
Dominant inheritance of cerebral gigantism
6
August 1977 The Journal of P E D I A T R I C S 251 Dominant inheritance of cerebral gigantism Cerebral gigantism is a syndrome consisting of characteristic dysmorphic features, accelerated growth in early childhood, and variable degrees of mental retardation. Its etiology and pathogenesis have not been defined. Three families are presented with multiple affected members. The vertical transmission of the trait and equal expression in both sexes in these families indicates a genetic etiology with a dominant pattern of inheritance, probably autosomal. As in previously reported cases, extensive endocrine evaluation failed to define the pathogenesis of the accelerated growth present in this disorder. Jonathan Zonana, M.D., Torrance, Calif., Juan F. Sotos, M.D., Torrance, Calif., Carolyn A. Romshe, M.D., Columbus, Ohio, Delbert A. Fisher, M.D., Torrance, Calif,, M. Jocelyn Eiders, M.D., Little Rock, Ark., and David L. Rimoin, M.D., Ph.D.,* Torrance, Calif. S I N c E the syndrome of cerebral gigantism was originally defined in 1964, over 80 cases, displaying similar physical From the Divisions of Medical Genetics and Perinatal Biology, Departments of Pediatrics and Medicine, UCLA School of Medicine-Harbor General Hospital," the Division of Endocrinology and Metabolism, Department of Pediatrics, College of Medicine, The Ohio State University and Children's Hospital Research Foundation; and Division of Endocrinology, Department of Pediatrics, University of A rkansas Medical Center. Presented in part at the 1975 Birth Defects Conference, Kansas City, Mo. Studies at Harbor General Hospital were supported by USPH RRO 0425 and HD-05624 and Birth Defects Center and research grants from the National Foundation~Mareh of Dimes, and the Easter Seal Research Foundation. Studies at the Children's Hospital, Columbus, Ohio were supported by the John W. Champion Center. Studies at the University of Arkansas were supported by United States Public Health Service grant A M 15901 and CA 13907. Dr. Zonana was supported by a National Foundation March of Dimes Postdoctoral Fellowship and United States Public Health Service Research Training Grants (5 TOI-HD 00417- 03 and 5 T22-DE 00102-02). *Reprint address: Division of Medical Genetics, Harbor General Hospital, 1000 West Carson St., Torrance, CA 90509. and developmental features, have appeared in the litera- ture. '< The characteristic developmental features include large size at birth, accelerated growth in early childhood with accompanying advanced bone age, early dental eruption, normal sexual development, and usually rela- tively normal final adult stature. Characteristic physical features are macrocrania with dolichocephaly, high- arched palate, prognathism, large hands and feet, and an increased arm span. Variable degrees of mental retarda- tion are present. Abnormal growth and/or mental retar- dation frequently are the presenting complaint. The etiology and pathogenesis of the disorder have not been defined. All previous cases, with the exception of single reported pairs of monozygotic twins, first cousins, and siblings, have apparently been sporadic?-:' The present report includes three families with multiple affected members, indicating that cerebral gigantism may be inherited as a dominant trait. CASE REPORTS Family A-(Fig. 1, A). Case A-1 (III-6), The proband, a 22-month-old black male, was the product of a normal term pregnancy in a gravida 5, para 2, aborti 3 41-year-old mother; the father was 39 years of age. Birth weight was 3,941 gm (97%); and length was 60 cm (> 94%) (+4.7 SD). Complications in the neonatal period included transient hypoglycemia and "jitteriness" of undetermined etiology. Hospitalization at one month of age for evaluation of Vol. 91, No. 2, pp. 251-256
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Dominant inheritance of cerebral gigantism

August 1977 The Journal o f P E D I A T R I C S 2 5 1
Dominant inheritance of cerebral gigantism
Cerebral gigantism is a syndrome consisting of characteristic dysmorphic features, accelerated growth in
early childhood, and variable degrees of mental retardation. Its etiology and pathogenesis have not been
defined. Three families are presented with multiple affected members. The vertical transmission of the
trait and equal expression in both sexes in these families indicates a genetic etiology with a dominant
pattern of inheritance, probably autosomal. As in previously reported cases, extensive endocrine
evaluation failed to define the pathogenesis of the accelerated growth present in this disorder.
Jonathan Zonana, M.D. , Torrance, Calif., Juan F. Sotos , M.D. , Torrance, Calif.,
Carolyn A. Romshe, M.D. , Columbus, Ohio, Delbert A. Fisher, M.D. ,
Torrance, Calif,, M. Jocelyn Eiders, M.D. , Little Rock, Ark., and
David L. Rimoin, M.D. , Ph.D.,* Torrance, Calif.
S I N c E the syndrome of cerebral gigantism was originally defined in 1964, over 80 cases, displaying similar physical
From the Divisions of Medical Genetics and Perinatal Biology, Departments of Pediatrics and Medicine, UCLA School of Medicine-Harbor General Hospital," the Division of Endocrinology and Metabolism, Department of Pediatrics, College of Medicine, The Ohio State University and Children's Hospital Research Foundation; and Division of Endocrinology, Department of Pediatrics, University of A rkansas Medical Center.
Presented in part at the 1975 Birth Defects Conference, Kansas City, Mo.
Studies at Harbor General Hospital were supported by USPH RRO 0425 and HD-05624 and Birth Defects Center and research grants from the National Foundation~Mareh of Dimes, and the Easter Seal Research Foundation.
Studies at the Children's Hospital, Columbus, Ohio were supported by the John W. Champion Center.
Studies at the University of Arkansas were supported by United States Public Health Service grant A M 15901 and CA 13907.
Dr. Zonana was supported by a National Foundation March of Dimes Postdoctoral Fellowship and United States Public Health Service Research Training Grants (5 TOI-HD 00417- 03 and 5 T22-DE 00102-02).
*Reprint address: Division of Medical Genetics, Harbor General Hospital, 1000 West Carson St., Torrance, CA 90509.
and developmental features, have appeared in the litera- ture. '< The characteristic developmental features include large size at birth, accelerated growth in early childhood with accompanying advanced bone age, early dental eruption, normal sexual development, and usually rela- tively normal final adult stature. Characteristic physical features are macrocrania with dolichocephaly, high- arched palate, prognathism, large hands and feet, and an increased arm span. Variable degrees of mental retarda- tion are present. Abnormal growth and/or mental retar- dation frequently are the presenting complaint. The etiology and pathogenesis of the disorder have not been
defined. All previous cases, with the exception of single
reported pairs of monozygotic twins, first cousins, and
siblings, have apparently been sporadic?-: ' The present
report includes three families with multiple affected members, indicating that cerebral gigantism may be
inherited as a dominant trait.
CASE REPORTS
Family A-(Fig. 1, A). Case A-1 (III-6), The proband, a 22-month-old black male,
was the product of a normal term pregnancy in a gravida 5, para 2, aborti 3 41-year-old mother; the father was 39 years of age. Birth weight was 3,941 gm (97%); and length was 60 cm (> 94%) (+4.7 SD). Complications in the neonatal period included transient hypoglycemia and "jitteriness" of undetermined etiology. Hospitalization at one month of age for evaluation of
Vol. 91, No. 2, pp. 251-256

2 5 2 Zonana et al. The Journal of Pediatrics August 1977
I
II
Ill
A
i ~ 3 4 J=5 ~
C
12
llI
�9 AFFECTED [ ] POSSIBLY AFFECTED
Fig. 1. Cerebral gigantism-pedigrees.
B
hypertonia failed to reveal a specific etiology. Subsequent devel- opment was characterized by delayed psychomotor milestones, early dental eruption, and accelerated growth.
Physical examination at age 22 months revealed a height of 95 cm ( > 94%) (+2.3 SD), an increased arm span of 106 cm, macrocephaly, with a head circumference of 57 cm ( > 97%) ( ~ + 6 SD), dolichocephaly with frontal bossing, prognathism, and a high-arched palate (Fig. 2, A). Hearing was decreased secondary to bilateral serous otitis media. There were large hands and feet, bilateral genu varum, and pes planus deformities. Neurologic examination was normal except for a developmental quotient of 50. Skeletal radiographs revealed a bone age of four years by Greulich-Pyle standards, and synostosis of the sagittal cranial suture. Computed tomography scan showed large lateral and third ventricles and prominent cortical sulci.
Case A-2 (11-3). The proband's 44-year-old mother, was noted to be similarly affected. (Fig. 2, B). She weighed approximately 5,000 gm ( > 97%) at birth, had delayed psychomotor develop- ment and early accelerated growth, and was always being taller than her peers in early childhood. Sexual development was normal, with menarche at age 11 years.
Height was 173 cm (97%), arm span 191 cm, and head circumference 58.5 cm ( > 97%). She also had dolichocephaly, prognathism, large hands and feet, increased arm span, as well as thoracic kyphosis. Her IQ was approximately 60.
Case A-3 (111-5). The proband's 17-year-old sister, evaluated at the University of Arkansas Medical Center, had been raised separately. At birth she had a weight of 4,810 gm ( > 97%) and a head circumference of 39.5 cm ( > 98%). She also displayed delayed psychomotor development, early dental eruption, and accelerated growth in early childhood.
She was hospitalized at age 71A years for evaluation of abnormal growth and surgical Correction of bilateral pes planus deformities. At that time her height was 146 cm ( > 97%) (~4.5 SD), weight 45 kg ( > 97%), and head circumference 58.5 cm ( > 97%) (+ 5 SD). She manifested frontal bossing, prognathism, large hands and feet, and bilateral pes planus. Bone age was 10 years.
She subsequently had normal sexual development with
menarche at age 10; her adult height is 167 cm (75%). Her IQ is approximately 67 and she attends a special education class. Pregnancy at age 18 resulted in an affected male infant (Case A- 4).
Peripheral blood karyotype with G-banding and extensive endocrinologic evaluation including serum growth hormone, thyroxine, thyroid-stimulating hormone, prolactin, cortisol, and adrenal and gonadal steroid profiles were normal in the proband, his mother, and sister.
Case A-4 (11/-1). This one-month-old-black male, the proband's nephew, was the product of a normal term pregnancy (to A-3). He was delivered by cesarean section due to his large size and macrocrania. Birth weight was 4,840 gm ( > 97%), length 56 cm ( > 98%) (~3.5 SD) and head circumference 39 cm ( > 98%) ( ~ 4 SD). He had large hands and feet and an increased arm span.
At one month of age his height was 61.5 cm ( > 98%), and head circumference 40 cm ( > 98%). Bone age was advanced approxi- mating six to nine months. A Single random serum growth hormone level and somatomedin activity were within normal limits. Serum glucose and insulin response to an oral glucose tolerance test was normal, except for a serum glucose concentra- tion of 40 mg/dl at three hours.
Family history. The maternal grandfather is stated to have craniofacial features similar to the affected family members, but confirmation is not possible. The father and maternal grand- mother were normal upon examination, and the're was no history of consanguinity.
Family B-(Fig. 1, B). Case B-1 (111-1). The proband, a 5-9/12-year-01d white female,
was referred to Columbus Children's Hospital for evaluation of accelerated growth, poor motor coordination, and slow mental development (Fig. 3, A). She was a term infant, delivered by cesarean section to a gravida 3, para 3 32-year-old white female and 30-year-old father. Birth weight was 4.1 kg (95%) and length 63 cm ( > 97%) ( + 7.5 SD). The neonatal course was complicated by Rh incompatibility requiring two exchange transfusions.
Physical examination revealed a height of 129 c m ( > 98%) (+3 SD), weight of 27 kg ( > 98%), and arm span of 129.5 cm. She had a large head circumference of 56 c m ( > 98%) ( ~ +4 SD), dolichocephaly, high forehead with frontal bossing, bilateral epicanthic folds, prognathism, high-arched palate, and large hands and feet. A grade 2/6 systolic ejection murmur was noted in the pulmonic area. Neurologic examination was normal except for clumsiness. Intellectual functioning was in the average range (IQ of 90). Perceptual and motor performance, however, were below the five-year level.
Skeletal radiographs revealed a bone age of 10 years by Greulich-Pyle standards; skull roentgenograms were normal. Peripheral blood karyotype and extensive metabolic and endo- crine evaluation, including human growth hormone, thyroxine, and ACTH reserve were normal.
Case B-2 (11-3). The proband's 35-year-old father was appar- ently a large child, though no records are available (Fig. 3, B). He was a slow learner and attended school only through the fourth grade. He has manifested a behavior disorder and becomes easily frustrated and violent at times.
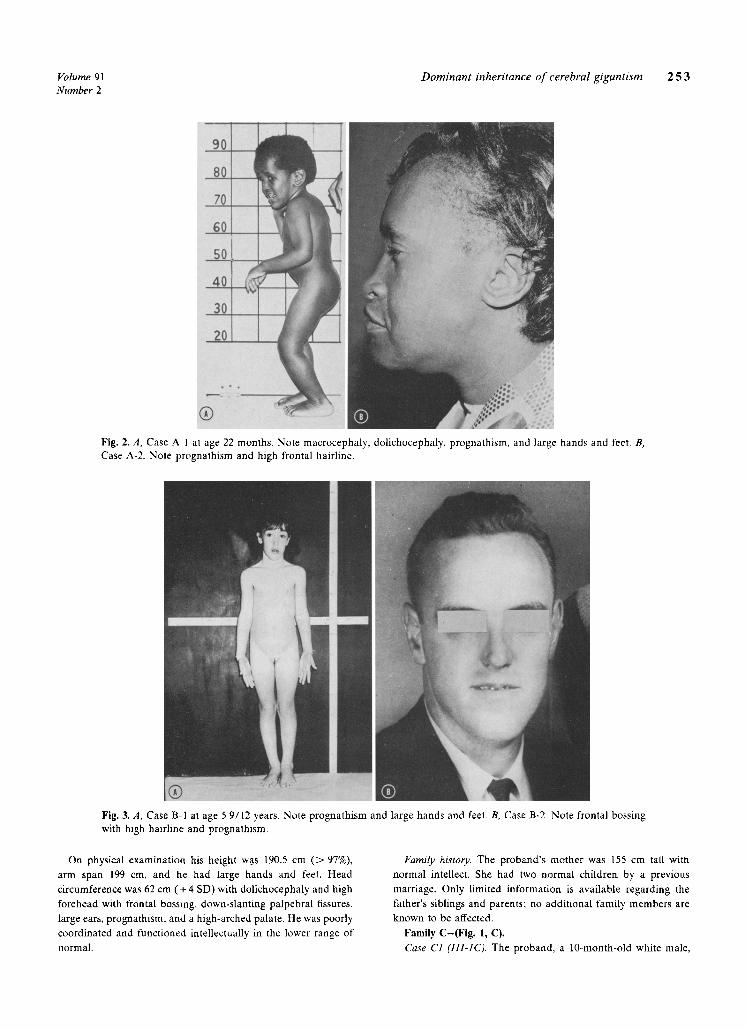
Volume 91 Dominant inheritance of cerebral gigantism 2 5 3 Number 2
c...
Fig. 2, A, Case A 1 at age 22 months, Note macrocephaly, dolichocephaly, prognathism, and large hands and feet, B, Case A-2. Note prognathism and high frontal hairline.
Fig. 3, A, Case B 1 at age 5 9/12 years, Note prognathism and large hands and feet. B, Case B-2. Note frontal bossing with high hairline and prognalhism,
On physical examination his height w.as 190.5 cm ( > 97%), arm span 199 cm, and he had large hands and feet. Head circumference was 62 cm (+ 4 SD) with dolichocephaly and high forehead with frontal bossing, down-slanting palpebral fissures, large ears, prognathism, and a high-arched palate. He was poorly coordinated and functioned intellectually in the lower range of normal.
Family history. The proband's mother was L55 cm tall with normal intellect. She had two normal children by a previous marriage. Only limited information is available regarding the father's siblings and parents; no additional family members are known to be affected,
Family C-(Fig. 1, C), Case C1 (III-1C). The proband, a 10-month-old white male,
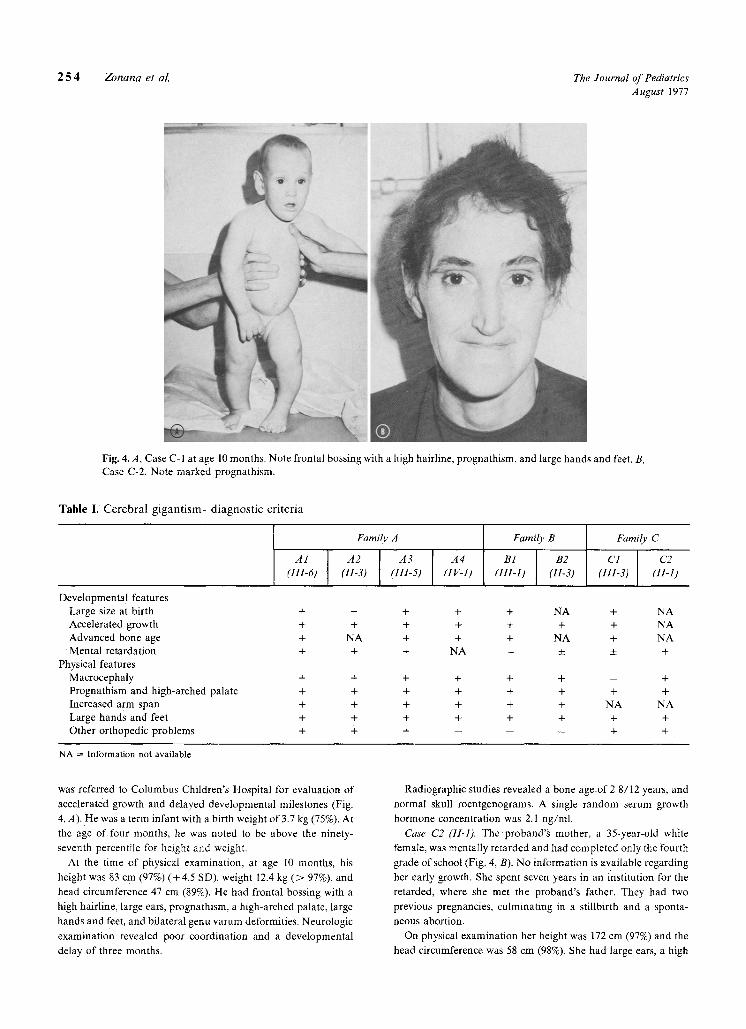
2 5 4 Zonana et al. The Journal of Pediatrics August 1977
Fig. 4.A, Case C-I at age 10 months. Note frontal bossing with a high hairline, prognathism, and large hands and feet. B, Case C-2. Note marked prognathism.
Tab l e I. C e r e b r a l g i g a n t i s m - d i a g n o s t i c cr i ter ia
A1 �9 (111-6)
Developmenta ! features Large size at birth + Accelerated growth + Advanced bone age + Mental retardation +
Physical features Macrocephaly + Prognathism and high-arehed palate + Increased arm span + Large hands a n d feet + Other orthopedic problems +
Family A
A2 A3 A4 (II-3) (III-5) (IV-I)
Family B
B1 [ B2 (IH-1) (II-3)
Family C
C1 C2 (111-3) (11-1)
+ + + + NA + NA + + + + + + NA
NA + + + NA + NA + + NA - • • +
+ + + + + - + + + + + + + +
+ + + + + NA NA + + + + + + + + -- _ _ + +
NA = Information not available.
was referred to Columbus Children's Hospital for evaluation of
accelerated growth and delayed developmental milestones (Fig.
4, A). He was a term infant with a birth weight of 3.7 kg (75%)i At
the age o f four months, he was noted to be above the ninety-
seventh percentile for height and weight.
At the time of physical examination, at age 10 months , his
height was 83 cm (97%) (+4 .5 SD), weight 12.4 kg ( > 97%), and
headci rcumference 47 cm (89%). He had frontal bossing with a
high hairline, large ears, prognathism, a high-arched palate, large
hands and feet, and bilateral genu varum deformities. Neurologic
examination revealed poor coordination and a developmental
delay of three months.
Radiographic studies revealed a bone age.of 2 8/12 years, and
normal skull roentgenograms. A single random serum growth
hormone concentration was 2.1 ng/ml .
Case C2 (11-1). The proband 's mother, a 35-year-old white
female , was mentally retarded and had completed only the fourth
grade o f school (Fig. 4, B). No information is available regarding
her early growth. She spent seven years in an institution for the
retarded, where she met the proband's father. They had two
previous pregnancies, culminating in a stillbirth and a sponta-
neous abortion.
On physical examination her height was 172 cm (97%) and the
head circumference was 58 cm (98%). She had large ears, a high

Volume 91 Dominant inheritance of cerebral gigantism 2 5 5 Number 2
frontal hairline, prognathism, large hands and feet, and moderate thoracic kyphoscoliosis. Neurologic examination revealed clum- siness, incoordination, and an IQ of approximately 70.
Family history. The 35-year-old father was mentally retarded, but had normal physical features. The mother had no siblings. No further history is available.
D I S C U S S I O N
These kindreds each demonstrate linear transmission of cerebral gigantism. Although there are no pathognomonic clinical or laboratory findings for the diagnosis of cerebral gigantism, sufficient characteristic physical and develop- "mental criteria are present in affected members in these three families to allow for an unequivocal diagnosis in each instance ~ (Table I). All of the prepubertal patients showed large size at birth, markedly accelerated early growth, and an advanced bone age. In two of the adult patients, history of a similar pattern of growth was obtained, although documentation is not available. The adult patients underwent normal pubertal development, and their final adult heights are al lhe upper end of the normal range, without striking gigantism.
Typical skeIetaI features including large hands and feet and an increased arm span were present. Kyphoscoliosis, present in the two adult women, has been a variable feature of this syndrome.: Additional orthopedic prob- lems of genu varum and pes planus deformities were present in three of the prepubertal cases. Macrocephaly, with a dolichocephalic configuration, a high frontal hair- line, prognathis m, and a high-arched palate are other characteristic dysmorphic features present in these patients.
The degree of mental deficit in cerebral gigantism has been variable.-' In our three families intelligence ranged from the lower range of normal to moderate mental retardation.
As in previous studies, growth hormone, insulin, thyroid, adrenal, gonadal, and pituitary functions appeared normal and no cause for the accelerated growth was apparent, ~ ...... " Despite earlier postulation and reports of abnormal growth hormone control or hypothal- amic dysfunction, the pathogenesis of this disorder remains unclear. ' "-~"
All three patients diagnosed as adults were ascertained through their affected children. Almost all previously reported cases have been in children, suggesting that affected parents in previously reported "sporadic" cases may not have been recognized. The apparent lack of affected adults is probably attributable to several factors. In the adult with this syndrome, final height is not strikingly increased and thus abnormal stature would not usually be a presenting feature. In the postpubertal
patient, with epiphyseal closure, advanced bone age would no longer be a helpful diagnostic aid. Intellectual deficits tend to be mild, if present, and would not necessarily provoke medical evaluation, Finally, their dysmorphic features are certainly far from striking and may be overlooked. For these reasons, and also perhaps because of a lack of awareness of this entity among internists, this entity may go undiagnosed.
The occurrence of three and perhaps four generation vertical transmission, with equal male and female involve- ment in family A, the affected mother and son in family B, and the father and daughter in family C, is suggestive of dominant inheritance of cerebral gigantism. Hansen and Friis 1'+ have recently reported an affected mother and son. They have been two previous suggestions of an affected parent and child, including a father and son; however, these cases have not been documented. '~- 1., Male-to-male transmission is not present in our pedigrees. Thus, an X- linked dominant mode of inheritance cannot be excluded. Only one affected male, however, is of reproductive age. Moreover, the equal sex ratio and similar phenotypic expression in affected males and females in the present, as welI as in previous cases, would favor autosomat domi- nant inheritance.
There have been a few previous reports of familial cases of cerebral gigantism. Concordant monozygotic twins were reported in one family with an otherwise negative family history. + Subsequently, two brothers with varying severity of the disorder were described; their maternal grandfather and maternal uncle were reported to be macrocephalic without mental retardation or gigantism. '~ Hooft and associates" also reported affected first cousins, with normal nonconsanguineous parents and an other- wise negative family history, except for a high frequency of stillbirths and spontaneous abortions.
Recently, autosomal recessive inheritance of cerebral gigantism has been postulated on the basis of a single highly inbred family. '~' The affected siblings and double first cousins are not, in our opinion, typical cases of cerebral gigantism because of the presence of atypical features such as edema at birth, undescended testis, bilateral wrist drop, dorsiflexion contractures of the feet, spindle-shaped fingers, rapidly advancing hydrocephalus with early death in one child, and relative microcephaly in another. This family probably represents a separate distinct genetic entity.
Most previously reported cases of cerebral gigantism have seemed sporadic. If the syndrome is inherited as an autosomal dominant trait, some of these cases may represent new mutations. Advanced paternal age has been found to be associated with new mutations in certain dominantly inherited syndromes. Paternal age was

2 5 6 Zonana et al. The Journal of Pediatrics August 1977
recently found to be borderl ine advanced in cerebral
gigantism, but identification o f new mutations in such
studies are dependent on close examinat ion o f the parents
to rule out minor manifestations of the disorder.'-'" Since
adults with cerebral gigantism may go undiagnosed, some
cases thought to be sporadic may in fact have had an
affected parent, potentially resulting in a spurious
decrease in the mean paternal age of "sporadic" cases. In
addition, if there is decreased biologic or social reproduc-
tive fitness in a dominant disorder, a greater percentage of
cases would be new mutations and therefore sporadic.
Two of our families suggest increased reproductive
wastage in affected women.
In conclusion, the families represented herein indicate
that cerebral gigantism may be inherited as a dominant
disorder, probably autosomal. There is increasing recog-
nition of genetic heterogeneity in many diseases, however,
and the possibility exists of heterogeneity in cerebral
gigantism as well. Fol low-up study of the reproductive
fitness and offspring of previously reported sporadic
cases, some of whom are now of reproductive age, and
continued family studies with close examinat ion of first
degree relatives will be necessary to definitively answer
these questions.
A D D E N D U M
We have recently encountered another family in which
a mother and son have typical cerebral gigantism.
We thank Dr. Guy Abraham for performing the adrenal and gonadal steroid profiles.
REFERENCES
1. Sotos JF, Dodge PR, Muirhead D, Crawford JD, and Talbot NB: Cerebral gigantism in childhood, N Engl J Med 271:109, 1964.
2. Jaeken J, Vander Schueren-Lodeweyckx M, and Eeckels R: Cerebral gigantism syndrome-a report of four cases and review of the literature, Z Kinderheilk 112:332, 1972.
3. Ott JE, and Robinson A: Cerebral gigantism, Am J Dis Child 117:357, 1969.
4. Hook EB, and Reynolds JW: Cerebral gigantism: Endocri-
nological and clinical observations of six patients including a congenital giant, concordant monozygotic twins, and a child who achieved adult gigantic size, J PI~DIATR 7:900, 1967.
5. Hooft C, Schotte H, and Van Horren G: Gigantism cerebral familial, Acta Paediatr Belg 22:173, 1968.
6. Bejar RL, Smith GF, Park S, Spellacy WN, Wolfson SL, and Nyhan WL: Cerebral gigantism-concentrations of amino acids in plasma and muscle, J PEDIATR 76:105, 1970.
7. Mace JW, and Gotlin RW: Cerebral gigantism--triad of findings helpful in diagnosis, Clin Pediatr 9:662, 1970.
8. Abraham JM, and Snodgrass GJ: Soto's syndrome of cerebral gigantism, Arch Dis Child 44:203, 1969.
9. Stephenson JN, Mellinger RC, and Manson G: Cerebral gigantism, Pediatrics 41:130, 1968.
10. Crardner-Medwin D: Cerebral gigantism? Dev Med Child Neurol 11:796, 1969.
11. Milunsky A, Cowie VA, and Donoghue EC: Cerebral gigantism in childhood. A report of two cases and a review of the literature, Pediatrics 40:395, 1967.
12. Kjellman B: Cerebral gigantism, Acta Paediatr Scand 54:603, 1965.
13. Sizonenko PC, Job JC, Sebouk S, and Rossier A: Gigan- tisme cerebral de L'enfant dosage de l'hormone de crois- sance dans le plasma, Arch Fr Pediatr 25:151, 1968.
14. Gardier B, Franchmont P, Ponte C, Nuyts JP, and Rycke- waert Ph.: Le gigantisme cerebral (Apropos d'une observa- tion avec etude de la fonction somatotrope), Pediatrie 24:325, 1969.
15. Butenandt VO, and Knorr D: Wachstum und Wachstum- shormon beim "zerebralen gigantismus," Munch Med Wochenschr 155:577, 1973.
16. Hansen FJ, and Friis B, Familial occurrence of cerebral gigantism, Sotos syndrome, Acta Paediatr Scand 65:387, 1976.
17. Weber B, Hirsch W, and Schibata K: Cerebrat gigantism: Hormonal studies and dermatoglyphic patterns, Acta Paediatr Scand 58:662, 1969.
18. McKusick VA: Cerebral gigantism (Sotos syndrome), in Mendelian inheritance in man, ed 4, Baltimore, 1975, The Johns Hopkins University Press, p 375.
19. Nevo S, Zeltzer M, Benderly, and Levy J: Evidence for autosomal recessive inheritance in cerebral gigantism, J Med Gen 11:158, 1974.
20. Jones KL, Smith DW, Harvey MAS, Hall BD, and Quan L: Older paternal age and fresh gene mutation: Date on additional disorders, J PEDIATR 86:84, 1975.




















