
Catalytic Oxidative Degradation of 17α-ethinylestradiol by FeIII-TAML/H2O2: Estrogenicities of the...
11
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/231611832 Catalytic oxidative degradation of 17α- ethinylestradiol by FeIII-TAML/H2O2: Estrogenicities of the... Article in Water Research · September 2012 DOI: 10.1016/j.watres.2012.09.012 · Source: PubMed CITATIONS 8 READS 197 5 authors, including: Jian Lin Chen City University of Hong Kong 18 PUBLICATIONS 192 CITATIONS SEE PROFILE L. James Wright University of Auckland 166 PUBLICATIONS 3,727 CITATIONS SEE PROFILE Naresh Singhal University of Auckland 99 PUBLICATIONS 835 CITATIONS SEE PROFILE All content following this page was uploaded by Naresh Singhal on 01 December 2016. The user has requested enhancement of the downloaded file. All in-text references underlined in blue are linked to publications on ResearchGate, letting you access and read them immediately.
Transcript of Catalytic Oxidative Degradation of 17α-ethinylestradiol by FeIII-TAML/H2O2: Estrogenicities of the...
Seediscussions,stats,andauthorprofilesforthispublicationat:https://www.researchgate.net/publication/231611832
Catalyticoxidativedegradationof17α-ethinylestradiolbyFeIII-TAML/H2O2:Estrogenicitiesofthe...
ArticleinWaterResearch·September2012
DOI:10.1016/j.watres.2012.09.012·Source:PubMed
CITATIONS
8
READS
197
5authors,including:
JianLinChen
CityUniversityofHongKong
18PUBLICATIONS192CITATIONS
SEEPROFILE
L.JamesWright
UniversityofAuckland
166PUBLICATIONS3,727CITATIONS
SEEPROFILE
NareshSinghal
UniversityofAuckland
99PUBLICATIONS835CITATIONS
SEEPROFILE
AllcontentfollowingthispagewasuploadedbyNareshSinghalon01December2016.
Theuserhasrequestedenhancementofthedownloadedfile.Allin-textreferencesunderlinedinblue
arelinkedtopublicationsonResearchGate,lettingyouaccessandreadthemimmediately.

ww.sciencedirect.com
wat e r r e s e a r c h 4 6 ( 2 0 1 2 ) 6 3 0 9e6 3 1 8
Available online at w
journal homepage: www.elsevier .com/locate/watres
Catalytic oxidative degradation of 17a-ethinylestradiol byFeIII-TAML/H2O2: Estrogenicities of the products of partial,and extensive oxidation
Jian Lin Chen a, Shanthinie Ravindran a, Simon Swift b, L. James Wright c,Naresh Singhal a,*aDepartment of Civil & Environmental Engineering, University of Auckland, Auckland 1142, New ZealandbMolecular Medicine and Pathology, University of Auckland, Auckland 1142, New ZealandcSchool of Chemical Sciences, University of Auckland, Auckland 1142, New Zealand
a r t i c l e i n f o
Article history:
Received 9 April 2012
Received in revised form
29 August 2012
Accepted 3 September 2012
Available online 12 September 2012
Keywords:
FeIII-TAML
FeIII-B*
EE2Estrogenicity
Catalysis
Oxidation
Daughter products
* Corresponding author. Department of CiviRoad, Auckland 1142, New Zealand. Tel.: þ6
E-mail address: [email protected]/$ e see front matter ª 2012 Elsevhttp://dx.doi.org/10.1016/j.watres.2012.09.012
a b s t r a c t
The oxidative degradation of the oral contraceptive 17a-ethinylestradiol (EE2) in water by
a new advanced catalytic oxidation process was investigated. The oxidant employed was
hydrogen peroxide in aqueous solution and the catalyst was the iron tetra-amido macro-
cyclic ligand (FeIII-TAML) complex that has been designated Na[Fe(H2O)(B*)] (FeIII-B*). EE2(10 mM) was oxidised rapidly by the FeIII-B*/H2O2 (5 nM/4 mM) catalytic oxidation system at
25 �C, and for reactions at pH 8.40e11.00, no unchanged EE2 was detected in the reaction
mixtures after 60 min. No oxidation of EE2 was detected in blank reactions using either
H2O2 or FeIII-B* alone. The maximum rate of EE2 loss occurred at pH 10.21. At this pH the
half-life of EE2 was 2.1 min and the oxidised products showed around 30% estrogenicity
removal, as determined by the yeast estrogen screen (YES) bioassay. At pH 11.00, partial
oxidation of EE2 by FeIII-B*/H2O2 (5 nM/4 mM) was studied (half-life of EE2 was 14.5 min) and
in this case the initial intermediates formed were a mixture of the epimers 17a-ethynyl-
1,4-estradiene-10a,17b-diol-3-one (1a) and 17a-ethynyl-1,4-estradiene-10b,17b-diol-3-one
(1b) (identified by LC-ToF-MS and 1H NMR spectroscopy). Significantly, this product
mixture displayed a slightly higher estrogenicity than EE2 itself, as determined by the YES
bioassay. Upon the addition of further aliquots of FeIII-B* (to give a FeIII-B* concentration of
500 nM) and H2O2 (to bring the concentration up to 4 mM assuming the final concentration
had dropped to zero) to this reaction mixture the amounts of 1a and 1b slowly decreased to
zero over a 60 min period as they were oxidised to unidentified products that showed no
estrogenicity. Thus, partial oxidation of EE2 gave products that have slightly increased
estrogenicity, whereas more extensive oxidation by the advanced catalytic oxidation
system completely removed all estrogenicity. These results underscore the importance of
controlling the level of oxidation during the removal of EE2 from water by oxidative
processes.
ª 2012 Elsevier Ltd. All rights reserved.
l & Environmental Engineering, The University of Auckland, Private Bag 92019, 3 Grafton4 9 923 4512; fax: þ64 9 373 7462.z (N. Singhal).ier Ltd. All rights reserved.

wat e r r e s e a r c h 4 6 ( 2 0 1 2 ) 6 3 0 9e6 3 1 86310
1. Introduction peroxide is a readily manufactured oxidant that is free of both
Endocrine disrupting compounds (EDCs) are subclass of
organic contaminants that have been widely detected in
wastewater and surface waters (Kolpin et al., 2002). Among
them, the synthetic estrogen 17a-ethinylestradiol (EE2),
a component of oral contraceptives since 1970s has been well
documented in the literature to cause endocrine disruption in
non-target species (Sumpter and Johnson, 2005). In vivo tests
show that EE2 is approximately 10-fold more potent than the
female sex hormone 17a-estradiol (E2) in inducing vitellogenin
in male sheepshead minnow (exposed for 16 days) (Thorpe
et al., 2003). On the basis of in vivo estrogenic potency on the
life-cycle of the fathead minnow (Pimephales promelas) (Lange
et al., 2001) EE2 was suggested as one of the most important
EDCs in effluents from wastewater treatment plants (WTPs)
(Johnson and Sumpter, 2001). The predicted no effect
concentration (PNEC) is proposed at 0.1 ng L�1 (0.1 ppt) for EE2based upon the fertilization success in full life cycle study
with zebra fish in a report commissioned by the Environment
Agency of England and Wales (Young et al., 2002). Effluents
from conventional WTPs often contain EE2 in excess of its
PNEC, e.g. typical sewage effluent EE2 concentrations were
found to be ca. 0.5 ng L�1 (Hashimoto et al., 2007).
Several studies have shown that both ozonation and
chlorination are useful technologies for the oxidation of
pharmaceuticals and EDCs during water treatment (Lee et al.,
2008; Lee and Gunten, 2008; Ternes et al., 2002). Ozone treat-
ment would be effective but expensive, whereas chlorination
would be cheaper but can potentially produce various unde-
sirable chlorinated disinfection by-products (DBPs)
(Crittenden et al. 2005). It has been demonstrated by E-screen
assays that showed oxidation with ozone resulted in the rapid
transformation of EE2, reaching a stabilized estrogenic level
(proliferative effect ¼ 2.5) in 10 min, whereas for chlorination
it tookmore than 120 min for elicited estrogenicity to stabilize
(proliferative effect¼ 8.2) (Alum et al., 2004). Potassium ferrate
(Fe(VI)) is an emerging water treatment chemical that can be
dosed directly into water and its effectiveness for the oxida-
tive removal of phenolic EDCs has been confirmed in both
natural water and wastewater (Lee et al., 2005). As a stoichio-
metric oxidant, potassium ferrate oxidizes reduced sulfur-
containing compounds, pharmaceuticals, and endocrine dis-
ruptors (He et al., 2009; Lee et al., 2009). However, because of
its low selectivity, for example it is also highly effective for
phosphate removal, Fe(VI) shows low efficiency overall for the
removal of organic pollutants when applied in the real
wastewater matrices (Lee et al., 2009). More recently it has
been demonstrated that iron complexes of specially designed
tetra-amido macrocyclic ligands (FeIII-TAMLs) can catalyse
oxidations by H2O2 in water and that the resulting oxidation
systems can deeply oxidize and significantly mineralize water
contaminants such as pentachlorophenol, trichlorophenol
(Gupta et al., 2002) and toxic thiophosphate pesticide hydro-
lyzates (Chanda et al., 2006a). Importantly, the degradation
products of these reactions do not appear to present toxicity
concerns. The FeIII-TAML/H2O2 oxidation system is regarded
as “green” because the catalysts themselves are not toxic and
they contain no inherently toxic elements. Also, hydrogen
chlorine and toxic heavy metals.
The FeIII-B*/H2O2 oxidation system has also been shown to
effectively eliminate estrogenicity fromwater contaminated by
natural and synthetic estrogens, which are environmental
persistent and resistant to bacterial degradation (Khunjar et al.,
2011; Snyder et al., 1999). In particular, treatment of an aqueous
solution of EE2 (80 mM) with the FeIII-TAML designated FeIII-B*
(83 nM) and H2O2 (4 mM) for 60 min at pH 10.0 has been shown
to result in >95% removal of EE2 with accompanying loss of
estrogenicity (ca. 95% reduction). In this study we have inves-
tigated the oxidative removal of EE2 by this method in more
detail, specifically the partial EE2 oxidation by FeIII-TAML to
intermediate products with the aim of assessing the change in
overall estrogenicity in sub-optimal degradation environments.
2. Experimental
2.1. Materials and reagents
EE2 (>98%) was obtained from Sapphire Bioscience (New
Zealand). The molecular structure of EE2 is shown in Fig. 1a.
Solvents were high-performance liquid chromatography
grade from Ajax FineChem (New Zealand). H2O2 (30%) was
purchased from Ajax FineChem (New Zealand). The synthesis
of FeIII-B* has been reported in the previous study (Horwitz
Colin et al., 2006) and the structure is shown in Fig. 1b.
2.2. Methodology
Triplicate experiments were performed for each reaction. The
initial conditions of reactions were: 10 mM EE2 dissolved in the
0.01 M Na2CO3/NaHCO3 buffer, 5 nM FeIII-B*, 4 mM H2O2,
shaking at 120 cycles min�1 in an incubation shaker at 25 �C.The degradation of EE2 in the FeIII-B*/H2O2 systemwas studied
at pH 8.40e11.00. The reactions were carried out for each time
point (0, 2, 5, 10, 15, 30 and 60 min) in 50 mL buffer in different
amber glass reagent bottles. Previous studies have shown that
the FeIII-TAML catalysts undergo self-destruction at pH 11 and
25 �C with half-lives of less than 30 min (Chanda et al., 2006b),
suggesting that the FeIII-TAML catalyst was exhausted after
1 h of reaction. Therefore, after the reaction at pH 11.00 had
continued for 60min (designated reaction I) an extra aliquot of
H2O2 and FeIII-B* in the same buffer solution was added to the
reaction in succession to make the [FeIII-B*/H2O2] either 5 nM/
4 mM (designated reaction II), 50 nM/4 mM (reaction III) or
500 nM/4 mM (reaction IV) (assuming the concentrations of
these reactants were zero before the addition). Samples were
taken from reactions II, III and IV at total times 65, 70, 90 and
120min. Triplicate reactionswere carried out for each of these
sampling time points. At the termination of reactions, cata-
lase (12,000 units which is 60 times the concentration capable
of destroying 4 mM H2O2 in 1 min) was added, and then the
sampleswere acidified to pH 3.5 by addition of 4MHCl and left
to stand a further 1 min. This destroyed residual hydrogen
peroxide and deactivated the FeIII-B* through acid-induced
demetalation (Shappell et al., 2008). Control reactions were
carried out in the absence of either H2O2 or FeIII-B* or both to

OH
C C
HO
12
3
45
67
8
910
1112
1314
15
16
17 H
H3C
H
H
H
a b
Fig. 1 e Chemical structures of a: EE2 and b: FeIII-B*.
wat e r r e s e a r c h 4 6 ( 2 0 1 2 ) 6 3 0 9e6 3 1 8 6311
quantify the background decay of EE2 and a blank reaction
(FeIII-B* plus H2O2 in the buffer but without EE2) was also
evaluated.
2.3. Analytical methods and characterization
2.3.1. Liquid chromatographyemass spectroscopy (LCeMS)Liquid chromatography and analysis were performed on
a Shimadzu Series LCeMS 2020 system equipped with
a degasser, a binary pump, an autosampler, and a column
oven. The detector of this system was a quadrupole mass
spectrometer. The ionization method used was electrospray
ionization (ESI). The analysis was carried on a C12 polar
reversed-phase column (Synergi MAX-RP, Phenomenex) with
the particle size of 4 mm, the length of 150 mm and inner
diameter of 2.0 mm. The column was guarded with a pre-
column with 4 mm � 3.0 mm C12 security guard cartridge
(Synergi MAX-RP, Phenomenex). The column oven tempera-
ture was set to 30 �C. The mobile phase was 65% organic
solvent (acetoniltrile/methanol, 2/3) in deionized water at
a flow rate of 0.2 mL min�1. Ten microliter samples were
infused into the LC column. Samples were analyzed by MS
under full scanmode (m/z 50e500). Based on the results of full
scans, negative selected ion monitoring (SIM) of m/z 295 and
311 were studied for the residual EE2 and the initially oxidised
intermediate, respectively. Meanwhile, the samples from the
control and blank reactions were also analyzed under the
negative SIM mode of m/z 295 and 311.
2.3.2. EstrogenicityThe estrogenicity of the samples from reactions I, II, III and IV,
as well the sample from the end of reaction at pH 10.21 were
assessed by a four-hour yeast estrogen screen (YES) bioassay
(Balsiger et al., 2010; Liu and Picard, 1998; Picard et al., 1990).
All samples and the blank reaction were evaluated by
measuring the hormone-induced chemiluminescent signal on
Xenogen IVIS-200 optical in vivo imaging system. A sequence
of EE2 solutions at different concentrations (ranging from
8 � 10�5 to 8 mM) was prepared to study the response of
estrogenicity to different EE2 concentration.
2.3.3. Liquid chromatographyetime of flight-massspectroscopy (LCeToF-MS)The solution at the end of reaction 1 and after addition of
catalase and acid as described above was filtered by GF 2 glass
fiber filter, and concentrated by solid phase extraction with
a 500-mg hydrophilicelipophilic balance (HLB) cartridge from
Waters Corp. The cartridges were preconditioned with 5 mL
methanol followed by 5 mL Milli-Q water. The acidified and
filtered solution was loaded onto the preconditioned cartridge
at a flow rate of 10.0 mL min�1. The cartridge was dried under
high vacuum and then eluted with 5 mL methanol/acetone (1/
1, v/v) at a flow rate of 4 mL min�1. The eluate was collected
and the organic solventwas evaporated to dryness by exposure
to a slow stream of nitrogen gas. Methanol (0.5mL) was used to
dissolve the residue and the solution made up to 1 mL with
deionized water. Ten microliters of this concentrated sample
was introduced using flow-injection (FIA) into an ESI source
(positive ionizationmode, capillary voltage�4500V) using pure
nitrogen as drying/nebulizer gas (180 �C, 6 L min�1 drying gas,
nebulizer pressure 3 bar) in a Bruker micro-ToF-QII (Bruker
Daltonics, Bremen, Germany) coupled with a Dionex Ultimate
3000 HPLC with autosampler (Dionex, Germering, Germany)
and the same column used in previous LCeMS analysis. The
mobile phase was 65% organic solvent (acetoniltrile/methanol,
2/3) in deionized water containing 0.1% formic acid at a flow
rate of 0.2 mL min�1. Data obtained for 1a/b are as follows: MS
(ESI, DCM) (m/z): calc. for C20H24NaO3þ, 335.1618, found
335.1616; calc. for C20H25O3þ, 313.1798, found 313.1811.
2.3.4. Nuclear magnetic resonance (NMR)Further structure confirmation of initial intermediates was
performed by Bruker AVIII-400 MHz spectrometer (Bruker
BioSpin), which provided 1H NMR analysis of pure EE2 dis-
solved in CDCl3. A portion of the same sample (dissolved in
methanol) that was used in the LC-ToF-MS analysis was
evaporated to dryness by a slow stream of nitrogen gas, dis-
solved in CDCl3 and the 1H NMR spectrum obtained. 1H NMR
(CDCl3, 400 MHz) data for 1a are: d (pprn) ¼ 7.17 (d, 1H, C1-H);
6.36 (d.d., lH, C2-H); 6.16 (t, lH, C4-H); for 1b are: d (pprn) ¼ 7.10
(d, 1H, C1-H); 6.16 (d.d., lH, C2-H); 6.01 (t, lH, C4-H).
2.4. Calculation of the frontier electron density
The Frontier Electron Density of different EE2 atoms was
calculated using Gaussian 03 program (Gaussian, Inc.) Opti-
mization of the EE2 structure was carried out with the STO-3G
basis set at level of Unrestricted Hartree�Fock (UHF) (Yi and
Harper, 2007). Based on this optimized structure, the highest
occupied molecular orbital (HOMO) was calculated using the
wat e r r e s e a r c h 4 6 ( 2 0 1 2 ) 6 3 0 9e6 3 1 86312
samemethod and basis set. The frontier electron density�c4r
can be calculated as � c4r ¼ ½2 �PðCriHOMOÞ2�, where r is the
number of carbon atoms in i: 2s, 2px, 2py, and 2pz.
3. Results and discussion
3.1. Effect of pH on EE2 degradation by FeIII-B*/H2O2
There were minimal changes in the EE2 concentrations in the
control reactions (0.5e0.9%), indicating that the degradation
of EE2 by either H2O2 or FeIII-B* alone was negligible. The time-
dependent degradation kinetics of EE2 when treated with the
FeIII-B*/H2O2 system was described using the first-order decay
kinetics Shappell et al., 2008, C/C0 ¼ S/S0 ¼ e�kt, where C0
(mg L�1) is the initial concentration, C is the concentration at
time t (min), S0 is the initial peak area corresponding to the
initial concentration C0, S is the peak area corresponding to
the concentration C at time t (min), and k (min�1) is the
apparent decay coefficient. Plots of residual EE2 (determined
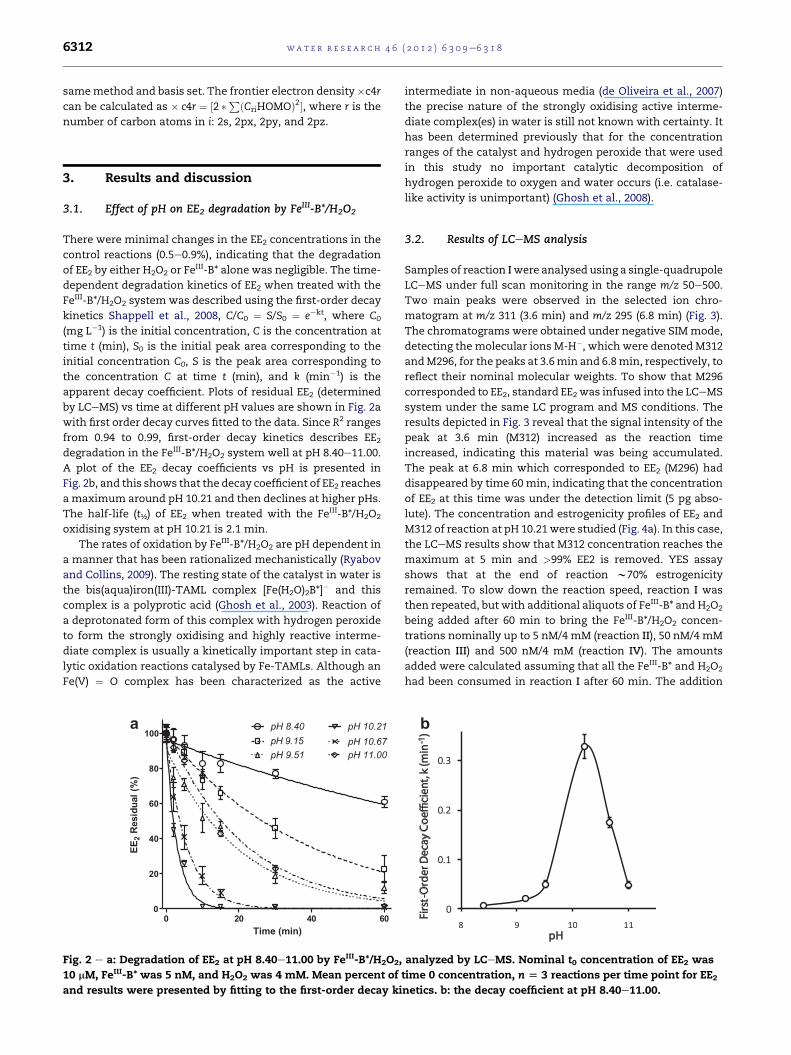
by LCeMS) vs time at different pH values are shown in Fig. 2a
with first order decay curves fitted to the data. Since R2 ranges
from 0.94 to 0.99, first-order decay kinetics describes EE2degradation in the FeIII-B*/H2O2 system well at pH 8.40e11.00.
A plot of the EE2 decay coefficients vs pH is presented in
Fig. 2b, and this shows that the decay coefficient of EE2 reaches
a maximum around pH 10.21 and then declines at higher pHs.
The half-life (t½) of EE2 when treated with the FeIII-B*/H2O2
oxidising system at pH 10.21 is 2.1 min.
The rates of oxidation by FeIII-B*/H2O2 are pH dependent in
a manner that has been rationalized mechanistically (Ryabov
and Collins, 2009). The resting state of the catalyst in water is
the bis(aqua)iron(III)-TAML complex [Fe(H2O)2B*]� and this
complex is a polyprotic acid (Ghosh et al., 2003). Reaction of
a deprotonated form of this complex with hydrogen peroxide
to form the strongly oxidising and highly reactive interme-
diate complex is usually a kinetically important step in cata-
lytic oxidation reactions catalysed by Fe-TAMLs. Although an
Fe(V) ¼ O complex has been characterized as the active
a
0 20 40 60
0
20
40
60
80
100
pH 8.40
pH 9.15
pH 9.51
pH 10.21
pH 11.00
pH 10.67
Time (min)
EE
2 R
esid
ual (%
)
Fig. 2 e a: Degradation of EE2 at pH 8.40e11.00 by FeIII-B*/H2O2,
10 mM, FeIII-B* was 5 nM, and H2O2 was 4 mM. Mean percent of
and results were presented by fitting to the first-order decay ki
intermediate in non-aqueous media (de Oliveira et al., 2007)
the precise nature of the strongly oxidising active interme-
diate complex(es) in water is still not known with certainty. It
has been determined previously that for the concentration
ranges of the catalyst and hydrogen peroxide that were used
in this study no important catalytic decomposition of
hydrogen peroxide to oxygen and water occurs (i.e. catalase-
like activity is unimportant) (Ghosh et al., 2008).
3.2. Results of LCeMS analysis
Samples of reaction Iwere analysed using a single-quadrupole
LCeMS under full scan monitoring in the range m/z 50e500.
Two main peaks were observed in the selected ion chro-
matogram at m/z 311 (3.6 min) and m/z 295 (6.8 min) (Fig. 3).
The chromatograms were obtained under negative SIMmode,
detecting the molecular ions M-H�, which were denoted M312
andM296, for the peaks at 3.6min and 6.8min, respectively, to
reflect their nominal molecular weights. To show that M296
corresponded to EE2, standard EE2 was infused into the LCeMS
system under the same LC program and MS conditions. The
results depicted in Fig. 3 reveal that the signal intensity of the
peak at 3.6 min (M312) increased as the reaction time
increased, indicating this material was being accumulated.
The peak at 6.8 min which corresponded to EE2 (M296) had
disappeared by time 60 min, indicating that the concentration
of EE2 at this time was under the detection limit (5 pg abso-
lute). The concentration and estrogenicity profiles of EE2 and
M312 of reaction at pH 10.21 were studied (Fig. 4a). In this case,
the LCeMS results show that M312 concentration reaches the
maximum at 5 min and >99% EE2 is removed. YES assay
shows that at the end of reaction w70% estrogenicity
remained. To slow down the reaction speed, reaction I was
then repeated, but with additional aliquots of FeIII-B* and H2O2
being added after 60 min to bring the FeIII-B*/H2O2 concen-
trations nominally up to 5 nM/4 mM (reaction II), 50 nM/4 mM
(reaction III) and 500 nM/4 mM (reaction IV). The amounts
added were calculated assuming that all the FeIII-B* and H2O2
had been consumed in reaction I after 60 min. The addition
b
analyzed by LCeMS. Nominal t0 concentration of EE2 was
time 0 concentration, n [ 3 reactions per time point for EE2netics. b: the decay coefficient at pH 8.40e11.00.
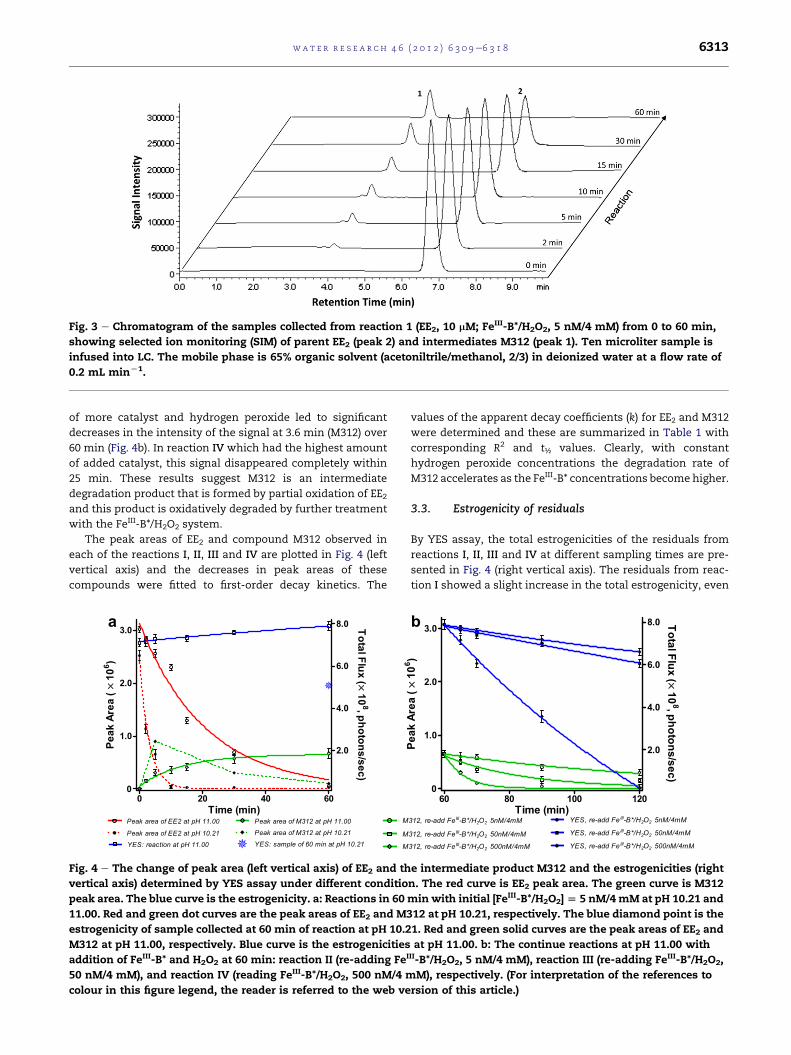
Fig. 3 e Chromatogram of the samples collected from reaction 1 (EE2, 10 mM; FeIII-B*/H2O2, 5 nM/4 mM) from 0 to 60 min,
showing selected ion monitoring (SIM) of parent EE2 (peak 2) and intermediates M312 (peak 1). Ten microliter sample is
infused into LC. The mobile phase is 65% organic solvent (acetoniltrile/methanol, 2/3) in deionized water at a flow rate of
0.2 mL minL1.
wat e r r e s e a r c h 4 6 ( 2 0 1 2 ) 6 3 0 9e6 3 1 8 6313
of more catalyst and hydrogen peroxide led to significant
decreases in the intensity of the signal at 3.6 min (M312) over
60 min (Fig. 4b). In reaction IV which had the highest amount
of added catalyst, this signal disappeared completely within
25 min. These results suggest M312 is an intermediate
degradation product that is formed by partial oxidation of EE2and this product is oxidatively degraded by further treatment
with the FeIII-B*/H2O2 system.
The peak areas of EE2 and compound M312 observed in
each of the reactions I, II, III and IV are plotted in Fig. 4 (left
vertical axis) and the decreases in peak areas of these
compounds were fitted to first-order decay kinetics. The
a
Fig. 4 e The change of peak area (left vertical axis) of EE2 and th
vertical axis) determined by YES assay under different conditio
peak area. The blue curve is the estrogenicity. a: Reactions in 60
11.00. Red and green dot curves are the peak areas of EE2 and M3
estrogenicity of sample collected at 60 min of reaction at pH 10.2
M312 at pH 11.00, respectively. Blue curve is the estrogenicities
addition of FeIII-B* and H2O2 at 60 min: reaction II (re-adding FeI
50 nM/4 mM), and reaction IV (reading FeIII-B*/H2O2, 500 nM/4 m
colour in this figure legend, the reader is referred to the web ve
values of the apparent decay coefficients (k) for EE2 and M312
were determined and these are summarized in Table 1 with
corresponding R2 and t½ values. Clearly, with constant
hydrogen peroxide concentrations the degradation rate of
M312 accelerates as the FeIII-B* concentrations become higher.
3.3. Estrogenicity of residuals
By YES assay, the total estrogenicities of the residuals from
reactions I, II, III and IV at different sampling times are pre-
sented in Fig. 4 (right vertical axis). The residuals from reac-
tion I showed a slight increase in the total estrogenicity, even
b
e intermediate product M312 and the estrogenicities (right
n. The red curve is EE2 peak area. The green curve is M312
min with initial [FeIII-B*/H2O2][ 5 nM/4mM at pH 10.21 and
12 at pH 10.21, respectively. The blue diamond point is the
1. Red and green solid curves are the peak areas of EE2 and
at pH 11.00. b: The continue reactions at pH 11.00 withII-B*/H2O2, 5 nM/4 mM), reaction III (re-adding FeIII-B*/H2O2,
M), respectively. (For interpretation of the references to
rsion of this article.)
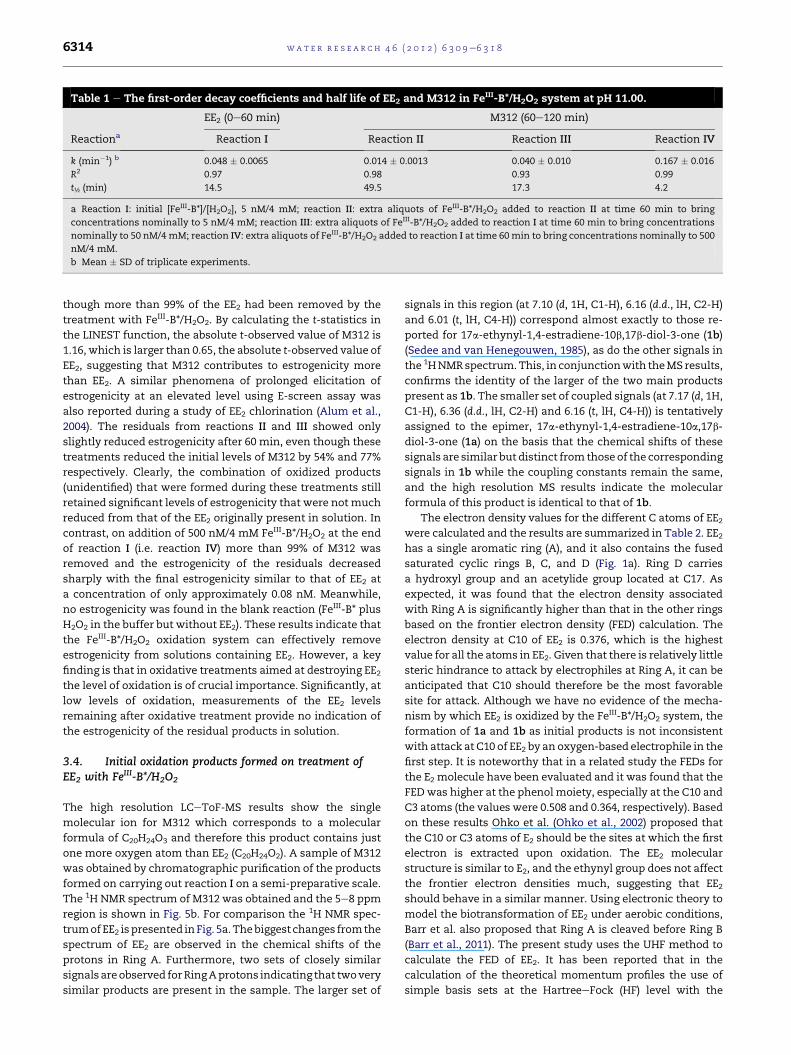
Table 1 e The first-order decay coefficients and half life of EE2 and M312 in FeIII-B*/H2O2 system at pH 11.00.
EE2 (0e60 min) M312 (60e120 min)
Reactiona Reaction I Reaction II Reaction III Reaction IV
k (min�1) b 0.048 � 0.0065 0.014 � 0.0013 0.040 � 0.010 0.167 � 0.016
R2 0.97 0.98 0.93 0.99
t½ (min) 14.5 49.5 17.3 4.2
a Reaction I: initial [FeIII-B*]/[H2O2], 5 nM/4 mM; reaction II: extra aliquots of FeIII-B*/H2O2 added to reaction II at time 60 min to bring
concentrations nominally to 5 nM/4 mM; reaction III: extra aliquots of FeIII-B*/H2O2 added to reaction I at time 60 min to bring concentrations
nominally to 50 nM/4 mM; reaction IV: extra aliquots of FeIII-B*/H2O2 added to reaction I at time 60min to bring concentrations nominally to 500
nM/4 mM.
b Mean � SD of triplicate experiments.
wat e r r e s e a r c h 4 6 ( 2 0 1 2 ) 6 3 0 9e6 3 1 86314
though more than 99% of the EE2 had been removed by the
treatment with FeIII-B*/H2O2. By calculating the t-statistics in
the LINEST function, the absolute t-observed value of M312 is
1.16, which is larger than 0.65, the absolute t-observed value of
EE2, suggesting that M312 contributes to estrogenicity more
than EE2. A similar phenomena of prolonged elicitation of
estrogenicity at an elevated level using E-screen assay was
also reported during a study of EE2 chlorination (Alum et al.,
2004). The residuals from reactions II and III showed only
slightly reduced estrogenicity after 60 min, even though these
treatments reduced the initial levels of M312 by 54% and 77%
respectively. Clearly, the combination of oxidized products
(unidentified) that were formed during these treatments still
retained significant levels of estrogenicity that were not much
reduced from that of the EE2 originally present in solution. In
contrast, on addition of 500 nM/4 mM FeIII-B*/H2O2 at the end
of reaction I (i.e. reaction IV) more than 99% of M312 was
removed and the estrogenicity of the residuals decreased
sharply with the final estrogenicity similar to that of EE2 at
a concentration of only approximately 0.08 nM. Meanwhile,
no estrogenicity was found in the blank reaction (FeIII-B* plus
H2O2 in the buffer but without EE2). These results indicate that
the FeIII-B*/H2O2 oxidation system can effectively remove
estrogenicity from solutions containing EE2. However, a key
finding is that in oxidative treatments aimed at destroying EE2the level of oxidation is of crucial importance. Significantly, at
low levels of oxidation, measurements of the EE2 levels
remaining after oxidative treatment provide no indication of
the estrogenicity of the residual products in solution.
3.4. Initial oxidation products formed on treatment ofEE2 with FeIII-B*/H2O2
The high resolution LCeToF-MS results show the single
molecular ion for M312 which corresponds to a molecular
formula of C20H24O3 and therefore this product contains just
one more oxygen atom than EE2 (C20H24O2). A sample of M312
was obtained by chromatographic purification of the products
formed on carrying out reaction I on a semi-preparative scale.
The 1H NMR spectrum of M312 was obtained and the 5e8 ppm
region is shown in Fig. 5b. For comparison the 1H NMR spec-
trumofEE2 is presented inFig. 5a.Thebiggest changes fromthe
spectrum of EE2 are observed in the chemical shifts of the
protons in Ring A. Furthermore, two sets of closely similar
signals areobserved forRingAprotons indicating that twovery
similar products are present in the sample. The larger set of
signals in this region (at 7.10 (d, 1H, C1-H), 6.16 (d.d., lH, C2-H)
and 6.01 (t, lH, C4-H)) correspond almost exactly to those re-
ported for 17a-ethynyl-1,4-estradiene-10b,17b-diol-3-one (1b)
(Sedee and van Henegouwen, 1985), as do the other signals in
the 1HNMRspectrum.This, in conjunctionwith theMS results,
confirms the identity of the larger of the two main products
present as 1b. The smaller set of coupled signals (at 7.17 (d, 1H,
C1-H), 6.36 (d.d., lH, C2-H) and 6.16 (t, lH, C4-H)) is tentatively
assigned to the epimer, 17a-ethynyl-1,4-estradiene-10a,17b-
diol-3-one (1a) on the basis that the chemical shifts of these
signals are similar but distinct from thoseof the corresponding
signals in 1b while the coupling constants remain the same,
and the high resolution MS results indicate the molecular
formula of this product is identical to that of 1b.
The electron density values for the different C atoms of EE2were calculated and the results are summarized in Table 2. EE2has a single aromatic ring (A), and it also contains the fused
saturated cyclic rings B, C, and D (Fig. 1a). Ring D carries
a hydroxyl group and an acetylide group located at C17. As
expected, it was found that the electron density associated
with Ring A is significantly higher than that in the other rings
based on the frontier electron density (FED) calculation. The
electron density at C10 of EE2 is 0.376, which is the highest
value for all the atoms in EE2. Given that there is relatively little
steric hindrance to attack by electrophiles at Ring A, it can be
anticipated that C10 should therefore be the most favorable
site for attack. Although we have no evidence of the mecha-
nism by which EE2 is oxidized by the FeIII-B*/H2O2 system, the
formation of 1a and 1b as initial products is not inconsistent
with attack at C10 of EE2 by an oxygen-based electrophile in the
first step. It is noteworthy that in a related study the FEDs for
the E2 molecule have been evaluated and it was found that the
FEDwas higher at the phenol moiety, especially at the C10 and
C3 atoms (the values were 0.508 and 0.364, respectively). Based
on these results Ohko et al. (Ohko et al., 2002) proposed that
the C10 or C3 atoms of E2 should be the sites at which the first
electron is extracted upon oxidation. The EE2 molecular
structure is similar to E2, and the ethynyl group does not affect
the frontier electron densities much, suggesting that EE2should behave in a similar manner. Using electronic theory to
model the biotransformation of EE2 under aerobic conditions,
Barr et al. also proposed that Ring A is cleaved before Ring B
(Barr et al., 2011). The present study uses the UHF method to
calculate the FED of EE2. It has been reported that in the
calculation of the theoretical momentum profiles the use of
simple basis sets at the HartreeeFock (HF) level with the
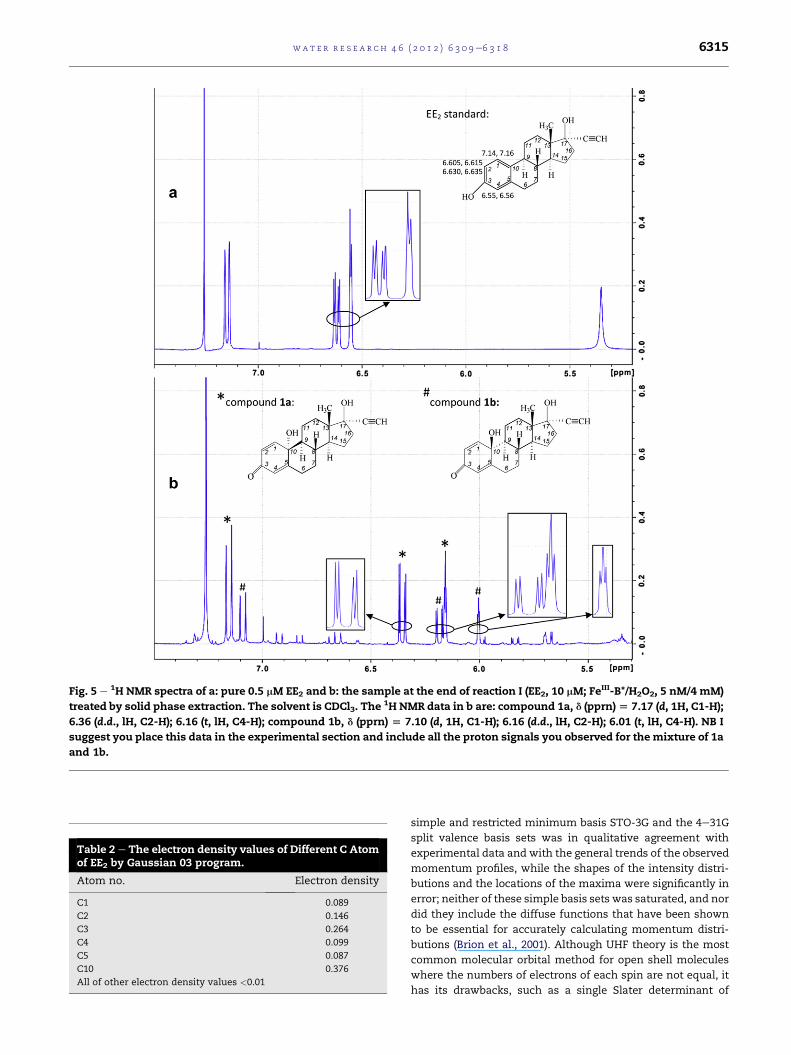
Fig. 5 e 1H NMR spectra of a: pure 0.5 mM EE2 and b: the sample at the end of reaction I (EE2, 10 mM; FeIII-B*/H2O2, 5 nM/4 mM)
treated by solid phase extraction. The solvent is CDCl3. The1H NMR data in b are: compound 1a, d (pprn)[ 7.17 (d, 1H, C1-H);
6.36 (d.d., lH, C2-H); 6.16 (t, lH, C4-H); compound 1b, d (pprn) [ 7.10 (d, 1H, C1-H); 6.16 (d.d., lH, C2-H); 6.01 (t, lH, C4-H). NB I
suggest you place this data in the experimental section and include all the proton signals you observed for themixture of 1a
and 1b.
Table 2 e The electron density values of Different C Atomof EE2 by Gaussian 03 program.
Atom no. Electron density
C1 0.089
C2 0.146
C3 0.264
C4 0.099
C5 0.087
C10 0.376
All of other electron density values <0.01
wat e r r e s e a r c h 4 6 ( 2 0 1 2 ) 6 3 0 9e6 3 1 8 6315
simple and restricted minimum basis STO-3G and the 4e31G
split valence basis sets was in qualitative agreement with
experimental data and with the general trends of the observed
momentum profiles, while the shapes of the intensity distri-
butions and the locations of the maxima were significantly in
error; neither of these simple basis sets was saturated, and nor
did they include the diffuse functions that have been shown
to be essential for accurately calculating momentum distri-
butions (Brion et al., 2001). Although UHF theory is the most
common molecular orbital method for open shell molecules
where the numbers of electrons of each spin are not equal, it
has its drawbacks, such as a single Slater determinant of

wat e r r e s e a r c h 4 6 ( 2 0 1 2 ) 6 3 0 9e6 3 1 86316
different orbitals for different spins is not a satisfactory
eigenfunction of the total spin operator-<S2> and that the
electronic state can be contaminated by excited states. Alter-
native methods for calculating the electronic spin density
ratios include the density functional theory (DFT) and second-
order MøllerePlesset perturbation (UMP) method. The unre-
stricted DFT methods (generalized gradient approximation or
hybrid functionals alike) are less affected by the phenomenon
of spin contamination than the HartreeeFock theory and also
give better predictions of the electron spin resonance spectra
of organic radicals (Boone et al., 2003).
Structure-activity relationships for natural, synthetic and
environmental estrogens indicate that the H-bonding ability
of the phenolic ring and the presence of a ring structure as
being the primary contributors to estrogenicity of compounds
(Fang et al., 2001). The results of 1H NMR in the present study
(Fig. 5) show that except for C3 and C10 there is no difference
between themain ring structure of EE2 and the intermediates.
The phenolic ring (Ring A) is therefore not destroyed and the
intermediates would be expected to show estrogenicity.
Investigations of the oxidation of EE2, e.g. by ozone (O3),
oxygen under photosensitizing conditions, hydroxyl radical
($OH), chlorine dioxide, ferrate (Fe(VI)), cytochrome P450,
chlorination and bromination, have all confirmed that reac-
tion initially occurs at Ring A (Alum et al., 2004; Lee et al., 2008;
Lee and Gunten, 2008; Skotnicka-Pitak et al., 2008; Wang et al.,
2004). Oxidative biodegradation of EE2 also proceeds via initial
oxidation of Ring A. Thus, in the biodegradation of EE2 by
Sphingobacterium sp. JCR5 oxidation to estrone (E1) initially
occurs via oxidation of Ring A and this is then followed by ring
opening oxidation reactions on Ring B (Haiyan et al., 2007; Ren
et al., 2007). Also, in studies of the link between nitrification
and biotransformation of EE2 it was shown that Ring A is the
site of electrophilic initiating reactions including conjugation
and hydroxylation (Yi and Harper, 2007). This appears to be
the first time that the epimers 1a and 1b have been isolated as
products obtained directly from the oxidation of EE2 although
compound 1b has been obtained from chemical reduction of
the corresponding hydroperoxide that in turnwas obtained by
the photosensitized oxidation of EE2 by oxygen (Sedee and van
Henegouwen, 1985). Compound 1a/b has been proposed as an
intermediate in the mineralization of EE2 by hydroxyl radicals
generated by the autocatalytic decomposition of ozone (Zhang
et al., 2006) and by microalgae, Scenedesmus quadricauda
(Della Greca et al., 2008).
It has been established that the FeIII-B* catalysed oxida-
tions by H2O2 do not proceed via $OH radicals, i.e. they are
“non-Fenton” oxidations (Ghosh et al., 2008). The Fenton
reaction (Kiwi et al., 1994; Yaping and Jiangyong, 2008) and
other catalysed oxidations by H2O2 (Szabo-Bardos et al., 2011;
Timofeeva et al., 2011) produce �OH radicals. Instead it is
proposed that in the FeIII-B*/H2O2 oxidation system, the active
oxidant is an iron-oxo-based intermediate that is formed
through the interaction of FeIII-B* with H2O2 in the presence of
base (Collins, 2002). This could react with EE2 via amechanism
that involves oxygen atom transfer in a key step, although
other mechanisms that involve electron transfer are also
possible. From our results it would appear that the species
that are formed very early on in the oxidation of EE2 by FeIII-B*/
H2O2 are 1a and 1b. Further oxidation of these intermediates
by this catalytic system probably leads to cleavage of Ring A
(and/or Ring B) with subsequent loss of estrogenicity. Under
the appropriate conditions the FeIII-B*/H2O2 oxidation system
could ultimately lead to mineralization of EE2.
3.5. Significance of FeIII-TAML
An essential green chemistry criterion for catalysts used in
water purification is they must not display any endocrine
disrupting properties themselves. A range of different FeIII-
TAML activators have been demonstrated not to alter tran-
scription mediated by mammalian thyroid, androgen, or
estrogen hormone receptors. This suggests that the FeIII-
TAMLs do not bind to these receptors, thereby reducing
concerns that these catalysts might have endocrine disrupt-
ing activity (Ellis et al., 2010). In the present study, the results
of the YES assay for the blank reaction with FeIII-B* confirmed
that this catalyst has no endocrine disrupting activity. Also,
FeIII-TAML catalysts are not persistent in water, at least under
catalytic operating conditions (Chanda et al., 2006b).
Although this study is conducted at the optimal pH of
10.21, the removal efficiency of EE2 by FeIII-B*/H2O2 system
continues to be high at pH w9 with a half-life of w30 min
(Fig. 2). While the present FeIII-B* functions best at high pH,
there are FeIII-TAML variants being developed that can func-
tion at neutral pH. These activators do not alter the general
mechanism of catalysis by FeIII-TAML activators of peroxides
(Ellis et al., 2010), which involves the interaction of FeIII-TAML
with hydrogen peroxide (H2O2) to form strongly oxidising,
high-valent iron intermediate complexes as the active
oxidants. Differences between the different FeIII-TAML vari-
ants lie in the “oxidising strengths” of the high-valent iron
intermediates, the rates at which these react with substrates
and the longevity of the catalysts during catalytic oxidations.
It is therefore expected that most of the FeIII-TAML variants
follow the same initial oxidative degradation pathways for
substrates such as EE2. The current findings will provide
valuable information relating to the optimization and opera-
tion of practical systems using such catalysts.
The present study highlights the need for integrating the
determination of intermediate metabolites and estrogenicity
assessment in treatment studies. Their significance is illus-
trated via the partial oxidation of EE2 with the FeIII-B*/H2O2
system which results in the formation of a mixture of prod-
ucts that show increased estrogenicity. The intermediates
1a/1b have been previously reported (without identification
of specific isomers) during EE2 degradation by hydroxyl radi-
cals produced via ozone decomposition (Zhang et al., 2006), by
microalgae S. quadricauda (Della Greca et al., 2008), and by
photosensitized decomposition (Sedee and van Henegouwen,
1985); however, these studies did not monitor the change in
estrogenicity of the treated solution.
4. Conclusion
It has been found that the FeIII-B*/H2O2 catalytic oxidation
system under suitable conditions (pH 10.21, [FeIII-B*] 5 nM,
[H2O2] 4 mM) can rapidly oxidize EE2 in aqueous solution (t½for loss of EE2 is 2.1 min) to products that after 1 h show

wat e r r e s e a r c h 4 6 ( 2 0 1 2 ) 6 3 0 9e6 3 1 8 6317
around 30% estrogenicity removal, as determined by the YES
bioassay. However, under low levels of oxidation (partial
oxidation) the epimers 17a-ethynyl-1,4-estradiene-10a,17b-
diol-3-one (1a) and 17a-ethynyl-1,4-estradiene-10b,17b-diol-3-
one (1b) are the dominant products formed (as determined by
LCeMS). Under such a condition (exact conditions and pH
perhaps not being that important) the mixture of products
shows increased estrogenic activity, despite the reduction in
the level of EE2; the estrogenicity of the solutions correlated
with the amount of these compounds present. Based on these
observations it is highly likely that partial oxidation by other
oxidation systems might also produce the same products and
hence also result in increased estrogenic activity. These
results emphasize the importance of avoiding low levels of
oxidation (partial oxidation) in any oxidative EE2 removal
protocols and also that reduction in EE2 levels in solution
during oxidative treatments does not necessarily correlate
with a reduction of estrogenic levels.
Acknowledgment
This study was supported by a grant from the University of
Auckland Faculty Research Development Fund. Yeast strains
for the YES assay were kindly provided by Dr Marc B. Cox,
University of Texas at El Paso. The authors thank Brendan
Harvey for assistance with the NMR analysis experiments and
helpful discussions; and Dr. Siouxsie Wiles and Priscila
Dauros for assistance with luminometry for the YES bioassay.
r e f e r e n c e s
Alum, A., Yoon, Y., Westerhoff, P., Abbaszadegan, M., 2004.Oxidation of bisphenol A, 17b-estradiol, and 17a-ethynylestradiol and byproduct estrogenicity. EnvironmentalToxicology 19 (3), 257e264.
Balsiger, H.A., de la Torre, R., Lee, W.-Y., Cox, M.B., 2010. A four-hour yeast bioassay for the direct measure of estrogenicactivity in wastewater without sample extraction,concentration, or sterilization. Science of the TotalEnvironment 408 (6), 1422e1429.
Barr, W.J., Yi, T., Aga, D., Acevedo, O., Harper, W.F., 2011. Usingelectronic theory to Identify metabolites present in 17a-Ethinylestradiol biotransformation pathways. EnvironmentalScience & Technology 46 (2), 760e768.
Boone, A.J., Chang, C.H., Greene, S.N., Herz, T., Richards, N.G.J.,2003. Modeling the spin-dependent properties of open-shellFe(III)-containing systems: towards a computationaldescription of nitrile hydratase. Coordination ChemistryReviews 238, 291e314.
Brion, C.E., Young, J.B., Litvinyuk, I.V., Cooper, G., 2001. Aninvestigation of the HOMO frontier orbital electron densitydistributions of NH(3), the methylamines and NF(3) using DFTand electron momentum spectroscopy. Chemical Physics 269(1e3), 101e106.
Chanda, A., Khetan, S.K., Banerjee, D., Ghosh, A., Collins, T.J.,2006a. Total degradation of fenitrothion and otherorganophosphorus pesticides by catalytic oxidationemploying Fe-TAML peroxide activators. Journal of theAmerican Chemical Society 128 (37), 12058e12059.
Chanda, A., Ryabov, A.D., Mondal, S., Alexandrova, L., Ghosh, A.,Hangun-Balkir, Y., Horwitz, C.P., Collins, T.J., 2006b. Activity-stability parameterization of homogeneous green oxidationcatalysts. Chemistry e A European Journal 12 (36), 9336e9345.
Collins, T.J., 2002. TAML oxidant activators: a new approach to theactivation of hydrogen peroxide for environmentallysignificant problems. Accounts of Chemical Research 35 (9),782e790.
Crittenden, J.C., Trussell, R.R., Hand, D.W., Howe, K.J.,Tchobanoglous, G., 2005. Water Treatment e Principles andDesign, second ed. John Wiley & Sons.
de Oliveira, F.T., Chanda, A., Banerjee, D., Shan, X.P., Mondal, S.,Que, L., Bominaar, E.L., Munck, E., Collins, T.J., 2007. Chemicaland spectroscopic evidence for an Fe-V-Oxo complex. Science315 (5813), 835e838.
Della Greca, M., Pinto, G., Pistillo, P., Pollio, A., Previtera, L.,Temussi, F., 2008. Biotransformation of ethinylestradiol bymicroalgae. Chemosphere 70 (11), 2047e2053.
Ellis, W.C., Tran, C.T., Roy, R., Rusten, M., Fischer, A.,Ryabov, A.D., Blumberg, B., Collins, T.J., 2010. Designinggreen oxidation catalysts for purifying environmentalwaters. Journal of the American Chemical Society 132 (28),9774e9781.
Fang, H., Tong, W.D., Shi, L.M., Blair, R., Perkins, R., Branham, W.,Hass, B.S., Xie, Q., Dial, S.L., Moland, C.L., Sheehan, D.M., 2001.Structure-activity relationships for a large diverse set ofnatural, synthetic, and environmental estrogens. ChemicalResearch in Toxicology 14 (3), 280e294.
Ghosh, A., Mitchell, D.A., Chanda, A., Ryabov, A.D., Popescu, D.L.,Upham, E.C., Collins, G.J., Collins, T.J., 2008.Catalase�peroxidase activity of Iron(III)�TAML activators ofhydrogen peroxide. Journal of the American Chemical Society130 (45), 15116e15126.
Ghosh, A., Ryabov, A.D., Mayer, S.M., Horner, D.C., Prasuhn, D.E.,Sen Gupta, S., Vuocolo, L., Culver, C., Hendrich, M.P.,Rickard, C.E.F., Norman, R.E., Horwitz, C.P., Collins, T.J., 2003.Understanding the mechanism of Hþ-induced demetalationas a design strategy for robust iron(III) peroxide-activatingcatalysts. Journal of the American Chemical Society 125 (41),12378e12379.
Gupta, S.S., Stadler, M., Noser, C.A., Ghosh, A., Steinhoff, B.,Lenoir, D., Horwitz, C.P., Schramm, K.-W., Collins, T.J., 2002.Rapid total destruction of chlorophenols by activatedhydrogen peroxide. Science 296 (5566), 326e328.
Haiyan, R., Shulan, J., ud din Ahmad, N., Dao, W., Chengwu, C.,2007. Degradation characteristics and metabolic pathway of17[alpha]-ethynylestradiol by Sphingobacterium sp. JCR5.Chemosphere 66 (2), 340e346.
Hashimoto, T., Onda, K., Nakamura, Y., Tada, K., Miya, A.,Murakami, T., 2007. Comparison of natural estrogen removalefficiency in the conventional activated sludge process andthe oxidation ditch process. Water Research 41 (10),2117e2126.
He, C., Li, X.Z., Sharma, V.K., Li, S.Y., 2009. Elimination of sludgeodor by oxidizing sulfur-containing compounds withferrate(VI). Environmental Science & Technology 43 (15),5890e5895.
Horwitz Colin, P., Collins Terrence, J., Spatz, J., Smith Hayden, J.,Wright, L.J., Stuthridge Trevor, R., Wingate Kathryn, G.,McGrouther, K., 2006. Feedstocks for the Future. AmericanChemical Society, pp. 156e169.
Johnson, A.C., Sumpter, J.P., 2001. Removal of endocrine-disrupting chemicals in activated sludge treatment works.Environmental Science & Technology 35 (24), 4697e4703.
Khunjar, W.O., Mackintosh, S.A., Skotnicka-Pitak, J., Baik, S.,Aga, D.S., Love, N.G., 2011. Elucidating the relative roles ofammonia oxidizing and heterotrophic bacteria during thebiotransformation of 17 alpha-ethinylestradiol and

wat e r r e s e a r c h 4 6 ( 2 0 1 2 ) 6 3 0 9e6 3 1 86318
Trimethoprim. Environmental Science & Technology 45 (8),3605e3612.
Kiwi, J., Pulgarin, C., Peringer, P., 1994. Effect of Fenton and photo-Fenton reactions on the degradation and biodegradability of 2and 4-nitrophenols in water treatment. Applied Catalysis BEnvironmental 3 (4), 335e350.
Kolpin, D.W., Furlong, E.T., Meyer, M.T., Thurman, E.M.,Zaugg, S.D., Barber, L.B., Buxton, H.T., 2002. Pharmaceuticals,hormones, and other organic wastewater contaminants in u.s.streams, 1999�2000: a national reconnaissance.Environmental Science & Technology 36 (6), 1202e1211.
Lange, R., Hutchinson, T.H., Croudace, C.P., Siegmund, F.,Schweinfurth, H., Hampe, P., Panter, G.H., Sumpter, J.P., 2001.Effects of the synthetic estrogen 17a-ethinylestradiol on thelife-cycle of the fathead minnow (Pimephales promelas).Environmental Toxicology and Chemistry 20 (6), 1216e1227.
Lee, Y., Escher, B.I., von Gunten, U., 2008. Efficient removal ofestrogenic activity during oxidative treatment of waterscontaining steroid estrogens. Environmental Science &Technology 42 (17), 6333e6339.
Lee, Y., Gunten, U.v, 2008. Transformation of 17a-ethinylestradiolduring water chlorination: effects of bromide on kinetics,products, and transformation pathways. EnvironmentalScience & Technology 43 (2), 480e487.
Lee, Y., Yoon, J., von Gunten, U., 2005. Kinetics of the oxidation ofphenols and phenolic endocrine disruptors during watertreatment with ferrate (Fe(VI)). Environmental Science &Technology 39 (22), 8978e8984.
Lee, Y., Zimmermann, S.G., Kieu, A.T., von Gunten, U., 2009.Ferrate (Fe(VI)) application for municipal wastewatertreatment: a novel process for simultaneous micropollutantoxidation and phosphate removal. Environmental Science &Technology 43 (10), 3831e3838.
Liu, J.W., Picard, D., 1998. Bioactive steroids as contaminants ofthe common carbon source galactose. Fems MicrobiologyLetters 159 (2), 167e171.
Ohko, Y., Iuchi, K.-i., Niwa, C., Tatsuma, T., Nakashima, T.,Iguchi, T., Kubota, Y., Fujishima, A., 2002. 17b-Estradioldegradation by TiO2 photocatalysis as a means of reducingestrogenic activity. Environmental Science & Technology 36(19), 4175e4181.
Picard, D., Khursheed, B., Garabedian, M.J., Fortin, M.G.,Lindquist, S., Yamamoto, K.R., 1990. Reduced levels of hsp90compromise steroid receptor action in vivo. Nature 348 (6297),166e168.
Ren, Y.-X., Nakano, K., Nomura, M., Chiba, N., Nishimura, O.,2007. Effects of bacterial activity on estrogen removal innitrifying activated sludge. Water Research 41 (14), 3089e3096.
Ryabov, A.D., Collins, T.J., 2009. Advances. In: Rudi van, E.,Colin, D.H. (Eds.), Inorganic Chemistry. Academic Press,pp. 471e521.
Sedee, A., van Henegouwen, G.B., 1985. Photosensitizeddecomposition of contraceptive steroids: a possibleexplanation for the observed (photo)allergy of the oralcontraceptive Pill. Archiv Der Pharmazie 318 (2), 111e119.
Shappell, N.W., Vrabel, M.A., Madsen, P.J., Harrington, G.,Billey, L.O., Hakk, H., Larsen, G.L., Beach, E.S., Horwitz, C.P.,
Ro, K., Hunt, P.G., Collins, T.J., 2008. Destruction of estrogensusing Fe-TAML/peroxide catalysis. Environmental Science &Technology 42 (4), 1296e1300.
Skotnicka-Pitak, J., Garcia, E.M., Pitak, M., Aga, D.S., 2008.Identification of the transformation products of 17 alpha-ethinylestradiol and 17 beta-estradiol by mass spectrometryand other instrumental techniques. Trac-trends in AnalyticalChemistry 27 (11), 1036e1052.
Snyder, S.A., Keith, T.L., Verbrugge, D.A., Snyder, E.M.,Gross, T.S., Kannan, K., Giesy, J.P., 1999. Analytical methodsfor detection of selected estrogenic compounds in aqueousmixtures. Environmental Science & Technology 33 (16),2814e2820.
Sumpter, J.P., Johnson, A.C., 2005. Lessons from endocrinedisruption and their application to other issues concerningtrace organics in the aquatic environment. EnvironmentalScience & Technology 39 (12), 4321e4332.
Szabo-Bardos, E., Somogyi, K., TorT, N., Kiss, G., Horvath, A., 2011.Photocatalytic decomposition of l-phenylalanine over TiO2:identification of intermediates and the mechanism ofphotodegradation. Applied Catalysis B: Environmental 101(3e4), 471e478.
Ternes, T.A., Meisenheimer, M., McDowell, D., Sacher, F.,Brauch, H.-J., Haist-Gulde, B., Preuss, G., Wilme, U., Zulei-Seibert, N., 2002. Removal of pharmaceuticals during drinkingwater treatment. Environmental Science & Technology 36 (17),3855e3863.
Thorpe, K.L., Cummings, R.I., Hutchinson, T.H., Scholze, M.,Brighty, G., Sumpter, J.P., Tyler, C.R., 2003. Relative potenciesand combination effects of steroidal estrogens in fish.Environmental Science & Technology 37 (6), 1142e1149.
Timofeeva, M.N., Hasan, Z., Orlov, A.Y., Panchenko, V.N.,Chesalov, Y.A., Soshnikov, I.E., Jhung, S.H., 2011. Fe-containing nickel phosphate molecular sieves asheterogeneous catalysts for phenol oxidation andhydroxylation with H2O2. Applied Catalysis B Environmental107 (1e2), 197e204.
Wang, B., Sanchez, R.I., Franklin, R.B., Evans, D.C., Huskey, S.-E.W., 2004. The involement of CYP3A4 and CYP2C9 in themetabolism of 17a-ethinylestradiol. Drug Metabolism andDisposition 32 (11), 1209e1212.
Yaping, Z., Jiangyong, H., 2008. Photo-Fenton degradation of 17b-estradiol in presence of a-FeOOHR and H2O2. AppliedCatalysis B Environmental 78 (3e4), 250e258.
Yi, T., Harper, W.F., 2007. The link between nitrification andbiotransformation of 17a-ethinylestradiol. EnvironmentalScience & Technology 41 (12), 4311e4316.
Young, W.F., Whitehouse, P., Johnson, I., Sorokin, N., 2002.Proposed Predicted-No-Effect-Concentrations (PNECs) forNatural and Synthetic Steroid Oestrogens in Surface Waters.Environment Agency R & D Technical Report P2-T04/1;England and Wales Environment Agency, Bristol, U.K., pp.93e95.
Zhang, X., Chen, P.Y., Wu, F., Deng, N.S., Liu, J.T., Fang, T., 2006.Degradation of 17 alpha-ethinylestradiol in aqueous solutionby ozonation. Journal of Hazardous Materials 133 (1e3),291e298.




















