BAFFR deficiency results in limited CD169 + macrophage function during viral infection
27
1 BAFFR deficiency results in limited CD169 + macrophage function during viral infection 1 2 Haifeng C. Xu 1,2,3,# , Jun Huang 2,# , Vishal Khairnar 3 , Vikas Duhan 3 , Aleksandra A. Pandyra 3 , Melanie 3 Grusdat 2 , Prashant Shinde 2 , David R. McIlwain 2,4 , Sathish Kumar Maney 2 , Jennifer Gommerman 5 , Max 4 Löhning 6,7 , Pamela S. Ohashi 1,5 , Tak W. Mak 1,5 , Kathrin Pieper 8 , Heiko Sic 8 , Matthaios Speletas 9 , 5 Hermann Eibel 8 , Carl F. Ware 10 , Alexei V. Tumanov 11 , Andrey A. Kruglov 6 , Sergei A. Nedospasov 6,14 , 6 Dieter Häusinger 2 , Mike Recher 12* , Karl S. Lang 3* , and Philipp A. Lang 1,2,13* 7 8 1 Campell Family Institute for Breast Cancer Research, Princess Margaret Cancer Centre, University 9 Health Network (UHN), 620 University Ave., Toronto, Ontario, Canada M5G 2C1 10 2 Department of Gastroenterology, Hepatology and Infectious Diseases, University of Düsseldorf, 11 Universitätsstr. 1, 40225 Düsseldorf, Germany 12 3 Institute of Immunology, Medical Faculty, University of Duisburg-Essen, Hufelandstr. 55, Essen 13 45147, Germany 14 4 Baxter Laboratory in Stem Cell Biology, Department of Microbiology and Immunology, Stanford 15 University, Stanford, California, 94305, USA 16 5 Department of Immunology, University of Toronto, 1 King´s Circle, Toronto, Ontario Canada, M5S 17 1A8 18 6 Experimental Immunology, Department of Rheumatology and Clinical Immunology, Charité University 19 Medicine Berlin, 10117 Berlin, Germany 20 7 German Rheumatism Research Center (DRFZ), a Leibniz Institute, 10117 Berlin, Germany 21 8 University Medical Centre Freiburg, Center of Chronic Immunodeficiency, Engesserstraße 4, D-79110 22 Freiburg, Germany 23 9 Department of Immunology & Histocompatibility, Faculty of Medicine, University of Thessaly, Larissa, 24 Greece 25 10 Sanford Burnham Medical Research Institute, 10901, La Jolla, CA 92037, USA 26 11 Trudeau Institute, 154 Algonquin Avenue, Saranac Lake, NY 12983 27 12 Primary Immunodeficiency Clinic, Medical Outpatient Division and Immunodeficiency Lab, 28 Department of Biomedicine, University Hospital Basel, Switzerland 29 13 Department of Molecular Medicine II, Universitatsstrasse 1, D-40225 Düsseldorf, Germany 30 14 Lomonosov Moscow State University, Moscow 119991 Russia. 31 32 #,* contributed equally to this work 33 34 35 Address correspondence to Philipp A. Lang MD, PhD, Department of Gastroenterology, Hepatology 36 and Infectious Diseases, Universitätsstrasse 1, Geb. 23.12.U1 Room 41, D-40225 Düsseldorf, 37 Germany, tel.: +49 211 8113580, fax.: +49 211 8113421, [email protected] 38 JVI Accepted Manuscript Posted Online 11 February 2015 J. Virol. doi:10.1128/JVI.02976-14 Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Transcript of BAFFR deficiency results in limited CD169 + macrophage function during viral infection

1
BAFFR deficiency results in limited CD169+ macrophage function during viral infection 1
2
Haifeng C. Xu1,2,3,#, Jun Huang2,#, Vishal Khairnar3, Vikas Duhan3, Aleksandra A. Pandyra3 , Melanie 3
Grusdat2, Prashant Shinde2, David R. McIlwain2,4, Sathish Kumar Maney2, Jennifer Gommerman5, Max 4
Löhning6,7, Pamela S. Ohashi1,5, Tak W. Mak1,5, Kathrin Pieper8, Heiko Sic8, Matthaios Speletas9, 5
Hermann Eibel8, Carl F. Ware10, Alexei V. Tumanov11, Andrey A. Kruglov6, Sergei A. Nedospasov6,14, 6
Dieter Häusinger2, Mike Recher12*, Karl S. Lang3*, and Philipp A. Lang1,2,13* 7
8 1Campell Family Institute for Breast Cancer Research, Princess Margaret Cancer Centre, University 9 Health Network (UHN), 620 University Ave., Toronto, Ontario, Canada M5G 2C1 10 2Department of Gastroenterology, Hepatology and Infectious Diseases, University of Düsseldorf, 11 Universitätsstr. 1, 40225 Düsseldorf, Germany 12 3Institute of Immunology, Medical Faculty, University of Duisburg-Essen, Hufelandstr. 55, Essen 13 45147, Germany 14 4Baxter Laboratory in Stem Cell Biology, Department of Microbiology and Immunology, Stanford 15 University, Stanford, California, 94305, USA 16 5Department of Immunology, University of Toronto, 1 King´s Circle, Toronto, Ontario Canada, M5S 17 1A8 18 6Experimental Immunology, Department of Rheumatology and Clinical Immunology, Charité University 19 Medicine Berlin, 10117 Berlin, Germany 20 7German Rheumatism Research Center (DRFZ), a Leibniz Institute, 10117 Berlin, Germany 21 8University Medical Centre Freiburg, Center of Chronic Immunodeficiency, Engesserstraße 4, D-79110 22 Freiburg, Germany 23 9Department of Immunology & Histocompatibility, Faculty of Medicine, University of Thessaly, Larissa, 24 Greece 25 10Sanford Burnham Medical Research Institute, 10901, La Jolla, CA 92037, USA 26 11Trudeau Institute, 154 Algonquin Avenue, Saranac Lake, NY 12983 27 12Primary Immunodeficiency Clinic, Medical Outpatient Division and Immunodeficiency Lab, 28 Department of Biomedicine, University Hospital Basel, Switzerland 29 13Department of Molecular Medicine II, Universitatsstrasse 1, D-40225 Düsseldorf, Germany 30 14Lomonosov Moscow State University, Moscow 119991 Russia. 31 32 #,* contributed equally to this work 33 34 35 Address correspondence to Philipp A. Lang MD, PhD, Department of Gastroenterology, Hepatology 36
and Infectious Diseases, Universitätsstrasse 1, Geb. 23.12.U1 Room 41, D-40225 Düsseldorf, 37
Germany, tel.: +49 211 8113580, fax.: +49 211 8113421, [email protected] 38
JVI Accepted Manuscript Posted Online 11 February 2015J. Virol. doi:10.1128/JVI.02976-14Copyright © 2015, American Society for Microbiology. All Rights Reserved.

2
Abstract 39
The B cell activating factor of the TNF family (BAFF) is critical for B-cell development and humoral 40
immunity in mice and humans. While the role of BAFF in B cells has been widely described, its role in 41
innate immunity remains unknown. Using BAFFR deficient mice we characterized BAFFR related 42
innate and adaptive immune functions following infection with vesicular stomatitis virus (VSV) and 43
lymphocytic choriomeningitis virus (LCMV). We identified a critical role for BAFFR signalling in the 44
generation and maintenance of the CD169+ macrophage compartment. Consequently, Baffr–/– mice 45
exhibited limited induction of innate type I interferon production after viral infection. Lack of BAFFR 46
signalling reduced virus amplification and presentation following viral infection, resulting in highly 47
reduced anti-viral adaptive immune responses. As a consequence, BAFFR deficient mice showed 48
exacerbated and fatal disease after viral infection. Mechanistically, transient lack of B cells in Baffr–/– 49
animals resulted in limited lymphotoxin expression, which was critical for maintenance of CD169+ cells. 50
In conclusion, BAFFR signalling affects both innate and adaptive immune activation during viral 51
infections. 52
53
Importance 54
Viruses cause acute and chronic infections in humans resulting in millions of deaths every year. Innate 55
immunity is critical for the outcome of a viral infection. Innate type I interferon production can limit viral 56
replication while adaptive immune priming by innate immune cells induces pathogen specific immunity 57
with long term protection. Here we show that BAFFR deficiency not only perturbed B cells, but also 58
resulted in limited CD169+ macrophages. These macrophages are critical in amplifying viral particles 59
to trigger type I interferon production and initiate adaptive immune priming. Consequently, BAFFR 60
deficiency resulted in reduced enforced viral replication, limited type I interferon production, and 61
reduced adaptive immunity when compared to BAFFR competent controls. As a result, BAFFR 62
deficient mice were predisposed to fatal viral infections. Thus, BAFFR expression is critical for innate 63
immune activation and anti-viral immunity. 64

3
Introduction 65
The B cell activating factor of the TNF family (BAFF) binding receptor (BAFFR) is critical for B 66
cell development(1, 2). Patients lacking the BAFFR have been identified within cohorts with common 67
variable immunodeficiency, the most prevalent symptomatic primary immunodeficiency in adult 68
patients. BAFFR deficient humans exhibit severe B cell lymphopenia and impaired immunglobulin 69
production(3). Similarly, the lack of BAFF signalling in Baffr–/– mice is also associated with severe B 70
cell lymphopenia(4). BAFF binds to three receptors, the BAFF receptor (BAFFR), transmembrane 71
activator and CAML interactor (TACI), and B cell maturation antigen (BCMA)(5). While TACI and 72
BCMA engage both BAFF and a proliferation inducing ligand (APRIL), BAFFR binds BAFF exclusively. 73
The BAFF – BAFFR association leads to recruitment and degradation of TRAF3(6). TRAF3 negatively 74
regulates NF-NB-inducing kinase (NIK), the upstream kinase for NF-NB2 activation(7). In the presence 75
of BAFF, degradation of TRAF3 leads to stabilization of NIK and activation NF-NB2, which triggers B 76
cell survival(8-11). Furthermore, BAFF triggers activation of Akt signalling pathways, which increase 77
the metabolic activity of B cells(12). Additionally, BAFF transmits survival signals via Erk activation, 78
which triggers phosphorylation and degradation of the pro-apoptotic molecule Bim(13-15). Together, 79
these signalling pathways promote BAFF-mediated survival of B cells. Whether innate immunity may 80
be abnormal in Baffr–/– mice has not yet been investigated. 81
Successful defense against viral infections relies on effective innate and adaptive immunity. 82
During infection, viral particles are captured in secondary lymphoid organs such as the lymph node or 83
the spleen(16). In mice, macrophages in the marginal sinus in the splenic white pulp or macrophages 84
located in the subcapsular space of the lymph node filter pathogens from the blood and the lymph 85
respectively(16-18). Metallophilic macrophages and subcapsular sinus macrophages are 86
characterized by expression of the C-type lectin CD169 (Siglec-1)(19). CD169+ cells are critical for 87
innate cytokine production and viral antigen presentation to B cells and represent an important link 88
between innate and adaptive immunity(18). In particular, Usp18 driven suppression of anti-viral type I 89
interferon signalling allows viral replication to occur in CD169+ cells in close proximity to marginal zone 90
B cells(20, 21). Consequently, large quantities of viral antigen are made available in order to induce 91
rapid and robust adaptive immunity(22-25). The rapid induction of adaptive immune responses as a 92
result of early viral replication in the spleen guarantees virus elimination and survival of the virus 93
infected host(20, 21). 94

4
B cells are important for the generation of the splenic architecture including the maintenance 95
of the marginal zone(26, 27). Following a recent report indicating that BAFF is produced by neutrophils 96
in the marginal zone of the spleen(28), we chose to investigate the impact of BAFFR signalling on 97
innate immune responses. We found that absence of BAFFR resulted in reduced lymphotoxin 98
expression, decreased presence of CD169+ cells, delayed and impaired innate and adaptive immune 99
activation, and consequently promotion of fatal disease development after viral infection. 100

5
Materials and Methods: 101
Mice: Mice were infected intravenously with VSV or LCMV at the indicated doses. Baffr–/– mice were 102
bred on a C57BL/6 genetic background(4). CD45.1+ mice were purchased from Jackson Laboratory. 103
Ltbfl/fl x CD19-Cre+ and Ifnar1-/- mice were previously described(29, 30). All experiments were 104
performed in single ventilated cages. During survival experiments, the health status of the mice was 105
checked twice daily. Upon the appearance of clinical signs of VSV replication in the central nervous 106
system (CNS), such as paralysis, mice were removed from the experiment. Animal experiments were 107
carried out with the authorization of the Veterinäramt of Nordrhein Westfalen, Germany, in accordance 108
with the German law for animal protection and the institutional guidelines of the Ontario Cancer 109
Institute. 110
111
Viruses: VSV, Indiana strain (VSV-IND, Mudd-Summers isolate), was originally obtained from Prof. D. 112
Kolakofsky (University of Geneva, Switzerland). Virus was propagated on BHK-21 cells at a multiplicity 113
of infection (MOI) of 0.01 and was then plaqued onto Vero cells. LCMV was used and virus titers 114
determined as previously described(21). Viruses were administrated to mice through intravenous 115
injection. 116
117
Poly I:C and recombinant interferon: poly I:C (GE Healthcare Life Sciences) and mouse 118
recombinant interferon alpha A (PBL Biosciences) were administrated to mice through intravenous 119
injection at indicated doses. 120
121
Neutralizing antibodies were determined by plaque reduction neutralization test (PRNT) as 122
previously described(21). Serum was prediluted (1:40). Complement system was inactivated (56°C for 123
30 min). Serum was titrated 1:2 over 12 steps and incubated with 1000 PFU of VSV. After 90 minutes 124
of incubation, the virus-serum mixture was plaqued on Vero cells. Overlay was added after one hour. 125
Plaques were counted 24 hours later by crystal violet staining. The cut of was 50% reduction plaques 126
when compare to serum free controls 127
128
ELISA: Interferon alpha ELISA (PBL Biosciences) was performed as instructed by the manufacturer. 129
130

6
Purification of B cells: For B cell purification, single cell suspended splenocytes were enriched 131
following the manufacturer’s instructions with the CD45R (B220) MicroBeads, mouse kit (Miltenyi) 132
133
RT-PCR analyses: RNA purification and RT-PCR analyses were performed as previously described 134
according to manufacturer’s instructions (Qiagen)(31). Gene expression of Isg15, Mx1, Ifit2, Lta, Ltb 135
and Gapdh was performed using kits from Applied Biosystems. For analysis, the expression levels of 136
all target genes were normalized to Gapdh expression levels (∆Ct). Gene expression values were then 137
calculated based on the ∆∆Ct method relative to naïve WT controls. Relative quantities (RQ) were 138
determined using the equation: RQ=2^-∆∆Ct. 139
140
Histology: Histological analyses were performed on snap-frozen tissue as described previously by 141
using self-made anti-VSV-G monoclonal antibody (clone Vi10)(21). CD90.2, CD4, CD8 and B220 142
antibody was purchased from eBioscience (San Diego, CA). CD169 (clone: MOMA-1) antibody was 143
obtained from Abcam (Cambridge, MA). H&E staining was previously described(32). Bars indicate 50 144
μm or 100 µm, as indicated in the corresponding figure legend. 145
146
Flow cytometry: Analyses were performed such as previously described for LCMV tetramer staining 147
and intracellular cytokine staining(33). Different spleen immune populations were identified using anti-148
B220 (RA3-6B2), anti-MHCII (M5/114.15.2), anti-CD11c (N418), anti-GR-1 (RB6-8C5), anti-F4/80 149
(BM8), anti-CD3 (145-2C11), anti-CD8 (52-6.7), anti-CD4 (GK1.5) antibodies (clone). Stem cell 150
analyses were performed as previously described(34). Lin– was considered negative staining for: CD3, 151
CD11b (M1/70), Ly6C, Ter119 (TER-119), CD19, CD11c, MHCII, IL7R (A7R34), and NK1.1. CMP and 152
GMP were determined using anti-CD34 (RAM34), anti-CD16/32 (93), anti-Sca1 (D7), and anti-CD117 153
(2B8) antibodies. All antibodies were obtained from eBioscience (San Diego CA), except anti-CD169 154
(3D6.112), which was obtained from AbD Serotec (Dusseldorf). 155
156
Statistical analysis: Data were expressed as means ± S.E.M. Student’s t-test was used to detect 157
statistically significant differences between two groups. Significant differences between several groups 158
were detected by one-way analysis of variance (ANOVA) with Bonferroni or Dunnett post hoc tests or 159
mentioned specifically in the corresponding figure legend. The level of statistical significance was set 160
at P < 0.05. 161

7
Results 162
BAFFR is critical in overcoming viral infection. 163
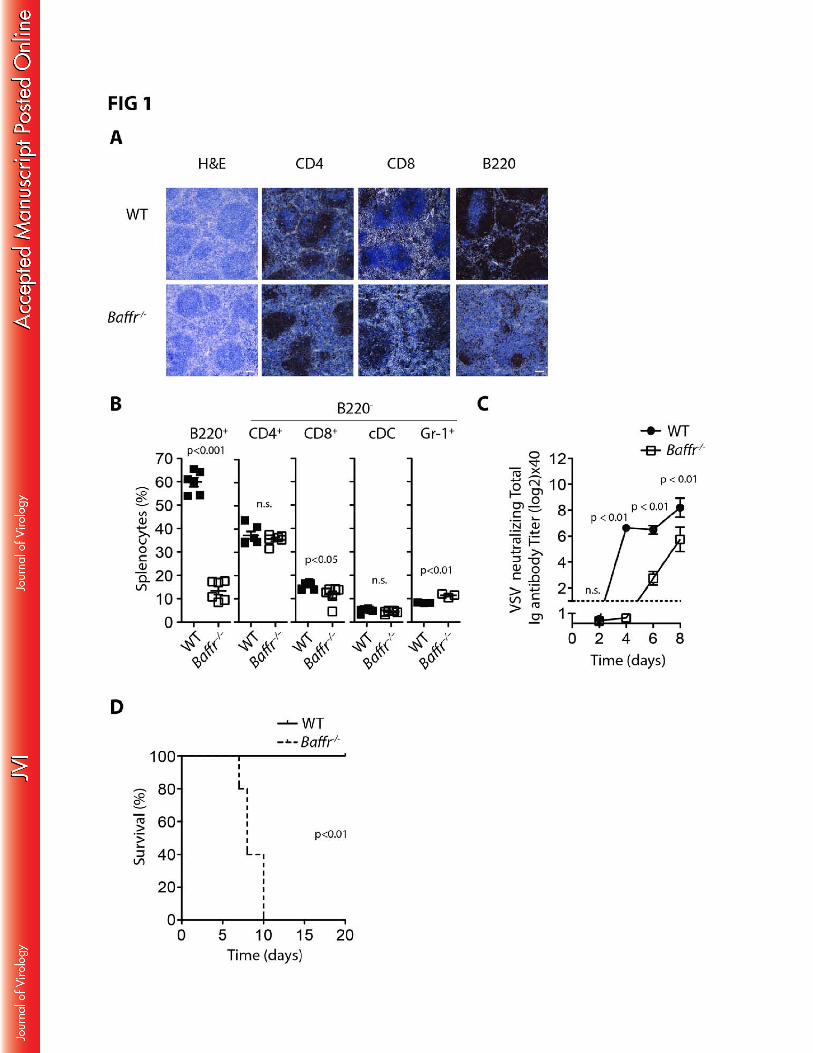
Murine BAFFR deficiency resulted in severe B cell lymphopenia but did not have a major 164
impact on T cell, dendritic cell or neutrophil numbers (FIG 1A-B). As expected, Baffr–/– mice exhibited 165
delayed virus neutralizing antibody production after infection with the cytolytic vesicular stomatitis virus 166
(VSV) (FIG 1C). Consistent with a requirement for rapid neutralizing antibody formation for elimination 167
of VSV(21), Baffr–/– mice succumbed to VSV infection, while wild-type (WT) animals overcame the 168
infection (FIG 1D). When neuronal tissue was harvested from the sick animals (day 7-10), the 169
presence of VSV could be detected (data not shown). These data suggest that absence of BAFFR 170
resulted in fatal disease development during infection with VSV. 171
172
BAFFR mediates enforced viral replication during viral infection 173
Despite detection of VSV replication in the CNS in the later phase of infection, VSV titers were 174
below detection limit in spleen tissue of Baffr-/- animals 8h and 24h after infection with VSV, while virus 175
was readily detectable in WT controls (FIG 2A). Overcoming VSV infection is not only dependent on 176
production of neutralizing antibodies, but also on innate type I interferon production(30, 35, 36). 177
Consistent with decreased replication of VSV in spleen tissue, Baffr–/– mice had reduced type I 178
interferon levels in the serum when compared to infected WT mice (Figure 2B). Moreover, the 179
increase of interferon regulated genes (IRGs) such as Isg15, Mx1, and Ifit2 24 hours after VSV 180
infection was reduced in brain tissue harvested from BAFFR deficient animals when compared to their 181
corresponding controls (FIG 2C). However, competent production of type I interferon was detectable in 182
Baffr–/– mice following injection of the TLR3 agonist poly I:C in vivo (FIG 2D). Furthermore, IRG 183
expression in the brain tissue from Baffr-/- mice and their WT controls following treatment with poly I:C 184
was similar (FIG 2E). This indicates that the capacity to induce type I interferon was normal in Baffr–/– 185
mice. Moreover when animals were treated with mouse recombinant IFN-α, similar brain IRG 186
expression was observed between BAFFR deficient and WT mice (FIG 2E). Collectively, reduced type 187
I interferon levels in Baffr–/– mice following virus infection, was likely explainable by a different 188
mechanism than defective pathogen recognition receptor (PRR) signalling. 189
190
BAFF signalling is required for maintenance of metallophilic macrophages in the spleen. 191

8
We have recently demonstrated that early virus replication in the spleen depends on CD169+ 192
metallophilic macrophages and is triggered by Usp18 mediated resistance to type I interferon in these 193
cells(21). In mice, CD169+ cells in the spleen are in direct contact with the blood stream and remove 194
virus particles and apoptotic cells from circulation(21, 37). Lack of CD169+ macrophages blocks virus 195
replication early after infection, causing limited antigen amplification and reduced virus induced 196
immune activation(21, 38). Since BAFFR deficient animals exhibited lower viral replication during early 197
infection, we investigated potential mechanisms by which BAFFR signalling might control enforced 198
viral replication. Myeloid progenitor populations in the bone marrow such as the common myeloid 199
progenitors (CMP: Lin–, Sca1–, IL7R–, CD117+, CD34+, CD16/32–) or the granulocyte and macrophage 200
progenitors (GMP: Lin–, Sca1–, IL7 R–, CD117+, CD34+, CD16/32+) did not differ between Baffr–/– and 201
WT mice (FIG 3A). However, CD169+ metallophilic macrophages which have a critical role during 202
early viral replication(21), were highly reduced in spleen tissue of Baffr–/– mice compared to WT 203
animals (FIG 3B), while red pulp macrophages were present at similar frequencies (FIG 3C). Taken 204
together, BAFFR appears to be required for maintenance of CD169+ cells. 205
Next, we investigated the presence of CD169+ cells following infection. CD169+ cells are 206
present at reduced numbers in BAFFR deficient animals, but a residual population is still detectable in 207
the naïve state (FIG 3D-E). However, shortly after infection with VSV, CD169+ cells rapidly disappear 208
in Baffr–/– mice in contrast to WT animals (FIG 3D). As a consequence of reduced CD169+ 209
macrophages, VSV protein (detected by the VSV-specific monoclonal antibody Vi10) was reduced in 210
Baffr–/– mice in spleen sections following VSV infection, while it was readily detectable in spleen 211
sections from WT animals (FIG 3E). Next, we wondered whether type I interferon directly affected 212
CD169+ cell survival. However, the distribution of CD169+ cells in spleen tissues harvested from 213
Ifnar1–/– mice and WT animals was similar (FIG 4). Taken together, these data indicate that lack of 214
BAFFR expression is associated with a decreased presence of splenic CD169+ cells after virus 215
infection, which consequently diminishes early viral replication. 216
217
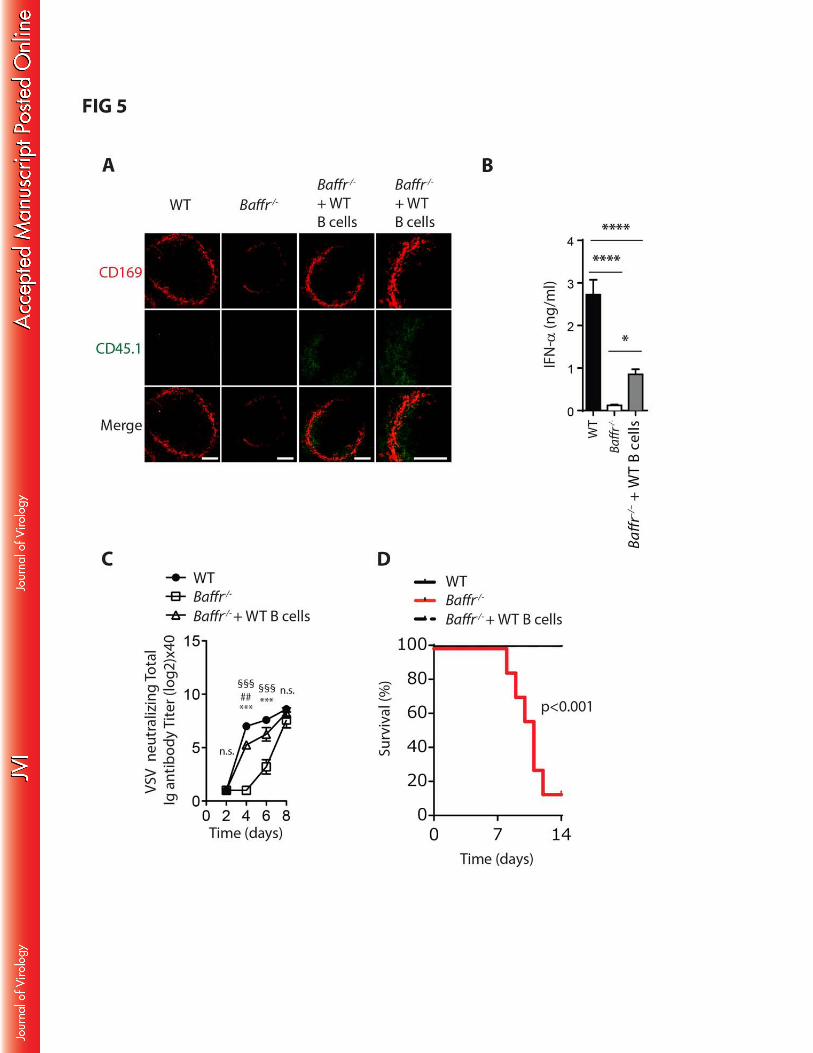
BAFFR deficiency results in reduced B cell mediated maintenance of CD169+ cells. 218
Next, we addressed whether the reduction of CD169+ cells in Baffr–/– mice occurred due to 219
severe B cell lymphopenia(39, 40). We transferred WT B cells into Baffr–/– mice and monitored 220
CD169+ cells in spleen tissue after 40 days. Interestingly, CD169+ cells were readily detectable in 221
spleen tissue of BAFFR deficient mice that were supplemented with B cells (FIG 5A). Furthermore, 222

9
type I interferon production following VSV infection could be partially rescued by the transfer of WT B 223
cells into Baffr–/– mice (FIG 5B). Moreover, neutralizing antibody titers of the Baffr–/– animals which 224
received WT B cells was increased when compared into Baffr–/– animals (FIG 5C), and animals could 225
overcome the VSV infection (FIG 5D). These data indicate that B cells mediate maintenance of 226
CD169+ cells and consequently contribute to innate immunity during infection. 227
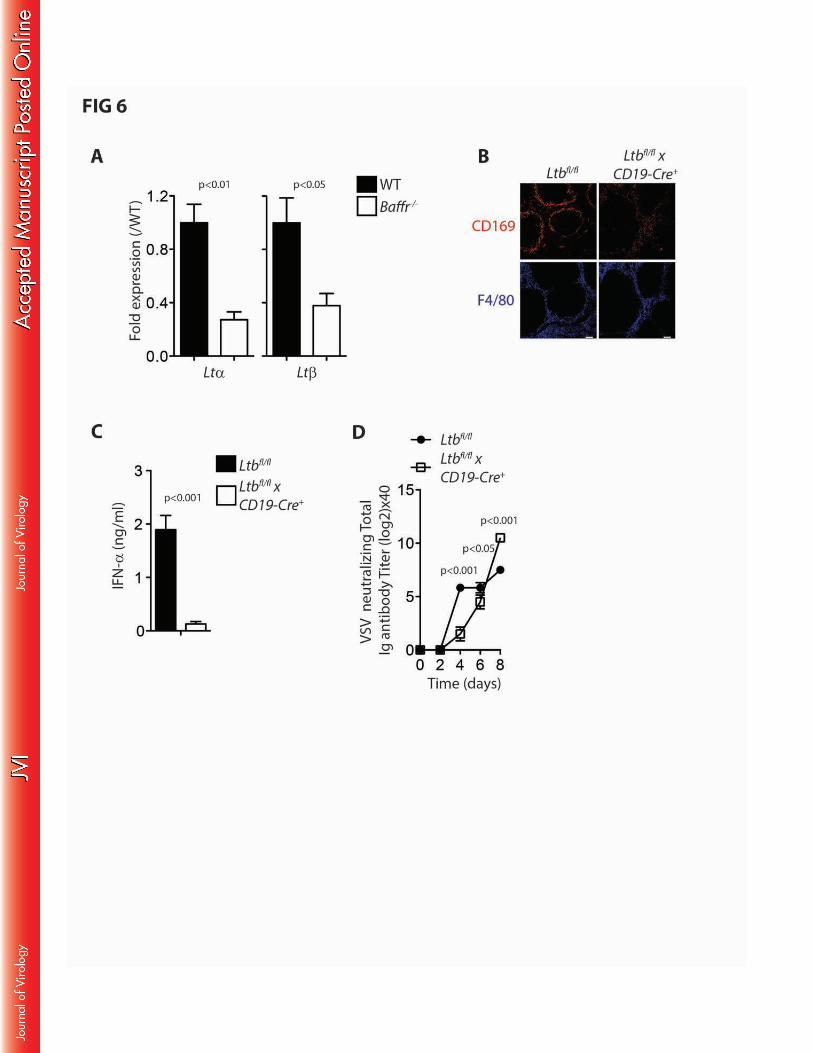
Lymphotoxin signalling is critical for CD169+ cell development in the spleen and lymph node 228
tissue(32, 38, 39, 41). Moreover, it has been shown that lymphotoxins derived from B cells which are 229
important for maintenance of CD169+ cells(29, 39). Consistently, BAFFR deficient animals exhibited 230
lower lymphotoxin alpha and lymphotoxin beta expression levels compared to their corresponding 231
controls (FIG 6A). These data suggest that impaired B cell numbers in Baffr–/– mice may contribute to 232
insufficient lymphotoxin expression to maintain normal levels of CD169+ cells(29, 38, 41, 42). To 233
investigate the role of lymphotoxin beta during enforced viral replication, we infected Ltbfl/flxCD19-Cre+ 234
animals and compared them to their corresponding controls. As expected, these animals exhibited 235
less CD169+ cells than the WT controls (FIG 6B) (29, 42). Consistent with previous reports and our 236
data obtained in Baffr–/– mice, we observed reduced type I interferon production shortly after infection 237
in Ltbfl/flxCD19-Cre+ when compared to Ltbfl/flxCD19-Cre- animals (FIG 6C) (39). Furthermore, low dose 238
infection resulted in reduced production of neutralizing antibodies (FIG 6D). These data indicate that 239
lack of B cells or B cell derived expression of LtE results in limited innate immune activation and 240
delayed adaptive immune priming. 241
242
BAFFR deficiency results in limited innate immune activation following LCMV infection. 243
To further analyze the importance of CD169+ macrophages in viral replication and the 244
induction of anti-viral immunity, we examined Baffr–/– mice following LCMV infection. Consistent with 245
the VSV infection, we observed reduced LCMV replication in the spleen tissue of Baffr-/- mice in 246
comparison to WT controls at 72 hours after infection (FIG 7A). Furthermore type I interferon levels 247
were highly reduced in the serum of Baffr-/- animals when compared to WT controls (FIG 7B). 248
Tetramer+ LCMV specific T cells were highly reduced following LCMV infection of Baffr–/– mice when 249
compared to WT mice (FIG 7C). Moreover, IFN-J production after in vitro re-stimulation with the 250
immunodominant LCMV peptides was reduced in both CD8+ and CD4+ T cells harvested from Baffr–/– 251
mice in comparison to WT controls (FIG 7D). When viral titers were determined 20 days after infection, 252
WT animals had eliminated the virus from all organs tested (FIG 7E). In sharp contrast, LCMV infected 253

10
Baffr–/– mice displayed high virus titers in all organs tested (FIG 7E). Collectively, these data suggest 254
that the absence of BAFFR signalling causes impaired generation of the marginal zone compartment, 255
and impaired induction of innate and adaptive immune responses during viral infection. 256

11
Discussion 257
In this study, we have identified a critical role for BAFFR in the maintenance of CD169+ 258
macrophages. Baffr–/– mice showed limited innate immune activation and reduced adaptive immune 259
priming associated with fatal disease outcome. Mechanistically, impaired B-cell development in Baffr–/– 260
mice resulted in limited lymphotoxin expression and likely as a consequence, reduced presence of 261
CD169+ cells. 262
BAFF can be produced by a variety of immune cells including dendritic cells, macrophages 263
and neutrophils(5). Interestingly, a recent report identified BAFF producing neutrophils to be located in 264
the marginal zone of the spleen(28). These neutrophil B helper cells contribute to marginal zone B cell 265
activation and antibody production against pathogens(28). Based on our results, BAFF production by 266
neutrophil B helper cells may, through promoting B cell mediated lymphotoxin production, also affect 267
CD169+ cell survival and subsequently enforce antigen amplification and presentation. Furthermore, 268
BAFF overexpression has been linked to a variety of autoimmune diseases such as rheumatoid 269
arthritis, lupus erythematosus and Sjögren syndrome(5, 43-45). A clinically used BAFF blocking 270
antibody, belimumab, is effective in treating some lupus patients (46) and potentially, some clinical 271
efficacy of BAFF neutralization in lupus patients may be due to effects on CD169+ macrophages. 272
Viral infections are potent activators of the immune system and can trigger autoimmunity(47) 273
through several mechanisms including molecular mimicry and bystander activation(48). Increased 274
BAFF levels may not only affect B cell mediated autoimmunity, but also B cell mediated effects on 275
CD169+ macrophages to increase bystander activation. Furthermore, replication of low affinity 276
antigens in CD169+ macrophages may contribute to development of virus mediated autoimmunity 277
induced by molecular mimicry(49). Considering our data, altered BAFF expression levels may lead to 278
increased immune responses during viral infections. These mechanisms could also contribute to 279
induction of autoantibodies observed during viral infections(50, 51). 280
As we show here, defects in BAFFR expression may limit innate immunity during infection. 281
This may be triggered by reduced lymphotoxin beta production of B cells, as lack of lymphotoxin beta 282
resulted in reduced presence of CD169+ macrophages (29, 38, 39). Furthermore, lymphotoxins trigger 283
innate type I interferon production during viral infection(52, 53). This may be in part triggered by 284
enforced viral replication in CD169+ cells, which is also critical for induction of adaptive immune 285
priming(21, 49). Considering our data, deletions in BAFFR may not only affect B cell driven immunity 286

12
but also trigger defects in innate immunity. Future studies may analyze the role of BAFFR deficiency 287
during innate immunity in human patients. 288
In conclusion we have identified that BAFFR deficiency mediates reduced presence of B cells 289
impacting the maintenance of CD169+ macrophages and innate immunity. 290

13
Acknowledgements: 291
The authors are grateful for the technical assistance of Eugene Bäcker, Konstanze Schättel, and 292
Stefanie Münch. MR holds a professorship from the Swiss National Science Foundation 293
(PP00P3_144863). This study was supported by the CIHR grant of Pamela S. Ohashi (CIHR- MOP-294
106529). PSO holds a Canada Research Chair in Autoimmunity and Tumor Immunity. Also, this study 295
was supported by a grant to JG (CIHR-MOP-67157). This research was funded in part by the Ontario 296
Ministry of Health and Long Term Care. The views expressed do not necessarily reflect those of the 297
OMOHLTC. Moreover, this study was supported by the Alexander von Humboldt Foundation 298
(SKA2008 and SKA2010), the German Research Council (CRC974, and LA2558/3-1/4-1/5-1). 299

14
References: 300
1. Schiemann, B., J. L. Gommerman, K. Vora, T. G. Cachero, S. Shulga-Morskaya, 301 M. Dobles, E. Frew, and M. L. Scott. 2001. An essential role for BAFF in the 302 normal development of B cells through a BCMA-independent pathway. Science 303 293:2111-2114. 304
2. Thompson, J. S., S. A. Bixler, F. Qian, K. Vora, M. L. Scott, T. G. Cachero, C. 305 Hession, P. Schneider, I. D. Sizing, C. Mullen, K. Strauch, M. Zafari, C. D. 306 Benjamin, J. Tschopp, J. L. Browning, and C. Ambrose. 2001. BAFF-R, a newly 307 identified TNF receptor that specifically interacts with BAFF. Science 293:2108-2111. 308
3. Warnatz, K., U. Salzer, M. Rizzi, B. Fischer, S. Gutenberger, J. Bohm, A. K. 309 Kienzler, Q. Pan-Hammarstrom, L. Hammarstrom, M. Rakhmanov, M. 310 Schlesier, B. Grimbacher, H. H. Peter, and H. Eibel. 2009. B-cell activating factor 311 receptor deficiency is associated with an adult-onset antibody deficiency syndrome in 312 humans. Proceedings of the National Academy of Sciences of the United States of 313 America 106:13945-13950. 314
4. Sasaki, Y., S. Casola, J. L. Kutok, K. Rajewsky, and M. Schmidt-Supprian. 2004. 315 TNF family member B cell-activating factor (BAFF) receptor-dependent and -316 independent roles for BAFF in B cell physiology. J Immunol 173:2245-2252. 317
5. Mackay, F., and P. Schneider. 2009. Cracking the BAFF code. Nat Rev Immunol 318 9:491-502. 319
6. Gardam, S., F. Sierro, A. Basten, F. Mackay, and R. Brink. 2008. TRAF2 and 320 TRAF3 signal adapters act cooperatively to control the maturation and survival signals 321 delivered to B cells by the BAFF receptor. Immunity 28:391-401. 322
7. Liao, G., M. Zhang, E. W. Harhaj, and S. C. Sun. 2004. Regulation of the NF-323 kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced 324 degradation. J Biol Chem 279:26243-26250. 325
8. Claudio, E., K. Brown, S. Park, H. Wang, and U. Siebenlist. 2002. BAFF-induced 326 NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol 327 3:958-965. 328
9. Sasaki, Y., E. Derudder, E. Hobeika, R. Pelanda, M. Reth, K. Rajewsky, and M. 329 Schmidt-Supprian. 2006. Canonical NF-kappaB activity, dispensable for B cell 330 development, replaces BAFF-receptor signals and promotes B cell proliferation upon 331 activation. Immunity 24:729-739. 332
10. Matsuzawa, A., P. H. Tseng, S. Vallabhapurapu, J. L. Luo, W. Zhang, H. Wang, 333 D. A. Vignali, E. Gallagher, and M. Karin. 2008. Essential cytoplasmic 334 translocation of a cytokine receptor-assembled signaling complex. Science 321:663-335 668. 336
11. Vallabhapurapu, S., A. Matsuzawa, W. Zhang, P. H. Tseng, J. J. Keats, H. Wang, 337 D. A. Vignali, P. L. Bergsagel, and M. Karin. 2008. Nonredundant and 338 complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that 339 activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol 9:1364-1370. 340
12. Patke, A., I. Mecklenbrauker, H. Erdjument-Bromage, P. Tempst, and A. 341 Tarakhovsky. 2006. BAFF controls B cell metabolic fitness through a PKC beta- and 342 Akt-dependent mechanism. The Journal of experimental medicine 203:2551-2562. 343
13. Craxton, A., K. E. Draves, A. Gruppi, and E. A. Clark. 2005. BAFF regulates B 344 cell survival by downregulating the BH3-only family member Bim via the ERK 345 pathway. The Journal of experimental medicine 202:1363-1374. 346
14. Otipoby, K. L., Y. Sasaki, M. Schmidt-Supprian, A. Patke, R. Gareus, M. 347 Pasparakis, A. Tarakhovsky, and K. Rajewsky. 2008. BAFF activates Akt and Erk 348 through BAFF-R in an IKK1-dependent manner in primary mouse B cells. 349

15
Proceedings of the National Academy of Sciences of the United States of America 350 105:12435-12438. 351
15. Schweighoffer, E., L. Vanes, J. Nys, D. Cantrell, S. McCleary, N. Smithers, and V. 352 L. Tybulewicz. 2013. The BAFF receptor transduces survival signals by co-opting the 353 B cell receptor signaling pathway. Immunity 38:475-488. 354
16. Junt, T., E. Scandella, and B. Ludewig. 2008. Form follows function: lymphoid 355 tissue microarchitecture in antimicrobial immune defence. Nat Rev Immunol 8:764-356 775. 357
17. Mebius, R. E., and G. Kraal. 2005. Structure and function of the spleen. Nat Rev 358 Immunol 5:606-616. 359
18. Junt, T., E. A. Moseman, M. Iannacone, S. Massberg, P. A. Lang, M. Boes, K. 360 Fink, S. E. Henrickson, D. M. Shayakhmetov, N. C. Di Paolo, N. van Rooijen, T. 361 R. Mempel, S. P. Whelan, and U. H. von Andrian. 2007. Subcapsular sinus 362 macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral 363 B cells. Nature 450:110-114. 364
19. Oetke, C., G. Kraal, and P. R. Crocker. 2006. The antigen recognized by MOMA-I 365 is sialoadhesin. Immunology letters 106:96-98. 366
20. Muller, S., L. Hunziker, S. Enzler, M. Buhler-Jungo, J. P. Di Santo, R. M. 367 Zinkernagel, and C. Mueller. 2002. Role of an Intact Splenic Microarchitecture in 368 Early Lymphocytic Choriomeningitis Virus Production. Journal of Virology 76:2375-369 2383. 370
21. Honke, N., N. Shaabani, G. Cadeddu, U. R. Sorg, D. E. Zhang, M. Trilling, K. 371 Klingel, M. Sauter, R. Kandolf, N. Gailus, N. van Rooijen, C. Burkart, S. E. 372 Baldus, M. Grusdat, M. Lohning, H. Hengel, K. Pfeffer, M. Tanaka, D. 373 Haussinger, M. Recher, P. A. Lang, and K. S. Lang. 2012. Enforced viral 374 replication activates adaptive immunity and is essential for the control of a cytopathic 375 virus. Nat Immunol 13:51-57. 376
22. Aichele, P., K. Brduscha-Riem, R. M. Zinkernagel, H. Hengartner, and H. 377 Pircher. 1995. T cell priming versus T cell tolerance induced by synthetic peptides. 378 The Journal of experimental medicine 182:261-266. 379
23. Iezzi, G., K. Karjalainen, and A. Lanzavecchia. 1998. The duration of antigenic 380 stimulation determines the fate of naive and effector T cells. Immunity 8:89-95. 381
24. Zinkernagel, R. M. 2000. Localization dose and time of antigens determine immune 382 reactivity. Seminars in immunology 12:163-171; discussion 257-344. 383
25. Lanzavecchia, A., and F. Sallusto. 2001. Antigen decoding by T lymphocytes: from 384 synapses to fate determination. Nat Immunol 2:487-492. 385
26. Karlsson, M. C., R. Guinamard, S. Bolland, M. Sankala, R. M. Steinman, and J. 386 V. Ravetch. 2003. Macrophages control the retention and trafficking of B 387 lymphocytes in the splenic marginal zone. The Journal of experimental medicine 388 198:333-340. 389
27. You, Y., R. C. Myers, L. Freeberg, J. Foote, J. F. Kearney, L. B. Justement, and 390 R. H. Carter. 2011. Marginal zone B cells regulate antigen capture by marginal zone 391 macrophages. J Immunol 186:2172-2181. 392
28. Puga, I., M. Cols, C. M. Barra, B. He, L. Cassis, M. Gentile, L. Comerma, A. 393 Chorny, M. Shan, W. Xu, G. Magri, D. M. Knowles, W. Tam, A. Chiu, J. B. 394 Bussel, S. Serrano, J. A. Lorente, B. Bellosillo, J. Lloreta, N. Juanpere, F. 395 Alameda, T. Baro, C. D. de Heredia, N. Toran, A. Catala, M. Torrebadell, C. 396 Fortuny, V. Cusi, C. Carreras, G. A. Diaz, J. M. Blander, C. M. Farber, G. 397 Silvestri, C. Cunningham-Rundles, M. Calvillo, C. Dufour, L. D. Notarangelo, V. 398 Lougaris, A. Plebani, J. L. Casanova, S. C. Ganal, A. Diefenbach, J. I. Arostegui, 399 M. Juan, J. Yague, N. Mahlaoui, J. Donadieu, K. Chen, and A. Cerutti. 2012. B 400

16
cell-helper neutrophils stimulate the diversification and production of immunoglobulin 401 in the marginal zone of the spleen. Nat Immunol 13:170-180. 402
29. Tumanov, A., D. Kuprash, M. Lagarkova, S. Grivennikov, K. Abe, A. Shakhov, L. 403 Drutskaya, C. Stewart, A. Chervonsky, and S. Nedospasov. 2002. Distinct role of 404 surface lymphotoxin expressed by B cells in the organization of secondary lymphoid 405 tissues. Immunity 17:239-250. 406
30. Muller, U., U. Steinhoff, L. F. Reis, S. Hemmi, J. Pavlovic, R. M. Zinkernagel, 407 and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral 408 defense. Science 264:1918-1921. 409
31. Lang, P. A., H. C. Xu, M. Grusdat, D. R. McIlwain, A. A. Pandyra, I. S. Harris, N. 410 Shaabani, N. Honke, S. Kumar Maney, E. Lang, V. I. Pozdeev, M. Recher, B. 411 Odermatt, D. Brenner, D. Haussinger, P. S. Ohashi, H. Hengartner, R. M. 412 Zinkernagel, T. W. Mak, and K. S. Lang. 2013. Reactive oxygen species delay 413 control of lymphocytic choriomeningitis virus. Cell death and differentiation. 414
32. Fu, Y. X., G. Huang, M. Matsumoto, H. Molina, and D. D. Chaplin. 1997. 415 Independent signals regulate development of primary and secondary follicle structure 416 in spleen and mesenteric lymph node. Proceedings of the National Academy of 417 Sciences of the United States of America 94:5739-5743. 418
33. Xu, H. C., M. Grusdat, A. A. Pandyra, R. Polz, J. Huang, P. Sharma, R. Deenen, 419 K. Kohrer, R. Rahbar, A. Diefenbach, K. Gibbert, M. Lohning, L. Hocker, Z. 420 Waibler, D. Haussinger, T. W. Mak, P. S. Ohashi, K. S. Lang, and P. A. Lang. 421 2014. Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. 422 Immunity 40:949-960. 423
34. Fogg, D. K., C. Sibon, C. Miled, S. Jung, P. Aucouturier, D. R. Littman, A. 424 Cumano, and F. Geissmann. 2006. A clonogenic bone marrow progenitor specific 425 for macrophages and dendritic cells. Science 311:83-87. 426
35. Fensterl, V., J. L. Wetzel, S. Ramachandran, T. Ogino, S. A. Stohlman, C. C. 427 Bergmann, M. S. Diamond, H. W. Virgin, and G. C. Sen. 2012. Interferon-induced 428 Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS pathogens 429 8:e1002712. 430
36. Nair, S., K. Michaelsen-Preusse, K. Finsterbusch, S. Stegemann-Koniszewski, D. 431 Bruder, M. Grashoff, M. Korte, M. Koster, U. Kalinke, H. Hauser, and A. 432 Kroger. 2014. Interferon regulatory factor-1 protects from fatal neurotropic infection 433 with vesicular stomatitis virus by specific inhibition of viral replication in neurons. 434 PLoS pathogens 10:e1003999. 435
37. Miyake, Y., K. Asano, H. Kaise, M. Uemura, M. Nakayama, and M. Tanaka. 436 2007. Critical role of macrophages in the marginal zone in the suppression of immune 437 responses to apoptotic cell-associated antigens. The Journal of clinical investigation 438 117:2268-2278. 439
38. Futterer, A., K. Mink, A. Luz, M. H. Kosco-Vilbois, and K. Pfeffer. 1998. The 440 lymphotoxin beta receptor controls organogenesis and affinity maturation in 441 peripheral lymphoid tissues. Immunity 9:59-70. 442
39. Moseman, E. A., M. Iannacone, L. Bosurgi, E. Tonti, N. Chevrier, A. Tumanov, 443 Y. X. Fu, N. Hacohen, and U. H. von Andrian. 2012. B cell maintenance of 444 subcapsular sinus macrophages protects against a fatal viral infection independent of 445 adaptive immunity. Immunity 36:415-426. 446
40. Nolte, M. A., R. Arens, M. Kraus, M. H. van Oers, G. Kraal, R. A. van Lier, and 447 R. E. Mebius. 2004. B cells are crucial for both development and maintenance of the 448 splenic marginal zone. J Immunol 172:3620-3627. 449
41. Hiemstra, I. H., M. R. Beijer, H. Veninga, K. Vrijland, E. G. F. Borg, B. J. 450 Olivier, R. E. Mebius, G. Kraal, and J. M. M. den Haan. 2014. The identification 451

17
and developmental requirements of colonic CD169+macrophages. Immunology 452 142:269-278. 453
42. Junt, T., A. V. Tumanov, N. Harris, M. Heikenwalder, N. Zeller, D. V. Kuprash, 454 A. Aguzzi, B. Ludewig, S. A. Nedospasov, and R. M. Zinkernagel. 2006. 455 Expression of lymphotoxin beta governs immunity at two distinct levels. Eur J 456 Immunol 36:2061-2075. 457
43. Mackay, F., P. A. Silveira, and R. Brink. 2007. B cells and the BAFF/APRIL axis: 458 fast-forward on autoimmunity and signaling. Curr Opin Immunol 19:327-336. 459
44. Cancro, M. P., D. P. D'Cruz, and M. A. Khamashta. 2009. The role of B 460 lymphocyte stimulator (BLyS) in systemic lupus erythematosus. The Journal of 461 clinical investigation 119:1066-1073. 462
45. McCarthy, D. D., J. Kujawa, C. Wilson, A. Papandile, U. Poreci, E. A. Porfilio, L. 463 Ward, M. A. Lawson, A. J. Macpherson, K. D. McCoy, Y. Pei, L. Novak, J. Y. 464 Lee, B. A. Julian, J. Novak, A. Ranger, J. L. Gommerman, and J. L. Browning. 465 2011. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-466 associated nephropathy. The Journal of clinical investigation 121:3991-4002. 467
46. Navarra, S. V., R. M. Guzman, A. E. Gallacher, S. Hall, R. A. Levy, R. E. 468 Jimenez, E. K. Li, M. Thomas, H. Y. Kim, M. G. Leon, C. Tanasescu, E. Nasonov, 469 J. L. Lan, L. Pineda, Z. J. Zhong, W. Freimuth, and M. A. Petri. 2011. Efficacy 470 and safety of belimumab in patients with active systemic lupus erythematosus: a 471 randomised, placebo-controlled, phase 3 trial. Lancet 377:721-731. 472
47. Getts, D. R., E. M. Chastain, R. L. Terry, and S. D. Miller. 2013. Virus infection, 473 antiviral immunity, and autoimmunity. Immunological reviews 255:197-209. 474
48. Munz, C., J. D. Lunemann, M. T. Getts, and S. D. Miller. 2009. Antiviral immune 475 responses: triggers of or triggered by autoimmunity? Nat Rev Immunol 9:246-258. 476
49. Honke, N., N. Shaabani, D. E. Zhang, G. Iliakis, H. C. Xu, D. Haussinger, M. 477 Recher, M. Lohning, P. A. Lang, and K. S. Lang. 2013. Usp18 driven enforced 478 viral replication in dendritic cells contributes to break of immunological tolerance in 479 autoimmune diabetes. PLoS pathogens 9:e1003650. 480
50. Hunziker, L., M. Recher, A. J. Macpherson, A. Ciurea, S. Freigang, H. 481 Hengartner, and R. M. Zinkernagel. 2003. Hypergammaglobulinemia and 482 autoantibody induction mechanisms in viral infections. Nat Immunol 4:343-349. 483
51. Barzilai, O., M. Ram, and Y. Shoenfeld. 2007. Viral infection can induce the 484 production of autoantibodies. Curr Opin Rheumatol 19:636-643. 485
52. Benedict, C. A., T. A. Banks, L. Senderowicz, M. Ko, W. J. Britt, A. Angulo, P. 486 Ghazal, and C. F. Ware. 2001. Lymphotoxins and cytomegalovirus cooperatively 487 induce interferon-beta, establishing host-virus detente. Immunity 15:617-626. 488
53. Schneider, K., A. Loewendorf, C. De Trez, J. Fulton, A. Rhode, H. Shumway, S. 489 Ha, G. Patterson, K. Pfeffer, S. A. Nedospasov, C. F. Ware, and C. A. Benedict. 490 2008. Lymphotoxin-mediated crosstalk between B cells and splenic stroma promotes 491 the initial type I interferon response to cytomegalovirus. Cell host & microbe 3:67-76. 492
493 494

18
Figure Legends: 495
FIG 1: BAFFR is essential for anti-viral immunity. 496
(A) Sections from snap frozen spleen tissue of WT and Baffr–/– mice were stained with H&E (scale bar 497
= 100µm) or stained with anti-CD4, anti-CD8, anti-B220 antibodies (scale bar = 50µm). (One 498
representative out of n=5 is shown). (B) Single cell suspensions from splenocytes derived from WT 499
and BAFFR deficient mice were analyzed for immune cell populations with monoclonal antibodies 500
specific for the indicated markers (n=6, n.s. indicates not significant; except for B220+ cells, % is 501
related to B220- splenocytes). (C-D) WT and Baffr–/– mice were infected with 105 pfu of VSV. (C) 502
Serum was taken at the indicated time points and analyzed for anti-viral neutralizing antibodies by 503
PRNT assay (starting with n=8). (D) Survival was monitored over the indicated time period (starting 504
with n=5). Error bars show SEM, n.s. indicates not significant. 505
506
FIG 2: BAFFR mediates enforced viral replication during viral infection 507
(A-C) WT and Baffr–/– mice were infected with 105 pfu of VSV. (A) Virus titers were measured in the 508
spleen 8h (left panel) and 24h (right panel) after infection (n=6) (B) IFN-D concentration was measured 509
12 h and 24 h after infection with 105 pfu of VSV in the serum of WT and BAFFR deficient mice (n=6). 510
(C) Isg15, Ifit2 and Mx1 mRNA expression was determined from brain tissue of infected WT and Baffr–511 /– mice after 24h p.i. (n=5). (D) IFN-D concentration was examined in the serum of WT and Baffr–/– 512
mice 3h after challenge with 25µg or 100µg poly I:C (n=6). (E) Isg15, Ifit2 and Mx1 mRNA expression 513
was determined from brain tissue of WT and Baffr-/- 6 hours after treatment of 10,000 units of mouse 514
recombinant IFN-α or 100 µg poly I:C (n=6). Error bars show SEM, n.s. indicates not significant. 515
516
FIG 3: BAFFR signals are critical for maintenance of CD169+ cells and viral replication in spleen 517
tissue early after infection. 518
(A) CMP and GMP in the bone marrow of WT and Baffr–/– mice were analyzed by flow cytometry (n=6, 519
n.s. indicates not significant). (B) Left panel, sections of snap frozen spleen tissue of WT and Baffr–/– 520
mice were stained with anti-CD169 and analyzed by fluorescence microscopy (clone: MOMA-1, one 521
representative of n=6 is shown, scale bar = 100µm). Right panel, CD169+ cells were measured in 522
splenocytes of WT vs. Baffr–/– mice by flow cytometry (n=6). (C) F4/80+ cells were analyzed in spleen 523
tissue from WT and BAFFR deficient animals by flow cytometry (n=6). (D) Snap frozen spleen 524
sections were stained with an anti-CD169 antibody 0, 3, 5, and 7 h after VSV infection of WT vs. 525

19
BAFFR deficient mice (One representative of n=6 is shown, scale bar = 100µm). (E) Sections from 526
snap frozen spleen tissue obtained from WT and Baffr–/– mice after the indicated time periods following 527
VSV infection were stained with anti-CD169 and anti-VSV-G-protein (clone: Vi10) (one representative 528
of n=6 is shown, scale bar = 100µm). 529
530
FIG 4: CD169+ cell survival following VSV infection in Ifnar1–/– mice. 531
Sections from snap frozen spleen tissue obtained from WT and Ifnar1–/– mice after the indicated time 532
periods following VSV infection were stained with anti-CD169 and anti-VSV-G-protein (clone: Vi10) 533
(one representative of n=6 is shown, scale bar = 100µm). 534
535
FIG 5: B cell dependent maintenance of CD169+ cells. 536
(A) Baffr–/– mice were reconstituted with sorted WT B cells. 40 days later, spleen tissue was harvested 537
and compared to WT and Baffr–/– mice. CD169+ (upper panels) and transferred B cells (CD45.1, 538
middle panels) are shown in WT, Baffr–/– and B cell transferred Baffr–/– mice (n=5, one representative 539
is shown, scale bars = 100µm). (B-D) Baffr–/– mice were reconstituted with sorted WT B cells. 40 days 540
later, animals were infected with 105 pfu of VSV and compared to WT and Baffr–/– mice. (B) IFN-D 541
concentration was measured 24h after infection in WT, Baffr–/– mice and WT B cell transferred Baffr–/–542
(* (p<0.05), **** (p<0.0001), Holm-Sidak test was used for post hoc test). (C) Neutralizing Ig titers 543
were determined at indicated time points after infection (n=4-5, *** (p<0.001) between WT and Baffr–/– 544
mice, ## (p<0.01), between WT and Baffr–/– mice after WT B cell transfer, §§§ (p<0.001) between 545
Baffr–/– and B cell transferred Baffr–/– mice). (D) Survival was monitored in WT, Baffr–/– mice and Baffr–546 /– mice 40 days after WT B cell transfer following infection with 105 pfu of VSV (n=7-8). Error bars 547
show SEM, n.s. indicates not significant. 548
549
FIG 6: B cell dependent maintenance of CD169+ cells depend on lymphotoxin beta expression. 550
(A) Lymphotoxin alpha (left panel) and beta (right panel) mRNA expression levels were analyzed from 551
spleen tissue harvested from WT and Baffr–/– mice (n=5-7). (B) Snap frozen spleen sections from Ltbfl/fl 552
x CD19-Cre+ mice and control animals were stained with anti-CD169 and F4/80 antibody (one 553
representative of n=3 is shown). (C) IFN-D concentration was determined 24 h after infection with 105 554
pfu of VSV from Ltbfl/fl x CD19-Cre+ and control animals (n=4-6). (D) Neutralizing antibody titers were 555

20
measured in sera harvested from Ltbfl/fl x CD19-Cre+ and control animals at the indicated time points 556
after infection (n=4-6). Error bars show SEM, n.s. indicates not significant. 557
558
FIG 7: Impaired innate and adaptive immunity in BAFFR deficient mice during LCMV infection. 559
WT and Baffr–/– were infected with 200 pfu of LCMV-Docile. (A) Virus titers were measured in spleen, 560
liver, lung, kidney, brain and spinal cord tissue 72 hours after infection (n=5-6). (B) IFN-D 561
concentration was determined in the serum of infected animals 72 hours following infection (n=5-6). (C) 562
20 days after infection virus specific CD8+ T cells were examined by tetramer staining in spleen tissue 563
of mice (n=5–6). (D) IFN-J production of T cells as assessed by intracellular cytokine staining and flow 564
cytromteric analysis was measured after in vitro re-stimulation with the MHCI peptides gp33 and 565
np396 (left panel) and the MHCII peptide gp61 (right panel) (n=5-6). (E) Virus titers were measured in 566
spleen, liver, lung, kidney, brain and spinal cord tissue 20 days after infection (n=5–6). Error bars 567
show SEM, n.s. indicates not significant. 568