIRAMANI AND SARASWATHI
process is more favorable under H,/CI. Diphenyl fumarate (17) and maleate (18) more or less behave in a similar way to this sequence of steps, and the elimination of phenol from (M + H)+ is a dominant process.
Wengang et al. reported differences in the abundances of (MH - H,O)+ and (MH + 28)+ ions in the methanol CI mass spectra of fumaric (21) and maleic (22) acids (40). The (MH + 28)+ ion is formed by diesterification in the source of the mass spectrometer under CI conditions. Isobutane CIMS of fumaric (21) and maleic (22) acids show extra peaks obtained by the reduction of the double bond by the hydrogen atoms present in the source prior to ionization of the sample molecule (41). The (M + 3)+ ion thus obtained is larger in the case of the (E)-isomer (21).
Ammonia CIMS of diethyl fumarate, maleate, mesaconate, and citraconates (3,4,35,36) produce significant differences in the abundances of (M + H)+ and (M + NH,)+ ions (42). The spectra of diethyl fumarate (3) and mesaconate (35) contain mainly (M+NH,)' ions, whereas those of diethyl maleate (4) and citraconate (36) contain abundant (M + H)+ and (M + NH,)' ions. This was explained in terms of higher proton affinity of the (Z)-isomers (4,36). In addition, reduction of the double bond is also observed under high ammonia pressure conditions; the (E)-isomers (3,35) underwent more facile reduction than the (Z)-isomers. The (E)-isomers also form (M + N,H,)+ adduct ions at higher pressures.
Under NHJ CI conditions hydrogenation of the double bond is also ob- served for fumaric (21) and maleic (22) acids, and this process favors the (E)-isomer (21) (43). Kostyanovskii ef al. studied the CIMS of dimethyl fumarate (1) and maleate (2) under a wide range of pressures (44). Under certain conditions it was found that 1 forms cluster ions, whereas 2 does not.
The dibromoethane CI mass spectra of two pairs of ( E ) - and (Z)-* isomers (Scheme XII) of unsaturated dicarboxylic acids (21-24) and their corresponding dimethyl esters 1, 2, 33, 34 show substantial differences in the relative abund- ances of (M + 107)+ and (M + 107 - H,O)+ ions (45). In the case of acids (21-24) the (E)-isomers (21-23) form stable (M + 107)+ adducts, but in the (Z)-isomers (22,24) the adducts are not found; instead (M + 107 - H,O)+ ions are abundant. For the dimethyl esters (1, 2, 33, 34), (M + 107)+ ions are more abundant for the (E)-isomers (1,33) than the (Z)-isomers (2,34). Also, the abundance of the (M + H)+ ion is higher for the (E)-isomers. Acrylonitrile CI mass spectra of fumaric (21), maleic (22), mesaconic (23), and citraconic (24) acids have abun- dant (M + H)+ ions for the (Z)-isomers (22,24) and abundant (M + 54)+ ions for the (E)-isomers (21,23) (46).
Budzikiewicz et al. reported the isobutane and nitric oxide CIMS of (E)- and (Z)-octadecenes (47-49). The (Z)-isomers, such as Z-A6-octadecene and 2-A7- octadecene, form relatively more abundant adducts with C,H,f and NO' ions, generated from isobutane and nitric oxide respectively, compared to the respec- tive (E)-isomers. The authors also found that the relative abundance of (M - H)+ and (M + X)' (X = C,H, and NO) ions is controlled by the steric hindrance across C=C. Dimethyl ether CIMS of (Z, Z)-2,4-hexadiene and (E, E)-2,4- hexadiene show a difference in the abundance of the (M + H)+ ion, which is greater for the (Z, Z)-isomer (50).

MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 505
II
+O - CH2CHpr II 0
II H -C -OH C%-CH2 + \ + / - 0
2 1 -
0 II
H
II 0
22 -
+ 0 - CH2CH2Br II
+ C' H Br I I
0 0 1 0
Scheme XI1
Iron [Fe(I)] chemical ionization mass spectrometry was shown to be useful in differentiating isomeric alkenes and alkynes (51,52). It was also found that the (E)- and (Z)-isomers can be differentiated by the collisionally activated dis- sociation spectra of the olefin Fe(1)-complexes. The (E)-5-decene complex with Fe(1) undergoes more facile isomerization than that of (Z)-5-decene and gives rise to more abundant ions corresponding to (FeC,H,,)' and (FeC,C,,)+ ions (Scheme XIII). A similar tendency is observed for ( E ) - and (Z)-2-octeneJ 3-octene, 4-octene, 3-heptene, and 4-decene complexes. The (E)- and (Z)- isomers of 2-pentene, 2-hexene, 2-decene, and 3-decene do not show significant differences.
The ethylene oxide (oxiran) CI mass spectra of 2-methyl-l-pyrrolidyl-l(E), 3(E)-pentadiene (70), and 2-methyl-l-pyrrolidyl-(lZ, 3E)-pentadiene-1,3 (71) show minor differences in the form of higher abundance of M+*, (M + 43)+, (M + 45)+, and (M + 57)+ ions for the (E, E)-isomer 70 (53).
Both the electron impact and methane CI mass spectra of (E)- and (Z)- isomers of 1,2,3-triaryl-2-propen-l-one are closely similar (54). Differences between the (E)- (72) and (Z)- (73) isomers are, however, observed under diethyl ether, ammonia and methylamine CI conditions (55). The ratio of (M+NH,)+/(M+H)+ is always higher for the (Z)-isomer (73). This was explained in terms of differences in the proton affinities [proton affinity of (E)-isomer > proton affinity of (Z)-isomer]. At source temperatures <lo0 "C,

506 VAIRAMANI AND SARASWATHI
I Fe C7 H T'
( 1 5 4 F= c8 ~ ; t 6 FeC6H&
(168) (1LO)
Scheme XI11
however, no differences between the (E)- and (Z)-isomers are observed (56). Reduction of the double bond by the hydrogen radicals present in the source is a prominent process under these conditions.
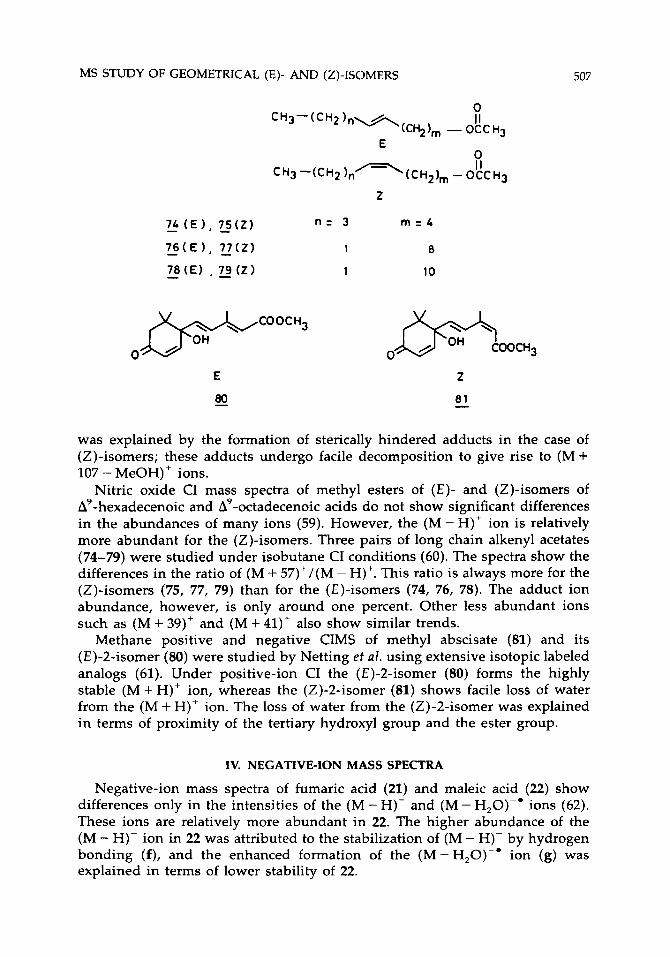
Methane CI mass spectra of ( E ) - and (Z)-cinnamic acids and their methyl and ethyl esters show differences in the abundance of the (M + C,H,)+ ion and the ion resulting from the loss of ROH (R = H, CH,, C,H,) from the (M + C,H,)' adduct ion (57). The loss of ROH from the (M+C,H,)+ adduct is observed only for the (Z)-isomers. The difference observed between the ( E ) - and (2)-isomers was explained in terms of adduct formation with the aromatic ring and the carbonyl group. Vairamani et al. studied the mass spectra of substituted ( E ) - and (Z)-methyl cinnamates under 1,2-dibromoethane CI condi- tions (58). The (E)-isomers show abundant adduct (M + 107)+ ions, whereas the (Z)-isomers exhibit abundant (M + 107 - MeOH)+ ions also. This difference
Q- c H j
E 7,o
z 71 -
E
7 2 - z
MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 507
0
E
eo z 81 -
was explained by the formation of sterically hindered adducts in the case of (2)-isomers; these adducts undergo facile decomposition to give rise to (M + 107 - MeOH)' ions.
Nitric oxide CI mass spectra of methyl esters of ( E ) - and (Z)-isomers of A'-hexadecenoic and A9-octadecenoic acids do not show significant differences in the abundances of many ions (59). However, the (M - H)' ion is relatively more abundant for the (Z)-isomers. Three pairs of long chain alkenyl acetates (74-79) were studied under isobutane CI conditions (60). The spectra show the differences in the ratio of (M + 57)+/(M - H)+. This ratio is always more for the (Z)-isomers (75, 77, 79) than for the (E)-isomers (74, 76, 78). The adduct ion abundance, however, is only around one percent. Other less abundant ions such as (M + 39)+ and (M + 41)' also show similar trends.
Methane positive and negative CIMS of methyl abscisate (81) and its (E)-2-isomer (80) were studied by Netting et al. using extensive isotopic labeled analogs (61). Under positive-ion CI the (E)-2-isomer (80) forms the highly stable (M+H)' ion, whereas the (Z)-2-isomer (81) shows facile loss of water from the (M + H)+ ion. The loss of water from the (Z)-2-isomer was explained in terms of proximity of the tertiary hydroxyl group and the ester group.
IV. NEGATIVE-ION MASS SPECTRA
Negative-ion mass spectra of fumaric acid (21) and maleic acid (22) show differences only in the intensities of the (M - H)- and (M - H,O)-* ions (62). These ions are relatively more abundant in 22. The higher abundance of the (M - H)- ion in 22 was attributed to the stabilization of (M - H)- by hydrogen bonding (f), and the enhanced formation of the (M-H,O)-' ion (g) was explained in terms of lower stability of 22.

508 VAIRAMANI AND SARASWATHI
0 II cC-) C- II O f
0 I 1
QC\0-. C'
II 0
9
Atmospheric pressure ionization mass spectra of fumaric (21) and maleic (22) acids do not show differences under negative conditions, except in the abund- ance of the molecular ion (63). Fumaric acid (21) shows a more abundant molecular ion than 22. Ion mobility data obtained for the above compounds with negative-ion plasma chromatography are also similar.
The negative-ion fast atom bombardment (FAB) mass spectra of fumaric (21), maleic (ZZ), mesaconic (23), and citraconic (24) acids show differences in the abundances of M-' and (M - CO,)-' ions, both being more abundant for the (€)-isomers (21,23) than for the (Z)-isomers (22,24) (64). In the (Z)-isomers the (M - H)- ion is formed preferentially. This was explained by an intramolecular hydrogen bonding that stabilizes ion f in the (Z)-isomer.
The Br- negative-ion chemical ionization (NICI) mass spectra of many saturated and unsaturated dicarboxylic acids have been studied. Maleic (22) and citraconic (24) acids form exclusively (M - H)- ions, whereas fumaric (21) and mesaconic (23) acids form (M+Br)- adduct ions. We attributed this phenomenon to the stabilization of (M - H)- ions by intramolecular hydrogen bonding, which is possible only in the (Z)-isomers (22,24) (65). The unsatu- rated carboxylic acids undergo hydrogenation in the source of the mass spectrometer in the presence of methanol prior to ionization of the sample molecule (66). The (Z)-isomers (22,24) undergo faster reduction than the (E)-isomers (21,23) and the chloride NICI technique was used to prove this phenomenon. The abundance of (M+H)- ions is always more for the ( Z ) - isomers. The study also shows that the reduction process is a metal surface catalyzed phenomenon. The (E)- and (Z)-isomers of A9- and All-octadecenoic acids do not show differences in their spectra, although these compounds also undergo reduction. The (E)- and (Z)-cinnamic acids show a difference in the form of a more abundant (M - H)- ion in the case of the (Z)-isomer, which we have attributed to the lower stability of the (M + C1)- adduct ion because of the steric hindrance.
Resonance electron capture (REC) mass spectra of (E)- and (Z)-a,/?- difluorocinnamic acids have been reported (31). The (E)-isomer has two addi- tional maxima on the effective yield curve at 4.2 and 4.8eV in contrast to the (Z)-isomer, and this difference was explained in terms of specific formation of the (M - CO0H)- ion. Collisional activated dissociation spectra of (M - H)- ions of unsaturated fatty acids are useful in locating the position of the double bond (67). However, the study of five pairs of (E)- and (Z)-isomers, namely, A'-hexadecenoic acids, A6-, A9, All-octadecenoic acids, and A13-dodecadecenoic acids showed that this method does not differentiate these (E)- and ( Z ) - isomers. Under Br- and C1- NICI conditions a difference between the ( E ) - and

MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 509
0 -7- M-'(m/z: 278)
81 - I C02 CH3
0 Jy m l z : 152
Scheme XIV
(Z)-isomers of 1,2,3-triaryl 2-propen-1-one (72,73), in the form of favorable halide adduct formation with the (Z)-isomers (73), is reported (55).
Under negative-ion CI methyl abscisate (81) forms a stable molecular ion (61). The (E)-2-isomer (80) gives abundant (M-H)- ions in addition to the molecular ion. The authors explained that the hydroxyl hydrogen is linked with the ester carbonyl group through hydrogen bonding in 81 and hence the removal of this hydrogen is not a favorable process. On the contrary, both C, hydrogen and the hydroxyl hydrogen are available in 80. Facile loss of the side chain is also observed in the (Z)-isomer, and this loss results in the formation of an ion at r n l z 152 (Scheme XIV). In a recent report, however, Heath et al. have shown that molecular oxygen plays an important role in fragmentation of methyl abscisate (81) under electron capture negative ionization (ECNI) (68). A detailed investigation utilizing "0, as a reagent showed that the abundances of ions at r n l z 141 and 152 are highly dependent on the presence of air as an impurity during the analysis. It is also reported that the ECNI spectra of (E)-2- and (Z)-2-isomers (80,81) are identical.
V. DESORPTION TECHNIQUES
Under positive-ion FAB conditions, differences in the abundances of the ions (M + H - H,)+, (M + H - CO,)+, and (M + H - H2C02)+ are noticed for the two pairs of ( E ) - and (Z)-dicarboxylic acids 21-24 (64). The (M+H)+ ions of maleic (22) and citraconic (24) acids are more stable because of the formation of a cyclic structure h. This proton bridged complex results in decreased fragmen- tation of the (Z)-isomers 22 and 24. The enhanced abundance of (M + H - H,)+, (M + H - H,CO,)+, and (M + H - CO,)' ions in the fumaric (21) and mesaconic (23) acids was explained by double bond protonation followed by elimination (Scheme X V ) of neutral molecules. The study of itaconic acid (82) and glutaconic acid (83) is also in support of the above results. Under field
OH h

510 VAIRAMANI AND SARASWATHI
OH I
OH /
, c ?o.. CH2COOH - H ~ c+ - c 0 2 __c H - R-CEC-C,, +
O H
protonation
oxygen HOOC, /H HOOC ,c ,H II C+
protonation \ Coo* ‘c’ H + - H20 C - co I1 - C R’ R’ ‘c+
II I I 0 0
0 I I
- c 0 2
iH2 R-C, +
C=OH I OH
( MH - C02)+ I
OH (MH)+
Scheme XV
H CHzCOOH / COOH \ /
c = c c =c, ”\ H’ ‘CH2COOH HOOC’ H
c2 ltaconic acid 82 Glutaconic acid
ionization (FI) conditions these compounds (21-24, 82, 83) undergo less frag- mentation. The (Z)-isomers (22,24) give abundant (M + H)’ ions relative to the (E)-isomers (21, 23, 82, 83). Another difference noticed is the enhanced loss of CO, from molecular ions of (Z)-isomers; and this result is similar to their electron-impact behavior (12).
Lehmann and Beckey studied the FI mass spectra of some (E)- and (Z)- isomers of 1,2-dibromoethylenes (69). These compounds show the difference in the loss of bromine from the molecular ion. Under FI conditions, when the tip emitter was used, the (E)-isomer lost bromine more easily than the (Z)-isomer. This was explained in terms of orientation of the molecules in a high electric field. The (E)-isomer has the preferred orientation for easy breaking of the C-Br bond, but the (Z)-isomer requires another orientation besides the preferred one if the bond is to break. The reverse trend is, however, observed when the spectra were recorded at 12.5 eV under electron ionization (69). The FI study was extended to (E)- and (Z)-isomers of substituted alkenes using a wire emitter. The (E)- and (Z)-isomers of 4,4-dimethyl-2-pentener 2,2-dimethyl-3-

MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 511
hexene, and 2,5-dimethyl-3-hexene do not show significant differences. It is also reported that the FI spectra of ( E ) - and (Z)-isomers of n-alkenes (-CJ are indistinguishable (70).
VI. METASTABLE AND COLLISIONALLY ACTIVATED DISSOCIATION
In 1968, Daly et al. developed a new detector system, later known as the Daly detector, for recording metastable ions (71). By using this detector, they showed that the metastable ion spectra of (E)- and (Z)-2-butenes are distinguishable.
The low-energy collisionally activated dissociation spectra of (M + H)' ions of diethyl fumarate, maleate, mesaconate, and citraconates (3, 4, 35, 36) show a high degree of stereospecificity among the isomeric compounds (72). The (Z)-isomers (4,36) give abundant (MH - EtOH)+ and (MH - EtOH - C,H,)+ ions. On the contrary, in the (E)-isomers (3,35), consecutive loss of two molecules of ethylene from the (M + H)+ ions is observed. Loss of ethanol from the (M + H)+ of 3 and 35 does not take place because it involves a symmetry- forbidden 1,3-hydrogen transfer, which requires high energy. In the absence of a competing C,H, loss, however, the (E)-isomers of dimethyl esters 1 and 33 lose methanol from the ( M + H)+ ions. The high energy required for this particular process is reflected in the formation of more abundant (MH- MeOH - CO)' ions in these isomers. These data were obtained by using a triple-stage quadrupole mass spectrometer.
Charge exchange (CE) spectra of dimethyl fumarate (1) and maleate (2) taken with (COS)" (11.9 eV) and N l (15.3 eV) show differences between the isomers similar to those observed under electron impact (73). Loss of methoxy radical is predominant in the (Z)-isomer (2), while the (E)-isomer (1) shows abundant peaks due to the loss of CH,O and CO,CH, (Scheme XVI). The differences between the isomers are more significant in the metastable ion spectra.
The metastable molecular ion of 2 undergoes enhanced loss of methoxy radical and gives rise to stable cyclic ion i, whereas the metastable molecular ion of 1 does not lose methoxy radical, because the resulting acyclic ion j is
+. 0 1
c r n l z 11L
0 +. II
+ - CH30CH=CH-C0 + [COZ+CH3] H3C0, r?r""'"" O 1 II rn lz 85
- Scheme XVI

512 VAIRAMANI AND SARASWATHI
+ OCH3 + I t ,CEO
HC HC/'\ II II 0 CH HC .C' H3CO-C'
I I 0
II 0
I j
much less stable. These results show that both high- and low-energy molecular ions of 1 and 2 do not interconvert.
The EI mass spectra of diethyl fumarate (3) and maleate (4) were studied in detail by Harrison et al. with the help of D-labeled compounds, metastable ion spectra, kinetic energy release measurements, and charge exchange spectra (73).
l+. 0 I I c I
+OH
- L m l z 127 m l z 99
- OC2 Hg I +
8
- co - t C2HgOCH=CKC:O
m l z 99
m l z 9 9 m l z 127
- (CH 3CH0 + CO 1 + 0 CH2-CH-CfO
m I z 55
0 Scheme XVII

MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 513
Based on these results, they concluded that the (M - OC,H,)+ ions produced by EI from the isomeric diethyl esters have different structures. The fragmenta- tion pathways given in Scheme XVII can explain the results. The metastable ion dissociation of di(ethy1-d,) maleate and fumarate molecular ions obtained by the mass analyzed ion kinetic energy (MIKE) technique showed the same fragmentation reactions with similar yields. The (COS)+' CE spectra of the two di(ethy1-d,) ester; are also quite similar, and these results indicate that the low-energy molecular ions of these isomers undergo extensive interconversion (Scheme XVIII). The CO" (-3.4eV internal energy) and N, (-4.7eV internal energy) CE spectra, however, have substantial differences showing that the high-energy molecular ions do not interconvert among themselves. Though the isomerization process has a lower critical reaction energy than the fragmenta- tion, it has a much less favorable frequency factor. Because of this, at higher internal energy, fragmentation is faster than isomerization.
These findings were later confirmed by Tajima et al. by deuterium labeling and MIKE studies (74). A remarkable inverse isotope effect was observed for the losses of COOCH, and COOCD, radicals from the metastable molecular ion of dimethyl fumarate-d,. This was explained by a hidden hydrogen transfer in the rate-determining step for the loss of the carbomethoxy group (Scheme XIX).
The isomeric methyl ethyl esters of phenyl fumaric and maleic acids were investigated under CI conditions for intramolecular hydrogen deuterium ex- change by low-energy collisionally activated dissociation and extensive deuterium-labeling studies (75). A preferential loss of the alkoxy group from
0 I 1 +
0 l+. II
0c20S I + c=oo C-OC2D5 - c-?D5 -
c - 0 C O C 0 ~ II 0
C-OC2 0 5 II 0
c- O C 0 2 t 0 2 I I 0 *I -'2O5
0 I1
C - O D c- OCD-CD2 II
+OD m l z 1L8 mlz 148
0 m l z 130
1 +. 0 I 1
* + L C ~ D S O C H = C H - C I O + CO + C 2 D 5 O
Ds c2 0, II m l z 10L 0
Scheme XVIII
c$3 O

514 VAIRAMANI AND SARASWATHI
0 0 I t
Ph-C-I
H- C-1 II
m / z 14L m l z 85
Scheme XIX
Major 7 (MD- MeOD)++ ( MD- MeOH 1' COOMe CD;
COOEt - MD' 0.9 0.1
m l z 236 (MD- EtOD)++ (MD-EtOH)'
0.68 0.32
Scheme XX
Ph-C-COOR C1 + C I D I I - MH - (MH-R'OH)+ ( MH-ROH)+ R'OOC- C- H mlz 235 Major Minor
OR'
Scrambling (& -. OR
H ',
R'O 4 0
(M D - R'OH) +
(M D- R '0 o)+ +
Scheme XXI
+
D
9 C O O R t
R'O 0
( MD - R'OH I+

MS STUDY OF GEOMETRICAL (E)- AND (Z)-ISOMERS 515
the carbon carrying the phenyl group is reported for the maleates. Considerable intramolecular hydrogen-deuterium exchange is observed for the loss of al- cohol involving the remote alkoxy group (Scheme XX). On the contrary, in the isomeric fumarates loss of alcohol incorporating the alkoxy group which is on the same side as the phenyl group is noticed.
This loss also involves extensive hydrogen-deuterium exchange of the external deuterium and the phenyl ring hydrogens. This is explained in Scheme XXI. This study was carried out by using CD, as well as ND, as CI reagents and also by comparison with a similar study of methyl ethyl t-butyl maleates and succinates (76,77).
The mass spectral fragmentation of the ( E ) - and (Z)-isomers of methyl 3-chloropropionate and ( E ) - and (Z)-methyl 2,3-dichloropropionate was dis- cussed with the help of D-labeled compounds and metastable ions (78). No significant differences were reported, however.
VII. CONCLUSIONS
The present survey of mass spectral study of geometrical isomers across the C=C bond brings out certain general modes of fragmentation for ( E ) - and (Z)-isomers. The (Z)-isomers prefer to form ions with cyclic structures because of the proximity of functional groups. This effect is more pronounced in 2-butene-1,4-dicarboxylic acids and their homologues and derivatives, which have been studied more extensively. More challenging cases like alkenes, unsaturated fatty acids, and their derivatives have not shown remarkable differences among the isomers. With the development of many new mass spectral techniques, there is more scope for exploring many more geometrically isomeric systems.
ACKNOWCEDGMENTS
The authors thank Dr. A. V. Rama Rao, Director of Indian Institute of Chemical Technology, for constant encouragement. One of the authors (M. S.) thanks CSIR, New Delhi, for the award of a fellowship. This article is IICT Communication No. 2947.
1.
2.
3. 4.
5. 6. 7. 8.
9.
REFERENCES
Silverstein, R. M.; Bassler, G. C.; Momll, T.C. Spectrometric Identification of Organic Compounds, 4th ed.; Wiley: New York, 1981. Ladd, M. F. C.; Palmer, R. A. Structure Determination by X-Ray Crystallography, 2nd ed; Plenum: New York, 1985. Meyerson, S.; Weitkamp, A. W. Org. Mass Specfrom. 1968, 1, 659. Green, M. M. In Tropics in Stereochemistry; Eliel, E. L.; Allinger, N. L., Eds.; Wiley: New York, 1976; pp 9, 35. Mandelbaum, A. In Stereochemisty, Kagan, H., Ed.; Thieme: Stuttgart, 1977; pp 1, 37. Mandelbaum, A. Mass Spectrom. Rev. 1983, 2, 223. Natalis, P. Mass Spectrometry; NATO Advanced Study Institute: Glasgow, 1965; p 379. Bowie, J. H.; Williams, D. H.; Madsen, P.; Schroll, G.; Lawesson, S. 0. Tetrahedron 1967, 23, 305. Meyerson, S.; Ihrig, P. J.; Hunter, T. L. J . Org. Chem. 197l, 36, 995.

516
10. 11. 12. 13. 14.
15. 16. 17. 18. 19. 20. 21. 22. 23. 24.
25.
26.
27.
28. 29. 30. 31.
32. 33. 34.
35. 36. 37. 38. 39. 40.
41. 42.
43. 44.
4s. 46.
47. 48. 49. 50. 51. 52. 53. 54.
55.
VAIRAMANI AND SARASWATHI
Harrison, A. G.; Kallury R. K. M. R. Org. Mass Spectrom. 1980, 15, 277. Large, R.; Saunders, K. J. Org. Mass Spectrom. 1973, 7, 291. Benoit, F.; Holmes, J. L.; Isaacs, N. S. Org. Mass Spectrom. 1969, 2, 591. Gil-Av, E.; Leftin, J. H.; Mandelbaum, A.; Weinstein, S. Org. Mass Spectrom. 1970, 4,475. Mandelbaum, A.; Weinstein, S.; Gil-Av, E.; Leftin, J. H. Org. Mass Spectrom. 1975, 10, 842. Thomas, A. F.; Willhalm, B. Org. Mass Spectrom. 1976, 11, 831. Liebich, H. M.; Pickert, A.; Stierle, U.; Woll, J. J. Chromatogr. 1980, 199, 181. Bornstein, D.; Weisz, A.; Mandelbaum, A. Org. Mass Spectrom. 1986, 21, 225. Cason, J.; Khodair, A. I. A. J . Org. Chem. 1966, 31, 3618. Merksammer, I.; Mandelbaum, A. Tetrahedron 1985, 41, 775. Natalis, P.; Franklin, J. L. Bull. SOC. Chim. Belg. 1966, 75, 328. Meisels, G. G.; Park, J. Y.; Giessner, B. G. J. Am. Chem. SOC. 1970, 92, 254. Sunner, J . lnt. J . Mass Spectrom. lon. Phys. 1980, 32, 285 (CA. 93: 7081f). Selim, E. T. M. Proc. lndian Acad. Sci. (Chem. Sci.) 1982, 91, 359. Polyakova, A. A.; Lukashenko, I. M.; Sosulina, L. N.; Rang, S.; Eisen, 0. Zh. Org. Khim. 1969, 5, 1720 (CA. 72: 16638m). Takakis, I. M.; Hayes, R. N.; Mylona, A.; Nikokavouras, J. Org. Mass Spectrom. 1989, 24, 841. Wageningen, A. V.; Cerfontain, H.; Nibbering, N. M. M. Rec. Trav. Chim. Pays-Bas Belg. 1974, 93, 43. Rosado, A.; Doerffel, K.; Schubert, D.; Tapanes, R. Rev. CENlC Cienc. Fis. 1979, 10, 83 (CA. 94: 103573j). Zaretskii, Z. V. I. Adv. Mass Spectrom. 1980, 8A, 590. Zaretskii, Z. V. I. Biomed. Mass Spectrom. 1978, 5, 576. Agrawal, V. P.; Poulose, A. J. J. Nepal Chem. SOC. 1981, I, 1 (CA. 103: 95582~). Parakhnenko, A. I.; Nekrsov, Yu. S.; Mazunov, V. A.; Khvostenko, V. I.; Kremlev, M. M.; Fialkov, Yu. A.; Yagupol'skii, L. M. lzv. Akad. Nauk. SSSR Ser. Khim, 1984, 1307. Hemnann, R.; Schwarz, H. Z. Naturforsch. 1976, 31b, 1013. Bowles, A. J.; Brittain, E. F. H.; George, W. 0. Org. Mass Spectrom. 1969, 2, 809. Holmes, J . L.; Terlouw, J, K.; Vijfhuizen, P. C.; ACampo, C. Org. Mass. Spectrom. 1979, 14, 204. Pohjala, E. K.; Nieminen, A. 0. K. Org. Mass Spectrom. 1981, 16, 519. Korhonen, I . 0. 0. Org. Mass Spectrom. 1988, 23, 751. Korhonen, I . 0. 0. Biomed. Environ. Mass Spectrom. 1990, 19, 143. Iwamura, J.; Hosotsubo, H.; Hirao, N. Shitsuryo Bunseki 1979, 27, 173 (CA. 94: 83153s). Fales, H. M.; Milne, G. W. A.; Nicholson, R. S. Anal. Chem. 1971, 43, 1785. Wengang, C.; Guanghui, W.; Zhiling, Zu.; Jiongguang, P.; Shumin. D. Org. Mass Spectrom. 1983, 18, 64. Issachar, D.; Yinon, J. Anal. Chem. 1980, 52, 49. Das, K. G.; Reddy, A. M.; Vairamani, M.; Madhusudanan, K. P.; Fraisse, D.; Tabet, J. C. Tetrahedron 1984, 40, 4085. Madhusudanan, K. P.; Murthy, V. S.; Fraisse, D. Org. Mass Spectrom. 1987, 22, 665. Kostyanovskii, R. G.; Revel'skii, I. A.; Voznesenskii, V. N.; Yashin, Yu. S. lzv . Akad. Nauk. S S S R Ser. Khim. 1985, 815 (CA. 103: 104370g). Vairamani, M.; Srinivas, R.; Mirza, U. A. Org. Mass Spectrom. 1988, 23, 620. Srinivas, R.; Vairamani, M.; Viswanadha Rao, G. K.; Mirza, U. A. Org. Mass. Spectrom. 1989, 24, 435. Budzikiewicz, H.; Busker, E. Tetrahedron 1980, 36, 255. Budzikiewicz, H.; Busker, E.; Brauner, A. Adv. Mass Spectrom. 1980, 8A, 713. Budzikiewicz, H. Fresenius, Z. Anal. Chem. 1985, 321, 150. Keough, T. Anal. Chem. 182, 54, 2540. Peake, D. A.; Gross, M. L.; Ridge, D. P. J. Am. Chem. SOC. 1984, 106, 4307. Peake, D. A.; Gross, M. L. Anal. Chem. 1985, 57, 115. Lange, C. Org. Mass Spectrom. 1987, 22, 55. Madhusudanan, K. P.; Mittal, S.; Durani, S.; Kapil, R. S. Org. Mass Spectrom. 1985, 20, 215. Madhusudanan, K. P.; Mittal, S.; Durani, S.; Kapil, R. S. Org. Mass Spectrom. 1985, 20, 323.

MS STUDY OF GEOMETRICAL (E)- AND (2)-ISOMERS 517
56. 57. 58.
59. 60. 61.
62. 63. 64.
65. 66.
67. 68.
69. 70.
71. 72.
73. 74. 75.
76. 77.
78.
Madhusudanan, K. P. Jain, R.; Durani, S.; Kapil, R. S. Zndian J. Chem. 1989, 28B, 1082. Anderson, G. B.; Gills, R. G.; Porter, Q. N. Org. Mass Spectrom. 1986, 21, 383. Vairamani, M.; Saraswathi, M.; Viswanadha Rao, G. K. Org. Mass. Spectrom. 1991, 26, 786. Brauner, A.; Budzikiewicz, H.; Francke, W. Org. Mass Specfrom, 1985, 20, 578. Vekey, K.; Baan, G.; Jennings, K. R. Biomed. Environ. Mass Spectrom. 1988, 16, 267. Netting, A. G.; Milborrow, B. V; Vaughan, G. T. Biomed. Environ. Mass Spectrom. 1988, 15, 375. Ito, A.; Matsumoto, K.; Takeuchi, T. Org. Mass Spectrom. 1973, 7, 1279. Kim, S. H.; Karasek, F. W. 1. Chrornatogr. 1982, 234, 13. Dallinga, J. W.; Nibbering, N. M. M.; van der Greef, J.; Ten Noever de Brauw, M. C. Org. Mass Spectrom. 1984, 19, 10. Vairamani, M.; Saraswathi, M. Org. Mass Spectrom. 1989, 24, 355. Vairamani, M.; Saraswathi, M.; Viswanadha Rao, G. K. Org. Mass Spectrom. 1990, 25, 274. Tomer, K. B.; Crow, F. W.; Gross, M. L. 1. Am. Chem. SOC. 1983, 105, 5487. Heath, T. G.; Gage, D. A.; Zeevaart, J. A. D.; Watson, J. T. Org. Mass Specfrom. 1990,25, 665. Lehmann, W. D.; Beckey, H. D. Org. Mass Spectrom. 1974, 9, 1086. Rang, S. A.; Muurisepp, A. M. A.; Liitmaa, M. M.; Eisen, 0. G. Org. Mass Spectrom. 1978, 13, 181. Daly, N. R.; McCormick, A.; Powell, R. E. Org. Mass Spectrom. 1968, 3, 167. Weisz, A.; Mandelbaum, A.; Shabanowitz, J.; Hunt, D. F. Org. Mass Spectrom. 1984, 29, 238. Harrison, A. G.; Nacson, S.; Mandelbaum, A. Org. Mass Spectrom. 1987, 22, 283. Tajima, S.; Ishiguro, A.; Tobita, S. Znt. J. Mass Spectrom. Zon. Process. 1987, 75, 147. Mandelbaum, A.; Muller, D. R.; Richter, W. J.; Vidavsky, I. Znt. J. Mass Spectrom. Zon. Process. 1990, 100, 565. Weisz, A.; Cojocaru, M.; Mandelbaum, A.J. Chem. SOC. Chem. Commun. 1989, 331. Mandelbaum, A.; Muller, D. R.; Richter, W. J.; Vidavsky, I.; Weisz, A. Org. Mass Spectrum. 1989, 24, 857. Korhonen, I . 0. 0. Org. Mass Spectrom. 1983, 18, 295.
Copyright © 2022 FDOKUMEN















![ChemInform Abstract: Ecofriendly Synthesis of Thieno[2,3-b]pyridines Derivatives](https://static.fdokumen.com/doc/165x107/632083a318429976e4063ccf/cheminform-abstract-ecofriendly-synthesis-of-thieno23-bpyridines-derivatives.jpg)




