
Archaeal Binding Protein-Dependent ABC Transporter: Molecular and Biochemical Analysis of the...
10
JOURNAL OF BACTERIOLOGY, 0021-9193/98/$04.0010 Feb. 1998, p. 680–689 Vol. 180, No. 3 Copyright © 1998, American Society for Microbiology Archaeal Binding Protein-Dependent ABC Transporter: Molecular and Biochemical Analysis of the Trehalose/Maltose Transport System of the Hyperthermophilic Archaeon Thermococcus litoralis REINHOLD HORLACHER, 1 KARINA B. XAVIER, 2 HELENA SANTOS, 2 JOCELYNE DIRUGGIERO, 3 MARINA KOSSMANN, 1 AND WINFRIED BOOS 1 * Department of Biology, University of Konstanz, D-78457 Konstanz, Germany 1 ; Instituto de Tecnologia Quı ´mica e Biolo ´gica UNL, 2780 Oeiras, Portugal 2 ; and Center of Marine Biotechnology, University of Maryland, Baltimore, Maryland 21202 3 Received 28 August 1997/Accepted 18 November 1997 We report the cloning and sequencing of a gene cluster encoding a maltose/trehalose transport system of the hyperthermophilic archaeon Thermococcus litoralis that is homologous to the malEFG cluster encoding the Escherichia coli maltose transport system. The deduced amino acid sequence of the malE product, the trehalose/ maltose-binding protein (TMBP), shows at its N terminus a signal sequence typical for bacterial secreted proteins containing a glyceride lipid modification at the N-terminal cysteine. The T. litoralis malE gene was expressed in E. coli under control of an inducible promoter with and without its natural signal sequence. In addition, in one construct the endogenous signal sequence was replaced by the E. coli MalE signal sequence. The secreted, soluble recombinant protein was analyzed for its binding activity towards trehalose and maltose. The protein bound both sugars at 85°C with a K d of 0.16 mM. Antibodies raised against the recombinant soluble TMBP recognized the detergent-soluble TMBP isolated from T. litoralis membranes as well as the products from all other DNA constructs expressed in E. coli. Transmembrane segments 1 and 2 as well as the N-terminal portion of the large periplasmic loop of the E. coli MalF protein are missing in the T. litoralis MalF. MalG is homologous throughout the entire sequence, including the six transmembrane segments. The conserved EAA loop is present in both proteins. The strong homology found between the components of this archaeal transport system and the bacterial systems is evidence for the evolutionary conservation of the binding protein-dependent ABC transport systems in these two phylogenetic branches. High-affinity binding protein-dependent ABC transporters were originally discovered in gram-negative bacteria. They consist of a high-affinity substrate-binding protein located in the periplasmic space as their major substrate recognition site, two hydrophobic membrane proteins forming the translocation pore, and two additional subunits peripherally associated with the membrane proteins at the inner face of the membrane. By ATP hydrolysis the last two subunits provide the energy for the accumulation of substrate against the concentration gradient (7). In the case of the Escherichia coli maltose/maltodextrin transport system, the periplasmic binding protein (maltose- binding protein or MalE) is encoded by malE, the membrane components MalF and MalG are encoded by the malF and malG genes, and the two ATP-hydrolyzing subunits of MalK are encoded by malK. These genes form a cluster on the E. coli chromosome in which malE, malF, and malG constitute an operon that is oriented divergently to malK (8). Recently, it has been recognized that binding protein-dependent ABC trans- porters are also present in gram-positive bacteria (20). In these cases, the soluble periplasmic binding proteins are anchored in the membrane by an N-terminal lipid modification consisting of a diglyceride connected to the N-terminal cysteine via a thioether bond (51). Binding protein-dependent ABC trans- porters have also been found in thermophilic bacteria (25, 41). Despite the large amount of information available on this type of transport system in bacteria, only one study of an archaeal ABC system, that of the hyperthermophile Thermococcus lito- ralis, has been reported so far (52). This transport system has several unusual properties: it shows an extremely high affinity (K m of about 20 nM) at 85°C, the optimum growth tempera- ture of this organism; it recognizes with equal affinity its very different substrates, maltose and trehalose; and it is not inhib- ited by maltodextrins. We undertook to further characterize this newly discovered transport system. Here we report on the purification of the native trehalose/maltose-binding protein (TMBP), the cloning and sequencing of the malEFG gene cluster, and the expression of the malE gene in E. coli as well as the purification and characterization of its encoded binding protein. The rationale for analyzing a binding protein-depen- dent transport system from a hyperthermophilic organism whose function is optimal at 85°C but is less than 5% at room temperature is the expectation that is conformation will be more rigid at room temperature and will become accessible to structural analysis under these conditions. In addition, evolu- tionary aspects and its unusual substrate specificity make it attractive for study. MATERIALS AND METHODS Cloning and sequencing. A DNA clone from Pyrococcus furiosus was se- quenced and shown to have high homology to the malG gene from Mycobacte- rium leprae by BLASTX analysis (9). PCR primers for the gene were designed from the DNA sequence and were used to amplify a 500-bp malG fragment from P. furiosus genomic DNA. A Lambda Zap mixed partial EcoRI genomic library of T. litoralis was screened by using this PCR fragment, which was labeled with [a- 32 P]dATP by random priming. Several positive plaques were rescued into the pBluescript KS 1 plasmid (Stratagene, La Jolla, Calif.) and were purified with cesium chloride gradients (2). The positive clones were sequenced by the dideoxy chain termination method with primer-walking methodology (2). Computer * Corresponding author. Mailing address: Department of Biology, University of Konstanz, D-78457 Konstanz, Germany. Phone: 49 7531- 88-2658. Fax: 49 7531-88-3356. E-mail: Winfried.Boos@uni-konstanz .de. 680 on January 18, 2015 by guest http://jb.asm.org/ Downloaded from
-
Upload
uni-konstanz -
Category
Documents
-
view
2 -
download
0
Transcript of Archaeal Binding Protein-Dependent ABC Transporter: Molecular and Biochemical Analysis of the...

JOURNAL OF BACTERIOLOGY,0021-9193/98/$04.0010
Feb. 1998, p. 680–689 Vol. 180, No. 3
Copyright © 1998, American Society for Microbiology
Archaeal Binding Protein-Dependent ABC Transporter: Molecular andBiochemical Analysis of the Trehalose/Maltose Transport System
of the Hyperthermophilic Archaeon Thermococcus litoralisREINHOLD HORLACHER,1 KARINA B. XAVIER,2 HELENA SANTOS,2 JOCELYNE DIRUGGIERO,3
MARINA KOSSMANN,1 AND WINFRIED BOOS1*
Department of Biology, University of Konstanz, D-78457 Konstanz, Germany1; Instituto de TecnologiaQuımica e Biologica UNL, 2780 Oeiras, Portugal2; and Center of Marine Biotechnology,
University of Maryland, Baltimore, Maryland 212023
Received 28 August 1997/Accepted 18 November 1997
We report the cloning and sequencing of a gene cluster encoding a maltose/trehalose transport system of thehyperthermophilic archaeon Thermococcus litoralis that is homologous to the malEFG cluster encoding theEscherichia coli maltose transport system. The deduced amino acid sequence of the malE product, the trehalose/maltose-binding protein (TMBP), shows at its N terminus a signal sequence typical for bacterial secretedproteins containing a glyceride lipid modification at the N-terminal cysteine. The T. litoralis malE gene wasexpressed in E. coli under control of an inducible promoter with and without its natural signal sequence. Inaddition, in one construct the endogenous signal sequence was replaced by the E. coli MalE signal sequence.The secreted, soluble recombinant protein was analyzed for its binding activity towards trehalose and maltose.The protein bound both sugars at 85°C with a Kd of 0.16 mM. Antibodies raised against the recombinant solubleTMBP recognized the detergent-soluble TMBP isolated from T. litoralis membranes as well as the productsfrom all other DNA constructs expressed in E. coli. Transmembrane segments 1 and 2 as well as the N-terminalportion of the large periplasmic loop of the E. coli MalF protein are missing in the T. litoralis MalF. MalG ishomologous throughout the entire sequence, including the six transmembrane segments. The conserved EAAloop is present in both proteins. The strong homology found between the components of this archaeal transportsystem and the bacterial systems is evidence for the evolutionary conservation of the binding protein-dependentABC transport systems in these two phylogenetic branches.
High-affinity binding protein-dependent ABC transporterswere originally discovered in gram-negative bacteria. Theyconsist of a high-affinity substrate-binding protein located inthe periplasmic space as their major substrate recognition site,two hydrophobic membrane proteins forming the translocationpore, and two additional subunits peripherally associated withthe membrane proteins at the inner face of the membrane. ByATP hydrolysis the last two subunits provide the energy for theaccumulation of substrate against the concentration gradient(7). In the case of the Escherichia coli maltose/maltodextrintransport system, the periplasmic binding protein (maltose-binding protein or MalE) is encoded by malE, the membranecomponents MalF and MalG are encoded by the malF andmalG genes, and the two ATP-hydrolyzing subunits of MalKare encoded by malK. These genes form a cluster on the E. colichromosome in which malE, malF, and malG constitute anoperon that is oriented divergently to malK (8). Recently, it hasbeen recognized that binding protein-dependent ABC trans-porters are also present in gram-positive bacteria (20). In thesecases, the soluble periplasmic binding proteins are anchored inthe membrane by an N-terminal lipid modification consistingof a diglyceride connected to the N-terminal cysteine via athioether bond (51). Binding protein-dependent ABC trans-porters have also been found in thermophilic bacteria (25, 41).Despite the large amount of information available on this typeof transport system in bacteria, only one study of an archaeal
ABC system, that of the hyperthermophile Thermococcus lito-ralis, has been reported so far (52). This transport system hasseveral unusual properties: it shows an extremely high affinity(Km of about 20 nM) at 85°C, the optimum growth tempera-ture of this organism; it recognizes with equal affinity its verydifferent substrates, maltose and trehalose; and it is not inhib-ited by maltodextrins. We undertook to further characterizethis newly discovered transport system. Here we report on thepurification of the native trehalose/maltose-binding protein(TMBP), the cloning and sequencing of the malEFG genecluster, and the expression of the malE gene in E. coli as wellas the purification and characterization of its encoded bindingprotein. The rationale for analyzing a binding protein-depen-dent transport system from a hyperthermophilic organismwhose function is optimal at 85°C but is less than 5% at roomtemperature is the expectation that is conformation will bemore rigid at room temperature and will become accessible tostructural analysis under these conditions. In addition, evolu-tionary aspects and its unusual substrate specificity make itattractive for study.
MATERIALS AND METHODS
Cloning and sequencing. A DNA clone from Pyrococcus furiosus was se-quenced and shown to have high homology to the malG gene from Mycobacte-rium leprae by BLASTX analysis (9). PCR primers for the gene were designedfrom the DNA sequence and were used to amplify a 500-bp malG fragment fromP. furiosus genomic DNA. A Lambda Zap mixed partial EcoRI genomic libraryof T. litoralis was screened by using this PCR fragment, which was labeled with[a-32P]dATP by random priming. Several positive plaques were rescued into thepBluescript KS1 plasmid (Stratagene, La Jolla, Calif.) and were purified withcesium chloride gradients (2). The positive clones were sequenced by the dideoxychain termination method with primer-walking methodology (2). Computer
* Corresponding author. Mailing address: Department of Biology,University of Konstanz, D-78457 Konstanz, Germany. Phone: 49 7531-88-2658. Fax: 49 7531-88-3356. E-mail: [email protected].
680
on January 18, 2015 by guesthttp://jb.asm
.org/D
ownloaded from

analysis of the DNA sequences was done with programs of the Wisconsin Pack-age, version 9.0 (Genetics Computer Group, Madison, Wis.) (15).
Organism and growth conditions. T. litoralis DSM5473 was obtained from theDeutsche Sammlung von Mikroorganismen und Zellkultur GmbH (Braun-schweig, Germany). Cells were cultured as previously described (52) with yeastextract (inducing conditions) and peptone as carbon sources. At the end of theexponential phase and at an optical density at 600 nm of 0.4, cells were harvestedby centrifugation (5,000 3 g for 15 min at 27°C) and washed once with a solutionof the same composition as the growth medium (pH 6.5) but without an addedcarbon source. The cells were then frozen and stored at 270°C until used.
Purification of TMBP from membranes of T. litoralis. Solubilized membraneextracts from T. litoralis cells were prepared as reported previously (52). Aftercells were harvested, all manipulations were done under aerobic conditions. Thecell paste was mixed with an equal volume of 50 mM Tris-HCl (pH 7.5) con-taining 1 mM MgCl2 and homogenized, and a small amount of DNase I wasadded. Ten-milliliter aliquots of the cell suspension were passed through aFrench pressure cell at 16,000 lb/in2 to break the cells. The suspension (10 ml)was clarified by centrifugation at 8,000 3 g for 20 min at 4°C. The supernatantwas then centrifuged at 100,000 3 g for 1 h at 4°C. The pellet was washed twicewith 10 ml of 50 mM Tris-HCl (pH 7.5) and resuspended in 10 ml of the samebuffer containing 1% octyl-b-glucoside. This suspension was stirred for 1 h at4°C. Insoluble material was removed by centrifugation (100,000 3 g for 1 h).Typically, the detergent was added to a solution with a protein concentration of2 to 4 mg/ml, and a clear solution containing 75% of the initial protein wasobtained. One hundred microliters of 10% dodecyl-b-maltoside was added, andthe solution was dialyzed twice against 3 liters of 50 mM Tris-HCl (pH 7.5)containing 0.01% dodecyl-b-maltoside. The solution was then applied to anHR5/5 MonoQ anion-exchange column (Pharmacia) that had been equilibratedwith 50 mM Tris-HCl (pH 7.5) containing 0.01% dodecyl-b-maltoside. Thecolumn was washed with the same buffer until the eluate (15 ml) was protein free,and then 12.5 ml of 50 mM Tris-HCl (pH 7.5) containing 0.6% lauryldimethyl-amine oxide (LDAO) was added. Fractions were assayed at 85°C for binding to[14C]trehalose or [14C]maltose by using binding assays with saturated ammoniumsulfate as described previously (52). Protein fractions were routinely analyzed bysodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as de-scribed previously (29). The TMBP was eluted under these conditions in onepeak without the application of a salt gradient. The fractions showing highbinding activity were pooled and used for SDS-PAGE analysis as well as fluo-rescence spectroscopy.
Fluorescence spectroscopy. All spectra were obtained at 55°C with an SPEXFluorolog 2002 spectrofluorometer equipped with a thermostated cuvette. Spec-tra were recorded at an excitation wavelength of 280 nm and an emissionwavelength of 328 nm. Emission scans were done from 280 to 400 nm. Measure-ments were done with 600 ml of 50 mM Tris-HCl (pH 7.5) containing 0.01%dodecyl-b-maltoside. Three microliters of the protein solution (0.2 mg/ml) wasadded to the prewarmed buffer, and the solution was mixed by inversion. Thesubstrate was added in 3-ml samples of concentrated solutions. The fluorescenceintensity was measured after 10 min of incubation to reach the desired temper-ature. The temperature was constant within 60.2°C.
Construction of plasmids. Chromosomal DNA from T. litoralis was isolated asdescribed previously (2). To replace the N-terminal part of TMBP with the signalsequence of E. coli MalE, PCR was performed with the isolated T. litoralischromosomal DNA and primers introducing a DraI site at the fusion point (Fig.1A) and a HindIII site after the coding sequence of malE. The E. coli part of therecombinant sequence (corresponding to the N terminus of the recombinantprotein) was amplified by PCR with plasmid pmalP2 (New England Biolabs) asthe template and primers introducing a StuI site at the fusion point (Fig. 1A) andan MluI site upstream of the promoter sequence. After digestion of the PCRfragments with the corresponding enzymes, they were ligated in one step intopmalP2 opened with MluI and HindIII, yielding plasmid pRHo1000. To clonethe entire T. litoralis malE gene (Fig. 1C), the gene was amplified from chromo-somal DNA by PCR with primers introducing a BspHI site at the start codon andan SphI site downstream of the stop codon. After digestion with both restrictionenzymes, the fragment was ligated into pJLA502 (43) opened with NcoI andSphI, yielding plasmid pRHo1001. The gene fusion with the signal sequencetruncated (Fig. 1B) was constructed by PCR with pRHo1000 as the template andthe following two primers: the 59 primer changed AAA (encoding K [Lys]) toATG (encoding M [Met]) and introduced a BspHI site; the 39 primer introducedan SphI site after the stop codon of malE. The PCR fragment was digested withthe corresponding enzymes and ligated into pJLA502 previously digested withNcoI and SphI, yielding plasmid pRHo1002. After all cloning steps for the PCRproducts, the correctness of the sequence was confirmed by sequencing thecloned fragment.
Purification of the recombinant protein. E. coli SF120 (3) was transformedwith plasmid pRHo1000 (Fig. 1A) and cultivated at 28°C in 10 liters of NZAmedium (10 g of NZ-amine A [Sheffield Products, Inc.], 5 g of yeast extract, and5 g of NaCl per liter) containing 200 mg of ampicillin per ml. After an opticaldensity at 600 nm of 0.7 was reached, the temperature was increased to 37°C andexpression of the hybrid malE gene was induced by adding IPTG (isopropyl-b-D-thiogalactopyranoside) to a final concentration of 100 mM. After 3 h of incu-bation, the culture was harvested and cold osmotic shock was performed by astandard protocol (34). The lyophilized periplasmic proteins were resuspendedin 4 ml of 30 mM Tris-HCl (pH 7.5) and dialyzed against the same buffer.Heat-labile E. coli proteins were denatured by heating the solution for 10 min to80°C. After centrifugation for 10 min at 18,000 3 g, aliquots of the clarifiedprotein solution were applied to a MonoQ column and eluted with a lineargradient of 0 to 1 M NaCl in 30 mM Tris-HCl (pH 7.5). Fractions containingpurified recombinant hybrid TMBP were pooled and stored at 220°C. Routinely,about 3 mg of periplasmic hybrid protein was obtained from 10 liters of culture.
For purification of the cytoplasmic form of the hybrid TMBP (Fig. 1A), thecells collected after cold osmotic shock were ruptured in a French pressure cellat 16,000 lb/in2 and centrifuged for 30 min at 35,000 3 g. Again, the heat-labileproteins were removed by heating the solution for 10 min to 80°C. After cen-trifugation (10 min at 18,000 3 g), the protein appeared to be homogeneous onSDS-PAGE (see Fig. 7B). The recombinant protein showed a tendency to ag-gregate, which was most pronounced in samples kept at 4°C. Even though theprotein appeared to be homogeneous on SDS-PAGE, the solution showed alarge absorption at 260 nm. After treatment with DNase I and RNase followed
FIG. 1. Constructs for the expression of malE from T. litoralis in E. coli. (A) pRHo1000. The cleavable signal sequence of E. coli MalE replaced the lipid anchorsequence of TMBP. Arrowhead 2 indicates where E. coli MalE is cleaved during secretion; arrowhead 1 shows the N terminus (determined by amino acid sequencing)of the periplasmic hybrid protein expressed in E. coli. The shaded sequence is that of the hybrid protein. (B) pRHo1002. The sequence of the hybrid protein from panelA has been shortened; its N terminus now is after the expected E. coli cleavage site SASALA. The N-terminal lysine (K) has been changed to methionine (M). Theprotein was soluble and remained in the cytoplasm. The shaded sequence is that of the hybrid protein. (C) pRHo1001. The intact malE gene of T. litoralis, includingits lipid anchor-encoding sequence, was cloned after the inducible E. coli promoter. The protein expressed in E. coli (shaded sequence) was lipid modified and tightlymembrane bound. The putative site of lipidation and its mature N terminus (cysteine) is indicated by arrowhead 3. Asterisks indicate identity; dots indicate homologousexchanges. Ec, E. coli; Tl, T. litoralis.
VOL. 180, 1998 T. LITORALIS TREHALOSE/MALTOSE TRANSPORT SYSTEM 681
on January 18, 2015 by guesthttp://jb.asm
.org/D
ownloaded from

by dialysis and DEAE chromatography, the protein was free of material absorb-ing at 260 nm. Simultaneously, it had lost the tendency to aggregate.
Preparation of antibodies and Western blot analysis. The recombinant hybridTMBP (Fig. 1A) was separated from other proteins by SDS-PAGE (29) andeluted from the gel as described previously (23). A chicken was immunized fivetimes with 50 mg of protein each. At 14 days after the last immunization,antibodies were prepared from 10 eggs (37). Western blot analysis was done asdescribed previously (23, 50), using the primary antibody (17 mg/ml) in a dilutionof 1:10,000.
Determination of the binding affinity (Kd). To measure the Kd of binding, amethod based on the retention of ligand by the binding proteins (1, 46) was used.A small dialysis tubing (Medicell International, Ltd., London, United Kingdom)(diameter, 1 cm) open on one end was tightly fit with its open end onto a bluntlycut plastic pipetting tip of a 1-ml automatic pipette. Four hundred microliters ofpure binding protein solution (0.41 mM in 50 mM Tris-HCl, pH 7.0) was intro-duced into the dialysis tubing and immersed in a 2-liter Erlenmeyer flask filledwith 50 mM Tris-HCl, pH 7.0. A final concentration of 135 nM [14C]maltose (630mCi/mmol) or 303 nM [14C]trehalose (500 mCi/mmol) (26) was added in 20 ml.The buffer in the Erlenmeyer flask was kept at 85°C and gently stirred during theassay. Aliquots of 20 ml were removed from the bag at different time intervals,and the radioactivity in 6 ml of toluene-based scintillation fluid was counted. Thesame procedure was repeated for both substrates but in the absence of protein.The time to release half of the substrate from the dialysis bag is greater by afactor of 1 1 (P/Kd) in the presence of molar concentrations of binding proteinP (assuming one binding site) than in its absence. Kd is obtained in molarconcentration.
Detection of binding activity in nondenaturing polyacrylamide gels. SDS-freegels (12% polyacrylamide) were prepared as described previously (53) exceptthat no pyridoxal phosphate was present in the gel solution and no activity stainwas added to the gel. Prior to electrophoresis 1 to 2 ml of [14C]trehalose (500mCi/mmol; about 30,000 cpm) (26) was added to the individual protein samplesand incubated for 5 min at 85°C. After electrophoresis at room temperature, thegel pockets were carefully rinsed, and the gel was dried and subjected to auto-radiography.
Nucleotide sequence accession number. The sequences reported in this paperhave been deposited in the GenBank database and assigned accession no.AF012836.
RESULTS
Purification of TMBP from membranes of T. litoralis. Mem-branes from trehalose-induced T. litoralis cells (with yeast ex-tract in the growth medium) were isolated by disruption in aFrench pressure cell and solubilized in 1% octyl-b-glucoside ata ratio of 0.2 to 0.4 mg of protein per mg of detergent. Initialattempts to purify the protein by affinity chromatographythrough an amylose column were unsuccessful, since the pro-tein failed to bind to this material. Therefore, we used anion-exchange chromatography. In the first attempts, the membraneextract (100 mg of total protein) in 1% octyl glucoside wasapplied to a Source 30Q anion-exchange column after equili-brating the column with 50 mM Tris-HCl (pH 7.5) and 0.8%octyl-b-glucoside. Under these conditions, more than 75% ofthe binding protein did not adsorb to the column and waseluted even before the application of an NaCl gradient. In-creasing the pH and reducing the ionic strength did not im-prove the adsorption properties. The protein remaining on thecolumn in some experiments could be eluted by a salt gradient(0 to 500 mM) and appeared at 150 mM NaCl but was not freeof contaminants. Adsorption of the protein on the column wasgreatly improved by replacing octyl-b-glucoside (after solubi-lization of the proteins from the membrane in this detergent)with dodecyl-b-maltoside, yet the protein could not be elutedfrom the column with a 0 to 500 mM NaCl gradient. We found,however, that a major portion of the protein eluted as a ratherhomogeneous preparation when the column was washed with50 mM Tris-HCl (pH 7.5) containing 0.6% of the detergentLDAO, even in the absence of a salt gradient. The proteinsample eluted in this way was analyzed by SDS-PAGE (Fig. 2,lane 3); its apparent molecular weight was 47,000. The proteinwas highly active as judged by a binding test involving precip-itation with ice-cold saturated ammonium sulfate of a sampleheated to 85°C in the presence of [14C]trehalose or [14C]mal-
tose, followed by filtration and measurement of the radioac-tivity of the filter (52). This test cannot be used to determinethe binding characteristics of the protein quantitatively eventhough it is very useful in qualitative assays during purification.Also, equilibrium dialysis at 85°C in the presence of detergentscould not be done, since the detergents precipitated at thistemperature and blocked the diffusion pores of the dialysistubing. In addition, we found that dodecyl-b-maltoside, thedetergent that allowed adsorption of the protein to the ion-exchange column, acted as a substrate, interfering with thebinding assay.
We used the change in the intrinsic fluorescence of theprotein upon binding the substrate as a method to monitor apossible conformational change resulting from substrate bind-ing. At 55°C the detergents did not precipitate, allowing thereading of the fluorescence spectra (excitation at 280 nm).Figure 3 shows that the emission decreased in the presence oftrehalose and increased in the presence of maltose. An excessof one substrate competes with the effect of the other. Thisdemonstrates that substrate binding elicits a conformationalchange of the protein and that the protein binds maltose andtrehalose in different conformations. Using different trehaloseconcentrations, we found that the half-maximal fluorescencechange occurred at concentrations of above 5 mM. From theKm of transport in intact cells, which is around 20 nM, we hadexpected a much better Kd for binding. Possibly, the presenceof dodecyl maltoside, which cannot be removed by dialysis,interferes by competitive binding. Therefore, binding assayshad to be carried out with a soluble derivative of the bindingprotein with which dodecyl-b-maltoside would not interfere.
Cloning and sequencing of the maltose/trehalose operon ofT. litoralis. A fragment of the P. furiosus malG gene has beenpreviously identified by random sequencing of a genomic DNAclone (9). A PCR product derived from this clone was used toscreen a lambda Zap library of T. litoralis. Several clones wereidentified by plaque hybridization and rescued into pBluescriptKS1. Sequencing of two of these clones, pJDR1.12 andpJDR6.22, gave the complete sequences of the T. litoralismalE, -F, and -G genes and their flanking regions. Part of thissequence, including the promoter region and the intergenicregions between malE and malF as well as between malF andmalG, is shown in Fig. 4. The sequence of malE consists of
FIG. 2. SDS-PAGE of the native binding protein as isolated from the mem-brane of T. litoralis. Lanes: 1, membrane preparation from the uninduced strain(no yeast extract in the medium); 2, membrane preparation from the inducedstrain (with yeast extract in the medium); 3, purified protein containing the lipidanchor; 4, water-soluble hybrid protein (the construct carries the cleavable signalsequence of E. coli at its N terminus) isolated from the E. coli periplasm (as areference); St, molecular mass standards (from top to bottom: 66, 45, 36, 29, 24,20.1, and 14.2 kDa).
682 HORLACHER ET AL. J. BACTERIOL.
on January 18, 2015 by guesthttp://jb.asm
.org/D
ownloaded from

1,353 nucleotides; hence, the putative binding protein is com-posed of 450 residues, i.e., is slightly larger than the E. coliMalE protein, which contains 396 residues. The deduced TMBPamino acid sequence shows 28 and 26% identity with the mal-tose-binding protein sequences of E. coli (16) and Streptococ-cus pneumoniae (39), respectively, and 24% identity with the
maltodextrin-binding protein of Thermoanaerobacterium ther-mosulfurigenes (41). The T. litoralis sequence is also 31% iden-tical with the product of a gene from M. leprae which is locatedupstream of malF on the chromosome of this organism (Gen-Bank accession no. U1756V). In addition, the TMBP aminoacid sequence showed high homology with a family of periplas-mic binding proteins, named cluster 1 binding proteins, formalto-oligosaccharides, multiple sugars, sn-glycerol-3-phos-phate, and iron transport systems (Fig. 5) (48). This shortsegment of sequence was derived from the alignment of theeight proteins constituting cluster 1 binding proteins and isspecific for this family of proteins (48). In gram-negative bac-teria, the binding proteins are located in the periplasm be-tween the inner and outer membranes and are water soluble.In gram-positive bacteria, which lack an outer membrane,binding proteins are soluble lipoproteins with an N-terminalglyceride-cysteine (51) which allows the anchorage of the bind-ing proteins to the external surface of the cell membrane.Archaeal membrane lipids are based on isopranyl glycerolethers (di- and tetraethers), and in hyperthermophiles themembrane is organized as a monolayer of lipids (27) with anS-layer on the outside (36). This is a situation similar to that ofgram-positive bacteria with the absence of a restricting outermembrane. We found that the first 27 amino acids of theputative T. litoralis TMBP show characteristics typical of signalpeptides of secretory precursor proteins: (i) a hydrophobiccore adjacent to the N terminus and (ii) the sequence VASGCIG corresponding to the consensus of lipoprotein signal pep-tidase cleavage sites (LAAGCSS) (48).
Sequences of the inner membrane proteins. In E. coli, MalFand MalG are hydrophobic inner membrane components me-diating the energy-dependent translocation of substrate intothe cytoplasm. Two T. litoralis genes for inner membrane com-
FIG. 3. Fluorescence changes of TMBP in the presence of trehalose andmaltose. The recordings were done consecutively. First, the middle trace wasobtained after temperature equilibration without addition of substrate. Theupper trace was then recorded after the addition of 1 mM (final concentration)maltose, followed by the lower trace after the addition of 100 mM trehalose.Similar tracings in the reverse order were obtained when the order of theadditions was reversed, i.e., first 1 mM trehalose and then 100 mM maltose (datanot shown). Conditions were as follows: temperature, 55°C; excitation, 280 nm;TMBP concentration, 16 mg/ml; buffer, 50 mM Tris-HCl (pH 7.5) containing0.6% LDAO.
FIG. 4. Partial nucleotide sequences of the T. litoralis malE, -F, and -G genes and flanking regions. The deduced amino acid sequences are given in one-letter codebelow the nucleotide sequences, and the nucleotide and amino acid numbers are given on the right. 300/13 indicates the last amino acid of the malF gene product (no.300) and last amino acid of MalG in this line (no. 13). In the nucleotide sequence, a putative boxA promoter element is boxed, the putative ribosome binding site isin boldface, and a putative terminator sequence is underlined. In the deduced amino acid sequence of TMBP (the malE gene product), the conserved recognitionsequence for the cleavage sites of lipoprotein signal peptidases is underlined and the presumably formed N-terminal cysteine of the mature protein is in boldface andmarked with an arrowhead. Most of the sequence within the structural genes is omitted, as indicated by points and deletion signs. The complete sequence is depositedin GenBank under accession no. AF012836.
VOL. 180, 1998 T. LITORALIS TREHALOSE/MALTOSE TRANSPORT SYSTEM 683
on January 18, 2015 by guesthttp://jb.asm
.org/D
ownloaded from

ponents have been sequenced, i.e., malF and malG, consistingof 903 and 837 bp, respectively. The deduced amino acid se-quences of the corresponding proteins are homologous to theMalF and MalG protein sequences of gram-positive and gram-negative bacteria (Table 1). Both the T. litoralis MalF andMalG proteins contain the sequence EAAX2DGAX8IXLP,which is homologous to the consensus sequence EAAX2L/DGAX8IXLP found in membrane components of ABC transport-ers (7, 42). In this conserved region, the amino acids are pre-dominantly hydrophobic, with a consistent location at the Cterminus. The exact function of the EAA loop is not yet clear;it might be involved in binding the ATP-hydrolyzing subunit ofthe transport system and thus be connected to the energytransduction process (33).
The topology of T. litoralis inner membrane protein MalG issimilar to that of E. coli MalG (11), with six membrane-span-ning segments (MSS) (Fig. 6B). However, the T. litoralis MalFprotein is missing MSS1 and MSS2 of E. coli MalF (10, 18),including the N-terminal portion of the large periplasmic loopthat is found in E. coli MalF between MSS3 and MSS4 (Fig.6A). T. litoralis MalF is composed of 300 amino acids, com-pared to 514 amino acids for the E. coli protein. The MSS ofthe T. litoralis proteins shown in Fig. 6 are based on computeranalysis. Preliminary experiments using malF-phoA fusions tothe N-terminal portion of MalF are in agreement with themodel shown in Fig. 6A (21), implying that both protein ter-mini are located in the cytoplasm. MalF homologs from bac-teria other than E. coli, for instance, from gram-positivebacteria, lack the same portion of the corresponding MalFprotein, in particular the large periplasmic loop. It seems as ifthis loop is a peculiarity of the maltose system in E. coli and itsvery close neighbors (12). The alignment in Fig. 6 shows noparticular enrichment of identity along the sequence except forthe EAA motif, which is common to all membrane componentsof binding protein-dependent ABC systems throughout thebacterial world.
Expression of T. litoralis malE in E. coli. In the first con-struct, pRHo1000 (Fig. 1A), the putative signal sequence ofthe T. litoralis malE gene was replaced with the signal sequenceof the E. coli malE gene contained in the IPTG-induciblevector pmalP2 (New England Biolabs). Upon induction withIPTG, the hybrid protein was expressed in E. coli. After ap-plication of the classical cold osmotic shock procedure of Neuand Heppel (34), less than 5% of this protein were recovered
in the periplasmic fraction (Fig. 7A, lanes 5 and 6); the bulkremained in the cytoplasm. The protein was purified from theperiplasmic fraction by heating to 80°C for 10 min, removingthe precipitate, and subjecting the supernatant to ion-exchangechromatography through a MonoQ column (Fig. 2, lane 4, and7A, lane 7). The protein thus obtained was used for measuringthe binding constant (see below).
When the cells harboring pRHo1000 were disrupted with aFrench pressure cell (after cold osmotic shock treatment),most of TMBP was found in the cellular extract. Heating theextract to 80°C for 10 min precipitated all other proteins andyielded homogeneous TMBP (Fig. 7A, lane 4). Thus, the con-taminating proteins of heat-treated periplasmic extracts (Fig.7A, lane 6) consisted of a few heat-resistant periplasmic E. coliproteins that were absent in heat-treated cytoplasmic extractsof cells from which the periplasmic proteins had been removedby the osmotic shock procedure (Fig. 7A, lane 4). In SDS-polyacrylamide gels, the periplasmic and the cytoplasmic formsof the protein were identical in size (45 kDa) and carriedthe same N-terminal amino acid sequence, SASALAKIEEGKIV. . ., indicating that they had not been processed at theexpected site during secretion. The expected cleavage site ob-served in E. coli MalE was after the third amino acid in theASA part of the sequence shown above (16). The hybrid pro-tein therefore lacks the first 20 amino acids of the maturenative protein as isolated from T. litoralis, which had beenreplaced by the E. coli N-terminal sequence SASALAKIEE.The yield of the periplasmic form of the protein was not in-creased when the construct was expressed in the prlA421 mu-tant WP794 (38), which allows the secretion in E. coli of MalEforms lacking a signal sequence (14). This may indicate that thefolding process of TMBP in E. coli at 37°C is aberrant andinterferes with the secretion process.
In the second construct, pRHo1002 (Fig. 1B), the DNA seg-ment encoding the E. coli signal sequence of the above-de-scribed hybrid protein was removed in order to express theprotein as a soluble cytoplasmic protein in E. coli. The ATGinitiation codon was positioned in such a way that the hybridprotein shown in Fig. 1A would begin directly after the pre-dicted cleavage site ALA. This construct, cloned in the tem-perature-inducible vector pJLA502 (43), formed soluble, ac-tive TMBP when expressed in E. coli. Heating crude extracts to80°C for 10 min resulted in considerable purification as judgedby SDS-PAGE analysis (Fig. 7B, lane 4). Heat treatment of thecell extracts again was more effective (and yielded homoge-neous protein) if, prior to disruption, the cells were subjectedto cold osmotic shock for the removal of heat-resistant peri-plasmic proteins (data not shown).
The cytoplasmic forms of the protein containing the E. colisignal sequence (Fig. 1A and 7A, lane 4) or lacking it (Fig. 1Band 7B, lane 4) both consisted of multimeric aggregates ofvarious compositions, containing up to five identical polypep-
FIG. 5. Alignment of the signature sequences of cluster 1 binding proteinswith TMBP of T. litoralis. The different amino acids possible for each position areindicated below the signature sequence. Numbers in parentheses indicate posi-tions. The highly conserved lysine residue (K) is boxed. Residues of T. litoralisthat match the signature sequence are in boldface, and residues conserved inother sequences are underlined. The signature sequence is from reference 48. Tl,T. litoralis; Ec, E. coli (16); St, Salmonella typhimurium (12); Sp, S. pneumoniae(39); Tt, T. thermosulfurigenes (41); MalE, MalX, and AmyE, maltose/maltodex-trin-binding proteins; UgpB (35), glycerol-3-phosphate-binding protein.
TABLE 1. Similarities and identities of inner membrane proteins ofbinding protein-dependent transport systems and the inner
membrane proteins of T. litoralis
T. litoralisprotein
% Similarity/% identity with homolog(s) ina:
M. leprae S. pneumoniae E. coli T. thermosulfurigenes
MalF 62/38 56/31 57/35, 57/33 59/26MalG 61/34 56/28 57/31, 56/29 56/27
a The homologs for MalF and MalG, respectively, are as follows: M. leprae,MalF and MalG; S. pneumoniae, MalC and MalD; E. coli, MalF, UgpA andMalG, UgpE; T. thermosulfurigenes, AmyD and AmyC.
684 HORLACHER ET AL. J. BACTERIOL.
on January 18, 2015 by guesthttp://jb.asm
.org/D
ownloaded from

tide chains. These aggregates could be seen clearly when ana-lyzed in nondenaturing polyacrylamide gels. When 14C-labeledtrehalose was added (and incubated at 80°C for 10 min) priorto electrophoresis, the label remained bound to the differentaggregates (Fig. 8, lane 1), demonstrating their binding activ-ity. We observed that the tendency to aggregate was associatedwith the presence of nucleic acid in the heat-treated sample.Treatment with DNase I and RNase followed by extensivedialysis and anion-exchange chromatography removed the ma-terial absorbing at 260 nm and the tendency to form multi-meric forms.
In the last construct (Fig. 1C), the entire open reading frameof the T. litoralis malE gene was cloned behind the tempera-ture-inducible promoter of plasmid pJLA502 (43). This con-struct (pRHo1001) was also expressed in E. coli and produceda membrane-bound protein. After solubilization in 1% octyl-b-glucoside, it was binding active. When tested in SDS-PAGE,the protein’s apparent molecular weight was similar to that ofthe protein which had been isolated from the membranes of T.
litoralis, but it formed a double band on these gels (Fig. 7C,lane 2), indicating heterogeneous lipid modification when ex-pressed in the heterologous host. Separation in nondenaturinggels supported this conclusion. On these gels the two formswere widely separated, but both were binding active (Fig. 8,lane 3).
All recombinant TMBPs produced by the different DNAconstructs as well as the protein contained in T. litoralis mem-branes cross-reacted by Western blot analysis (Fig. 7D) withantibodies that were raised in chicken against the cytoplasmicform of the recombinant TMBP containing the E. coli MalEsignal sequence (Fig. 1A). The most important point is thatthese antibodies recognize a trehalose-inducible protein in themembranes of T. litoralis (Fig. 7D, lanes 4 and 5) and thepurified recombinant TMBP proteins produced from all T. lito-ralis malE-harboring constructs. Thus, it is clear that the genemalE encodes the protein purified from the T. litoralis mem-branes.
The hybrid protein carrying the cleavable signal sequence
FIG. 6. Putative MSS in inner membrane proteins. (A) T. litoralis and E. coli MalF proteins. In contrast to E. coli MalF, the T. litoralis protein has only six putativeMSS (there are eight in E. coli MalF) and no large periplasmic loop between MSS3 and MSS4. MSS1 and MSS2 from E. coli MalF are missing in T. litoralis MalF.Since the numbering of the MSS corresponds to the E. coli sequence, the most N-terminal MSS of TMBP is termed MSS3. (B) T. litoralis and E. coli MalG proteins.The positions of the MSS appear to be conserved; the numbering is identical to that of the E. coli sequence. Tl, T. litoralis; Ec, E. coli. Asterisks indicate identicalresidues; dots indicate homologous residues.
VOL. 180, 1998 T. LITORALIS TREHALOSE/MALTOSE TRANSPORT SYSTEM 685
on January 18, 2015 by guesthttp://jb.asm
.org/D
ownloaded from

from E. coli was clearly synthesized as a precursor (Fig. 7D,lanes 1 and 6) which was no longer seen after preparation of acellular extract (Fig. 7D, lane 7). Thus, the cytoplasmicallylocalized precursor in E. coli apparently gained access to thesignal sequence peptidase and matured mainly after rupturingthe cells. The TMBP antiserum was raised in chicken andcontained antibodies against a few E. coli proteins but notagainst any protein from T. litoralis other than TMBP. Theantibody did not recognize E. coli MalE. Likewise, antibodiesagainst E. coli MalE did not recognize TMBP (data notshown).
Binding affinity of the periplasmic form of TMBP. Of theseveral possible ways to measure the Kd of binding, only theuse of the retention behavior (1, 46) is insensitive to the pres-ence of bound unlabeled substrate. Since the assay could notbe done with the native protein isolated from the membranesof T. litoralis (see above), we used the recombinant proteinfrom a DNA construct in which the natural signal sequencewas replaced by the E. coli MalE signal sequence. The proteinisolated from the periplasmic fraction was monomeric in solu-tion (Fig. 8, lane 2). At a concentration of 0.2 mg/ml, it was
dialyzed against 2 liters of Tris-HCl (pH 7.0) at 85°C. Smallamounts of 14C-labeled trehalose or 14C-labeled maltose wereadded into the dialysis tubing, and the rate of substrate exit wasmeasured over time. By comparing the rates of substrate exit inthe presence and absence of binding protein (Fig. 9) and withthe knowledge of the amount of binding protein present (as-suming one binding site), the Kd of binding can be calculated.It was determined to be 160 nM for trehalose and maltose.
DISCUSSION
The organization of the T. litoralis trehalose/maltose trans-port operon (Fig. 4) is very similar to that of E. coli and otherbacterial binding protein-dependent ABC transport systems(7). These transport systems usually have an outer membranediffusion pore, a soluble periplasmic substrate-binding proteinof high affinity, two integral membrane proteins, and one ortwo energy-transducing and ATP-hydrolyzing polypeptides lo-cated at the cytoplasmic side of the membrane. In the ar-chaeon T. litoralis, we found two genes, malF and malG, withstrong homology to the genes of two membrane components of
FIG. 7. SDS-PAGE analysis of TMBP after expression in E. coli. (A) TMBP with the N-terminal cleavable signal sequence of E. coli (construct A in Fig. 1). Lanes:1, uninduced E. coli cells harboring pRHo1000; 2, induced cells; 3, cytoplasmic extract of induced cells that were treated by cold osmotic shock; 4, same as lane 3 butthe sample was heated to 80°C for 10 min; 5, periplasmic shock proteins of induced cells; 6, same as lane 5 but the sample was heated to 80°C for 10 min; 7, TMBPpurified from the sample shown in lane 6; 8, TMBP isolated from T. litoralis membranes; St, molecular mass standards (from top to bottom: 94, 67, 43, and 30 kDa).(B) Cytoplasmic TMBP; its N terminus is the expected cleavage site after the E. coli signal sequence (construct B in Fig. 1). Lanes: 1, uninduced E. coli cells harboringpRHo1002; 2, induced E. coli cells; 3, cytoplasmic extract of induced cells; 4, same as lane 3 but the sample was heated to 80°C for 10 min; 5, purified periplasmic TMBP(construct A in Fig. 1); 6, TMBP isolated from T. litoralis; St, as for panel A. (C) Native precursor TMBP from T. litoralis. The protein was lipid modified and membranebound (construct C in Fig. 1). Lanes: 1, membranes from E. coli harboring pRHo1001 solubilized in octyl-b-glucoside and induced for the expression of the nativeprecursor TMBP; 2, same as lane 1 but the sample was heated for 10 min to 80°C; 3, purified TMBP from T. litoralis; 4, purified periplasmic TMBP endowed with theE. coli signal sequence; St, as for panel A. (D) Western blot of TMBP from T. litoralis membranes and from the different constructs expressed in E. coli. Lanes: 1, wholeE. coli cells harboring pRHo1000 encoding the hybrid protein with the E. coli signal sequence; 2, whole cells harboring pRHo1002 encoding the hybrid protein withoutthe E. coli signal sequence; 3, whole cells harboring pRHo1001 encoding the intact T. litoralis protein; 4, membrane preparations of T. litoralis uninduced for thetransport system; 5, membrane preparations of T. litoralis induced for the transport system; St, protein standards; 6, same as lane 1 (whole cells harboring pRHo1000);7, French pressure cell extract of cells harboring pRHo1000; 8, periplasmic proteins isolated from cells harboring pRHo1000.
686 HORLACHER ET AL. J. BACTERIOL.
on January 18, 2015 by guesthttp://jb.asm
.org/D
ownloaded from
the maltose (13, 19) and glycerol-3-phosphate (35) transportsystems of E. coli, both of which are members of the bindingprotein-dependent transport system family. Upstream of themalF and malG genes we found malE, with the characteristicsignature sequences of binding proteins (48) (Fig. 5). Thesethree genes constitute an operon with a putative archaeal pro-moter sequence (22, 24) and a putative prokaryotic ribosome-binding site upstream of malE (Fig. 4). Only 26 bp separatesthe malE and malF genes, and 1 bp, creating a frameshift,separates malF and malG (Fig. 4). An oligo(dT) sequencedetected at the end of malE resembles transcription termina-tion sequences described for archaea (49). The gene clustercontaining malEFG did not contain the E. coli malK analog (4,45). This has been observed with other binding protein-depen-dent ABC transport systems in gram-positive bacteria as wellas in the thermophilic bacterium T. thermosulfurigenes (41).The possibility that T. litoralis has an ATP-hydrolyzing subunitfor more than one transport system (44) should also be con-sidered.
Binding protein-dependent transport systems must have ap-peared early in evolution. A malE-like gene has also beenfound in Thermotoga maritima, which, like T. litoralis for ar-chaea, is one of the most deeply branched bacteria, with amaximum growth temperature of 90°C (31).
It was important to demonstrate that the malEFG genecluster of T. litoralis was indeed encoding the transport system(including the membrane-bound binding protein) that we havediscovered in this organism as a high-affinity (Km 5 20 nM)and trehalose-inducible system for the uptake of trehalose andmaltose (52). This was corroborated by the observation thatafter expression in E. coli, the gene product of malE showedthe same binding specificity as the binding protein isolatedfrom trehalose-induced T. litoralis cells. Moreover, antibodiesraised against the protein expressed in E. coli cross-reactedwith the protein isolated from T. litoralis membranes, either inpure form or when present in detergent-solubilized mem-branes.
The finding that the T. litoralis system which is obviously
related to the E. coli maltose system also recognizes trehalose(and is even induced by it) is rather surprising. In E. colitrehalose utilization depends on a different type of transportsystem belonging to the large group of sugar phosphotransfer-ase systems (28). In addition, the enzymes metabolizing treha-lose in E. coli are different from those which degrade maltose(40). Since trehalose can accumulate in T. litoralis under stressconditions (high salt) (30) but only when it is present in themedium, this sugar might possibly not be metabolized, and itsacquisition as a substrate of the maltose system might be partof an osmoprotecting mechanism. Yet, the transport systemdescribed here is also peculiar with respect to maltose trans-port. In E. coli the maltose transport system is in fact a mal-todextrin transport system geared for the utilization of maltoseas well as larger maltodextrins. This can be deduced from thefunction of the outer membrane l receptor as a diffusion porefor maltodextrins (17) and the binding specificity of maltose-binding protein, as well as the characteristics of the maltodex-trin-degradative enzymes (8). In contrast, the T. litoralis uptakesystem recognizes only maltose and not longer maltodextrins.FIG. 8. PAGE under nondenaturing conditions and in the presence of ra-
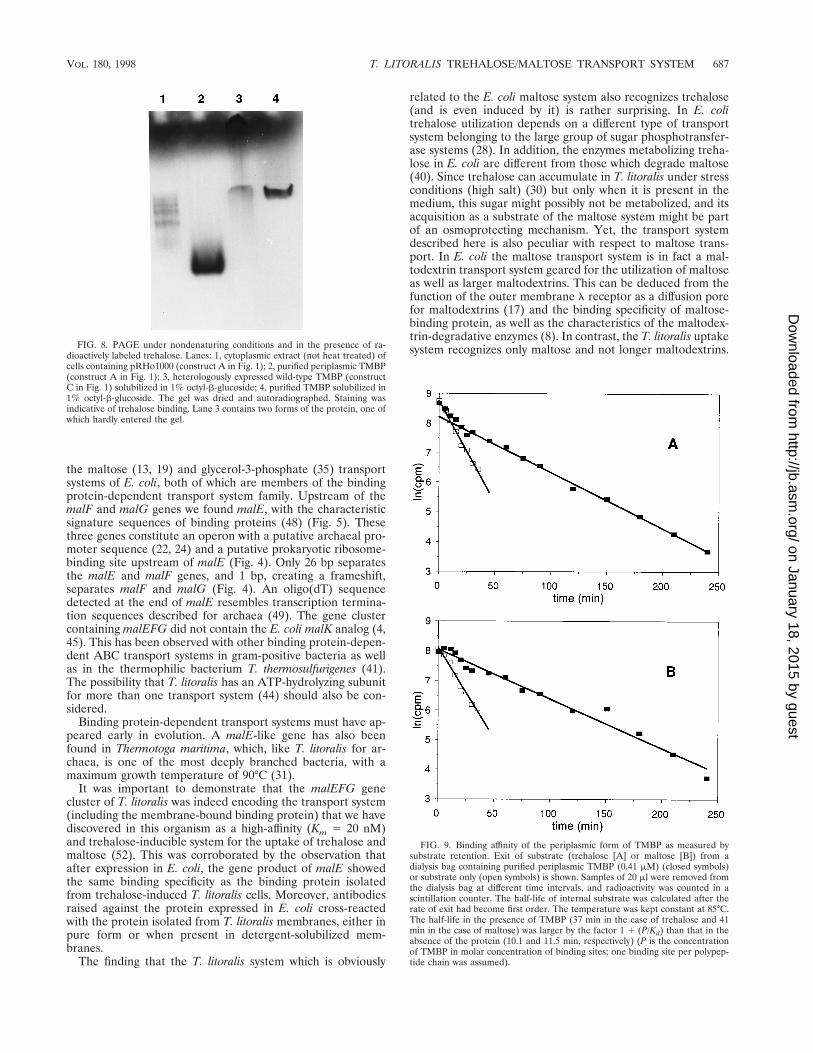
dioactively labeled trehalose. Lanes: 1, cytoplasmic extract (not heat treated) ofcells containing pRHo1000 (construct A in Fig. 1); 2, purified periplasmic TMBP(construct A in Fig. 1); 3, heterologously expressed wild-type TMBP (constructC in Fig. 1) solubilized in 1% octyl-b-glucoside; 4, purified TMBP solubilized in1% octyl-b-glucoside. The gel was dried and autoradiographed. Staining wasindicative of trehalose binding. Lane 3 contains two forms of the protein, one ofwhich hardly entered the gel.
FIG. 9. Binding affinity of the periplasmic form of TMBP as measured bysubstrate retention. Exit of substrate (trehalose [A] or maltose [B]) from adialysis bag containing purified periplasmic TMBP (0.41 mM) (closed symbols)or substrate only (open symbols) is shown. Samples of 20 ml were removed fromthe dialysis bag at different time intervals, and radioactivity was counted in ascintillation counter. The half-life of internal substrate was calculated after therate of exit had become first order. The temperature was kept constant at 85°C.The half-life in the presence of TMBP (37 min in the case of trehalose and 41min in the case of maltose) was larger by the factor 1 1 (P/Kd) than that in theabsence of the protein (10.1 and 11.5 min, respectively) (P is the concentrationof TMBP in molar concentration of binding sites; one binding site per polypep-tide chain was assumed).
VOL. 180, 1998 T. LITORALIS TREHALOSE/MALTOSE TRANSPORT SYSTEM 687
on January 18, 2015 by guesthttp://jb.asm
.org/D
ownloaded from

It will be interesting to analyze the maltose-degradative en-zymes in T. litoralis and to determine whether these enzymesare related to trehalose metabolism as well.
The determination of the binding activity of the recombinanthybrid TMBP gave a Kd of binding of 0.16 mM. This is surpris-ing in view of the fact that the apparent Km of uptake in intactcells was 1 order of magnitude lower. One could argue that theprotein without its lipid anchor exhibits a lower binding affinitythan the native protein. This appears unlikely to us, sincebinding is brought about (at least in E. coli MalE) by themovement of the two soluble lobes forming the binding sitebetween them (47). Thus, the lipid anchor should not influencebinding affinity. Therefore, the transport in intact cells mayexhibit a low Km (20 nM) despite a higher Kd of its majorsubstrate recognition site. Considering models for the mecha-nism of binding protein-dependent transport systems (5, 6),one may argue that in the case of the T. litoralis trehalose/maltose system, only the substrate-loaded binding protein isable to interact with the membrane components (model 1).This would be in contrast to the E. coli maltose system, whereboth the substrate-loaded and substrate-free binding proteincan interact with the membrane components (model 2) (32).Mathematical analysis for model 1 predicts that the Km oftransport becomes infinitely low (increasing affinity) with in-creasing amounts of binding protein, whereas in model 2 theKm of transport approaches the Kd of the binding protein atincreasing binding protein concentrations. Clearly, the amountof binding protein in the cell envelope of T. litoralis afterinduction by trehalose is substantial, and the transport mech-anism may in fact follow the predictions of model 1. Theputative transcription termination site downstream of themalE gene (Fig. 4) indicates that the amount of TMBP is farlarger than that of the membrane components.
ACKNOWLEDGMENTS
We thank M. Ehrmann for his help in analyzing the two-dimensionaltopology of MalF. We are indebted to A. Macanita and J. C. Lima,who helped us measure the intrinsic fluorescence of TMBP.
This research was supported by grants from the Deutsche For-schungsgemeinschaft, Forschergruppe: Struktur und Funktionssteu-erung an zellularen Oberflachen (to W.B.), by the Department ofEnergy (DE-F902-92ER20083) (to J.D.), and by the PRAXIS XXIprogramme, contract no. PRAXIS/2/2.1/BIO/1109/95 (to H.S.). K. B.Xavier acknowledges a Ph.D. grant from Praxis XXI, Portugal (BD/2760/94). The collaboration between the two European laboratorieswas supported by the Deutsche Akademische Austauschdienst and theConselho de Reitores das Universidades Portuguesas, Proc. AI-A/96.
REFERENCES1. Argast, M., and W. Boos. 1979. Purification and properties of sn-glycerol
3-phosphate-binding protein of Escherichia coli. J. Biol. Chem. 254:10931–10935.
2. Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. A. Smith, J. G.Seidman, and K. Struhl. 1987. Current protocols in molecular biology.Greene Publishing Associates, New York, N.Y.
3. Baneyx, F., and G. Georgiou. 1991. Construction and characterization ofEscherichia coli strains deficient in multiple secreted proteases: protease IIIdegrades high-molecular-weight substrates in vivo. J. Bacteriol. 173:2696–2703.
4. Bavoil, P., M. Hofnung, and H. Nikaido. 1980. Identification of a cytoplasmicmembrane-associated component of the maltose transport system of Esch-erichia coli. J. Biol. Chem. 255:8366–8369.
5. Bohl, E., and W. Boos. 1995. Binding protein-dependent transporters: ananswer of mathematics to biology. J. Comput. Appl. Math. 63:11–25.
6. Bohl, E., and W. Boos. 1997. Quantitative analysis of binding protein-medi-ated ABC transport systems. J. Theor. Biol. 186:65–74.
7. Boos, W., and J. M. Lucht. 1996. Periplasmic binding protein-dependentABC transporters, p. 1175–1209. In F. C. Neidhardt, R. Curtiss III, J. L.Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley,M. Schaechter and H. E. Umbarger (ed.), Escherichia coli and Salmonella:cellular and molecular biology. American Society for Microbiology. Wash-ington, D.C.
8. Boos, W., R. Peist, K. Decker, and E. Zdych. 1996. The maltose system ofEscherichia coli, p. 201–229. In E. C. C. Lin and A. S. Lynch (ed.), Regulationof gene expression in Escherichia coli. R. G. Landes Company, Austin, Tex.
9. Borges, K. M., S. R. Brummet, A. Bogert, M. C. Davis, K. M. Hujer, S. T.Domke, J. Szasz, J. Ravel, J. DiRuggiero, J. W. Chase, and F. T. Robb. 1996.A survey of the genome of the hyperthermophilic archaeon, Pyrococcusfuriosus. Genome Sci. Tech. 1:37–46.
10. Boyd, D., C. Manoil, and J. Beckwith. 1987. Determinants of membraneprotein topology. Proc. Natl. Acad. Sci. USA 84:8525–8529.
11. Boyd, D., B. Traxler, and J. Beckwith. 1993. Analysis of the topology of amembrane protein by using a minimum number of alkaline phosphatasefusions. J. Bacteriol. 175:553–556.
12. Dahl, M. K., E. Francoz, W. Saurin, W. Boos, M. D. Manson, and M.Hofnung. 1989. Comparison of sequences from the malB regions of Salmo-nella typhimurium and Enterobacter aerogenes with Escherichia coli K12: apotential new regulatory site in the intergenic region. Mol. Gen. Genet.218:199–207.
13. Dassa, E. 1993. Sequence-function relationships in MalG, an inner mem-brane protein from the maltose transport system in Escherichia coli. Mol.Microbiol. 7:39–47.
14. Derman, A. I., J. W. Puziss, P. J. Bassford, and J. Beckwith. 1993. A signalsequence is not required for protein export in prlA mutants of Escherichiacoli. EMBO J. 12:879–888.
15. Devereux, J., P. Haeberli, and O. Smithies. 1984. A comprehensive set ofsequence analysis programs for the VAX. Nucleic Acids Res. 12:387–395.
16. Duplay, P., H. Bedouelle, A. Fowler, I. Zabin, W. Saurin, and M. Hofnung.1984. Sequences of the malE gene and of its product, the maltose-bindingprotein of Escherichia coli K12. J. Biol. Chem. 259:10606–10613.
17. Dutzler, R., Y.-F. Wang, P. J. Rizkallah, J. P. Rosenbusch, and T. Schirmer.1996. Crystal structures of various maltooligosaccharides bound to malto-porin reveal a specific sugar translocation pathway. Structure 4:127–134.
18. Ehrmann, M., D. Boyd, and J. Beckwith. 1990. Genetic analysis of mem-brane protein topology by a sandwich gene fusion approach. Proc. Natl.Acad. Sci. USA 87:7574–7578.
19. Froshauer, S., and J. Beckwith. 1984. The nucleotide sequence of the genefor MalF protein, an inner membrane component of the maltose transportsystem of Escherichia coli. Repeated DNA sequences are found in the malE-malF intercistronic region. J. Biol. Chem. 259:10896–10903.
20. Gilson, E., G. Alloing, T. Schmidt, J. P. Claverys, R. Dudler, and M. Hof-nung. 1988. Evidence for high affinity binding-protein dependent transportsystems in gram-positive bacteria and in Mycoplasma. EMBO J. 7:3971–3974.
21. Gottermann, S., R. Horlacher, J. DiRuggiero, and W. Boos. 1997. Unpub-lished observations.
22. Hain, J., W. Reiter, U. Hudepohl, and W. Zillig. 1992. Elements of anarchaeal promoter defined by mutational analysis. Nucleic Acids Res. 20:5423–5428.
23. Harlow, E., and D. Lane. 1988. Antibodies: a laboratory manual. Cold SpringHarbor Laboratory, Cold Spring Harbor, N.Y.
24. Hausner, W., G. Frey, and M. Thomm. 1991. Control regions of an archaealgene. A TATA box and an initiator element promote cell-free transcriptionof the tRNA(Val) gene of Methanococcus vannielii. J. Mol. Biol. 222:495–508.
25. Herrmann, A., A. Schlosser, R. Schmid, and E. Schneider. 1996. Biochemicalidentification of a lipoprotein with maltose-binding activity in the thermoaci-dophilic Gram-positive bacterium Alicyclobacillus acidocaldarius. Res. Mi-crobiol. 147:733–737.
26. Horlacher, R., R. Peist, and W. Boos. 1996. Improved method for the pre-parative synthesis of labeled trehalose of high specific activity by Escherichiacoli. Appl. Environ. Microbiol. 62:3861–3863.
27. Kates, M. 1993. Membrane lipids of archaea, p. 261–295. In M. Kates et al.(ed.), The biochemistry of archaea (archaebacteria). Elsevier Science Pub-lishers B.V., Amsterdam, The Netherlands.
28. Klein, W., R. Horlacher, and W. Boos. 1995. Molecular analysis of treBencoding the Escherichia coli enzyme II specific for trehalose. J. Bacteriol.177:4043–4052.
29. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4. Nature 227:680–685.
30. Lamosa, P., L. O. Martins, M. da Costa, and H. Santos. 1996. Unpublishedresults.
31. Liebl, W., I. Stemplinger, and P. Ruile. 1997. Properties and gene structureof the Thermotoga maritima a-amylase AmyA, a putative lipoprotein of ahyperthermopholic bacterium. J. Bacteriol. 179:941–948.
32. Merino, G., W. Boos, H. A. Shuman, and E. Bohl. 1995. The inhibition ofmaltose transport by the unliganded form of the maltose-binding protein ofEscherichia coli: experimental findings and mathematical treatment. J.Theor. Biol. 177:171–179.
33. Mourez, M., N. Hofnung, and E. Dassa. 1997. Subunit interactions in ABCtransporters: a conserved sequence in hydrophobic membrane proteins ofperiplasmic permeases defines an important site of interaction with theATPase subunits. EMBO J. 16:3066–3077.
34. Neu, H. C., and L. A. Heppel. 1965. The release of enzymes from Escherichia
688 HORLACHER ET AL. J. BACTERIOL.
on January 18, 2015 by guesthttp://jb.asm
.org/D
ownloaded from

coli by osmotic shock and during the formation of spheroplasts. J. Biol.Chem. 240:3685–3692.
35. Overduin, P., W. Boos, and J. Tommassen. 1988. Nucleotide sequence of theugp genes of Escherichia coli K-12: homology to the maltose system. Mol.Microbiol. 2:767–775.
36. Peters, J., W. Baumeister, and A. Lupas. 1996. Hyperthermostable surfacelayer protein tetrabrachion from the archaebacterium Staphylothermus ma-rinus: evidence for the presence of a right-handed coiled coil derived fromthe primary structure. J. Mol. Biol. 257:1031–1041.
37. Polson, A., M. B. von Wechmar, and M. H. van Regenmortel. 1980. Isolationof viral IgY antibodies from yolks of immunized hens. Immunol. Commun.9:475–493.
38. Prinz, W. A., C. Spiess, M. Ehrmann, C. Schierle, and J. Beckwith. 1996.Targeting of signal sequenceless proteins for export in Escherichia coli withaltered protein translocase. EMBO J. 15:5209–5217.
39. Puyet, A., and M. Espinosa. 1993. Structure of the maltodextrin-uptake locusof Streptococcus pneumoniae. Correlation to the Escherichia coli maltoseregulon. J. Mol. Biol. 230:800–811.
40. Rimmele, M., and W. Boos. 1994. Trehalose-6-phosphate hydrolase of Esch-erichia coli. J. Bacteriol. 176:5654–5664.
41. Sahm, K., M. Matuschek, H. Muller, W. J. Mitchell, and H. Bahl. 1996.Molecular analysis of the amy gene locus of Thermoanaerobacterium thermo-sulfurigenes EM1 encoding starch-degrading enzymes and a binding protein-dependent maltose transport system. J. Bacteriol. 178:1039–1046.
42. Saurin, W., W. Koster, and E. Dassa. 1994. Bacterial binding protein-de-pendent permeases: characterization of distinctive signatures for functionallyrelated integral cytoplasmic membrane proteins. Mol. Microbiol. 12:993–1004.
43. Schauder, B., H. Blocker, R. Frank, and J. E. McCarthy. 1987. Inducibleexpression vectors incorporating the Escherichia coli atpE translational ini-tiation region. Gene 52:279–283.
44. Schlosser, A., T. Kampers, and H. Schrempf. 1997. The Streptomyces ATP-
binding component MsiK assists in cellobiose and maltose transport. J.Bacteriol. 179:2092–2095.
45. Shuman, H. A., and T. J. Silhavy. 1981. Identification of the malK geneproduct. A peripheral membrane component of the Escherichia coli maltosetransport system. J. Biol. Chem. 256:560–562.
46. Silhavy, T. J., W. Boos, S. Szmelcman, and M. Schwartz. 1975. On thesignificance of retention of ligand by protein. Proc. Natl. Acad. Sci. USA 72:2120–2124.
47. Spurlino, J. C., L. E. Rodseth, and F. A. Quiocho. 1992. Atomic interactionsin protein-carbohydrate complexes—tryptophan residues in the periplasmicmaltodextrin receptor for active transport and chemotaxis. J. Mol. Biol.226:15–22.
48. Tam, R., and M. H. Saier. 1993. Structural, functional, and evolutionaryrelationships among extracellular solute-binding receptors of bacteria. Mi-crobiol. Rev. 57:320–346.
49. Thomm, M., W. Hausner, and C. Hethke. 1994. Transcription factors andtermination of transcription in Methanococcus. Appl. Microbiol. 16:648–655.
50. Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer ofproteins from polyacrylamide gels to nitrocellulose sheets: procedure andsome applications. Proc. Natl. Acad. Sci. USA 76:4350–4354.
51. Wu, H. C. 1996. Biosynthesis of lipoproteins, p. 1005–1014. In F. C. Neid-hardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik,W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Esch-erichia coli and Salmonella: cellular and molecular biology. American Societyfor Microbiology, Washington, D.C.
52. Xavier, K. B., L. O. Martins, R. Peist, M. Kossmann, W. Boos, and H.Santos. 1996. High-affinity maltose/trehalose transport system in the hyper-thermophilic archaeon Thermococcus litoralis. J. Bacteriol. 178:4773–4777.
53. Zdych, E., R. Peist, J. Reidl, and W. Boos. 1995. MalY of Escherichia coli isan enzyme with the activity of a bC-S lyase (cystathionase). J. Bacteriol.177:5035–5039.
VOL. 180, 1998 T. LITORALIS TREHALOSE/MALTOSE TRANSPORT SYSTEM 689
on January 18, 2015 by guesthttp://jb.asm
.org/D
ownloaded from




















