
Functional Assembly of Intrinsic Coagulation Proteases on ...

0011-9164/04/$– See front matter © 2004 Elsevier B.V. All rights reserved
Desalination 169 (2004) 231–244
Arsenic removal by coagulation and filtration: comparison ofgroundwaters from the United States and Bangladesh
S.R. Wickramasinghea*, Binbing Hana, J. Zimbronb, Z. Shenc, M.N. Karima
aDepartment of Chemical Engineering, Colorado State University, Fort Collins, CO 80523-1370, USATel. +1 (970) 491-5276; Fax: +1 (970) 491-7369; email: [email protected]
bMFG/Shepherd Miller Inc., 3801 Automation Way Suite 100, Fort Collins, CO 80525, USAcJiangsu Institute of Microbiology, 7 Qian Rong Road, Wuxi, Jiangsu, 214063, P.R. China
Received 18 March 2003; accepted 8 March 2004
Abstract
Arsenic contamination of drinking water is a concern in many parts of the world. In the United States, theEnvironmental Protection Agency recently reduced the maximum contaminant level of arsenic in drinking water from50 to 10 µg/L (ppb). In Bangladesh the arsenic concentration in drinking water can be as high as hundreds of parts perbillion while the maximum contaminant level is 50 ppb. Consequently, there is a great need for new cost-effectivemethods to remove arsenic from drinking water. Here arsenic removal by coagulation and filtration was investigatedusing groundwater from a city in southern Colorado in the United States and from Sonargaon in Bangladesh. Theresults of the bench-scale experiments conducted indicate that coagulation with ferric ions followed by filtration iseffective in reducing arsenic concentration in the water tested. However, the actual efficiency of removal is highlydependent on the raw water quality. Further, addition of a polyelectrolyte coagulant aid may lead to improved permeatefluxes during tangential flow microfiltration but has little effect on the residual arsenic concentration.
Keywords: Arsenic removal; Groundwater; Coagulation; Iron hydroxide sorption; Filtration; Microfiltration
1. Introduction
Arsenic occurs naturally in water in manyparts of the world [1,2]. Arsenic levels of over60 mg/L (ppm) are lethal for human consump-tion. In the US, the maximum contaminant level
*Corresponding author.
(MCL) in drinking water used to be 50 µg/L(ppb) [3,4]. However, the US EnvironmentalProtection Agency (USEPA) recently finalized anew MCL of 10 µg/L [5,6]. This new MCL isthe same as stated by the World Health Organi-zation guidelines [7]. The USEPA estimates thatadopting this lower arsenic standard will require3,000 community water systems, serving 11

S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244232
million people, to take corrective action to lowerthe current levels of arsenic in their drinkingwater [3].
Millions of wells were drilled into the Gangesand Bhramaputra basins in order to supply drink-ing water for Bangladesh and West Bengal(India) [8,9]. The release of arsenic from thearsenic-bearing aquifer sediments may be respon-sible for polluting more than 2 million of theapproximately 5 million tubewells in Bangladesh,affecting up to 70 million people [10]. The BritishGeological Survey (BGS) reports that out of9,307 tube wells tested, 22 had arsenic concen-trations in the range 100 to 250 µg/L, muchhigher than the 50 µg/L limit set by the Govern-ment of the People’s Republic of Bangladesh[11,12]. Though there remains some uncertaintyas to the actual level of arsenic contamination inBangladesh, there is no doubt that this is a veryserious problem.
Arsenic removal from groundwater in the USand Bangladesh represents major challenges. Inboth cases cost-effective treatment steps arerequired that are easily implementable. However,the challenges in these two countries are verydifferent. In the US the focus is on reducingarsenic in drinking water from less than 50 ppb toless than 10 ppb. In Bangladesh the required levelof removal is much greater and varies dependingon the location of the well. For example, Mengand Korfiatis [8] reported arsenic concentrationsvarying from 280 to 600 ppb in water obtainedfrom wells in the Kishoreganj, Munshiganj andChandpur districts in Bangladesh. Treatmentprocesses are required that will reduce the arsenicconcentration to less than 50 ppb.
There are many methods to remove arsenicfrom drinking water [4]. The USEPA has identi-fied seven technologies as the best availabletechnologies (BATs), which are given in Table 1.Modified coagulation followed by sand or gravityfiltration is already classified as a BAT [4]. Thecombination of enhanced coagulation and micro-
Table 1Best available technologies and their arsenic removalefficiency [4]. Percentage removal figures are for arsenic(V) removal. Preoxidation may be required to convert As(III) to As (V)
Treatment technology Maximumremoval, %
Ion exchange (sulfate 50 mg/L) 95Activated alumina 95Reverse osmosis >95Modified coagulation/filtration 95Modified lime softening (pH >10.5) 90Electrodialysis reversal 85Oxidation/filtration (iron:arsenic = 20:1) 80
filtration (MF) is not included among these BATsdue to a lack of sufficient pilot plant data, al-though the USEPA has noted that this technologydoes meet the criteria for BAT designation [4].This work presents an evaluation of enhancedcoagulation and MF for arsenic removal in twovery different groundwaters, characteristic of twolarge regions that represent a large-scale arsenicproblem. MF was tested using batch, vacuum-driven, dead-end, filtration and also under a con-tinuous system using tangential flow MF.
The detailed chemical reactions involved inthe adsorption of arsenic species by iron oxideshave been studied by previous investigators usingcarefully prepared solutions [13–15]. The aim ofthis work was to determine the efficiency of ferricion coagulation using real groundwater from theUS and Bangladesh. In particular, the effect offerric ion dose (added as ferric chloride or ferricsulfate) and initial raw water pH on the arsenicconcentration and turbidity of the treated waterwere determined. The addition of a small amountof polyelectrolyte as a coagulant aid was alsoinvestigated. The results for real groundwaterwere then compared to the predictions obtainedfrom studying carefully prepared solutions.

S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244 233
2. TheoryArsenic adsorption by iron oxides has been
studied by a number of investigators [13–15]. Inthese studies, As(III) or As(V) stock solutionswere used. Arsenic adsorption was reported to bemore dependent on its oxidation state than pHwithin the pH range 5.5–7.5 [15]. In the studiesreported here, groundwater was obtained from asmall city in southern Colorado in the US andfrom a tubewell in the courtyard of a mosqueabout 3 km from Sonargaon, Bangladesh. Sonar-gaon is a small town about 50 km outside Dhaka,the capital of Bangladesh. Here only As(V) wasconsidered since this is the dominant oxidationstate of arsenic in the groundwater investigated inthis study (this was observed by speciation testson the US groundwater and achieved by oxida-tion of the Bangladesh groundwater).
The acid base dissociation reactions of arsenicacid can be described as [16]:
H3AsO4 : H+ + H2AsO4! pKa = 2.20 (1)
H2AsO4! : H+ + HAsO4
2! pKa = 6.97 (2)
HAsO42! : H+ + AsO4
3! pKa = 11.53 (3)
Since the pH of the groundwater used here ishigher than 7 both for the US and Bangladeshwater (see Table 2), it was expected that HAsO4
2!
would be the dominant species present. In pastexperimental studies [16–19], since the pH of thesolutions used was carefully controlled, the num-ber of dissolved arsenic containing species wasaccurately known.
Ferrihydrite (Fe5HO8•4H2O) was synthesizedusing documented procedures [20]. Ferrihydriteis the initial precipitate that results from the rapidhydrolysis of Fe(III) solutions. By using anaccurately known amount of ferrihydrite, theadsorption of various arsenic containing speciesper kg ferrihydrite can be determined. ExtendedX-ray absorption fine structure (EXAFS) and
Fourier transform infrared (FTIR) spectroscopywere used to study the reaction of arsenic con-taining species with iron oxides [21,22]. Thesestudies provided strong evidence that arsenate isadsorbed onto iron oxide surfaces by formingbidentate binuclear complexes. It has been shownthat arsenate adsorption increases with decreasingpH in the range 4–9 [16–19]. Raven et al. [16]have shown that the overall adsorption of arsenatecan be best described by a Freundlich isotherm.
The effect of competing anions on the adsorp-tion of arsenate has been studied using carefullyprepared solutions. Jain and Loeppert [17] foundthat the presence of sulfate did not influencearsenate adsorption in the pH range 4–9. How-ever, Wilkie and Hering [23] did observe adecrease in arsenate adsorption in the presence ofsulfate ions. It is noticeable that the experimentalconditions of these two studies were quite differ-ent. This result highlights the complex nature ofthe adsorption reactions that occur. Meng et al.[18] found that the presence of silicates lead todecreased arsenate removal.
In this work arsenate removal from twosources of groundwater is investigated. Thesewaters are not as well characterized as the idealsolutions tested in previous studies. Nevertheless,the experimental results will be compared toprevious theoretical results for model solutions.
3. Experimental methods
3.1. US groundwater
To generate reliable test results effectively atlow arsenic concentrations, the groundwater wasstored in a controlled environment at 22°C underhigh-purity argon gas. The groundwater wasfrequently monitored to ensure that there was nochange in the chemical analysis. Six wells areused to supply drinking water to the city in south-ern Colorado. Water tested from all six wells wasfound to have a high silicate concentration
S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244234
varying from 110 to 140 mg/L while the pHvaried from 8.2 to 8.7. The arsenic concentrationvaried from 20 to 70 µg/L. At present, in order toensure an arsenic concentration less than 50 µg/L,water from the six wells was blended.
Speciation tests were conducted on water fromthe six wells that supply the city using ion-exchange columns according to a field speciationprotocol [24]. These tests indicated that practic-ally all the arsenic was present in the As(V) form.The water collected for the bench-scale studiesdescribed here was obtained from a well, which
Table 2Results of analysis of US and Bangladesh water samples
Analyte Concentration, mg/L
US water Bangladeshwater
Alkalinity total (as CaCO3) 92.5 407Aluminum <0.02 0.03Arsenic 0.068 0.138Calcium 6.5 108.8Chloride 0.77 23.6Chromium <0.006 0.02EMF (mV) 135.3 NAFluoride 1.07 NAHCO3
! NA 497Iron 0.035 1.7Magnesium 0.21 26.5Manganese 0.003 1.89Nitrate (as N) 0.1 <0.1Nitrite (as N) <0.05 <0.1Orthophosphate as P 0.180 0.34pH 8.7 7.5Potassium 6.6 2.7Silica 141.1 NASodium 52.9 33.0Sulfate 9.2 0.3TSS 0.2 691.7Turbidity, NTU 0.13 45.5
NA: Data not available.
previously showed the highest arsenic concen-tration among the six wells. Table 2 gives theresults of a chemical analysis of this water. Thehigh pH of this water and the arsenic speciationtests [24] indicate that this water is typical ofmany groundwaters in the US containing higharsenic concentrations, in which the oxidizingconditions and the high pH result in arsenic oxi-dation from the groundwater matrix and subse-quent mobilization [2].
3.2. Bangladesh water
Bangladesh water was obtained from a tube-well in Sonargaon that was marked with red paintindicating that the arsenic concentration washigher than 50 ppb. The water was sealed in anairtight container and shipped to the US. It hasbeen reported that about 50% of the total arsenicin groundwaters from various parts of Bangladeshexists as As(III) [2,24]. The Bangladesh waterwas oxidized prior to testing to assure that all thearsenic would be in its oxidized state [As(V)].Table 2 gives the results of a chemical analysis ofthis water.
3.3. Test methods
All glass and plastic equipment used in bench-scale testing was cleaned and acid washed using0.1 mol/L nitric acid, triple-rinsed with deio-nized water, then triple-rinsed with an aliquot oftest solution prior to contacting the samplesolution. Samples of raw US groundwater, 1.0 Lin volume, pH 8.7, were dosed with ferric ionspresent either as ferric chloride or ferric sulfateand stirred for 2.0 min at 300 rpm using amagnetic stirrer. After stirring the samples wereimmediately vacuum filtered using a 0.22 µmpore size membrane (mixed ester of celluloseacetate and cellulose nitrate) discs obtained fromWhatman (Maidstone, UK). A second series ofexperiments was conducted where the pH of thegroundwater was adjusted prior to ferric ion

S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244 235
dosing. Two pH values were tested: 6.2 and 6.8.For samples dosed with ferric chloride, sulfuricacid was added to adjust the pH while for samplesdosed with ferric sulfate, hydrochloric acid wasadded. In these experiments the residual arsenicconcentration and turbidity were measured.Table 3 summarizes the initial pH values of thewater before addition of ferric chloride or ferricsulfate.
The same procedure was used for Bangladeshwater. However, due to a limited supply of thegroundwater, sample volumes tested were 50 mLrather than 1 L. Further, based on the results ob-tained for US water, the pH of the Bangladesh
Table 3Summary of initial water pH before addition of coagu-lant. For US water three pH values were tested usingferric chloride or ferric sulfate. For Bangladesh wateronly pH 6.8 using ferric chloride or ferric sulfate and pH6.2 using ferric sulfate were tested
pH US water Bangladesh water
8.7 FeCl3, Fe2(SO4)3 —6.8 FeCl3, Fe2(SO4)3 FeCl3, Fe2(SO4)3
6.2 FeCl3, Fe2(SO4)3 Fe2(SO4)3
groundwater was adjusted to either 6.8 or 6.2. AtpH 6.8, ferric chloride and ferric sulfate weretested as coagulants. However, at pH 6.2 onlyferric sulfate was used.
MF experiments were conducted using the ex-perimental set-up shown in Fig. 1. Feed streamsconsisting of 500 mL of US or Bangladesh waterwere coagulated using 3.4 ppm ferric ion addedas ferric chloride (10 ppm FeCl3). In addition, acationic polyelectrolyte (CY 2461, Cytec Indus-tries, Stamford, CT, molecular weight 8–12×106,charge density 40%) was added as a coagulant aidto some of the feed streams. The polyelectrolytedose were 0.02 and 0.3 ppm. The suspension wasstirred at 300 rpm for 1 min and 70 rpm for10 min before commencing MF. A/G Technology(Needham, MA) hollow-fiber modules were used.These modules contain six hollow fibers, 10.9 cmin length and 1 mm ID. The nominal pore size ofhollow fibers is 0.1 µm. The feed stream waspumped from the feed reservoir using a WatsonMarlow 504U peristaltic pump (Watson Marlow,Wilmington, MA). After flowing through thelumen of the hollow fibers, the retentate wasrecycled back to the feed reservoir while thepermeate was withdrawn from the shell side andcollected in the permeate reservoir. The permeate
Fig. 1. Experimental set-up.
S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244236
was also returned to the feed reservoir intermit-tently in order to ensure a constant floc concen-tration. The feed flow rate was 138 mL/min andwall shear rate was about 4,000 s!1. The trans-membrane pressure drop was 34.5 kPa (5 psi).
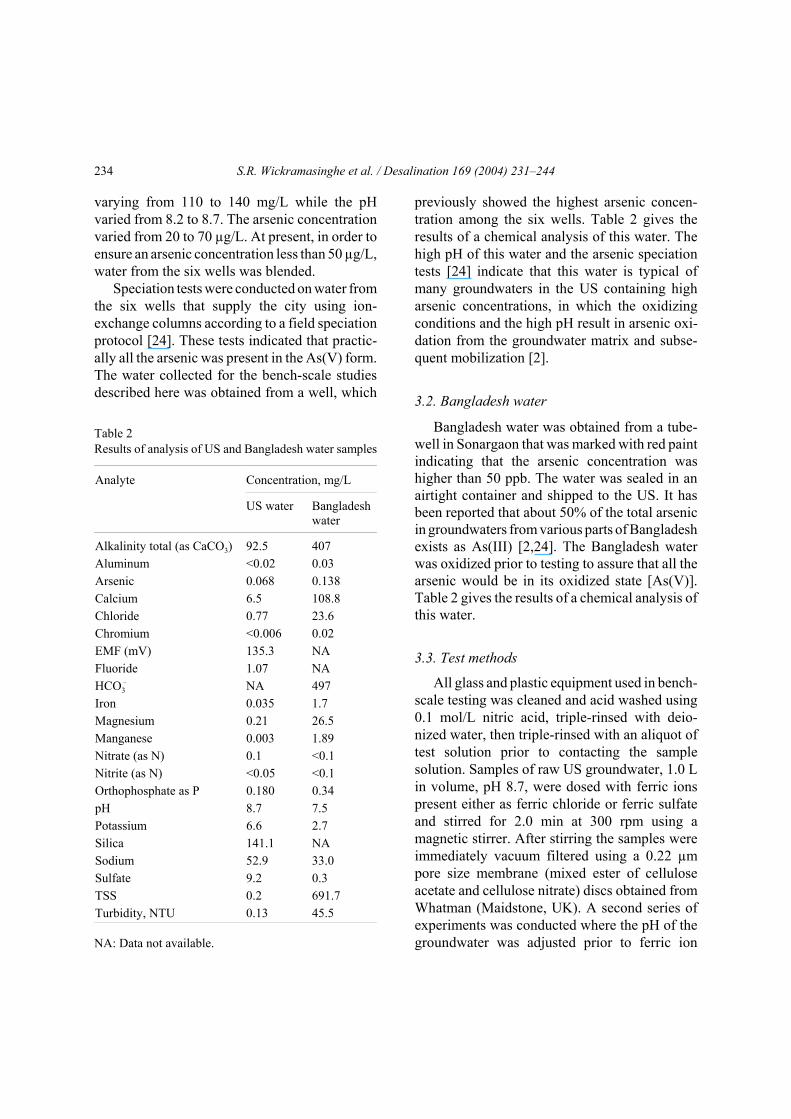
4. ResultsFig. 2 shows the variation of the residual
arsenic concentration with ferric ion dose addedeither as ferric chloride or ferric sulfate for USand Bangladesh groundwaters. For US water,three different pH values were tested: pH 8.7 (pHof raw water), 6.8 and 6.2. For each initial rawwater pH, ferric ions were added as ferric chlo-ride or ferric sulfate. The suspension was vacuumfiltered without floc aging using a 0.22 µm filterdisk. As can be seen, if the residual arsenicconcentration is plotted as a function of the ferric
ion dose, the presence of sulfate ions has littleeffect on the efficiency of arsenic removal.Further, the lower the initial raw water pH, thelower the residual arsenic concentration for agiven ferric ion dose. These results are in agree-ment with an earlier study [25]. Fig. 2 also showsthe variation of the residual arsenic concentrationwith ferric ion dose added as either ferric chlorideor ferric sulfate for Bangladesh water. However,as only a limited amount of Bangladesh waterwas available, only two pH values, 6.8 and 6.2,were tested. Since these two pH values were alsoused for US water, the results for the twogroundwaters can be compared directly. Further,results for US water and previous studies [16–19]indicated that arsenate adsorption by ferric ionsincreases with decreasing pH in the range 4–9.Consequently, for Bangladesh water without pHadjustment (pH 7.5), it is expected that the
Fig. 2. Effect of raw water pH on the variation of residual arsenic concentration with ferric ion dose. After coagulation,the suspension was vacuum filtered using 0.22 µm pore size membrane disk. O + thin solid line: US water, pH 8.7,Fe2(SO4)3 as coagulant; — + thin solid line: US water, pH 6.8, Fe2(SO4)3 as coagulant; • + thin solid line: US water,pH 6.2, Fe2(SO4)3 as coagulant; Q + thick solid line: US water, pH 8.7, FeCl3 as coagulant; " + thick solid line: US water,pH 6.8, FeCl3 as coagulant; ) + thick solid line: US water, pH 6.2, FeCl3 as coagulant; ": unfiltered Bangladesh water,pH 7.5; " + dashed line: Bangladesh water, pH 6.8, Fe2(SO4)3 as coagulant; ) + dashed line: Bangladesh water, pH 6.2,Fe2(SO4)3 as coagulant; G + dashed line: Bangladesh water, pH 6.8, FeCl3 as coagulant.

S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244 237
amount of arsenate adsorbed will be less than atpH 6.8 and 6.2. As was the case for the US water,if the residual arsenic concentration is plotted asa function of the ferric ion dose, the presence ofsulfate ions has little effect on the efficiency ofarsenic removal. Fig. 2 also indicates that there islittle difference in the residual arsenic concen-tration for an initial water pH of 6.8 and 6.2 forBangladesh water.
Table 2 indicates that the initial turbidity ofthe Bangladesh water was 45.5 NTU while theUS water was only 0.13 NTU. Vacuum filtrationof the Bangladesh water without the addition offerric ions resulted in a decrease in the arsenicconcentration from 138 to 64 ppb, while for US
water no decrease in the arsenic concentration inthe absence of ferric ion was observed. Conse-quently in presenting the experimental results, theresidual arsenic concentration for 0 ppm ferric ionaddition followed by vacuum filtration is given as64 ppb for Bangladesh water. The initial arsenicconcentration in the Bangladesh water prior tovacuum filtration (138 ppm) is included in Fig. 2as a point on the y-axis.
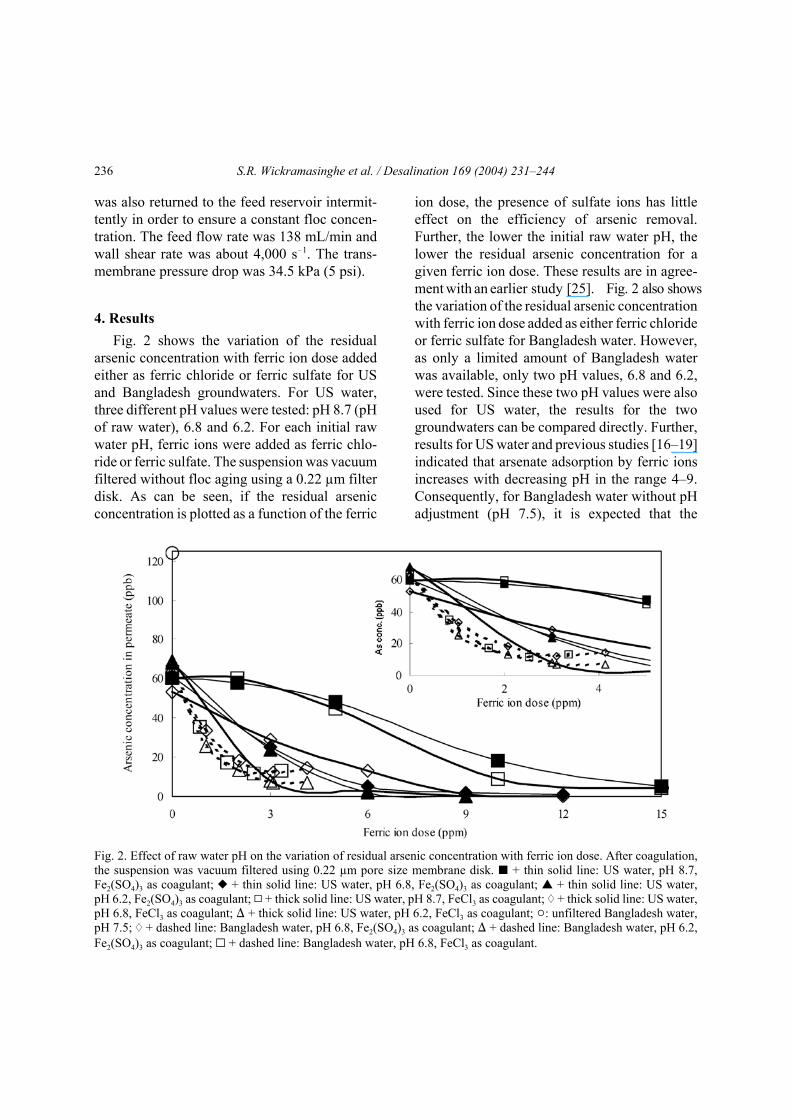
The variation of residual turbidity with ferricion dose for the US and Bangladesh ground-waters is shown in Fig. 3. For US water, three pHvalues, pH 8.7, 6.8 and 6.2, were tested. Theresidual turbidity is significantly less for ferricsulfate compared to ferric chloride. However, for
Fig. 3. Effect of initial raw water pH on the variation of the residual turbidity with ferric ion dose. After coagulation, thesuspension was vacuum filtered using a 0.22 µm pore size membrane disk. O + thin solid line: US water, pH 8.7, Fe2(SO4)3as coagulant; — + thin solid line: US water, pH 6.8, Fe2(SO4)3 as coagulant; • + thin solid line: US water, pH 6.2,Fe2(SO4)3 as coagulant; Q + thick solid line: US water, pH 8.7, FeCl3 as coagulant; " + thick solid line: US water, pH 6.8,FeCl3 as coagulant; ) + thick solid line: US water, pH 6.2, FeCl3 as coagulant; ": unfiltered Bangladesh water, pH 7.5;" + dashed line: Bangladesh water, pH 6.8, Fe2(SO4)3 as coagulant; ) + dashed line: Bangladesh water, pH 6.2, Fe2(SO4)3
as coagulant; G + dashed line: Bangladesh water, pH 6.8, FeCl3 as coagulant.
S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244238
both ferric chloride and ferric sulfate the turbidityincreases at first and then decreases with anincreasing ferric ion dose.
Fig. 3 also gives the variation of residualturbidity with ferric ion dose for Bangladeshwater. As already noted, Table 2 indicates that theinitial turbidity of the Bangladesh water was45.5 NTU, while the US water was only0.13 NTU. Vacuum filtration of the Bangladeshwater without the addition of ferric ions resultedin a decrease in the residual turbidity from 45.5 to0.1 NTU. However, for the US water, only asmall decrease (0.13 to 0.08 NTU) in the turbiditywas observed after vacuum filtration without theaddition of ferric ions. Consequently, in present-ing the experimental results the residual turbidityfor 0 ppm ferric ion addition followed by vacuumfiltration is given as 0.1 NTU for the Bangladeshwater. The turbidity in the initial Bangladeshwater prior to vacuum filtration (45.5 NTU) isincluded in Fig. 3 as a point on the y-axis. Theresidual turbidity at the same ferric ion dose and
initial water pH is significantly less for theBangladesh water compared to the US water. Fur-ther, little difference is seen between the residualturbidity for ferric sulfate and ferric chloride forBangladesh water.
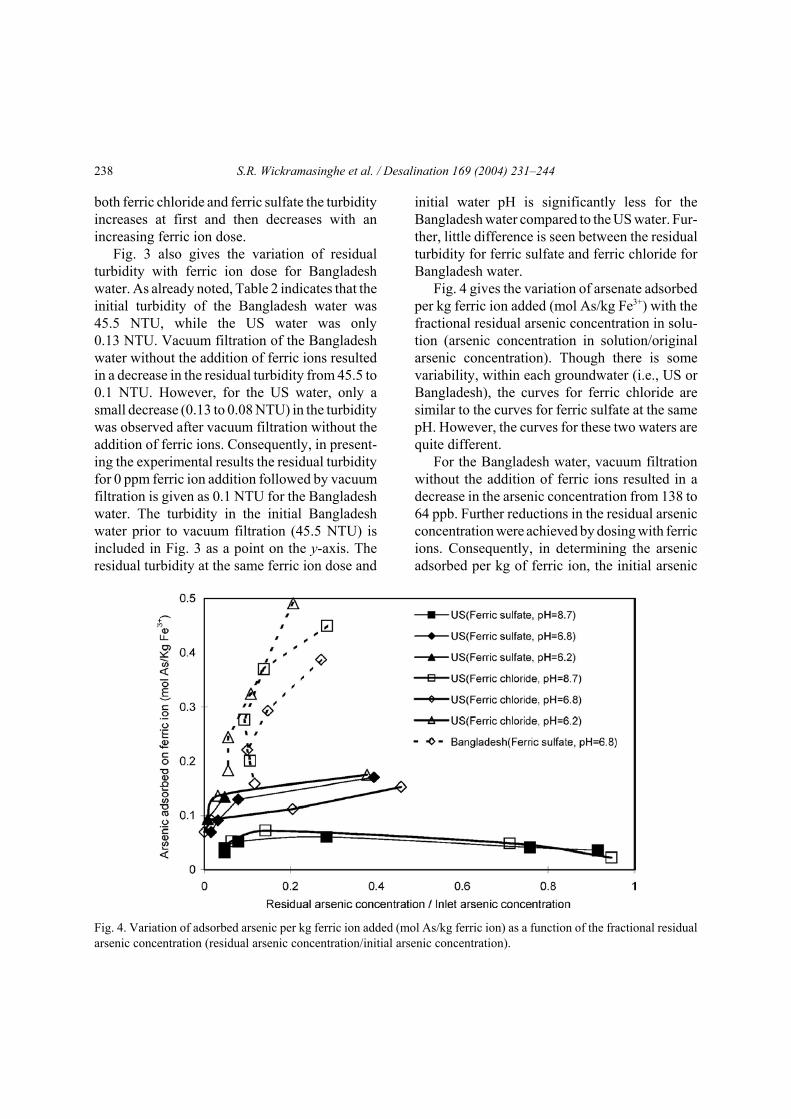
Fig. 4 gives the variation of arsenate adsorbedper kg ferric ion added (mol As/kg Fe3+) with thefractional residual arsenic concentration in solu-tion (arsenic concentration in solution/originalarsenic concentration). Though there is somevariability, within each groundwater (i.e., US orBangladesh), the curves for ferric chloride aresimilar to the curves for ferric sulfate at the samepH. However, the curves for these two waters arequite different.
For the Bangladesh water, vacuum filtrationwithout the addition of ferric ions resulted in adecrease in the arsenic concentration from 138 to64 ppb. Further reductions in the residual arsenicconcentration were achieved by dosing with ferricions. Consequently, in determining the arsenicadsorbed per kg of ferric ion, the initial arsenic
Fig. 4. Variation of adsorbed arsenic per kg ferric ion added (mol As/kg ferric ion) as a function of the fractional residualarsenic concentration (residual arsenic concentration/initial arsenic concentration).

S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244 239
concentration is taken as 64 ppb since this repre-sents the initial concentration of arsenic thatwould be available for adsorption on to addedferric ions after vacuum filtration of the rawwater. In the case of US water, no arsenic remov-al occurred by vacuum filtration alone. As can beseen, even after adjusting the initial arsenicconcentration in the Bangladesh water to accountfor arsenic removal by filtration alone, the arsenicremoved per kg ferric ions is much greater for theBangladesh water.
The MF results are given in Fig. 5a and 5b forUS and Bangladesh waters, respectively. In thesefigures, the permeate flux divided by the perme-ate flux for distilled water at the same wall shearrate and transmembrane pressure drop, expressedas a percentage, is plotted as a function of time.In these experiments, 3.4 ppm ferric ion (added asferric chloride) was added to the water. For USwater, in the absence of the cationic polyelec-trolyte the permeate flux falls to about 60% of thepermeate flux for distilled water after 30 min.However, the addition of 0.3 ppm polyelectrolytehas a dramatic effect. No decrease in the
Fig. 5a. Microfiltration results for US water. Permeateflux divided by the permeate flux for distilled waterexpressed as a percentage is plotted as a function of time.The wall shear rate was 4,000 s!1 and the transmembranepressure drop was 34.5 kPa. The pH of the water was notadjusted (pH 8.7). All feeds streams were coagulated with3.4 ppm ferric ions added as ferric chloride. Addition ofa small amount of cationic polyelectrolyte had a signifi-cant effect on the permeate flux.
permeate flux is observed for 2.5 h. For Bangla-desh water in the absence of cationic polyelectro-lyte, there is no decrease in the permeate flux forover 1 h. Consequently, addition of the cationicpolyelectrolyte has little effect on the permeateflux for Bangladesh water.
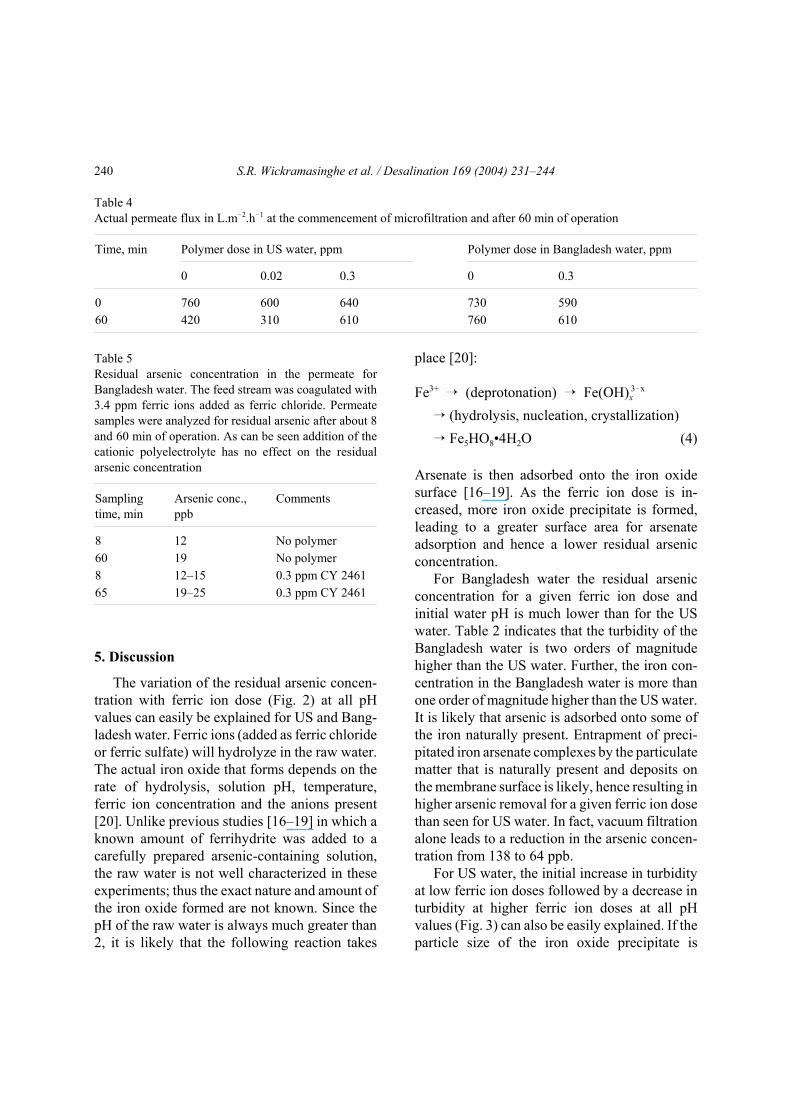
Table 4 compares the actual permeate flux inL.m!2.h!1 at the commencement of MF or timezero (i.e., permeate flux of DI water) and after60 min of operation. Within experimental error,the initial permeate fluxes for US and Bangladeshwater are similar.
Table 5 gives the residual arsenic concen-tration in the permeate as it exits the module afterabout 8 and 60 min of operation for Bangladeshwater. As can be seen, addition of the cationicpolyelectrolyte has little effect on the residualarsenic concentration. Comparing Table 5 andFig. 2, the residual arsenic concentration in thepermeate after tangential flow MF using a 0.1 µmpore size membrane is in agreement with theresidual arsenic in the filtrate after vacuum fil-tration using 0.22 µm pore sized filter discs. ForUS water it was also observed that addition of thecationic polyelectrolyte had little effect on theresidual arsenic concentration.
Fig. 5b. Microfiltration results for Bangladesh water.Permeate flux divided by the permeate flux for distilledwater expressed as a percentage is plotted as a function oftime. The wall shear rate was 4,000 s!1 and the trans-membrane pressure drop was 34.5 kPa. The pH of thewater was not adjusted (pH 7.5). All feeds streams werecoagulated with 3.4 ppm ferric ions added as ferricchloride.
S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244240
Table 4Actual permeate flux in L.m!2.h!1 at the commencement of microfiltration and after 60 min of operation
Time, min Polymer dose in US water, ppm Polymer dose in Bangladesh water, ppm
0 0.02 0.3 0 0.3
0 760 600 640 730 59060 420 310 610 760 610
Table 5Residual arsenic concentration in the permeate forBangladesh water. The feed stream was coagulated with3.4 ppm ferric ions added as ferric chloride. Permeatesamples were analyzed for residual arsenic after about 8and 60 min of operation. As can be seen addition of thecationic polyelectrolyte has no effect on the residualarsenic concentration
Samplingtime, min
Arsenic conc.,ppb
Comments
8 12 No polymer60 19 No polymer8 12–15 0.3 ppm CY 246165 19–25 0.3 ppm CY 2461
5. Discussion
The variation of the residual arsenic concen-tration with ferric ion dose (Fig. 2) at all pHvalues can easily be explained for US and Bang-ladesh water. Ferric ions (added as ferric chlorideor ferric sulfate) will hydrolyze in the raw water.The actual iron oxide that forms depends on therate of hydrolysis, solution pH, temperature,ferric ion concentration and the anions present[20]. Unlike previous studies [16–19] in which aknown amount of ferrihydrite was added to acarefully prepared arsenic-containing solution,the raw water is not well characterized in theseexperiments; thus the exact nature and amount ofthe iron oxide formed are not known. Since thepH of the raw water is always much greater than2, it is likely that the following reaction takes
place [20]:
Fe3+ ÷ (deprotonation) ÷ Fe(OH)x3!x
÷ (hydrolysis, nucleation, crystallization)÷ Fe5HO8•4H2O (4)
Arsenate is then adsorbed onto the iron oxidesurface [16–19]. As the ferric ion dose is in-creased, more iron oxide precipitate is formed,leading to a greater surface area for arsenateadsorption and hence a lower residual arsenicconcentration.
For Bangladesh water the residual arsenicconcentration for a given ferric ion dose andinitial water pH is much lower than for the USwater. Table 2 indicates that the turbidity of theBangladesh water is two orders of magnitudehigher than the US water. Further, the iron con-centration in the Bangladesh water is more thanone order of magnitude higher than the US water.It is likely that arsenic is adsorbed onto some ofthe iron naturally present. Entrapment of preci-pitated iron arsenate complexes by the particulatematter that is naturally present and deposits onthe membrane surface is likely, hence resulting inhigher arsenic removal for a given ferric ion dosethan seen for US water. In fact, vacuum filtrationalone leads to a reduction in the arsenic concen-tration from 138 to 64 ppb.
For US water, the initial increase in turbidityat low ferric ion doses followed by a decrease inturbidity at higher ferric ion doses at all pHvalues (Fig. 3) can also be easily explained. If theparticle size of the iron oxide precipitate is

S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244 241
sufficiently large, the precipitate containingadsorbed arsenic will be rejected by the mem-brane disc. At low ferric ion doses, the totalnumber of insoluble precipitate particles presentwill be low. Consequently, growth of largerparticles by collisions between precipitate parti-cles will be limited. Thus fewer particles will berejected by the membrane and the decrease inresidual arsenic concentration will be limited.Compared to the original raw water, the presenceof precipitated iron oxide will lead to an increasein turbidity. As the ferric ion dose is increased thenumber of precipitate particles increases andgrowth of larger particles by collisions is morelikely. Consequently, more particles will berejected by the membrane and the turbidity of thesolution will decrease. Fig. 3 shows that with anincreasing ferric ion dose, the turbidity reaches amaximum value and then decreases.
Unlike the US water, there is little variation inthe residual turbidity for the Bangladesh waterwith ferric ion dose. This is most likely due to thevery high initial turbidity of the water. In fact,significant removal of turbidity occurs withoutaddition of ferric ions. Thus as small precipitateparticles containing adsorbed arsenate form, theyare easily trapped by the naturally occurring par-ticulate matter that is rejected by the membrane.
The effect of pH on the residual arsenicconcentration and turbidity may also be explainedbased on previous studies. Numerous investi-gators have shown that as pH decreases, arsenateadsorption increases [16–19]. Increased arsenateadsorption will lead to increased particle size.Thus the degree of rejection of precipitate parti-cles by the membrane at a given ferric ion doseshould increase with decreasing pH. Fig. 3 showsthat as the pH of the US water is decreased, themaximum residual turbidity occurs at a lowerferric ion dose as expected. Fig. 3 also shows thatthe actual value of the maximum turbidity chan-ges with pH. As the pH of the raw water islowered, not only will the amount of arsenic ad-sorbed onto the iron oxide increases, but also the
amount and structure of the iron oxide precipitateis likely to change [20]. The actual value of theturbidity will be affected by the amount andstructure of the iron oxide precipitate formed.
Unlike the US water, Bangladesh water showslittle dependence of the residual arsenic concen-tration on the initial pH of the water. It is likelythat the very high initial turbidity and the largedecrease in both residual arsenic concentrationand turbidity that occur in the absence of ferricion coagulation mask any pH effects that exist.
In this study the concentration of silicates wasnot varied. However, as shown in Table 2, thetotal silica present is high for the US water. Thepresence of silicates is known to reduce arsenateadsorption by ferric oxides [18]. When the ferricion dose vs. residual arsenic concentration for USwater obtained here is compared to previousresults [26,27], the doses used here are higher,suggesting competition by the silicates present.Though not measured in this study, the silicaconcentration in the Bangladesh water fromSonargaon is much lower [28]. The presence ofsilicates in the US water may also explain themuch lower amounts of arsenate adsorbed per kgferric ion compared to Bangladesh water (Fig. 4).
The effect of sulfates on the adsorption ofarsenate depends on the experimental conditions[17,23]. In this study when ferric sulfate was usedas the coagulant, the pH of the raw water wasadjusted using hydrochloric acid in order to mini-mize the concentration of sulfates present. Inthese experiments sulfates were not found toaffect the adsorption of arsenate. For the Bangla-desh water, Table 2 indicates that the naturalsulfate concentration is very low. Further, thevery high initial turbidity and the large decreasein both residual arsenic concentration and tur-bidity that occur in the absence of ferric ioncoagulation may mask any effects due to thepresence of sulfates.
The sulfate concentration in the US water ismore than one order of magnitude higher than theBangladesh water. It is possible that the reason

S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244242
sulfates were found not to affect arsenate adsorp-tion for US water was due to the fact that the totalsilicates present is much higher than the sulfatespresent; thus competition by sulfates for bindingsites on the iron oxide precipitate was negligiblecompared to competition by the silicates present.However, Fig. 3 does indicate that for the USwater, coagulation in the presence of sulfatesleads to a lower residual turbidity, suggesting thatthe sulfate groups are involved in the coagulationreactions. The presence of sulfate groups couldlead to larger flocs that are more easily rejectedby the membrane, compared to chloride ions.
Fig. 4 gives the variation of the concentrationof adsorbed arsenate as a function of residualarsenate in solution. Previous investigators havedetermined adsorption isotherms for arsenateadsorption by ferrihydrite [16]. These isothermsare very different to Fig. 4. Adsorption isothermsgive the amount of arsenate as a function of arse-nate concentration in solution at equilibrium.Raven et al. [16] ran their adsorption experimentsfor 24 h in order to ensure equilibrium conditionsexisted. Even though much shorter, Meng et al.[18] ran their experiments for 1 h to ensure equili-brium had been reached. In an actual process thiswould be impractical. In the experiments de-scribed here contact between the arsenate solutionand iron oxide precipitate was for 2 min withrapid mixing (300 rpm).
Raven et al. [16] knew the amount of ferri-hydrite added; thus the amount of adsorbentpresent was accurately known. In a commercialsystem, ferric ions will be added, not ferrihydrate.Not all these ferric ions will form precipitate.Further, the actual structure of the precipitate isunknown. Thus the results obtained here areexpressed in terms of arsenate adsorbed per kgferric ions added. Competition between arsenateand other anions will also affect the results. Final-ly, since groundwater is used here, the arsenateconcentration in solution is the concentration thatoccurs naturally. In most model equilibrium
studies, in order to determine the adsorptionisotherm, the arsenate concentration in solution ismuch higher than would be found in naturallyoccurring groundwater.
The amount of arsenate adsorbed onto ironoxide is a function of the solution pH. Duringprecipitation of ferric oxide and adsorption ofarsenate, the pH of the system will change [16].In studies that focus on determining adsorptionisotherms pH is carefully monitored and acid orbase added in order to ensure a constant pH.However, in a commercial coagulation and filtra-tion system, during coagulation the pH will beallowed to drift. Consequently, in the experimentspresented here, the pH was allowed to drift. Forthese reasons comparison of Fig. 4 with theresults of ideal equilibrium studies is not justified.
Fig. 4 is, however, very useful in presentingthe experimental data. As can be seen withinexperimental uncertainty, there is no difference inthe amount of arsenate adsorbed by ferricchloride or ferric sulfate. Further, as the initial pHof the raw water is decreased, the amount ofarsenate adsorbed increases. Fig. 4 may be usedto predict the residual arsenate concentration fora specific groundwater under the experimentalconditions tested here. Fig. 4 also highlights thelarge differences that may exist for the efficiencyof arsenate adsorption from different ground-waters.
Table 5 indicates that addition of a cationicpolyelectrolyte as a coagulant aid has little effecton the residual arsenic concentration. However, itmay lead to improved permeate fluxes. It is likelythat the cationic polyelectrolyte forms bridgesbetween precipitated ferric particles in the USwater. Thus plugging of the membrane pores byprecipitate particles is suppressed, resulting in themaintenance of higher permeate fluxes. ForBangladesh water smaller ferric precipitate parti-cles may adsorb on to the large amount of parti-culate matter that is naturally present in the water.Consequently, a coagulant aid is not required.

S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244 243
Fig. 2 and Table 5 indicate that the residual arse-nic concentration after vacuum filtration using0.22-µm membrane discs is in good agreementwith the residual arsenic concentration in thepermeate during tangential flow MF using 0.1 µmpore size membranes. However, vacuum filtrationexperiments are much easier to run and providequick results when testing different ferric iondoses and initial water pH.
Fig. 2 indicates that vacuum filtration alone,without coagulation using ferric ions, reduces thearsenic concentration in Bangladesh water from138 ppb to 64 ppb. Thus addition of ferric ionswill be required in order to meet the maximumcontaminant level for arsenic set by the Bangla-desh Government. Further, MF appears to be verysuitable for this water. For US water, coagulationwith ferric ions followed by filtration is success-ful in reducing the arsenic concentration below10 ppb. However, if tangential flow MF is to beused, addition of a coagulant aid may lead to asignificant increase in the permeate flux.
While coagulation and filtration are an effi-cient arsenic removal technology, the results pre-sented here show that design of a commercialsystem will be very dependent on the water qual-ity. Since a lower pH leads to improved arsenateremoval, pH adjustment may be necessary inorder to reduce the ferric ion dose required.However, the degree of pH adjustment will bedetermined by the competing requirements of thevolume of acid required and the savings in ferricchloride solution. Besides technical feasibility,further geographical and economical factorsinherent to each site must also be consideredbefore large-scale testing and design of a facilityfor arsenic removal.
6. Conclusions
The feasibility of arsenic removal by coagu-lation followed by filtration for groundwater from
a city in southern Colorado in the US and fromSonargaon in Bangladesh was investigated usingferric chloride and ferric sulfate as coagulants.The results obtained here suggest that using eithercoagulant will lead to a practical process. Coagu-lation with ferric sulfate, however, leads to alower residual turbidity for the US water. It islikely that pH adjustment of the groundwaterprior to coagulation will be required. The actualefficiency of arsenic removal is highly dependenton the quality of the raw water. Use of polyelec-trolytes as coagulant aids may lead to enhancedpermeate fluxes; however, the polyelectrolyte hadno effect on the residual arsenic concentration.
AcknowledgementsThe authors would like to acknowledge
Prof. A.K.M.A. Quader, Department of ChemicalEngineering, Bangladesh University of Engineer-ing and Technology, Dhaka, Bangladesh, forproviding the Bangladesh water and for his manyhelpful suggestions. Phipps & Bird (Richmond,VA, USA) is thanked for providing the jar-testerused in this work. Financial support was providedby MFG/Shepherd Miller Inc., Fort Collins, CO,USA, and the National Science Foundation ofUSA (INT-0108257).
References[1] H.W. Chen, M.M. Frey, D. Clifford, L.S. McNeill
and M. Edwards, Arsenic treatment considerations,J. AWWA, 91 (1999) 74–85.
[2] P.L. Smedley and D.G. Kinniburgh, A review of thesource, behaviour and distribution of arsenic innatural waters, Appl. Geochem., 17 (2002) 517–568.
[3] USEPA, Arsenic in Drinking Water Rule: EconomicAnalysis, EPA/815/R-00/026, Washington, 2000.
[4] USEPA, Technologies and Costs for Removal ofArsenic from Drinking Water, EPA/815/R-00/028,Washington, 2000.
[5] National Research Council, Arsenic in DrinkingWater, National Academy Press, Washington, 1999.

S.R. Wickramasinghe et al. / Desalination 169 (2004) 231–244244
[6] USEPA, National Primary Drinking Water Regula-tions: Arsenic and Clarifications to Compliance andNew Source Contaminant Monitoring, Washington,2001.
[7] Guidelines for Drinking Water Quality, Vol. 2:Health Criteria and other Supporting Information,2nd ed., World Health Organization, Geneva, Switz-erland, 1993.
[8] X. Meng and G.P. Korfiatis, Removal of arsenicfrom Bangladesh well water using a householdfiltration system, in: M.F. Ahmed, M.A. Ali and Z.Adeel, eds., Technologies for Arsenic Removal fromDrinking Water, Bangladesh University of Engi-neering and Technology (Dhaka) and the UnitedNations University (Tokyo), Dhaka, Bangladesh,2001, pp. 121–130.
[9] A.K.M. Munir, S.B. Rasul, M. Habibuddowla,M. Alauddin, A. Hussam and A.H. Khan, Evaluationof the performance of Sono 3-Kolshi filter for arsenicremoval from groundwater using zero valent ironthrough laboratory and field studies, in: M.F. Ahmed,M.A. Ali and Z. Adeel, eds., Technologies forArsenic Removal from Drinking Water, BangladeshUniversity of Engineering and Technology (Dhaka)and the United Nations University (Tokyo), Dhaka,Bangladesh, 2001, pp. 171–189.
[10] W. Lepkowski, Arsenic crisis spurs scientists —Bangladesh-born US scientists press hard for policyinfluence in the arsenic contamination crisis in theirnative country, Chem. Eng. News, 77 (1999) 45–49.
[11] British Geological Survey (BGS), GroundwaterStudies for Arsenic Contamination in Bangladesh,British Geological Survey and Mott and MacDonald,UK, 1999.
[12] Environment Conservation Regulation 1997, Gov-ernment of Peoples’ Republic of Bangladesh, Dhaka,Bangladesh, 1997.
[13] M.L. Pierce and C.B. Moore, Adsorption of arseniteand arsenate on amorphous iron hydroxide, WaterRes., 16 (1982) 1247–1253.
[14] M.L. Pierce and C.B. Moore, Adsorption of arseniteon amorphous iron hydroxide from dilute aqueoussolution, Envir. Sci. Technol., 14 (1980) 214–216.
[15] J.F. Ferguson and M.A. Anderson, Chemical formsof arsenic in water supplies and their removal, in:A.J. Rubin, ed., Chemistry of Water Supply,Treatment and Distribution, Ann Arbor Science, AnnArbor, MI, 1974, pp. 137–158.
[16] K.P. Raven, A. Jain and R.H. Loeppert, Arsenite andarsenate adsorption on ferrihydrite: kinetics, equili-brium and adsorption envelopes, Envir. Sci.Technol., 32 (1998) 344–349.
[17] A. Jain and R.H. Loeppert, Effect of competinganions on the adsorption of arsenate and arsenite byferri-hydrite, J. Envir. Qual., 29 (2000) 1422–1430.
[18] X. Meng, S. Bang and G.P. Korfiatis, Effects of sili-cate, sulfate and carbonate on arsenic removal byferric chloride, Water Res., 34 (2000) 1255–1261.
[19] A. Jain, K.P. Raven and R.H. Loeppert, Arsenite andarsenate adsorption on ferrihydrite: surface chargereduction and net OH- release stoichiometry, Envir.Sci. Technol., 33 (1999) 1179–1184.
[20] U. Schwertmann and R.M. Cornell, Iron oxides inthe laboratory — preparation and characterization,2nd ed., Wiley-VCH, Weinheim, Germany, 2000.
[21] S. Fendorf, M.J. Eick, P. Grossl and D.L. Sparks,Arsenate and chromate retention mechanisms ongoethite .1. Surface structure, Envir. Sci. Technol.,31 (1997) 315–320.
[22] D.G. Lumsdon, A.R. Fraser, J.D. Russell and N.T.Livesey, New infrared band assignments for the arse-nate ion adsorbed on synthetic goethite ("-FeOOH),J. Soil. Sci., 35 (1984) 381–386.
[23] J.A. Wilkie and J.G. Hering, Adsorption of arseniconto hydrous ferric oxide: effects of adsorbate/adsorbent ratios and co-occurring solutes, ColloidSurf. A, 107 (1996) 97–110.
[24] M. Edwards, S. Patel, L. McNeill, H.W. Chen, M.Frey, A.D. Eaton, R.C. Antweiler and H.E. Taylor,Considerations in As analysis and speciation, J.AWWA, 90 (1998) 103–113.
[25] B. Han, T. Runnells, J. Zimbron and S.R. Wickrama-singhe, Arsenic removal from drinking water byflocculation and microfiltration, Desalination, 145(2002) 293–298.
[26] J.D. Chwirka, B.M. Thomson and J.M. Stomp,Removing arsenic from groundwater, J. AWWA, 92(2000) 79–88.
[27] J.G. Hering, P.Y. Chen, J.A. Wilkie, M. Elimelechand S. Liang, Arsenic removal by ferric chloride, J.AWWA, 88 (1996) 155–167.
[28] A.K.M.A. Quader, Personal communication, Bangla-desh University of Engineering and Technology,Dhaka, Bangladesh, 2001.
Copyright © 2022 FDOKUMEN




















