
Monoclonal antibodies discriminating netrin-G1 and netrin-G2 neuronal pathways

Arsenite Delays Progression through Each Cell Cycle Phaseand Induces Apoptosis following G2/M Arrest in U937 MyeloidLeukemia Cells
Geniece McCollum, Peter C. Keng, J. Christopher States, and Michael J. McCabe, Jr.Departments of Environmental Medicine (G.M., M.J.M.) and Radiation Oncology (P.C.K.), School of Medicine and Dentistry,University of Rochester, Rochester, New York; and Department of Pharmacology and Toxicology, University of Louisville,Louisville, Kentucky (J.C.S.)
Received November 12, 2004; accepted February 17, 2005
ABSTRACTArsenic is a well known toxicant and carcinogen that is alsoeffective as a chemotherapeutic in the treatment of acute pro-myelocytic leukemia. Although its effects on humans are welldocumented, arsenic’s mechanism of action is not well under-stood. Its ability to act as a carcinogen and as a chemothera-peutic seems paradoxical. However, cancer cell transformationand cancer cell destruction can both occur through perturba-tions of the cell cycle machinery, making cell cycle function alikely target of arsenic action. Arsenic has previously beenshown to inhibit cancer cell cycle progression, but the targetedcell cycle phase has been debated. This study was designed toidentify the cell cycle phase at which U937 cells are mostsensitive to arsenite-induced growth inhibition. Centrifugal elu-triation was used to divide asynchronous cell cultures into
specific cell cycle phase-enriched fractions. These fractionswere monitored for cell cycle phase progression in the pres-ence and absence of sodium arsenite. We found an overallreduction in cell cycle progression rather than induction ofarrest at one specific checkpoint. G2/M is the phase mostsensitive to arsenite-induced apoptosis. However, arsenite pro-foundly affects U937 cell growth by increasing the length oftime it takes cells to transit each phase of the cell cycle. Futurestudy of cell cycle inhibition by arsenic should consider that theeffect may not be mediated by the major cell cycle checkpoints.Arsenic’s ability to inhibit growth in any cell cycle phase mayincrease its value as a chemotherapeutic used together withother, more phase-selective agents, such as camptothecin.
Despite its well known toxicity and carcinogenicity, arsenicis approved by the Food and Drug Administration as a che-motherapeutic agent for the treatment of acute promyelo-cytic leukemia (APL) (Antman, 2001). Treatment with ar-senic trioxide, a source of trivalent, inorganic arsenic,otherwise known as arsenite, produces complete remission inmore than 50% of APL patients (Miller et al., 2002). Chemo-therapeutic blood plasma levels of arsenic trioxide corre-sponding to 1 to 4 �M arsenite lead to apoptosis of leukemiccells, whereas lower levels (0.2–1 �M arsenite) lead to differ-entiation (Chen et al., 2001). The large body of researchdirected at understanding the mechanism through which
arsenic acts to eliminate APL cells is summarized by Milleret al. (2002). Various mechanisms have been proposed, but nounified agreement has been reached. Some examples of pro-posed mechanisms include: degradation of the PML-RAR�fusion oncoprotein characteristic of APL cells, influence onsignaling pathways (such as activation of mitogen-activatedprotein kinase), and induction of oxidative stress (Huang etal., 1999; Jing et al., 1999; Cai et al., 2000; Zhu et al., 2001).Arsenite reacts with biomolecules through binding to vicinalthiol groups, resulting in disruption of protein activity (Reg-linski, 1998; Miller et al., 2002). It has been shown to inhibitenzymes such as succinic dehydrogenase and acetylcholineesterase (Reglinski, 1998; Chou et al., 2000). These targetsare affected by arsenite at very high concentrations and maybe more relevant to overt arsenite toxicity than to chemother-apeutic effects. The specific proteins targeted by more clini-cally relevant concentrations of arsenite are unknown.
To date, most study of the chemotherapeutic mechanism ofarsenite has concentrated on experiments with the NB4 cell
This work was supported by National Institutes of Health Grants P30ES01247 and R01 ES011314 (to J.C.S.). During the course of this study, G.M.was supported by National Institutes of Health Grant T32 ES07026 and by aPredoctoral Fellowship in Pharmacology/Toxicology from the PharmaceuticalResearch and Manufacturers of America Foundation.
Article, publication date, and citation information can be found athttp://jpet.aspetjournals.org.
doi:10.1124/jpet.104.080713.
ABBREVIATIONS: APL, acute promyelocytic leukemia; PML, promyelocytic leukemia protein; RAR�, retinoic acid receptor-�; BrdU, 5-bromo-2�-deoxyuridine; BSA, bovine serum albumin; PI, propidium iodide; Z-VAD-FMK, N-CBZ-Val-Ala-Asp(O-Me) fluoromethyl ketone; FITC, fluoresceinisothiocyanate; APC, allophycocyanin; PBS, phosphate-buffered saline; FBS, fetal bovine serum.
0022-3565/05/3132-877–887$20.00THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 313, No. 2Copyright © 2005 by The American Society for Pharmacology and Experimental Therapeutics 80713/1198082JPET 313:877–887, 2005 Printed in U.S.A.
877
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from

line (Wang et al., 1998; Jing et al., 1999; Cai et al., 2000).NB4 cells were derived from an APL patient, and they con-tain the PML-RAR� fusion oncoprotein (de The and Chelbi-Alix, 2001). The U937 cell line is another model system forstudying the effects of arsenite (McCabe et al., 2000). U937was derived from a non-Hodgkin’s lymphoma associated witha block in differentiation of monocyte progenitors (Sund-strom and Nilsson, 1976). U937 does not contain the PML-RAR� fusion protein, yet arsenite inhibits its growth andenhances its differentiation (McCabe et al., 2000). Therefore,U937 is a good model system for the study of arsenite’s effectson myeloid leukemia cells outside of its effects on PML-RAR�. Because U937 cells respond to arsenite similarly toAPL cells, arsenite’s mechanism of action in this context willshed light on its chemotherapeutic mechanism.
Sodium arsenite, another source of trivalent, inorganicarsenic, can cause a growth delay in U937 cells at concentra-tions below 10 �M (McCabe et al., 2000). However, the na-ture of this delay is unknown. Cell growth is dependent onthe efficient passage of cells through all phases of the cellcycle (Murray and Hunt, 1993). During times of stress, it iscommonly held that cells may pause in their cell cycle pro-gression at one of two major checkpoints. One checkpoint liesat the end of G1 as a cell prepares to enter S phase. The othercan be triggered during G2, before a cell commits to mitosis.However, cell cycle progression can be altered outside ofthese two best studied checkpoints. In response to DNA dam-age, not only do cells arrest in G2 phase, but S phase transitis also slowed to allow for DNA repair to occur in parallelwith its replication (Abraham, 2001). Agents that interferewith mitotic spindle function induce a metaphase arrest,preventing asymmetrical distribution of chromosomes (How-ell et al., 2000). Many proteins are involved in the regulationof each of these cell cycle transitions. To narrow the field ofpossible molecular targets of arsenite, it is desirable to definethe cell cycle phase during which cells are most sensitive.Here, we present evidence that supports previous findingsthat G2/M cells are most sensitive to arsenite-induced cellcycle arrest leading to apoptosis (Li and Broome, 1999; Parket al., 2001; Halicka et al., 2002; Ling et al., 2002; States etal., 2002; Cai et al., 2003). However, another dramatic effectof arsenite is an induction of nonphase-specific cell cycleprolongation (i.e., arsenite increases the length of time ittakes a cell to transit each phase of the cell cycle). Theprolongation of cell cycle transit time has the potential tocontribute greatly to growth suppression by arsenite, be-cause at any given time, the percentage of cells susceptible toG2/M arrest and apoptosis is small compared with the per-centage of cells susceptible to G1 or S phase delay.
Materials and MethodsReagents. Sodium m-arsenite (i.e., NaAsO2) was obtained from
Sigma-Aldrich (St. Louis, MO). Fresh stock solutions of sodium ar-senite (2 mM in Hanks’ balanced salt solution) were prepared beforeevery experiment and filter sterilized using a 0.2-�m syringe filter.The following reagents were also purchased from Sigma-Aldrich:5-bromo-2�-deoxyuridine (BrdU), pepsin, bovine serum albumin(BSA), Tween 20, propidium iodide (PI), RNase A, N-CBZ-Val-Ala-Asp(O-Me) fluoromethyl ketone (Z-VAD-FMK), and camptothecin.Concentrated HCl was purchased from Fisher Scientific Co. (Pitts-burgh, PA). Purified mouse �-bromodeoxyuridine, fluorescein iso-thiocyanate (FITC)-conjugated rat anti-mouse IgG1 specific monoclo-
nal antibody, 1� cell lysis buffer, and Ac-DEVD-7-amino-4-methylcoumarin substrate were purchased from BD BiosciencesPharMingen (San Diego, CA). Allophycocyanin (APC)-conjugatedgoat anti-mouse IgM antibody was purchased from Caltag Labora-tories (Burlingame, CA). The TG-3 antibody (in hybridoma culturesupernatant) was a generous gift from Dr. Peter Davies (AlbertEinstein College of Medicine, New York, NY). HEPES solution (1 M)and 1� PBS were purchased from Invitrogen (Carlsbad, CA). Glyc-erol and dithiothreitol were purchased from J. T. Baker (Phillips-burg, NJ).
Cell Culture. U937 cells were obtained from the American TypeCulture Collection (Manassas, VA) and maintained in RPMI 1640supplemented with 5% fetal bovine serum (FBS), 1% L-glutamine,and 0.1% gentamicin. RPMI 1640, FBS, glutamine, and gentamicinwere obtained from Invitrogen. Cells were maintained in logarithmicgrowth at a density between 0.1 and 1 � 106 cells/ml at 37°C in ahumidified atmosphere consisting of 5% CO2. For most experiments,the seeding cell density was 2.0 � 105 cells/ml.
PI Exclusion Assay. U937 cells (2.7 � 106 cells/10 ml) werecultured for up to 72 h with or without arsenite. Cells were washedtwice with 1� PBS, then 1 � 106 cells in 1 ml of PBS were stainedwith 1.67 �g of propidium iodide. PI fluorescence was determined byflow cytometry on a FACScalibur (BD Biosciences, San Jose, CA).Cells with intact membranes excluded PI and were counted as via-ble. The percentage of PI-excluding cells in each sample was normal-ized to the percentage of such cells in the control sample. A minimumof 10,000 cells/sample were analyzed. Data were collected and ana-lyzed using CellQuest software.
Flow Cytometric Analysis of Cell Cycle Progression. U937cells were enriched for cells in various cell cycle phases via centri-fugal elutriation as described by Donaldson et al. (1997). Cells (2.0 �106 cells/10 ml) were exposed to 0 to 5 �M arsenite and/or 0 to 50 �MZ-VAD-FMK for 0 to 24 h and pulse-labeled by incubation with 10�M BrdU for 15 min at 37°C in a humidified atmosphere with 5%CO2. After being washed with 10 ml of 1� PBS, the cells were fixedwith 70% ethanol overnight at 4°C. Fixed cells were rinsed with PBSand incubated with 0.2 mg/ml pepsin/2 N HCl/1� PBS for 20 min at37°C. Following two washes with 1� PBS/0.5% BSA/0.5% Tween 20,cells were treated with 1� PBS/2% FBS for 20 min at room temper-ature. Cells were incubated with 0.5 �g of purified mouse �-bromode-oxyuridine and 1 �l of TG-3 hybridoma culture supernatant/10 �l inthe dark overnight at 4°C and washed in 1� PBS/0.5% BSA/0.5%Tween 20. Cells were incubated with 0.5 �g of FITC-conjugated ratanti-mouse IgG1 monoclonal antibody and 0.1 �g of APC-conjugatedgoat anti-mouse IgM antibody/10 �l in the dark for 2 h at 4°C andwashed in 1� PBS/0.5% BSA/0.5% Tween 20. They were then incu-bated in 500 �l of PBS containing 10 �g/ml PI in the presence ofRNase A (100 U/ml final) for at least 30 min at room temperature. PIfluorescence (i.e., DNA content), FITC fluorescence (BrdU incorpo-ration), and APC fluorescence (TG-3 labeling) were determined byflow cytometry on a FACScalibur (BD Biosciences). A minimum of20,000 cells/sample were analyzed. Data were collected and analyzedusing CellQuest software.
Flow Cytometric Analysis of Cell Cycle Kinetics. U937 cells(2.0 � 106 cells/10 ml) were pulse labeled by incubation with 10 �MBrdU for 15 min at 37°C in a humidified atmosphere with 5% CO2.They were then washed once with 1� PBS and cultured for 4 h withor without arsenite. After being washed with 10 ml of PBS, the cellswere fixed with 70% ethanol overnight at 4°C. Fixed cells werelabeled with anti-BrdU and TG-3 antibody, stained with PI, andanalyzed as described above.
Caspase Activity Assay. Elutriated U937 cells were exposed to0 to 5 �M arsenite and/or 0 to 50 �M Z-VAD-FMK for 8 or 12 h. Cellswere harvested and washed in ice-cold PBS. Cell pellets were snap-frozen in dry ice bath to preserve protein integrity and stored at�80°C. Pellets were lysed with 1� cell lysis buffer (10 mM Tris-HCl/10 mM NaH2PO4/NaHPO4, pH 7.5/130 mM NaCl/1% TritonX-100/10 mM sodium pyrophosphate). Cell lysate was added to 1�
878 McCollum et al.
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from
HEPES buffer (20 mM HEPES, pH 7.5, 10% glycerol, and 2 mMdithiothreitol) containing Ac-DEVD-7-amino-4-methylcoumarin sub-strate. The fluorescence emission yielded by 7-amino-4-methylcou-marin release was monitored every 45 s for 1 h at a temperature of37°C in a SPECTRAmax GEMINI XS Dual-Scanning MicroplateSpectrofluorometer from Molecular Devices (Sunnyvale, CA). Theprotein concentration of each lysate was determined by bicinchoninicacid protein assay using a kit from Pierce Chemical (Rockford, IL).Caspase activity was expressed in terms of arbitrary fluorescenceunits released per mg protein per second.
ResultsArsenite (5 �M) Inhibits U937 Cell Growth without
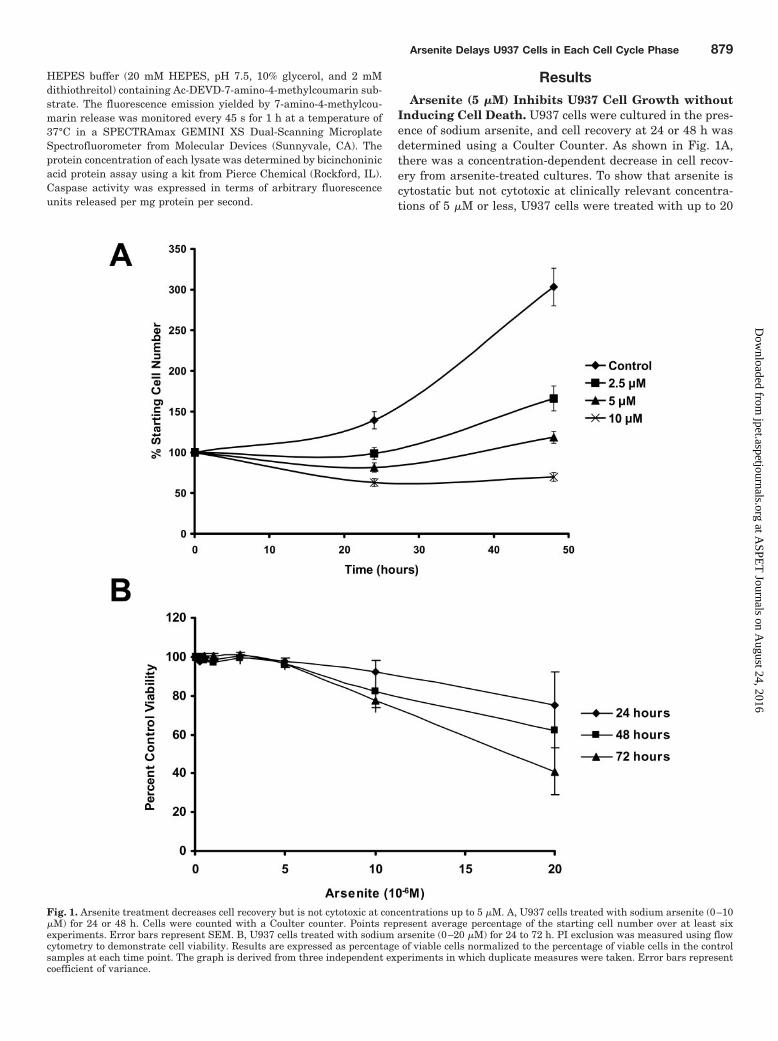
Inducing Cell Death. U937 cells were cultured in the pres-ence of sodium arsenite, and cell recovery at 24 or 48 h wasdetermined using a Coulter Counter. As shown in Fig. 1A,there was a concentration-dependent decrease in cell recov-ery from arsenite-treated cultures. To show that arsenite iscytostatic but not cytotoxic at clinically relevant concentra-tions of 5 �M or less, U937 cells were treated with up to 20
Fig. 1. Arsenite treatment decreases cell recovery but is not cytotoxic at concentrations up to 5 �M. A, U937 cells treated with sodium arsenite (0–10�M) for 24 or 48 h. Cells were counted with a Coulter counter. Points represent average percentage of the starting cell number over at least sixexperiments. Error bars represent SEM. B, U937 cells treated with sodium arsenite (0–20 �M) for 24 to 72 h. PI exclusion was measured using flowcytometry to demonstrate cell viability. Results are expressed as percentage of viable cells normalized to the percentage of viable cells in the controlsamples at each time point. The graph is derived from three independent experiments in which duplicate measures were taken. Error bars representcoefficient of variance.
Arsenite Delays U937 Cells in Each Cell Cycle Phase 879
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from
�M arsenite for 24 to 72 h, and cell viability based on PIexclusion was assessed using flow cytometry (Fig. 1B). Theability of the PI exclusion assay to detect cell death is dem-onstrated by the loss of viability seen with exposure tohigher, cytotoxic concentrations of arsenite (10–20 �M).However, the majority of U937 cells are viable with arsenitetreatments up to 5 �M for 72 h. Therefore, lower concentra-tions of arsenite (�5 �M) inhibit cell growth but do notimmediately induce cell death. Arsenic trioxide (As2O3),rather than sodium arsenite (NaAsO2), is the chemical formof the drug approved by the Food and Drug Administrationfor the treatment of APL. Both are sources of trivalent inor-ganic arsenic, differing mainly in their stoichiometry. Theexperiments shown in Fig. 1 were repeated using 0.5 to 5 �Marsenic trioxide to establish that the effect is similar to thatseen with sodium arsenite. The results were virtually iden-tical to those shown in Fig. 1 except that arsenic trioxideinhibits growth and reduces viability at lower concentrations(data not shown), a difference predicted by the fact thatarsenic trioxide contains more arsenic per mole than doessodium arsenite. We chose to use sodium arsenite in theremaining experiments because it is far more soluble in aque-ous solution than is arsenic trioxide, which must be solubi-lized in 1 M sodium hydroxide prior to use.
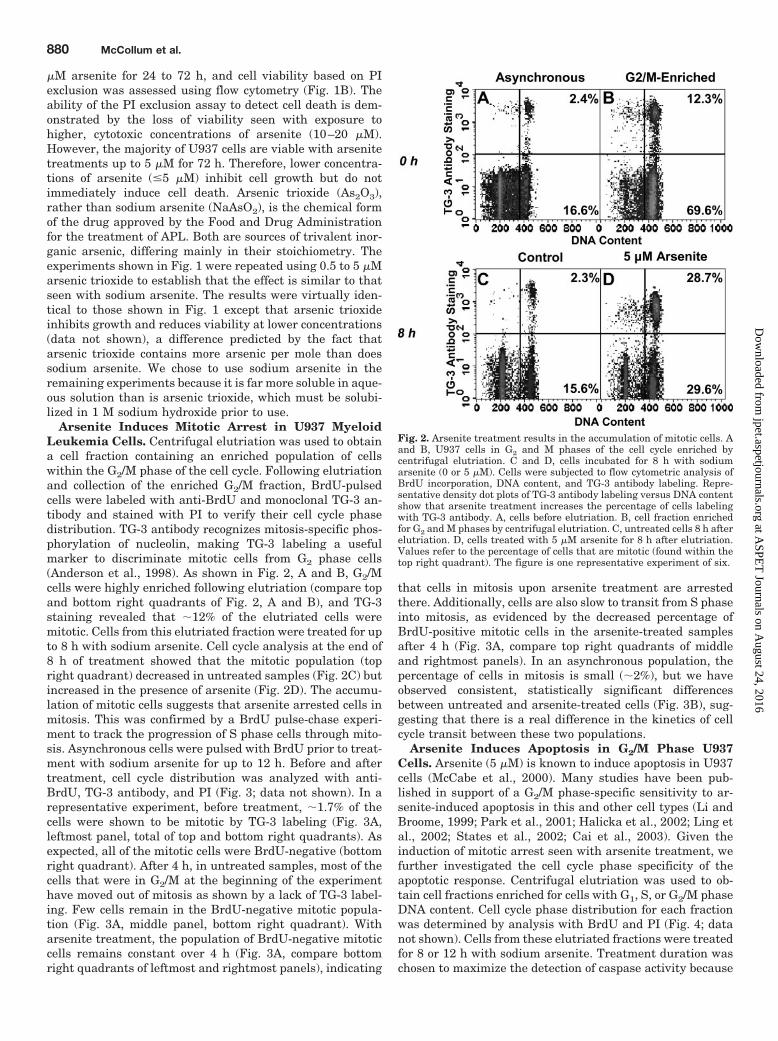
Arsenite Induces Mitotic Arrest in U937 MyeloidLeukemia Cells. Centrifugal elutriation was used to obtaina cell fraction containing an enriched population of cellswithin the G2/M phase of the cell cycle. Following elutriationand collection of the enriched G2/M fraction, BrdU-pulsedcells were labeled with anti-BrdU and monoclonal TG-3 an-tibody and stained with PI to verify their cell cycle phasedistribution. TG-3 antibody recognizes mitosis-specific phos-phorylation of nucleolin, making TG-3 labeling a usefulmarker to discriminate mitotic cells from G2 phase cells(Anderson et al., 1998). As shown in Fig. 2, A and B, G2/Mcells were highly enriched following elutriation (compare topand bottom right quadrants of Fig. 2, A and B), and TG-3staining revealed that �12% of the elutriated cells weremitotic. Cells from this elutriated fraction were treated for upto 8 h with sodium arsenite. Cell cycle analysis at the end of8 h of treatment showed that the mitotic population (topright quadrant) decreased in untreated samples (Fig. 2C) butincreased in the presence of arsenite (Fig. 2D). The accumu-lation of mitotic cells suggests that arsenite arrested cells inmitosis. This was confirmed by a BrdU pulse-chase experi-ment to track the progression of S phase cells through mito-sis. Asynchronous cells were pulsed with BrdU prior to treat-ment with sodium arsenite for up to 12 h. Before and aftertreatment, cell cycle distribution was analyzed with anti-BrdU, TG-3 antibody, and PI (Fig. 3; data not shown). In arepresentative experiment, before treatment, �1.7% of thecells were shown to be mitotic by TG-3 labeling (Fig. 3A,leftmost panel, total of top and bottom right quadrants). Asexpected, all of the mitotic cells were BrdU-negative (bottomright quadrant). After 4 h, in untreated samples, most of thecells that were in G2/M at the beginning of the experimenthave moved out of mitosis as shown by a lack of TG-3 label-ing. Few cells remain in the BrdU-negative mitotic popula-tion (Fig. 3A, middle panel, bottom right quadrant). Witharsenite treatment, the population of BrdU-negative mitoticcells remains constant over 4 h (Fig. 3A, compare bottomright quadrants of leftmost and rightmost panels), indicating
that cells in mitosis upon arsenite treatment are arrestedthere. Additionally, cells are also slow to transit from S phaseinto mitosis, as evidenced by the decreased percentage ofBrdU-positive mitotic cells in the arsenite-treated samplesafter 4 h (Fig. 3A, compare top right quadrants of middleand rightmost panels). In an asynchronous population, thepercentage of cells in mitosis is small (�2%), but we haveobserved consistent, statistically significant differencesbetween untreated and arsenite-treated cells (Fig. 3B), sug-gesting that there is a real difference in the kinetics of cellcycle transit between these two populations.
Arsenite Induces Apoptosis in G2/M Phase U937Cells. Arsenite (5 �M) is known to induce apoptosis in U937cells (McCabe et al., 2000). Many studies have been pub-lished in support of a G2/M phase-specific sensitivity to ar-senite-induced apoptosis in this and other cell types (Li andBroome, 1999; Park et al., 2001; Halicka et al., 2002; Ling etal., 2002; States et al., 2002; Cai et al., 2003). Given theinduction of mitotic arrest seen with arsenite treatment, wefurther investigated the cell cycle phase specificity of theapoptotic response. Centrifugal elutriation was used to ob-tain cell fractions enriched for cells with G1, S, or G2/M phaseDNA content. Cell cycle phase distribution for each fractionwas determined by analysis with BrdU and PI (Fig. 4; datanot shown). Cells from these elutriated fractions were treatedfor 8 or 12 h with sodium arsenite. Treatment duration waschosen to maximize the detection of caspase activity because
Fig. 2. Arsenite treatment results in the accumulation of mitotic cells. Aand B, U937 cells in G2 and M phases of the cell cycle enriched bycentrifugal elutriation. C and D, cells incubated for 8 h with sodiumarsenite (0 or 5 �M). Cells were subjected to flow cytometric analysis ofBrdU incorporation, DNA content, and TG-3 antibody labeling. Repre-sentative density dot plots of TG-3 antibody labeling versus DNA contentshow that arsenite treatment increases the percentage of cells labelingwith TG-3 antibody. A, cells before elutriation. B, cell fraction enrichedfor G2 and M phases by centrifugal elutriation. C, untreated cells 8 h afterelutriation. D, cells treated with 5 �M arsenite for 8 h after elutriation.Values refer to the percentage of cells that are mitotic (found within thetop right quadrant). The figure is one representative experiment of six.
880 McCollum et al.
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from

the relatively small number of cells obtained in each fractionof a centrifugal elutriation limits the number of samples thatcan be generated. Caspase activity was detected only in ar-senite-treated samples with G2/M phase populations largerthan those in the corresponding untreated samples (Fig. 4).An untreated population enriched for G2/M phase cellslargely progressed into G1 phase over 8 h. When treated with5 �M arsenite, the same population was delayed in G2/Mphase and contained activated caspases (Fig. 4B). Caspaseactivation was also detected in arsenite-treated S phase-enriched cells that were delayed in G2/M following incubationfor 12 h (Fig. 4C). On the other hand, arsenite treatmentdelays G1 phase-enriched cells in G1 phase, but caspase ac-tivation is largely absent (Fig. 4A). This suggests, in agree-ment with previous studies, that arsenite induces apoptosisspecifically in delayed G2/M phase cell populations (Li andBroome, 1999; Park et al., 2001; Halicka et al., 2002; Ling etal., 2002; States et al., 2002; Cai et al., 2003). Experimentsusing the general caspase inhibitor, Z-VAD-FMK, furthersupported that G2/M phase is the specific cell cycle phase atwhich cells are vulnerable to apoptosis induction by arsenite.
A cell fraction enriched for cells with S phase DNA contentwas obtained by centrifugal elutriation, and its cell cycledistribution was confirmed by analysis with BrdU and PI(Fig. 5A). Cells from this S phase-enriched fraction weretreated for 16 h with sodium arsenite, with Z-VAD-FMK, orwith both arsenite and the inhibitor. Once again, the treat-ment time was chosen to maximize the effect to most effi-ciently use the elutriated cells. Z-VAD-FMK treatment,alone, had no effect on cell cycle distribution. Over the courseof 16 h, most of the cells in the control sample move from Sphase to G1 or S phase of the next cell cycle (Fig. 5B). Theseresults are not unexpected because cell cycle kinetics analy-ses have shown that untreated U937 cells complete S phasein approximately 10 to 12 h and G2/M phase in approxi-mately 4 to 8 h (data not shown). In the experiment depictedin Fig. 5, many of the cells are already near the end of Sphase at elutriation, allowing plenty of time for them to entera subsequent S phase by 16 h. With arsenite treatment,however, 16.9% of cells are arrested in mitosis, and a popu-lation of BrdU-negative cells with S phase DNA contentappears (Fig. 5, A and C). The addition of Z-VAD-FMK to the
Fig. 3. Arsenite treatment results in mitotic arrest. An asynchronous population of U937 cells was labeled with BrdU for 15 min. Cells were thenincubated for 4 h with 0 or 5 �M sodium arsenite. After treatment, they were subjected to flow cytometric analysis of BrdU incorporation and TG-3antibody labeling. A, representative contour plots of BrdU incorporation versus TG-3 antibody labeling. Values refer to the percentage of cells in thetop or bottom right quadrants. The bottom right quadrant contains BrdU-negative mitotic cells, and the top right quadrant contains cells that havemoved from S phase into mitosis during the 4-h incubation. The figure is one representative experiment of three. B, compilation of the data from thethree experiments represented in A. Results are presented in a bar graph as average cell percentage (white, control; black, arsenite-treated). Errorbars � S.D. �, P � 0.008; ��, P � 0.0002, arsenite-treated compared with control according to Student’s paired t test with a two-tailed distribution.
Arsenite Delays U937 Cells in Each Cell Cycle Phase 881
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from

culture results in the complete inhibition of arsenite-inducedcaspase activity (Fig. 4). Also, more cells are arrested inmitosis in response to arsenite (24.5%), and fewer cells ap-pear in the BrdU-negative population with S phase DNAcontent (Fig. 5, A and D). Because a portion of this populationdepends on caspase activity, it is likely that this portionrepresents G2/M phase cells in which caspase-dependentDNA degradation is occurring. It is appropriate to say thatthis population has sub-G2 DNA content because one wouldsay that cells undergoing apoptosis out of G1 phase developsub-G1 DNA content (Nicoletti et al., 1991).
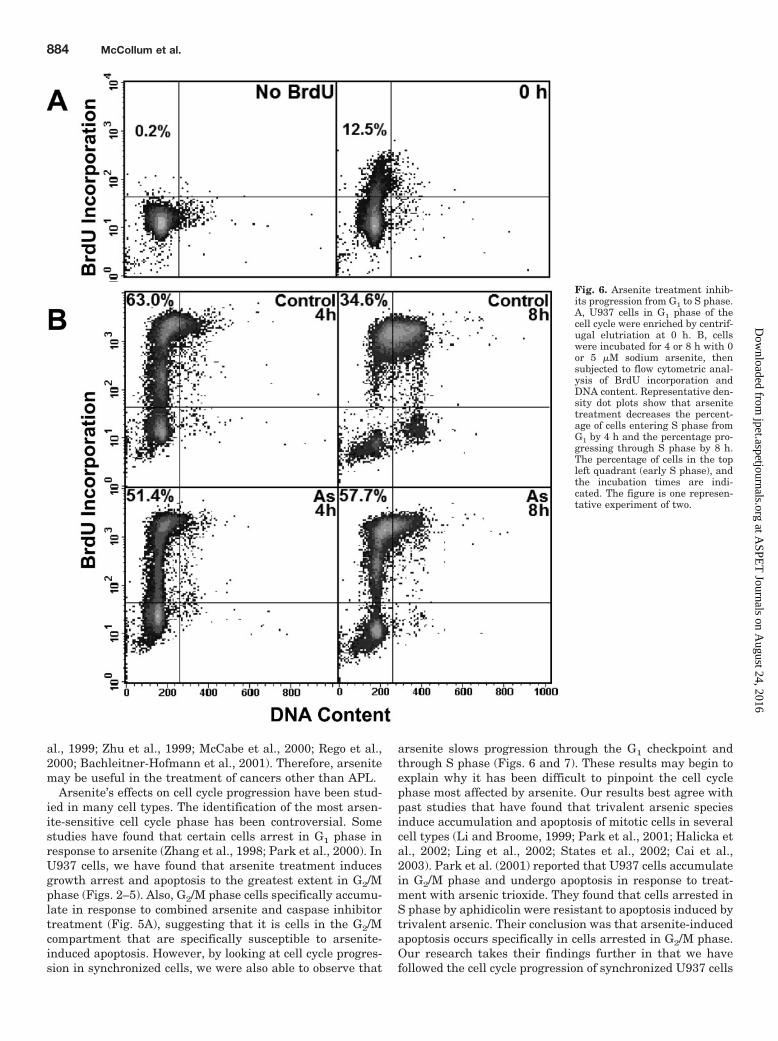
Arsenite Inhibits Cell Cycle Progression through AllPhases of the Cell Cycle. It has been established that ar-senite induces mitotic arrest and apoptosis of mitotic cells (Liand Broome, 1999; Ling et al., 2002). However, it was importantto examine the effect of arsenite on other phases of the cell cyclebecause �98% of an asynchronous population is made up ofnonmitotic cells, making it likely that effects on these cells havean important impact on overall growth inhibition. We havelooked at progression of arsenite-treated cells from each cellcycle stage to the next and have found that arsenite slows cellgrowth in every phase of the cycle. Cells elutriated in G1 phaseenter S phase more slowly in the presence of arsenite (Fig. 6B,compare left panels). Once in early S phase, arsenite-treatedcells are slow to progress to late S phase (Fig. 6B, compare rightpanels). Analysis of U937 cell cycle kinetics using the methoddescribed by Begg et al. (1985) reveals that the DNA synthesis
time in asynchronous populations of untreated cells is between10 and 12 h (data not shown). When cells are treated with 5 �Msodium arsenite, the DNA synthesis time increases to 16 h,indicating that transit through S phase is affected. We notedearlier that cell movement from S phase to mitosis is slowed inthe presence of arsenite (Fig. 3, right panel, compare top rightquadrants). We wanted to determine whether it was the S to G2
phase or the G2 to M phase transition that was more sensitiveto arsenite. We used centrifugal elutriation to collect fractions ofcells excluding those in G1 phase to ensure that the cell popu-lations under study contained only actively cycling cells. Thesefractions were allowed to grow in the presence or absence ofarsenite for 8 h. An 8-h treatment allowed enough time for mostcells in the untreated samples to progress out of S phase but notenough time for mitotic cells to enter a subsequent cell cycle.After treatment, the samples were analyzed by BrdU, TG-3,and PI staining. The results clearly show that cell passage fromany cell cycle phase to the next is inhibited by arsenite (Fig. 7).At elutriation, �49% of cells are in late S phase (Fig. 7A). Incontrol samples after 8 h, �44% of cells have moved on from Sphase to later phases of the cell cycle (Fig. 7A, compare leftand top right panels). In arsenite-treated samples, however,only 38% are able to exit S phase during the 8 h treatment (Fig.7A, compare left and bottom right panels). Similar cell cyclephase transition inhibition is seen for movement from G2 to Mphase and, as established earlier, for movement out of mitosis(Fig. 7B).
Fig. 4. Arsenite induces caspase activation in cycling cells. U937 cells in individual phases of the cell cycle (A, G1 phase-enriched cells; B, G2/Mphase-enriched cells; C, S phase-enriched cells) were enriched by centrifugal elutriation. Cells were incubated for 8 or 12 h with 0 or 5 �M sodiumarsenite and/or 0 or 50 �M Z-VAD-FMK. Cell lysates were subjected to caspase activity and protein concentration analyses. Results are presented inbar graphs as average fluorescence units released per second by each sample normalized to protein concentration (white, control; black, arsenite-treated). Up to five independent experiments in which triplicate measures were taken were performed. Error bars � S.D. Cell cycle phase distributionsdetermined by flow cytometric analysis of BrdU incorporation and DNA content are shown as pie charts.
882 McCollum et al.
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from

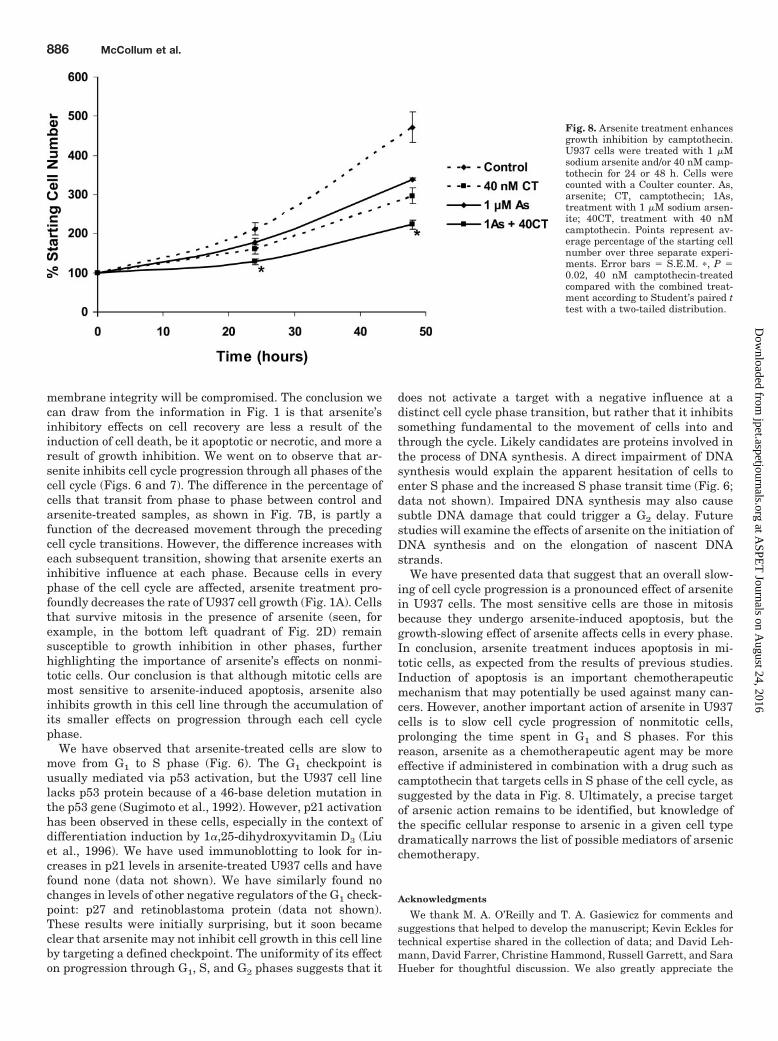
Since an important action of arsenite in U937 cells seems tobe slowing of S phase progression, we thought that perhapsarsenite as a chemotherapeutic agent may be more effective ifadministered in combination with a drug that targets S phasecells. Such a drug is the topoisomerase inhibitor camptothecin.U937 cells were cultured in the presence of 40 nM camptothecintogether with 1 �M sodium arsenite, and cell recovery at 24 or48 h was determined using a Coulter counter. As shown in Fig.8, the decrease in cell recovery from camptothecin-treated cul-tures was enhanced by the addition of only 1 �M sodium arsen-ite. It is possible that both arsenite and camptothecin may beeffective chemotherapeutic agents at lower concentrations ifused in combination.
DiscussionArsenite has potential as an effective chemotherapeutic
agent in the treatment of a variety of cancers (Wang et al.,1998; Lu et al., 1999; Rousselot et al., 1999; Seol et al., 1999;
Zhang et al., 1999; Zhu et al., 1999; Rego et al., 2000; Ant-man, 2001; Bachleitner-Hofmann et al., 2001; Chen et al.,2001; de The and Chelbi-Alix, 2001; Wang, 2001; Miller et al.,2002). Arsenite’s chemotherapeutic mechanism is not wellunderstood. Arsenic trioxide is Food and Drug Administra-tion-approved for the treatment of APL, which is most oftencharacterized by the presence of the oncogenic fusion proteinPML-RAR� within the leukemic cells (Chen et al., 2001; deThe and Chelbi-Alix, 2001; Soignet, 2001). PML-RAR� exertsa double dominant-negative effect on the function of bothPML and RAR� proteins, blocking both apoptosis and differ-entiation. It is thought that arsenite exerts its apoptotic anddifferentiative effects on these cells by inducing PML-RAR�
degradation (Zhu et al., 2001). However, degradation of theoncoprotein is not the sole mechanism of arsenite actionbecause arsenite does induce apoptosis and differentiation incell lines that lack the fusion protein (Wang et al., 1998; Luet al., 1999; Rousselot et al., 1999; Seol et al., 1999; Zhang et
Fig. 5. Arsenite treatment results in apoptosis of mitotic cells. U937 cells in S phase of the cell cycle were enriched by centrifugal elutriation (noteS phase percentage at elutriation in A). Cells were cultured in the presence or absence of 5 �M sodium arsenite and 50 �M Z-VAD-FMK for 16 h, atwhich time they were harvested for cell cycle analysis. A, representative cell cycle distributions, determined by flow cytometric analysis of BrdUincorporation and DNA content, are shown for each treatment group. Mitotic cell percentage was determined from TG-3 labeling. Arsenite-treated cellpopulations total to less than 100% because of the presence of BrdU-negative cells with S phase DNA content that fall within the gates drawn in Cand D. B, gates used to determine the percentage of cells in G1, S, and G2/M phase are indicated on a representative contour plot of BrdU incorporationversus DNA content for the untreated control sample 16 h postelutriation. C and D, representative contour plots of BrdU incorporation versus DNAcontent for samples treated with 5 �M arsenite (C) or with 5 �M arsenite and 50 �M Z-VAD-FMK (D). Values refer to the percentage of cells withinthe gates, which contain BrdU-negative cells with sub-G2 (or S phase) DNA content. The figure is one representative experiment of three.
Arsenite Delays U937 Cells in Each Cell Cycle Phase 883
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from
al., 1999; Zhu et al., 1999; McCabe et al., 2000; Rego et al.,2000; Bachleitner-Hofmann et al., 2001). Therefore, arsenitemay be useful in the treatment of cancers other than APL.
Arsenite’s effects on cell cycle progression have been stud-ied in many cell types. The identification of the most arsen-ite-sensitive cell cycle phase has been controversial. Somestudies have found that certain cells arrest in G1 phase inresponse to arsenite (Zhang et al., 1998; Park et al., 2000). InU937 cells, we have found that arsenite treatment inducesgrowth arrest and apoptosis to the greatest extent in G2/Mphase (Figs. 2–5). Also, G2/M phase cells specifically accumu-late in response to combined arsenite and caspase inhibitortreatment (Fig. 5A), suggesting that it is cells in the G2/Mcompartment that are specifically susceptible to arsenite-induced apoptosis. However, by looking at cell cycle progres-sion in synchronized cells, we were also able to observe that
arsenite slows progression through the G1 checkpoint andthrough S phase (Figs. 6 and 7). These results may begin toexplain why it has been difficult to pinpoint the cell cyclephase most affected by arsenite. Our results best agree withpast studies that have found that trivalent arsenic speciesinduce accumulation and apoptosis of mitotic cells in severalcell types (Li and Broome, 1999; Park et al., 2001; Halicka etal., 2002; Ling et al., 2002; States et al., 2002; Cai et al.,2003). Park et al. (2001) reported that U937 cells accumulatein G2/M phase and undergo apoptosis in response to treat-ment with arsenic trioxide. They found that cells arrested inS phase by aphidicolin were resistant to apoptosis induced bytrivalent arsenic. Their conclusion was that arsenite-inducedapoptosis occurs specifically in cells arrested in G2/M phase.Our research takes their findings further in that we havefollowed the cell cycle progression of synchronized U937 cells
Fig. 6. Arsenite treatment inhib-its progression from G1 to S phase.A, U937 cells in G1 phase of thecell cycle were enriched by centrif-ugal elutriation at 0 h. B, cellswere incubated for 4 or 8 h with 0or 5 �M sodium arsenite, thensubjected to flow cytometric anal-ysis of BrdU incorporation andDNA content. Representative den-sity dot plots show that arsenitetreatment decreases the percent-age of cells entering S phase fromG1 by 4 h and the percentage pro-gressing through S phase by 8 h.The percentage of cells in the topleft quadrant (early S phase), andthe incubation times are indi-cated. The figure is one represen-tative experiment of two.
884 McCollum et al.
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from

in the absence of chemical manipulation, allowing us to elim-inate any unexpected effects that may result from treatingcells with two different cell cycle inhibitory agents simulta-neously (i.e., trivalent arsenic and aphidicolin). Also, we wereable to separate effects on G2 phase cells from those onmitotic cells by use of the TG-3 antibody. As a result, wefound that sodium arsenite induces arrest followed by apo-ptosis in both of these distinct cell cycle compartments. Linget al. (2002) used a panel of various cancer cell lines andfound that all of them underwent mitotic arrest in responseto arsenite. Along with mitotic arrest, the cells also displayedcharacteristics of apoptosis [activation of caspases, poly-(ADP-ribose) polymerase cleavage] that were specific to mi-totic cells. Similarly, we found that arsenite-induced caspaseactivation occurs primarily in cells outside of G1 phase (Fig.4) and that caspase inhibition exacerbates arsenite-inducedG2/M phase arrest (Fig. 5). We used a terminal deoxynucle-
otidyltransferase dUTP nick end labeling assay to detectcaspase-induced DNA strand breaks in elutriated cell popu-lations and found that with 8 h of arsenite treatment, suchbreaks could only be detected in cells with S or G2/M phaseDNA content (data not shown). It is important to understandthe mechanism by which arsenite induces arrest and apopto-sis in M phase because this may be an important way inwhich arsenic can target cancer cells.
In the U937 cell model, however, it seems that the effectsof arsenite on mitotic cells, although severe, may be out-weighed by its ability to stall cell cycle progression in otherphases of the cell cycle. For instance, it is interesting that noappreciable cell death is observed even at 72 h of arsenitetreatment at a concentration known to induce apoptosis inthe U937 cell line (Fig. 1B). Although apoptotic cells willmaintain membrane integrity longer than necrotic cells, un-der cell culture conditions, at late stages, even apoptotic cell
Fig. 7. Arsenite treatment decreases transit through S, G2, and M phases. A, actively cycling U937 cells enriched by centrifugal elutriation. Cells wereincubated for 8 h with 0 or 5 �M sodium arsenite and then subjected to flow cytometric analysis of BrdU incorporation, DNA content, and TG-3antibody labeling. Representative contour plots of BrdU incorporation versus DNA content are shown. Values for M phase are taken from analysis ofTG-3 antibody labeling. B, cell cycle progression determined from the values in A depicted as a flow chart for both control and arsenite-treated cells.The top arrows contain values representing the percentage of cells moving between the indicated cell cycle phases in control samples over 8 h. Thebottom arrows contain the same types of values for arsenite-treated cells. The values between the top and bottom arrows represent the differencebetween control and arsenite-treated samples. The figure is one representative experiment of four.
Arsenite Delays U937 Cells in Each Cell Cycle Phase 885
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from
membrane integrity will be compromised. The conclusion wecan draw from the information in Fig. 1 is that arsenite’sinhibitory effects on cell recovery are less a result of theinduction of cell death, be it apoptotic or necrotic, and more aresult of growth inhibition. We went on to observe that ar-senite inhibits cell cycle progression through all phases of thecell cycle (Figs. 6 and 7). The difference in the percentage ofcells that transit from phase to phase between control andarsenite-treated samples, as shown in Fig. 7B, is partly afunction of the decreased movement through the precedingcell cycle transitions. However, the difference increases witheach subsequent transition, showing that arsenite exerts aninhibitive influence at each phase. Because cells in everyphase of the cell cycle are affected, arsenite treatment pro-foundly decreases the rate of U937 cell growth (Fig. 1A). Cellsthat survive mitosis in the presence of arsenite (seen, forexample, in the bottom left quadrant of Fig. 2D) remainsusceptible to growth inhibition in other phases, furtherhighlighting the importance of arsenite’s effects on nonmi-totic cells. Our conclusion is that although mitotic cells aremost sensitive to arsenite-induced apoptosis, arsenite alsoinhibits growth in this cell line through the accumulation ofits smaller effects on progression through each cell cyclephase.
We have observed that arsenite-treated cells are slow tomove from G1 to S phase (Fig. 6). The G1 checkpoint isusually mediated via p53 activation, but the U937 cell linelacks p53 protein because of a 46-base deletion mutation inthe p53 gene (Sugimoto et al., 1992). However, p21 activationhas been observed in these cells, especially in the context ofdifferentiation induction by 1�,25-dihydroxyvitamin D3 (Liuet al., 1996). We have used immunoblotting to look for in-creases in p21 levels in arsenite-treated U937 cells and havefound none (data not shown). We have similarly found nochanges in levels of other negative regulators of the G1 check-point: p27 and retinoblastoma protein (data not shown).These results were initially surprising, but it soon becameclear that arsenite may not inhibit cell growth in this cell lineby targeting a defined checkpoint. The uniformity of its effecton progression through G1, S, and G2 phases suggests that it
does not activate a target with a negative influence at adistinct cell cycle phase transition, but rather that it inhibitssomething fundamental to the movement of cells into andthrough the cycle. Likely candidates are proteins involved inthe process of DNA synthesis. A direct impairment of DNAsynthesis would explain the apparent hesitation of cells toenter S phase and the increased S phase transit time (Fig. 6;data not shown). Impaired DNA synthesis may also causesubtle DNA damage that could trigger a G2 delay. Futurestudies will examine the effects of arsenite on the initiation ofDNA synthesis and on the elongation of nascent DNAstrands.
We have presented data that suggest that an overall slow-ing of cell cycle progression is a pronounced effect of arsenitein U937 cells. The most sensitive cells are those in mitosisbecause they undergo arsenite-induced apoptosis, but thegrowth-slowing effect of arsenite affects cells in every phase.In conclusion, arsenite treatment induces apoptosis in mi-totic cells, as expected from the results of previous studies.Induction of apoptosis is an important chemotherapeuticmechanism that may potentially be used against many can-cers. However, another important action of arsenite in U937cells is to slow cell cycle progression of nonmitotic cells,prolonging the time spent in G1 and S phases. For thisreason, arsenite as a chemotherapeutic agent may be moreeffective if administered in combination with a drug such ascamptothecin that targets cells in S phase of the cell cycle, assuggested by the data in Fig. 8. Ultimately, a precise targetof arsenic action remains to be identified, but knowledge ofthe specific cellular response to arsenic in a given cell typedramatically narrows the list of possible mediators of arsenicchemotherapy.
Acknowledgments
We thank M. A. O’Reilly and T. A. Gasiewicz for comments andsuggestions that helped to develop the manuscript; Kevin Eckles fortechnical expertise shared in the collection of data; and David Leh-mann, David Farrer, Christine Hammond, Russell Garrett, and SaraHueber for thoughtful discussion. We also greatly appreciate the
Fig. 8. Arsenite treatment enhancesgrowth inhibition by camptothecin.U937 cells were treated with 1 �Msodium arsenite and/or 40 nM camp-tothecin for 24 or 48 h. Cells werecounted with a Coulter counter. As,arsenite; CT, camptothecin; 1As,treatment with 1 �M sodium arsen-ite; 40CT, treatment with 40 nMcamptothecin. Points represent av-erage percentage of the starting cellnumber over three separate experi-ments. Error bars � S.E.M. �, P �0.02, 40 nM camptothecin-treatedcompared with the combined treat-ment according to Student’s paired ttest with a two-tailed distribution.
886 McCollum et al.
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from

generous donation of the TG-3 antibody by Peter Davies of the AlbertEinstein College of Medicine.
ReferencesAbraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR
kinases. Genes Dev 15:2177–2196.Anderson HJ, de Jong G, Vincent I, and Roberge M (1998) Flow cytometry of mitotic
cells. Exp Cell Res 238:498–502.Antman KH (2001) Introduction: the history of arsenic trioxide in cancer therapy.
Oncologist 6 (Suppl 2):1–2.Bachleitner-Hofmann T, Gisslinger B, Grumbeck E, and Gisslinger H (2001) Arsenic
trioxide and ascorbic acid: synergy with potential implications for the treatment ofacute myeloid leukaemia? Br J Haematol 112:783–786.
Begg AC, McNally NJ, Shrieve DC, and Karcher H (1985) A method to measure theduration of DNA synthesis and the potential doubling time from a single sample.Cytometry 6:620–626.
Cai X, Shen YL, Zhu Q, Jia PM, Yu Y, Zhou L, Huang Y, Zhang JW, Xiong SM, ChenSJ, et al. (2000) Arsenic trioxide-induced apoptosis and differentiation are associ-ated respectively with mitochondrial transmembrane potential collapse and reti-noic acid signaling pathways in acute promyelocytic leukemia. Leukemia 14:262–270.
Cai X, Yu Y, Huang Y, Zhang L, Jia PM, Zhao Q, Chen Z, Tong JH, Dai W, and ChenGQ (2003) Arsenic trioxide-induced mitotic arrest and apoptosis in acute promy-elocytic leukemia cells. Leukemia 17:1333–1337.
Chen Z, Chen GQ, Shen ZX, Chen SJ, and Wang ZY (2001) Treatment of acutepromyelocytic leukemia with arsenic compounds: in vitro and in vivo studies.Semin Hematol 38:26–36.
Chou S, Odin M, Sage GW, and Little S (2000) Toxicological profile for arsenic, inAgency for Toxic Substances and Disease Registry, Atlanta, GA.
de The H and Chelbi-Alix MK (2001) APL, a model disease for cancer therapies?Oncogene 20:7136–7139.
Donaldson KL, McShea A, Wahl AF (1997) Separation by counterflow centrifugalelutriation and analysis of T- and B-lymphocytic cell lines in progressive stages ofcell division cycle. J Immunol Meth 203:25–33.
Halicka HD, Smolewski P, Darzynkiewicz Z, Dai W, and Traganos F (2002) Arsenictrioxide arrests cells early in mitosis leading to apoptosis. Cell Cycle 1:201–209.
Howell BJ, Hoffman DB, Fang G, Murray AW, and Salmon ED (2000) Visualizationof Mad2 dynamics at kinetochores, along spindle fibers and at spindle poles inliving cells. J Cell Biol 150:1233–1250.
Huang C, Ma WY, Li J, and Dong Z (1999) Arsenic induces apoptosis through a c-JunNH2-terminal kinase-dependent, p53-independent pathway. Cancer Res 59:3053–3058.
Jing Y, Dai J, Chalmers-Redman RM, Tatton WG, and Waxman S (1999) Arsenictrioxide selectively induces acute promyelocytic leukemia cell apoptosis via ahydrogen peroxide-dependent pathway. Blood 94:2102–2111.
Li YM and Broome JD (1999) Arsenic targets tubulins to induce apoptosis in myeloidleukemia cells. Cancer Res 59:776–780.
Ling YH, Jiang JD, Holland JF, and Perez-Soler R (2002) Arsenic trioxide producespolymerization of microtubules and mitotic arrest before apoptosis in humantumor cell lines. Mol Pharmacol 62:529–538.
Liu M, Lee M-H, Cohen M, Bommakanti M, and Freedman LP (1996) Transcrip-tional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induceddifferentiation of the myelomonocytic cell line U937. Genes Dev 10:142–153.
Lu M, Levin J, Sulpice E, Sequeira-Le Grand A, Alemany M, Caen JP, and Han ZC(1999) Effect of arsenic trioxide on viability, proliferation and apoptosis in humanmegakaryocytic leukemia cell lines. Exp Hematol 27:845–852.
McCabe MJ Jr, Singh KP, Reddy SA, Chelladurai B, Pounds JG, Reiners JJ Jr, andStates JC (2000) Sensitivity of myelomonocytic leukemia cells to arsenite-inducedcell cycle disruption, apoptosis, and enhanced differentiation is dependent on theinter-relationship between arsenic concentration, duration of treatment and cellcycle phase. J Pharmacol Exp Ther 295:724–733.
Miller WH Jr, Schipper HM, Lee JS, Singer J, and Waxman S (2002) Mechanisms ofaction of arsenic trioxide. Cancer Res 62:3893–3903.
Murray A and Hunt T (1993) The Cell Cycle. Oxford University Press, Oxford, UK.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, and Riccardi C (1991) A rapid and
simple method for measuring thymocyte apoptosis by propidium iodide stainingand flow cytometry. J Immunol Methods 139:271–279.
Park JW, Choi YJ, Jang MA, Baek SH, Lim JH, Passaniti T, and Kwon TK (2001)Arsenic trioxide induces G2/M growth arrest and apoptosis after caspase-3 activa-tion and bcl-2 phosphorylation in promonocytic U937 cells. Biochem Biophys ResCommun 286:726–734.
Park WH, Seol JG, Kim ES, Hyun JM, Jung CW, Lee CC, Kim BK, and Lee YY (2000)Arsenic trioxide-mediated growth inhibition in MC/CAR myeloma cells via cellcycle arrest in association with induction of cyclin-dependent kinase inhibitor, p21and apoptosis. Cancer Res 60:3065–3071.
Reglinski J (1998) Environmental and medicinal chemistry of arsenic, antimony andbismuth, in Chemistry of Arsenic, Antimony, and Bismuth (Norman NC ed) pp403–440, Blackie Academic and Professional, London, UK.
Rego EM, He LZ, Warrell RP Jr, Wang ZG, and Pandolfi PP (2000) Retinoic acid (RA)and As2O3 treatment in transgenic models of acute promyelocytic leukemia (APL)unravel the distinct nature of the leukemogenic process induced by the PML-RARalpha and PLZF-RARalpha oncoproteins. Proc Natl Acad Sci USA 97:10173–10178.
Rousselot P, Labaume S, Marolleau JP, Larghero J, Noguera MH, Brouet JC, andFermand JP (1999) Arsenic trioxide and melarsoprol induce apoptosis in plasmacell lines and in plasma cells from myeloma patients. Cancer Res 59:1041–1048.
Seol JG, Park WH, Kim ES, Jung CW, Hyun JM, Kim BK, and Lee YY (1999) Effectof arsenic trioxide on cell cycle arrest in head and neck cancer cell line PCI-1.Biochem Biophys Res Commun 265:400–404.
Soignet SL (2001) Clinical experience of arsenic trioxide in relapsed acute promy-elocytic leukemia. Oncologist 6 (Suppl 2):11–16.
States JC, Reiners JJ Jr, Pounds JG, Kaplan DJ, Beauerle BD, McNeely SC,Mathieu P, and McCabe MJ Jr (2002) Arsenite disrupts mitosis and inducesapoptosis in SV40-transformed human skin fibroblasts. Toxicol Appl Pharmacol180:83–91.
Sugimoto K, Toyoshima H, Sakai R, Miyagawa K, Hagiwara K, Ishikawa F, TakakuF, Yazaki Y, and Hirai H (1992) Frequent mutations in the p53 gene in humanmyeloid leukemia cell lines. Blood 79:2378–2383.
Sundstrom C and Nilsson K (1976) Establishment and characterization of a humanhistiocytic lymphoma cell line (U-937). Int J Cancer 17:565–577.
Wang Z-G, Rivi R, Delva L, Konig A, Scheinberg DA, Gambacorti-Passerini C,Gabrilove JL, Warrell RP Jr, and Pandolfi PP (1998) Arsenic trioxide and melar-soprol induce programmed cell death in myeloid leukemia cell lines and functionin a PML and PML-RARalpha independent manner. Blood 92:1497–1504.
Wang ZY (2001) Arsenic compounds as anticancer agents. Cancer Chemother Phar-macol 48 (Suppl 1):S72–76.
Zhang TC, Cao EH, Li JF, Ma W, and Qin JF (1999) Induction of apoptosis andinhibition of human gastric cancer MGC-803 cell growth by arsenic trioxide. EurJ Cancer 35:1258–1263.
Zhang W, Ohnishi K, Shigeno K, Fujisawa S, Naito K, Nakamura S, Takeshita K,Takeshita A, and Ohno R (1998) The induction of apoptosis and cell cycle arrest byarsenic trioxide in lymphoid neoplasms. Leukemia 12:1383–1391.
Zhu J, Lallemand-Breitenbach V, and de The H (2001) Pathways of retinoic acid- orarsenic trioxide-induced PML/RARalpha catabolism, role of oncogene degradationin disease remission. Oncogene 20:7257–7265.
Zhu XH, Shen YL, Jing YK, Cai X, Jia PM, Huang Y, Tang W, Shi GY, Sun YP, DaiJ, et al. (1999) Apoptosis and growth inhibition in malignant lymphocytes aftertreatment with arsenic trioxide at clinically achievable concentrations. J NatlCancer Inst 91:772–778.
Address correspondence to: Dr. Michael J. McCabe, Jr., Department ofEnvironmental Medicine, University of Rochester School of Medicine andDentistry, 575 Elmwood Ave., Rochester, NY 14642. E-mail: [email protected]
Arsenite Delays U937 Cells in Each Cell Cycle Phase 887
at ASPE
T Journals on A
ugust 24, 2016jpet.aspetjournals.org
Dow
nloaded from
Copyright © 2022 FDOKUMEN







![G2 8`ge Z_ TcZdZd Rd DYZ_UV deVR]d $! >=2d - Daily Pioneer](https://static.fdokumen.com/doc/165x107/63358f97d2b728420307f868/g2-8ge-z-tczdzd-rd-dyzuv-devrd-2d-daily-pioneer.jpg)












