Changes of the corrosion potential of iron in stagnation and flow conditions and their relationship...
11
Changes of the corrosion potential of iron in stagnation and flow conditions and their relationship with metal release Massimiliano Fabbricino a,* , Gregory V. Korshin b a Department of Civil, Architectural and Environmental Engineering, University of Naples Federico II, Via Claudio 21, 80125 Naples, Italy b Department of Civil and Environmental Engineering, University of Washington, Seattle, WA 98195-2700, United States article info Article history: Received 7 March 2014 Received in revised form 25 May 2014 Accepted 29 May 2014 Available online 7 June 2014 Keywords: Iron Metal release Corrosion potential Sulfate Chloride Online monitoring abstract This study examined the behavior of corrosion potential (E corr ) of iron exposed to drinking water during episodes of stagnation and flow. These measurements showed that during stagnation episodes, E corr values decrease prominently and consistently. This decrease is initially rapid but it becomes slower as the stagnation time increases. During flow episodes, the E corr values increase and reach a quasi-steady state. Experiments with varying con- centrations of dissolved oxygen showed that the decrease of E corr values characteristic for stagnation is likely to be associated with the consumption of dissolved oxygen by the exposed metal. The corrosion potential of iron and its changes during stagnation were sensitive to the concentrations of sulfate and chloride ions. Measurements of iron release showed that both the absolute values of E corr measured prior to or after stagnation episodes were well correlated with the logarithms of concentrations of total iron. The slope of this dependence showed that the observed correlations between E corr values and Fe concen- trations corresponded to the coupling between the oxidant consumption and changes of Fe redox status. These results demonstrate that in situ E corr measurements can be a sensitive method with which to ascertain effects of hydrodynamic conditions and short-term vari- ations of water chemistry on metal release and corrosion in drinking water. This approach is valuable practically because E corr measurements are precise, can be carried out in situ with any desired time resolution, do not affect the state of exposed surface in any extent and can be carried out with readily available equipment. © 2014 Elsevier Ltd. All rights reserved. 1. Introduction Corrosion of metals causes massive damage to water distri- bution networks and deterioration of drinking water quality (Ahammed and Melchers, 1996; Ahammed, 1998; Aleksyuk, 2002; Caleyo et al., 2002; Netto et al., 2005; Cosham et al., 2007; Netto et al., 2007; Teixeria et al., 2008; Li et al., 2009; Nuhi et al., 2011). The latter process is associated with the release of solutes and particles that affect aesthetic and * Corresponding author. E-mail addresses: [email protected] (M. Fabbricino), [email protected] (G.V. Korshin). Available online at www.sciencedirect.com ScienceDirect journal homepage: www.elsevier.com/locate/watres water research 62 (2014) 136 e146 http://dx.doi.org/10.1016/j.watres.2014.05.053 0043-1354/© 2014 Elsevier Ltd. All rights reserved.
Transcript of Changes of the corrosion potential of iron in stagnation and flow conditions and their relationship...

ww.sciencedirect.com
wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6
Available online at w
ScienceDirect
journal homepage: www.elsevier .com/locate /watres
Changes of the corrosion potential of iron instagnation and flow conditions and theirrelationship with metal release
Massimiliano Fabbricino a,*, Gregory V. Korshin b
a Department of Civil, Architectural and Environmental Engineering, University of Naples Federico II, Via Claudio 21,
80125 Naples, Italyb Department of Civil and Environmental Engineering, University of Washington, Seattle, WA 98195-2700, United
States
a r t i c l e i n f o
Article history:
Received 7 March 2014
Received in revised form
25 May 2014
Accepted 29 May 2014
Available online 7 June 2014
Keywords:
Iron
Metal release
Corrosion potential
Sulfate
Chloride
Online monitoring
* Corresponding author.E-mail addresses: [email protected] (M. F
http://dx.doi.org/10.1016/j.watres.2014.05.0530043-1354/© 2014 Elsevier Ltd. All rights rese
a b s t r a c t
This study examined the behavior of corrosion potential (Ecorr) of iron exposed to drinking
water during episodes of stagnation and flow. These measurements showed that during
stagnation episodes, Ecorr values decrease prominently and consistently. This decrease is
initially rapid but it becomes slower as the stagnation time increases. During flow episodes,
the Ecorr values increase and reach a quasi-steady state. Experiments with varying con-
centrations of dissolved oxygen showed that the decrease of Ecorr values characteristic for
stagnation is likely to be associated with the consumption of dissolved oxygen by the
exposed metal. The corrosion potential of iron and its changes during stagnation were
sensitive to the concentrations of sulfate and chloride ions. Measurements of iron release
showed that both the absolute values of Ecorr measured prior to or after stagnation episodes
were well correlated with the logarithms of concentrations of total iron. The slope of this
dependence showed that the observed correlations between Ecorr values and Fe concen-
trations corresponded to the coupling between the oxidant consumption and changes of Fe
redox status. These results demonstrate that in situ Ecorr measurements can be a sensitive
method with which to ascertain effects of hydrodynamic conditions and short-term vari-
ations of water chemistry on metal release and corrosion in drinking water. This approach
is valuable practically because Ecorr measurements are precise, can be carried out in situ
with any desired time resolution, do not affect the state of exposed surface in any extent
and can be carried out with readily available equipment.
© 2014 Elsevier Ltd. All rights reserved.
1. Introduction
Corrosion of metals causes massive damage to water distri-
bution networks and deterioration of drinking water quality
abbricino), [email protected]
rved.
(Ahammed and Melchers, 1996; Ahammed, 1998; Aleksyuk,
2002; Caleyo et al., 2002; Netto et al., 2005; Cosham et al.,
2007; Netto et al., 2007; Teixeria et al., 2008; Li et al., 2009;
Nuhi et al., 2011). The latter process is associated with the
release of solutes and particles that affect aesthetic and
shington.edu (G.V. Korshin).

wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6 137
organoleptic water characteristics (Lin et al., 2001; Critchley
et al., 2004; Dietrich et al., 2004; Rogers et al., 2004) while the
leaching of heavy metals such as copper and lead from
household plumbing, service lines and other components
exposed to drinking water has been associated with adverse
health effects in exposed populations (Korshin et al., 2000;
Zietz et al., 2001, 2003; Dietrich et al., 2004; Edwards, 2004;
Kim and Herrera, 2010; Peng et al., 2010; Gonzalez et al.,
2013). In addition, trace-level contaminants such as arsenic,
vanadium and others tend to accumulate in corrosion scales
where their concentrations are greatly increased compared to
those in ambient water. As a result, instability of the corrosion
solids and their colloidal mobilization can result in exposures
of water consumers to increased levels of these contaminants
(Schock et al., 2008; Gerke et al., 2010; Peng et al., 2010; Peng
and Korshin, 2011; Yang et al., 2012).
The fundamental need to maintain the integrity of water
distribution networks and ensure the safety of drinking water
provided by them necessitates that corrosion and metal
release in drinking water be suppressed and responses of
these processes to changes of water chemistry and other
conditions in the systems (e.g., variations of temperature, flow
rates) be understood and controlled. Given the inherent
complexity of corrosion and metal release processes in com-
bination with diverse water chemistries and variable expo-
sure conditions typical for drinking water distribution
systems and household plumbing, meeting these goals re-
quires that approaches to quantify corrosion and metal
release processes in realistic conditions be employed.
In principle, overall rates of corrosion can be obtained via
weight loss or electrochemical (EC) measurements. The
complication with weight loss measurements is that while
they may be adequate for iron-based materials, they require
long exposure periods, cannot be used to examine effects of
short-term water chemistry variations and have a limited
applicability in the case of corrosion of copper- and lead-
containing materials.
Alternatively, overall corrosion rates can be estimated by
means of fitting, typically using the ButlereVolmer equation
or its variants (Flitt and Schweinsberg, 2005; McCafferty, 2005;
Mansfeld, 2005; Zhang et al., 2009), relationships between
experimentallymeasured currents through the exposedmetal
surface and its electrochemical (EC) potential which in these
measurements is varied using an external device (a poten-
tiostat). While EC measurements can be done in situ, external
polarization of the metal surfaces that is necessary for
potentiodynamic experiments may alter properties of the
examined surfaces. Even a larger complication is that the
interpretation of the data obtained in sweeps of EC potential
or other EC methods such EC impedance spectroscopy (EIS)
may be ambiguous due to the occurrence of processes unac-
counted for in the ButlereVolmer equation, for instance mass
transfer limitations, multiple parallel EC and chemical re-
actions and others. Per se, the EC measurements are also not
useful for predicting rates of metal release (they determine
only those of the electrochemical oxidation of the substrate)
from exposed surfaces, nor can they ascertain short-term
changes of metal release associated with, for instance, water
stagnation in water distribution networks or household
plumbing.
Another limitation of EC measurements of corrosion rates
in drinking water is that they do not provide unambiguous
information concerning the stability of surface-scales accu-
mulated as a result of metal corrosion on the exposed sur-
faces. These scales tend to have complex mineralogical
characteristics and are spatially non-uniform, especially in
the case of corrosion of iron and lead. Because physico-
chemical properties of these scales are strongly affected by
the chemistry of ambient water, they can become unstable in
response to changes of its properties (Sander et al., 1996, 1997;
Broo et al., 1997; Volk et al., 2000; Boulay and Edwards, 2001;
Sarin et al., 2001, 2004; D’Antonio et al., 2008; Nawrocki
et al., 2010).
Given the complexity of predicting responses of corrosion
processes and metal release to long- and short-term varia-
tions of drinking water properties and flow conditions, it is
critical to monitor these responses in real time (Volk et al.,
2000; Sancy et al., 2010; Ishii and Boyer, 2011). In principle,
this can be done by water sampling using designated sam-
pling locations in a water distribution system or using corro-
sion rigs of varying complexities. These options provide data
that can be interpreted in the context of applicable regulations
but their use requires complicated exposure and sampling
systems and ex situ analyses for metals of interest.
The extent of these limitations supports a notion that at
least ideally, metal release and corrosion processes should be
monitored and quantified using an approach based on in situ
measurements of a surrogate parameter or a system of pa-
rameters that can be measured with high precision and suf-
ficient ease in real time without use of expensive equipment
or ambiguous data processing techniques. Such parameter(s)
should be also intrinsically tied to (rather than to be purely
statistically correlated with) the occurrence and intensity of
processes that play a fundamental role in corrosion andmetal
release.
Below, we present data demonstrating that in situ moni-
toring of changes of the corrosion potential (otherwise
referred to as open circuit potential, as it is measured in the
absence of any external EC polarization) of a metal surface
exposed to drinking water and its changes in response to
variations of exposure conditions may be a viable alternative
approach for onlinemonitoring ofmetal release and corrosion
phenomena. To demonstrate this, we examined the behavior
of the corrosion potential (denoted henceforth as Ecorr) of iron
exposed to water containing varying concentrations of back-
ground salts. We also correlate phenomena observed during
episodes of water stagnation and flowwith the extent ofmetal
release in the examined systems.
This study was focused on the corrosion of iron due to
several reasons. First, iron is one of the most important ma-
terials present in drinking water distribution systems (Lin
et al., 2001; Tang et al., 2006; Sancy et al., 2010; �Swietlik
et al., 2012; Yang et al., 2012). The corrosion of iron results in
massive amounts of lost non-revenue water and damage to
the infrastructure that amount to up to several billions dollars
per year. Iron release affects taste and color of drinking water
and excessive exposure to it can be harmful (Brittenham, 2003;
Mora et al., 2009). Solid iron corrosion products can be rich in
toxic elements such as arsenic (Copeland et al., 2007; Kim
et al., 2011; Peng and Korshin, 2011) and release of such

wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6138
products can lead to adverse health effects. Finally, scales
formed on exposed iron surfaces are colonized by microor-
ganisms forming biofilms whose properties are critical in
controlling the microbiological stability of drinking water
(Volk et al., 2000; McNeill and Edwards, 2001; Lee and
Newman, 2003; Lehtola et al., 2004; Little et al., 2007.)
The corrosion of iron, formation of surface solids and iron
release are sensitive to multiple water chemistry parameters
such as concentrations of dissolved oxygen, chloride, sulfate,
carbonate, natural organic matter and others (Sander et al.,
1996, 1997; Broo et al., 1999; Shi et al., 2000; Volk et al., 2000;
Appenzeller et al., 2001; Mc Neil and Edwards, 2001; Sarin
et al., 2004; Choi et al., 2005; Tang et al., 2006; Shams El Din,
2009; Nawrocki et al., 2010; Peng et al., 2010; Yang et al.,
2012; Liang et al., 2013; Liu et al., 2013; Peng et al., 2013).
Corrosion scales formed on iron exposed to drinking water
tend to form several distinct layers whose structure and
composition is affected both by the chemistry of ambient
water and diffusion processes at the involved interfaces and
through the solid phases (Sander et al., 1997). Some compo-
nents of corrosion scales formed on iron surfaces, notably
green rusts, are particularly sensitive to variations of levels of
common background anions such as chloride and sulfate
(Refait et al., 2003; Sarin et al., 2004; Nawrocki et al., 2010;�Swietlik et al., 2012). Green rusts are mixed layered double
hydroxides in which positive charges of the layers formed by
Fe2þ and Fe3þ ions are balanced by negative charges of the
interlayer anions. The sensitivity of green rusts to the type of
the intercalated anions results in their potentially strong
response to variations of sulfate and chloride concentrations.
Sulfate and chloride ions can also form soluble complexes
with Fe(II) and Fe(III) ions thus affecting the stability of the
surface solids (Lasheen et al., 2008). In general, higher levels of
sulfate and chloride tend to result in increased rates of iron
corrosion and release (Hedberg and Johansson, 1987; Sander
et al., 1996; Veleva et al., 1998; Lin et al., 2001; Refait et al.,
2003; Sarin et al., 2004; Imran et al., 2005, 2006; Nawrocki
et al., 2010; Yang et al., 2012) although more complex trends
have also been observed (Bondietti et al., 1993; McNeill and
Edwards, 2001).
These observations support the point that the occurrence
and significance of potentially rapidly changing concentra-
tions of sulfate, chloride and other water components need to
be monitored to elucidate the response of iron corrosion and
release to water quality variations. Below, we present
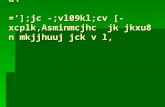
Corrosion ciron electr
centrifugal pump
Ag/Age
recirculation tankperistaltic
pump
sampler
Fig. 1 e General schematic of th
experimental datasets that provide information concerning
effects of some of these water chemistry parameters of the
values of Ecorr of iron exposed to drinking water.
2. Materials and methods
The study used two corrosion cells exposed to water whose
composition underwent a series of short-term changes during
the experiments. Main elements of the experimental system
are shown in Fig. 1. Each of the two setups employed in this
study contained a flow-through corrosion cell equippedwith a
two parallel iron plates, a reference Ag/AgCl electrode and a 2-
L polycarbonate recirculation tank which was open to the air.
The water was recirculated using a centrifugal pump. The
recirculation loop contained a flow meter and a valve to con-
trol the flow rate. All elements of the system were connected
using Tygon tubing. Periodic sampling of water in the recir-
culation tank was done using a peristaltic pump that with-
drew small water aliquots from the recirculation tank. The
centrifugal and peristaltic pumps were controlled by a pro-
grammable timer that used to run a predetermined sequence
of flow and stagnation conditions in the cell and to withdraw
water samples with desired frequency. Water samples with-
drawn from the system using the peristaltic pump were
acidified with high purity nitric acid and analyzed for iron
concentration using a PerkinElmer DRC-e Elan ICP/MS spec-
trometer. The calibration of the ICP/MS instrument and other
related procedures were carried as described in Peng and
Korshin 2011.
The iron cells had two rectangular 50 � 140 mm grey cast
iron plates with a 16 mm (5/800) thickness. This material was
purchased from McMaster-Carr. The plates were separated
with a 2 mm rubber seal. Each plate had a 28.2 cm2 area
exposed to ambient water, and the total exposed iron surface
was 56.4 cm2. A gel-filled Ag/AgCl reference electrode (model
A57193, Beckman Coulter Inc.) was placed in the center of the
upper plate. The tip of the reference electrode was positioned
flush with the metal surface. The potential of the surface vs.
the reference electrode was recorded every 10 s using a high
impedance multi-channel digital data acquisition unit (Agi-
lent Technologies Model 34970A) connected to a computer
that was used to store and process the data.
Initial exposures to Seattle tap water were performed
to allow the metal surfaces to develop surface scales.
Flow meter
ell with odes
PC
High impedance digital multimeter
Cl reference lectrode
e experimental apparatus.

wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6 139
The exposures of the two cells were initiated at different times
(ca. 2 months difference) to allow the metal surfaces in one of
the cells to develop more extensive scales than those in the
other. Thus the two corrosion cells allowed comparing re-
sponses of relatively freshly exposed and more extensively
corroded surfaces.
The initial exposureswere followed by tests to examine the
generate data concerning effects of flow and stagnation con-
ditions of the corrosion potential of iron and examine the
reproducibility of the observed effects. These tests allowed
establishing a 4 h flow followed by a 4 h stagnation sequence
that was deemed to be adequately representative of condi-
tions in drinking water distribution systems or household
plumbing.
All experiments were carried out with Seattle tap water
that was recirculated in the system. The water initially con-
tained ca. 0.9 mg L�1 active chlorine but this oxidant dis-
appeared quickly after the beginning of each recirculation
test. Water in the recirculation tanks was exchanged daily.
Seattle tap water has lowmineralization, with background
sulfate and chloride concentrations of 3 and 2 mg L�1,
respectively. The alkalinity and hardness of the water are ca.
20 and 25 mg L�1 as CaCO3, respectively. The baseline water
conductivity is ca. 60 mS cm�1.
Further experiments addressed effects of variation of
concentrations of dissolved oxygen, chloride and sulfate.
Concentrations of NaCl or Na2SO4, used in the experiments
are listed in Table 1. In some experiments, the concentration
of dissolved oxygen in the ambient water was increased by
bubbling oxygen gas through the recirculation tank. In other
experiments the water was deoxygenated by bubbling nitro-
gen or adding requisite amounts of 0.1 mol L�1 sodium sulfite
stock solution to the recirculation tank. The pH of the water
was checked during recirculation and was found to be always
stable.
Between any successive tests that employed a different
water chemistry, the cells were flushed with the tap water for
2 h and then for 1 h by water with the desired chemical
composition. The water used for flushing was discarded.
Following this, measurements of Ecorr values and metal
release were resumed. When experiments with variations of
any controlled water chemistry parameter were finished and
before the commencement of a next series of experiments, all
plastic tubing, fittings and flow-meters were replaced and the
Table 1 e Characteristics of used solutions.
NaCl Na2SO4
mol L�1 mS cm�1 pH mol L�1 mS cm�1 pH
0.00005 73 6.5 0.00001 73 6.5
0.00068 153 6.5 0.00010 89 6.5
0.00102 197 6.5 0.00021 111 6.5
0.00136 230 6.5 0.00032 126 6.5
0.00170 261 6.5 0.00042 154 6.5
0.00205 287 6.5 0.00052 179 6.5
0.00273 395 6.5 0.00074 191 6.5
0.00341 456 6.6 0.00095 212 6.6
0.00411 492 6.6 0.00116 273 6.6
0.00479 565 6.7 0.00137 311 6.7
recirculation tankswere acidwashed and rinsed several times
with tap water.
3. Results and discussion
3.1. Effect of hydrodynamic conditions
Measurements of the corrosion potential of iron exposed to
the baseline water quality indicated that Ecorr values exhibited
pronounced and reproducible changes during episodes of
stagnation and flow (Fig. 2). This figure demonstrates that
during flow episodes, the corrosion potential rose gradually
from its value measured at the end of a preceding stagnation
episode. That increase wasmost prominent during first one to
2 h of flow. Following this, Ecorr values stabilized although they
continued to increase slowly until the end of flow episode
which was 4 h in our experiments. The absolute values of the
observed potentials were somewhat higher than those re-
ported for Evian water with varying concentrations of free
chlorine (e.g., Frateur et al., 1999) or Karlsruhe drinking water
with varying concentration of dissolved oxygen (Kuch, 1988).
In general, the observed Ecorr valueswere close to those largely
dominated by redox transition involving carbonate-
containing green rusts (their general formula is FeII4-FeIII2(OH)12CO3) formed on the electrode surface (G�enin et al.,
2006).
When the flow of water through the system was inter-
rupted, the corrosion potential decreased almost immediately
by ca. 20 mV. This was likely to be caused by nearly-
instantaneous consumption of oxygen in the layer of solu-
tion adjacent to the metal surface. After this initial rapid
change, the corrosion potential continued decreasing more
gradually as shown in Fig. 2. In the baseline water quality, the
overall decrease of the corrosion potential during a 4-h stag-
nation episode was ca. 90 mV.
This trend was similar for both cells used in this study
although the absolute values of their corrosion potentials
measured, for instance, during flow episodes were different
(�0.05 V and �0.17 V). The existence of differences of the ab-
solute values of Ecorr measured for the cells was not surprising
since the two cells had different amounts and, potentially,
properties of corrosion solids formed on the surface of iron.
Notwithstanding the observed differences in the absolute Ecorrvalues, their changes induced by alternating stagnation and
flow conditions were practically identical for both cells and
highly reproducible from one experiment to another as
demonstrated in Fig. 2.
3.2. Effect of sulfate and chloride concentration
Furthermeasurements showed that the corrosion potential of
iron was sensitive to variations of concentrations of sulfate
and chloride. As mentioned in the introductory section, such
variations are likely to occur in any distribution system
especially in those that use water sources with different
background chemistries (e.g., groundwater and surface water,
or their blends with desalinated water) (Broo et al., 1999;
Appenzeller et al., 2001; Choi et al., 2005; Tang et al., 2006;

Fig. 2 e Behavior of the corrosion potential of iron in Seattle tap water during episodes of stagnation and flow.
wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6140
Shams El Din, 2009; Peng et al., 2010; Liang et al., 2013; Liu
et al., 2013; Peng et al., 2013).
Figs. 3e5 demonstrate that increases of NaCl or Na2SO4
concentrations led to several effects. The absolute values of
the corrosion potential of iron during both flowand stagnation
conditions decreased as the concentration of sodium chloride
increased (Fig. 3). The observed decreases of the corrosion
potential in the presence of either chloride or sulfate are
consistent with the notion that the redox properties of the
exposed surfaces are controlled by carbonate-, chloride- and
sulfate-containing green rusts (G�enin et al., 2006); the hy-
pothesis concerning the presence of these solids will
Fig. 3 e Effects of sodium chloride concentration on the corrosion
ultimately need to be confirmed by means of relevant struc-
tural measurements.
While the behavior of relative changes of Ecorr values dur-
ing the flow conditions did not undergo considerable changes
at varying NaCl or Na2SO4 levels, the time profiles of Ecorrduring stagnation were affected by the concentrations of
these salts. The decrease of the corrosion potential (calculated
vs. its value at the end of the preceding flow episode) within
less than a minute after the interruption of flow was most
prominent in the baseline water chemistry without additions
of NaCl or sodium sulfate (Figs. 4A and 5A). Increases of NaCl
concentration lead to a slower change of Ecorr values for
of potential of iron during episodes of stagnation and flow.

Fig. 4 e Effects of sodium chloride concentration on
changes of corrosion potential of iron during stagnation
episodes. (A) stagnation time up to 5 min and (B)
stagnation time up to 60 min.Fig. 5 e Effects of sodium sulfate concentration on changes
of corrosion potential of iron during stagnation episodes.
(A) stagnation time up to 5 min and (B) stagnation time up
to 60 min.
wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6 141
stagnation times less than 2 min but for longer stagnation
times the decrease of Ecorr wasmore prominent at higher NaCl
concentrations. This effect was evident for stagnation times
up to ca. 60 min (Figs. 4B and 5B). For longer stagnation times,
changes of Ecorr vs. time tended to be close for NaCl concen-
trations above ca. 0.0014 mol L�1. For sulfate, trends of the
time profiles of Ecorr values at increasing Na2SO4 concentra-
tions were similar compared to those observed for sodium
chloride.
As mentioned above, the observed decreases of the Ecorrvalues of iron at increasing concentrations of chloride and
sulfate are consistent with the presence of green rusts on the
surface processes but the nature and overall significance of
these effects need to be studied in more detail in experiments
with varying concentrations of carbonate, pH and other pa-
rameters of the solution chemistry that are supposed to cause
theoretically predictable changes on the composition of sur-
face scales.
3.3. Effect of dissolved oxygen
On the other hand, changes of the Ecorr values consistently
observed during stagnation episodes may be hypothesized to
reflect the consumption of dissolved oxygen as a result of
oxidation of the exposed metal, similarly to what was
reported in Kuch 1988. While presenting a detailed theoretical
discussion concerning the effects that take place in the
ambient water and on the electrode surface during each
stagnation episode goes beyond the scope of this paper, suf-
fice it mention here that, fundamentally, the oxidation of iron
surface is coupled with the reduction of dissolved oxygen
present in the ambient water in the cell and/or in the pores of
the corrosion solids that cover the exposed surface. If the rate
of corrosion is sufficiently high, the dissolved oxygen in these
compartments will be noticeably reduced and the values of
Ecorr that are defined by the EC properties of the surface per se
and the concentration of dissolved oxygen at the surface may
exhibit changes reflecting the consumption of the oxidant and
other concurrent processes.
To test this hypothesis, measurements of Ecorr values at
varying levels of dissolved oxygen in the ambient water were
carried out. In one of the experiments, the water that was
recirculated through the cells was saturated with oxygen
which was continuously bubbled through the water reservoir.
This resulted in a consistent increase of the corrosion poten-
tials of iron during both flow and stagnation conditions (Fig. 6).
The behavior of Ecorr during stagnationwas in this case similar
to that observed to the background oxygen concentration but

Fig. 6 e Effects of oxygen and sulfite of the corrosion potential of iron exposed to Seattle tap water.
wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6142
during the first 2 min of stagnation the corrosion potential in
water saturated with oxygen decreased more rapidly
compared with its change at the ambient dissolved oxygen
concentration.
The corrosion potential of iron strongly responded to the
removal of dissolved oxygen caused by the addition of the
oxygen scavenger sodium sulfite (Fig. 6). When the amount of
sodium sulfite corresponding to a 50% removal of the dis-
solved oxygen present in the recirculating water was added,
the corrosion potential measured during a flow episode
decreased by ca. 20mVwithin 10min after the injection of the
oxygen scavenger. Increase of sodium sulfite concentration to
a level corresponding to a 100% removal of the dissolved ox-
ygen resulted in a rapid decrease of the corrosion potential
that continued throughout the flow episode. The extent of the
changes of Ecorr values in these conditions was comparable to
that observed during flow conditions in the experiments re-
ported by Kuch 1988 for Kalsruhe drinking water in which the
concentration of oxygen was varied from close to zero to ca.
5 mg L�1.
Further increase of sodium sulfite concentrations to a level
corresponding to 200% of the expected dissolved oxygen con-
centration resultedonly in a relatively small transient changeof
Ecorr values which continued to decrease following the trend
established by the injection of the amount of sulfite equivalent
to a 100% of dissolved oxygen. The interruption of the flow and
onset of stagnation resulted in a small transient of Ecorr values
which continued changing like it did during the flow of oxygen-
free water. The experiment with addition of sulfite was then
interrupted to prevent a potentially massive reduction of iron
corrosion products that may occur in strongly reducing condi-
tions.Recirculationwithwatercontainingdissolvedoxygenwas
resumed and after two sequences of flow/stagnation the
corrosionpotential of iron returned to its valueobservedprior to
the introduction of Na2SO3.
These results confirm the hypothesis that the changes of
Ecorr values observed during the stagnation episodes can be
attributed to the consumption of oxygen in the corrosion cell
albeit that process may not be the only mechanism that
manifests itself as changes of Ecorr during stagnation. For
instance, the corrosion potential of iron is likely to be sensitive
to the redox state of solid phases, e.g., Fe2þ and Fe3þ con-
taining solids such as ferrous hydroxide, ferrihydrate, lep-
idocrosite, magnetite, siderite and potentially green rusts
(G�enin et al., 2006; Nawrocki et al., 2010) that may respond to
changes of near-surface concentrations of oxygen as well as
those of chloride, sulfate and other solution components. EC
potentials of metals are also sensitive to the concentration of
the same ion in the solution. Theoretically, the Nernst equa-
tionwhose general form is predicts that the EC potential of the
relevant metal (e.g., Fe) is linearly correlated with the loga-
rithm of activity of the free form of the oxidized form of that
metal (e.g., Fe2þ) but in the case of iron surfaces or generally
metal surface covered with scales, this dependence may not
necessarily be observed. Another complication factor is that
Ecorr values are defined by the EC potentials of both iron and
oxygen couples as well as on the intrinsic EC parameters such
as Tafel slopes that characterize the dependence of the
oxidation and reduction currents vs. the actual potential of
the surface. As a result, the observed Ecorr changes can be
indicative of several concurrent processes, including both the
accumulation of iron oxidation products and also the con-
sumption of dissolved oxygen, as shown in Fig. 6.
3.4. Relationships among corrosion potential and metalrelease
To examine relationships between changes of Ecorr and metal
release, concentrations of total iron found in the recirculation
reservoir immediately after the end of each stagnation

Fig. 8 e Correlations between iron concentrations and
wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6 143
episode were measured. These measurements showed that
the total Fe concentrations in the recirculation reservoir ten-
ded to increase with those of chloride or sulfate (Fig. 7)
although incremental increases of the Fe concentrations were
small for Cl and sulfate concentrations exceeding 0.0018 and
0.0007 mol L�1, respectively. The extent of these effects was
comparable for the two examined inorganic ions, and the
observed Fe concentrations were close for the two cells used
in the experiments.
Comparisons of the total Fe concentrations and Ecorr values
or their changes during the stagnation episodes showed that
the absolute values of Ecorr measured either during flow con-
ditions or in the end of the stagnation episode after which Fe
concentrations were measured are were reasonably strongly
correlated (R2 values from 0.72 to 0.79) with logarithms of the
observed Fe concentrations (Fig. 8).
The data shown in Fig. 8 combine the results of measure-
ments carried out for variations of chloride and sulfate ions.
These results show that for Ecorr values measured in the flow
conditions preceding stagnation episodes after which the Fe
concentrations were determined, the correlations between
Ecorr values and Fe release are linear but different for the
examined cells, apparently due to their different histories and
properties of the scales on their surfaces. However, these
correlations become stronger and closer to each other for Ecorrvalues measured at the end of the stagnation episodes after
which Fe concentrations in the recirculation reservoir were
measured.
This appears to indicate that the decrease of Ecorr and Fe
release are intrinsically correlated, which is consistent with
Fig. 7 e Total iron concentrations measured during flow
periods as function of concentration of (A) chloride and (B)
sulfate.
corrosion potentials measured in (A) flow conditions and
(B) stagnation conditions.
the view that Fe release is enhanced in conditions when the
dissolved oxygen at the surface of exposed iron surface or
within the surface scales is consumed. The latter process
drives the redox status of Fe solids on the surface towards
higher prevalence of ferrous iron that has higher solubility
and is released during the stagnation episodes. The actual
mechanisms that define the extent and kinetics of changes of
Ecorr values of iron during stagnation may be more complex
and involve the decrease of dissolved oxygen concentrations
at the surface, possible changes of local pH and concentra-
tions of chloride, sulfate and other ions in the reactive zone.
These processes will also strongly depend on the nature of
solid phases formed on exposed iron surface; the properties of
these solids and their sensitivity to long- and short-term
changes of water chemistry are likely to be site specific. The
extent and significance of such site-specificity as well as
intrinsic mechanisms defining the behavior of Ecorr values
presented in this paper need to be addressed in future
research.
4. Conclusions
This study examined the behavior of corrosion potential (Ecorr)
of iron exposed to drinking water with varying composition.

wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6144
Measurements of corrosion potential of iron during episodes
of stagnation and flow showed that Ecorr value exhibit com-
plex but highly reproducible changes. During stagnation epi-
sodes, the corrosion potential of iron decreases noticeably.
This decrease is initially rapid but it becomes progressively
slower as the stagnation time increases. During flow episodes,
the Ecorr values increase and reach a quasi-steady state. Ex-
periments with varying concentrations of dissolved oxygen in
the water showed that the decrease of Ecorr values character-
istic for stagnation conditions may be at least partially asso-
ciated with the consumption of dissolved oxygen by the
exposedmetal. The corrosion potential of iron and its changes
during stagnation episodes were also determined to be sen-
sitive to the concentrations of sulfate and chloride ions.
Measurements of iron release showed that both the absolute
values of Ecorr measured prior to or after stagnation episodes
were well correlated with the logarithms of concentrations of
total iron. The slope of this dependence showed that the
observed correlations between Ecorr values and total Fe con-
centrations were not reflective of the applicability of the
Nernst-type relationship between the electrochemical po-
tentials and metal concentrations but rather they corre-
sponded to the coupling between the oxidant consumption
and metal oxidation which is fundamental for corrosion pro-
cesses at open circuit potentials.
While detailed further examination of the effects of tem-
perature, flow conditions, solution chemistry, for instance pH,
carbonate concentration, NOM, hardness cations etc. is
necessary to gainmore insight into the nature of the observed
phenomena, the data generated in this study demonstrate
that in situ Ecorr measurements can be a sensitive and ulti-
mately powerful method with which to ascertain effects of
hydrodynamic conditions and short-term variations of water
chemistry on metal release and corrosion in drinking water.
This approach can be all the more valuable given that Ecorrmeasurements are precise, can be carried out in situ with any
desired time resolution, do not affect the state of exposed
surface in any extent and do not require any equipment other
than a standard reference electrode and a high-impedance
voltmeter both of which are readily available. This enables
this type monitoring to be carried out at virtually any site in a
drinking water distribution system or in household plumbing.
The implementation of this approach is contingent on the
data of further experiments with iron, its alloys and other
metals typical for drinking water systems, notably copper-
and lead-containing materials, stainless steel and results of
relevant monitoring for systems experiencing variations of
water chemistry, temperature and other conditions affecting
rates of corrosion and metal release.
Acknowledgements
This research was supported by the U.S. National Science
Foundation (grant 0931676). Dr. Fabbricino expresses his deep
gratitude to the Department of Civil and Environmental En-
gineering of the University of Washington and especially to
Prof. Stephen Burgess for the June 2004 Endowed Visiting
Professorship that supported his long-term visit that allowed
carrying out experiments reported in this paper. Views and
conclusions presented in this study do not necessary express
those of the funding agencies.
r e f e r e n c e s
Ahammed, M., 1998. Probabilistic estimation of remaining life of apipeline in the presence of active corrosion defects. Int. J.Press. Vessels Pip. 75 (4), 321e329.
Ahammed, M., Melchers, R.E., 1996. Reliability estimation ofpressurised pipelines subject to localised corrosion defects.Int. J. Press. Vessels Pip. 69 (3), 267e272.
Aleksyuk, M.M., 2002. Prediction of the strength of steel pipesdamaged by aqueous corrosion. Strength Mater. 34 (2),200e205.
Appenzeller, B.M.R., Batt�e, M., Mathieu, L., Block, J.C.,Lahoussine, V., Cavard, J., Gatel, D., 2001. Effect of addingphosphate to drinking water on bacterial growth in slightlyand highly corroded pipes. Water Res. 35 (4), 1100e1105.
Bondietti, G., Sinniger, J., Stumm, W., 1993. The reactivity of FE(III)(hydr)oxides: effects of ligands in inhibiting the dissolution.Colloids and Surfaces A: Physicochem. Eng. Aspects 79 (2e3),157e167.
Boulay, N., Edwards, M., 2001. Role of temperature, chlorine, andorganic matter in copper corrosion by-product release in softwater. Water Res. 35 (3), 683e690.
Brittenham, G.M., 2003. Iron chelators and iron toxicity. Alcohol30 (2), 151e158.
Broo, A.E., Berghult, B., Hedberg, T., 1997. Copper corrosion indrinking water distribution systems e the influence of waterquality. Corros. Sci. 39 (6), 1119e1132.
Broo, A.E., Berghult, B., Hedberg, T., 1999. Drinking waterdistribution e the effect of natural organic matter (NOM) onthe corrosion of iron and copper. Water Sci. Technol. 40 (9),17e24.
Caleyo, F., Gonzalez, J.L., Hallen, J.M., 2002. A study on thereliability assessment methodology for pipelines with activecorrosion defects. Int. J. Press. Vessels Pip. 79 (1), 77e86.
Choi, Y.S., Shim, J.J., Kim, J.G., 2005. Effects of Cr, Cu, Ni and Ca onthe corrosion behavior of low carbon steel in synthetic tapwater. J. Alloys Compd. 391 (1), 162e169.
Copeland, R.C., Lyte, D.A., Dionysiou, D.D., 2007. Desorption ofarsenic from drinking water distribution system solids.Environ. Monit. Assess. 127 (1e3), 523e535.
Cosham, A., Hopkins, P., Macdonald, K.A., 2007. Best practice forthe assessment of defects in pipelines e corrosion. Eng. Fail.Anal. 14 (7), 1245e1265.
Critchley, M.M., Pasetto, R., O‘Halloran, R.J., 2004. Microbiologicalinfluences in “blue water” copper corrosion. J. Appl. Microbiol.97 (3), 590e597.
D’Antonio, L., Fabbricino, M., Nasso, M., Trifuoggi, M., 2008.Copper release in low and high alkaline water. Environ.Technol. 29 (4), 473e478.
Dietrich, A.M., Glindemann, D., Pizarro, F., Gidi, V., Olivares, M.,Araya, M., Camper, A., Duncan, S., Dwyer, S., Whelton, A.J.,2004. Health and aesthetic impacts of copper corrosion ondrinking water. Water Sci. Technol. 49 (2), 55e62.
Edwards, M., 2004. Controlling corrosion in drinking waterdistribution systems: a grand challenge for the 21st century.Water Sci. Technol. 49 (2), 1e8.
Flitt, H.J., Schweinsberg, D.P., 2005. Evaluation of corrosion ratefrom polarisation curves not exhibiting a Tafel region. Corros.Sci. 47 (12), 3034e3052.
Frateur, I., Deslouis, C., Kiene, L., Levi, Y., Tribollet, B., 1999. Freechlorine consumption induced by cast iron corrosion in

wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6 145
drinking water distribution systems. Water Res. 33 (8),1781e1790.
G�enin, J.-M.R., Ruby, C., G�ehina, A., Refait, P., 2006. Synthesis ofgreen rusts by oxidation of Fe(OH)2, their products of oxidationand reduction of ferric oxyhydroxides; EhepH Pourbaixdiagrams. Comptes Rendues Geosci. 338 (6), 433e446.
Gerke, T.L., Scheckel, K.G., Maynard, J.B., 2010. Speciation anddistribution of vanadium in drinking water iron pipe corrosionby-products. Sci. Total Environ. 408 (23), 5845e5853.
Gonzalez, S., Lopez-Roldan,R.,Cortina, J.-L., 2013.Presenceofmetalsin drinking water distribution networks due to pipe materialleaching: a review. Toxicol. Environ. Chem. 95 (6), 870e889.
Hedberg, T., Johansson, E., 1987. Protection of pipes againstcorrosion. Water Supply 5 (3e4), SS20e21.
Imran, S.A., Dietz, J.D., Mutoti, G., Taylor, J.S., Randall, A.A., 2005.Modified Larsons ratio incorporating temperature, water age,and electroneutrality effects on red water release. J. Environ.Eng. 131 (11), 1514e1520.
Imran, S.A., Dietz, J.D., Mutoti, G., Xiao, W., Taylor, J.S., Desai, V.,2006. Optimizing source water blends for corrosion and residualcontrol in distribution systems. J. AWWA 98 (5), 107e115.
Ishii, S.K., Boyer, T.H., 2011. Evaluating the secondary effects ofmagnetic ion exchange: focus on corrosion potential in thedistribution system. Desalination 274 (1), 31e38.
Kim, E.J., Herrera, J.E., 2010. Characteristics of lead corrosionscales formed during drinking water distribution and theirpotential influence on the release of lead and othercontaminants. Environ. Sci. Technol. 44 (16), 6054e6061.
Kim, E.J., Herrera, J.E., Huggins, D., Braam, J., Koshowski, S., 2011.Effect of pH on the concentrations of lead and tracecontaminants in drinking water: a combined batch, pipe loopand sentinel home study. Water Res. 45 (9), 2763e2774.
Korshin, G.V., Ferguson, J.F., Lancaster, A.N., 2000. Influence ofnatural organic matter on the corrosion of leaded brass inpotable water. Corros. Sci. 42 (1), 53e66.
Kuch, A., 1988. Investigation of the reduction and re-oxidationkinetics of iron(III) oxide scales formed in waters. Corros. Sci.28 (3), 221e231.
Lasheen, M.R., Sharaby, C.M., El-Kholy, N.G., Elsherif, I.Y., El-Wakeel, S.T., 2008. Factors influencing lead and iron releasefrom some Egyptian drinking water pipes. J. Hazard. Mater.160 (2e3), 675e680.
Lee, A.K., Newman, D.K., 2003. Microbial iron respiration: impactson corrosion processes. Appl. Microbiol. Biotechnol. 62 (2e3),134e139.
Lehtola, M.J., Nissinen, T.K., Miettinen, I.T., Martikainen, P.J.,Vartiainen, T., 2004. Removal of soft deposits from thedistribution system improves the drinking water quality.Water Res. 38 (3), 601e610.
Li, S.-X., Yu, S.-R., Zeng, H.-L., Li, J.-H., Liang, R., 2009. Predictingcorrosion remaining life of underground pipelines with amechanically-based probabilistic model. J. Petrol. Sci. Eng. 65(3e4), 162e166.
Liang, J., Deng, A., Xie, R., Gomez, M., Hu, J., Zhang, J., Ong, C.N.,Adin, A., 2013. Impact of flow rate on corrosion of cast ironand quality of re-mineralized seawater reverse osmosis(SWRO) membrane product water. Desalination 322, 76e83.
Lin, J., Ellaway, M., Adrien, R., 2001. Study of corrosion materialaccumulated on the inner wall of steel water pipe. Corros. Sci.43 (11), 2065e2081.
Little, B.J., Mansfeld, F.B., Arps, P.J., Earthman, J.C., 2007.Microbiologically Influenced Corrosion. Wiley-VCH VerlagGmbH & Co. KGaA.
Liu, H., Schonberger, K.D., Peng, C.Y., Ferguson, J.F.,Desormeaux, E., Meyerhofer, P., Luckenback, H., Korshin, G.V.,2013. Effects of blending of desalinated and conventionallytreated surface water on iron corrosion and its release from
corroding surfaces and pre-existing scales. Water Res. 47 (11),3817e3826.
Mansfeld, F., 2005. Tafel slopes and corrosion rates obtained inthe pre-Tafel region of polarization curves. Corros. Sci. 47 (12),3178e3186.
McCafferty, E., 2005. Validation of corrosion rates measured bythe Tafel extrapolation method. Corros. Sci. 47 (12),3178e3186.
McNeill, L.S., Edwards, M., 2001. Review of iron pipe corrosion indrinking water distribution systems. J. AWWA 93 (7), 88e100.
Mora, A., Mac-Quhae, C., Calzadilla, M., S�anchez, L., 2009. Surveyof trace metals in drinking water supplied to rural populationsin the eastern Llanos of Venezuela. J. Environ. Manag. 90 (2),752e759.
Nawrocki, J., Raczyk-Stanislawiak, U., �Swietlik, J., Olejnik, A.,Sroka, M.J., 2010. Corrosion in a distribution system: steadywater and its composition. Water Res. 44 (6), 1863e1872.
Netto, T.A., Ferraz, U.S., Estefen, S.F., 2005. The effect of corrosiondefects on the burst pressure of pipelines. J. Constr. Steel Res.61 (8), 1185e1204.
Netto, T.A., Ferraz, U.S., Botto, A., 2007. On the effect of corrosiondefects on the collapse pressure of pipelines. Int. J. SolidsStruct. 44 (22e23), 7597e7614.
Nuhi, M., Seer, T.A., Al Tamimi, A.M., Modarres, M., Seibi, A., 2011.reliability analysis for degradation effects of pitting corrosionin carbon steel pipes. Procedia Eng. 10, 1930e1935.
Peng, C.-Y., Korshin, G.V., Valentine, R.L., Hill, A.S.,Friedman, M.J., Reiber, S.H., 2010. Characterization ofelemental and structural composition of corrosion scales anddeposits formed in drinking water distribution systems. WaterRes. 44 (15), 4570e4580.
Peng, C.-Y., Korshin, G.V., 2011. Speciation of trace inorganiccontaminants in corrosion scales and deposits formed indrinking water distribution systems. Water Res. 45 (17),5553e5563.
Peng, C.Y., Ferguson, J.F., Korshin, G.V., 2013. Effects of chloride,sulfate and natural organic matter (NOM) on the accumulationand release of trace-level inorganic contaminants fromcorroding iron. Water Res. 47 (14), 5257e5269.
Refait, P., G�ehin, A., Abdelmoula, M., G�enin, J.-M., 2003.Coprecipitation thermodynamics of iron(IIeIII)hydroxysulphate green rust from Fe(II) and Fe(III) salts. Corros.Sci. 45 (4), 659e676.
Rogers, H.R., Norris, M.W., James, H.A., 2004. Effects of materialsof construction on tastes and odours in drinking water. Rev.Environ. Sci. Biotechnol. 3 (1), 23e32.
Sancy, M., Gourbeyre, Y., Sutter, E.M.M., Tribollet, B., 2010.Mechanism of corrosion of cast iron covered by aged corrosionproducts: application of electrochemical impedancespectrometry. Corros. Sci. 52 (4), 1222e1227.
Sander, A., Berghult, B., Broo, A.E., Johansson, E.L., Hedberg, T.,1996. Iron corrosion in drinking water distributionsystemsdThe effect of pH, calcium and hydrogen carbonate.Corros. Sci. 38 (3), 443e455.
Sander, A., Berghult, B., Ahlberg, E., Broo, A.E., Johansson, E.L.,Hedberg, T., 1997. Iron corrosion in drinking water distributionsystems e surface complexation aspects. Corros. Sci. 39 (1),77e93.
Sarin, P., Snoeyink, V.L., Bebee, J., Kriven, W.M., Clement, J.A.,2001. Physico-chemical characteristics of corrosion scales inold iron pipes. Water Res. 35 (12), 2961e2969.
Sarin, P., Snoeyink, V.L., Lytle, D.A., Kriven, W.M., 2004. Ironcorrosion scales: model for scale growth, iron release, andcolored water formation. J. Environ. Eng. 130 (4), 364e373.
Shams El Din, A.M., 2009. Three strategies for combating thecorrosion of steel pipes carrying desalinated potable water.Desalination 238 (1), 166e173.

wat e r r e s e a r c h 6 2 ( 2 0 1 4 ) 1 3 6e1 4 6146
Shi, Z., Liu, M., Atrens, A., 2000. Measurement of the corrosionrate of magnesium alloys using Tafel extrapolation. Corros.Sci. 52 (2), 579e588.
Schock, M.R., Hyland, R.N., Welch, M.M., 2008. Occurrence ofcontaminant accumulation in lead pipe scales from domesticdrinking-water distribution systems. Environ. Sci. Technol. 42(12), 4285e4291.
�Swietlik, J., Raczyk-Stanislawiak, U., Piszora, P., Nawrocki, J., 2012.Corrosion in drinking water pipes: the importance of greenrusts. Water Res. 46 (1), 1e10.
Tang, Z., Hong, S., Xiao, W., Taylor, J., 2006. Impacts of blendingground, surface, and saline waters on lead release in drinkingwater distribution systems. Water Res. 40 (5), 943e950.
Teixeria, A.P., GuedesSoares, C., Netto, T.A., Estefen, S.F., 2008.Reliability of pipelines withcorrosion defects. Int. J. Press.Vessels Pip. 85 (4), 228e237.
Veleva, L., Castro, P., Hernandez-Duque, G., Schorr, M., 1998. Thecorrosion performance of steel and reinforced concrete in atropical humid climate. A review. Corros. Rev. 16 (3), 235e284.
Volk, C., Dundore, E., Schiermann, J., LeChevallier, M., 2000.Practical evaluation of iron corrosion control in a drinkingwater distribution system. Water Res. 34 (6), 1967e1974.
Yang, F., Shi, B., Gu, J., Wang, D., Yang, M., 2012. Morphologicaland physicochemical characteristics of iron corrosion scalesformed under different water source histories in a drinkingwater distribution system. Water Res. 46 (16), 5423e5433.
Zhang, X.L., Jiang, ZhH., Yao, ZhP., Song, Y., Wu, ZhD., 2009.Effects of scan rate on the potentiodynamic polarization curveobtained to determine the Tafel slopes and corrosion currentdensity. Corros. Sci. 51 (3), 581e587.
Zietz, B., Dassel de Vergara, J., Kevekordes, S., Dunkelberg, H., 2001.Lead contamination in tap water of households with children inLower Saxony, Germany. Sci. Total Environ. 275 (1), 19e26.
Zietz, B.P., Dieter, H.H., Lakomek, M., Schneider, H., Keßler-Gaedtke, B., Dunkelberg, H., 2003. Epidemiologicalinvestigation on chronic copper toxicity to children exposedvia the public drinking water supply. Sci. Total Environ. 302(1), 127e144.