
Cerebral spinal fluid dynamics: effect of hypoxia and ...
12
HIGHLIGHTED TOPIC Hypoxia 2015 Cerebral spinal fluid dynamics: effect of hypoxia and implications for high-altitude illness X Justin S. Lawley, 1,2 Benjamin D. Levine, 1,2 Michael A. Williams, 3 Jon Malm, 4 Anders Eklund, 5 David M. Polaner, 6 Andrew W. Subudhi, 7,8 Peter H. Hackett, 9 and Robert C. Roach 8 1 Institute for Exercise and Environmental Medicine, Presbyterian Hospital of Dallas, Dallas, Texas; 2 UT Southwestern Medical Center, Dallas, Texas; 3 Sandra and Malcolm Berman Brain & Spine Institute, Dept. of Neurology, Sinai Hospital, Baltimore, Maryland; 4 Department of Clinical Neuroscience, Umeå University, Umeå, Sweden; 5 Department of Radiation Sciences, Umeå University, Umeå, Sweden; 6 Departments of Anesthesiology and Pediatrics, University of Colorado School of Medicine and Children’s Hospital Colorado, Aurora, Colorado; 7 Department of Biology, University of Colorado, Colorado Springs, Colorado; 8 Altitude Research Center, Department of Emergency Medicine, University of Colorado Anschutz Medical Campus, Aurora, Colorado; and 9 Institute for Altitude Medicine, Telluride, Colorado Submitted 7 May 2015; accepted in final form 17 August 2015 Lawley JS, Levine BD, Williams MA, Malm J, Eklund A, Polaner DM, Subudhi AW, Hackett PH, Roach RC. Cerebral spinal fluid dynamics: effect of hypoxia and implications for high-altitude illness. J Appl Physiol 120: 251–262, 2016. First published October 22, 2015; doi:10.1152/japplphysiol.00370.2015.— The pathophysiology of acute mountain sickness and high-altitude cerebral edema, the cerebral forms of high-altitude illness, remain uncertain and controversial. Persistently elevated or pathological fluctuations in intracranial pressure are thought to cause symptoms similar to those reported by individuals suffering cerebral forms of high-altitude illness. This review first focuses on the basic physiology of the craniospinal system, including a detailed discussion of the long-term and dynamic regulation of intracranial pressure. Thereafter, we critically examine the available literature, based primarily on invasive pressure monitoring, that suggests intracranial pressure is acutely elevated at altitude due to brain swelling and/or elevated sagittal sinus pressure, but normalizes over time. We hypothesize that fluctuations in intracranial pressure occur around a slightly elevated or normal mean intracranial pressure, in conjunction with oscillations in arterial PO 2 and arterial blood pressure. Then these modest fluctuations in intracra- nial pressure, in concert with direct vascular stretch due to dilatation and/or increased blood pressure transmission, activate the trigeminal vascular system and cause symptoms of acute mountain sickness. Elevated brain water (vasogenic edema) may be due to breakdown of the blood-brain barrier. However, new information suggests cerebral spinal fluid flux into the brain may be an important factor. Regardless of the source (or mechanisms responsible) for the excess brain water, brain swelling occurs, and a “tight fit” brain would be a major risk factor to produce symptoms; activities that produce large changes in brain volume and cause fluctuations in blood pressure are likely contributing factors. acute mountain sickness; high-altitude cerebral edema; headache and intracranial pressure ACUTE MOUNTAIN SICKNESS (AMS) is characterized by headache, fatigue, dizziness, and nausea and vomiting, whereas hallmarks of high-altitude cerebral edema (HACE) are ataxic gait, severe lassitude, and altered consciousness, including confusion and impaired mentation. AMS typically occurs when the onset of hypoxemia is rapid and severe i.e., 4,500 m or oxygen saturation 80%. Under these circumstances, mild symptoms such as headache occur in most individuals (15), with 40-65% of headache suffers being diagnosed with AMS, but astonish- ingly some remain completely symptom free. In contrast, HACE is rare (0.5–1.8%), even with rapid ascent to altitudes 4,000 m (11, 31). Despite decades of investigation, the pathophysiology of AMS and HACE remains uncertain and controversial. An enticing hypothesis of the cause of AMS and HACE is that persons with smaller intracranial and intraspinal cerebral Address for reprint requests and other correspondence: J. S. Lawley, Dept. of Internal Medicine, Univ. of Texas Southwestern Medical Center, Institute for Exercise and Environmental Medicine, Texas Health Presby- terian Hospital Dallas, 7232 Greenville Ave., Ste. 435, Dallas, TX 75231 (e-mail: [email protected]). J Appl Physiol 120: 251–262, 2016. First published October 22, 2015; doi:10.1152/japplphysiol.00370.2015. Review 8750-7587/16 Copyright © 2016 the American Physiological Society http://www.jappl.org 251
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Cerebral spinal fluid dynamics: effect of hypoxia and ...

HIGHLIGHTED TOPIC Hypoxia 2015
Cerebral spinal fluid dynamics: effect of hypoxia and implications forhigh-altitude illness
X Justin S. Lawley,1,2 Benjamin D. Levine,1,2 Michael A. Williams,3 Jon Malm,4 Anders Eklund,5
David M. Polaner,6 Andrew W. Subudhi,7,8 Peter H. Hackett,9 and Robert C. Roach8
1Institute for Exercise and Environmental Medicine, Presbyterian Hospital of Dallas, Dallas, Texas; 2UT SouthwesternMedical Center, Dallas, Texas; 3Sandra and Malcolm Berman Brain & Spine Institute, Dept. of Neurology, Sinai Hospital,Baltimore, Maryland; 4Department of Clinical Neuroscience, Umeå University, Umeå, Sweden; 5Department of RadiationSciences, Umeå University, Umeå, Sweden; 6Departments of Anesthesiology and Pediatrics, University of Colorado School ofMedicine and Children’s Hospital Colorado, Aurora, Colorado; 7Department of Biology, University of Colorado, ColoradoSprings, Colorado; 8Altitude Research Center, Department of Emergency Medicine, University of Colorado Anschutz MedicalCampus, Aurora, Colorado; and 9Institute for Altitude Medicine, Telluride, Colorado
Submitted 7 May 2015; accepted in final form 17 August 2015
Lawley JS, Levine BD, Williams MA, Malm J, Eklund A, Polaner DM,Subudhi AW, Hackett PH, Roach RC. Cerebral spinal fluid dynamics: effect ofhypoxia and implications for high-altitude illness. J Appl Physiol 120: 251–262,2016. First published October 22, 2015; doi:10.1152/japplphysiol.00370.2015.—The pathophysiology of acute mountain sickness and high-altitude cerebral edema,the cerebral forms of high-altitude illness, remain uncertain and controversial.Persistently elevated or pathological fluctuations in intracranial pressure arethought to cause symptoms similar to those reported by individuals sufferingcerebral forms of high-altitude illness. This review first focuses on the basicphysiology of the craniospinal system, including a detailed discussion of thelong-term and dynamic regulation of intracranial pressure. Thereafter, we criticallyexamine the available literature, based primarily on invasive pressure monitoring,that suggests intracranial pressure is acutely elevated at altitude due to brainswelling and/or elevated sagittal sinus pressure, but normalizes over time. Wehypothesize that fluctuations in intracranial pressure occur around a slightlyelevated or normal mean intracranial pressure, in conjunction with oscillations inarterial PO2 and arterial blood pressure. Then these modest fluctuations in intracra-nial pressure, in concert with direct vascular stretch due to dilatation and/orincreased blood pressure transmission, activate the trigeminal vascular system andcause symptoms of acute mountain sickness. Elevated brain water (vasogenicedema) may be due to breakdown of the blood-brain barrier. However, newinformation suggests cerebral spinal fluid flux into the brain may be an importantfactor. Regardless of the source (or mechanisms responsible) for the excess brainwater, brain swelling occurs, and a “tight fit” brain would be a major risk factor toproduce symptoms; activities that produce large changes in brain volume and causefluctuations in blood pressure are likely contributing factors.
acute mountain sickness; high-altitude cerebral edema; headache and intracranialpressure
ACUTE MOUNTAIN SICKNESS (AMS) is characterized by headache,fatigue, dizziness, and nausea and vomiting, whereas hallmarksof high-altitude cerebral edema (HACE) are ataxic gait, severelassitude, and altered consciousness, including confusion andimpaired mentation. AMS typically occurs when the onset of
hypoxemia is rapid and severe i.e., �4,500 m or oxygensaturation �80%. Under these circumstances, mild symptomssuch as headache occur in most individuals (15), with 40-65%of headache suffers being diagnosed with AMS, but astonish-ingly some remain completely symptom free. In contrast,HACE is rare (0.5–1.8%), even with rapid ascent to altitudes�4,000 m (11, 31). Despite decades of investigation, thepathophysiology of AMS and HACE remains uncertain andcontroversial.
An enticing hypothesis of the cause of AMS and HACE isthat persons with smaller intracranial and intraspinal cerebral
Address for reprint requests and other correspondence: J. S. Lawley,Dept. of Internal Medicine, Univ. of Texas Southwestern Medical Center,Institute for Exercise and Environmental Medicine, Texas Health Presby-terian Hospital Dallas, 7232 Greenville Ave., Ste. 435, Dallas, TX 75231(e-mail: [email protected]).
J Appl Physiol 120: 251–262, 2016.First published October 22, 2015; doi:10.1152/japplphysiol.00370.2015. Review
8750-7587/16 Copyright © 2016 the American Physiological Societyhttp://www.jappl.org 251

spinal fluid (CSF) capacity, a “tight fit” brain, would bedisposed to develop high-altitude illness because they wouldnot tolerate brain swelling as well as those with more “room”in the craniospinal axis (74). In our opinion, this hypothesis hasnot received adequate investigation. To reinvigorate scientificenthusiasm for this hypothesis and suggest future researchdirections, this review will 1) summarize the basic physiologyof the craniospinal system focusing on the regulatory mecha-nisms that control intracranial volume and pressure homeosta-sis over short and long periods of time; 2) evaluate the effectof hypoxia on brain swelling and CSF dynamics, includingintracranial pressure (ICP); and 3) link these observations toAMS and HACE.
PHYSIOLOGY OF THE CRANIOSPINAL SYSTEM
CSF secretion and reabsorption. CSF is secreted predomi-nantly by the choroid plexus at a rate of �0.4 ml/min (24), whichaccounts for a total daily production of �600 ml in the adult. Giventhat the total volume of CSF, located cranially (�80%) and spinally(�20%), equals only �300 ml (39), a constant and equivalent rate ofCSF reabsorption (CSFreabsorption) is required to maintain volumeequilibrium and thus ICP.
Regulation of CSF secretion is highly complex and modu-lated indirectly by factors such as choroid plexus blood flow[change (�) in substrate delivery], ventilation (�PCO2), atrialnatriuretic peptide release, and carbonic anhydrase activity(19). CSF outflow (CSFoutflow or CSFreabsorption) is predomi-nantly determined by the difference between ICP and sagittalsinus pressure (Psag) relative to the resistance of CSF move-ment from its site of production to its site of absorption (Rout,Davson equation, Eq. 1). Put simply, ICP must exceed Psag forCSFoutflow to occur. Rout is the sum of total resistance ofCSFoutflow of the system, most importantly at the site of thearachnoid granulations. Nonetheless, the intracranial-venouspressure gradient dominates CSF volume homeostasis overprolonged periods of time, as Rout has been shown to remainstable within the same individual, despite modest elevations inICP from baseline (3).
CSFoutflow�ml ⁄ min� � �ICP�mmHg�� Psag�mmHg�� ⁄ Rout�mmHg.ml.min-1� (1)
The primary route of CSFreabsorption under steady-state su-pine conditions is through the arachnoid granulations into thesagittal sinus (see Fig. 1H) (20). Simple provocations thatobstruct venous outflow, such as jugular venous compressionand head turning, provide empirical support for this proposi-tion; elevated cerebral venous blood volume and Psag cause areduction in the transmural pressure gradient (ICP � Psag) and
increased ICP (41, 70). Other routes of CSFreabsorption exist,including arachnoid villi coursing along spinal nerve roots[draining into the spinal venous plexus (22, 50)] and extracra-nial lymphatic vessels (47, 89) into the perivascular space (29).These alternate CSFoutflow pathways may become importantunder pathological conditions and even during normal dailyactivities. Indeed, data suggest that as much as �60% of spinalCSFoutflow is through spinal roots when standing upright, com-pared with only �20% when supine (22) (see Fig. 1). Further-more, animal studies suggest that CSF-interstitial flux (gli-olymphatic drainage; see Fig. 1I) may be predominantly dor-mant during waking hours, but substantial while asleep (96).
ICP. Contents of the craniospinal compartment includebrain, spinal cord, arterial and venous blood, and CSF. Understeady-state resting conditions, brain, spinal cord, and bloodvolumes are essentially constant. Therefore, solving Eq. 1 forICP highlights that the mean static ICP operating point isgoverned by the rate of CSF production, Rout and Psag. As CSFproduction equals CSFoutflow in the steady-state condition, ICPtypically reflects changes in Psag over prolonged periods.
A typical supine resting ICP in healthy young individuals is�11 mmHg. However, there is considerable interindividualvariability in the “normal” ICP, ranging from 7 to 15 mmHg(24, 60). Moreover, hydrostatic pressure gradients affect ICP,whereby ICP with reference to the head is progressivelylowered with increasing upright tilt angles (72) and typically�5 to �10 mmHg (4, 5, 25) while in the upright posture. Toobtain accurate measurements, clinical and/or experimentalcontrol is essential, as simple maneuvers that affect Psag in thesupine posture, such as simply elevating the head or legs on apillow, can either reduce (�5 mmHg; Ref. 26) or increase (�3mmHg) ICP (personal observations).
Around the static operating point, ICP varies with changes inperipheral and central vasomotor tone, respiration, and in-trathoracic pressure and with the cardiac cycle (36, 71, 86) (seeFig. 1G). Each contraction of the left cardiac ventricle pro-duces a small (1.5 to 2.0 ml; Ref. 85) increase in arterialcerebral blood volume (see Fig. 1F), expansion of the brainparenchyma, a rise in ICP and translocation of CSF though theforamen magnum into the spinal thecal sac (87). Duringdiastole the reverse is observed, including declining ICP and areversal of CSF flow into the cranium (see Fig. 1F).
Intracranial compartment volumes. In the context of thepresent review, two intracranial compartments are of interestfor alterations in CSF dynamics: cerebral blood volume andwater content. Two likely causes of elevated cerebral arterialand venous blood volume are active dilation or passive disten-sion. Venous volume may also increase due to restricted
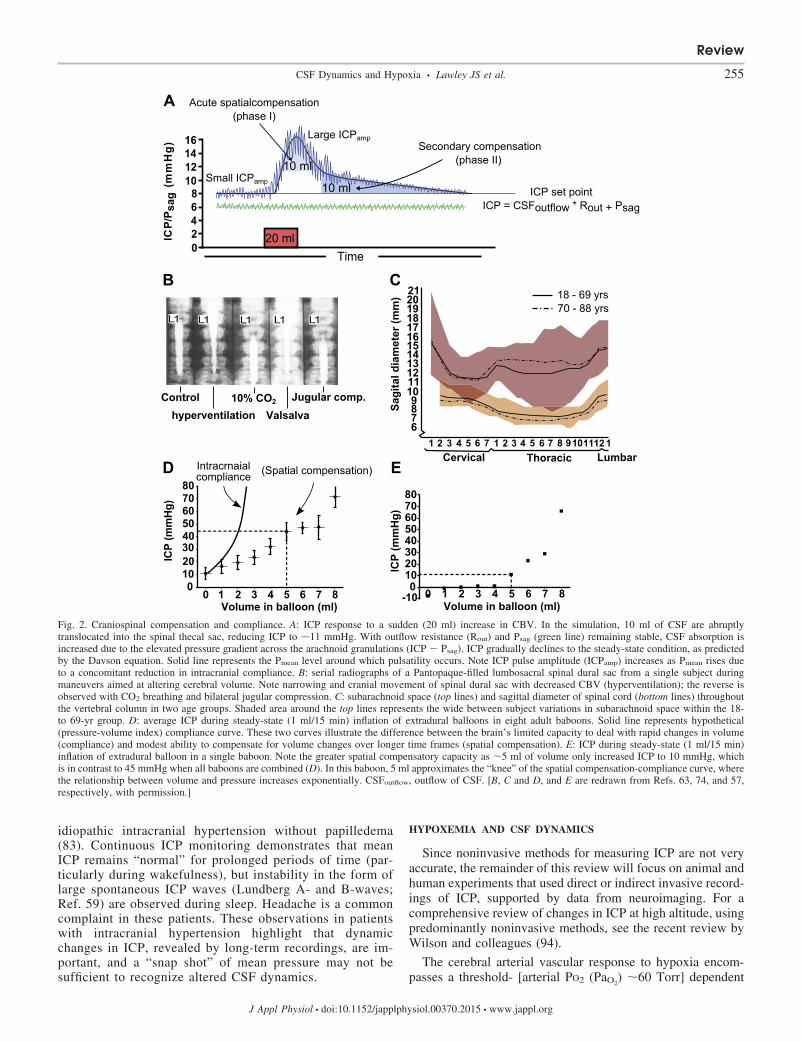
Fig. 1. Cerebral spinal fluid (CSF) dynamics. Blood flow over a cardiac cycle within the internal carotid and vertebral arteries (A), sagittal sinus (B), and jugularveins (C) is shown. To and fro flushing of CSF through the Sylvius aqueduct (D) and spinal canal at C2–C3 (E) is shown. F: pulsatile arterial flow (internal andvertebral arteries) over the cardiac cycle leads to a small (1.5–2.0 ml) increase in cerebral blood volume (CBV). G: intracranial pressure (ICP) pulsatility (cardiacpulsations) represents the contribution of change in CBV and craniospinal compliance, whereas respiratory oscillations are due to changes in central venouspressure (CVP) and sagittal sinus pressure (Psag). H: CSF absorption. Primary site of CSF absorption through arachnoid granulations into the sagittal sinus, whichis driven by the ICP-Psag pressure gradient. I: glymphatic system. Interstitial solute and fluid clearance from the brain is facilitated by para-arterial influx of CSFand paravenous clearance. Convective bulk interstitial fluid flow is facilitated via aquaporin-4 (AQP-4) studded on astrocyte end-feet. J: greater spinal fluidabsorption in the active (1-min walking) upright compared with resting supine posture (left), measured by the radionuclide clearance from sacral/lumbar tothoracic/cervical level immediately (0) and 20, 40, and 60 min after injection (right). a–e, Lowercase letters identify the location of blood or CSF flow in A–E,respectively. ECG, electrocardiogram; BP, arterial blood pressure; �V, change in volume; CBVmean, mean CBV; Pmax, Pmin, and Pmean: maximum, minimum,and mean pressure, respectively. [J and G are redrawn from Refs. 22, and 36, respectively, with permission.]
Review
252 CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org

venous outflow. Importantly, this is not due to a mismatchbetween blood inflow and outflow: if inflow exceeds outflowby even 0.1% (0.7 ml/min, assuming a total inflow of 700ml/min), ICP would increase uncontrollably, and death wouldfollow. Instead, focal narrowing of the intracranial transverse
sinus causes elevated venous pressure proximal to the stenosis;Psag and thus ICP are proposed to rise in proportion to thedegree of stenosis and increased resistance, if the sinus werethe only outflow pathway. This mechanism has been advocatedin patients with idiopathic intracranial hypertension, but a
Review
253CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org

number of questions remain, including 1) identifying causationbetween elevated ICP and venous stenosis; and 2) explainingthe lack of collateral venous drainage that is effective in theupright posture. Finally, increased brain water in the form ofvasogenic brain edema causes increased intracranial volume,but fluid shifts between compartments do not.
Craniospinal axis compensation and compliance. The brainparenchyma, spinal cord, arterial and venous blood, and CSFare encased within a rigid container (skull and vertebral col-umn). Intracranially, the meninges (dura mater, arachnoidspace, and pia mater) separate the brain from the inner surfaceof the skull. The meninges also separate the spinal cord fromthe vertebral column, although the lumbar dura and thecal sacare less restrained as the dura mater is separated from thevertebral lining by fatty tissue and a venous plexus (63). CSFforms a freely communicating fluid column between the cra-nium and spinal thecal sac. Thus craniospinal axis compensa-tion (see below) and compliance include the interaction be-tween the physical (anatomical) and physiological (hemo- andhydrodynamic) properties of the cranial vault and vertebralcolumn (58, 62, 82). The ability of the craniospinal system toaccommodate changes in volume can be divided into spatialcompensation and craniospinal compliance (�volume/�pressure) (7).
Spatial compensation represents the degree to which volumeincrements can be accommodated by compensatory reductionsin the volume of other intracranial compartments. First, inter-nal spatial compensation represents the translocation of CSFfrom the intracranial to the spinal thecal sac. Internal spatialcompensation is determined by the magnitude of the internalpressure gradient between the cranium and spinal thecal sacand the compliance of the spinal dura or more likely the spinalepidural venous plexus. As such, the spinal dura sac acts as areservoir for changing cerebral volume with a finite ability tomaintain normal ICP (Ref. 63; see Fig. 2B). The secondcomponent is spatial compensation via quasi-static reductionsin craniospinal CSF volume through increased CSFreabsorption
(CSFoutflow). As long as Psag remains normal, this compensa-tion will occur over time, whereby ICP will gradually return tothe steady-state condition, as predicted by CSF formation rate,Rout (see Fig. 2A). The spinal subarachnoid CSF volume, andthus the potential spinal compensatory capacity, varies consid-erably between individuals (Fig. 2C). Once spatial compensa-tion is exhausted, further increases in volume result in sus-tained intracranial hypertension. The capacity of craniospinalspatial compensation is dominated by CSF volume, whereasthe time constant (delay) is determined by the increase in brainvolume, the shape of the compliance curve, the ICP-Psag
gradient, and Rout. The variability of spatial compensation canbe appreciated by examination of Fig. 2. Figure 2D shows themean response of ICP to slow inflation of an extradural balloonin a group of nonhuman primates, whereby 5 ml of volumeincreases ICP to 45 mmHg. In contrast, in a single nonhumanprimate, 5 ml of volume only increased ICP to 10 mmHg (Fig.2E) (57).
Craniospinal compliance typically reflects the elastic prop-erties of the craniospinal system and reflects the acute volume-pressure (�volume/�pressure) relationship at any specificpoint in time. It is worth noting that the pressure-volume index(volume required to increase ICP by a factor of 10) exemplifiesthe finite craniospinal compliance; 25 ml would increase ICP
from 10 to 100 mmHg, if not for spatial compensatory mech-anisms. As noted above, small volume increments occur duringeach heartbeat and give rise to the CSF pulse pressure wave-form. This phenomenon is synonymous with continuous, smallvolume-pressure tests, whereby the ICP pulse amplitude in-creases linearly with increasing ICP (reduced compliance, seeFig. 2A) above an individual’s specific ICP threshold point(71), therein reflecting the compliance of the craniospinalsystem. Craniospinal axis compliance and thus “bufferingcapacity” dictate the slope of the volume-pressure curve andthus the rate of rise in ICP. This capacity depends on theoverall biomechanical properties of the cranium and spinalcanal (61, 62), including the rigidity of the brain, spinal cord,meninges, spinal dura, and the tone and collapsibility of thevascular bed, specifically the low-capacitance venous seg-ments.
Impact of reduced spatial compensation and craniospinalcompliance. As volume is added to the intracranial space, fourstages can be identified. Stage 1 is initial spatial compensation,whereby ICP rises very modestly, or not at all, despite addedvolume. As spatial compensation becomes exhausted and thespinal intradural volume reaches its capacity, free movement ofCSF is hindered, and its pulse dampening (windkessel) effectreduced, and, as a result, intracranial pulse transmission isenhanced. In stage 2, spatial compensation is exhausted, andpatients suffer headache and drowsiness. Spontaneous patho-logical oscillations in arterial blood pressure and ICP Lundbergpressure waves (59) are observed, especially with the additionof hypercapnia or hypoxia. At this stage, further increments involume lead to exponential elevations in ICP. In stage 3,autoregulatory capacity is absent, and cerebral vasomotor pa-ralysis occurs with a flattening of the volume-pressure curveand stabilization of high ICP. At this stage, patients experiencefleeting episodes of altered consciousness, and fluctuations inarterial blood pressure cause reciprocal changes in ICP. Instage 4, the patient is unconscious, intracranial hypertension isirreversible, arterial and cerebral perfusion pressure fall, anddeath follows (54).
What constitutes altered CSF dynamics? A central questionis exactly when do altered CSF dynamics cause pathologicalsymptoms? Clinical guidelines regard steady-state supine ICPto be elevated when it rises above �20–25 mmHg (27). Giventhe wide interindividual variation in resting steady-state supineICP (�7–15 mmHg), it seems unlikely that a single absolutemean pressure adequately describes pathologically elevatedICP in all individuals. In contrast to mean pressure, increasesin the ICP pulse amplitude may be important. In the supineposition, intracranial pulsatility increases linearly beyond anindividual’s ICP operating point (71), usually close to thepatient’s normal ICP at rest. Therefore, an increase in meanICP from 10 to 20 mmHg in one individual would result intwice the amplitude gain, and theoretically increased pathol-ogy, compared with another individual whose ICP rose from15 to 20 mmHg. Interestingly, patients diagnosed withidiopathic intracranial hypertension, visual disturbances,and headache often present with elevated intracranial pul-satility, despite over 50% having normal mean ICP duringwaking hours (23).
Intracranial instability and the occurrence of pressurewaves are also of pathological concern. Perhaps the bestillustration of such a phenomena occurs in patients with
Review
254 CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org
idiopathic intracranial hypertension without papilledema(83). Continuous ICP monitoring demonstrates that meanICP remains “normal” for prolonged periods of time (par-ticularly during wakefulness), but instability in the form oflarge spontaneous ICP waves (Lundberg A- and B-waves;Ref. 59) are observed during sleep. Headache is a commoncomplaint in these patients. These observations in patientswith intracranial hypertension highlight that dynamicchanges in ICP, revealed by long-term recordings, are im-portant, and a “snap shot” of mean pressure may not besufficient to recognize altered CSF dynamics.
HYPOXEMIA AND CSF DYNAMICS
Since noninvasive methods for measuring ICP are not veryaccurate, the remainder of this review will focus on animal andhuman experiments that used direct or indirect invasive record-ings of ICP, supported by data from neuroimaging. For acomprehensive review of changes in ICP at high altitude, usingpredominantly noninvasive methods, see the recent review byWilson and colleagues (94).
The cerebral arterial vascular response to hypoxia encom-passes a threshold- [arterial PO2 (PaO2
) �60 Torr] dependent
Fig. 2. Craniospinal compensation and compliance. A: ICP response to a sudden (20 ml) increase in CBV. In the simulation, 10 ml of CSF are abruptlytranslocated into the spinal thecal sac, reducing ICP to �11 mmHg. With outflow resistance (Rout) and Psag (green line) remaining stable, CSF absorption isincreased due to the elevated pressure gradient across the arachnoid granulations (ICP � Psag). ICP gradually declines to the steady-state condition, as predictedby the Davson equation. Solid line represents the Pmean level around which pulsatility occurs. Note ICP pulse amplitude (ICPamp) increases as Pmean rises dueto a concomitant reduction in intracranial compliance. B: serial radiographs of a Pantopaque-filled lumbosacral spinal dural sac from a single subject duringmaneuvers aimed at altering cerebral volume. Note narrowing and cranial movement of spinal dural sac with decreased CBV (hyperventilation); the reverse isobserved with CO2 breathing and bilateral jugular compression. C: subarachnoid space (top lines) and sagittal diameter of spinal cord (bottom lines) throughoutthe vertebral column in two age groups. Shaded area around the top lines represents the wide between subject variations in subarachnoid space within the 18-to 69-yr group. D: average ICP during steady-state (1 ml/15 min) inflation of extradural balloons in eight adult baboons. Solid line represents hypothetical(pressure-volume index) compliance curve. These two curves illustrate the difference between the brain’s limited capacity to deal with rapid changes in volume(compliance) and modest ability to compensate for volume changes over longer time frames (spatial compensation). E: ICP during steady-state (1 ml/15 min)inflation of extradural balloon in a single baboon. Note the greater spatial compensatory capacity as �5 ml of volume only increased ICP to 10 mmHg, whichis in contrast to 45 mmHg when all baboons are combined (D). In this baboon, 5 ml approximates the “knee” of the spatial compensation-compliance curve, wherethe relationship between volume and pressure increases exponentially. CSFoutflow, outflow of CSF. [B, C and D, and E are redrawn from Refs. 63, 74, and 57,respectively, with permission.]
Review
255CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org
increase in cerebral blood flow and pial artery and arteriolardilatation (i.e., a reduction in cerebrovascular resistance andincreased arterial blood volume; Refs. 6, 17, 48, 77, 90).Venous capacitance vessels will also dilate (55, 88, 91), thusalso increasing the cerebral venous blood volume pool.
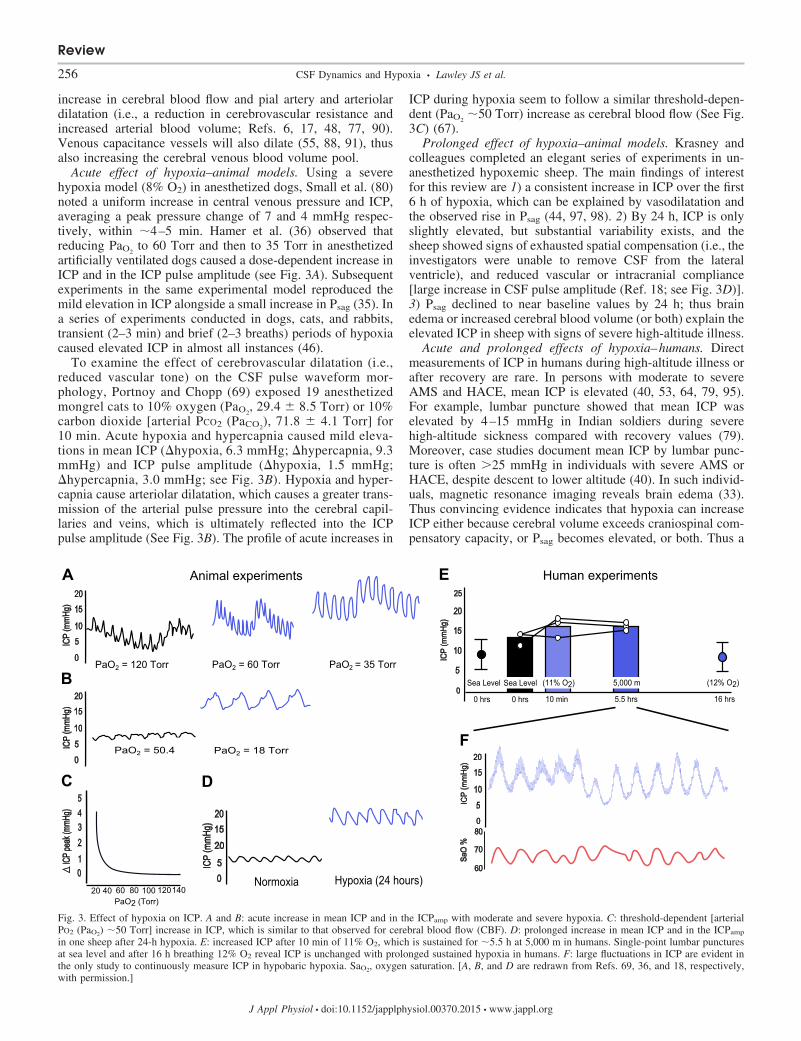
Acute effect of hypoxia–animal models. Using a severehypoxia model (8% O2) in anesthetized dogs, Small et al. (80)noted a uniform increase in central venous pressure and ICP,averaging a peak pressure change of 7 and 4 mmHg respec-tively, within �4–5 min. Hamer et al. (36) observed thatreducing PaO2
to 60 Torr and then to 35 Torr in anesthetizedartificially ventilated dogs caused a dose-dependent increase inICP and in the ICP pulse amplitude (see Fig. 3A). Subsequentexperiments in the same experimental model reproduced themild elevation in ICP alongside a small increase in Psag (35). Ina series of experiments conducted in dogs, cats, and rabbits,transient (2–3 min) and brief (2–3 breaths) periods of hypoxiacaused elevated ICP in almost all instances (46).
To examine the effect of cerebrovascular dilatation (i.e.,reduced vascular tone) on the CSF pulse waveform mor-phology, Portnoy and Chopp (69) exposed 19 anesthetizedmongrel cats to 10% oxygen (PaO2
, 29.4 � 8.5 Torr) or 10%carbon dioxide [arterial PCO2 (PaCO2
), 71.8 � 4.1 Torr] for10 min. Acute hypoxia and hypercapnia caused mild eleva-tions in mean ICP (�hypoxia, 6.3 mmHg; �hypercapnia, 9.3mmHg) and ICP pulse amplitude (�hypoxia, 1.5 mmHg;�hypercapnia, 3.0 mmHg; see Fig. 3B). Hypoxia and hyper-capnia cause arteriolar dilatation, which causes a greater trans-mission of the arterial pulse pressure into the cerebral capil-laries and veins, which is ultimately reflected into the ICPpulse amplitude (See Fig. 3B). The profile of acute increases in
ICP during hypoxia seem to follow a similar threshold-depen-dent (PaO2
�50 Torr) increase as cerebral blood flow (See Fig.3C) (67).
Prolonged effect of hypoxia–animal models. Krasney andcolleagues completed an elegant series of experiments in un-anesthetized hypoxemic sheep. The main findings of interestfor this review are 1) a consistent increase in ICP over the first6 h of hypoxia, which can be explained by vasodilatation andthe observed rise in Psag (44, 97, 98). 2) By 24 h, ICP is onlyslightly elevated, but substantial variability exists, and thesheep showed signs of exhausted spatial compensation (i.e., theinvestigators were unable to remove CSF from the lateralventricle), and reduced vascular or intracranial compliance[large increase in CSF pulse amplitude (Ref. 18; see Fig. 3D)].3) Psag declined to near baseline values by 24 h; thus brainedema or increased cerebral blood volume (or both) explain theelevated ICP in sheep with signs of severe high-altitude illness.
Acute and prolonged effects of hypoxia–humans. Directmeasurements of ICP in humans during high-altitude illness orafter recovery are rare. In persons with moderate to severeAMS and HACE, mean ICP is elevated (40, 53, 64, 79, 95).For example, lumbar puncture showed that mean ICP waselevated by 4–15 mmHg in Indian soldiers during severehigh-altitude sickness compared with recovery values (79).Moreover, case studies document mean ICP by lumbar punc-ture is often �25 mmHg in individuals with severe AMS orHACE, despite descent to lower altitude (40). In such individ-uals, magnetic resonance imaging reveals brain edema (33).Thus convincing evidence indicates that hypoxia can increaseICP either because cerebral volume exceeds craniospinal com-pensatory capacity, or Psag becomes elevated, or both. Thus a
Fig. 3. Effect of hypoxia on ICP. A and B: acute increase in mean ICP and in the ICPamp with moderate and severe hypoxia. C: threshold-dependent [arterialPO2 (PaO2) �50 Torr] increase in ICP, which is similar to that observed for cerebral blood flow (CBF). D: prolonged increase in mean ICP and in the ICPamp
in one sheep after 24-h hypoxia. E: increased ICP after 10 min of 11% O2, which is sustained for �5.5 h at 5,000 m in humans. Single-point lumbar puncturesat sea level and after 16 h breathing 12% O2 reveal ICP is unchanged with prolonged sustained hypoxia in humans. F: large fluctuations in ICP are evident inthe only study to continuously measure ICP in hypobaric hypoxia. SaO2, oxygen saturation. [A, B, and D are redrawn from Refs. 69, 36, and 18, respectively,with permission.]
Review
256 CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org

limited compensatory capacity “tight fit” (74) is a rationalexplanation for a predisposition to severe AMS and HACE.
In a well-designed series of experiments, Schaltenbrand (75)performed continuous measurements of ICP by lumbar punc-ture during acute reductions in atmospheric pressure. Althoughthe threshold altitude varied between patients, CSF pressurerose consistently, especially above 3,000 m. In one subjectexposed to 4,500 m for �10 min, ICP rose from 11 to 20mmHg, gradually decreasing to settle at 18 mmHg. Adminis-tration of oxygen aborted the rise in ICP or restored ICP tonormal in most cases. Hartig and Hackett (37) conducted apilot study with three subjects whereby ICP was measured vialumbar catheter. Subjects breathed hypoxic gas acutely (11%O2, 10 min) and were exposed to a hypobaric altitude of 5,000m for around 6 h. ICP increased in two of the three subjectsduring acute hypoxic gas breathing (see Fig. 3E) and remainedelevated after 6 h (see Fig. 3E). Over this time period, two ofthe three subjects reported moderate headache, but only onehad slightly elevated mean ICP. Similar to the data obtained inunanesthetized sheep (18, 97), lumbar punctures obtained inhealthy volunteers before and after 16-h exposure to simulatedaltitude of 4,500 m documented no change in mean ICP (10)(see Fig. 3E). Perhaps the greatest endeavor to measure ICPduring gradual exposure to hypoxia (similar to trekking withpartial acclimatization) was undertaken by Dr. Brian Cummins,a British neurosurgeon (93). Transdural pressure was measuredby implanting invasive telemetric monitoring devices into threeindividuals, including himself. At 5,029 m in the resting supineposition, ICP was normal in one individual and rose by �7mmHg and �5 mmHg in the other two. The same investigationalso noted that individuals with the smallest intracranial ven-tricles at sea level, and thus intraventricular CSF volume,suffered the worst headache (93).
Up to this point, we have discussed either spot measure-ments of ICP or continuous data reported as mean values.Given the reduction in vascular tone and in CSF volume, anda slight increase in mean resting ICP, craniospinal dynamicsare likely altered in many individuals at high altitude. Indeed,in the only study to continuously measure ICP, Hartig andHackett (37) noted that ICP became markedly elevated duringexertion and spontaneous periodic breathing while awake. Oneof their subjects with periodic breathing demonstrated a re-markable threefold increase in CSF pressure from 10 to 30mmHg in phase with the nadir of the oscillating oxygensaturation (See Fig. 3F). Furthermore, in these subjects, hy-poxic gas breathing produced a greater increase in ICP at highaltitude than sea level. No other investigations have performedprolonged continuous ICP monitoring in hypoxic subjects.
These invasive data are generally consistent with mostneuroimaging studies. Using high-resolution magnetic reso-nance imagining, Dubowitz et al. (21) noted a mean increase inbrain volume of 3 and 8 ml after 20 and 40 min at a simulatedaltitude of 3,800 m (12.5% oxygen). Spatial compensation wasapparent, as CSF volume decreased by a similar amount. Withemployment of a similar magnetic resonance imaging tech-nique, brain volume increased by 7 and 59 ml after 2 and 10 hat a simulated altitude of 4,500 m (12% oxygen) (55). Again,evidence for spatial compensation was noted by reductions inventricular and subarachnoid CSF volume. Finally, in the samesubjects who underwent lumbar punctures (10), average brainvolume was only slightly increased (7 � 4.8 ml) after 16 h at
4,500 m, although substantial variability existed; subjects suf-fering the worst symptoms had the largest increase in brainvolume (10), but CSF volume was not reported. Interestingly,under these acute hypoxic conditions, increased brain waterhas rarely been noteworthy, but fluid shifts within white matterare observed consistently (42, 49, 56, 66, 76).
Summary. The available animal and human literature sug-gests that acute moderate to severe hypoxia causes profoundcerebral vasodilatation and an increase in cerebral arterial andvenous blood volume. ICP is acutely elevated in animal mod-els; in the few humans who have been studied, ICP seems to bemildly elevated acutely and remains elevated at least over thefirst 6 h. The initial rise in ICP may be explained by elevatedcerebral blood volume, whereas the maintenance of elevatedICP can be explained by elevated Psag, as seen in animalmodels. What causes an acute increase in Psag is unknown, butheterogeneous intracranial venous anatomy or collapsibility ofthe nondominant transverse sinus (12, 55, 91, 92) are possibleexplanations. In some animals, ICP remains elevated for 24 h,associated with brain edema; spatial compensation is evident.In others, ICP returns toward normal values, which is consis-tent with data obtained in humans. However, in the onlyexperiment to continuously measure ICP, large dynamic fluc-tuations were observed after 6 h in hypobaric hypoxia.
PERSPECTIVES
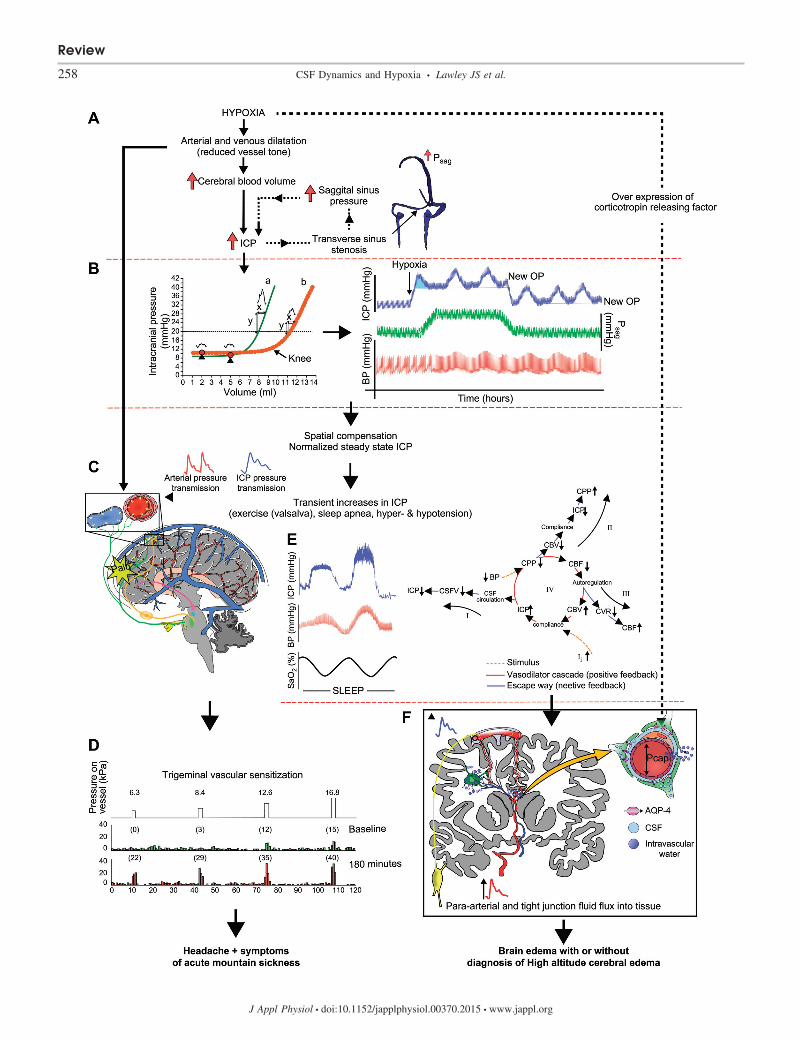
High-altitude headache and AMS. Investigations on themechanisms of high-altitude headache are to some extentstudies of AMS. One could even argue that it is the headacheitself that causes other symptoms, such as anorexia, nausea,lassitude, and insomnia, as is commonly seen in migraine ortension headaches, and that mild AMS is essentially due toheadache.
Headache is generally caused by activation of the trigeminalvascular system (28). High-altitude headache occurs in mostindividuals after rapid ascent to high altitude (15), is often dull,confined to the front of the head (78), and takes a number ofhours to develop (15). Although speculative, these anatomicaland physiological features suggest a possible role for pro-longed low-grade activation of trigeminal afferents innervatingdura or pial arteries or tributary veins that pass into the venoussinus (73). Theoretically, chronic or transient elevations in ICPcould activate trigeminal afferents. Alternatively, as arterialand venous vessel tone is reduced, greater transmission of thearterial pressure into the arteries and veins would be sensedduring every heartbeat in multiple receptive fields and couldproduce summation of afferent recruitment, which exceed thethreshold for pain sensation. Note that the latter situation doesnot necessitate constantly elevated ICP. With either scenario,subsequent central or peripheral sensitization (14, 68) in com-bination with feed-forward parasympathetic activation and/ordiffuse inflammatory mediator release could be responsible forfurther vasodilatation, sensitization, and expansion of trigem-inal nociceptive fields and enhanced mechanosensitivity topreviously nonpainful stimuli, as is observed clinically, i.e.,coughing, bending, and exercising; see Fig. 4 (78). Nausea andvomiting may also be due to vagal activation or transientreductions in blood flow to vomiting centers within the lateralreticular formation and/or the chemoreceptor trigger zonearound the fourth ventricle (13), although at present this is
Review
257CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org
Review
258 CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org

speculative. If, similar to animal models, human Psag normal-izes over time, facilitated CSFoutflow should also normalizeICP. However, ICP may titrate the “knee” of the spatialcompensation-compliance curve (see Fig. 4B), and large fluc-tuations around a normal mean pressure will be observed inconjunction with fluctuation in PaO2
and arterial blood pres-sure. Reductions in symptom intensity will result from theinteraction between numerous factors, including decreasedcerebral blood flow and cerebral blood volume with ventilatoryacclimatization (77), the time constant for spatial compensa-tion, and central/peripheral trigeminal sensitization.
Acute hypoxia causes increased accumulation of intracellu-lar water within astrocytic cells (49, 56, 76) that seems moresevere in individuals with established AMS (49, 56, 76). Fluidshifts within white and gray matter are also related to illnessseverity with prolonged hypoxia (2–7 days; Ref. 42). A com-mon explanation for these observations is disruption of cellularmembrane Na�-K�-ATPase. At present, no evidence of globalor localized ischemia or hypometabolism exists, but elevationsin ICP have been shown to disturb energy metabolism in theperiventricular white matter, independent of PaO2
(1). Alterna-tively, hypoxia has been shown to upregulate the expression ofneuropeptide corticotrophin releasing factor, which activatesthe water channel aquaporin-4 and facilitates water (from CSF)influx into glial cells (16). Indeed, aquaporin-4 located at theterminal end-feet of astrocytes are the most abundant waterchannels in the brain, giving glia approximately four timesgreater water permeability (34). In addition, Iliff and col-leagues (43) have shown that CSF-interstitial fluid flux (theglymphatic system) is driven by cerebral arterial pulsations.We propose that hypoxia increases arterial pressure transmis-sion within the intracranial space, which is more severe inthose with poor craniospinal compensatory capacity and AMS(see Fig. 4). Therefore, enhanced CSF-interstitial fluid fluxmay partly explain altered white matter water mobility inhypoxic individuals with AMS.
HACE. Clinically diagnosed HACE occurs in only a fewindividuals, despite rapid ascent to very high altitudes (11, 31,32). Therefore, anatomical and pathological interactions that
result in overt symptomology (ataxic gait, altered conscious-ness, etc.) are apparently rare. Since the main pathologicalprocess is brain swelling, an anatomically poor spatial com-pensatory capacity (“tight fit”) would be a major risk factor.Poor compensatory capacity, craniospinal axis compliance, andhigh Rout likely govern a general neural intolerance to highaltitude, but a specific predisposition to HACE. However, itmust be emphasized that anatomical differences are only riskfactors: although HACE is rare with current ascent profiles,brain edema may develop in many individuals if they ascendtoo high too fast. Moreover, despite an anatomical predisposi-tion, extreme altitudes can be achieved by most individualswith slow ascent.
Potential mechanisms for vasogenic edema have been re-viewed previously and mainly encompass mechanical break-down and increased leakiness of the blood-brain barrier (8, 30,32, 52). At high altitude, arterial blood pressure is elevatedtogether with pronounced low-frequency (0.05 Hz) sympa-thetic vasomotor oscillations, which are transmitted into theintracranial space due, in part, to reduced vasomotor tone andimpaired autoregulation (9, 45, 81). Brain regions with poorsympathetically mediated vasoconstriction would be particu-larly susceptible (38). In line with this rationale, the tendencyfor vasogenic edema to occur within the splenium of the corpuscallosum could be explained by its vascular pattern (65) andadrenergic innervation (51) that favors arterial pressure trans-mission, increased capillary pressure, and fluid filtration (28).An alternative or contributing factor may be paravascular CSFflux into the brain via the low-resistance glymphatic pathwaynoted above. During AMS, fluid redistribution seems benign;however, over time, Na� influx into the brain may set up anoncotic potential to further potentiate brain edema. In fact, theinteraction between transient rises in capillary pressure relativeto brain tissue pressure (ICP) and an elevated oncotic potential(Starling forces) will substantially dictate fluid filtration acrossthe blood-brain barrier. Factors that likely accelerate vasogenicbrain edema include classic risk factors that increase cerebralblood volume too quickly (fast and high ascent) and activitiesthat produce large fluctuations in blood pressure, such as
Fig. 4. A: pathophysiology of acute mountain sickness and high-altitude cerebral edema. Hypoxia causes elevated CBF, CBV, and a reduction in vascular tone,which causes a rise in ICP. However, if transverse sinus stenosis and elevated Psag are present, ICP remains elevated (ICP must be higher than Psag to maintainCSF absorption). B, left: two curves highlight extremes in spatial compensation capacity and craniospinal elasticity. In a person with anatomical/physiologicalcharacteristics typical of curve a, an additional 3 ml would increase ICP to �20 mmHg; no change in ICP would occur for the same volume change in curveb. Once entering the steep part of the compliance curve, a 1-ml �V (during each heartbeat) causes a much larger increase in ICP pulsatility in curve a than curveb. Red circles identify the ICP operating points (OPs); note the close proximity to the “knee” of the compensatory-compliance curve in a but not b. Right:hypothetical model of dynamic pressure fluctuations over the first few hours at high altitude. Due to reduced vessel tone and altered intracranial compartmentvolumes, ICP waves are observed during every heartbeat and low-frequency BP oscillations. Furthermore, evidence from animal models suggests Psag isincreased; thus ICP is persistently elevated. Over time, mean Psag decreases and ICP normalizes due to both translocation of CSF into the spinal canal andincreased CSF absorption (spatial compensation), but pathological ICP waves are persistently observed. Vascular stretch due to vasodilatation and/or increasedpressure transmission causes activation of trigeminal afferents (green lines) innervating dura or pial arteries or tributary veins that pass into the venous sinus,causing headache. Thereafter, feedforward parasympathetic activation (orange lines) causes the release of potent vasodilators and further vasodilatation. C:central or peripheral sensitization causes a reduced threshold and greater firing rate of meningeal nociceptors, which causes expansion of trigeminal nociceptivefields and enhanced mechanosensitivity to previously nonpainful stimuli, as is observed clinically, i.e., coughing, bending, and exercising. D: although dependenton ascent rate and altitude gain, persons resistant to acute mountain sickness may exhibit an advantageous anatomical/physiological response at any stage in thisschema (i.e., greater ventilatory drive and PaO2 at any given altitude, lower cerebral vascular reactivity to hypoxia, greater spatial compensatory capacity,intracranial compliance, or a high nociceptive threshold for trigeminal activation or pain processing). E: at high altitude, apnea and arousals during sleep causelarge fluctuations in SaO2, PaCO2, and BP. ICP passively rises with BP. With the fall in BP, autoregulation increases CBV, leading to a vasodilatory cascade andthe appearance of large pressure waves. Several negative feedback loops are included that stabilize ICP. F: elevated brain water occurs due to para-arterial influxof CSF, which is facilitated by greater arterial pulsations and expression of corticotrophin releasing factor. Increased leakiness of endothelial tight junctions dueto circulating inflammatory mediators, in concert with BP-dependent opening of the blood-brain barrier, may lead to fluid flux into the cerebral parenchyma fromthe vascular space. CSFV, cerebrospinal fluid volume; CPP, cerebral perfusion pressure; CVR, cerebrovascular resistance; Pcap, capillary pressure. [D and E areredrawn from Refs. 68 and 84, respectively, with permission.]
Review
259CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org

prolonged strenuous mountaineering and sleep at high altitude(2). Sleep at high altitude likely presents a “perfect storm” andthe cumulating event to the development of ICP waves andvasogenic brain edema in most but not all individuals, i.e.,fluctuating PaO2
, PaCO2, thoracic [i.e., central and sagittal ve-
nous pressure, and arterial blood pressure (84) and enhancedpara-arterial (glymphatic) fluid transport (43); see Fig. 4].HACE is most common after a night or two sleeping at highaltitude.
FUTURE RESEARCH DIRECTIONS
The major challenge to test the hypotheses put forward inthis review will be to obtain continuous direct measurements ofICP in humans during prolonged periods of hypoxemia, withand without pharmacological treatment for high-altitude ill-ness. Careful documentation of pathological pressure wavesand the impact of changes in posture, sleep, and exercise willbe critical. In this regard, the combination of advanced neuro-imaging and high-resolution hemodynamic monitoring willlead to a greater understanding of the pressure-flow dynamicsboth globally and regionally within the hypoxic brain. Ex-tended physiological monitoring during sleep at high altitudewill also substantially advance this field. Clearly, some indi-viduals seem more susceptible to high-altitude illness thanothers. Identifying which anatomical factors, if any, predisposeindividuals to high-altitude illness, especially the life-threaten-ing condition HACE, will be valuable. Future research focus-ing on interindividual variations in spatial compensatory ca-pacity, cranial and spinal compliance, Rout, and intracranialvenous anatomy is warranted. Finally, new experimental mod-els that reproducibly cause brain edema (increased multiechoT2 relaxation on magnetic resonance imaging) in human sub-jects are required; at present, frank evidence of brain edema inany short-term human investigation is marginal at best. Oncedeveloped, experimental models aimed at modulating waterflux through both endothelial and glial blood-brain interfaceswill be important.
CONCLUSIONS
As hypothesized by Ross (74), interindividual variability incraniospinal compensatory capacity is a rational explanationfor the individual susceptibility to cerebral manifestations ofhigh-altitude illness. We hypothesize that 1) individuals whoare asymptomatic, despite rapid ascent to high altitude, possessa compensatory capacity (large spinal compliance and/or lowRout) that outweighs changes in brain volume, whereby arteri-olar tone is not exhausted and pressure transmission is notfacilitated. Importantly, the compensatory capacity of the spi-nal thecal sac, which is a central tenant of the tight-fit hypoth-esis, has never been assessed in individuals with and withouthigh-altitude illness. 2) Acute increases in ICP will be ob-served in individuals with poor spinal compliance (acute spa-tial compensatory capacity) relative to large increases in brainvolume; ICP will return toward the normal operating pointslower in individuals with a concomitant high Rout. Con-versely, ICP will remain elevated if Psag is elevated. 3) If meanICP returns to near normal, it will titrate the “knee” of thespatial compensation-compliance curve, and large fluctuationswill be observed in conjunction with fluctuations in PaO2
andarterial blood pressure, which contribute to symptoms of high-
altitude headache and AMS. Direct arterial or venous dilatationor increased arterial pressure transmission, independent of ICP,could also cause AMS symptoms. 4) Continual acute eleva-tions in ICP will be observed with gains in altitude, but, ifgradual, will return to normal on every occasion due to in-creased CSFoutflow and normalization of Psag. However, atsome point, each individual will reach their compensatorycapacity for a given altitude gain and ascent rate. At this point,despite the appearance of acclimatization, further ascent, ex-ercise, and sleep will be disastrous, brain edema will develop,A- and B-waves will be observed, and HACE will occur.
GRANTS
This work was supported by the National Space Biomedical ResearchInstitute through NASA NCC 9-58.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.S.L., B.D.L., M.A.W., J.M., A.E., D.M.P., A.W.S.,P.H.H., and R.C.R. conception and design of research; J.S.L. prepared figures;J.S.L. drafted manuscript; J.S.L., B.D.L., M.A.W., J.M., A.E., D.M.P., A.W.S.,P.H.H., and R.C.R. edited and revised manuscript; J.S.L., B.D.L., M.A.W.,J.M., A.E., D.M.P., A.W.S., P.H.H., and R.C.R. approved final version ofmanuscript.
REFERENCES
1. Agren-Wilsson A, Eklund A, Koskinen LO, Bergenheim AT, Malm J.Brain energy metabolism and intracranial pressure in idiopathic adulthydrocephalus syndrome. J Neurol Neurosurg Psychiatry 76: 1088–1093,2005.
2. Ainslie PN, Burgess K, Subedi P, Burgess KR. Alterations in cerebraldynamics at high altitude following partial acclimatization in humans:wakefulness and sleep. J Appl Physiol (1985) 102: 658–664, 2007.
3. Andersson N, Malm J, Eklund A. Dependency of cerebrospinal fluidoutflow resistance on intracranial pressure. J Neurosurg 109: 918–922,2008.
4. Andresen M, Hadi A, Petersen LG, Juhler M. Effect of posturalchanges on ICP in healthy and ill subjects. Acta Neurochir (Wien) 157:109–113, 2015.
5. Andresen M, Juhler M. Intracranial pressure following complete re-moval of a small demarcated brain tumor: a model for normal intracranialpressure in humans. J Neurosurg 121: 797–801, 2014.
6. Armstead WM. Role of nitric oxide, cyclic nucleotides, and the activationof ATP-sensitive K� channels in the contribution of adenosine to hypoxia-induced pial artery dilation. J Cereb Blood Flow Metab 17: 100–108,1997.
7. Avezaat CJJ, van Eijndhoven JHM. Cerebrospinal Fluid Pulse Pres-sure and Craniospinal Dynamics: A Theoretical Clinical and Experimen-tal Study. The Hague: Jongbloeden Zoon, 1984.
8. Bailey DM, Bartsch P, Knauth M, Baumgartner RW. Emerging con-cepts in acute mountain sickness and high-altitude cerebral edema: fromthe molecular to the morphological. Cell Mol Life Sci 66: 3583–3594,2009.
9. Bailey DM, Evans KA, James PE, McEneny J, Young IS, Fall L,Gutowski M, Kewley E, McCord JM, Moller K, Ainslie PN. Alteredfree radical metabolism in acute mountain sickness: implications fordynamic cerebral autoregulation and blood-brain barrier function. JPhysiol 587: 73–85, 2009.
10. Bailey DM, Roukens R, Knauth M, Kallenberg K, Christ S, Mohr A,Genius J, Storch-Hagenlocher B, Meisel F, McEneny J, Young IS,Steiner T, Hess K, Bartsch P. Free radical-mediated damage to barrierfunction is not associated with altered brain morphology in high-altitudeheadache. J Cereb Blood Flow Metab 26: 99–111, 2006.
11. Bärtsch P, Roach RC. Acute mountain sickness and high-altitude cere-bral edema. In: High Altitude: An Exploration of Human Adaptation,edited by Hornbein TF, Schoene RB. New York: Dekker, 2001, p.731–776.
Review
260 CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org

12. Bateman GA, Stevens SA, Stimpson J. A mathematical model ofidiopathic intracranial hypertension incorporating increased arterial inflowand variable venous outflow collapsibility. J Neurosurg 110: 446–456,2009.
13. Borison HL, Wang SC. Physiology and pharmacology of vomiting.Pharmacol Rev 5: 193–230, 1953.
14. Burstein R, Jakubowski M. Analgesic triptan action in an animal modelof intracranial pain: a race against the development of central sensitization.Ann Neurol 55: 27–36, 2004.
15. Burtscher M, Wille M, Menz V, Faulhaber M, Gatterer H. Symptomprogression in acute mountain sickness during a 12-hour exposure tonormobaric hypoxia equivalent to 4500 m. High Alt Med Biol 15: 446–451, 2014.
16. Chen SJ, Yang JF, Kong FP, Ren JL, Hao K, Li M, Yuan Y, Chen XC,Yu RS, Li JF, Leng G, Chen XQ, Du JZ. Overactivation of corticotro-phin-releasing factor receptor type 1 and aquaporin-4 by hypoxia inducescerebral edema. Proc Natl Acad Sci U S A 111: 13199–13204, 2014.
17. Craigen ML, Jennett S. Pial arterial response to systemic hypoxia inanaesthetised cats. J Cereb Blood Flow Metab 1: 285–296, 1981.
18. Curran-Everett DC, Iwamoto J, Meredith MP, Krasney JA. Intracra-nial pressures and O2 extraction in conscious sheep during 72 h ofhypoxia. Am J Physiol Heart Circ Physiol 261: H103–H109, 1991.
19. Damkier HH, Brown PD, Praetorius J. Cerebrospinal fluid secretion bythe choroid plexus. Physiol Rev 93: 1847–1892, 2013.
20. Davson H, Domer FR, Hollingsworth JR. The mechanism of drainage ofthe cerebrospinal fluid. Brain 96: 329–336, 1973.
21. Dubowitz DJ, Dyer EA, Theilmann RJ, Buxton RB, Hopkins SR. Earlybrain swelling in acute hypoxia. J Appl Physiol (1985) 107: 244–252,2009.
22. Edsbagge M, Tisell M, Jacobsson L, Wikkelso C. Spinal CSF absorp-tion in healthy individuals. Am J Physiol Regul Integr Comp Physiol 287:R1450–R1455, 2004.
23. Eide PK, Kerty E. Static and pulsatile intracranial pressure in idiopathicintracranial hypertension. Clin Neurol Neurosurg 113: 123–128, 2011.
24. Ekstedt J. CSF hydrodynamic studies in man. 2. Normal hydrodynamicvariables related to CSF pressure and flow. J Neurol Neurosurg Psychiatry41: 345–353, 1978.
25. Farahmand D, Qvarlander S, Malm J, Wikkelso C, Eklund A, TisellM. Intracranial pressure in hydrocephalus: impact of shunt adjustmentsand body positions. J Neurol Neurosurg Psychiatry 86: 222–228, 2015.
26. Feldman Z, Kanter MJ, Robertson CS, Contant CF, Hayes C, Shei-nberg MA, Villareal CA, Narayan RK, Grossman RG. Effect of headelevation on intracranial pressure, cerebral perfusion pressure, and cere-bral blood flow in head-injured patients. J Neurosurg 76: 207–211, 1992.
27. Ghajar J. Traumatic brain injury. Lancet 356: 923–929, 2000.28. Goadsby PJ, Charbit AR, Andreou AP, Akerman S, Holland PR.
Neurobiology of migraine. Neuroscience 161: 327–341, 2009.29. Greitz D, Hannerz J. A proposed model of cerebrospinal fluid circula-
tion: observations with radionuclide cisternography. AJNR Am J Neuro-radiol 17: 431–438, 1996.
30. Hackett PH. The cerebral etiology of high-altitude cerebral edema andacute mountain sickness. Wilderness Environ Med 10: 97–109, 1999.
31. Hackett PH, Rennie D, Levine HD. The incidence, importance, andprophylaxis of acute mountain sickness. Lancet 2: 1149–1155, 1976.
32. Hackett PH, Roach RC. High altitude cerebral edema. High Alt Med Biol5: 136–146, 2004.
33. Hackett PH, Yarnell PR, Hill R, Reynard K, Heit J, McCormick J.High-altitude cerebral edema evaluated with magnetic resonance imaging:clinical correlation and pathophysiology. JAMA 280: 1920–1925, 1998.
34. Haj-Yasein NN, Vindedal GF, Eilert-Olsen M, Gundersen GA, SkareO, Laake P, Klungland A, Thoren AE, Burkhardt JM, Ottersen OP,Nagelhus EA. Glial-conditional deletion of aquaporin-4 (Aqp4) reducesblood-brain water uptake and confers barrier function on perivascularastrocyte endfeet. Proc Natl Acad Sci U S A 108: 17815–17820, 2011.
35. Hamer J, Alberti E, Hoyer S. Effects of arterial hypoxaemia, hypercap-nia, and changes in cerebral perfusion pressure on mean cerebrospinalfluid and sagittal sinus pressure. Acta Neurochir (Wien) 30: 167–179,1974.
36. Hamer J, Alberti E, Hoyer S, Wiedemann K. Influence of systemic andcerebral vascular factors on the cerebrospinal fluid pulse waves. J Neuro-surg 46: 36–45, 1977.
37. Hartig GS, Hackett PH. Cerebral spinal fluid pressure and cerebral bloodvelocity in acute mountain sickness. In: Hypoxia and Mountain Medicine,
edited by Sutton JR, Coates G, and Houston CS. Burlington, VT: QueenCity Press, 1992, p. 260–265.
38. Heistad DD, Marcus ML. Effect of sympathetic stimulation on perme-ability of the blood-brain barrier to albumin during acute hypertension incats. Circ Res 45: 331–338, 1979.
39. Hodel J, Lebret A, Petit E, Leclerc X, Zins M, Vignaud A, Decq P,Rahmouni A. Imaging of the entire cerebrospinal fluid volume with amultistation 3D SPACE MR sequence: feasibility study in patients withhydrocephalus. Eur Radiol 23: 1450–1458, 2013.
40. Houston CS, Dickinson J. Cerebral form of high-altitude illness. Lancet2: 758–761, 1975.
41. Hulme A, Cooper R. The effects of head position and jugular veincompression (JVC) on intracranial pressure (ICP). A clinical study. In:Intracranial Pressure III, edited by Beks JF, Bosch DA, and Brock M.Berlin: Springer, 1976, p. 259–263.
42. Hunt JS Jr, Theilmann RJ, Smith ZM, Scadeng M, Dubowitz DJ.Cerebral diffusion and T(2): MRI predictors of acute mountain sicknessduring sustained high-altitude hypoxia. J Cereb Blood Flow Metab 33:372–380, 2013.
43. Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y,Deane R, Nedergaard M. Cerebral arterial pulsation drives paravascularCSF-interstitial fluid exchange in the murine brain. J Neurosci 33: 18190–18199, 2013.
44. Iwamoto J, Curran-Everett DC, Krasney E, Krasney JA. Cerebralmetabolic and pressure-flow responses during sustained hypoxia in awakesheep. J Appl Physiol (1985) 71: 1447–1453, 1991.
45. Iwasaki K, Zhang R, Zuckerman JH, Ogawa Y, Hansen LH, LevineBD. Impaired dynamic cerebral autoregulation at extreme high altitudeeven after acclimatization. J Cereb Blood Flow Metab 31: 283–292, 2011.
46. Jennett S, Pitts LH, North JB. Rapid cerebral vasodilatation in briefhypoxia in anaesthetized animals. Q J Exp Physiol 66: 447–463, 1981.
47. Johnston M, Papaiconomou C. Cerebrospinal fluid transport: a lym-phatic perspective. News Physiol Sci 17: 227–230, 2002.
48. Julien-Dolbec C, Tropres I, Montigon O, Reutenauer H, Ziegler A,Decorps M, Payen JF. Regional response of cerebral blood volume tograded hypoxic hypoxia in rat brain. Br J Anaesth 89: 287–293, 2002.
49. Kallenberg K, Bailey DM, Christ S, Mohr A, Roukens R, Menold E,Steiner T, Bartsch P, Knauth M. Magnetic resonance imaging evidenceof cytotoxic cerebral edema in acute mountain sickness. J Cereb BloodFlow Metab 27: 1064–1071, 2007.
50. Kido DK, Gomez DG, Pavese AM Jr, Potts DG. Human spinal arach-noid villi and granulations. Neuroradiology 11: 221–228, 1976.
51. Kobayashi S, Tsukahara S, Sugita K, Nagata T. Adrenergic andcholinergic innervation of rat cerebral arteries. Consecutive demonstrationon whole mount preparations. Histochemistry 70: 129–138, 1981.
52. Krasney JA. A neurogenic basis for acute altitude illness. Med Sci SportsExerc 26: 195–208, 1994.
53. Kronenberg RS, Safar P, Leej Wright F, Noble W, Wahrenbrock E,Hickey R, Nemoto E, Severinghaus JW. Pulmonary artery pressure andalveolar gas exchange in man during acclimatization to 12,470 ft. J ClinInvest 50: 827–837, 1971.
54. Langfitt TW, Weinstein JD, Kassell NF. Cerebral vasomotor paralysisproduced by intracranial hypertension. Neurology 15: 622–641, 1965.
55. Lawley JS, Alperin N, Bagci AM, Lee SH, Mullins PG, Oliver SJ,Macdonald JH. Normobaric hypoxia and symptoms of acute mountainsickness: Elevated brain volume and intracranial hypertension. Ann Neurol75: 890–898, 2014.
56. Lawley JS, Oliver SJ, Mullins PG, Macdonald JH. Investigation ofwhole-brain white matter identifies altered water mobility in the patho-genesis of high-altitude headache. J Cereb Blood Flow Metab 33: 1286–1294, 2013.
57. Leech P, Miller JD. Intracranial volume–pressure relationships duringexperimental brain compression in primates. 1. Pressure responses tochanges in ventricular volume. J Neurol Neurosurg Psychiatry 37: 1093–1098, 1974.
58. Lofgren J, Zwetnow NN. Cranial and spinal components of the cerebro-spinal fluid pressure-volume curve. Acta Neurol Scand 49: 575–585, 1973.
59. Lundberg N. Continuous recording and control of ventricular fluidpressure in neurosurgical practice. Acta Psychiatr Scand Suppl 36: 1–193,1960.
60. Malm J, Jacobsson J, Birgander R, Eklund A. Reference values forCSF outflow resistance and intracranial pressure in healthy elderly. Neu-rology 76: 903–909, 2011.
Review
261CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org

61. Marmarou A, Maset AL, Ward JD, Choi S, Brooks D, Lutz HA,Moulton RJ, Muizelaar JP, DeSalles A, Young HF. Contribution ofCSF and vascular factors to elevation of ICP in severely head-injuredpatients. J Neurosurg 66: 883–890, 1987.
62. Marmarou A, Shulman K, LaMorgese J. Compartmental analysis ofcompliance and outflow resistance of the cerebrospinal fluid system. JNeurosurg 43: 523–534, 1975.
63. Martins AN, Wiley JK, Myers PW. Dynamics of the cerebrospinal fluidand the spinal dura mater. J Neurol Neurosurg Psychiatry 35: 468–473,1972.
64. Matsuzawa Y, Kobayashi T, Fujimoto K, Shinozaki S, Yoshikawa S.Cerebral edema in acute mountain sickness. In: High Altitude Medicine,edited by Ueda G, Reeves JT, Sekiguchi M. Matsumoto, Japan: ShinshuUniversity Press, 1992, p. 300–304.
65. Moody DM, Bell MA, Challa VR. Features of the cerebral vascularpattern that predict vulnerability to perfusion or oxygenation deficiency:an anatomic study. AJNR Am J Neuroradiol 11: 431–439, 1990.
66. Morocz IA, Zientara GP, Gudbjartsson H, Muza S, Lyons T, RockPB, Kikinis R, Jolesz FA. Volumetric quantification of brain swellingafter hypobaric hypoxia exposure. Exp Neurol 168: 96–104, 2001.
67. North JBR, Gorman PL, Grant D, C, Ludbrook GL. The effect ofhypoxia on intracranial pressure and cerebral blood flow. In: IntracranialPressure VIII, edited by. Avezaat CJJ, van Eijndhoven JHM, Maas AIR,Tans JTJ. Berlin: Springer, 1993, p. 238–243.
68. Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain inmigraine: evidence for peripheral sensitisation. Lancet Neurol 8: 679–690,2009.
69. Portnoy HD, Chopp M. Cerebrospinal fluid pulse wave form analysisduring hypercapnia and hypoxia. Neurosurgery 9: 14–27, 1981.
70. Potts DG, Deonarine V. Effect of positional changes and jugular veincompression on the pressure gradient across the arachnoid villi andgranulations of the dog. J Neurosurg 38: 722–728, 1973.
71. Qvarlander S, Malm J, Eklund A. The pulsatility curve-the relationshipbetween mean intracranial pressure and pulsation amplitude. Physiol Meas31: 1517–1528, 2010.
72. Qvarlander S, Sundstrom N, Malm J, Eklund A. Postural effects onintracranial pressure: modeling and clinical evaluation. J Appl Physiol(1985) 115: 1474–1480, 2013.
73. Ray BS, Wolff HG. Experimental studies on headache: pain-sensitivestructures of the head and their significance in headache. Arch Surg 41:813–856, 1940.
74. Ross RT. The random nature of cerebral mountain sickness. Lancet 1:990–991, 1985.
75. Schaltenbrand G. Atmospheric pressure, circulation, respiration andcerebrospinal fluid pressure. Acta Aerophysiol 1: 65–78, 1933.
76. Schoonman GG, Sandor PS, Nirkko AC, Lange T, Jaermann T,Dydak U, Kremer C, Ferrari MD, Boesiger P, Baumgartner RW.Hypoxia-induced acute mountain sickness is associated with intracellularcerebral edema: a 3 T magnetic resonance imaging study. J Cereb BloodFlow Metab 28: 198–206, 2008.
77. Severinghaus JW, Chiodi H, Eger EI 2nd, Brandstater B, HornbeinTF. Cerebral blood flow in man at high altitude. Role of cerebrospinalfluid pH in normalization of flow in chronic hypocapnia. Circ Res 19:274–282, 1966.
78. Silber E, Sonnenberg P, Collier DJ, Pollard AJ, Murdoch DR,Goadsby PJ. Clinical features of headache at altitude: a prospective study.Neurology 60: 1167–1171, 2003.
79. Singh I, Khanna PK, Srivastava MC, Lal M, Roy SB, SubramanyamCS. Acute mountain sickness. N Engl J Med 280: 175–184, 1969.
80. Small HS, Weitzner SW, Nahas GG. Cerebrospinal fluid pressuresduring hypercapnia and hypoxia in dogs. Am J Physiol 198: 704–708,1960.
81. Subudhi AW, Panerai RB, Roach RC. Acute hypoxia impairs dynamiccerebral autoregulation: results from two independent techniques. J ApplPhysiol (1985) 107: 1165–1171, 2009.
82. Tain RW, Bagci AM, Lam BL, Sklar EM, Ertl-Wagner B, Alperin N.Determination of cranio-spinal canal compliance distribution by MRI:methodology and early application in idiopathic intracranial hypertension.J Magn Reson Imaging 34: 1397–1404, 2011.
83. Torbey MT, Geocadin RG, Razumovsky AY, Rigamonti D, WilliamsMA. Utility of CSF pressure monitoring to identify idiopathic intracranialhypertension without papilledema in patients with chronic daily headache.Cephalalgia 24: 495–502, 2004.
84. Ursino M, Lodi CA. A simple mathematical model of the interactionbetween intracranial pressure and cerebral hemodynamics. J Appl Physiol(1985) 82: 1256–1269, 1997.
85. Wahlin A, Ambarki K, Birgander R, Alperin N, Malm J, Eklund A.Assessment of craniospinal pressure-volume indices. AJNR Am J Neuro-radiol 31: 1645–1650, 2010.
86. Wahlin A, Ambarki K, Birgander R, Malm J, Eklund A. Intracranialpulsatility is associated with regional brain volume in elderly individuals.Neurobiol Aging 35: 365–372, 2014.
87. Wahlin A, Ambarki K, Hauksson J, Birgander R, Malm J, Eklund A.Phase contrast MRI quantification of pulsatile volumes of brain arteries,veins, and cerebrospinal fluids compartments: repeatability and physio-logical interactions. J Magn Reson Imaging 35: 1055–1062, 2012.
88. Weinbrecht PT, Johnson LC, Longmuir IS, Knopp JA. Influence ofPaO2 on cerebral macro- and microcirculation as observed by light reflec-tion: time course of changes. Adv Exp Med Biol 200: 131–136, 1986.
89. Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage ofthe brain and the pathophysiology of neurological disease. Acta Neuro-pathol (Berl) 117: 1–14, 2009.
90. Wilderman MJ, Armstead WM. Role of neuronal NO synthase inrelationship between NO and opioids in hypoxia-induced pial arterydilation. Am J Physiol Heart Circ Physiol 273: H1807–H1815, 1997.
91. Wilson MH, Davagnanam I, Holland G, Dattani RS, Tamm A, HiraniSP, Kolfschoten N, Strycharczuk L, Green C, Thornton JS, Wright A,Edsell M, Kitchen ND, Sharp DJ, Ham TE, Murray A, Holloway CJ,Clarke K, Grocott MP, Montgomery H, Imray C. Cerebral venoussystem and anatomical predisposition to high-altitude headache. AnnNeurol 73: 381–389, 2013.
92. Wilson MH, Imray CH, Hargens AR. The headache of high altitude andmicrogravity–similarities with clinical syndromes of cerebral venous hy-pertension. High Alt Med Biol 12: 379–386, 2011.
93. Wilson MH, Milledge J. Direct measurement of intracranial pressure athigh altitude and correlation of ventricular size with acute mountainsickness: Brian Cummins’ results from the 1985 Kishtwar expedition.Neurosurgery 63: 970–974; discussion 974–975, 2008.
94. Wilson MH, Wright A, Imray CH. Intracranial pressure at altitude. HighAlt Med Biol 15: 123–132, 2014.
95. Wilson R. Acute high-altitude illness in mountaineers and problems ofrescue. Ann Intern Med 78: 421–428, 1973.
96. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’DonnellJ, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Neder-gaard M. Sleep drives metabolite clearance from the adult brain. Science342: 373–377, 2013.
97. Yang SP, Bergo GW, Krasney E, Krasney JA. Cerebral pressure-flowand metabolic responses to sustained hypoxia: effect of CO2. J ApplPhysiol (1985) 76: 303–313, 1994.
98. Yang YB, Sun B, Yang Z, Wang J, Pong Y. Effects of acute hypoxia onintracranial dynamics in unanesthetized goats. J Appl Physiol (1985) 74:2067–2071, 1993.
Review
262 CSF Dynamics and Hypoxia • Lawley JS et al.
J Appl Physiol • doi:10.1152/japplphysiol.00370.2015 • www.jappl.org




















