
Artificial Binding Proteins (Affitins) as Probes for Conformational Changes in Secretin PulD
11
Artificial Binding Proteins (Affitins) as Probes for Conformational Changes in Secretin PulD Martin Krehenbrink 1 , Mohamed Chami 2 , Ingrid Guilvout 1 , Pedro M. Alzari 3 , Frédéric Pécorari 3,4 and Anthony P. Pugsley 1 ⁎ 1 Institut Pasteur, Unité de Génétique moléculaire, CNRS URA2172, 25, rue du Dr. Roux, 75724 Paris Cedex 15, France 2 M.E. Müller Institute for Structural Biology, Biozentrum University of Basel, CH 4056 Basel, Switzerland 3 Institut Pasteur, Unité de Biochimie structurale, CNRS URA2185, 25, rue du Dr. Roux, 75724 Paris Cedex 15, France 4 Université de Nantes, CNRS UMR6204, 2 rue de la Houssinière, BP 92208, Nantes 44322 Cedex 3, France Received 13 May 2008; received in revised form 26 August 2008; accepted 2 September 2008 Available online 16 September 2008 The DNA-binding protein Sac7d was previously modified to bind with high affinity to the N domain of the outer membrane secretin PulD from the bacterium Klebsiella oxytoca. Here, we show that binding of the Sac7d derivatives (affitins) to PulD is sensitive to conformational changes caused by denaturant and by the zwitterionic detergent Zwittergent 3–14 routinely used to extract secretins from outer membranes. This sensitivity to the conformational state of PulD allowed us to use the affitins as probes for the native structure of PulD and to devise protocols for examining in vitro synthesized protein in nonionic detergent and for the affinity purification of native PulD using affitins as ligands. When fused to periplasmic PhoA, three affitins inhibited PulD multimerization in vivo and caused loss of function. In two cases, this was likely to be due to dimerization of the affitin by the bound PhoA, as the effect was absent when the affitins were fused to monomeric MalE. In the third case, the MalE and PhoA moieties probably interfered sterically with PulD protomer interactions and, thereby, inhibited multimerization. None of the affitins tested interacted with PulD at sites of protomer interaction or blocked the secretin channel through which exoproteins cross the outer membrane in the Type II secretion pathway of which PulD is a key component. © 2008 Elsevier Ltd. All rights reserved. Edited by I. B. Holland Keywords: Sac7d; outer membrane protein; secretin; Type II protein secretion; protein folding Introduction Type II protein secretion systems (T2SSs) are widespread in Gram-negative bacteria, where they facilitate the movement of specific proteins from the periplasm to outside of the cell. The T2SS is composed of 12–15 different proteins that are associated with either the inner or outer membrane and that function together as a membrane potential- and ATP-dependent 1,2 protein secretion machinery, the secreton. 3 Plant and animal pathogens such as Xanthomonas campestris, Erwinia spp., and Vibrio cholerae make extensive use of the T2SS to secrete hydrolytic enzymes and virulence factors essential for pathogenesis. 4 Common to all studied T2SSs is a homomultimeric outer membrane protein complex (secretin) that probably represents the channel through which exoproteins are secreted. 5 The Erwinia chrysanthemi secretin OutD binds the exoprotein pectate lyase directly, 6 suggesting an additional role in substrate recognition. Secretins also play major roles in the elaboration of other bacterial outer-membrane-spanning structures such as those of the Type III injectisome, 7 Type IV pili, 8,9 and the filamentous bacteriophage secretion apparatus. 10 The pullulanase (PulA)-specific T2SS of Klebsiella oxytoca is one of the most extensively studied secretons. Cryo-electron microscopy (EM) of purified protein solubilized in detergent revealed that the *Corresponding author. E-mail address: [email protected] . Abbreviations used: T2SS, Type II protein secretion system; EM, electron microscopy; NRMSD, normalized root-mean-square deviation; POE, polyoxyethylene; EDTA, ethylenediaminetetraacetic acid. doi:10.1016/j.jmb.2008.09.016 J. Mol. Biol. (2008) 383, 1058–1068 Available online at www.sciencedirect.com 0022-2836/$ - see front matter © 2008 Elsevier Ltd. All rights reserved.
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Artificial Binding Proteins (Affitins) as Probes for Conformational Changes in Secretin PulD

doi:10.1016/j.jmb.2008.09.016 J. Mol. Biol. (2008) 383, 1058–1068
Available online at www.sciencedirect.com
Artificial Binding Proteins (Affitins) as Probes forConformational Changes in Secretin PulD
Martin Krehenbrink1, Mohamed Chami2, Ingrid Guilvout1,Pedro M. Alzari3, Frédéric Pécorari3,4 and Anthony P. Pugsley1⁎
1Institut Pasteur, Unité deGénétique moléculaire, CNRSURA2172, 25, rue du Dr. Roux,75724 Paris Cedex 15, France2M.E. Müller Institute forStructural Biology, BiozentrumUniversity of Basel, CH 4056Basel, Switzerland3Institut Pasteur, Unité deBiochimie structurale, CNRSURA2185, 25, rue du Dr. Roux,75724 Paris Cedex 15, France4Université de Nantes, CNRSUMR6204, 2 rue de laHoussinière, BP 92208, Nantes44322 Cedex 3, FranceReceived 13 May 2008;received in revised form26 August 2008;accepted 2 September 2008Available online16 September 2008
*Corresponding author. E-mail addrAbbreviations used: T2SS, Type II
system; EM, electron microscopy; Nroot-mean-square deviation; POE, pEDTA, ethylenediaminetetraacetic a
0022-2836/$ - see front matter © 2008 E
The DNA-binding protein Sac7d was previously modified to bind with highaffinity to the N domain of the outer membrane secretin PulD from thebacterium Klebsiella oxytoca. Here, we show that binding of the Sac7dderivatives (affitins) to PulD is sensitive to conformational changes causedby denaturant and by the zwitterionic detergent Zwittergent 3–14 routinelyused to extract secretins from outer membranes. This sensitivity to theconformational state of PulD allowed us to use the affitins as probes for thenative structure of PulD and to devise protocols for examining in vitrosynthesized protein in nonionic detergent and for the affinity purification ofnative PulD using affitins as ligands. When fused to periplasmic PhoA,three affitins inhibited PulD multimerization in vivo and caused loss offunction. In two cases, this was likely to be due to dimerization of the affitinby the bound PhoA, as the effect was absent when the affitins were fused tomonomeric MalE. In the third case, the MalE and PhoA moieties probablyinterfered sterically with PulD protomer interactions and, thereby, inhibitedmultimerization. None of the affitins tested interacted with PulD at sites ofprotomer interaction or blocked the secretin channel through whichexoproteins cross the outer membrane in the Type II secretion pathway ofwhich PulD is a key component.
© 2008 Elsevier Ltd. All rights reserved.
Keywords: Sac7d; outer membrane protein; secretin; Type II proteinsecretion; protein folding
Edited by I. B. HollandIntroduction
Type II protein secretion systems (T2SSs) arewidespread in Gram-negative bacteria, where theyfacilitate the movement of specific proteins from theperiplasm to outside of the cell. The T2SS iscomposed of 12–15 different proteins that areassociated with either the inner or outer membraneand that function together as a membrane potential-and ATP-dependent1,2 protein secretion machinery,the secreton.3 Plant and animal pathogens such as
ess: [email protected] secretionRMSD, normalizedolyoxyethylene;cid.
lsevier Ltd. All rights reserve
Xanthomonas campestris, Erwinia spp., and Vibriocholerae make extensive use of the T2SS to secretehydrolytic enzymes and virulence factors essentialfor pathogenesis.4 Common to all studied T2SSs is ahomomultimeric outer membrane protein complex(secretin) that probably represents the channelthrough which exoproteins are secreted.5 TheErwinia chrysanthemi secretin OutD binds theexoprotein pectate lyase directly,6 suggesting anadditional role in substrate recognition. Secretinsalso play major roles in the elaboration of otherbacterial outer-membrane-spanning structures suchas those of the Type III injectisome,7 Type IV pili,8,9
and the filamentous bacteriophage secretionapparatus.10
The pullulanase (PulA)-specific T2SS of Klebsiellaoxytoca is one of the most extensively studiedsecretons. Cryo-electron microscopy (EM) of purifiedprotein solubilized in detergent revealed that the
d.

1059Affitins as Probes for PulD Conformational Changes
secretin (PulD) of this system assembles into adodecameric structure composed of two rings sand-wiching a central disk.11 Tryptic digestion showedthat the PulD polypeptide has two major domains: atrypsin-sensitive N domain and a trypsin-resistant Cdomain. While the C domain can assemble intomembrane-associated dodecamers, the N domain,which forms part of the periplasmic region of thePulD multimer,11 remains soluble and monomericwhen produced separately.12 The related secretinOutD from E. chrysanthemi interacts with the inner-membrane secreton component OutC and containsspecificity determinants for some, but not all, secretedproteins,6,13 but an equivalent interaction of PulDwith either the secreted protein PulA or the OutChomologue PulC could not be demonstrated (unpub-lished data). Despite extensive studies, neither thehigh-resolution structure nor the specific function ofPulD, and specifically its N domain, is known indetail.To overcome problems inherent in using antibody
fragments as inhibitors of cellular processes, aspromoters of protein crystallization, and as probesfor changes in protein conformation,we recently useda mutagenesis and selection approach to alter thespecificity of the small (66 amino acids), highly stableDNA-binding protein (Sac7d) from the hyperthermo-philic archaeon Sulfolobus acidocaldarius14 to enable itto bind to the PulD N domain.15 Sac7d has a well-characterized oligonucleotide/oligosaccharide-bind-ing fold comprising a five-stranded β-barrel cappedby anα-helix. Proteins containing this structuralmotifspecifically recognize a wide range of substrates suchas oligosaccharides, oligonucleotides, proteins, andmetal ions.16 Specificity is mediated by variations ofspecific amino acid residues located at conservedpositions on the binding face, which result in apotential binding area of around 1200 Å2. Sac7dvariants, hereafter called affitins, which bind to the Ndomain, were previously selected by ribosome dis-play from a combinatorial library of variants inwhich14 residues exposed on the original DNA bindingface17,18 were permutated by gene synthesis. Thisresulted in a large collection of affitins (Sac7⁎) that allbound specifically to the PulD N domain with highaffinity and with different amino acids at most ofthese 14 positions. When fused to alkaline phospha-tase and exported to the periplasm, these affitinsinhibited PulA secretion and PulDmultimerization tovarying extents.15 The present study further exploresthe PulD-N/affitin interaction and demonstrates theuse of affitins as valuable tools for probing proteinstructure in vivo and in vitro.
Results
In vivo effects of monomeric affitins on PulDmultimerization
Coexpression of the genes encoding any of threedifferent PhoA-Sac7⁎ chimeras (Sac7⁎6, Sac7⁎33,
and Sac7⁎40) with the K. oxytoca T2SS (pul) genesin Escherichia coli inhibits PulD multimerization and,consequently, PulA secretion, but the chimerasnevertheless bound to full-length PulD dodecamersin Far Western blot experiments.15 Since PhoA isdimeric, PulD multimerization could also have beenaffected by simultaneous binding of two PulDmonomers, rather than by occlusion of sites requiredfor PulD multimerization. Previously, PulD wasonly detected in strains producing periplasmicSac7⁎6-, Sac7⁎33-, or Sac7⁎40-PhoA chimeras in theabsence of cell envelope proteases, suggesting thatPulD bound by these bifunctional affitins wasdegraded by periplasmic proteases. In the absenceof these proteases, more monomeric PulD wasdetected than in the wild-type strain, but very littlemultimer was detected.15 The form of PulD targetedfor degradation was therefore likely to be the affitin-bound monomers, rather than the fully assembledmultimer.To study the in vivo binding of affitins to PulD
further and to avoid possible complications caused byPhoA protein dimerization, chimeras of monomericmaltose binding protein (MalE) and Sac7⁎6, Sac7⁎33,or Sac7⁎40 were targeted to the periplasm of E. colicarrying theK. oxytocaT2SS genes on the chromosome(PAP7232). Only the MalE-Sac7⁎40 chimera inhibitedPulD multimerization and PulA secretion. The levelsof PulD monomer were higher in strains expressingthis construct than in the wild-type strain (or strainsproducing MalE-Sac7⁎ constructs that did not showdecreased levels ofmultimer). The total levels of PulD,estimated from the amount of monomer after phenoldissociation of the multimers, were comparable in allstrains (Fig. 1a). Incubating purified MalE-Sac7⁎40with membranes from strain PAP7232 allowedcosedimentation of MalE-Sac7⁎40 with the mem-branes, while no such cosedimentation occurredwhen membranes lacking PulD (from strainPAP7447) were used (Fig. 1b), indicating that at leastpart of the binding site remains accessible to MalE-Sac7⁎40 in the PulD multimer.The Sac7⁎40 protein was produced as a periplas-
mic construct carrying only a short eight-amino-acidextension (Strep-tag) at the C-terminus to test thepossibility that MalE-Sac7⁎40 inhibited multimer-ization by steric hindrance by the bulky MalEprotein. This construct did not affect multimeriza-tion or secretion in strain PAP7232 (Fig. 1c) but stillbound to the membrane-inserted PulD multimer inthese cells, as indicated by the specific cosedimenta-tion of Sac7⁎40-Strep with membranes derived fromthem (Fig. 1b).
PulD-derived peptides are not recognized bySac7⁎40
The PulD-N2 protein used as bait to select affitinsrecognizing the PulD N domain comprised residues28–262 (EEFSAS-KQLDRQ) of the PulD preproteinfollowing the hexahistidine tag and thrombincleavage site of pET15b (MGSSHHHHHHSSGL-VPRGSHM). Partial proteolysis of the PulD-N2
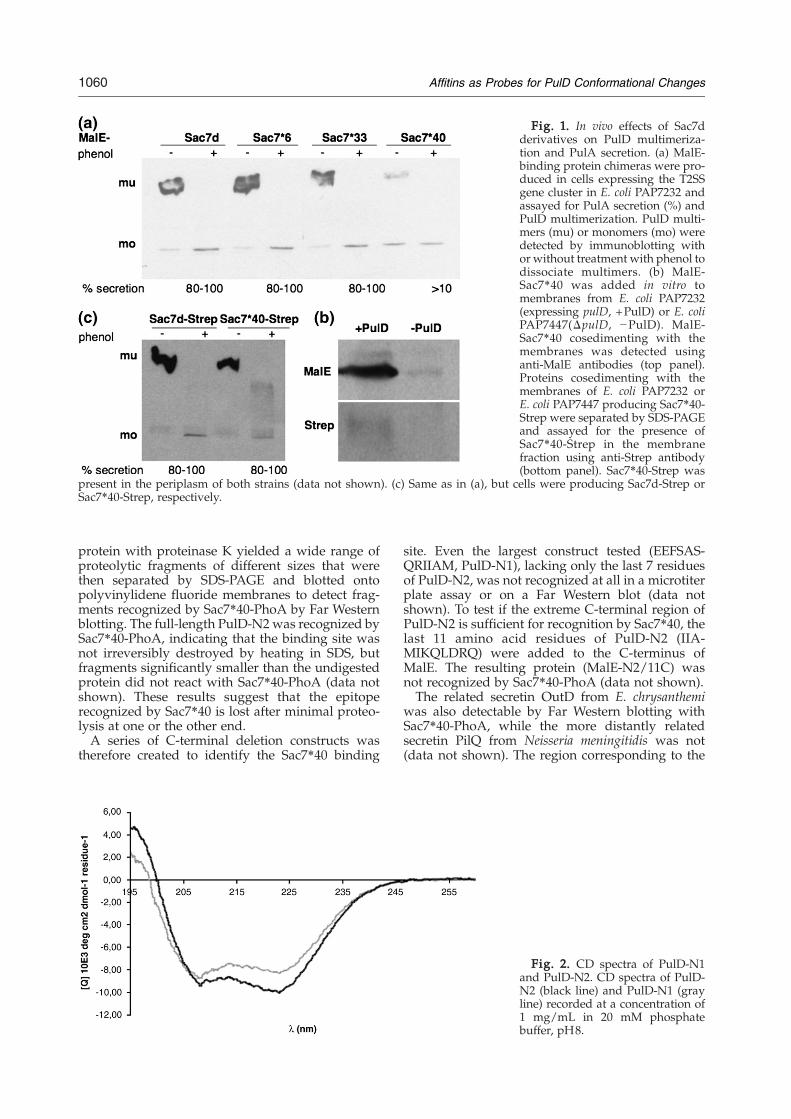
Fig. 1. In vivo effects of Sac7dderivatives on PulD multimeriza-tion and PulA secretion. (a) MalE-binding protein chimeras were pro-duced in cells expressing the T2SSgene cluster in E. coli PAP7232 andassayed for PulA secretion (%) andPulD multimerization. PulD multi-mers (mu) or monomers (mo) weredetected by immunoblotting withor without treatment with phenol todissociate multimers. (b) MalE-Sac7⁎40 was added in vitro tomembranes from E. coli PAP7232(expressing pulD, +PulD) or E. coliPAP7447(ΔpulD, −PulD). MalE-Sac7⁎40 cosedimenting with themembranes was detected usinganti-MalE antibodies (top panel).Proteins cosedimenting with themembranes of E. coli PAP7232 orE. coli PAP7447 producing Sac7⁎40-Strep were separated by SDS-PAGEand assayed for the presence ofSac7⁎40-Strep in the membranefraction using anti-Strep antibody(bottom panel). Sac7⁎40-Strep was
present in the periplasm of both strains (data not shown). (c) Same as in (a), but cells were producing Sac7d-Strep orSac7⁎40-Strep, respectively.
1060 Affitins as Probes for PulD Conformational Changes
protein with proteinase K yielded a wide range ofproteolytic fragments of different sizes that werethen separated by SDS-PAGE and blotted ontopolyvinylidene fluoride membranes to detect frag-ments recognized by Sac7⁎40-PhoA by Far Westernblotting. The full-length PulD-N2 was recognized bySac7⁎40-PhoA, indicating that the binding site wasnot irreversibly destroyed by heating in SDS, butfragments significantly smaller than the undigestedprotein did not react with Sac7⁎40-PhoA (data notshown). These results suggest that the epitoperecognized by Sac7⁎40 is lost after minimal proteo-lysis at one or the other end.A series of C-terminal deletion constructs was
therefore created to identify the Sac7⁎40 binding
site. Even the largest construct tested (EEFSAS-QRIIAM, PulD-N1), lacking only the last 7 residuesof PulD-N2, was not recognized at all in a microtiterplate assay or on a Far Western blot (data notshown). To test if the extreme C-terminal region ofPulD-N2 is sufficient for recognition by Sac7⁎40, thelast 11 amino acid residues of PulD-N2 (IIA-MIKQLDRQ) were added to the C-terminus ofMalE. The resulting protein (MalE-N2/11C) wasnot recognized by Sac7⁎40-PhoA (data not shown).The related secretin OutD from E. chrysanthemi
was also detectable by Far Western blotting withSac7⁎40-PhoA, while the more distantly relatedsecretin PilQ from Neisseria meningitidis was not(data not shown). The region corresponding to the
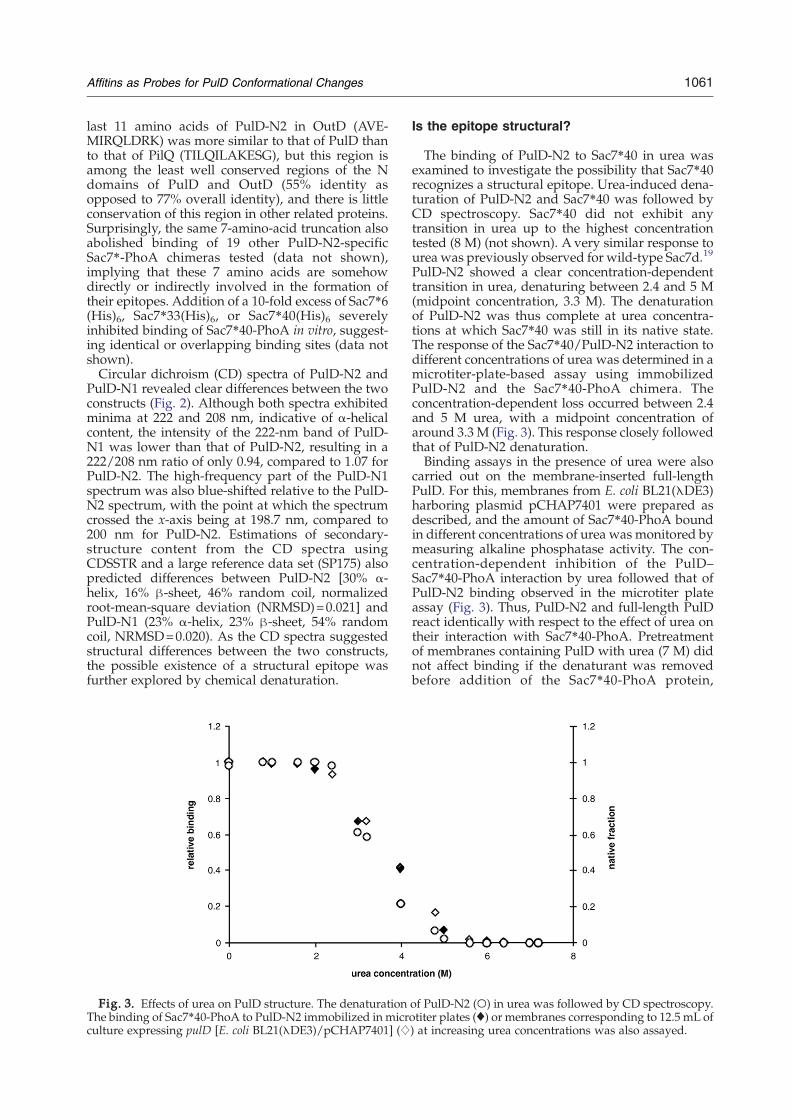
Fig. 2. CD spectra of PulD-N1and PulD-N2. CD spectra of PulD-N2 (black line) and PulD-N1 (grayline) recorded at a concentration of1 mg/mL in 20 mM phosphatebuffer, pH8.
1061Affitins as Probes for PulD Conformational Changes
last 11 amino acids of PulD-N2 in OutD (AVE-MIRQLDRK) was more similar to that of PulD thanto that of PilQ (TILQILAKESG), but this region isamong the least well conserved regions of the Ndomains of PulD and OutD (55% identity asopposed to 77% overall identity), and there is littleconservation of this region in other related proteins.Surprisingly, the same 7-amino-acid truncation alsoabolished binding of 19 other PulD-N2-specificSac7⁎-PhoA chimeras tested (data not shown),implying that these 7 amino acids are somehowdirectly or indirectly involved in the formation oftheir epitopes. Addition of a 10-fold excess of Sac7⁎6(His)6, Sac7⁎33(His)6, or Sac7⁎40(His)6 severelyinhibited binding of Sac7⁎40-PhoA in vitro, suggest-ing identical or overlapping binding sites (data notshown).Circular dichroism (CD) spectra of PulD-N2 and
PulD-N1 revealed clear differences between the twoconstructs (Fig. 2). Although both spectra exhibitedminima at 222 and 208 nm, indicative of α-helicalcontent, the intensity of the 222-nm band of PulD-N1 was lower than that of PulD-N2, resulting in a222/208 nm ratio of only 0.94, compared to 1.07 forPulD-N2. The high-frequency part of the PulD-N1spectrum was also blue-shifted relative to the PulD-N2 spectrum, with the point at which the spectrumcrossed the x-axis being at 198.7 nm, compared to200 nm for PulD-N2. Estimations of secondary-structure content from the CD spectra usingCDSSTR and a large reference data set (SP175) alsopredicted differences between PulD-N2 [30% α-helix, 16% β-sheet, 46% random coil, normalizedroot-mean-square deviation (NRMSD)=0.021] andPulD-N1 (23% α-helix, 23% β-sheet, 54% randomcoil, NRMSD=0.020). As the CD spectra suggestedstructural differences between the two constructs,the possible existence of a structural epitope wasfurther explored by chemical denaturation.
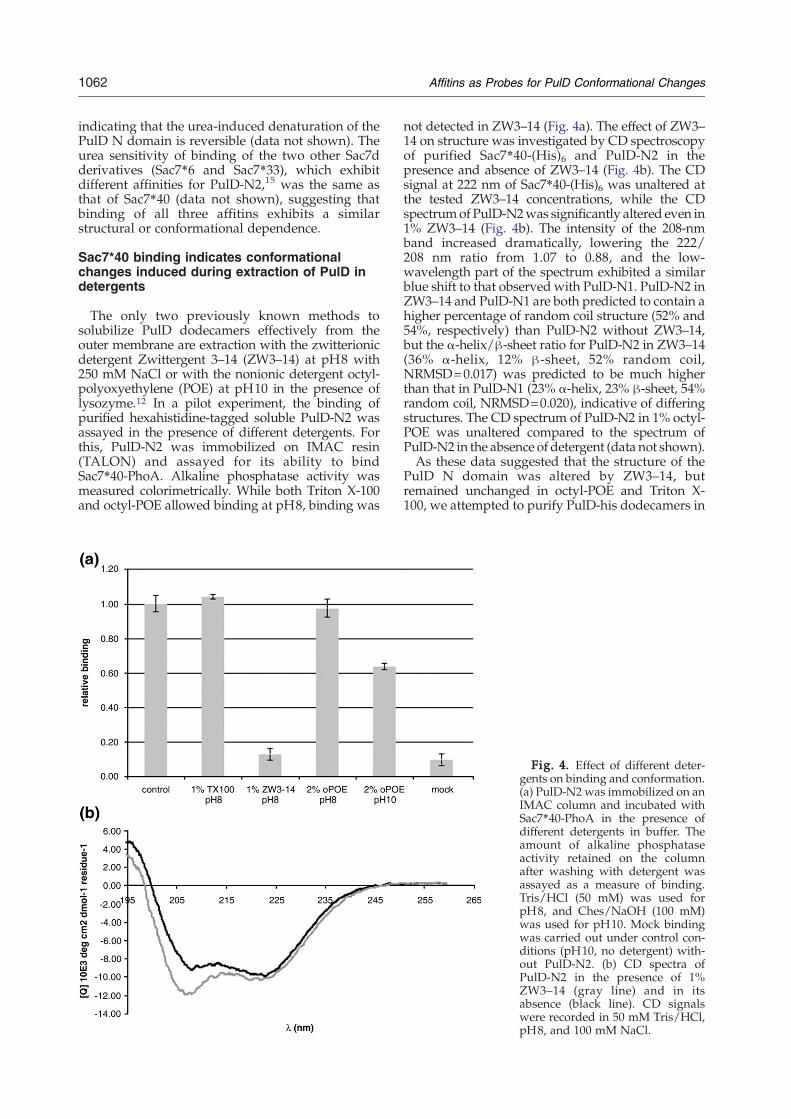
Fig. 3. Effects of urea on PulD structure. The denaturationThe binding of Sac7⁎40-PhoA to PulD-N2 immobilized in micrculture expressing pulD [E. coli BL21(λDE3)/pCHAP7401] (⋄
Is the epitope structural?
The binding of PulD-N2 to Sac7⁎40 in urea wasexamined to investigate the possibility that Sac7⁎40recognizes a structural epitope. Urea-induced dena-turation of PulD-N2 and Sac7⁎40 was followed byCD spectroscopy. Sac7⁎40 did not exhibit anytransition in urea up to the highest concentrationtested (8 M) (not shown). A very similar response tourea was previously observed for wild-type Sac7d.19
PulD-N2 showed a clear concentration-dependenttransition in urea, denaturing between 2.4 and 5 M(midpoint concentration, 3.3 M). The denaturationof PulD-N2 was thus complete at urea concentra-tions at which Sac7⁎40 was still in its native state.The response of the Sac7⁎40/PulD-N2 interaction todifferent concentrations of urea was determined in amicrotiter-plate-based assay using immobilizedPulD-N2 and the Sac7⁎40-PhoA chimera. Theconcentration-dependent loss occurred between 2.4and 5 M urea, with a midpoint concentration ofaround 3.3 M (Fig. 3). This response closely followedthat of PulD-N2 denaturation.Binding assays in the presence of urea were also
carried out on the membrane-inserted full-lengthPulD. For this, membranes from E. coli BL21(λDE3)harboring plasmid pCHAP7401 were prepared asdescribed, and the amount of Sac7⁎40-PhoA boundin different concentrations of urea was monitored bymeasuring alkaline phosphatase activity. The con-centration-dependent inhibition of the PulD–Sac7⁎40-PhoA interaction by urea followed that ofPulD-N2 binding observed in the microtiter plateassay (Fig. 3). Thus, PulD-N2 and full-length PulDreact identically with respect to the effect of urea ontheir interaction with Sac7⁎40-PhoA. Pretreatmentof membranes containing PulD with urea (7 M) didnot affect binding if the denaturant was removedbefore addition of the Sac7⁎40-PhoA protein,
of PulD-N2 (○) in urea was followed by CD spectroscopy.otiter plates (♦) or membranes corresponding to 12.5 mL of) at increasing urea concentrations was also assayed.
1062 Affitins as Probes for PulD Conformational Changes
indicating that the urea-induced denaturation of thePulD N domain is reversible (data not shown). Theurea sensitivity of binding of the two other Sac7dderivatives (Sac7⁎6 and Sac7⁎33), which exhibitdifferent affinities for PulD-N2,15 was the same asthat of Sac7⁎40 (data not shown), suggesting thatbinding of all three affitins exhibits a similarstructural or conformational dependence.
Sac7⁎40 binding indicates conformationalchanges induced during extraction of PulD indetergents
The only two previously known methods tosolubilize PulD dodecamers effectively from theouter membrane are extraction with the zwitterionicdetergent Zwittergent 3–14 (ZW3–14) at pH8 with250 mM NaCl or with the nonionic detergent octyl-polyoxyethylene (POE) at pH10 in the presence oflysozyme.12 In a pilot experiment, the binding ofpurified hexahistidine-tagged soluble PulD-N2 wasassayed in the presence of different detergents. Forthis, PulD-N2 was immobilized on IMAC resin(TALON) and assayed for its ability to bindSac7⁎40-PhoA. Alkaline phosphatase activity wasmeasured colorimetrically. While both Triton X-100and octyl-POE allowed binding at pH8, binding was
not detected in ZW3–14 (Fig. 4a). The effect of ZW3–14 on structure was investigated by CD spectroscopyof purified Sac7⁎40-(His)6 and PulD-N2 in thepresence and absence of ZW3–14 (Fig. 4b). The CDsignal at 222 nm of Sac7⁎40-(His)6 was unaltered atthe tested ZW3–14 concentrations, while the CDspectrumof PulD-N2was significantly altered even in1% ZW3–14 (Fig. 4b). The intensity of the 208-nmband increased dramatically, lowering the 222/208 nm ratio from 1.07 to 0.88, and the low-wavelength part of the spectrum exhibited a similarblue shift to that observed with PulD-N1. PulD-N2 inZW3–14 and PulD-N1 are both predicted to contain ahigher percentage of random coil structure (52% and54%, respectively) than PulD-N2 without ZW3–14,but the α-helix/β-sheet ratio for PulD-N2 in ZW3–14(36% α-helix, 12% β-sheet, 52% random coil,NRMSD=0.017) was predicted to be much higherthan that in PulD-N1 (23% α-helix, 23% β-sheet, 54%random coil, NRMSD=0.020), indicative of differingstructures. The CD spectrum of PulD-N2 in 1% octyl-POE was unaltered compared to the spectrum ofPulD-N2 in the absence of detergent (data not shown).As these data suggested that the structure of the
PulD N domain was altered by ZW3–14, butremained unchanged in octyl-POE and Triton X-100, we attempted to purify PulD-his dodecamers in
Fig. 4. Effect of different deter-gents on binding and conformation.(a) PulD-N2 was immobilized on anIMAC column and incubated withSac7⁎40-PhoA in the presence ofdifferent detergents in buffer. Theamount of alkaline phosphataseactivity retained on the columnafter washing with detergent wasassayed as a measure of binding.Tris/HCl (50 mM) was used forpH8, and Ches/NaOH (100 mM)was used for pH10. Mock bindingwas carried out under control con-ditions (pH10, no detergent) with-out PulD-N2. (b) CD spectra ofPulD-N2 in the presence of 1%ZW3–14 (gray line) and in itsabsence (black line). CD signalswere recorded in 50 mM Tris/HCl,pH8, and 100 mM NaCl.

Fig. 5. Affinity purification of PulD using Sac7⁎40-Strep. The DTT-treated membrane fraction of E. coli BL21(λDE3)/pCHAP7401 was solubilized in 50 mM Tris/HCl,pH8, 2% Triton X-100, 250 mM KCl, and 10 mM EDTA inthe presence of 100 μg/mL lysozyme and then allowed tobind to purified Sac7⁎40-Strep immobilized on Streptactin(IBA) resin in a column for 1 h. The column was washedwith 5 bed volumes of 50 mM Tris/HCl, pH8, 2% TritonX-100, and 250 mM KCl. Elution was performed in thesame buffer containing 2.5 mM desthiobiotin. After SDS-PAGE, proteins were detected by Coomassie Blue Rstaining (left lane), and PulD was identified by immuno-blotting (right lane).
1063Affitins as Probes for PulD Conformational Changes
these nonionic detergents. While octyl-POE does notefficiently extract PulD multimers from membranesat pH values below pH10,12 PulD dodecamersextracted in octyl-POE at pH10 remained solublewhen the pH was lowered from 10 to 8 by theaddition of appropriate buffer (data not shown).Preincubation of PulD-N2 at pH10 did not have anyeffect on Sac7⁎40-PhoA binding at pH8 in an ELISAplate assay (data not shown), suggesting thatbinding to the PulD dodecamer should also befully restored at pH8. Another protocol was devisedfor the solubilization of PulD dodecamers frommembrane fractions by extraction with Triton X-100at pH8 in the presence of ethylenediaminetetraaceticacid (EDTA) and lysozyme. EDTA and lysozymewere only required for the extraction step and werenot necessary to retain PulD multimers in solution(data not shown).Although PulD-his binds only weakly to IMAC
columns (TALON) and elutes at low imidazoleconcentrations (20 mM) in ZW3–14, it is never-theless possible to purify sufficient amounts of theprotein for structural analysis.11 Contrary to this,PulD-his solubilized as described above in octyl-POE or Triton X-100 did not bind to IMAC columnseven in the absence of imidazole and, therefore,could not be purified using this method. Takingadvantage of the affinity of Sac7⁎40 for the PulD Ndomain, we devised a protocol for the purification ofPulD by affinity chromatography using purifiedSac7⁎40-Strep bound to a Streptactin matrix (IBA).The Triton X-100-solubilized membrane fraction ofE. coli BL21 (λDE3) (pCHAP7401) was added to theStreptactin/Sac7⁎40-Strep slurry and left to bind for1 h. The membrane fraction was treated with 10 mMdithiothreitol (DTT) prior to solubilization in orderto remove PulD-associated PulS.11,20 After washing,resin-bound Sac7⁎40-Strep and associated proteinswere eluted and separated by SDS-PAGE. Coomas-sie staining and Western blotting revealed that PulDcoeluted with Sac7⁎40-Strep (Fig. 5). This resultdemonstrates that the Sac7d derivatives can be usedas immobilizable ligands for the affinity purificationof PulD in its native conformation. In the proceduredescribed here, the ligand (Sac7⁎40) remains boundto the eluted and single-step purified PulD dodeca-mers and, therefore, the method is not ideally suitedfor the purification of PulD multimers for structuralstudies. It does, however, open up the possibility ofpurifying PulD from bacterial membranes undernative conditions. This will be used in furtherstudies aiming to identify proteins interacting withPulD by copurification. Copurification of PulDmultimers solubilized in nonionic detergent alsoserved to confirm the native state of the PulD Ndomain under these conditions.
Characterization of the PulD multimerssolubilized in nonionic detergent by EM
To determine whether the PulD multimers areindeed solubilized as single particles, and not asaggregated material, by nonionic detergents, we
examined PulD multimers solubilized in Triton X-100 by EM. Since the structure of the PulDN domainis altered in ZW3–14 (see above), the examination ofPulD solubilized under native conditions alsopromised new insights into the structure of thisprotein. As discussed above, under native condi-tions, PulD can only be purified from bacterial cellenvelopes as a complex with Sac7⁎40 derivatives,but it was desirable to also examine PulD in theabsence of Sac7⁎40 to determine the structure ofnative PulD dodecamers alone. Instead of purifyingPulD from bacterial membranes, the protein wasproduced using an in vitro transcription/translationprotocol in the presence of artificial liposomes.21
Purified liposomes from this experiment yieldedPulD that was sufficiently concentrated and pure fornegative staining and EM analysis. Liposomescontaining PulD were then extracted with TritonX-100 and urea, and the urea was subsequentlyremoved by dialysis before examination of nega-tively stained protein by EM.PulD multimers appeared mostly as individual
particles in both Triton X-100 and in ZW3–14,although occasional aggregates comprising two tofive multimers were observed in both samples. PulDmultimers were clearly recognizable in side andaxial views in both cases (Fig. 6). Markedly, sideviews were more abundant than top views in theTriton X-100 preparation than in the ZW3–14preparation (90% of 871 analyzed particles wereside views in Triton X-100 versus 10% of 586 particles

Fig. 6. EM of negatively stainedPulD purified in ZW3–14 (a) andTriton X-100 (b). PulD multimerswere produced and inserted intoliposomes by in vitro translation/transcription and extracted frompurified liposomes with either Tri-ton X-100 or ZW3–14. The purifiedprotein was negatively stained andobserved by EM. Black and whitearrows indicate top and side viewsof secretin PulD, respectively. Theinsets are averaged images of twomajor classes: Top views [left,n=117 in (a), n=95 in (b)] and sideviews [right, n=94 in (a), n=170 in(b)]. The periplasmic side is facingdownwards in the averaged sideviews. The scale bar represents50 nm, and the inset baseline is17 nm.
1064 Affitins as Probes for PulD Conformational Changes
in ZW3–14). Averaging of particle images failed toreveal any major difference in overall shape in thetwo detergents (Fig. 6). Thus, the structural changesthat lead to loss of affitin binding in ZW3–14 do notappear to lead to a major reorganization of thesecretin complex. Nevertheless, closer examinationof single particles revealed additional electron-densematerial consistently present on the presumedperiplasmic side of side views in Triton X-100 thatwas absent from the particles examined in ZW3–14.However, this could not be recognized as a definedstructure and was not resolved by averaging,possibly indicating a domain that is flexible inTriton X-100 and even less structured in ZW3–14.Further EM studies will aim at stabilizing thestructure of this unresolved portion of PulD andmapping of the affitin binding site on the 3Dstructure, leading to an enhanced understanding ofsecretin structure.
Discussion
The results presented indicate that the binding ofpreviously described affitins to the N domain ofsecretin PulD15 is exquisitely sensitive to conforma-tional changes in the target protein. For example, theloss of binding in urea closely follows the denatura-tion of the isolated N domain fragment PulD-N2,and binding is also abolished in the detergent ZW3–14, which induces a structural change in PulD-N2.Removal of as few as seven amino acids from the C-terminus of PulD-N2, which was used as the bait inthe ribosome-display-based selection of the Sac7d
derivatives, also leads to a complete loss of recogni-tion. This deletion induces a considerable conforma-tional change in the N domain, as revealed by thealtered CD spectrum, suggesting that the last sevenamino acids are required for the correct folding ofdifferent conformational epitopes recognized by thedifferent affitins. However, we cannot formallyexclude that all of the tested Sac7d derivatives bindto clustered conformational epitopes inwhich criticalamino acids are always contributed by the last sevenresidues of PulD-N2. Competition experimentsindicate that the three best characterized affitins,Sac7⁎6, Sac7⁎33, and Sac7⁎40, do indeed haveoverlapping binding sites, despite the fact that theyhave different effects on PulDmultimerization15 (seebelow). The reason why all of the 19 tested affitinsselected using the PulD-N2 fragment as bait in theribosome display system depend directly or indir-ectly for their binding on the last seven amino acidsof this PulD fragment remains unclear, but the datasuggest that the selection systemwe used introduceda strong bias (possibly the requirement for highaffinity) that favored these derivatives. It is surpris-ing that this bias nevertheless allowed the selectionof Sac7d derivatives with different sequences on thebinding face.15 Unfortunately, none of the PulD-N2/affitin complexes purified and analyzed to dateproduces diffracting crystals (unpublished data). Weare currently analyzing the properties of a newcollection of Sac7d derivatives generated by ribo-some display with different segments of the Ndomain as the bait.The extreme sensitivity of Sac7⁎ binding to
conformational changes in the N domain of PulD

1065Affitins as Probes for PulD Conformational Changes
opens up their use as conformational probes and asligands for affinity purification. For example, theloss of Sac7⁎40 binding to PulD in the zwitterionicdetergent ZW3–14 reflects changes in the CDspectrum induced by the detergent in the N domainand indicates that the conformation of at least a partof the binding site in this domain is altered by thedetergent. Consequently, the 3D structure deter-mined for PulD solubilized in this detergent11 mightbe influenced by the presence of the detergent.Fortunately, the conformational changes induced byZW3–14 are reversible, at least as measured bySac7⁎40 binding, and we describe here methods toreplace this detergent by others that do not affect thePulD-N2 structure and, thus, allow Sac7⁎40 bindingand the use of Sac7⁎40 as an affinity purificationligand. This opens up the possibility of futurecopurification studies and of refining the currentstructure of PulD by cryo-EM and single-particleanalysis.11 The preliminary negative-stain EMimages presented here already point towards thepresence of additional structure in the presumablyperiplasmic region of PulD in nonionic detergent.This finding corresponds well with the observationthat the PulD-N2 protein, which represents thisregion of full-length PulD, is structurally altered inthe detergent used in previous structural studies,ZW3–14. Further studies might resolve the differ-ences between the current 3D structures determinedby EM for PulD11 and the closely related secretinPilQ from N. meningitidis.22
One of the most intriguing properties of the PulD-specific affitins is their ability to prevent PulDmultimerization when fused to PhoA.15 We showhere that this property of Sac7⁎6 and Sac7⁎33 isdependent on the presence of the PhoA part of thechimera, which presumably causes the chimera todimerize, bind two PulD protomers, and preventtheir full oligomerization. Sac7⁎40 prevented PulDmultimerization when fused to monomeric MalEbut not when extended by a small epitope, suggest-ing that the bulky MalE prevents the intimatecontact of PulD protomers required for their multi-merization. The fate of the PulD monomers boundto MalE-Sac7⁎40 remains to be determined. The factthat MalE-Sac7⁎40 binds to the PulD monomer andprevents its multimerization indicates that theepitope recognized by this Sac7d derivative ispresent in the unassembled protomer, which hastherefore acquired at least part of its final conforma-tion prior to multimerization and insertion into theouter membrane.
Materials and Methods
Strains, plasmid construction, growth conditions, andsecretion assays
E. coli strain PAP105 [Δ(lac-pro) F′ (lacIq1 ΔlacZM15proAB+ Tn10)] was the transformation recipient for allcloning experiments. PulA secretion and PulD multi-merization were assayed as described before23 in E. coli K-
12 PAP7232, which contains the K. oxytoca T2SS genecluster on the chromosome.24 Strain PAP7447 is aderivative of PAP7232 that lacks pulD.24 The constructionof Sac7⁎-PhoA fusions in pQUANTagen (Qbiogene,Carlsbad, CA) was described previously,15 as was theconstruction of PulD-N212 and of pCHAP710, whichcarries the entire T2SS gene cluster.25
Plasmids for the production of MalE-Sac7⁎ proteinswere constructed by amplifying sac7⁎ by PCR usingprimers P1 (5′-ATTTCTAGAACCGGATCCGTGGAA-3′)and P2 (5′-TTTCTGCAGTTATTGAGCTCTAAGCTTTTT-3′). This introduces an XbaI and PstI site, respectively,which was used to ligate the amplified fragment intopMALp2X (New England Biolabs). The sac7⁎-strep con-structs were made by amplifying sac7⁎ using primers P3(5′-ATATGAATTCGTGAAGGTGAAA-3′) and P4 (5′-TAATTGACCATGGAGCTTTTTTTCACG-3′) to introducean EcoRI and a PstI site and cloning into pASK IBA2C(IBA). A plasmid encoding MalE-N2/11C was made byligating an oligonucleotide dimer of P5 (5′-AATTGAT-CATCGCCATGATTAAGCAGCTCGACCGCCAGTAG-3′) and P6 (5′-AGCTCTACTGGCGGTCGAGCTGCT-TAATCATGGCGATGATC-3′) into pMALp2X cut withEcoRI and HindIII. Plasmid pCHAP7401 was constructedby amplifying pulD(his)6 from plasmid pCHAP350423
using primers P7 (5′-TAACCCTTACCGTTGAAC-3′) andP8 (5′-CCGTAGTCATCAGAAAATACGACGTTG-TAAAACGAC-3) and amplifying pulS from PAP7232genomic DNA using primers P9 (5′-GGACCTCACCG-TATTTAG-3′) and P10 (5′-GTCGTTTTACAACGTCG-TATTTTCTGATGACTACGG-3′). Both fragments werepurified and used as a template for another round ofPCR using P7 and P9 as primers. This amplified afragment fused via the complementary sequences in P8and P10, which was cloned into pCR2.1-TOPO (Invitro-gen). The orientation of the fragment was checked by PCRand the plasmid used to transform E. coli BL21(λDE3)(Novagen).Bacteria were grown at 30 °C on LB agar or in LB broth.
Ampicillin was used at 100 μg/mL, chloramphenicol at25 μg/mL, and IPTG at 50 μM.
In vitro binding assays
For binding assays in microtiter plate format, purifiedprotein was absorbed overnight at 4 °C to Maxisorp 96-well plates (Nunc) in 100-μL aliquots per well at aconcentration of 20 μg/mL in 50 mM Tris/HCl, pH8,and 100 mM NaCl. Blocking was done with 5% (w/v)skimmed milk for 1 h. Binding reactions were allowed tocontinue for 1 h, after which the wells were washed threetimes with 50 mM Tris/HCl, pH8, and 100 mM NaCl. Ifthe binding buffer differed from this in its composition, thefirst wash was carried out using the appropriate buffer.Alkaline phosphatase activity was detected by adding100 μL/well of chromogenic substrate solution (1 M Tris/HCl, pH8, 0.04% dinitrophenyl phosphate, 10 mMMgCl2,and 5 mM ZnCl2). The optical density at 420 nm wasmeasured after 15 min at room temperature.To assay binding tomembrane-inserted PulD,we isolated
the membrane fraction of cells expressing pulD as describedpreviously.15 Membranes were resuspended in the appro-priate buffer before the addition of the required Sac7dderivative. After incubation at room temperature for 1 hwith gentle agitation, membranes were pelleted by centri-fugation at 55,000g for 20 min and washed once. For thedetection of bound MalE-Sac7⁎40, the pellet was resus-pended in SDS-PAGE sample buffer and further processedby electrophoresis and immunoblotting. For the detection of

1066 Affitins as Probes for PulD Conformational Changes
Sac7⁎40-PhoA, the pellet was resuspended in 50 mM Tris/HCl, pH8, and aliquots were assayed for alkaline phospha-tase activity as described for the microtiter plate assay.To rule out any effects of denaturant or detergents on
the alkaline phosphatase activity used as a reporter, weincubated Sac7⁎40-PhoA with urea or denaturant for 1 h.After dilution, the alkaline phosphatase activity wasunaltered by this pretreatment.
SDS-PAGE and immunoblotting
Proteins were separated by SDS-PAGE in gels containing10% or 12% acrylamide using a Tris/glycine buffer system.The separated proteins were either stained with CoomassieBrilliant Blue or transferred onto nitrocellulose membranesby semidry blotting. Specific proteins were detected bychemiluminescence (Pierce ECL) using horseradish-perox-idase-coupled anti-rabbit immunoglobulinG as a secondaryantibody and specific primary antibodies [anti-PulD-N,12anti-MalE (New England Biolabs), and anti-Strep (GEHealthcare)]. Far Western blots using Sac7⁎40-PhoA werecarried out as previously described.15
Partial proteolysis of PulD-N2
Purified PulD-N2 (250 μg/mL) was incubated at 100 °Cfor 5 min in 50 mM Tris/HCl, pH8, 100 mM NaCl, and0.5% (w/v) SDS. After cooling in a 37 °C water bath,proteinase K was added to 100-μL aliquots to a finalconcentration between 0 and 33.33 μg/mL. After incuba-tion at 37 °C for 5 min, the reaction was stopped byheating at 100 °C for 10 min, addition of SDS-PAGEsample buffer, and heating to 100 °C for a further 5 min.Proteolytic fragments were then separated by SDS-PAGE.
Purification of tagged proteins
All tagged proteins were purified using commercialsystems according to the manufacturer's instructions.His6-tagged proteins (PulD-N1, PulD-N2, and His6-tagged Sac7d derivatives) were purified by IMAC onTALON resin (Clontech) and further polished by gelfiltration chromatography on a Sephacryl 200 column (GEHealthcare). MalE chimeras were purified in a single stepon amylose resin (New England Biolabs). Strep-taggedproteins were also purified in a single step on Strep-Tactincolumns (IBA).
Solubilization and purification of PulD
The membrane fraction of E. coli was prepared asdescribed before.12 Solubilization of PulD using ZW3–14and octyl-POE has also been described previously. The pHof preparations of membrane proteins extracted withoctyl-POE at pH10 was adjusted to pH8 by the addition of1 M Tris/HCl, pH8. For the extraction of PulD frombacterial membranes using Triton X-100, membranes weresuspended in extraction buffer (50 mM Tris/HCl, pH8, 2%Triton X-100, 250 mM KCl, 10 mM EDTA, and 0.1 mg/mLlysozyme) to a final concentration of 1 mg/mL proteinand gently agitated at room temperature for 3 h. Insolublematerial was sedimented by centrifugation at 150,000g for20 min at 18 °C. For purification using Sac7⁎40, thesupernatant of this centrifugation was added to a slurry ofStreptactin matrix preloaded with Sac7⁎40-Strep andincubated for 1 h with gentle agitation. The membraneswere pretreated with 10 mM DTT in 50 mM Tris/HCl,pH8, and 250 mM KCl for 30 min at room temperature
before membrane solubilization to reduce intramoleculardisulfide bonds in PulS to prevent copurification of thisprotein with PulD. The column was then washed with 5column volumes of wash buffer (50 mM Tris/HCl, pH8,1% Triton X-100, and 250mMKCl). Elution was done with2 column volumes of wash buffer containing 2.5 mMdesthiobiotin.In vitro synthesis of PulD in the presence of soybean
lecithin liposomes was carried out in 50-μL reactions aspreviously described.21 Liposomes containing PulD werepelleted by centrifugation at 55,000g for 10 min andwashed with 100 μL of 50 mM Tris/HCl, pH8, 250 mMNaCl, and 4 M urea before pelleting again to removeprotein peripherally associated with the liposomes. Thedenaturation of the PulD N domain in urea is reversible,and pretreatment of liposomes containing PulD did notaffect Sac7⁎40 binding if the denaturant was removed bywashing in urea-free buffer. To extract PulD dodecamersfrom the liposomes, we extracted the pellet with mildagitation for 2 h in 50 μL of 50 mM Tris/HCl, pH8,250 mM KCl, 4 M urea, and 2% Triton X-100. Urea wasremoved by dialysis against 50 mM Tris/HCl, pH8, and250 mM KCl using a 10,000-Da cutoff membrane to retainprotein and Triton X-100 micelles. Samples were centri-fuged again to sediment aggregated protein. Controlextractions were carried out in 50 μL of 50 mM Tris/HCl,pH8, 250 mM NaCl, and 1% ZW3–14; the dialysis stepwas omitted in this case. If required, detergent concentra-tions were lowered by dilution in the appropriate bufferwithout detergent.
Electron microscopy
For microscopy, 5 μL of purified PulD was adsorbedonto glow-discharged 200-mesh carbon-coated grid andstained with 2% (w/v) uranyl acetate. The micrographwas recorded at an accelerating voltage of 100 kV and amagnification of 52,000×, using a Philips CM100 electronmicroscope. All micrographs were recorded on Kodak SO-163 films.Reference-free alignment was performed on manually
selected particles from digitized electron micrographsusing the EMAN image processing package.26 After usinga reference-free alignment procedure, particle projectionswere classified by multivariant statistical analysis. Theclass averages with the best signal-to-noise ratio wereselected.
CD spectroscopy
CD was measured on a JASCO J-810 instrument (Jasco,Japan), using a quartz cell with a path length of 0.2 or0.02 cm (Hellma, Germany). The temperature of thesample was maintained at 20 °C with a programmablePeltier single cell holder. Spectra were recorded in stepmode (data pitch, 0.1 nm; bandwidth, 2 nm; responsetime, 0.125 s) and averaged over three scans. Proteins werediluted to a final concentration of 1 mg/mL (PulD-N1,PulD-N2) or 0.33 mg/mL (Sac7⁎40) in 20 mM phosphatebuffer, pH8.0. The influence of the detergent ZW3–14 wasdetermined by recording spectra after 2 h equilibration atroom temperature. A baseline correction was made withthe buffer.Urea denaturation transitions were assessed by mon-
itoring the CD signal at 222 nm after overnight incubationof samples at room temperature to reach equilibrium.These experiments were done with the proteins diluted toa final concentration of 0.1 mg/mL (PulD-N2) or 0.33 mg/mL (Sac7⁎40) in a 50-mM Tris buffer, pH8.0, containing

1067Affitins as Probes for PulD Conformational Changes
100 mM NaCl. CD signal was converted to mean residueellipticity.Secondary-structure predictions from CD spectra were
done using the Dichroweb software27,28 using the SP175reference data set29 and the CDSSTR analysis algorithm.30
Acknowledgements
We are grateful to our colleagues in the StructuralBiology and Molecular Genetics Units of theInstitut Pasteur for their help and interest and toPatrick England and Andreas Engel for discussionsconcerning conformational changes in PulD. Wethank Masayuki Takahashi and Fabrice Fleury(Université de Nantes, UMR6204) for their helpand for the use of the CD instrument. The workwas financed in part by a grant from the FrenchNational Research Agency (ANR-05-0307-01), bySwiss National Science Foundation grant 3100A-108299, and by the M.E. Müller Foundation ofSwitzerland.
References
1. Letellier, L., Howard, S. P. & Buckley, T. J. (1997).Studies on the energetics of proaerolysin secretionacross of the outer membrane of Aeromonas spp:evidence for requirement for both the protonmotiveforce and ATP. J. Biol. Chem. 272, 11109–11113.
2. Possot, O., Letellier, L. & Pugsley, A. P. (1997). Energyrequirement for pullulanase secretion by the mainterminal branch of the general secretory pathway.Mol.Microbiol. 24, 457–464.
3. Johnson, T. L., Abendroth, J., Hol, W. G. & Sandkvist,M. (2006). Type II secretion: from structure to function.FEMS Microbiol. Lett. 255, 175–186.
4. Sandkvist, M. (2001). Type II secretion and pathogen-esis. Infect. Immun. 69, 3523–3535.
5. Sandkvist, M. (2001). Biology of type II secretion. Mol.Microbiol. 40, 271–283.
6. Shevchik, V. E., Robert-Badouy, J. & Condemine, G.(1997). Specific interaction between OutD, an Erwiniachrysanthemi outer membrane protein of the generalsecretory pathway, and secreted proteins. EMBO J. 16,3007–3016.
7. Koster, M., Bitter, W., de Cock, H., Allaoui, A.,Cornelis, G. & Tommassen, J. (1997). The outermembrane component, YscC, of the Yop secretionmachinery of Yersinia enterocolitica forms a ring-shaped multimeric complex. Mol. Microbiol. 26,789–797.
8. Collins, R. F., Frye, S. A., Balasingha, S., Ford, R. C.,Tonjum, T. & Derrick, J. P. (2005). Interaction with typeIV pili induces structural changes in the bacterial outermembrane secretin PilQ. J. Biol. Chem. 280,18923–18930.
9. Wolfgang, M., van Putten, J. P. M., Hayes, S. F.,Dorward, D. & Koomey, M. (2000). Components anddynamics of fiber formation define a ubiquitousbiogenesis pathway for bacterial pili. EMBO J. 19,6408–6418.
10. Opalka, N., Beckmann, R., Boisset, N., Sion, M. N.,Russel, M. & Darst, S. (2003). Structure of the
filamentous phage pIV multimer by cryo-electronmicroscopy. J. Mol. Biol. 325, 461–470.
11. Chami, M., Guilvout, I., Gregorini, M., Rémigy, H.,Müller, S. A., Valerio, M. et al. (2005). Structuralinsights into the secretin PulD and its trypsin resistantcore. J. Biol. Chem. 280, 37732–37741.
12. Guilvout, I., Chami, M., Engel, A., Pugsley, A. P. &Bayan, N. (2006). Bacterial outer membrane secretinPulD assembles and inserts into the inner mem-brane in the absence of its pilotin. EMBO J. 25,5241–5249.
13. Bouley, J., Condemine, G. & Shevchik, V. E. (2001). ThePDZ domain of OutC and the N-terminal region ofOutD determine the secretion specificity of the type IIout pathway of Erwinia chrysanthemi. J. Mol. Biol. 27,205–219.
14. Peters, W. B., Edmondson, S. P. & Shriver, J. W. (2004).Thermodynamics of DNA binding and distortion bythe hyperthermophile chromatin protein Sac7d. J. Mol.Biol. 343, 339–360.
15. Mouratou, B., Schaeffer, F., Guilvout, I., Tello-Manigne,D., Pugsley, A. P., Alzari, P. M. & Pecorari, F. (2007).Remodeling a DNA binding protein as a specific invivo inhibitor of bacterial secretin PulD. Proc. NatlAcad. Sci. USA, 104, 17983–17988.
16. Arcus, V. (2002). OB-fold domains: a snapshot of theevolution of sequence, structure and function. Curr.Opin. Struct. Biol. 12, 794–801.
17. Gao, Y. G., Su, S. Y., Robinson, H., Padmanabhan, S.,Lim, L., McCrary, B. S. et al. (1998). The crystalstructure of the hyperthermophile chromosomalprotein Sso7d bound to DNA. Nat. Struct. Biol. 5,782–786.
18. Su, S., Gao, Y. G., Robinson, H., Liaw, Y. C.,Edmondson, S. P., Shriver, J. W. & Wang, A. H.(2000). Crystal structures of the chromosomal proteinsSso7d/Sac7d bound to DNA containing T–G mis-matched base-pairs. J. Mol. Biol. 303, 395–403.
19. McCrary, B. S., Edmondson, S. P. & Shriver, J. W.(1996). Hyperthermophile protein folding thermody-namics: differential scanning calorimetry and chemi-cal denaturation of Sac7d. J. Mol. Biol. 264, 784–805.
20. Pugsley, A. P., Bayan, N. & Sauvonnet, N. (2001).Disulphide bond formation in secreton componentPulK provides a possible explanation for the role ofDsbA in pullulanase secretion. J. Bacteriol. 183,1312–1319.
21. Guilvout, I., Chami, M., Berrier, C., Ghazi, A., Engel,A., Pugsley, A. P. & Bayan, N. (2008). Secretinmultimers insert into liposome membranes in vitro. J.Mol. Biol. 382, 13–23.
22. Collins, R. F., Frye, S. A., Kitmitto, A., Ford, R. C.,Tonjum, T. & Derrick, J. P. (2004). Structure of theNeisseria meningitidis outer membrane PilQ secretincomplex at 12 Å resolution. J. Biol. Chem. 279,39750–39756.
23. Hardie, K. R., Lory, S. & Pugsley, A. P. (1996). Insertionof an outer membrane protein in Escherichia colirequires a chaperone-like protein. EMBO J. 15,978–988.
24. Possot, O., Vignon, G., Bomchil, N., Ebel, F. & Pugsley,A. P. (2000). Multiple interactions between pull-ulanase secreton components involved in stabilizationand cytoplasmic membrane association of PulE. J.Bacteriol. 182, 2142–2152.
25. Kornacker, M. G. & Pugsley, A. P. (1989). Molecularcharacterization of pulA and its product, pullulanase,a secreted enzyme of Klebsiella pneumoniae UNF5023.Mol. Microbiol. 4, 73–85.

1068 Affitins as Probes for PulD Conformational Changes
26. Ludtke, S. J., Baldwin, P. R. & Chiu, W. (1999).EMAN: semiautomated software for high resolutionsingle-particle reconstructions. J. Struct. Biol. 128,82–97.
27. Whitmore, L. & Wallace, B. A. (2004). DICHROWEB,an online server for protein secondary structureanalyses from circular dichroism spectroscopic data.Nucleic Acids Res. 32, W668–W673.
28. Whitmore, L. & Wallace, B. A. (2008). Proteinsecondary structure analyses from circular dichroism
spectroscopy: methods and reference databases.Biopolymers, 89, 392–400.
29. Lees, J. G., Miles, A. J., Wien, F. &Wallace, B. A. (2006).A reference database for circular dichroism spectro-scopy covering fold and secondary structure space.Bioinformatics, 22, 1955–1962.
30. Compton, L. A. & Johnson, W. C., Jr (1986). Analysisof protein circular dichroism spectra for secondarystructure using a simple matrix multiplication. Anal.Biochem. 155, 155–167.




















