
Analisi modale, polifonia e teoria musicale tardo-medievale: un approccio storico-critico
Upload
khangminh22Category
view
56download
0
1
APPROCCIO A UN PAZIENTE NEUROLOGICO ANAMNESI Ricordarsi di iniziare sempre con: nome, cognome e data di nascita.
L’anamnesi neurologica può essere difficoltosa, perché spesso il paziente non è in grado di fornirla (se comatoso, afasico,
…). Bisogna accertarsi che ci sia un testimone presente quando si è verificato l’evento neurologico stesso. Se manca anche
il testimone (il paziente è stato trovato solo dopo un periodo indefinito di tempo) è molto difficile ricostruire quello che è
successo, bisogna dedurlo. Qualora il paziente sia in grado di esprimersi, bisogna sempre dargli modo di esprimersi
liberamente e non condurre l’anamnesi secondo quello che noi pensiamo→ è meglio che usi un termine come
“mancamento” piuttosto che “assenza”, che può essere usato impropriamente deviando la diagnosi.
Indagare sempre la predisposizione genetica: malattie in famiglia, morti, …
ANAMNESI FISIOLOGICA: elementi fondamentali sono età (indirizza già verso una diagnosi), occupazione (importante sia
per esposizione a tossici ambientali, se lavora durante la notte, …).
ANAMNESI PATOLOGICA PROSSIMA: si possono chiedere più pareri, si può rivalutare l’anamnesi anche nei giorni successivi
con la testimonianza di più persone possibili aggiungendo quante più versioni (ma non cancellandole!).
ESAME OBIETTIVO NEUROLOGICO Si fa sempre anche l’ESAME OBIETTIVO GENERALE: sempre utile fare esame cardiovascolare, polmonare, …
Per l’EO neurologico non esiste uno schema universale.
MOTRICITÀ Destrimane o mancino Stabilire se il paziente è DESTRIMANE O MANCINO è importante per sapere la localizzazione del centro del linguaggio: la
maggior parte dei destrimani ha la predominanza dell’area del linguaggio a sinistra; nel mancino invece il 60% ce l’ha sempre
a sinistra, il 20% a destra e il 30% bilaterale. Questo è importante perché:
- A parità di lesione, una lesione a sinistra è più probabile che colpisca l’area del linguaggio.
- In ambito chirurgico, in una lesione a destra è più facile in quanto meno frequentemente verrà toccata l’area del
linguaggio mentre in uni intervento a sinistra bisogna assumere delle precauzioni maggiori.
Come si individua l’area del linguaggio? Iniettare dei barbiturici in carotide destra o sinistra e osservare se segue una fase
di afasia indotta.
Stazione eretta Lo si mette con piedi uniti, mani lungo i fianchi, capo eretto e sguardo avanti. Questo può già slatentizzare alcune patologie:
lesione delle vie somato-sensitive propriocettive (mielopatia con affezione del midollo che leda i cordoni posteriori).
Cosa c’è nei cordoni posteriori? Sensibilità tattile fine, propriocettiva, termico-dolorifica→ fascicolo gracile e cuneato,
lemnischi mediale e laterale, ….
Viene valutata:
o Ad occhi aperti
o Ad occhi chiusi: in una lesione delle vie propriocettive si può assistere a oscillazione e caduta→positiva al segno di
Romberg definisce una ATASSIA SENSITIVA (video). Le cause più comuni sono:
▪ Neuropatia periferica sia motoria (atrofia dei muscoli) sia sensitiva→ sono le fibre dei nervi periferici a essere
danneggiate, non quelle centrali.
▪ Lesioni che ledono i cordoni posteriori, come per una placca da sclerosi multipla.
Come si studia la sensibilità propriocettiva? Con il diapason: evoca una vibrazione (Do basso) che viene percepita
attraverso le vie propriocettive.
ATASSIA CEREBELLARE: causata da una lesione mediale del cervelletto (del verme, paleocerebello), che si esprime
con l’allargamento della base d’appoggio e oscillazioni in senso indifferente, presenti anche ad occhi aperti. Prove
che si possono fare:
- Durante la camminata, c’è un “lancio” dl tallone in avanti e le braccia vengono tenute larghe, a bilanciere
(tallonamento).
- Far mettere un piede avanti all’altro: sarà difficoltoso se già non sono capaci di camminare normalmente.

2
Deambulazione Cambia molto tra la camminata di un giovane e di un anziano. L’anziano assomiglia a un soggetto con malattia di Parkinson,
infatti ci sono elementi in comune→ con l’età si perde normalmente dopamina, nel Parkinson si ha una perdita maggiore
del 70%.
ANDATURA PARKINSONIANA:
▪ Lunghezza ridotta del passo
▪ Riduzione delle sincinesie pendolari: sono i normali movimenti delle braccia che accompagnano la camminata.
▪ Incurvamento in avanti: il baricentro si sposta in avanti→ atteggiamento camptocormico. Questo nella prima fase
del passo si accompagna a passettini per cercare di compensare il baricentro troppo avanti, e si dice che è come
se si affrettino, ma non è che corrono
→Sia nell’anziano che nella malattia di Parkinson avremo passi piccoli, con scarse sincinesie pendolari, schiena curva,
estrema difficoltà nel fare dietro-front.
ANDATURA FALCIANTE: è la camminata tipica di pazienti con ictus, è la circonduzione dall’indietro in avanti dell’arto del
lato affetto per poter esplicare il passo; il piede è cadente verso il basso. Indica un parziale recupero del danno ischemico,
soprattutto nell’arto inferiore; solitamente l’arto superiore recupera molto meno, ci vogliono molti più neuroni per far
muovere la mano finemente. In seguito a un danno delle vie motorie, i muscoli dell’arto sono in preda a una ipertonia
(SPASTICITÀ): lavorano patologicamente troppo secondo un certo criterio: nell’arto inferiore lavorano gli estensori,
nell’arto superiore lavorano i flessori.
Se si prova il riflesso rotuleo sull’arto del lato affetto cosa si verifica? Una ipereflessività→ una lesione delle vie motorie
centrali discendenti (primo motoneurone) porta a una esaltazione dei riflessi, mentre una lesione delle vie motorie
periferiche (secondo motoneurone) porta una riduzione dei riflessi.
ANDATURA STEPPANTE: è una situazione in cui c’è una paresi/plegia dei muscoli della loggia anterolaterale della gamba
8tibiale anteriore), cioè i muscoli che dorsiflettono il piede→ il piede non può essere dorsiflesso, l’avampiede cade e
costituisce un ostacolo al passo, si rischia di inciampare sulla superficie di appoggio. Il paziente dunque alza maggiormente
la gamba (e il piede) per poter procedere in avanti senza inciampare.
Qual è il nervo che serve questo gruppo muscolare? Rami terminali del nervo sciatico, precisamente il nervo peroneo
comune (SPE: sciatico popliteo esterno)→ si dirama a livello del poplite e passa esternamente. Non si sa perché, ma questo
nervo è il più comunemente lesionato.

3
Qual è un’altra causa comune di andatura steppante? Una lesione radicolare di L3-
L4-L5 (soprattutto L4), dovuto da un’ernia discale.
Qual è il territorio di distribuzione del nervo sciatico? Coscia posteriore, 1 dito del
piede.
Coordinazione In caso di una lesione cerebellare laterale, si avranno dei difetti di coordinazione
motoria, come il raggiungimento di un determinato bersaglio con le dita, …. Lo stesso
effetto è causato da una intossicazione da alcol.
In una lesione emisferica cerebellare, il soggetto avrà difficoltà a raggiungere un
oggetto dal lato coinvolto; il dito, cercando di raggiungere un oggetto, perderà
velocità quando si avvicina→ si parla di dismetria o frenage o tremore cerebellare.
Due prove per verificarlo sono la prova indice-naso (per arto superiore) o calcagno-
ginocchio (per arto inferiore).
Adiadococinesia: incapacità di svolgere movimenti alternati e rapidi (“prova delle lampadine”).
Prova del rimbalzo: si fa flettere l’avambraccio contro resistenza e si lascia andare di colpo; il soggetto normale è capace di
controllare il rimbalzo e di non picchiarsi da solo, mentre un soggetto con lesione cerebellare emisferica non riesce e si
picchia da solo.
Ipotonia muscolare: tipica di lesione cerebellare emisferica, si valuta con la resistenza passiva al movimento (difficile da
valutare).
Forza muscolare globale Parlando di forza globale, si testa l’innervazione contemporanea di molti muscoli che dipendono da muscoli e nervi diversi→
in comune hanno il comando motorio volontario. Le popolazioni di neuroni nella corteccia motoria primaria (area 4, davanti
al solco di Rolando nel polo frontale) coinvolte nel mantenimento di quella posizione, scaricheranno impulsi che
percorrendo il midollo spinale lungo il fascio piramidale (o cortico-spinale).
Le prove di forza muscolare globale sono dunque volte a saggiare l’integrità delle vie motorie centrali discendenti (cortico-
spinali, cioè il primo motoneurone). Un danno di queste vie si manifesta come una paresi, ossia una perdita parziale di
forza.
PARESI: perdita di forza parziale.
PLEGIA: perdita di forza totale.
PARAPLEGIA: perdita di forza totale degli arti inferiori.
TETRAPLEGIA: danno bilaterale a livello encefalico.
PROVA DI MINGAZZINI: in caso di lesione nella porzione sinistra della corteccia, l’arto controlaterale non è in grado di
mantenere la posizione e si avrà una asimmetria più o meno grave (fino alla plegia completa), che si manifesta come la
caduta della mano (che è la porzione più corticalizzata secondo l’homunculus) o del braccio intero.
PROVA DI STRUMPELL: come Mingazzini ma con gli arti superiori supinati; è leggermente più sensibile, l’arto
controlaterale rispetto al lato affetto tenderà a pronare.
Isteria: sono pazienti che inconsapevolmente sviluppano sintomi simil-neurologici (come paralisi, plegia, sordità, crisi simil-
epilettiche, …). Quando si effettuano queste manovre sorge il dubbio che si tratti di una
manifestazione psicogena, si ricorre a delle prove di distrazione (prova di deviazione attentiva):
con una penna si tracciano dei numeri sulla superficie cutanea e si invita il paziente a leggere
questi simboli solo con il tatto; il paziente si concentra su questo e si dimentica di mimare il
sintomo.

4
PROVA DELLA MANO CAVA: posizione difficile da mantenere, con le braccia alzate.
PROVA DI MINGAZZINI ARTI INFERIORI: DAL LATO CONTROLATERALE rispetto alla lesione
delle vie motorie discendenti c’è una caduta tentando di mantenere le gambe a squadra a 90°.
PROVA DI BARRET: si suddivide in 2 tipi:
- A paziente prono, si cerca di mantenere le gambe flesse a 90° sulla coscia e si osserva se vi è
una caduta.
- Si porta il tallone verso le natiche (ossia flessione massima) con la maggior forza possibile
tentando di contrastare la mano dell’operatore.
Come saranno i ROT? Ci sarà una asimmetria per una maggior vivacità sul lato paretico.
Questo si manifesta dopo un certo tempo, almeno qualche giorno; in fase iperacuta questo
reperto di ipereflessia può anche non esserci.
Forza muscolare segmentaria Non ha senso valutare tutti i muscoli del corpo, si vanno a valutare solo i principali. Bisognerebbe dare una quantificazione
della forza muscolare secondo la scala MRC (Medical Research Council):
• 5/5 alla scala MRC: movimento possibile contro resistenza massima;
• 4/5 alla scala MRC: movimento possibile solo contro resistenza minima;
• 3/5 alla scala MRC: movimento possibile solo contro gravità;
• 2/5 alla scala MRC: movimento possibile solo in assenza di gravità; Lo si valuta ponendo l’arto perpendicolare
all’asse di gravità
• 1/5 alla scala MRC: accenno al movimento;
• 0/5 alla scala MRC: assenza di movimento;
Movimenti involontari Si valuta la presenza di:
- Mioclonie: scatti involontari.
- Atetosi: movimenti involontari vermicolari, sinuosi.
- Distonia fasica (improvvisa) come il torcicollo: improvvise e abnormi torsioni causate dalla contrazione di gruppi di
muscoli antagonisti.
Attività muscolare intrinseca Fascicolazioni: sono delle ondulazioni della superficie del muscolo involontarie che però non causano il movimento dell’arto
perché coinvolgono una singola unità motoria (quindi non sono considerate dei movimenti involontari).
Trofismo
Tono muscolare L’esame del tono è empirico: si pone una mano sul muscolo e si effettua un movimento passivo con l’altra mano, tale da
stirarlo.
Es: si mette una mano sul muscolo e si estende l’avambraccio sul braccio; il bicipite darà una certa sensazione di resistenza
allo stiramento. È difficile da valutare.
• IPERTONO PLASTICO: lesione delle vie extrapiramidali, tipico dei soggetti con Parkinson; determina la contrazione
contemporanea di agonisti e antagonisti. Il soggetto parkinsoniano si presenta con arti superiori e inferiori flessi,
busto flesso in avanti→ predomina la muscolatura flessoria di tutto il corpo (atteggiamento posturale
camptocormico). Ha caratteristica di cedere a scatti allo stiramento passivo. Veniva chiamato anche segno della
troclea (o ruota dentata) perché simula il movimento di un ingranaggio.
• IPERTONO SPASTICO: lesione delle vie piramidali, si ha un aumento del tono progressivamente all’aumentare della
velocità del movimento passivo, fino a cedere di scatto (coltello a serramanico); colpisce soprattutto muscoli
antigravitari quindi si presenterà con un’ipertonia dei flessori degli arti superiori e una ipertonia degli estensori
degli arti inferiori (muscolatura antigravitaria)→ una emisindrome motoria può esitare in andatura falciante. È
connessa con l’iperreflessia OT in pazienti con danno del primo motoneurone.
• IPOTONO: si può valutare anche perturbando il busto scuotendolo e poi osservando l’escursione degli arti: se sono
aumentati (continua l’oscillazione a lungo) è segno di ipotonia.

5
RIFLESSI Riflessi propriocettivi/profondi Si tratta di contrazioni muscolari involontarie/automatiche.
Si percuote il tendine (del quadricipite sotto la rotula) costringendolo ad allungarsi
leggermente e quindi anche il muscolo→ questo provoca un allungamento di fusi
neuromuscolari: sono organelli propriocettivi sensibili alle variazioni di lunghezza delle
fibre muscolari; avvertono anche piccole variazioni di lunghezza e le trasmettono verso il
midollo spinale attraverso il nervo sensitivo. Il neurone sensitivo ha sede nel ganglio
paravertebrale e con un prolungamento raggiunge il midollo; qui si collega direttamente
al motoneurone alpha tramite una sinapsi eccitatoria: il motoneurone scorre nel nervo
motore e scaricherà sul muscolo causando una contrazione momentanea.
• RIFLESSO BICIPITALE: valuta il nervo muscolo-cutaneo.
• RIFLESSO STILORADIALE: nervo radiale (C5-C6).
• RIFLESSO TRICIPITALE: valuta il nervo radiale (C7).
• RIFLESSO ROTULEO: Le fibre sensitive e motorie decorrono nello stesso nervo
femorale, poiché si tratta di un nervo misto. Fa parte del plesso lombare. I neuroni motori sono collocati nelle corna
anteriori a livello L2-L3-L4.
• RIFLESSO ACHILLEO: nervo tibiale, a livello (S1-L4-L5).
Una lesione che intacchi uno dei nervi periferici coinvolti, può causare una iporeflessia o una areflessia. Esempio classico è
un’ernia del disco S1, che provoca un dolore violento (sciatica)→
LOMBOSCIATALGIA: dolore localizzato a coscia posteriore, poplite, pianta del
piede e alluce.
Un dolore localizzato alla coscia anteriore fa pensare invece alla
LOMBOCRURALGIA, cioè un danno a livello di L3-L4.
Riflessi cutanei o esterocettivi/superficiali RIFLESSO CUTANEO PLANTARE: allo strofinamento della pianta del piede, si
ha un riflesso flessorio. Dove c’è una lesione delle via extrapiramidali, si ha invece
l’estensione→ SEGNO DI BABINSKI.
RIFLESSO ADDOMINALE: si strofina un oggetto a punta smussa sulla cavità
addominale, causando una contrazione involontaria. Si divide in: superiori, medi
e inferiori. Da T6 a T12.
Ci interessano perché in caso di lesione delle vie motorie discendenti, essi si
attenuano o scompaiono.
RIFLESSO CREMASTERICO: non si fa più molto.
CONSIDERAZIONI SUL MIDOLLO SPINALE DA TENERE A MENTE: Il midollo finisce prima della colonna vertebrale, finisce a livello di L2. Sotto c’è la
cauda equina, cioè tutti i nervi già formati ma che aspettano di uscire dalla colonna. Facendo una puntura lombare sotto
L2, mal che vada si punge una radice nervosa ma non si danneggia il midollo.
Discrepanza tra il numero delle vertebre cervicali (7) e il numero delle radici nervose cervicali, che invece sono 8; la prima
radice fuoriesce tra la base del cranio e l’atlante.
Riflesso nocicettivo: è sempre un riflesso ma è più complesso perché non è più monosinaptico.
I riflessi hanno due applicazioni:
- per individuare un danno a livello periferico di un nervo che trasporta una delle branche del riflesso; in questo caso
sarà una iporeflessia.
- Ci forniscono informazioni anche sul controllo che i centri superiori hanno sul midollo→ possono essere rivelatori delle
vie motorie discendenti. In questo caso ci sarà iperrefessie.

6
In alcuni casi si possono avere delle plurireflessie (?) fino al clono: è la scarica continua del riflesso che può essere esauribile
(se termina da solo) o non esauribile (fino a che non si cambia posizione). Il clono si può evocare prendendo la rotula e
tirandola con forza verso il piede in modo da evocare sempre un allungamento del tendine. È segno di spasticità.
→ SINDROME PIRAMIDALE o DEL PRIMO MOTONEURONE: è quella sindrome caratterizzata da paralisi iperreflessiva,
Babinski, spasticità. È causata dalla lesione del primo motoneurone corticale.
SENSIBILITÀ Suddividiamo sensibilità in:
• ELEMENTARI o SUPERFICIALI→ condotte dal sistema spino-talamico.
▪ Tattile
▪ Termica-dolorifica
o PROFONDE→ condotte dal sistema lemniscale.
▪ Statochinestesia (batiestesia): senso di posizione del corpo
▪ Baroestesia: valutazione del peso degli oggetti.
▪ Pallestesica/vibratoria: la esaminiamo con il diapason.
• COMPLESSE o COMBINATE→ alta componente corticale, l’omuncolo
sensitivo deve agire per portare al risultato finale.
o Sensibilità discriminativa: serve per discriminare, cioè percepire come
due punti distinti sulla cute due punti vicini. Oltre un certo limite, i due punti vengono percepiti come uno solo.
Per rendere possibile questo, serve il sistema delle colonne dorsali e l’omuncolo in corteccia. Dove c’è una
maggior capacità discriminativa ci sarà una maggior quantità di recettori, che fanno riferimento a più neuroni,
i quali percorrono le vie dorsali e raggiungono la corteccia.
o Topoestesia: distinguere a occhi chiusi dove si è stati toccati; lesioni corticali parietali possono ridurre o
completamente annullare tale capacità.
o Grafestesia: riconoscere una parola o numero scritto sulla pelle; usata anche nelle prove di deviazione
attentiva→ infatti è implicato un lavorio corticale che distrae dal resto.
o Stereoestesia: capacità di riconoscere gli oggetti con il tatto. In alcuni casi la mano non è anestesica, ma se si
pone un oggetto non riuscirà a capire che oggetto è→ viene meno la rappresentazione corticale di tale oggetto
che permette di associare le caratteristiche tattili all’oggetto stesso.
• VISCERALI: trasmissione lenta e mal definita, spesso con localizzazione non precisa (si parla di dolore riferito).
RECETTORI SENSITIVI: abbiamo diversi tipi di
recettori, ognuno deputato a un tipo di sensibilità
diversa.
Il dolore è percepito dalle terminazioni nervose
libere.
SISTEMA LEMNISCALE Gli stimoli viaggiano lungo fibre mieliniche veloci Abeta (di grande diametro). Il primo neurone sensitivo alloggia nel ganglio
spinale della radice posteriore: tramite il dendrite raccoglie informazioni dai recettori e tramite l’assone entra nel midollo
spinale, senza fare altre sinapsi→ decorre nei cordoni dorsali nei fascicoli gracile e cuneato, arriva a livello del bulbo nei
nuclei di Goll e Burdach dove fa sinapsi.
Quasi subito qui avviene l’incrociamento (decussazione) delle vie; percorrono poi il tronco encefalico controlateralmente
e raggiunge i nuclei ventro-postero-laterali del talamo. Nel contesto della sostanza bianca emisferica l’assone del terzo
neurone poi si fa strada fino alla corteccia sensitiva→ qui la periferia è rappresentata ordinatamente secondo l’homunculus
sensitivo.
→ RAPPRESENTAZIONE SOMATOTOPICA: principio per cui la periferia è rappresentata ordinatamente nei livelli superiori
e soprattutto a livello corticale.

7
Homunculus: rappresenta il fatto che vi sono più neuroni sensitivi a livello della mano, delle labbra e della lingua rispetto
ad altre aree meno rappresentate.
SISTEMA SPINO-TALAMICO Trasporta la sensibilità termico-dolorifica.
Le fibre C sono piccole e amieliniche, quindi sono lente. Entrano nel midollo spinale e fanno sinapsi nella zona gelatinosa
di Rolando con un secondo neurone spino-talamico, da cui parte un assone che attraversa la linea mediana del midollo,
davanti alla commissura della sostanza grigia. Si portano quindi controlateralmente nel cordone laterale della sostanza
bianca e salgono fino all’encefalo; le fibre hanno destinazioni diverse:
- Alcune fanno sinapsi nel tronco (paleo-spinotalamico)
- Alcune nel talamo (neo-spinotalamico)
- Alcune interessano l’homunculus, altre invece interessano altre aree ancora→ l’animale decorticato infatti è in grado
di percepire il dolore (perché ci sono altre aree sottocorticali deputate alla sensibilità termico-dolorifica).
IL SISTEMA TRIGEMINALE I neuroni sensitivi abitano il ganglio di Gasser. I
prolungamenti centrifughi dei neuroni sensitivi
danno vita alle tre branche del trigemino: oftalmica,
mascellare e mandibolare; i prolungamenti
centripeti raggiungono i nuclei sensitivi del
trigemino, che sono sparsi ad occupare l’intera
lunghezza del tronco (mesencefalo ponte e bulbo) e
anche i primi segmenti del midollo spinale
cervicale→ nucleo della radice discendente (da
mesencefalo a midollo cervicale).

8
Le fibre sensitive propriocettive si distribuiscono ai nuclei mesencefalici, la tattile fine e la termico-dolorifica raggiunge la
parte spinale del trigemino.
Nevralgia del trigemino: è un accesso di dolore, una crisi dolorosa violenta di breve durata (da 30 secondi ad alcuni minuti)
simile a una scossa elettrica, scatenata dalla pressione su determinati punti, come i punti di emergenza delle branche del
trigemino. Tutti gli altri dolori al capo che non hanno queste caratteristiche non devono essere definiti nevralgie (se durano
qualche giorno, sono altro!).
ALTERAZIONI DELLA SENSIBILITÀ In clinica dobbiamo imparare a descrivere le alterazioni della sensibilità con diverse modalità: di fronte a una ipo/anestesia
bisogna chiedersi che territorio rappresenta→ ci sono 3 possibilità:
1) Disturbo della sensibilità di tipo tronculare→ es. tunnel carpale: il nervo mediano è schiacciato. Il nervo mediano
innerva I, II, III e IV dito (il IV è condiviso con l’ulnare). Bisogna quindi sapere i territori innervati dai singoli nervi.
2) Disturbo della sensibilità di tipo radicolare: la zona di cute che fa capo a una radice dorsale si chiama dermatomo; in
cartella neurologica c’è allegata una mappa dermatomerica. C’è un certo overlapping: la separazione tra un livello e
l’altro non è così netta. È impossibile saperle tutte con precisione, si può fare affidamento alla mappa.
I segmenti sacrali più caudali si riferiscono alla zona perineale, verso l’antica “coda”.
Es: disturbo di sensibilità del nervo sciatico. Zoster: si dispone a occupare uno o più dermatomeri o una branca del
trigemino.
Polineuropatia: disposizione a calza o a guanto. Es: SINDROME DI GUILLEN-BARRET. Alcune polineuropatie si
esprimono tanto più gravemente quanto più distale è il distretto coinvolto→ gradiente prossimo-distale di gravità: 8
neuropatie su 10 colpiscono piedi e mani. Tutti i nervi si ammalano e gli effetti più precoci e gravi si vedono nelle sedi
più lontane; la causa di questo è che il sistema di trasporto assonale viene messo alla prova dalla distanza, è più difficile
arrivare lontano.
1) Tronculare 2) Radicolare
3) Disturbo di sensibilità di tipo centrale: lesioni di midollo ed encefalo.
a. Lesione a livello encefalico: causa una ipo/anestesia faccio-brachio-crurale di un emisoma completo (emisoma ed
emivolto). La lesione è controlaterale rispetto all’emisoma coinvolto.
b. Lesione bulbare: viene compromessa la sensibilità dell’emisoma controlaterale (che si sono già incrociate) e il
sistema trigeminale omolaterale (i cui nuclei sono nel bulbo); sono danni per di più di base vascolare, dove si
danneggia la base del bulbo→ SINDROME DI WALLEMBERG: ipoestesia alterna, accompagnato da altri fenomeni.
Fa parte delle SINDROMI ALTERNE del tronco dove a livello dei nervi cranici la manifestazione è omolaterale
mentre a livello del soma la manifestazione è controlaterale.
c. Sezione trasversa midollare completa: livello orizzontale sotto cui la sensibilità è assente.
Dopo un trauma che causa frantumazione completa di C8, come si presenta il paziente? Ritenzione sfinterica (non
rilascio solitamente!), paraplegico→ PARAPLEGIA: paralisi totale degli arti inferiori.
d. Emisezione trasversa midollare: causa danno delle colonne posteriori dallo stesso lato della emisezione (perdita
di sensibilità profonda omolaterale e danno motorio), danno delle vie spinotalamiche controlaterali (perdita di
sensibilità termico-dolorifica e tattile fine controlaterale perché decussano subito)→ SINDROME DI BROWN-
SEQUARD.
e. Sindrome delle colonne posteriori: dissociazione tabetica tipica della neurosifilide, che ha un particolare tropismo
per le colonne posteriori. Si ha la perdita solo della sensibilità profonda di midollo interessato.

9
Un quadro simile che coinvolge le colonne posteriori è quello associato a ipovitaminosi B12 (MIELOSI
FUNICOLARE): tipica di soggetti gastroresecati.
f. Sindrome del cordone centrale: lesioni della parte centrale del midollo, che interessano una certa lunghezza del
midollo: dove si incrociano le fibre termico-dolorifiche, causa una perdita di sensibilità con disposizione “a
mantellina” in quanto coinvolgono principalmente il livello cervicale. Si parla di SIRINGOMIELIA: è una cavità che
si è venuta a creare a livello del canale centrale del midollo.
a. Lesione centrale b. Lesione bulbare c. Sezione trasversa midollare completa d. Emisezione trasversa midollare
e. Sindrome delle colonne posteriori f. Sindrome del cordone centrale

10
NEUROPSICOLOGIA E DEFICIT DELLE FUNZIONI
COGNITIVE Si occupa della parte cognitiva dell’approccio al paziente. Consiste prima di tutto nella valutazione della collaborazione del
paziente; deve essere vigile e collaborante per poter valutare tutte le funzioni cognitive superiori. Si parla di funzioni della
corteccia cerebrale che distinguono l’uomo dagli animali meno evoluti, che non sono in grado di comunicare e di fare
associazioni di stimoli: area visiva, area sensoriale del linguaggio, area motoria del linguaggio.
DEFICIT DI LINGUAGGIO: AFASIA Incapacità di parlare verbalmente. È diversa dalla disartria, che è una difficoltà a produrre linguaggio meccanicamente e
deriva da un deficit più basso sia a livello anatomico (danno a livello del nervo cranico) sia a livello gerarchico.
Esistono diversi sottotipi di afasia, sono state individuate nell’1800 da Broca e Wernicke:
1) Afasia sensoriale o di Wernicke: deficit sia della produzione del linguaggio che della comprensione.
2) Afasia di Broca: la comprensione è intatta ma la produzione no.
→ l’afasia comprende diversi tipi di disfunzioni.
Attenzione: per valutare un deficit di questo tipo, serve che prima il paziente fosse in grado di esprimersi normalmente,
non può essere valutato in soggetti con un deficit iniziale o nei bambini che ancora non si sanno esprimere. Il soggetto deve
essere integro, non deve esserci una deprivazione sensoriale prima dell’afasia: un sordo non avrà afasia, non comprende
perché non arriva il suono, o un danno alle corde vocali non parla perché non riesce, non perché ha un danno corticale.
Nella maggior parte dei casi, l’afasia è dovuta a un danno dell’emisfero sinistro: nel 90% delle persone l’area del linguaggio
si trova a sinistra.
L’Afasia è un disturbo acquisito che interessa i processi centrali di elaborazione linguistica, può coinvolgere una o più
componenti del processo di:
- Comprensione sia orale che scritta
- Produzione sia orale che scritta
- Ripetizione: in alcuni soggetti si riscontra solo un problema di ripetizione delle parole dette dall’esaminatore.
Una caratteristica dell’eloquio importante da analizzare per individuare un’afasia è la FLUENZA VERBALE: è la capacità di
produrre un linguaggio con un ritmo e una intonazione emotiva particolare.
Il termine fluenza indica un certo numero di caratteristiche quali:
a. Prosodia (ritmo e pattern di intonazione); cambia anche da lingua a lingua e da dialetto a dialetto.
b. Articolazione (sforzo, inceppi vs fluidità)
c. Abbondanza dell’eloquio: operativamente la più lunga sequenza di parole prodotta nel linguaggio spontaneo. Da
eloquio povero (nel deficit motorio) a eloquio più abbondante (nell’afasia sensitiva).
In base a questo distinguiamo in:
- AFASIA FLUENTE:
o Prosodia conservata
o Articolazione non difficoltosa
o Abbondanza dell’eloquio: 6-7 parole per sequenza
o Tipica di lesioni posteriori, temporo-parietali sinistre (area di Wernicke).
- AFASIA NON FLUENTE:
o Prosodia alterata
o Difficoltà articolatorie
o Abbondanza dell’eloquio: max 4 parole per sequenza
o Lesioni anteriori (aree frontali) dell’emisfero SN
ANATOMIA DELLE FUNZIONI LINGUISTICHE Esistono dei collegamenti stretti tra corteccia, talamo e nuclei della base, molto vicini tra loro: il danno quindi può essere
sia corticale che sottocorticali (disconnessione):

11
❖ Livello corticale: area perisilviana
o Settore anteriore: area di Broca (44), area associativa (45)
o Settore posteriore: area di Wernicke (22), aree associative (39 e 37)
Con aree associative si intendono le aree che mettono insieme i significati più complessi alla parola
(permette di associare una parola all’idea dell’oggetto che essa rappresenta, esempio la parola microfono
mi fa venire in mente l’immagine del microfono solo grazie all’area associativa).
❖ Livello sottocorticale:
o Talamo
o Nuclei della base
AFASIA DI BROCA È la più comune perché l’infarto cerebrale nell’emisfero di sinistra dà quasi sempre un’afasia motoria. Se è coinvolta anche
l’area di Wernicke sarà un’afasia globale.
20% di frequenza.
LINGUAGGIO ORALE:
• Produzione:
o Nei casi più gravi non parla proprio, è incapace di comunicare verbalmente.
o Se il disturbo è meno grave, si avvertono parole o fonemi isolati e il soggetto cerca di utilizzarle con grande
frustrazione. Ha una intonazione piatta (mancanza di prosodia) e uno stile telegrafico (agrammatismo).
• Ripetizione: non riesce a ripetere una parola.
• Comprensione: riesce a comprendere tutto quello che gli viene detto.
SCRITTURA: riesce al massimo a firmare e a scrivere qualche parola copiata.
LETTURA: riesce a leggere ma a fatica.
DENOMINAZIONE: Non riesce a nominare gli oggetti una volta mostrati.
SINTOMI ASSOCIATI: molto spesso si manifesta emiplegia destra, perché se riguarda il lobo sinistro può coinvolgere anche
l’aria motoria primaria.
Si rende conto del disturbo, ne ha consapevolezza e questo gli causa frustrazione e depressione.
AFASIA DI WERNICKE Riguarda il versante sensoriale.
20% di frequenza.
LINGUAGGIO ORALE:
• Produzione: eloquio fluente con normale intonazione emotiva (prosodia conservata e adeguata). Poiché non
comprende, utilizza parole senza senso→ parlata tanto ma non si capisce niente.
Parafrasia: invertono parole nella frase.
Loquacità: parlano tanto fino a diventare logorroici.
Neologismi.
• Comprensione: compromessa.
• Ripetizione: compromessa, molti errori fonemici.
SCRITTURA E LETTURA: compromesse.
Non è consapevole del suo deficit, continua a parlare e si stupisce del fatto che l’interlocutore non capisce ma non ci dà
molto peso. Non ha consapevolezza della malattia→ ANOSOGNOSIA.
SINTOMI ASSOCIATI: aprassia ideomotoria o ideativa e disturbo del campo visivo, data la zona di interesse→ dal corpo
cingolato laterale del talamo, le radiazioni ottiche salgono per raggiungere la corteccia occipitale; la parte inferiore della
radiazione ottica passa proprio nella parte profonda dell’area 22→ emianopsia laterale omonima.
AFASIA GLOBALE Combinazione di afasia di Wernicke + Broca. Non capisce ordini semplici, non parla→ a seconda della combinazione
possono avere o meno consapevolezza del problema.
AFASIA DI CONDUZIONE 4% di frequenza.
Lesione del fascio che collega le due aree 44 e 22; è un caso di disconnessione.
ELOQUIO: il soggetto riesce a comunicare con eloquio fluente ma fa parecchi errori.

12
COMPRENSIONE: comprende abbastanza bene, ma quando il segnale di entrata deve raggiungere l’area di elaborazione
del segnale di uscita, si blocca→ smascherato chiedendo di ripetere: fa molti più errori quando ripete che quando parla da
solo.
RIPETIZIONE: compromessa.
AFASIA TRANSCORTICALE - Motoria: simile alla Broca ma è in grado di ripetere.
- sensitiva: simile a Wernicke ma è in grado di ripetere. 2%.
- mista: ancora più rara
AFASIA ANOMICA È un deficit selettivo del denominare: parla e si esprime bene ma non riesce a trovare il nome giusto da dare alla parole
quindi usa parole simili di suono o di significato. 8%.
Segno precoce di malattia di Alzheimer.
TIPI DI ERRORI NELLE AFASIE STEREOTIPIE (ST): sono parole semplici (mamma) o parole costruite accostando sillabe semplici, come il linguaggio dei
bambini.
DIFFICOLTA’ ARTICOLATORIE (DA): Emissione esplosiva o scandita, inceppi, elisione e/o sostituzioni di fonemi secondo
tendenze ben definite.
ANOMIE: difficoltà a trovare le parole esatte.
PARAFASIE SEMANTICHE (PS): Parole semanticamente correlate alla parola bersaglio, simili per significato. (“frutta” per
ciliegia, “cane” per gatto).
PARAFASIE VERBALI: parole che non sono semanticamente e/o fonologicamente correlate alla parola (“cappello” per
/sedia/).
PARAFASIE FONEMICHE: Elisione, sostituzione, inserzione o trasposizione di un fonema di una parola che però resta
riconoscibile, cambio di pezzi di parole (“obrello” per ombrello; “telepono” per telefono; “automiobile” per automobile).
GERGO PARAFASICO: Successione fluente di parole in sé significative, ma semanticamente inappropriate emesse con
normale prosodia e integrate in frasi sintatticamente ben strutturate ma inutili alla comunicazione in quanto globalmente
incomprensibili (“ieri il tavolo con mio marito andremo più tardi” per /ombrello/). Talvolta si può rintracciare in questa
“insalata di parole” qualche nucleo di significato affine a ciò che è stato richiesto.
NEOLOGISMI (N): Non parole che non hanno somiglianza fonologica con la parola bersaglio (“pertina” per /barca/).
GERGO FONEMICO-NEOLOGISTICO (GNF): Serie sillabiche senza senso, emesse con scioltezza e normale prosodia, così da
dare l’impressione di un eloquio normale in idioma sconosciuto (“este ni falino pesso tone” per /orologio/); a volte il gergo
può essere costituito da un miscuglio di parole reali e sillabe senza senso.
ESAME OBIETTIVO Si costruisce uno schema in modo da indagare tutto:
1) Capacità di comprendere
2) Capacità di eloquio spontaneo
3) Capacità di ripetere
4) Qualità del linguaggio
5) Quantità del linguaggio
DEFICIT DI RICONOSCIMENTO: AGNOSIA Deficit del riconoscimento di stimoli, qualunque sia il canale centrale, è detto AGNOSIA: visiva, tattile, uditiva, …→ una per
qualsiasi via sensoriale (in realtà la forma gustativa e olfattiva non vengono esplorate). In teoria si possono avere tante
forme di agnosia quante sono le sorgenti dell’esperienza sensoriale.
È necessaria una esperienza percettiva di base normale. Per ogni canale sensoriale ci sono gradi di complessità diversi:
- FORMA APPERCETTIVA: incapacità di discriminare le qualità percettive elementari di un oggetto. Il soggetto agnosico
non sa riconoscere né descrivere le caratteristiche dell’oggetto: forma, dimensione, contorni, luminosità.
Es: se agnosia appercettiva tattile, non sa dire che è tondo, piatto, con scanalature.

13
- FORMA ASSOCIATIVA: incapacità di attribuire un significato e un nome a oggetti e figure. Il soggetto agnosico non sa
richiamare l’immagine mentale relativa all’oggetto ma sa descrivere le sue caratteristiche.
Es: se agnosia associativa tattile, sa dirvi tutte le caratteristiche ma non riesce a dire la parola moneta (si parla in
questo caso di asimbolia tattile).
AGNOSIA TATTILE (STEREOAGNOSIA) ❖ Appercettiva: ASTEROANESTESIA→ incapacità di discriminare forma, dimensioni, consistenza, superficie
dell’oggetto.
Lesione del lobo parietale, giro postcentrale.
❖ Associativa: ASIMBOLIA TATTILE→ Incapacità di concettualizzazione dello stimolo. Il soggetto non identifica l’oggetto
pur riconoscendone le caratteristiche formali.
Lesione del lobo parietale aree 5 e 7.
Possono esserci associati anche deficit di:
o Grafestesia: disegnare sul corpo del paziente una lettera o un numero.
o Somatoagnosia: capacità di riconoscere la disposizione di parti del proprio corpo. I disturbi iniziano con
una negligenza di una parte del corpo, fino al mancato riconoscimento della parte del corpo o addirittura
della malattia. I quadri sono diversi a seconda dell’emisfero colpito:
▪ Se lesione a destra, ci sarà tendenza a trascurare l’emisoma di sinistra, non percezione degli
stimoli da sinistra, non riconoscimento dell’emisoma, non riconoscimento della malattia.
▪ Se lesione a sinistra, ci sarà confusione destra-sinistra, incapacità di localizzazione dello stimolo
(topoagnosia) e incapacità di riconoscimento delle dita (sindrome di Gertsmann, agnosia
digitale).
AGNOSIA VISIVA ❖ Appercettiva: incapacità di discriminare le qualità formali dell’oggetto. Non sa dire se un oggetto è quadrato, tondo,
sferico, ….
❖ Associativa: mancato riconoscimento (o confusione) dell’oggetto, che però viene correttamente descritto.
PROSOPAGNOSIA: è una forma particolare di agnosia visiva legato all’incapacità di riconoscere i volti.
Due forme:
- Appercettiva: i volti sembrano tutti uguali.
- Associativa: incapacità a riconoscere i volti noti: tipico della demenza (non riconosce il figlio o il marito) ma riconosce
i volti nuovi.
CECITÀ VISIVA: forma più grave di agnosia visiva, causata da una lesione bilaterale della corteccia visiva; non è impossibile
in quanto sono vascolarizzate dalle cerebrali posteriori che derivano dalla basilare→ una lesione all’apice della basilare può
causare una occlusione bilaterale. Si perde totalmente la visione dell’oggetto e il soggetto è consapevole.
Esiste una forma di anosognosia di cecità quando vengono lesionate anche aree associative→ SINDROME DI ANTON-
BABINSKI.
AGNOSIE UDITIVE ❖ Appercettiva: non riesce a comprendere i suoni. La lesione deve essere bilaterale, perché le vie acustiche hanno una
rappresentazione bilaterale: questo deriva dal fatto che per comprendere la direzione del suono, serve che il suono
arrivi nel nucleo del tronco in tempi diversi→ questa differenza serve per individuare la direzione del suono. Dopo di
che le vie salgono in parallelo e comunicano solo in due punti: nel tronco e più in alto nel diencefalo→ questa
comunicazione fa sì che se si ha una lesione monolaterale non si perde totalmente l’udito, ma si ha una incapacità a
comprendere la qualità del suono (esempio riconoscimento della musica)→ AMUSIA SENSORIALE.
❖ Associativa.

14
DEFICIT DI GESTUALITÀ: APRASSIA
Le PRASSIE non sono semplici movimenti, ma sistemi coordinati di movimenti in funzione di un’intenzione o un risultato,
non sono senza scopo.
Un’aprassia è l’incapacità di compiere un gesto intenzionale (volontariamente, su richiesta) in assenza di deficit di moto, di
senso e di coordinazione, che giustifichino tale fallimento.
N.B.: sono conservati i movimenti spontanei! Si riesce a spazzolare senza problemi, ma se gli si chiede di spazzolarsi, non è
in grado di farlo→ quando si dà a un movimento un’intenzione o un risultato, non riesce più a svolgerlo.
Il disturbo aprassico non è un disturbo di tipo motorio ma del livello di ideazione, scelta o organizzazione del
comportamento motorio.
L’aprassia è caratterizzata dalla DISSOCIAZIONE AUTOMATICO-VOLONTARIA: durante la vita quotidiana il paziente
aprassico non è particolarmente limitato dal suo disturbo. Al contrario, quando si trova in una situazione non pragmatica
ed altamente intenzionale, come durante un test a tavolino affiora la rottura dell'organizzazione gestuale.
CLASSIFICAZIONE DEI GESTI Prima di parlare di aprassia bisogna capire cosa sono i gesti:
• Gesti mimici, espressivi dello stato d’animo, come minaccia, saluto, ecc.
• Gesti transitivi, rivolti agli oggetti da usare.
• Gesti intransitivi o simbolici, come fare il segno della croce, il segno della vittoria, il pollice verso, il saluto militare;
oppure mimare l’uso di alcuni oggetti in assenza dell’oggetto.
APRASSIE PRINCIPALI Due aprassie più importanti:
❖ IDEATORIA: il soggetto non sa cosa deve fare, non è in grado di ideare il gesto, è un disturbo a monte. Lo si distingue
perché quando si fa vedere il gesto, poi riesce a compierlo→ riesce a produrre il gesto su imitazione. Es: non sa come
tagliare una bistecca o allacciarsi la camicia, ma se lo si fa vedere poi riesce a farlo. A volte fa gli atti ma in ordine
sbagliato. Riguarda entrambe le mani.
Sono legate a una lesione emisferica sinistra del lobo parietale solitamente; spesso sono associati a disturbi afasici.
❖ IDEO-MOTORIA: il programma (conservazione dello schema generale) c’è ma il soggetto non riesce a metterlo in atto,
è consapevole di quello che deve fare ma non sa come farlo. È un danno più a valle del primo. Non è in grado di
riprodurlo su imitazione.
Il movimento non ha un significato, non è mirato e corretto; oppure ha una esecuzione simile al normale ma
lievemente sbagliata→ fa il saluto militare mettendo la mano sulla bocca anziché sulla testa (in quella ideatoria
non riesce nemmeno a capire che gesto deve fare) oppure inizia facendo il saluto militare poi ci infila elementi di
altri gesti.
Causato da un danno a emisfero sinistro lobo parietale e temporale.
VALUTAZIONE DI UN PZ APRASSICO: si chiede di svolgere un gesto complesso oppure lo si fa vedere e si chiede di ripeterlo.
PARAPRASSIE Sono i tipi di errori che si riscontrano nelle aprassie:
- Perplessità, il paziente non sa cosa fare
- Perseverazione, il paziente non riesce a portare a termine il movimento perché persevera nella riproduzione di un
elemento del gesto
- Maldestrezza, il paziente esegue l'azione in modo rozzo e inefficace
- Omissioni, il paziente salta uno o più passaggi nell'azione
- Errori di localizzazione, l'azione è appropriata ma eseguita nel luogo errato
- Uso erroneo, l'azione è concettualmente inappropriata (usa il cacciavite per tagliare la carne).
- Errori di sequenza, il soggetto compie singole azioni corrette, ma nella sequenza sbagliata (mette le calze sopra le
scarpe).
- Pantomima: si osserva spesso l’uso di parti del corpo come se fossero oggetti (es. alla richiesta di mostrare come si
usa uno spazzolino da denti il paziente userà il dito come se fosse lo spazzolino stesso invece di produrre il gesto con
cui viene usato).

15
ALTRE APRASSIE SPECIALIZZATE APRASSIA COSTRUTTIVA: Disturbo nel disegnare, costruire, pur non essendoci aprassia nei singoli movimenti. Viene
disegnata o costruita una forma errata dal punto di vista spaziale.
APRASSIA DELL’ABBIGLIAMENTO: Incapacità a eseguire correttamente gli atti corretti per vestirsi, in assenza di aprassia
ideatoria o ideomotoria. Il paziente infila gli indumenti alla rovescia, mette la maglia sopra la camicia, le scarpe al contrario,
ecc.
APRASSIA DELLA MARCIA: Il paziente non è in grado di utilizzare correttamente gli arti inferiori. Il paziente è in grado di
muovere gli arti in condizione supina, ma non è in grado di iniziare la marcia, resta come attaccato al suolo→ ha perso lo
schema motorio del cammino. Lesone della prima circonvoluzione frontale.

16
DEMENZE
Pag. 303
Compromissione intellettiva acquisita e persistente con alterazione di molteplici funzioni cognitive (memoria, linguaggio,
capacità visuo-spaziali…); quando il deficit cognitivo non impatta sulla funzionalità del soggetto non si parla di demente.
È un fenomeno in crescita.
Forme di demenza:
- Circa il 50% è causato dall’Alzheimer (includendo anche le forme miste).
- Demenze fronto-temporali
- Demenze vascolari
MCI: DETERIORAMENTO COGNITIVO LIEVE Negli ultimi 20 anni è emerso un concetto definito deterioramento cognitivo lieve: è la presenza di un deficit cognitivo a
carico della memoria in assenza di altre disfunzioni; non può essere definito demenza perché deve esserci un impatto
sociale, lavorativo e quotidiano. Non tutte le persone con questo deterioramento passano poi all’Alzheimer, perché è solo
un dato clinico.
C’è interesse verso questa forma di decadimento perché quando si troverà il modo per rallentare la demenza si dovrà agire
già nelle prime fasi (secondo il prof il decadimento cognitivo inizia già intorno ai 20 anni).
Criteri diagnostici:
1) Disturbo di memoria definito come la presenza di almeno uno dei seguenti:
a. Riferito direttamente
b. Riferito dal famigliare
c. Riferito dal medico curante
2) Presenza di tutte le seguenti caratteristiche:
a. Assenza di impatto funzionale
b. Test di cognitivista globale normali
c. Test di memoria anormali per età
d. Assenza di demenza.
3) Diagnosi mediante:
a. Valutazione clinica
b. Valutazione neuropsicologica
c. Esami di laboratorio
d. Esami strumentali: TC, RM encefalica, EEG, analisi del liquido cerebrospinale.
Il tasso di conversione da disturbo lieve a demenza è di circa la metà in 4 anni, quindi non tanto alto.
La demenza per essere definita tale deve coinvolgere tutti questi tre ambiti.
MANIFESTAZIONI PRINCIPALI DI DEMENZA DISTURBI DI MEMORIA:

17
• Incapacità a recuperare ricordi acquisiti e ad immagazzinare nuove informazioni: la metà dei casi di demenza iniziano
con disturbi di memoria procedurale attiva, cioè al capacità di immagazzinare nuove informazioni→ difficoltà a
ricordarsi nomi in sequenza quando viene richiesto di farlo. È diverso dal semplice dimenticarsi le cose, che invece è
solo distrazione→ alterazione della memoria a breve termine, solo in un secondo momento di quella a lungo termine.
• Alterazioni della memoria diacronica (difficoltà a collocare gli eventi nella giusta sequenza temporale): confondimento
dell’ordine cronologico degli eventi.
• Paramnesie (difficoltà a riconoscere i ricordi come personali): racconta un determinato ricordo come se fosse stato
vissuto in prima persona.
• Amnesia globale progressivamente ingravescente.
MANCANZA DI CONSAPEVOLEZZA (anosognosia): la si acquisisce dopo un po’, perché le prime volte si rende conto e ha un
impatto negativo sull’umore; ha consapevolezza che sta diventando demente, ma presto poi viene persa→ la demenza
infatti è più fastidiosa per i famigliari che per il malato stesso.
DISORIENTAMENTO:
• Spaziale: conservata più a lungo.
• Temporale: viene perso prima di quello spaziale. Alla domanda “che giorno è oggi” quasi tutti rispondono sbagliato.
COMPROMISSIONE DELL’ATTENZIONE:
• Difficoltà di concentrazione
• Facile distraibilità
COMPROMISSIONE DELLA CAPACITÀ D’ASTRAZIONE:
• Perdita capacità di critica e di giudizio
• Alterazione del comportamento sociale (disinibizione sessuale): spogliarsi in pubblico.
ALTERAZIONI DEL CARATTERE E DELL’UMORE:
- Accentuazione/esasperazione dei tratti caratteriali o comparsa di caratteristiche opposte.
- Generosità/prodigalità fino allo sperpero
- Parsimoniosità/avarizia
- Rispettosità delle regole/comportamenti asociali fino a scoppi di ira e manifestazioni di violenza
SINDROME ALOGICA DI REICH: combinazione di:
• Afasia: disturbi del linguaggio (incoerenza, confabulazioni, progressivo impoverimento, neologismi...)
• Aprassia: incapacità ad eseguire attività motorie nonostante l’integrità della comprensione e della motricità.
• Agnosia: incapacità a riconoscere oggetti, nonostante l’integrità delle funzioni sensoriali.
CLASSIFICAZIONE DELLE DEMENZE PRIMARIE: sono neurodegenerative
- Alzheimer
- Degenerazione fronto-temporale
- Parkinson
SECONDARIE:
- Demenza vascolare ischemica: è causata dal ripetersi di eventi vascolari acuti che causano decadimento cognitivo.
- Stati carenziali: può essere una condizione parzialmente reversibile.
- Idrocefalo normoteso
- Disturbi endocrini e metabolici
- Malattie metaboliche ereditarie
- Malattie infettive ed infiammatorie del SNC
- Sostanze tossiche
- Processi espansivi intracranici
- Altre
Tra queste alcune sono reversibili, ma quella che è veramente reversibile è quella da DEPRESSIONE.

18
MALATTIA DI ALZHEIMER Il primo caso fu una donna di circa 50 anni nei primi anni del 1900.
È presente anche in soggetti in età presenile ma l’età è il fattore di rischio principale quindi si manifesta pienamente in età
senile.
Esistono anche alcune forme monogeniche, ma sono rarissime, sono soprattutto casi sporadici.
La neuro degenerazione può estendersi da una forma temporale come l’Alzheimer o che parte come frontale e poi si
estende al temporale.
I neuroni devono poter comunicare tra loro, mantenere un certo equilibrio metabolico, mantenere una certa plasticità (di
riparazione); nell’A. queste tre funzioni vengono perse: degenerazione sinaptico, alterazioni a livello intracellulare e definit
nella capacità di riparazione.
Si è molto studiato il meccanismo di degenerazione, per l’enorme impatto epidemiologico→ due fondamentali dati:
1) A livello extracellulare si accumulano proteine mal tagliate e indigeribili, che sono frammenti lunghi della proteina
Beta-Amiloide. Si formano fibrille di oligomeri che interferiscono con la trasmissione sinaptica. Gli enzimi sono 3,
alfa beta e gamma secretasi che tagliano i frammenti proteici e possono o digerire la proteina AB o causare questi
frammenti lunghi che si accumulano.
2) A livello intracellulare le proteine tubulari Tau che costituiscono l’impalcatura del neurone vanno incontro a
fosforilazione che ne causa il collasso.
→ non è chiaro se l’amiloide si accumuli prima della degenerazione neuronale o dopo o insieme. È difficile interrompere
questi meccanismi una volta che si sono già attivati.
Dal punto di vista topografico, è più facile associare la demenza alla presenza di Tau: infatti essa si trova sempre in cervelli
dementi, ma invece si ritrova la beta-amiloide anche in cervelli normali, quindi si pensa che forse tau sia una conseguenza
che causa demenza.
Le donne sono più colpite degli uomini e i soggetti con sindrome di Down hanno una amiloidopatia molto marcata.
Molto controversa l’assunzione di farmaci antiossidanti.
Quello che si sa per certo è che i grovigli neurofibrillari sono tossici e inducono una reazione gliale infiammatoria che può
agire in senso negativo o in senso positivo (infatti in alcuni casi può riuscire a rimuovere alcuni grovigli).
In generale si ha una degenerazione con atrofia→ riduzione del volume cerebrale molto marcata, soprattutto nelle aree di
memoria e del linguaggio→ le aree limbiche e ippocampali iniziano già a degenerare anche 10 anni prima della
manifestazione della malattia.
Il soggetto diventa anche più fragile: perde peso, è più soggetto a infezioni, non cammina più, ….

19
Storia naturale della malattia - Inizia con disturbo della memoria
- Perdita di autosufficienza: in media avviene dopo 5 anni.
- Ricovero in struttura dopo 6-7 anni
- Morte dopo 10 anni circa.
Diagnosi Nella forma conclamata è semplice per accumulo di disturbi cognitivi; si fa uno studio per imaging per escludere altre cause
vascolari o strutturali. Se avanzata è anche inutile fare esame del liquor.
Nella fase di disturbo cognitivo lieve può avere senso fare indagine biochimica del liquor.
Criteri clinici per la diagnosi: fondamentale è la clinica, ultimamente a questi criteri clinici si aggiungono neuroimmagine,
biochimica e PET funzionale per arrivare prima alla diangosi.
Che esami si fanno?
- Esami che escludano altre cause: carenze nutrizionali (B12 e folati), infezioni, ipotiroidismo.
- Test neuropsicologici
- TAC o RM
- Esame del liquido cerebrospinale
Si stanno cercando marcatori che permettano una diangosi precoce:
- Stadio preclinico: se presente una mutazione monogenica ad altra espressività si può riconoscere già in età giovanile
(ma in Italia il numero di casi è molto basso).
- Stadio di MCI: PET e TAC diventano positivi già prima di questo stadio.
- Stadio di demenza: positività a RM
Trattamento Ha l’obiettivo di cercare una riduzione del peggioramento, deve essere precoce, gestire le complicanze e informare
adeguatamente i caregivers.
I farmaci più utilizzati sono quelli che aumentano il tono dei neurotrasmettitori→ anticolinesterasici che aumentano la
quantità di Ach; i risultati sono modesti.
- Tacrina (abbandonata)
- Rivastigmina
- Memantina: indicazione anche nella fase moderata, agisce a livello di NT bloccando la trasmissione del NMDA (canale
del glutammato), ha un ruolo marginale.
Immunoterapia: idea di contrastare i depositi proteici usando una sorta di “vaccinazione”: si induce nel soggetto una
risposta contro queste proteine o si danno passivamente anticorpi. Forse sono stati somministrati erroneamente in pazienti
troppo avanzati, non hanno ottenuto grossi risultati.
Altri tipi di demenze degenerative:
- FRONTO-TEMPORALI: la più impattante dopo Alzheimer, che ha molti sottotipi.

20
- DEMENZA A CORPI DI LEWY: convivono una forma di disturbo motorio tipo Parkinson associata a una demenza.
- Corea di Huntington
DEMENZE FRONTO-TEMPORALI Sottotipo più frequente è la VARIANTE COMPORTAMENTALE→ disinibizione in alcuni casi o profonda apatia e anedonia in
altri casi con deficit cognitivi che compaiono molto più tardi. Sempre un disturbo psichiatrico bipolare.
Età di esordio più precoce rispetto ad Alzheimer.
Criteri diagnostici • Compromissione cognitiva con prevalente perdita di capacità di giudizio con:
o Disinibizione e impulsività→ regrediscono quasi ad un stadio infantile.
o Alterazione della condotta sociale, perdita dell’identità sociale.
o Ritiro sociale.
• Una o più delle seguenti condotte: continua esplorazione orale e manuale, iperfagia, ecolalia, attività impetuose,
eccessiva giovialità, azioni o linguaggi inappropriati o ad argomento sessuale
• Deficit del giudizio o alterazione del comportamento sproporzionato al deficit mnesico.
Il disturbo comportamentale è molto di più rispetto a deficit cognitivo, cosa che invece è più presente nell’Alzheimer→
nelle prime fasi sono ben distinguibili, nelle fasi successive si sovrappongono.
Visibile degenerazione alle PET
Terapia Sono farmaci simili a psichiatria: stabilizzatore di umore per controllare fasi maniacali (acido valproico, litio), insieme a
farmaci neurobloccanti (aloperidolo).
Nel fenotipo opposto invece si usano anti-depressivi, che però possono slatentizzare un disturbo maniacale.
Patogenesi Non è stato trovato un punto debole, si tratta comunque di una malattia dei microtubuli, è sempre una malattia della
proteina Tau.
DEMENZA A CORPI DI LEWY Malattia della proteina alfa-sinnucleina; proteina che si accumula nel neurone e forma fibrille di oligomeri. Ha un effetto
tossico sui neuroni. Non è così chiaro il motivo per cui in alcuni soggetti si accumuli anche indipendentemente dalla
presenza di mutazioni del gene.
Si accumulano corpi di Lewy si a livello troncoencefalico, sia a livello corticale frontale e parieto-occipitale.
Caratteristiche cliniche ➢ Copresenza di sindrome parkinsoniana e demenza nel primo anno dalla diagnosi→ fondamentale che ci sia subito
demenza, perché nel Parkinson idiopatico può esserci demenza ma si manifesta molto dopo→ il periodo di insorgenza
di demenza è l’unica caratteristica che permette di distinguere le due forme, che altrimenti sono praticamente
identiche (ONE YEAR RULE).
➢ Coinvolge le capacità visuospaziali e cognitive: hanno difficoltà a trovare la via di casa, ma la memoria è conservata.
Per slatentizzare queste caratteristiche si possono fare test con sequenze motorie definite (pugno-taglio-piatto con
la mano velocemente: si confondono subito).
➢ Imponenti sindromi da allucinazioni visive→ succede nel Parkinson avanzato, è come se fosse un forma di Parkinson
accelerato (si vedono entro un anno anziché entro 10 anni).
Sono manifestazioni fluttuanti: non hanno una capacità percettiva costante, spesso verso sera peggiorano le
allucinazioni e vengono descritti degli sbalzi.
➢ Disautonomia: le alfa-sinnucleopatie sono caratterizzate anche da una degenerazione del SNA (pelle, sistema
enterico, …).
DD con forme vascolari→ alcune lesioni ischemiche in zone critiche simulano un quadro di demenza a corpi di Lewy.

21
Può manifestarsi come una degenerazione improvvisa: infatti anche se è già iniziata una degenerazione, spesso si mettono
in atto dei meccanismi di compenso che fanno sembrare il soggetto normale. Basta poi un evento scatenante (intervento,
infezione, trauma, …) che slatentizza tutto, ma era già presente da tempo.
Esame: SPECT→ analizza la capacità di trasportare dopamina, permette di spostarsi verso questa forma di demenza anziché
Alzheimer→ degenerazione nigrostriatale.
Terapia Sono stati proposti con discreti risultati gli stessi farmaci anticolienesterasi idi Alzheimer, ma non cambi in modo così
radicale la malattia→ la mancanza di Ach è comune a tutte le demenze, poco specifico.
Effetto paradosso con farmaci neurolettici (per schizofrenia) antidopaminergici: nella corpi di Lewy a volte possono dare
un peggioramento.
DEMENZA VASCOLARE Data la frequenza di ipertensione, ipercolesterolemia, fumo, …. Spesso la popolazione anziana ha quadri di vasculopatia
cerebrale→ arteriosclerosi accelera anche l’insorgenza di demenze neurodegenerative. Non sempre quindi demenza
vascolare e neuronale pura sono due entità diverse.
Si parla di demenza vascolare quando c’è una chiara evidenza di eventi ischemici→ con TAC o quando il decadimento segue
temporalmente un evento ischemico chiaramente identificato: es. il paziente arriva con ischemia cerebrale e poi manifesta
demenza.
Classificazione • Infarti lacunari
o Assenza di storia di stroke
o Demenza progressiva con deficit focali o demenza a tipo frontale
• Infarto singolo strategico
o Improvvisa sintomatologia afasica, agnosica, amnestica o frontale
• Infarti multipli che si accumulano nel tempo→ deficit cognitivi e motori progressivi ‘a scalini’
Malattia di Binswanger: rarefazione di sostanza bianca dovuta a infarti multipli, ora non più chiamata così
(demenza, apatia, agitazione, segni cortico-spinali/bulbari).
→ deve essere chiara la correlazione di infarti ischemici e demenza!
Diagnosi Anamnesi importante, ma spesso porta fuori strada.
Ideale per un soggetto demente sono le cure domiciliari, in cui cioè il paziente sta in casa sua, con i suoi riferimenti→
portarlo in ospedale non è la cosa ideale!
Analizzare malattie congenite come forale ovale pervio, che può predisporre a infarti cerebrali multipli.
Terapia Ridurre il rischio CV.

22
EPILESSIE Pag. 137
Si parla di “epilessie” in quanto esistono molte forme diverse.
Il nome deriva dal greco e significa “colti di sorpresa” dalla crisi epilettica; è infatti un evento parossistico, improvviso, che
irrompe e interrompe la vita normale.
Nell’antica Grecia veniva considerato un segno infausto se qualcuno durante un comizio veniva preso da un attacco
epilettico, e bisogna chiudere il comizio→ per questo motivo a volte viene chiamata crisi comiziale.
È uno stigma sociale, viene nascosto con vergogna.
Esistono rari casi di morte improvvisa inattesa in soggetto con epilessia, soprattutto se con crisi convulsive.
Il 30% dei casi è farmaco-resistente.
Epidemiologia. 50 milioni di malati nel mondo nel 1997, ora sono aumentati. La prevalenza è di 2/100 in Italia.
L’età più colpita non è l’età infantile, ma quella anziana; in età infantile c’è un picco di crisi epilettiche ma l’età più colpita
è quella avanzata→ con l’età aumentano le malattie neurologiche e ciascuna malattia (tra cui quelle degenerative) può
essere causa di epilessia.
Come mai anche nei bambini c’è una frequenza maggiore?
- Malformazioni dello sviluppo cerebrale
- Parto
- Infezioni
Vi è una lieve cunetta nell’età adolescenziale.
TERMINOLOGIA CRISI EPILETTICA: è una manifestazione clinica
parossistica dovuta ad una scarica anomala ipersincrona di una popolazione più o meno estesa di neuroni corticali (in realtà
non solo corticali). Può anche essere unica e può essere provocata (es. da una crisi ipoglicemica da eccessiva
somministrazione di insulina in un diabetico)→ non è sinonimo di convulsioni, ma si può manifestare in vari modi.
Il concetto di crisi epilettica come scarica elettrica parossistica è stato concepito prima che fosse inventato l’EEG (da
Jackson). L’EEG è stato scoperto nel 1929.
EPILESSIA: ripetersi spontaneo delle crisi, senza un evidente fattore che le provoca.
Se si cerca di situare la scarica anomala in determinate aree del cervello, si riesce a giustificare gli effetti di tale scarica; le
strutture a cui parlano i neuroni (che scaricano troppo) saranno stimolate durante la crisi: Es: se “lavora” troppo la corteccia
motoria pre-Rolandica, come manifestazione avremo movimenti violenti, involontari→ clonie. Se “lavora” troppo la
corteccia post-Rolandica sensitiva, come manifestazione avremo parestesia di vario tipo, nell’emisfero controlaterale.
Rare volte, l’iperfunzione parossistica di un gruppo neuronale produce un effetto inibitorio→ le crisi di ipoestesia sono più
frequenti, ma sono invece molto rare crisi di inibizione motoria, di paresi.
CLASSIFICAZIONE DELLE CRISI EPILETTICHE Una crisi epilettogena che parte dall’omuncolo
pre-rolandico genera una CRISI PARZIALE: è
l’attivazione solo di un’area della corteccia.
Se la crisi coinvolge tutta la corteccia avremo
CRISI GENERALIZZATE (la tipica convulsione).
Una crisi che iniziata come parziale veda una
diffusione delle attività elettriche patologiche
tale per cui in un secondo momento diventa
generalizzata→ CRISI PARZIALE SECONDARIAMENTE GENERALIZZATA.
Sono più frequenti le crisi parziali.

23
CRISI GENERALIZZATE Scarica elettrica anomala che coinvolge tutto l’encefalo. Ce ne sono di vari tipi ma dobbiamo conoscerne (almeno) 3:
1) CRISI TONICO-CLONICA GENERALIZZATA o CONVULSIVA
2) ASSENZE
3) MIOCLONIE
CRISI TONICO-CLONICA GENERALIZZATA Una volta veniva definita GRANDE MALE.
• Fase tonica: arti estesi, ipertonia.
• Fase clonica: scosse che seguono l’ipertonia, sembra risolversi ma poi compaiono le tonie.
Possibili manifestazioni:
- Spesso il paziente diventa cianotico, anche perché nella fase tonica l’irrigidimento muscolare è generalizzato, non
si respira correttamente.
- Non si deglutisce la saliva quindi si manifesta scialorrea.
- La lingua può rimanere intrappolata nell’arcata dentaria.
- Urlo: verso l’inizio della crisi può esserci un’emissione vocale incontrollata.
- Incontinenza sfinterica
- Perdita di coscienza (sempre).
Durata: 4-5 minuti massimo, poi tende a terminare da sola. Il paziente non si riprende subito, per diverse decine di minuti
sarà confuso e disorientato, soporoso. La fase post-critica dura più della crisi stessa ed è un carattere distintivo della crisi
tonico-clonica, permette di distinguerla da altre crisi che possono mimarla, come le crisi psicogene→ in queste crisi, al
risveglio il paziente lamenta affaticamento, cefalea, all’esame della CK si troverà un valore aumentato.
Assistenza in una crisi convulsiva: si può far ben poco:
• Allontanare gli oggetti che possono essere fonte di trauma
• Mettere qualcosa di morbido sotto la testa
• Non mettere niente in bocca, può causare traumi perché le contrazioni muscolari che coinvolgono anche i muscoli
masticatori sono molto forti. A volte il paziente subisce fratture dovute alla violenta contrattura muscolare.
• Se in ospedale: si può fare iniezione di 10 mg di diazepam in 1 minuto (bolo).
I problemi iniziano quando la crisi non passa: bisogna misurare il tempo, se sconfina oltre i 5 minuti si parla di STATO DI
MALE EPILETTICO; a volte sembra finire ma poi ricomincia con violenza analoga alla precedente. È una vera emergenza
medico-neurologica→ portarlo subito in rianimazione.
Se il soggetto è un noto epilettico con crisi frequenti, alla fine di una crisi non serve nemmeno portarlo in PS; se invece si
tratta di una prima crisi è sempre da portare in PS.
ASSENZA SEMPLICE Una volta chiamata PICCOLO MALE.
Sono eventi che colpiscono maggiormente l’età infantile.
Video-EEG: studio combinato di EEG in simultanea all’evento clinico.
Si tratta di crisi molto brevi (pochi secondi) in cui il soggetto perde il contatto con l’ambiente e sospende l’attività motoria
improvvisamente.
Il segno EEG classico è la scarica di punta-onda alla frequenza di 3/4/5 cicli al secondo, sincrona e simmetrica.
Per indurla si può indurre un’alcalosi tramite iperpnea (soffiare una girandola)→ iperpnea è un potente evento scatenante
delle assenze.
Procedure di attivazione della crisi epilettica, valido in molte forme di epilessia (in alcune più efficaci, in altre meno):
1) Deprivazione di sonno
2) Stimolazione luminosa intermittente
3) Iperpnea: molto potente nelle assenze semplici.
Si ritrovano in diverse sindromi epilettiche; il modello principale in cui si trovano assenze è la EPILESSIA DELL’INFANZIA
CON ASSENZE SEMPLICI: caratteristiche:

24
→ colpisce bambini nella prima età scolare.
→ Frequenza delle crisi: sono tantissime, anche 50-100 al giorno, brevissime.
→ Alterano il comportamento del bambino soprattutto in fase di apprendimento: infatti si perdono dei pezzi della
comunicazione che stano ricevendo. Di solito sono le maestre che si accorgono che qualcosa non va.
→ L’esame neurologo è normale.
→ Necessitano di terapia, di solito molto efficace.
→ Al sopraggiungere dell’età adulta nella maggior parte dei casi sarà senza crisi, quindi si può pensare di scalare la
dose del farmaco fino a sospenderlo→ nella maggior parte dei casi guarisce.
CRISI MIOCLONICA Sono mioclonie: scatti violenti che coinvolgono i segmenti corporei; più facili da vedere agli arti superiori.
Una singola mioclonia ha una durata brevissima (frazioni di secondo), possono susseguirsi anche più mioclonie.
Tipiche nella SINDROME MIOCLONICA GIOVANILE:
→ Si verificano tipicamente dopo il risveglio
→ Più colpite le femmine intorno all’età adolescenziale.
→ In questa sindrome epilettica si ritrovano tutte e 3 le forme di crisi generalizzate: tonico-cloniche, assenze e
miocloniche.
→ Si riconosce un’eziopatogenesi genetica: sono stati identificati molti geni candidati. Non vi è un’eredità mendeliana
pura, è più multifattoriale, a penetranza incompleta.
→ Rispondono alle terapie me la terapia è a vita: se la cessano, il rischio di recidive è molto elevato. Farmaco principale
prima era il valproato di sodio, ora invece gli sono stati imputati effetti teratogenici quindi è stato sostituito.
Queste forme con queste caratteristiche si raggruppano sotto il nome di EPILESSIE GENERALIZZATE IDIOPATICHE.
Fattori scatenanti le crisi miocloniche:
- Esposizione a luci intermittenti→ sono pazienti fotosensibili. Le sale degli EEG sono tutte dotati di lampada
stroboscopica (15-20 Hz). Infatti le crisi si manifestano in discoteca, durante la guida su rettilinei con alberi, per cui
si verifica un’alternanza di luce e ombra ritmica. Schermi televisivi: l’immagine dei display in realtà oscilla, non è
fissa→ fotosensibilità degli schermi televisivi.
- Visione di forme o colori: sono condizioni complesse e particolari (riguarda più altre epilessie).
- Deprivazione di sonno: sono soggetti che devono cercare di dormire il giusto.

25
CRISI PARZIALI Se il paziente è perfettamente vigile e responsivo durante le clonie, si parla di crisi parziali semplici. Se invece manifesta
una compromissione della coscienza si parla di crisi parziali complesse.
Le modificazioni della coscienza sono spesso graduate, non è detto che la coscienza è integra o è perduta, esistono molte
sfumature in mezzo.
Alcune crisi parziali sono facili da capire (motorie semplici, somato-sensoriali semplici), altre sono più difficili da individuare;
normalmente però il soggetto rimane cosciente.
CRISI DEL LOBO TEMPORALE È un quadro complesso dovuto alla complessità del lobo temporale stesso; non c’è univocità di funzione (come per
homunculus sensitivo e motorio), fa molte cose diverse. La scarica elettrica, alla base della crisi, può interessare anche altre
aree dando una manifestazione clinica variabile da un soggetto all’altro, con diversa gamma di estensione: la crisi tipica di
una sclerosi ippocampale può avere crisi dismnesiche, cioè manifestare un vissuto laddove non c’era (ha la netta sensazione
di aver già vissuto quella esperienza)→ CRISI PARZIALE SEMPLICE DI TIPO DISMNESICO. Sono eventi che possono succedere
anche in soggetti sani, ma devono preoccupare quando sono più frequenti o cominciano a comparire quando prima non
avvenivano.
Estensione della crisi. Spesso ci possono essere altre manifestazioni che causano una estensione della crisi, che in
ordine possono essere:
1. AURA EPIGASTRICA: può precedere il dejà-vu, ed è una sensazione sgradevole e ingiustificata di violento disagio che
dalla regione epigastrica tende a salire verso il capo, come una nausea. È già una crisi vegetativa.
2. Già vissuto
3. TURBA DELLA COSCIENZA: seguono il già vissuto, è un restringimento cognitivo con distacco dall’ambiente per
qualche secondo o pochi minuti, si ha un arresto psicomotorio: se chiamato non risponde (se alla guida, si schianta)→
crisi parziale complessa a contenuto vegetativo e dismnesico. “Complesso” indica il coinvolgimento della coscienza
in una crisi parziale; non deve per forza essere perduta la coscienza, può essere apparentemente cosciente ma è
irraggiungibile, è una diminuzione di coscienza. In alcuni casi il soggetto è consapevole del suo stato di
irraggiungibilità, altre volte no. Un testimone in questi casi è indispensabile per riuscire a identificare questa
condizione.
4. CRISI TONICO-CLONICA GENERALIZZATA SECONDARIA
→ 4 stadi, che possono manifestarsi tutti oppure no. A volte questi stadi possono essere saltati.
Possono associarsi anche ALLUCINOSI:
- Allucinosi olfattivo-gustative: nel lobo temporale mesiale vi è la percezione olfattiva, quindi ci possono essere
allucinosi olfatto-gustative→ cacosmia: sensazione di gusti e odori improvvisi cattivi.
- Allucinosi acustiche, infatti nel temporale laterale vi è l’area acustica. Si può manifestare come una voce, una musica,
spesso non distinguibile ma stereotipato, sempre uguale a se stesso (a differenza della schizofrenia dove vi sono voci
a contenuto persecutorio) e di breve durata. Es: sente un frammento di inno nazionale, una parola, ma è sempre
quella per lo stesso paziente.
- Allucinosi visive complesse: infatti dalle aree visive occipitali partono proiezioni verso il lobo temporale. Sono sempre
di breve durata e stereotipate nella presentazione.
Le regioni temporo-mesiali sono molto vicine ai gangli della base: strutture grigie profonde (nucleo pallido, putamen,
caudato, claustro, …) che hanno prima di tutto un significato motorio, della motricità schematica innata o appresa. Possono
quindi esserci anche MANIFESTAZIONI DI TIPO MOTORIO→ non sono scosse, riguardano questi schemi motori primitivi;
• alcune crisi possono essere caratterizzate da masticazione, succhiamento (automatismi oro-alimentari), a mimare
l’atto di alimentarsi o di cucciare latte.
• Strofinio di parti del viso o del corpo (tipico è del naso).
• Automatismi gestuali di vestizione o manipolazione dei vestiti (es: non risponde più e continua a sistemarsi i
bottoni della camicia).
Anche la SFERA AFFETTIVA può essere un bersaglio della crisi del lobo temporale:
- Si manifesta come una ingiustificata sensazione di estremo benessere e felicità, come un flash da stupefacenti→
estasi.

26
- Può manifestarsi anche come panico epilettico, come la paura di morire.
TURBE DEL LINGUAGGIO: l’area di Wernicke (22) si trova qui quindi può presentare deficit anche del linguaggio; sbaglia le
parole e le sillabe, capisce ma non riesce a esprimersi. Non è un’afasia che dura ore, dura solo pochi minuti o secondi.
→ Distinguere la crisi del lobo temporale è molto difficile se non si conosce l’anatomia e la fisiologia del lobo temporale.
In rarissimi casi, durante una crisi del lobo temporale si può manifestare una DEAMBULAZIONE involontaria e ingiustificata
(si alza e se ne va). Spesso questa cosa è stata usata in medicina legale per tentare di discolpare un imputato.
EEG: Onde cerebrali: la prima metà superiore indica il lobo destra, la seconda metà inferiore indica il lobo sinistro.
Spesso l’EEG causa artefatti in quanto è uno strumento molto sensibile; in alcuni casi però anche cercando di cancellarli non vanno via,
spesso sono dovuti ai muscoli masticatori.
È raro vedere tante manifestazioni in uno stesso soggetto in più a coscienza piena; se non conosciute bene o senza EEG possono essere
facilmente confuse con un disturbo psichiatrico.
CLASSIFICAZIONE DELLE EPILESSIE Le epilessie si possono classificare come:
- Idiopatiche
- Criptogeniche: da causa ignota.
- Sintomatiche: spesso le epilessia tradiscono la presenza di un processo patologico.
Epilessie idiopatiche. Sono quelle epilessie in cui il fattore genetico ha un ruolo determinante, sono genetiche (ma
non eredo-familiari!). Sono causate da difetti genetici che causano uno scorretto funzionamento dei canali ionici (di sodio,
calcio, potassio) a livello cerebrale. Sono età-dipendenti, si localizzano tra infanzia e adolescenza:
- Assenze: si manifestano come il bambino che rimane indietro a scuola ogni tanto
- Mioclonica giovanile: la ragazzina che passa la notte insonne in discoteca e il giorno dopo si manifesta la crisi.
- Sindrome con crisi di grande male soltanto: convulsione pura durante la notte o al risveglio.
Che farmaco diamo? Se è maschio, Valproato.
Epilessie sintomatiche. Sono chiamate anche strutturali (dovute a una alterazione di struttura) o metaboliche
(dovute ad alterazioni metaboliche); può essere suggerita da:
- Segno patologico all’esame obiettivo neurologico: ad esempio sfumata emiparesi sinistra.
- Segno radiologico
Epilessie criptogeniche. Epilessie di cui, pur essendo sospettabile in base ai dati clinici una lesione sottostante, essa
non è documentabile mediante le indagini diagnostiche (es: neuro-imaging) disponibili. Potrebbero essere definite come
“probabilmente sintomatiche”.
DIAGNOSI Ci sono dei confondenti: alcuni escono con diagnosi di epilessia ma non lo sono. L’errore più comune è prendere per
epilettiche delle crisi che non lo sono; per evitare questo bisogna fare una corretta anamnesi:
• Descrizione della crisi, il più possibile attendibile→ servono TESTIMONI, soprattutto se non è una crisi convulsiva
(che lascia dei segni come morsus, rilascio sfinterico con vestiti bagnati, cefalea persistente post-crisi, aumento
della CK) e se il paziente non rimane vigile così da poterla descrivere.
• Sintomi prima, durante e dopo
• Durata crisi

27
• Pronta ripresa di coscienza
• Circostanze scatenanti (alcool, farmaci, traumi cranici deprivazione sonno)
• Morsus, incontinenza, traumi.
DIAGNOSI DIFFERENZIALE Può succedere che sia confusa per epilessia una SINCOPE, dovuta a un ipoafflusso di sangue al tessuto cerebrale. Si
manifesta con perdita coscienza e tono posturale, caduta, di solito autolimitante: pronta ripresa spontanea.
Si crede che quando si perde la coscienza per via di una sincope, si rimanga immobili; in realtà succede l’opposto, nel 90%
dei casi c’è una attività motoria con clonie non così tanto evidenti o evidenti di vario genere→ non sta fermo, si muove
anche se impercettibilmente. In questi casi, se non si riconosce che è una sincope si tende a far fare l’EEG: il problema è
che però anche in persone normali ci possono essere delle attività simili a quelle epilettiche (soprattutto in chi soffre di
cefalee ed emicrania): si tende quindi a fare diangosi in condizioni normali.
→ tenere presente che in una sincope SI POSSONO AVERE MOVIMENTI CLONICI, che non sono indicazione di
epilessia!
→ se si sospetta una sincope, NON SI RICHIEDE EEG!
È stato fatto un esperimento in cui si induceva sincope in un gruppo di studenti, inducendo bradicardia tramite
iperventilazione e abbassando la pressione tramite esercizio fisico.
Morsus: nella crisi epilettica è più comune e avviene di solito a lato, mentre nella sincope è più raro e avviene più sulla
punta.
CRISI PSICOGENE Sono manifestazioni funzionali, isteriche. Chiamate anche di conversione.
• La stereotipia è un marcatore delle crisi epilettiche, cioè che all’interno dello stesso soggetto sono sempre uguali; nelle
crisi psicogene manca la stereotipia, questo ripetersi sempre uguale viene a mancare→ ogni volta una crisi diversa
(clonie diverse, a volte assenze). Oppure spesso vengono descritte clonie che non seguono i normali schemi anatomici
(es. arto superiore destro e arto inferiore sinistro).
• Durata: possono essere anche molto più lunghe di 5 minuti→ in epilessia questo significherebbe uno status di male
epilettico.
• Crisi epilettiche e pseudocrisi possono coesistere: sono spesso presentii in pazienti con epilessia.
• Sensibili alla suggestione, è facile indurle tramite inganno al paziente: bisogna fargli credere che una determinata
procedura scatenerà una crisi e che poi verrà messa in atto una contro-procedura per farla smettere.

28
Es: flebo fisiologica che viene fatta credere essere epilettogena→ si bypassa il consenso del paziente, quindi in realtà
è una procedura molto discussa. Si può fare la stessa cosa ma con due cerotti colorati diversi (meno invasiva e non ha
il problema del consenso).
Semiologia dell’evento. - Scarsa stereotipia comportamento
- Scosse diffuse, spesso a coscienza integra
- Scosse alternanti, asincrone
- Scosse agli arti con ipotonia
- Scosse pelviche, inarcamento del tronco
- Chiusura forzata occhi
- Improbabili prolungata confusione, dolenzia muscolare ed elevazione CK postictale
Diagnosi. Si ricorre alla video-EEG→ si registra la crisi tipica del paziente (e non una nuova bizzarra) e vedere che nel
corso della crisi l’EEG è assolutamente normale.
Elemento cruciale: mancanza di correlato EEG a fronte di una crisi con caratteristiche “atipiche”, abituale per il paziente.
STATO DI MALE EPILETTICO È una crisi epilettica che non finisce entro 4-5 minuti normali; se si tratta di una crisi convulsiva, dopo i 5 minuti bisogna
entrare in allarme, infatti i modelli animali di questa condizione indicano che trascorsi 20 minuti (30 per il libro) c’è il pericolo
che si verifichino lesioni cerebrali legate all’anossia.
Bisogna subito trasportare in un ospedale con una rianimazione.
Terapia. Il primo approccio è la somministrazione di benzodiazepine ev in bolo (massimo in 1 minuto di infusione), come
Diazepam (Valium) o Lorazepam o Midazolam (per via endonasale, ha un alto assorbimento attraverso la mucosa).
Se non funziona, c’è una seconda fase farmacologica, dove il protagonista è la fenitoina: viene somministrato velocemente
ma non tanto quanto il lorazepam. Oppure dopo le BDZ si può usare Levetiracetam. Un altro nuovo farmaco è la
Lacosamide.
→ riassumendo, ci sono due stadi farmacologici:
1) BDZ, anche ripetute
2) si passa a Fenitoina o Lebetiracetam o Lacosamide (uno solo).
Se non passa nemmeno con questi altri farmaci, ci si inizia a preoccupare. Interviene il rianimatore, si seda, si intuba e si
somministrano altri farmaci ancora: Barbiturici, anestetici (Propofol)→ mettere il paziente in narcosi, in cui rimane per un
tempo indefinito, durante il quale si fanno EEG per stabilire se persiste l’attività epilettica o no; se a un certo punto l’attività
epilettica diminuisce si alleggerisce la narcosi. Ovviamente non si può rimanere intubati per un tempo troppo lungo, ci sono
effetti collaterali già dopo qualche ora.
Ci sono diversi tipi di stati di male, esiste praticamente uno stato di male epilettico per ogni tipo di crisi epilettica:
• Esiste la versione “lunga” dell’assenza, in cui si manifesta un blocco psico-motorio indefinito, non più solo di qualche
secondo. È meno grave di uno convulsivo.
• È possibile anche uno stato parziale epilettico, come ad esempio una crisi del lobo temporale prolungata→ si manifesta
come uno stato confusionale acuto (detto anche DELIRIUM), agitato o soporoso; deve essere differenziato da altri tipi
di stato confusionale.
Ci sono tantissimi tipi di delirium, come si differenziano? Con un EEG: diremo che si tratta di uno stato di male epilettico
solo se ci sono manifestazioni sull’EEG; un delirium non epilettico non dà alterazioni all’EEG ma dà solo un
rallentamento (un delirium tremens ad esempio avrà solo un rallentamento all’EEG, non un’alterazione).
Patente di guida. Si può rinnovare la patente A e B se non ci sono state crisi per un anno (indipendentemente da
farmaci). Ci sono alcune eccezioni→ Es: sta bene da tanti anni, per cui si è deciso di scalare la terapia o sospenderla del
tutto; se dopo la sospensione compare una nuova crisi questa non conta ai fini della patente (quindi può essere rinnovata)
perché può essere considerata iatrogena (ovviamente si ripristina la terapia iniziale).
Patente C viene tolta anche per un’unica crisi epilettica o dubbia crisi.

29
Gravidanza. Non serve nessun intervento medico quando la paziente si presenta al sesto mese di gravidanza senza
nessun problema; infatti dopo i 3 mesi iniziali non c’è più rischio di malformazione fetale dovuta dai farmaci.
Il problema è se si vuole programmare una gravidanza laddove c’è una terapia con acido valproico→ è responsabile di
malformazioni e alterazioni di sviluppo mentale. Bisogna quindi cercare di ridurre il numero di farmaci e cercare di ridurre
la dose il più possibile. Si somministra acido folico e si avverte che alcuni farmaci passano nel latte quindi bisogna stare
attenti durante l’allattamento (fenobarbital passa, il levetiracetam no).
La probabilità di malformazioni fetali nella donna non epilettica è di circa 1%; nella donna con epilessia questa percentuale
raddoppia.
→ non si sospende mai la terapia del tutto perché se dovesse sopraggiungere uno stato di male epilettico in gravidanza
questo sarebbe molto più grave per il feto che i farmaci stessi.

30
ICTUS In Italia ci sono circa 200-300 nuovi casi di ictus ogni 100.000 abitanti all’anno.
La trombolisi è la terapia nuova più utilizzata ora, che può essere sia interventistica che farmacologica; questo ha rilanciato
molto il problema ictus, prima veniva disperso un po’ in tutti i reparti mentre ora è un caso trattato prettamente dal
neurologo→ le neurologia dovrebbero tutte avere la stroke unit semi-intensiva.
Uno dei problemi alla base dell’ictus è differenziare se sia ischemico o emorragico, e non è tanto difficile farlo.
Il sangue si vede bene alla TC perché appare bianco nel parenchima nero; se non si vede un’emorragia sospetto
un’ischemia→ l’ischemia non si vede subito con TC basale, molto spesso si vedono solo dei segni che lasciano presagire al
presenza di ischemia, dovrei aspettare ore/giorni prima di vedere qualcosa alla TC. Quindi una TC pulita permette
soprattutto di escludere un’emorragia.
EMORRAGIE INTRACRANICHE Comprendono sia le emorragie intracerebrali, le subaracnoidee e quelle di origine traumatica.
Noi parliamo specialmente di emorragie intracerebrali e subaracnoidee.
INTRACEREBRALI Ce ne sono di diversi tipi, almeno 2:
1. Primaria o “a sede tipica”: talamo, gangli della base (nucleo pallido, capsula interna, nucleo caudato, talamo), ponte
e cervelletto (strutture profonde).
2. Secondaria o “a sede atipica”: sostanza bianca nella profondità degli emisferi, nel lobo frontale (sede più lobare).
INTRACEREBRALI A SEDE TIPICA È tipica di chi soffre di ipertensione: è dovuta alla rottura di determinate arteriole (chiamate perforanti) che hanno la
particolarità di nascere con una direzione perpendicolare rispetto all’arteria più grande di origine; questo assetto determina
peculiari turbolenze nel flusso arterioso, che nel contesto di un quadro ipertensivo cronico portano a una degenerazione
della parete arteriolare. Si generano dei fisiologici sfondamenti del profilo di questi vasi (microsacche aneurismatiche)
destinate a rompersi in un dato momento per motivi imprevedibili.
Se il sangue sfonda la parete del ventricolo si parla di emoventricolo; in più la linea mediana può “arcuarsi”, dovuto
all’ingrossamento di tutto l’emisfero con una spinta in senso controlaterale→ arriva a premere sulle strutture mediane.
Intorno all’emorragia ci sarà di sicuro un alone di edema, che aiuta in questo processo di spinta→ si parla di EFFETTO
MASSA, con spostamento delle strutture della linea mediana. È un segno prognostico negativo, perché può annunciare una
compressione sulle strutture del tronco cerebrale.
Questo effetto massa si ritrova nei tumori o negli ascessi cerebrali (tutte condizioni che generano edema).
Quadro clinico Si manifesta come un evento improvviso, tanto da essere difficilmente accettata. Avrà un difetto a seconda della zona
colpita:
➢ In caso di emorragia capsulare si avrà come esito un’emiparesi o emiplegia facio-brachio-crurale. L’emiplegia sarà
dell’emivolto nella zona inferiore.
PARALISI DEL NERVO FACCIALE vs PARALISI CENTRALE
In caso di ictus centrale, avremo la paresi della parte inferiore del volto (solo della bocca ma non dell’occhio) e degli
arti di un lato solo. In caso di paralisi del facciale invece viene compromesso tutto l’emivolto ma non degli arti.
Questo avviene perché i muscoli dell’emivolto superiore sono innervati anche dal nervo controlaterale, quindi nel
momento in cui un lato viene danneggiato, il movimento vinee vicariato dal sistema cortico-spinale controlaterale.
➢ Emianestesia della parte controlaterale.
➢ Emianopsia laterale omonima: in caso di danno a destra, si avrà la perdita del campo laterale dello stesso lato
(guardare bene questione di campi visivi e incrociamento ottico).
➢ Afasia, se coinvolge il lato sinistro.
➢ Disturbi della vigilanza: spesso hanno un certo grado di sopore, o sopore misto a coma, o agitazione.
➢ Cefalea, vomito→ segni di ipertensione endocranica.

31
➢ Molte volte c’è una deviazione del capo e dello sguardo del paziente: il capo sarà ruotato preferenzialmente come
a distogliere lo sguardo dagli arti plegici, quindi verso la lesione.
➢ Crisi epilettiche: il sangue inoltre è un elemento molto irritante dal punto di vista elettrico, sia in fase acuta ce in fase
cronica (anche i suoi prodotti di degradazione), uqindi è molto epilettogeno.
➢ Segni meningei: se l’emorragia lesiona il sistema ventriclare e il sangue si infiltra negli spazi subaracnoidei (si parla di
emorragia cerebromeningea), genera dei segni affini alle meningiti.
Domanda d’esame: cosa ti interessa sapere in un paziente in coma con emiplegia?
1) Da che lato è?
2) Se il paziente è destro o mancino, perché posso predire se ci sono o meno disturbi del linguaggio→ prima riga dell’esame
obiettivo! È sempre meglio che ce lo abbia a destra, perché più del 90% ha la sede del linguaggio a sinistra.
Il quadro clinico ovviamente dipende dalle dimensioni dell’emorragia.
Se è piccola e limitata a un solo distretto, ad esempio il talamo, ci saranno alcune particolarità legate proprio alla sede
colpita.
Neurochirurgia Se si tratta di una grande emorragia capsulare, non si opera; si è visto infatti che agire chirurgicamente sulle grandi
emorragie intracerebrali profonde non ha un rapporto costo/beneficio tale da giustificare la chirurgia. Casi in cui la chirurgia
può essere un’opzione:
- Emorragie atipiche di piccole dimensioni più vicine alla corteccia (non indicato per le sedi tipiche-profonde).
- Emorragie cerebellari in rapida espansione superiori a 3 cm del cervelletto con un rapido deterioramento delle
condizioni cliniche del malato (per cui si tenta tutto il possibile)→ vedi tabella sul libro.
In caso di lesione massiva, non si può fare altro se non ricoverare in stroke unit, dare antiedemigeni, mantenere sotto
controllo la pressione arteriosa cranica e aspettare.
GESTI TRANSITIVI: sono gesti che interagiscono con il mondo esteriore attraverso un oggetto.
INTRACEREBRALI A SEDE ATIPICA Sostanza bianca nella profondità degli emisferi. La causa principale sottostante non è più l’ipertensione, ma una miscellanea
di tante condizioni (non tutte da sapere):
1. Malformazioni artero-venose (MAV) e aneurismi: gomitoli vascolari anomali in cui si instaura uno shunt precoce
tra il circolo arterioso e venoso → si passa da una parete atta a contenere la pressione del sangue a una che non
lo è senza passare dai capillari, quindi c’è possibilità di rottura.
2. Angiopatia amiloide dell’anziano: sede di accumulo di sostanze patologiche anomale come amiloide.
3. Abuso di sostanze simpaticomimetiche: come MDMA, alto rischio di emorragia cerebrale per una serie di motivi:
a. Aumentano pressione arteriosa
b. Azione tossica diretta sulla parete vasale
4. Trombolisi per stroke ischemico: il principale effetto collaterale di una trombolisi è la trasformazione emorragica
di un infarto ischemico; a volte questa trasformazione avviene spontaneamente.
5. Tumori: alcuni possono sanguinare. Si nota una sostanza disomogenea al suo interno, con aspetto cistico.
6. Vasculiti: connettiviti come il LES, PAN, arterite temporale, infatti la flogosi dà indebolimento del vaso con possibile
spandimento di sangue. Questa flogosi genera anche trombosi, per cui può dar vita anche a un ictus ischemico.
7. Trombosi seni venosi
Angiopatia amiloide cerebrale Emorragie cerebrali a sede atipica, per lo più coesistenti con multiple ischemie. Infatti i vasi sono alterati dall’accumulo di
amiloide quindi all’ischemia, ma sono più suscettibili alla rottura.
Tipica di soggetti con decadimento cognitivo; nell’iter diagnostico della demenza infatti si arriva a un certo punto in cui si
fa RMN→ evidenzierà segni di ischemie e di emorragie. Questa diagnosi è importante perché prevedere la
controindicazione all’acido acetilsalicilico.
Già nella popolazione generale, in caso di profilassi con ASA c’è un aumento di rischio del 10% di avere emorragie cerebrali.

32
Trombosi seni venosi Il circolo venoso fa capo alla giugulare interna, a cui le vene
encefaliche si collegano tramite dei seni venosi (RIPASSARE QUALI
SONO). Questi seni possono trombizzarsi, questo quadro clinico è
tipico di pazienti femminili, in età fertile, durante il puerperio (quindi
subito dopo il parto).
Ci saranno manifestazioni come crisi epilettiche prima mai
manifestatesi, segni focali (segni neurologici relativi a una
determinata sede: variano a seconda della sede occlusa -afasia,
emianopsia, emiparesi), turbe della vigilanza, cefalea più o meno
violenta.
Una ischemia venosa è chiamata anche stagnante perché impedisce
al sangue di refluire via; quindi pur essendo una ISCHEMIA venosa si
tratta di un infarto rosso, quindi si configura come una EMORRAGIA.
Magari poi il quadro si stabilizza un po’.
L’ostacolato deflusso venoso si traduce in un aumento della pressione endocranica secondaria; una delle prime “vittime”
è la papilla ottica→ edema della papilla che se non contrastato porta i un tempo relativamente breve a difetti del campo
visivo fino alla cecità.
Come si interviene? Con anticoagulanti, nonostante sembra una emorragia.
Come si dimostra questa trombosi, al di là dei dati clinici e anamnestici? Angio-RMN→ sono sequenze speciali che
consentono di vedere l’albero vascolare.
EMORRAGIA SUBARACNOIDEA Tre strati di meningi:
- Dura madre
- Aracnoide: ragnatela connettivale che collega i due
- Pia madre
Gli spazi che si formano nell’aracnoide contengono normalmente il liquor, ma in
senso patologico possono ospitare anche sangue in caso di rottura delle arterie che
decorrono qui dentro.
Nella maggior parte dei casi si rompono per presenza di aneurismi, che una volta si
definivano “congeniti”: in realtà non è vero, perché non ci si nasce ma sono
acquisiti. Accrescono nel corso della vita per manifestarsi poi più frequentemente
verso i 55-60 anni. In una minoranza di casi, sono a base eredo-familiare nel
contesto del rene policistico (ma non solo). Hanno una forma regolare, quasi
sferica, per cui sono chiamati SACCULATI.
Li si trova a carico di grossi vasi arteriosi del circolo del Willis, più frequentemente
nell’arteria cerebrale anteriore specialmente nella comunicante. A volte
continuano a ingrandirsi senza rompersi fino ad assumere dimensioni notevoli tali
da dislocare le strutture vicine e avere un effetto massa→ capacità di dislocare il
parenchima circostante a una determinata lesione, come veri e propri tumori.
Altri tipi di aneurismi:
- MICOTICI: sono sfiancamenti dovuti all’arrivo sulla parete arteriosa di emboli settici.
- ATEROSCLEROTICI:
- SINE MATERIA: a volte non si trova l’aneurisma, perché essendo talmente piccolo dopo essersi rotto non è più
riconoscibile.
Come si vedono? Con angio-TC, con una TC normale non si vedono a meno che non siano enormi.
Sintomi • Esordio repentino con violentissima cefalea: è una cefalea mai sperimentata prima, localizzata posteriormente (nuca,
collo, rachide cervico-toracico), a “colpo di pugnale” alla nuca. Solitamente c’è un evento scatenante, che deve aver
aumentato la pressione intracranica: torchio, esercizio fisico intensissimo, coito, tosse….

33
• Sintomi sentinella (warning signs): cefalea violenta e impressiva che insorge per la prima volta in età adulta, poi
regredisce.
• Alterazione dello stato di coscienza: soporosa, priva di coscienza, a volte in coma. Se non è in coma sussulterà a ogni
minio rumore, vomiterà, fotofobico.
• Segni meningei: il segno più affidabile è la rigidità nucale: in caso di paziente in coma bisogna comunque fare l’EO e si
può valutare la rigidità.
Diagnosi strumentale TC basale è un ottimo mezzo per individuare emorragia subaracnoidea: il sangue infatti si vede bene, è bianco dove non
dovrebbe.
A questa seguirà un angio-TC per dimostrare immediatamente un aneurisma.
In qualche raro caso, la TC non dimostra sangue: è poco oppure l’operatore che referta non è familiare (es il sangue non è
evidente).
Se la TC non dimostra nulla, si pensa alla meningite (DD). Nei casi di TC negativa ma rigidità nucale si preleva il liquor: in
caso di Emorragia non sarà limpido, ma apparirà diluito, rossiccio, mentre nei giorni successivi diventerà giallino.
Cosa si indaga? Esame chimico-fisico: quantità di proteine, di glucosio, globuli rossi, granulociti.
→ i segni meningei sono comuni a meningite e emorragia subaracnoidea.
Complicanze Non sono quadri benigni; a volte si risolvono a volte no.
o Sanguinamento che sembrava cessato può riprendere; avviene in fase abbastanza precoce e ce ne si accorge per il
deterioramento cognitivo del paziente.
o Vasospasmo: spasmo a carico delle grande arterie cerebrali legato alla presenza di sostanze chimicamente attiva che
derivano dalla lisi del sangue extravasato→ coesistenza paradossale di ischemia e emorragia.
o Idrocefalo
o Cardiologiche:
▪ Aritmie
▪ Danno miocardico di tipo ischemico (fino a infarti cardiaci a coronarie indenni)
▪ Edema polmonare.
o Iposodiemia (SIADH)
o Epilessia e stato di male
(no prognosi e scala di Hunt)
Terapia Quando le condizioni del paziente lo consentono, gli approcci possibili sono 2:
- radiologico interventistico: molto meno invasivo e meglio sopportato. Consiste in una procedura angiografica (pungendo
arteria femorale), si risale e si arriva in vicinanza di aneurisma: si utilizzano particolari dispositivi con cui si riempie
l’aneurisma e si esclude dal circolo.
- Neurochirurgico tradizionale: si raggiunge il poligono del Willis e si chiude l’aneurisma con una clip metallica, che viene
incarcerata proprio alla base dell’aneurisma (ovviamente si spera che l’aneurisma abbia una forma definita e il colletto ci
sia).
Gestione del paziente: sono importanti i calcio-antagonisti, in quanto sono gli unici farmaci che contrastano la tendenza al
vasospasmo.
(TUTTO QUELLO CHE C’è DA SAPERE SULLE EMORRAGIE SUBARACNOIDEE!!)

34
STROKE ISCHEMICO EPIDEMIOLOGIA Più frequente in Asia nord-orientale e in Russia europea. Ci sono circa
400 nuovi casi di ictus per 100.000 abitanti l’anno. In Piemonte la
mortalità per ictus è superiore alla media nazionale.
PATOGENESI Alla base c’è un’ischemia che genera anossia e necrosi (con apoptosi)
dei tessuti a valle. Non tutto il distretto ischemico però si comporta nello stesso modo, ma con diverse tempistiche e diversi
modi:
- Core ischemico: distretto centrale, che va rapidamente incontro a necrosi.
- Zona di penombra ischemica: periferia, va in necrosi più lentamente perché riceve magari irrorazione residua da rami
arteriosi non occlusi. È il motivo per cui bisogna intervenire velocemente per curare ischemia cerebrale, per salvare la
penombra.
La morte cellulare avviene per (meccanismi precoci e tardivi):
• Eccitotossicità (Glu)
• Attivazione di enzimi proteolitici
• Stresso ossidativo
• Disfunzione mitocondriale
• Infiammazione
• Apoptosi
CAUSE CLASSIFICAZIONE TOAST:
1. Aterosclerosi dei grossi vasi
2. Occlusione di piccole arterie
3. Cardioembolismo
4. Altre cause documentate
5. Cause indeterminate: non si riesce a risalire alla causa.
Cambia anche la frequenza: ultimamente si sta dando sempre più importanza alla causa autoimmunologica, soprattutto
nei giovani.
Aterosclerosi. Distinguiamo in:

35
• Aterosclerosi dei grandi vasi:
o Placche aterosclerotiche: si formano di solito all’origine
di carotide interna, arteria vertebrale, arteria basilare,
cerebrale media.
o Stenosi
o Trombosi in situ
o Embolismo arterioso
• Aterosclerosi dei piccoli vasi:
o Lipoialinosi
o Occlusione
Rami perforanti: sono zone tipiche dove arterie di piccolo calibro
si staccano ad angolo retto dall’arteria cerebrale anteriore
(sostanza perforata alla base del cranio).
Nell’iperteso cronico danno microaneurismi ed essere alla base di una emorragia dei gangli della base, ma allo
stesso tempo però la necrosi fibrinoide (ialinosi) può portare anche a occlusione con ischemia!!
→ risultati analoghi possono dare due outcome diversi!
ANGIOPATIA AMILOIDE: dà quadri contemporanei di ischemie e microemorragie diffuse.
Cardioembolismo. Più frequenti cause:
• Fibrillazione atriale
• Protesi valvolari o valvulopatia→ la FA da vizio mitralico è molto frequente; sono causa di embolismo anche in
assenza di FA perché si perde tessuto a livello dei lembi valvolari e si possono formare trombi.
Ci sono anche valvulopatie non reumatiche, come l’endocardite→ di causa batterica ma anche autoimmunitaria.
• Cardiomiopatie: anche malattie neurologiche possono presentarsi con una cardiomiopatia dilatativa, es. quelle
geneticamente trasmesse (X-linked, mitocondriali, …).
• PFO: è una questione controversa. Si diagnostica con ECO transesofageo o transcranico doppler dopo iniezione di
microbolle. È presente in 1/3 della popolazione, ma solo in rari casi si manifesta con ischemie cerebrali. Quando si
occludeva con un piccolo intervento.
DISSECAZIONE ARTERIOSA Tipico di giovani. Si verifica uno slaminamento dell’intima, per cui
il sangue si infiltra sotto la tonaca intima scompagina la parete
arteriosa, creando un falso lume. Traumatismi anche di entità
lieve possono, in alcuni soggetti predisposti, determinare una
dissecazione carotidea: incidenti stradali, attività sportive
violente.
La cefalea non è una presentazione comune nell’ictus, ma può
essere presente se la causa è la dissecazione.
Che segno cerco in PS per avere la quasi certezza che si tratti di dissezione? Il sistema oculo-simpatico per un tratto decorre
nella parte della carotide comune e interna: in caso di dissezione devo sempre cercare i segni della sindrome di Claude-
Bernard→ MIOSI, PTOSI E ANIDROSI.
MALATTIE EMATOLOGICHE Ipercoagulabilità (deficit di proteina C, deficit di proteina S, deficit di ATIII, resistenza alla proteina C attivata con mutazione
del FV Leiden, mutazione della protrombina, aumento del FVIII, iperomocisteinemia con mutazione MTHFR)
Sindrome da anticorpi anti-fosfolipidi
CID
Malattie mieloproliferative
Anemia falciforme: potente fattore ematologico per le ischemie distrettuali periferiche ma anche cerebrale.
FATTORI DI RISCHIO GENERALI Non modificabili: età, sesso, etnia.
Modificabili: fumo, alcol, obesità addominale, pressione arteriosa, diabete.

36
ICTUS GIOVANILE CON GIOVANE SI INTENDE SOTTO I 45-50 ANNI.
Un terzo dei casi rimane senza una diagnosi eziologica; si riesce quasi sempre a capire se si tratta di un embolo ma non si
sa da dove arrivi.
Sono ictus caratterizzati da minore mortalità a 30 giorni e minore disabilità.
Cause principali
• Disse(ca)zione arteriosa
• Stati di ipercoagulabilità
• 25-30% criptogenici
ICTUS DA CAUSE RARE Vasculiti:
- Arterite temporale di Horton: è da ricordare perché può causare cecità: il processo arteritico si trasmette all’arteria
oftalmica→ neuropatia ottica ischemica.
- PAN
- AR
Vasculopatie non infiammatorie: Marphan, Eglers Danlos, Moya-Moya: malattia genetica che genera ischemia cronica già
da età precoce; questo determina una intensa rete di piccoli rami collaterali delle arterie rimaste sane, che in angiografia
dava una immagine a “nuvola di fumo”.
Malattie genetiche: CADASIL.
SINDROMI CLINICHE Non chiedono le sigle delle varie sindromi cliniche, ma bisogna sapere cosa succede quando si ha ischemia di cerebrale media, anteriore
e posteriore→ sapere territorio di irrorazione delle principali arterie.
CIRCOLO ANTERIORE: si intende l’asse carotideo.
CIRCOLO POSTERIORE: l’asse vertebro-basilare.
Sindromi complete del circolo anteriore. Cerebrale anteriore: irrora la faccia mesiale degli emisferi, all’interno della scissura interemisferica→ corteccia motoria e
sensitiva dell’arto inferiore, quindi può dare paresi arti inferiori (l’homunculus è rappresentato con gli arti nella porzione
mesiale).
• Emiplegia controlaterale
• Emianestesia controlaterale
• Emianopsia laterale omonima: significa non vedere metà del mondo, ma bisogna vedere quale: se è destra non vede
più la destra.
• Disfunzione corticale superiore sinistra:
o Afasia
o Aprassia ideomotoria, bucco-facciale
o SINDROME DI GERSTMANN: possibile sindrome da lesione del lobo parietale sinistro:
acalculia, agrafia, agnosia digitale (non riconoscono le dita, né proprie né quelle di
altri), confusione mentale.
• Disfunzione corticale superiore destra:

37
o Aprassia costruttiva/abbigliamento
o Agitazione
o Emi-inattenzione (o somatoagnosia o neglect)→ in vari gradi: anosodiaforia (sa di essere malato ma non se ne
cura), somatoparanefria (grado massimo di neglect, in cui crede che quegli arti paretici siano del vicino di letto
ma non suoi).
o Prosopagnosia
→ + TURBE DI VIGILANZA! si verificano frequentemente in cado di aumento della pressione endocranica fino al coma.
CONSEGUENZE: cisti e pseudocisti→ porencefalia.
Edema cerebrale (non si vedono le anse corticali).
Decompressione tramite rimozione della calotta. Se non si fosse decompresso si sarebbe potuta avere un’erniazione dell’ippocampo
attraverso il tentorio in zona sottotentoriale).
Sindromi parziali del circolo anteriore. Una forma tra:
• emiparesi/plegia/anestesia + emianopsia l.o.
• emiparesi/plegia/anestesia+ compromissione corticale superiore
• compromissione corticale superiore + emianopsia l.o.
• deficit funzione corticale superiore isolata
Angio-TC: stop a livello della cerebrale media, ma il
mancato deflusso viene comunque comperato con
altri circoli.
RM: con sequenze DWI (diffusione) si possono
apprezzare le ischemie già in fasi molto precoci.
Appaiono iperintense. Non è un mezzo diagnostico di
urgenza, non è richiesta una RM in caso di sospetto di
ictus (nemmeno prima della trombolisi è richiesta).
Sindromi del circolo posteriore. Se completa, può essere data da ostruzione a livello dell’arteria basilare (il circolo del Willis è supportato solo da arterie
carotidi). Se monoletarale, può essere dovuta da occlusione arterie vertebrali, di arterie cerebrali posteriori, occlusione
arterie cerebellari.
Caratterizzato da:
• Sindrome alterna: paralisi di almeno un nervo cranico omolaterale ed emisindrome motoria/sensitiva controlaterale.
• Tetraparesi-plegia controlaterale: di solito si tratta di una occlusione incompleta di una sola vertebrale, quindi si
risparmia un lato del tronco.

38
• Disturbo coniugato di sguardo: aree 17, 18 e 19 sono vicine (intorno a scissura calcarina) quindi un danno ischemico
può danneggiarle tutte e tre.
• Sindromi cerebellari
Sindromi lacunari. ➢ No compromissione corticale
➢ emiparesi isolata
➢ emianestesia isolata
➢ emiparesi+emianestesia isolata
➢ emiparesi atassica
TIA Fenomeno così transitorio che si può valutare solo quando è già esaurito; quando arrivano in PS spesso i sintomi sono già
spariti. Bisognerebbe dimostrare che non vi è nessun segno di ischemia persistente, il che però si può fare solo tramite RM,
ma come abbiamo detto non si fa in tutti i casi di ischemia!
→ tutto ciò che on provoca un danno ischemico permanente.
NON DIRE CHE è UN INFARTO, perché NON LASCIA DANNO TESSUTALE.
Può dare una amnesia globale transitoria: può succedere che da un momento all’altro il paziente si perda, non sappia più
tornare a casa. Ha il pregio che tendono a non ripetersi. Stati convulsivi del lobo temporale possono mimare l’amnesia
globale.
DIAGNOSI STRUMENTALE TC BASALE: non fa vedere ischemia (negativa nelle prime 24 ore) ma l’importante è che non dimostri emorragia.
- Iperdensità dell’arteria cerebrale media (segno dell’arteria densa)→ trombosi piuttosto estesa di cerebrale media.
- Mancata separazione tra sostanza grigia e bianca, difficile da vedere ma evolverà in infarto.
RM e angio-TC non si fanno in urgenza.
TERAPIA
Trombolisi endovenosa. Una volta stabilito che si tratta di ictus ischemico, si può impostare una terapia trombolitica
se priva di controindicazioni assolute: piastrine sotto i 100.00, post-chirurgico (le controindicazioni sono tantissime ma non
sono da sapere).
Per la trombolisi non serve per forza angioTC, basta anche solo la TC.
Alteplase 0,9 mg/kg fino a un massimo di 90 mg.
Limite di tempo: 4,5 ore dall’evento ischemico. Prima che posso è meglio!
Trombolisi meccanica. Nei centri specializzati si può fare, previa una angio-TC, una tromboarteriectomia→ trombolisi
meccanica o endovascolare o intrarteriosa, passando da arteria femorale. Arrivati al trombo si asporta tramite una rete
metallica o si aspira.
Limite temporale: 6 ore.
I tempi della trombolisi meccanica possono esser anche dilatati oltre le 6 ore, a patto di dimostrare che ci sono ancora zona
di penombra, cioè non è ancora tutto core necrotico→ tramite TC perfusionale e varie tecniche di RM.
Si può fare prima trombolisi endovenosa e poi, se si individua il trombo, di può fare anche quella meccanica, oppure si può
fare direttamente la meccanica.
Complicanze: possono trasformare l’ischemia da bianca a rossa, causando quindi un emorragia.

39
NEUROFISIOLOGIA
ELETTROMIOGRAFIA L’elettromiografia classica in senso stretto ci introduce alla fisiopatologia
dei muscoli; si avvale di aghi elettrodi che penetrano nei muscoli. Serve
quando si è di fronte a un gruppo di muscoli debole o ipotrofico e si vuole
conoscere la causa.
Es: il tibiale anteriore è danneggiato perché c’è un processo patologico
proprio del muscolo (miosite, distrofia muscolare, …) o per via di un danno
nervoso (lesione di un nervo)?→ serve per scoprire se il danno di un
muscolo è di tipo miogeno (deriva dalla sua struttura) o neurogeno
(deriva dalla sua innervazione).
Non si tratta di un esame standard.
Si valutano solo i muscoli interessati dal quadro clinico, eventualmente confrontati con il lato sano.
È un esame che si valuta su due canali, visivo e acustico→ è importante apprezzare anche il suono.
Con gli aghi si possono registrare due tipi di variabili:
1. ATTIVITÀ ELETTRICA del muscolo durante la CONTRAZIONE VOLONTARIA: si chiede di contrarre il muscolo in
esame. I potenziali che si vedono in questo caso prendono il nome di Potenziali di Unità Motoria (PUM): è il
potenziale di un motoneurone con tutte le fibre innervate dalle sue terminazioni nervose→ ricordiamoci che è
essenziale per la graduazione dell’attività motoria: per passare da una forza minima a una massima bisogna
chiamare a raccolta il maggior numero di unità motorie e farle scaricare con una maggiore frequenza.
2. ATTIVITÀ SPONTANEA: se si mette un ago in un muscolo che non si contrae, si registra un’attività dovuta a un
minimo rilascio di NT a livello delle placche neuromuscolari anche a riposo. Questo vale per il muscolo normale, si
registra pressoché una isoelettrica (a essere fini in realtà non è proprio così). In condizioni patologiche, si possono
registrare altri tipi di attività elettriche che indicano una sofferenza neurogena.
ATTIVITÀ SPONTANEA PATOLOGICA
Fascicolazioni. Sono delle vermicolazioni del muscolo che a volte sono visibili a occhio nudo (e non serve l’esame in
questo caso) ma a volte sono profonde e non si vedono. Esiste tutto un argomento parafisiologico delle fascicolazioni (non
sempre si associato a patologia), ma ora non ci interessa.
Se si registrano a un’elettromiografia si associano sempre a una sofferenza muscolare secondaria a danno neurogeno
(nervi, radici o corpi dei motoneuroni). Una sofferenza propria del muscolo invece non dà fascicolazioni.
All’elettromiografia, danno un suono simile ai popcorn nel microonde.
Patogenesi: sono molto simili a dei potenziali di unità motoria: sono una scarica eccessiva di una unità motoria o di gran
parte di essa; c’è uno stato di ipereccitabilità delle fibre nervose danneggiate→ quando le fibre nervose sono danneggiate,
da una parte possono mostrare uno stato di ipofunzione (perché sono lese), da una parte invece possono mostrare uno
stato di iperfunzione.
→ testimoniano danno neurogeno, assenti in miopatie primitive.
Fibrillazioni. Sono un’altra attività spontanea patologica e non si vedono mai a occhio nudo, serve per forza l’esame
sterminale. Chiamate anche onde aguzze positive; in realtà non sono proprio sinonimi ma sono lo stesso fenomeno visto
da due punti diversi a seconda della posizione dell’ago.
RICORDARSI CHE “POSITIVO” IN NEUROFISIOLOGIA È QUALCOSA CHE VA VERSO IL BASSO, al di sotto dell’isoelettrica.
Sono brevissimi e piccoli potenziali intermittenti, simili al ticchettio dell’orologio; sono molto rade e piuttosto ritmiche.
Quando le fibrillazioni sono tantissime, si sovrappongono tra loro e ricordano il rumore della pioggia.

40
Questo reperto è utile ma non ha quell’univocità che invece hanno le fascicolazioni: non indica né una sofferenza neurogena
né una sofferenza miogena primitiva; è più probabile vederle in caso di sofferenza neurogena ma non è detto→ non
discriminano così bene.
Sono dei potenziali di fibra muscolare: se essa è staccata dai terminali nervosi diventa per un periodo ipereccitabile→
ipereccitabilità da denervazione. I quanti di acetilcolina rilasciati dalle fibre intatte sono sufficienti per far scaricare le fibre
nervose ipereccitabili.
Un processo patologico muscolare a carattere necrotico (come una miosite acuta nel contesto di una polimiosite) può avere
una violenza tale da ledere anche i terminali intramuscolari delle fibre nervose, portando a una separazione di alcune fibre
dai loro terminali senza che il nervo sia leso→ in alcuni processi miopatici primitivi si manifestano comunque le fibrillazioni.
Scariche miotoniche. Un certo numero di miopatie primitive o distrofie muscolari (come la distrofia miotonica)
presentano il fenomeno della MIOTONIA: è una forma di ipereccitabilità muscolare per cui il muscolo presenta una grande
facilità alla contrazione e una estrema difficoltà alla decontrazione→ descritto come “difficoltà a manipolare gli oggetti
perché ho fatica a rilasciare la muscolatura”, “prendo in mano un oggetto e faccio fatica a rilasciarlo”. Il neurologo allora
prende il martello di Dejerin e percuote il muscolo: la semplice percussione provoca la contrazione del muscolo o di una
sua gran parte → miotonia meccanica.
Non sempre i sintomi sono così evidenti, quindi si cercano scariche all’elettromiografia: sono potenziali di fibra muscolare
che si scatenano con alta frequenza, che aumenta all’inizio della scarica e poi diminuisce. Sono paragonati al rumore di un
motore a due tempi.
Si possono dare terapie farmacologiche per diminuire questa eccitabilità muscolare: si usano farmaci antiepilettici come
carbamazepina o antiaritmici (come mexidetina) che agiscono su canali ionici.
→ tipiche di miopatie primitive.
Quindi:
- Neuropatie: a riposo, fibrillazioni e fascicolazioni.
- Miopatie: a riposo, scariche miotoniche.
ATTIVITÀ VOLONTARIA Quando il muscolo si contrae, l’ago-elettrodo registra potenziali di unità motoria;
a contrazione debole ci saranno poche unità motorie in funzione e che scaricano
con una frequenza lenta, a contrazione massima ci saranno tante unità motorie in
funzione con una frequenza di scarica maggiore→ reclutamento delle unità
motorie e aumento della frequenza di scarica.
Si mette il paziente sul lettino e inserendo l’ago sul muscolo (es. bicipite brachiale)
si chiede di iniziare una contrazione volontaria; successivamente si chiede di
incrementare la forza fino al massimale. Per una contrazione debole, entrano in
gioco poche unità motorie (o a volte solo una in prossimità dell’ago); quando la
contrazione è forte il tracciato presenta una notevole sovrapposizione delle tante
unità motorie reclutate. Per una contrazione massimale si ottiene un tracciato complesso:
• Il tracciato durante la contrazione debole prende il nome di singole oscillazioni
• Tra le due fasi si parla di tracciato di transizione.
• Il tracciato complesso durante la contrazione massimale prende il nome di TRACCIATO INTERFERENZIALE (o
INTERFERENZA).
Dal punto di vista auscultatorio, si sente un ticchettio che piano piano aumenta sempre di più fino a non distinguersi più.

41
Danno neurogeno. Ad esempio un danno al nervo muscolo-cutaneo. Come fa a raggiungere l’interferenza se c’è stato
un danno di metà assoni (dunque funzionano solo metà delle unità motorie)? Il paziente cerca di produrre una contrazione
massimale ma non riesce a raggiungere l’interferenza→ tracciati incompleti, di transizione. Nei casi più gravi, se è
danneggiato quasi tutto il nervo, avremo in contrazione massimale un tracciato di singole oscillazioni. Dal punto di vista
obiettivo, questo si associa a un muscolo ipostenico/astenico.
I segnali appaiono nella schermata all’interno di un sistema di assi cartesiani dove le x sono il tempo (ms) e le y il voltaggio
(mV); la schermata può essere di 100 ms x 10 mV ma in alcuni casi gli impulsi sono più ampi, quindi si possono cambiare
tali valori in modo da farli rientrare nel grafico→ Questo aumento dell’ampiezza e della durata degli impulsi si osserva
nelle sofferenze neurogeniche croniche: è un esito della rigenerazione delle fibre nervose (proprio solo del SNP, non del
centrale), può essere infatti che una fibra lasciata orfana dalla terminazione nervosa danneggiata venga “adottata” dal
processo rigenerativo di un’altra terminazione nervosa→ REINNERVAZIONE COLLATERALE. Di conseguenza i pochi impulsi
nervosi che ci sono, sono più grandi.
In caso di neurotmesi, gli assoni non possono rigenerare, perché viene a mancare il connettivo che deve dare ordine al
nervo: infatti ci sono diverse guaine connettivali che sepimentano i tronchi nervosi e li rendono simili a un alveare. Se si
distrugge il sistema di sepimentazione connettivale, l’assone danneggiato non sa più dove andare, quindi magari rigenera
ma in modo disordinato.
Quindi: nel pattern neurologico, si hanno difetti di reclutamento volontario, che non raggiungono quindi l’interferenza
perché vengono reclutati meno PUM; possono essere di varie forme di gravità (dalla singola oscillazione nello sforzo
massimale fino a una interferenza lieve). Nel caso ci sia un fenomeno di reinnervazione collaterale, ci sono modifiche del
potenziale che lo fa ingigantire.
Danno miopatico. Nelle miopatie è più frequenze che le unità motorie siano preservate almeno nelle fasi iniziali della
malattia, cioè ci sono ancora ma sono difettose (nelle fasi avanzate di malattia invece sarebbero totalmente assenti perché
sostituite da grasso). Le unità motorie malate dunque produrranno meno forza, non sono morte ma sono ammalate quindi
avranno una intensità minore.
Quando il SNC invia il comando per lo sforzo massimale, in termini di forza il risultato è al di sotto dell’atteso rapidamente
si attiva il meccanismo del reclutamento, per cui si vede una facile interferenza→ non c’è più sequenza in crescendo tipica
del soggetto normale, ma si arriva rapidamente all’interferenza saltando tutti gli stadi intermedi→ A un muscolo debole
corrisponde un tracciato interferenziale.
Il suono è crepitante, assomigliano alle fibrillazioni.
NEUROGRAFIA Noi dobbiamo sapere le tecniche di routine, per cui si può stabilire qual è la velocità di conduzione dei nostri più grandi
nervi. Si tratta solitamente i nervi misti (fibre sensitivi e motorie); noi però studieremo separatamente le fibre sensitive e
motorie.
Bisogna sapere come si fa a misurare la velocità di conduzione sensitiva e quella motoria.
VELOCITÀ DI CONDUZIONE MOTORIA/MUSCOLARE Il nervo mediano dell’avambraccio conduce normalmente a 70 m/s. Vengono messi degli elettrodi al di fuori (non più aghi
dentro) che coprono tutta la grandezza del muscolo. Rileva un’onda M, onda muscolare o POTENZIALE DI AZIONE
MUSCOLARE COMPOSTO→ è composto da tutte le unità motorie di quel muscolo.
Si posiziona lo stimolatore dotato di anodo e catodo (attraverso cui passa una corrente brevissima, di 0,1 ms) vicino al
muscolo interessato (distanza polso-eminenza tenar); si inizia con stimoli di basso voltaggio e poi si aumenta sempre di
più. Pian piano inizia a contrarsi il muscolo e sullo schermo compare un’onda. Ci sarà:
- Latenza
- Onda sovramassimale: cioè all’aumentare dello stimolo l’onda non aumenta più. Perché c’è bisogno di onde
sovramassimali? Il nervo è composto da decine di fibre diverse; non tutte hanno lo stesso diametro e la stessa velocità;
per determinate condizioni di stimolo, si può misurare una velocità che non è massima→ noi dobbiamo essere certi di
aver stimolato tutte le fibre più veloci. Questo si capisce quando la latenza delle risposte è minima, e per avere una
latenza minima l’onda deve essere sovramassimale.

42
Dopo di che lo stimolatore viene allontanato dal muscolo e messo più prossimalmente sul passaggio del nervo (distanza
gomito-eminenza tenar); si ottiene un’altra onda M che sarà sempre sovramassimale ma la latenza sarà maggiore, perché
l’impulso deve prima percorrere un certo percorso. Questa latenza la chiamiamo LATENZA PROSSIMALE a differenza della
prima che la chiamiamo LATENZA DISTALE.
Con tutti questi dati, si può calcolare la velocità di conduzione: infatti ho lo spazio percorso e il tempo sarà dato dalla
differenza tra i due tempi di latenza.
Nell’arto superiore di solito si valutano: mediano e ulnare.
Nell’arto inferiore di solito si valutano: peroneo comune e tibiale.
VELOCITÀ DI CONDUZIONE SENSITIVA
Sindrome del tunnel carpale. Il neurologo è chiamato in causa soprattutto per la diagnosi. È una condizione molto
frequente che consiste nella comparsa di formicolii soprattutto notturni nel territorio di pertinenza del nervo mediano→
I-II-III e metà del IV dito; difficilmente verrà riferito dolore solo in metà del quarto dito, quindi è una cosa da indagare.
Con il passare del tempo, si può sviluppare una ipotrofia dell’eminenza tenar e vengono meno alcuni movimenti di destrezza
motoria fine (come inserire un filo in un ago); a lungo termine quindi è anche invalidante.
Due tipi di terapie: cortisonici o chirurgia→ consiste nel dare spazio al nervo nel suo canale ligamentoso.
Può essere utile valutare la velocità di conduzione sensitiva dei nervi digitali utilizzando degli elettrodi ad anello. Nelle dita
non ci sono muscoli, ma solo tendini, dunque tutte le fibre che ci sono nelle dita sono solo fibre sensitive. Posizionando gli
elettrodi sull’anulare, si valuta sia il nervo mediano che l’ulnare.
Si inizia a stimolare il nervo mediano al polso; si registrano varie onde sovramassimali, si registrano e si calcola la media→
rapporto segnale-rumore: quando si cercano segnali piccoli, si amplifica la sensibilità degli strumenti quindi appaiono
facilmente artefatti. Facendo invece la media di tanti stimoli diversi, si esalta la parte del segnale che si ripresenta sempre
nello stesso istante. Si ottengono delle onde nell’ordine di microV, sono molto più piccole rispetto alle onde M.
Si fa la stessa cosa per il nervo ulnare. Si osserva poi la differenza tra le onde del nervo mediano e del nervo ulnare; la cosa
che può cambia è il tempo di latenza, che nel caso del nervo mediano compromesso sarà più lunga con un’onda successiva
più piccola. Si tollera una differenza di latenza fino a 0.6 ms.
→ chi ha la sindrome del tunnel carpale ha anche un aumento significativo della latenza distale (nella velocità di
conduzione muscolare) oltre a quella sensitiva.
Come mai si stimola il nervo con un verso contrario? Infatti la sensibilità viaggia dalla periferia al centro, ma noi valutiamo
lo stimolo dal polso alle dita. Si fa così perché è teoricamente più semplice da rilevare→ si valuta quindi la VELOCITÀ
SENSITIVA ANTIDROMICA (in verso opposto) anziché quella ORTODROMICA (normale).
TEST NEUROFISIOLOGICI NELLA DIAGNOSI DELLA MIASTENIA GRAVE Domanda molto frequente sulla neurofisiologia.
Nella miastenia grave c’è un difetto della giunzione neuromuscolare o placca motrice (è un tipo di sinapsi colinergica, con
Ach come trasmettitore principale). Ci sono tanti possibili fenomeni autoimmunitari alla base; una volta si conosceva solo
la comparsa di anticorpi anti-recettore dell’Ach→ l’Ach viene liberata ma non può agire perché i suoi recettori sono
“occupati” in parte dagli anticorpi.

43
1) Test di Desmet in neurofisiologia
2) Ricerca di anticorpi antirecettore
Ora si conoscono anche altri meccanismi, che portano a una minor funzionamento della placca neuromuscolare, che
rientrano un fenomeno chiamato ESAURIBILITÀ DELLA TRASMISSIONE NEUROMUSCOLARE→ con l’esercizio, l’efficienza
della trasmissione neuromuscolare diminuisce molto rapidamente.
Es: inizia a masticare una cicca energicamente, ma al 4/5 morso, questa energia diminuisce.
Test di Desmedt Il test più utilizzato si basa proprio su questa esauribilità neuromuscolare ed è un test di conduzione di velocità muscolare;
invece di un singolo stimolo si danno una serie di stimoli in rapida successione (3 Hz): in un soggetto normale le onde M
mantengono la stessa ampiezza anche nel contesto di questa stimolazione ripetitiva sovramassimale. Nella miastenia grave
invece (non in tutte ma nelle forme più gravi), man mano che si procede con la stimolazione la risposta diminuisce; la prima
onda rispetto alla quinta è molto diminuita→ è la controparte elettrica di un fenomeno che si può osservare anche
clinicamente. Questa caduta di risposta solitamente si calcola tra la quinta onda della serie e la prima; non dovrebbe
esserci nessun decremento; tuttavia considerando possibili interferenze, si tollera una variazione fino al 10%, se è superiore
al 10% significa che c’è una riduzione effettiva dell’intensità.
In soggetto normale In miastenia gravis

44
EEG Berger: inventore dell’EEG.
QUALI ATTIVITÀ ELETTRICHE GENERANO L’EEG? Si tratta della sommazione di milioni di potenziali post-sinaptici (PSP). Possono essere di due tipi, eccitatori o inibitori.
Sono i potenziali generati dai dendriti apicali delle cellule della corteccia, in particolare da terzo, quarto e quinto strato
(rivedere anatomia -strati corteccia- e fisiologia).
La cosa importante è la sommazione dei potenziali sinaptici, che presuppone una sincronizzazione di eventi elettrici che
accadono nello stesso istante (e quindi risultare più grandi). Esistono dei “direttori” che sincronizzano una certa area di
corteccia a pulsare nello stesso momento; La struttura che organizza l’attività della corteccia è il TALAMO: è l’organo di
ritrasmissione delle vie sensitive e motorie e si interfaccia con la corteccia, è il pacemaker.
→ SOMMAZIONE DOVUTA ALLA SINCRONIZZAZIONE DEI POTENZIALI SINAPTICI.
COME SI REGISTRANO? Si mettono elettrodi sullo scalpo con un certo criterio topografico. Gli elettrodi hanno un nome che indica anche la loro
posizione secondo il SISTEMA INTERNAZIONALE 10-20: ha a che vedere con la distanza inter-elettrodica, cioè in questo
sistema sono sempre pari, o al 10 o al 20% della distanza tra nasion (attaccatura superiore del naso) e inion (protuberanza
occipitale) in senso sagittale, e della distanza tra i de punti
preauricolari in senso trasversale. Esistono delle cuffie
elastiche già predisposte per riprodurre questo schema.
Gli elettrodi con numeri pari sono sull’emicranio destro,
gli elettrodi con numeri dispari sono sull’emicranico di
sinistra; la Z indica la linea mediana.
- C: centrale, cioè al di sopra del solco di Rolando.
- F: frontale
- P: parietale
- O: occipitale
- T: temporale
Amplificazione del segnale. Il segnale che arriva chiaramente è debole, quindi gli apparecchi EEG sono degli
amplificatori. In routine, si usa una scala calibrata su µV/mm→ 1 µV corrisponde a 1 mm. Una pagina di EEG (30 cm, poco
più di un foglio) sono 20 secondi, 1 cm di onda sono 70 µV. A volte si usa una scala diversa, cioè 2 cm che corrispondono a
70 µV→ si amplifica il segnale per ricercare una eventuale morte cerebrale.
Filtri. Non tutte le onde che oscillano servono, di solito si tengono solo quelle che oscillano tra 1 e 70 Hz (cicli al secondo),
quindi si guardano solo quelle onde. Servono anodo e catodo per vedere differenze di potenziali (all’esame non chiede questo concetto).
Solitamente, in una pagina di EEG abbia metà dei canali pari (in alto) e metà dei canali dispari (in basso)→ idea sia di destra
che di sinistra.

45
TIPI DI ONDE Prima di tutto si guarda la frequenza delle onde, per cui si distinguono in:
- Delta: attività più lenta, da 1 a 4 Hz.
- Theta: tra 4 e 8 Hz.
- Alfa: tra 8 e 13 Hz. Vengono definite oscillazioni fusiformi.
- Beta: sopra 13 fino a 40 Hz, attività più rapida, le onde sono più ravvicinate.
Nell’adulto sveglio, l’attività delta non deve esserci; processi patologici come tumori, ascessi, ischemie, emorragie danno
un rallentamento dell’attività.
Ritmo alfa. Le alfa si trovano in soggetto fermo con occhi chiusi in stato di riposo psico-sensoriale, senza svolgere attività mentale che
richiedano concentrazione; quando apre gli occhi le alfa scompaiono. Sono quindi più rappresentate nelle porzioni
posteriori (occipitale e parieto-occipitale), ma in realtà si vedono in tutto l’encefalo.
Per essere alfa, devono scomparire con l’apertura degli occhi.
Non è detto che si trovino in tutti, ad esempio in soggetti nervosi e stressati non si trovano ma non per questo è patologico;
infatti la corteccia visiva si attiva anche solo con l’immaginazione, ad esempio immaginando un movimento, quindi bisogna
riuscire a fermare anche ogni processo di immaginazione.
Quando si valuta un EEG di un adulto come prima cosa si guarda se ha o no l’alfa.
Significati:
• “idling” (= girare al minimo) della corteccia visiva
• Attività inibitorie in una corteccia visiva non “in uso”
• Coordinamento e comunicazione tra reti neurali più complesse, con ruolo nella memoria a breve termine ed
attenzione
Reattività:
- All’apertura degli occhi deve verificarsi una reazione d’arresto (da sapere).
- Aumento di ampiezza alla chiusura degli occhi (reazione di rinforzo)
- Fenomeno dello “squeak” (squittìo): frequenza più rapida a chiusura OO.
RITMO DI FREQUENZA ALFA: in alcuni casi si ha un ritmo di frequenza come le onde alfa, ma differisocno per reattività (non
reagisce all’apertura degli occhi) e per topografia (prevalenza anteriore), quindi non sono propriamente alfa. Li si ritrova in
alcune forme di coma grave in alcune lesioni del tronco; non ha più una localizzazione posteriore e non è reattivo.
Ritmo mu. Assomiglia a attività alpha e si presenta in giovane adulto in veglia: onde con frequenza simile a alpha tipo 7-11 Hz. È
un’attività che si trova a sede centrale, vicino all’homunculus (corteccia sensitivo-motoria). Sparisce quando il soggetto
compie movimenti con arto controlaterale o immagina di muovere arti controlaterali, o se viene stimolata cute di emicorpo
controlaterale (quindi se si attiva l’homunculus sensistico e motorio).
Rispetto a alpha cambia che qui è implicata corteccia sensitivo-motoria mentre in alpha era corteccia visiva.

46
Si ottiene variazione di segnale elettrico cerebrale se si fa immaginare un movimento al pz. In un pz paralizzato posso
sfruttare la sua capacità di immaginare il movimento e attivare questa area per ad esempio attivare interruttore di braccio
robotico. Si può usare anche il movimento degli occhi perché sono elettricamente carichi.
Ritmo beta. Onde piccolissime e velocissime e si trovano in corteccia rolandica e tendono a scomparire con movimento o stimolazione
sensoriale controlaterale, oppure si trova incrementato durante attività mentali. In PS puoi vedere pz soporoso e poco
responsivo quasi in coma con TC negativa; tramite il riscontro all’EEG di abbondante attività beta si individua che è
un danno iatrogeno: tutti gli stimolanti del sistema GABA aumentano molto l’attività beta, quindi si procede con una
determinazione dei metaboliti di BDZ nelle urine, perché potrebbe essere un caso di sovradosaggio (per atto
anticonservativo). Per terapia allora devo antagonizzare le BDZ con flumazenil (anexate).
Ritmo theta. Modesto nell’adulto in veglia fisiologica, abbondante in sonnolenza e sonno. Dominante nel neonato. Localizzato nelle
regioni temporali. Dalla veglia verso il sonno infatti (nel sonno NREM 1) si ha un passaggio da onde alpha a theta.
Ritmo delta. Non deve esserci nell’adulto sano durante la veglia. Dominante nel neonato e nel sonno NREM. (no onde lambda)
FREQUENZA AMPIEZZA LOCALIZZAZIONE PRESENTE IN:
DELTA 1-4 Hz 150 µV Sonno NREM e neonato.
Anestesia generale.
THETA 4-8 Hz 75 µV Temporale Sonno NREM e neonato.
ALFA 8-13 Hz 30 µV Occipitale-parietale Veglia con occhi chiusi e riposo mentale.
Sonno REM
BETA 13-40 Hz 20 µV Corteccia sensitiva-
motoria rolandica.
Veglia con occhi aperti.
Sonno REM.
ARTEFATTI Sono molto probabili perché si lavora con segnali molto piccoli che vanno amplificati: saremo quindi alle prese anche con
onde da sorgenti extra cerebrale che possono derivare dal nostro corpo o dall’ambiente come:
❖ Ammiccamento degli occhi: vanno assolutamente tenuti fermi durante EEG. I canali 1 e 5 (frontali) risentono di più di
artefatti da movimenti oculari.
❖ Muscoli del volto.
❖ Anche i segnali elettrici cardiaci causano artefatti all’EEG.
Come faccio a discriminare? Si misura insieme anche ECG e
si vede dove cade il complesso QRS.
❖ Sudorazione e respirazione possono interferire.
❖ Interferenze ambientali.
→ In situazione di morte cerebrale il tracciato piatto diventa
terreno fertile per gli artefatti.
ESEMPI PATOLOGICI DI EEG Situazioni patologiche dove EEG ha una forte validità clinica:
❖ EPILESSIE: possiamo osservare elementi epilettiformi:
o Punte o spike di meno di 70 msec (brevi deflessioni
improvvise del tracciato)
o Onda puntuta o aguzza o sharp wave: onda più slargata, che dura di più.
Attenzione che “epilettiforme” richiama epilessia ma non significa epilessia: in pz che ha emicrania e che per sbaglio fa
EEG, può aver fuorvianti alterazioni epilettiformi.
Complessi epilettiformi:
o Complessi punta-onda lenta a 3-4 Hz diffusi a tutto lo scalpo sincroni e simmetrici, che è tipico di assenza
semplice (domanda frequente!).

47
o Complesso polipunta-onda dopo stimolazione luminosa intermittente, tipico di epilessia mioclonica giovanile.
o Complesso polipunta (PP)
o Complesso onda aguzza-onda lenta
❖ SONNO E POLISONNOGRAFIE
❖ COMA E MORTE CEREBRALE
❖ ENCEFALOPATIE METABOLICHE (epatica): non dà alterazioni a imaging perché è solo quadro disfunzionale che causa
segni all’EEG: si vedono rallentamenti diffusi che possono diventare onde trifasiche ad elevata ampiezza (3
deflessioni). Quando le encefalopatie diventano molto gravi si può arrivare anche a silenzio elettrico, ma prima avremo
abbondante attività lenta: sono epoche di tracciato di silenzio alternate a scoppi di attività lenta più o meno aguzza→
alternanza silenzio-scoppio (burst-suppression).
Anche l’encefalopatia uremica è caratterizzata da rallentamenti.
❖ PATOLOGIE INFETTIVE: encefaliti nei primissimi stadi, dove nei primi momenti le immagini possono essere negative.
Prima che le immagini (RMN, TC) si positivizzino, l’EEG può essere d’aiuto, possono presentarsi rallentamenti anche
clamorosi. Si può pensare a rachicentesi in sospetto di malattia infettiva.
Tra le encefaliti, la HSV (herpes simplex) è la più comune e di solito nasce da un lobo temporale, asimmetrica! Anche
EEG mostrerà alterazioni asimmetriche, con scariche epilettiche lateralizzate periodiche (a intervalli regolari), chiamate
PLEDs.
MALATTIE DA PRIONI: gravi e incurabili, tumultuose, si muore in poco tempo: in EEG c’è rallentamento come al solito,
ma la cosa importante è il realizzarsi di un tracciato periodico perché ci sono complessi di onda trifasica diffusi
abbastanza omogeneamente a tutto lo scalpo e che si ripetono a intervalli costanti.
Essendo malattie trasmissibili, è necessario che siano dichiarate con certezza, ed è sempre richiesto anche un EEG con
tacciato periodico.

48
TRAUMI CRANICI Prima causa di morte prima dei 45 anni. Circa metà dei decessi per cause traumatiche sono dovuti ai traumi cranio-
encefalici, la più frequente delle affezioni del SNC.
Cause: Incidenti stradali, infortuni sul lavoro, cadute, eventi sportivi, eventi bellici.
La gravità di un trauma cranio-encefalico dipende da numerosi fattori:
- Fisici: violenza del trauma, onda d’urto, natura dell’agente causale (ad es. arma da fuoco)
- Anatomici (sede della lesione, a livello del punto d’impatto e soprattutto in profondità).
Si possono verificare nel momento del trauma cranico lesioni del cuoio capelluto, lesioni ossee e durali, lesioni extradurali,
lesioni sottodurali, lesioni subaracnoidee, lesioni del parenchima encefalico. Nella maggior parte dei traumi cranici gravi
queste lesioni sono diversamente associate.
GLASGOW COMA SCALE (GCS) Un sistema semplice ma valido per “stratificare” la gravità dei traumi cranici è quello basato sulla GLASGOW COMA SCALE
che prevede una classificazione in:
- Trauma cranico lieve GCS 14-15
- Trauma cranico moderato GCS 9-13
- Trauma cranico severo GCS 3-8
È una scala di vigilanza (pertanto in presenza di eventuale deficit motorio la risposta motoria sarà quantificata sugli arti
non paralizzati) che si basa sulla valutazione di: apertura degli occhi (E), espressione verbale (V), risposta motoria (M) a
ordini semplici o alla stimolazione nocicettiva.
Ha il vantaggio di essere semplice da effettuare, facilmente riproducibile e alla portata anche di osservatori non medici: un
linguaggio comune può essere così ottenuto fra i diversi soccorritori (luogo del trauma, primo intervento, ospedale,
neurorianimazione) e l’evoluzione dello stato di coscienza risulta più chiaramente apprezzato.
Criterio fondante nella valutazione del trauma cranico è dunque lo stato di coscienza: si va dalla semplice concussione fino
alla perdita protratta della coscienza e al coma.
La CONCUSSIONE è una transitoria alterazione della coscienza che si manifesta con confusione, amnesia, breve perdita di
coscienza, in assenza di danni parenchimali alla TAC (al più lieve edema cerebrale da iperemia); la RMN evidenzia però
minime anomalie nel 25% dei casi. È dovuta ad un transitorio disturbo nella funzione neuronale: i livelli di glutammato
(neurotrasmettitore attivatore) aumentano e il cervello entra in uno stato iperglicolitico e ipermetabolico che può
persistere fino a 7-10 giorni dopo il trauma. In questa fase il cervello risulta più suscettibile ad un eventuale secondo trauma
che, in parte a causa di un difetto nell’autoregolazione cerebrale, può provocare molto più gravi sequele incluso un possibile
edema cerebrale maligno.

49
VALUTAZIONE RADIOLOGICA
TAC encefalica. Indicazioni alla TAC iniziale:
➢ Presenza di uno o più fattori di rischio moderato o elevato per lesione intracranica [GCS ≤ 14, assenza di
responsività, deficit neurologico focale, amnesia del trauma, alterato stato mentale (incluso quello secondario ad
abuso di alcool e droghe), deterioramento neurologico, cefalea ingravescente e vomito, crisi epilettiche, segni di
frattura affondata].
➢ Completamento delle indagini diagnostiche prima di una anestesia generale per altre procedure durante la quale
non sarebbe possibile un monitoraggio neurologico.
La TAC, senza contrasto, consente di evidenziare:
• Emorragia (extradurale, sottodurale, subaracnoidea, intraparenchimale, intraventricolare)
• Edema cerebrale (obliterazione delle cisterne della base, compressione dei ventricoli e dei solchi della convessità)
• Ernia cerebrale interna (compressione, dislocamento del tronco encefalico)
• Anossia cerebrale (scomparsa dell’interfaccia sostanza grigia/bianca)
• Fratture craniche (possono talora non apparire fratture lineari composte)
• Pneumocefalo (può indicare fratture della base o fratture aperte della convessità)
Nei casi in cui la dinamica del trauma non sia chiara, in presenza di emorragia subaracnoidea si impone l’angioTAC.
Indicazioni per la TAC nel follow-up:
• trauma cranico severo: la TAC viene ripetuta
o alcune ore dopo la prima, per evidenziare la comparsa tardiva o il peggioramento di un ematoma
extradurale, o sottodurale o di un focolaio lacero-contusivo
o nei pazienti stabili, nei giorni 1, 3, 5 e poi fra la 10^ e 14^ giornata
• trauma cranico lieve o moderato: la TAC viene ripetuta
o prima delle dimissioni nei pazienti con una TAC iniziale patologica
o no nei pazienti stabili con TAC iniziale normale
RMN encefalica. Di solito inappropriata nel trauma cranico in fase acuta, può essere utile nel paziente stabilizzato per
valutare lesioni del tronco encefalico e piccole alterazioni nella sostanza bianca (ad esempio emorragie puntiformi nel corpo
calloso come in caso di danno assonale diffuso).
La RMN spinale è invece indicata nei pazienti con lesioni vertebro-midollari.
RX del cranio. Può essere utile qualora non sia disponibile la TAC, potendo evidenziare lo shift della pineale, uno
pneumocefalo, livelli idro-aerei nei seni paranasali, fratture craniche (affondate, lineari,). Comunque, la sua affidabilità
nell’evidenziare lesioni intracraniche è veramente bassa, dello 0,4-2%. Aiuta nel visualizzare frammenti metallici nei traumi
penetranti.
RX vertebrale. Rachide cervicale: in tutti i gravi traumi cranici (in cui non sia disponibile una TAC a strato sottile con
ricostruzioni assiali e sagittali) deve essere radiologicamente evidenziato dalla giunzione cranio-cervicale al passaggio C7-
T1; fin tanto che non è studiato si devono rispettare tutte le precauzioni in atto per il trauma cervicale (collare cervicale).
Rachide dorsale e lombare: studiati in base all’esame neurologico e al meccanismo del trauma.
LESIONI CUTANEE Sono sempre presenti nel punto d’impatto; dalla semplice ecchimosi all’abrasione ed escoriazione del cuoio capelluto, alla
contusione, alle ferite da taglio (lineare, a stella, a lembo, con perdita di sostanza, scotennamento); nel bambino possono
essere responsabili di importanti emorragie che in breve conducono allo shock emorragico.
LESIONI OSSEE E DURALI A seconda della natura e dell’energia dell’agente contundente si possono avere fratture:
- Lineari
- Fratture radiali o comminute o a mappamondo
- Fratture affondate (dove un frammento osseo è proiettato verso l’interno della cavità cranica).
Squama temporale, squama occipitale e seni frontali rappresentano le zone di maggior debolezza del cranio.
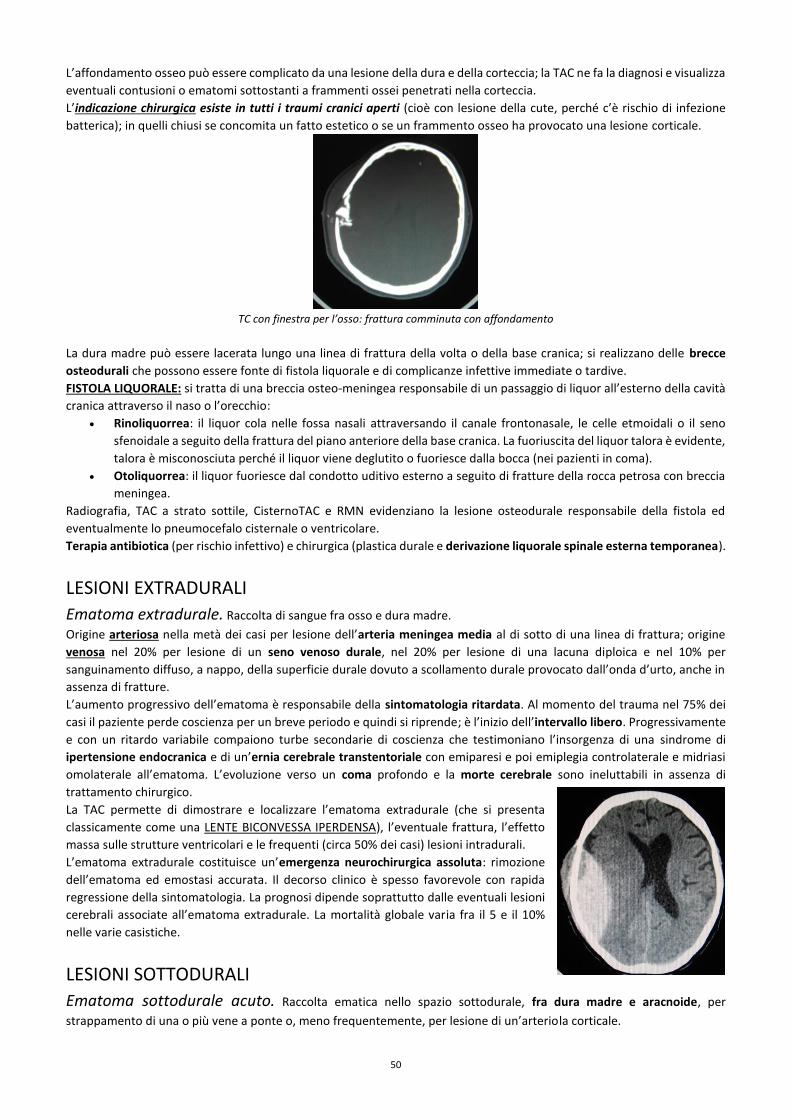
50
L’affondamento osseo può essere complicato da una lesione della dura e della corteccia; la TAC ne fa la diagnosi e visualizza
eventuali contusioni o ematomi sottostanti a frammenti ossei penetrati nella corteccia.
L’indicazione chirurgica esiste in tutti i traumi cranici aperti (cioè con lesione della cute, perché c’è rischio di infezione
batterica); in quelli chiusi se concomita un fatto estetico o se un frammento osseo ha provocato una lesione corticale.
TC con finestra per l’osso: frattura comminuta con affondamento
La dura madre può essere lacerata lungo una linea di frattura della volta o della base cranica; si realizzano delle brecce
osteodurali che possono essere fonte di fistola liquorale e di complicanze infettive immediate o tardive.
FISTOLA LIQUORALE: si tratta di una breccia osteo-meningea responsabile di un passaggio di liquor all’esterno della cavità
cranica attraverso il naso o l’orecchio:
• Rinoliquorrea: il liquor cola nelle fossa nasali attraversando il canale frontonasale, le celle etmoidali o il seno
sfenoidale a seguito della frattura del piano anteriore della base cranica. La fuoriuscita del liquor talora è evidente,
talora è misconosciuta perché il liquor viene deglutito o fuoriesce dalla bocca (nei pazienti in coma).
• Otoliquorrea: il liquor fuoriesce dal condotto uditivo esterno a seguito di fratture della rocca petrosa con breccia
meningea.
Radiografia, TAC a strato sottile, CisternoTAC e RMN evidenziano la lesione osteodurale responsabile della fistola ed
eventualmente lo pneumocefalo cisternale o ventricolare.
Terapia antibiotica (per rischio infettivo) e chirurgica (plastica durale e derivazione liquorale spinale esterna temporanea).
LESIONI EXTRADURALI
Ematoma extradurale. Raccolta di sangue fra osso e dura madre.
Origine arteriosa nella metà dei casi per lesione dell’arteria meningea media al di sotto di una linea di frattura; origine
venosa nel 20% per lesione di un seno venoso durale, nel 20% per lesione di una lacuna diploica e nel 10% per
sanguinamento diffuso, a nappo, della superficie durale dovuto a scollamento durale provocato dall’onda d’urto, anche in
assenza di fratture.
L’aumento progressivo dell’ematoma è responsabile della sintomatologia ritardata. Al momento del trauma nel 75% dei
casi il paziente perde coscienza per un breve periodo e quindi si riprende; è l’inizio dell’intervallo libero. Progressivamente
e con un ritardo variabile compaiono turbe secondarie di coscienza che testimoniano l’insorgenza di una sindrome di
ipertensione endocranica e di un’ernia cerebrale transtentoriale con emiparesi e poi emiplegia controlaterale e midriasi
omolaterale all’ematoma. L’evoluzione verso un coma profondo e la morte cerebrale sono ineluttabili in assenza di
trattamento chirurgico.
La TAC permette di dimostrare e localizzare l’ematoma extradurale (che si presenta
classicamente come una LENTE BICONVESSA IPERDENSA), l’eventuale frattura, l’effetto
massa sulle strutture ventricolari e le frequenti (circa 50% dei casi) lesioni intradurali.
L’ematoma extradurale costituisce un’emergenza neurochirurgica assoluta: rimozione
dell’ematoma ed emostasi accurata. Il decorso clinico è spesso favorevole con rapida
regressione della sintomatologia. La prognosi dipende soprattutto dalle eventuali lesioni
cerebrali associate all’ematoma extradurale. La mortalità globale varia fra il 5 e il 10%
nelle varie casistiche.
LESIONI SOTTODURALI
Ematoma sottodurale acuto. Raccolta ematica nello spazio sottodurale, fra dura madre e aracnoide, per
strappamento di una o più vene a ponte o, meno frequentemente, per lesione di un’arteriola corticale.

51
Si verifica per lo più dopo un trauma violento. Molto rapidamente evolutivo, è responsabile di quadri clinici gravi.
Raramente isolato, è frequentemente associato a lesioni corticali o a lesioni della sostanza bianca e a un edema cerebrale
diffuso che contribuiscono ad aggravare l’ipertensione endocranica (questa limita per sé stessa lo sviluppo dell’ematoma
che infatti può anche essere di spessore talora modesto al riguardo della gravità clinica) e portano rapidamente alla
formazione di ernie cerebrali interne (temporale transtentoriale) spiegando l’alta mortalità (30 - 90% nelle varie casistiche).
Gravi alterazioni della coscienza sopraggiungono repentinamente simulando un coma d’emblée. L’evoluzione potrà essere
stazionaria durante i primi 3 giorni, ma di solito è caratterizzata da un rapido aggravamento evolvente in un coma profondo.
Un’emiplegia, crisi comiziali e segni di impegno temporale (crisi di decerebrazione, anisocoria) sono manifestazioni usuali.
La TAC evidenzia una raccolta a SEMILUNA IPERDENSA, concava sopra la convessità emisferica (talora anche interemisferica,
raramente in fossa posteriore) e con importante effetto massa sulle strutture mediane; talora sono dimostrabili già in fase
acuta aree contusive ed edema cerebrale.
Il trattamento chirurgico si impone d’urgenza, il più rapidamente possibile: ampia craniotomia, evacuazione dell’ematoma,
emostasi della/e vena/e a ponte o dell’arteriola corticale sanguinanti, plastica durale, talora sutura cutanea senza
riposizionamento dell’opercolo osseo (craniotomia decompressiva). Monitoraggio della PIC e misure rianimatorie sono
indispensabili.
La mortalità rimane elevata; l’età, il quadro clinico iniziale, l’effetto massa alla TAC, la PIC pre e postoperatoria sono
attualmente considerati i principali fattori predittivi della mortalità e morbidità.
Craniotomia decompressiva con successivo riposizionamento dell’opercolo
LESIONI SUBARACNOIDEE Si tratta di emorragie subaracnoidee, sovente diffuse, di origine abitualmente venosa; espongono al rischio di idrocefalo
acuto o più spesso tardivo (per ostacolo al riassorbimento liquorale).
LESIONI ENCEFALICHE Comprendono le lesioni cortico-sottocorticali e le lesioni della sostanza bianca. Sono provocate da differenti meccanismi,
più o meno associati, di dislocamento e cavitazione (per compressione/decompressione) del parenchima encefalico
secondari alla brusca, improvvisa decelerazione della testa nel momento dell’impatto. Nel loro insieme costituiscono le
lesioni encefaliche “primarie” del trauma cranico.
Lesioni cortico-sottocorticali o contusioni. Sono frequenti nei traumi senza frattura: in questi casi la quasi
totalità dell’energia cinetica è trasmessa all’encefalo. Localizzate in corrispondenza del punto di impatto o in sede opposta
(“lesioni da contraccolpo”), uniche o multiple, risultano dallo schiacciamento dell’encefalo contro le strutture ossee. Si
tratta di lesioni parenchimali emorragiche lacerative necrotiche, talora veri ematomi intracerebrali, che inducono edema
cerebrale causa a sua volta di ipossia, ipercapnia e congestione venosa perifocali, fattori che favoriscono la diapedesi in
seno al tessuto lacerato e il suo progressivo accrescimento.
Alla TAC si presentano per lo più eterogenee (per associazione di fenomeni emorragici, edematosi, ischemici) come AREE
IPERDENSE (emorragiche) circondate da un ALONE IPODENSO DI EDEMA REATTIVO.
Talora si complicano con ipertensione endocranica e evolvono verso la comparsa o l’aggravamento del coma. Quando non
rispondono alla terapia antiedemigena vanno trattate con la rimozione chirurgica.
Lesioni della sostanza bianca o lesioni assonali diffuse. Sono legate alle differenze di densità e di
citoarchitettura delle strutture encefaliche che comportano delle velocità di dislocamento variabili fra le diverse strutture
nervose (mesencefalo-diencefalo, diencefalo-emisferi cerebrali) a seguito dell’accelerazione/decelerazione rotazionale che
si verifica in occasione di un grave impatto traumatico. A livello microscopico consistono in rotture assonali.
Si evidenziano alla RMN come microscopici focolai emorragici, diffusi, soprattutto nel corpo calloso e nella porzione
rostrale e dorsale del tronco encefalico. Si associano spesso ad un grave edema cerebrale che ne aggrava le conseguenze.

52
Sono responsabili di un coma grave “d’emblée”. Possono essere diagnosticate clinicamente quando la perdita di coscienza
persiste oltre le 6 ore in assenza di lesioni occupanti spazio alla TAC (sebbene possano essere concomitanti ad un ematoma
sottodurale o extradurale acuto).
La lesione encefalica iniziale/primaria può condizionare la comparsa e l’evoluzione di lesioni encefaliche secondarie
(necrosi, edema cerebrale con possibile ernia cerebrale interna) con due meccanismi:
• Il coma iniziale comporta una disregolazione delle funzioni ventilatoria (insufficienza), emodinamica (ipotensione) e
termica (ipertermia) il cui risultato finale è l’ipossia e nei casi più gravi l’anossia cerebrale;
• La lesione primaria determina una dissociazione fra il flusso ematico cerebrale e il consumo cerebrale d’ossigeno.
La perdita energetica che subisce la cellula neuronale induce una invasione calcica intracellulare, la distruzione di
membrana, la produzione di radicali liberi, la necrosi cellulare.
In assenza di trattamento precoce le lesioni primarie e secondarie comportano una ipertensione endocranica che può
esitare nell’arresto circolatorio cerebrale.
La NEURORIANIMAZIONE ha lo scopo di prevenire la comparsa e l’estensione della lesione encefalica secondaria:
- Sul luogo dell’incidente, in caso di coma, mediante intubazione-ventilazione (per contrastare l’anossia), sedazione con
farmaci a emivita corta, riempimento vascolare limitato allo stretto necessario (per contrastare ipovolemia ma anche
ipervolemia che favorirebbe l’edema cerebrale)
- In ambiente ospedaliero mediante stretta sorveglianza clinica, terapia farmacologica, ventilazione meccanica,
monitoraggio della PIC (con trasduttore intraventricolare o intraparenchimale) e della Saturazione d’Ossigeno.
Monitoraggio della PIC e della SO2 in particolare consentono di adattare tempestivamente la terapia e contrastare gli
aggravamenti acuti.
Contro l’Ipertensione Endocranica:
➢ Testa sollevata di 30° per agevolare il drenaggio venoso cerebrale;
➢ Iperventilazione assistita mantenendo una capnia di 25-30 mmHg;
➢ Restrizione idrosodica;
➢ Mantenimento di una normale pressione arteriosa sistolica;
➢ Terapia osmotica (Mannitolo);
➢ Coma barbiturico e ipotermia moderata (34-35° C) per ridurre il metabolismo e quindi l’attività neuronale;
➢ Derivazione ventricolare esterna per sottrarre liquor (quando possibile);
➢ Craniotomia decompressiva.
COMPLICANZE POST-TRAUMATICHE TARDIVE
Epilessia post-traumatica. Può comparire uno-due mesi dopo il trauma, tempo necessario per la costituzione di
una cicatrice gliale a livello della lesione corticale. Terapia anticomiziale (non profilattica).
Idrocefalo. Per lo più secondario a turbe del riassorbimento liquorale specie dopo un trauma che abbia comportato
un’emorragia subaracnoidea; talora secondario ad alterazioni della circolazione liquorale a causa di adesioni reattive delle
cisterne della base encefalica.
Infezioni. Ascesso cerebrale, meningite post-traumatica, tromboflebite settica di un seno venoso durale, empiema
sottodurale.
Sindrome post-traumatica soggettiva. A seguito di traumi minori possono comparire cefalea resistente agli
antalgici, vertigini o sensazioni vertiginose ai cambiamenti di posizione, turbe neuropsichiche (irritabilità, tendenza alla
depressione, astenia fisica, turbe mnesiche). Evoluzione favorevole in pochi mesi spontaneamente.
Ematoma sottodurale cronico. Raccolta liquida o mista (con qualche coagulo) nello spazio sottodurale, delimitata
da due membrane di cui l’esterna è riccamente vascolarizzata. Si manifesta dopo un intervallo variabile da 15 giorni ad
alcuni mesi successivo ad un trauma cranico, frequentemente lieve, che comporti un modesto sanguinamento nello spazio
sottodurale. La cefalea è frequente e così pure le turbe cognitive (rallentamento ideativo, disturbi della memoria, sindrome
confusionale); talora compaiono crisi comiziali parziali; col tempo si instaura una sindrome di ipertensione endocranica. La
TAC evidenzia una raccolta iso-ipodensa sottodurale con effetto massa sulle strutture mediane.
Terapia chirurgica con craniostomia e drenaggio dell’ematoma. Prognosi buona nell’80% dei casi; fattori sfavorevoli
l’etilismo cronico e l’età avanzata

53
Igroma sottodurale. Raccolta liquorale nello spazio sottodurale, ipodensa alla TAC. Segue di solito un trauma cranico
grave con lesione aracnoidale e passaggio liquorale nello spazio sottodurale. Per lo più sparisce spontaneamente o dopo
una breve terapia cortisonica. Richiede un drenaggio chirurgico solo se provoca deficit neurologici.
FATTORI CHE INFLUENZANO LA PROGNOSI NEI TRAUMI CRANICI - Prime misure terapeutiche sul luogo dell’incidente e poi in Ospedale
- Età: recupero migliore nelle persone giovani; negli adulti la mortalità aumenta con l’età, nei bambini tende piuttosto a
diminuire con l’età
- Stato clinico iniziale (GCS): nei traumi cranici gravi con GCS ≤ 8 la mortalità è in media del 43%, con un massimo del
94% per un GCS di 3 e un minimo del 16% per un GCS di 8; analogamente la qualità di sopravvivenza (reinserimento
sociale, dipendenza, stato vegetativo) è correlata al GCS iniziale.
- Tipo di lesioni cerebrali: mortalità del 62% (elevata) nei traumi con ematoma sottodurale acuto, del 52% nelle lesioni
assonali diffuse, del 44% nelle contusioni.
- Associazione ad un politraumatismo: nel 56-60% dei pazienti con GCS ≤ 8 si riscontra la lesione di uno o più altri organi;
nel 25% queste lesioni sono “chirurgiche”; nel 4-5% è presente una frattura vertebrale (soprattutto da C1 a C3). È
assolutamente importante la tempestività di diagnosi e di trattamento, anche in considerazione della comparsa e
dell’aggravamento delle lesioni encefaliche secondarie (indotte da anossia e/o ipotensione).

54
TRAUMI SPINALI Trauma spinale grave: 20% dei casi arrivano nell’ambito di un politrauma.
Nei bambini in età pediatrica c’è una prevalenza a carico della colonna cervicale probabilmente per una disposizione dei
pesi e del carico strutturale a carico del tratto cervicale.
Cause. Eventi traumatici: traumi stradali, cadute, tuffi, aggressioni→Leggera
prevalenza negli uomini.
Eventi non traumatici: osteoporosi, tumori infiltranti, infezioni (ascesso o
problematica di natura infettiva).
Esame principale per la diagnostica di lesioni midollari è la RMN: canale
bianco dietro le vertebre è il canale midollare, con il midollo che appare
grigio all’interno.
Prima però si fanno Rx e TC per valutare lesione vertebrale.
TRAUMA SPINALE • Livello della lesione:
o Motorio: motilità segmentaria o riflessi
o Sensitivo
o In caso di lesione cervico-bulbare (C2-C3) si ha tetraplegia, insufficienza respiratoria e arresto cardiaco.
• Tipo di lesione:
o Amielica
o Mielo-radicolare:
▪ Completa: deficit completo, motorio e sensitivo. Si può trattare di una lesione puramente midollare o
puramente radicolare→ se interessa L5, non c’è più midollo quindi il problema è radicolare (sindrome della
cauda equina, è più considerata una lesione incompleta).
▪ Incompleta: a seconda della porzione di midollo colpita (cordone centrale, anteriore, posteriore o
emimiidollare -sdr. Di Brown-Sequard).
LESIONE MIDOLLARE COMPLETA Quadro clinico:
• Subito dopo il trauma shock midollare
• Abolizione (totale) della motilità volontaria
• Atonia muscolare
• Areflessia profonda e superficiale

55
• Anestesia sottolesionale
• Alterazioni neurovegetative
Shock spinale. Consiste nella perdita temporanea delle funzioni spinali e dei riflessi al di sotto del livello della lesione
(trauma acuto con successiva ripresa della funzione):
• PARALISI FLACCIDA
• ANESTESIA
• AREFLESSIA: bulbo-cavernoso assente: consiste nella contrazione dello sfintere anale in risposta allo
schiacciamento del glande o del clitoride. Possono avere bradicardia e ipotensione (priapismo).
Timing: variabile, si risolve in genere entro 48 ore.
NB: Lo shock spinale si vede al di sotto del livello della lesione. In caso di lesione organica (ematoma, trauma, ferita
penetrante...) la perdita non è TEMPORANEA, ma evolve in un quadro di spasticità piramidale.
LESIONE MIDOLLARE INCOMPLETA
Sdr. midollare anteriore. Condizione acuta, può essere causata da
compressione diretta da trauma per movimento di flessione e compressione o da
occlusione dell'arteria spinale anteriore, che irrora i 2/3 anteriori del midollo.
Solitamente a livello cervicale.
(NB: cause possibili→ insufficienza aortica e aterosclerosi).
- Paralisi flaccida
- con anestesia termodolorifica, sotto il livello della lesione,
- risparmiati tatto, vibrazione, propriocezione.
È la sindrome a prognosi peggiore, può evolvere a sindrome completa. 10-20% di
chance di recupero motori.
Sdr. midollare posteriore. - Compromissione sensibilità tattile e profonda→ atassia sensitiva.
- Raramente paraplegia o paraparesi
- Livello cervicale: possibile lieve paresi AASS
Sdr. emimidollare. O emisezione di Brown-Sequard: Rara e di solito incompleta, tipiche
cause traumi penetranti o per trauma in rotazione.
- Paralisi flaccida omolaterale (che poi diventa spastica con iperreflessia, Babinski e
disfunzioni vasomotorie);
- Perdita omolaterale di vibrazione, discriminazione e propriocezione
- Anestesia controlaterale termodolorifica, sotto il livello della lesione.
Il tatto non è perso dato che la emisezione lascia da un lato le colonne posteriori intatte,
dall’altro il fascio spinotalamico anteriore.
Sdr. della cauda. - Lombalgia/lombosciatalgia
- Anestesia a sella
- Disturbi sfinterici con ritenzione urinaria
- Paraparesi/plegia
- Iporeflessia achillea
- Disfunzioni sessuali (deficit erezione)
- Riflesso bulbocavernoso spesso assente
MANAGEMENT INIZIALE ➢ Valutazione neurologica→ sempre controllo PA e volemia (possibile ipotensione e bradicardia).
➢ Accertamenti radiologici:
o RX: non sempre è facile fare diagnosi con RX perché no è chiara la distinzione tra le vertebre.
o TC: si distnignuono bene le vertebre.

56
o RM: non serve tanto per vedere il tipo di frattura (basta la TC per quello) ma per vedere bene il midollo e
come è messo: se è compresso, se è sofferente. Serve anche per vedere se una lesione è recente o
pregressa (un osso cioè deformato ma già guarito).
➢ Presidi medici per neuroprotezione: infusione continua di METIL-PREDNISOLONE→ ha un effetto neuroprotettivo
sulle lesioni midollari, soprattutto se non gravissime (se non richiedono decompressione o intervento).
Le fratture si distinguono in stabili o instabili: queste ultime si possono muovere causando danno.
COMPRESSIONE DI STRUTTURE NERVOSE Può avvenire per:
➢ Lussazione
➢ Frammenti ossei endocanalari
➢ Ernia discale
➢ Ematomi extra o sottodurali
DECOMPRESSIONE CHIRURGICA: dà dei benefici in termini di recupero clinico. Dal punto di vista teorico, si dà il limite di
48 ore, in realtà dopo le 24 ore di una lesione le possibilità di recupero del paziente sono quasi inesistenti.
• Indicata e urgente se lesione midollare incompleta:
o lussazione non riducibile
o progressione neurologica: se la compressione è ingravescente e se c’è la possibilità di un recupero, deve
essere fatta nel minor tempo possibile
o compressione mielo-radicolare (osso, disco)
• Non indicata se lesione midollare completa dopo 24h (non è dimostrato miglioramento neurologico dopo
decompressione) o se lesione midollare centrale.
• Controindicata se instabilità medica generale.
Es. laminectomia cervicale: asportazione ella lamina vertebrale in modo da ottenere un’apertura.
LESIONI TRAUMATICHE DELLA COLONNA CERVICALE
Cerniera atlanto-occipitale (C0-C1). Costituisce l’1% delle lesioni cervicali.
Può avvenire per lussazione (anteriore, posteriore o assiale -distrazione longitudinale-), per ffratture dei condili occipitali o
per fratture dell’atlante. Possono essere da paucisintomatiche a grave lesione cervico-bulbare.
No trazione in attesa del trattamento (peggioramento!).
Valutazione stabilità: condili occipitali, masse laterali di C2, dente C2, legamenti:
- Se stabile: immobilizzazione con collare per 3 mesi.
- Se instabile: stabilizzazione chirurgica con fusione occipito-cervicale (C0-C2-C3) o C1-C2.

57
Lesioni dell’atlante ed epistrofeo (C1-C2). Possono essere:
❖ Isolate C1: infrequenti deficit neurologici. Possono coinvolgere arco anteriore o posteriori mono o bilaterali (più
frequenti), o le masse laterali.
Trattamento: immobilizzazione esterna con collare o Halo-Vest.
❖ Combinate C1-C2 (atlanto-epistrofee): da sublussazione rotatoria atlo-assiale→ si interviene chirurgicamente con una
legatura posteriore di C1-C2.
Lesione di C1 isolata, con frattura in arco anteriore e posteriore. Intervento di legatura posteriore di C1-C2.
Lesioni dell’epistrofeo. ➢ Frattura istmica bilaterale di C2 (frattura di Hangman): nel 95% dei casi è senza deficit neurologici; in caso di
lussazione o angolazione di C2-C3 si ha instabilità:
o Tipo I: Stabile: sublussazione C2-C3 minore di 3 mm→ immobilizzazione con collare Philadelphia.
o Tipo II: Instabile: sublussazione C2-C3 maggiore di 3 mm o angolazione tra corpo e dente maggiore di 15° (con
rottura del legamento longitudinale posteriore)→ chirurgia e immobilizzazione.
o Tipo III: Instabile, lussazione anteriore di C2 (possibili deficit neurologici)→ chirurgia e immobilizzazione ( no
trazione!).
Stabilizzazione chirurgica consiste in 3 approcci:
- Sintesi istmica C2 per via posteriore: inserimento di due viti in senso postero-anteriore.
- Fusione anteriore C2-C3 per via combinata (anteriore e posteriore)
- Sintesi posteriore C1-C2.
Sintesi istmica per via posteriore
➢ Frattura del dente dell’epistrofeo: senza lussazione C1-C2 né frattura di C1.
o Tipo I: stabile, apice del dente, sopra legamento trasverso. Spesso si associa a dislocazione atlanto-occipitale.
o Tipo II: instabile, alla base del dente.
o Tipo III: instabile, con frattura del corpo di C2.

58
Trattamento:
- Chirurgico (osteosintesi anteriore del dente o artrodesi con fusione di C1-C2, soprattutto
se lussazione/frattura) e immobilizzazione.
- Conservativo per pazienti anziani.
Valutare sempre se mancata fusione a distanza; se instabilità dopo immobilizzazione esterna
(nonunion) si procede con chirurgia.
APPROCCIO CHIRURGICO TRANSORALE: permette l’asportazione del dente dell’epistrofeo
(decompressione) e fissazione di C1-C2 o occipito-cerviacale, in caso di:
- Compressione di midollo e trocno per arretramento e cranializzazione del dente.
- Instabilità con sublussazione C1-C2 per rotazione o spostamento antero-posteriore.
Fratture cervicali C3-C7. ❖ Distrazioni gravi da iperflessione: non si tratta di frattura ossea solo di rottura del
legamento longitudinale posteriore, che però può causare lesioni midollari severe. SI valuta
instabilità alla RM prevede immobilizzazione o stabilizzazione chirurgica.
❖ Fratture o lussazioni: possono essere classificate in base al meccanismo del trauma (flessione,
estensione, distrazione o compresisone). Si valuta instabilità con la teoria delle 3 colonne.
Trattamento:
o Stabilizzazione chirurgica in caso di Fratture del corpo o lussazioni
o Trattamento conservativo con immobilizzazione esterna in caso di sublussazioni e frattura
stabile teardrop somatica.
❖ Dislocazione delle faccette articolari: per distrazione, flessione o rotazione (se monolaterale, è per
rotazione dal lato opposto). Quelle bilaterali sono associate a danno del legamento posteriore. Si
possono associare frequentemente a danno midollare completo, incompleto o radicolare→
trattamento chirurgico:
o Approccio posteriore: posizionamento di viti articolari/ e peduncolari (fino alle masse laterali), con eventuale
decompressione posteriore.
o Via anteriore: usata soprattutto per lussazione con fratture somatiche (anche fratture a scoppio). Innesto
intersomatico, protesi somatica, placche e viti somatiche. Se compressione midollare, somectomia.
Viti somatiche e placche somectomia C5 con protesi somatica Innesti intersomatici
o Approccio combinato anteriore/posteriore: in caso di grave lesione somatica e lussazione di faccette articolari.

59
Lesione di giunzione cervico-toracica (C7-T1). È una zona sensibile, di transizione anatomica e biomeccanica.
Stabilizzazione posteriore preferibile per difficoltà accesso anatomico per via anteriore
Comunque criticità di livello per forze elevate sulle masse laterali di C7: viti peduncolari anche in C7
Sola stabilizzazione anteriore: 50% failure.
TEORIA DELLE 3 COLONNE Vale per fratture cervicali (da C3), dorsali e lombari:
1. Colonna anteriore: metà anteriore disco e corpo, legamento longitudinale anteriore.
2. Colonna media: metà posteriore disco e corpo, legamento longitudinale posteriore.
3. Colonna posteriore: arco posteriore, legamenti e processi articolari.
Una frattura si dice:
STABILE: se lesione di 1 colonna.
INSTABILE: se lesione di 2 colonne.
INSTABILE GRAVE: se lesione di 3 colonne.
FRATTURE DORSO-LOMBARI
Classificazione secondo Magerl. - Tipo A: colonna anteriore: frattura predominante del corpo, compressione somatica pura.
- Tipo B: colonna anteriore+posteriore: flessione-distrazione.
- Tipo C: tutte e 3 le colonne: dislocazione in rotazione e/o traslazione fino ad interessamento di tutte e tre le
colonne.
Trattamento. • Lesioni stabili: sono quelle con muro posteriore intatto e lieve compressione somatica→ conservativo e
immobilizzazione.

60
• Lesioni instabili: sono quelle con interessamento anche del muro posteriore: arretramento muro posteriore, fratture
da scoppio, frattura-lussazione. Hanno una progressione neurologica→ stabilizzazione e decompressione.
Trattamento chirurgico. - Approccio posteriore:
o Pro:
✓ Atto chirurgico più rapido, meno complesso, conservativo
✓ Migliore controllo durale e midollare, esclusivo radicolare
✓ Minori complicanze (generali, settiche, liquorali, ecc.)
✓ Possibilità di rimuovere la sintesi metallica dopo 8-10 mesi
o Contro:
Stabilizzazione meno solida sui pilastri anteriore e medio
Rischio di rottura della sintesi (viti peduncolari)
Prolungata necessità di busto
o Procedura:
1. Inserimento viti peduncolari
2. Moderata distrazione
3. Decompressione
4. Rimodellamento del muro posteriore
5. Immobilizzazione esterna (busto ortopedico, Jewitt)
- Approccio anteriore (toraco-lombo-laparotomico):
o Pro:
✓ Carico più resistente sulle colonne anteriore e media
✓ Via più diretta al midollo per la decompressione somatica
✓ Accesso diretto al disco
✓ Indicazione assoluta: deformità somatica a distanza
o Contro:
Complessità dell’atto chirurgico e composizione del team
Frequente controindicazione per gravità clinica (politrauma)
Più elevato rischio di complicanze (Fistola liquorale pleurica!)
Necessità frequente di approccio combinato (ant. + post.)
MIELOPATIA SPONDILOGENA CERVICALE E SPONDILOSI CERVICALE Talvolta usata come sinonimo di stenosi cervicale, è un processo degenerativo che si associa spesso all’ invecchiamento.
Cause. 1. Congenita (the "shallow cervical canal")
2. Degenerazione del disco intervertebrale (ernia molle e/o osteofiti)
3. Ipertrofia di uno dei seguenti componenti:
- Lamina
- Dura
- Faccette articolari
- Legamenti: LLP, legamento giallo
4. Sublussazione: degenerazione di disco e faccette articolari
5. Alterazione delle normali curvature del rachide (riduzione/inversione della normale curva lordotica fino alla cifosi;
iperlordosi)
La spondilosi cervicale è la più comune causa di mielopatia cervicale in pazienti di età > 55 anni.
La Mielopatia spondilogena cervicale si sviluppa in quasi tutti i pazienti con una stenosi del canale cervicale ≥ del 30% del
canale vertebrale.

61
Fase spondilogena senza mielopatia Fase mielopatica
Storia naturale. L’andamento dei sintomi è altamente variabile e imprevedibile.
Nel 75% dei casi di mielopatia spondilogena la patologia tende a progredire bruscamente (un terzo dei casi) o in modo
graduale (due terzi).
Pazienti con stenosi cervicale senza segni di mielopatia alla RM ma che presentano anomalie ai test neurofisiologici (PESS-
PEM) o che presentano radicolopatia, sono a rischio di sviluppare anche mielopatia (Classe I).
Stenosi midollari severe di lungo corso (anni), possono associarsi a deficit neurologici irreversibili inducendo necrosi della
sostanza bianca e grigia midollare (Classe III).
Sintomi motori. Associati a compressione midollare o radicolare. Sintomi motori più precoci sono tipicamente
debolezza dei m. tricipiti e dei m. intrinseci della mano associati o meno a ipotrofia. Potranno associarsi lentezza, rigidità
nell’apertura e chiusura dei pugni. È comune l’impaccio motorio nell’esecuzione dei movimenti fini della mano (scrivere,
allacciarsi i bottoni)
Spesso è presente anche debolezza dei muscoli prossimali degli arti inferiori (lieve- moderata ipostenia dell’ileopsoas è
presente nel 54% dei casi) e spasticità.
Sintomi sensitivi. Possono essere minimi, e quando presenti non hanno generalmente distribuzione radicolare. Può
esserci una perdita della sensibilità con distribuzione a guanto a livello delle mani. Livelli di alterazione sensitiva possono
essere presenti anche al di sotto della stenosi. Agli arti inferiori spesso è presente perdita della pallestesia (82%). La
compressione del tratto spinocerebellare può causare difficoltà nella corsa. Alcuni pazienti possono presentarsi con
disfunzione prevalente a carico dei cordoni posteriori (alterata propriocezione e difficoltà nel distinguere 2 punti come
separati).
Riflessi. Nel 72-87 % i riflessi sono iperelicitabili a distanza variabile al di sotto del livello della stenosi. Possono essere
presenti clono, segno di Babinski.
Sfinteri. Aumentata frequenza e urgenza minzionale sono comuni nella MSC. Incontinenza urinaria è rara, così come i
disturbi sfinterici anali.

62
MALATTIE DEL SONNO È uno stato comportamentale caratterizzato soprattutto da:
- Disimpegno e non responsività verso l’ambiente esterno
- Reversibilità→ se manca il riorno alla veglia si tratta di coma.
- Ridotta attività motoria
- Posture stereotipate
Come si studia? - Polisonnografia è l’esame principale
- ECG
- Rssamento
- Ossimetria
- Spirogramma toracico
- Actigrafia
- Posizionimetria
POLISONNOGRAFIA Consiste nel far dormire il paziente in studio con videocamere e registrazione di 3 parametri poligrafici:
• EEG
• EOG (oculo)
• EMG (mio): si registra dai muscoli del pavimento della bocca; è molto sensibile ai cambiamenti del tono.
Esiste anche una forma ridotta di tipo respiratorio, in cui si rinuncia all’EEG; il soggetto non viene inserito in un laboratorio
del sonno, ma sta a casa con un registratore portatile tipo Holter.
Fisiologia. Ritmo circadiano nictemerale del sonno: teoria che si basa sui nuclei ipotalamici (nucleo sovrachiasmatico):
consiste in un orologio biologico che dà il ritmo. Questo ritmo si matura nel corso dell’età: i bambini piccoli non hanno un
ritmo circadiano, dormono in maniera simile durante la notte e il giorno. Solo nell’adulto si raggiunge il pieno ritmo
circadiano, dalle 11 alle 7. Nell’anziano ritorna a esserci una fase di sonno post-prandiale.
Quante ore di sonno sono necessarie per un adulto? Circa 7.30 ore. Tende a rimanere costante in un dato individuo, eccezion
fatta per l’età. L’unico fattore in grado di aumentare il tempo del sonno è la deprivazione: se si depriva il sonno per un certo
periodo, questo deficit verrà compensato prima o poi con un aumento di ore del sonno.
Scopo del sonno: riportarci alle condizioni perfette di energie tipiche della veglia.
FASI DEL SONNO Il sonno non è uniforme, si divide in FASI REM (rapid eye movements) e FASI NREM. Sono state individuate solo tramite
EOG: solo così si seppe che si trattava di sonno, mentre all’osservazione poteva sembrare un movimento dato da un parziale
risveglio.

63
Sonno NREM. Caratterizzato da una diminuzione del tono ortosimpatico: si riduce la gittata cardiaca, la pressione, la
frequenta; il tono muscolare e i riflessi sono conservati. Si tratta di un sonno LENTO e RISTORATORE, perché il ritmo all’EEG
è lento. Si divide in 3 stadi:
1. STADIO 1, ADDORMENTAMENTO:
a. EEG: si passa da un’attività di base alfa, che pian piano si spezzetta e progressivamente si passa a un ritmo theta.
b. EOG: gli occhi hanno movimenti lenti, definiti di rotolamento.
c. EMG: non cambia particolarmente.
2. STADIO 2:
a. EEG: attività di fondo theta, dove compaiono i FUSI DEL SONNO: sono
oscillazioni ritmiche con frequenza 12-14 Hz. Compaiono anche COMPLESSI K:
sono onde lente aguzze, con una durata di almeno 0,5 s, che si inseriscono sul
ritmo theta→ fusi del sonno e complessi K su uno sfondo theta.
3. STADIO 3, SONNO LENTO: è il sonno ristoratore, profondo, quello in cui è più difficile svegliare.
a. EEG: dominato da attività delta. Prima si definiva stadio 3 fino al 50% di delta e stadio 4 dopo il 50% di delta, ora
la presenza di ritmo delta si parla solo di stadio 3.
b. EOG: assente
c. EMG: decremento rispetto alla veglia.
NREM Stadio 1
NREM Stadio 2
NREM Stadio 3
Sonno REM. • EEG: tracciato a frequenze miste di basso voltaggio, molto simile a quello del soggetto sveglio con gli occhi aperti
(come dopo la fase di apertura degli occhi). Ecco perché senza l’EOG si poteva pensare che in questa fase si fosse
risvegliato.
• EOG: si è scoperto che in realtà si hanno dei movimenti oculari rapidissimi e transitori (fasico).
• EMG: Drastica riduzione del tono muscolare antigravitario: diminuisce molto, o si abolisce, su cui si possono
innestare alcune scariche fasiche: l’apparato locomotore è praticamente inibito→ si parla di ATONIA MUSCOLARE
(o DISCONNESSIONE MOTORIA o PARALISI DEL REM): complesse catene neuronali, facendo sinapsi nel bulbo, si
approfondano nel midollo per dare forza all’inibizione→aumenta il tono inibitorio durante il sonno REM. (vedi
immagine slide).
Tumescenza dei genitali esterni: si sfrutta nella diagnostica neuro-urologica e andrologica nella diagnostica delle
disfunzioni rettili e impotenze. In presenza di una normale tumescenza dei genitali esterni, si assume che non ci siano danni
neurologici e che quindi la causa di disfunzione sia di altra natura.
IPNOGRAMMA È la rappresentazione grafica del ciclo del sonno con:
- Asse x: ore del sonno.
- Asse y: stadi del sonno.
Si vede una ripetizione ciclica di fenomeni abbastanza uguali: dallo stato di sveglia, si passa a stadio 1, 2, 3 poi risale e una
volta ritornato allo stadio 2 si passa alla fase REM. Questo ciclo si ripete per 4-5 volte per notte, con alcune differenze:
- Nelle prime parti della notte c’è più sonno profondo, quindi il sonno del mattino è più leggero.
- Le fasi REM aumentano di durata con la seconda parte della notte. In questa fase avvengono i sogni vividi.

64
Nell’anziano, manca il sonno profondo all’inizio della notte ed è un sonno molto più spezzettato, soprattutto al mattino
dove ci sono molto risvegli.
MICROSCRUTTURA DEL SONNO NREM Per molta parte degli stati NREM, anziché esserci una stabilità fisiologica, c’è una attività oscillatoria ulteriore: si distinguono
fasi stabili (N-CAP) dell’EEG e stadi chiamati CAP (Pattern Ciclico Alternante)→ fasi distinte di attivazione del trattato, dove
compaiono complesso K, scoppi di delta, epoche di alfa, contrapposte a fasi di quiescenza.
L’importante è sapere solo che nelle fasi NREM ci sono fasi di quiescenza alternate a fasi di attivazione del tracciato, non
dobbiamo sapere altro.
SOGNO Si sogna in TUTTE le fasi del sogno, non solo nella fase REM.
Dipende se si ratta di un sogno con alto contenuto visivo, con spostamento violento, con tono vivido e altamente pregnante,
spesso si tratta di qualcosa di illogico, bizzarro, incoerente, ansioso→ si tratta di un sogno della fase REM.
Ci sono però anche attività mentali meno intense, meno illogiche, meno vivide; sono più rimuginazioni, pensieri quasi
coscienti, ragionamenti→si tratta di sogni delle fasi NREM: è un pensiero più o meno logico, una previsione, una
rievocazione di quello che è successo.
SIGNIFICATO DEL SONNO 1. Ristoro psicofisico: alcune attività scadono se non si dorme.
2. Conservazione dell’omeostasi termoregolatoria
3. Attivazione ripetuta
DISTURBI DEL SONNO CLASSIFICAZIONE
1. Insonnie
2. Ipersonnie di origine centrale
3. Disturbi della respirazione sonno-correlati→ apnee ostruttive (OSAS)
4. Disturbi circadiani del sonno
5. Disturbi del movimento sonno-correlati
6. Parasonnie
→ le più frequenti sono insonnia e apnee ostruttive.
INSONNIA Negli stati uniti un terzo di popolazione ha insonnia. Se dura per più di un mese si definisce insonnia cronica. Percezione di
un sonno insufficiente, disturbato, non ristoratore.
Eziologia. • Medica: ogni malattia che causa dolore, dispnea, brivido, nausea, sudorazione→ ha un impatto sgradevole.
• Malattie psichiatriche: sindromi ansiose, disturbo bipolare (non si dorme nella fase maniacale, è instancabile e non
dorme mai; il depresso grave invece tende a dormire tanto ma la mattina molto presto si sveglia in preda ai pensieri),
disturbo affettivo→ Il 40% degli insonni ricevono una diagnosi psichiatrica.
• Tabacco e sostanze eccitanti.
• Alcol: da un sonno profondo a cui segue però un risveglio precoce.
• Disordini primitivi, senza una causa apparente:

65
o Insonnia psicofisiologica: anche in individui sani privi di abitudini tossiche, a seguito di una esperienza
psicologicamente traumatica: lutto, divorzio, separazioni, matrimonio, trasloco, esame, perdita al gioco. Con
il passare del tempo, non recupera più il sonno perduto→ inizia un circolo vizioso per cui questa insonnia
perdura anche oltre l’evento stesso per parecchio tempo (l’evento stesso di andare a dormire causa
preoccupazione)→ cronicizzazione.
o Errata percezione dello stato di sonno: il paziente si lamenta di insonnia e continua a consultare specialisti,
fino a quando non gli fanno una polisonnografia e si trova che in realtà dorme normalmente.
o Inadeguata igiene del sonno: stile di vita incompatibile con il mantenimento di una buona qualità di sonno e
con un pieno livello di vigilanza. Tutto il personale che lavora la notte a lungo andare avrà problemi del sonno,
si guastano i meccanismi fisiologici del sonno.
• Genetica: INSONNIA FATALE FAMILIARE: malattia autosomica dominante con accumulo di prioni a livello talamico. È
fatale perché i pazienti non riescono più a dormire, per cui perdono controllo delle funzioni fisiologiche e vanno
incontro a morte (nel giro di 7-37 mesi).
FUNZIONE DEL TALAMO: ha un ruolo importante nella gestione di tantissime funzioni, tra cui il sonno; riceve stimoli visivi
e condiziona il ritmo sonno-veglia, c’entra anche con la cognizione temporale (speleologi al buio possono stare svegli un
sacco di tempo ma prima o poi crollano).
Divisione degli individui in cronotipi:
- Cronotipo gufo: predisposizione ad andare a dormire tardi
- Cronotipo allodola: svegliarsi presto o dormire tardi e svegliarsi presto
Igiene del sonno. Porre limiti alla permanenza a letto
Evitare:
- Sonnellini
- Tentativi di dormire senza sonno
- Esercizio fisico o mentale prima del sonno: su base statistica, fanno male al sonno.
- Non mangiare, bere, guardare la TV a letto.
Trattamento. Ipnoinducenti:
o Benzodiazepine
o Imidazopiridina (zolpidem)
o Ciclopirroloni.
A volte si possono usare anche veri e propri psico-farmaci.
Non dovrebbero essere prescritte così facilmente, e non dovrebbero essere dati senza ricetta; il problema è che si instaura
una dipendenza ed è difficile poi rimuoverli dalla quotidianità.
Controindicazioni a ipnoinducenti: alcol, gravidanza, guida, miastenia, OSAS.
IPERSONNIE DI ORIGINE CENTRALE
Narcolessia. Malattia piuttosto rara, caratterizzata da 4 elementi:
1. Ripetuti attacchi di sonno incoercibili: sono alterazioni del sonno REM, il paziente cade addormentato in un sonno
REM.
2. Paralisi del sonno: si verifica un prolungamento del sonno REM da sveglio, quindi ha atonia, è sveglio e non può
muoversi.
3. Allucinazioni per lo più visive quando si svegliano e quando si addormentano.
4. Cataplessia: perdita improvvisa del tono muscolare da svegli, che a volte li fa cadere, spesso in risposta a stimoli
emotivi come sorpresa, risa, cattive notizie.
Esistono due tipi di narcolessia: in una abbiamo diminuzione di ipocretina, è una malattia classificata tra le patologie
autoimmuni→ causa una alterazione dei meccanismi di controllo tra veglia, sonno NREM e sonno.

66
DISTURBI RESPIRATORI SONNO-CORRELATI Ostruzione temporanea delle vie superiore, laringe, tipica di persone obese, grandi russatori, con BPCO. Il partner di solito
si accorge di questo problema, paziente che smette di respirare, e poi ha una ripresa dall’apnea che è molto rumorosa, fa
un grande respiro che è molto avvertibile nelle vicinanze. Persona che si sveglia la notte con fame d’aria, il sonno e la qualità
del sonno saranno compromesse, in più avrà una mala ossigenazione degli organi, perché può arrivare a tantissime apnee
in una notte.
La terapia è spesso la NIV (ventilazione meccanica non invasiva).
DISTURBI DEL MOVIMENTO SONNO-CORRELATI
Sindrome delle gambe senza riposo. Pz che va a dormire e inizia a sentire un fastidio alle gambe, perdurante. La
forma idiopatica risponde a farmaci anti-parkinson come i dopamino-agonisti (bromocriptina ecc..), non si sa perché
funzionano ma funzionano, lavoreranno sui gangli della base.
PARASONNIE Sono fenomeni indesiderati che si possono verificare, ma non sono un vero e proprio disturbo patologico.
Esistono parasonnie del sonno NREM (pavor nocturnus nel bambino e incubo nell’adulto) e del sonno REM.
Tra quelle del sonno REM, ricordiamo i DISTURBI COMPORTAMENTALI DEL SONNO REM: sono sintomo d’esordio o sono
parte integrante di patologie neurodegenerative soprattutto di sinnucleinopatie, cioè le Parkinson-affini. Pazienti che
sognano di fare a botte con qualcuno e lo fa davvero durante il sonno nel letto (es. lancia oggetti verso il mattino o inizia a
tirare calci).
(Studiare bene questi disturbi, sono domande che può fare)

67
MALATTIE DEI MOTONEURONI
Termine generale per indicare le malattie neurodegenerative ad andamento progressivo che colpiscono principalmente i
neuroni motori della corteccia, del tronco cerebrale e del midollo spinale.
• Primo motoneurone: parte dalla corteccia e arriva fino alle corna anteriori del midollo spinale.
• Secondo motoneurone: parte dalle corna anteriori del midollo spinale e l’assone arriva fino alle muscolatura
periferica.
SCLEROSI LATERALE AMIOTROFICA O malattia di Charcot (scopritore) o di Lou Gehrig.
Atrofia della via discendente motoria e delle corna anteriori del midollo→ sia di primo che di secondo motoneurone.
• Sclerosi: è una atrofia gliotica delle fibre.
• Laterale: interessa la via motoria, che passa nei fasci laterali.
• Amiotrofica: denerva il muscolo e causa una riduzione della massa muscolare.
Risparmio di:
• Sistemi sensitivi
• Sistemi della coordinazione motoria.
La selettività interessa anche il sistema motorio, infatti sono generalmente risparmiati:
o Motoneuroni che controllano la motilità oculare
o Neuroni motori degli sfinteri striati anale ed uretrale, localizzati nel midollo spinale sacrale, nucleo di Onuf, a
livello S2-S4.
→ non è chiaro il motivo, forse perché sono motoneuroni più grandi che
vengono conservati per più tempo.
Rapidità di decorso. La perdita di neuroni nella SLA comparata con altre
patologie (come il Parkinson o Alzheimer) è molto più rapida→ in 3-5 anni si
ha una degenerazione completa con conservazione solo di neuroni della
motilità oculare e sfinterica.
Neuropatologia. Il referto istopatologico era molto aspecifico, con
accumuli di ubiquitina come in tutti i processi neurodegenerativi. Dal 2006,
partendo dalla considerazione della mutazione del gene TDP-43 nelle
malattie ereditarie, si è scoperto che era accumulato in tutte le forme di
SLA. È una proteina che si accumula anche nella demenza fronto-
temporale→ quadri di overlapping.
• Perdita dei motoneuroni corticali e gliosi reattiva
• Vari pattern di degenerazione dei tratti corticospinali
• Atrofia del giro precentrale (corteccia motoria)
Epidemiologia. L’incidenza ha un trend in aumento negli ultimi 10 anni→ questo aumento interessa soprattutto le
donne nate negli anni ‘30, cioè nella generazione che ha iniziato a lavorare e quindi erano più esposte ad agenti esterni che
potrebbero aver scatenato la malattia (in più le misure di sicurezza valevano solo per gli uomini, le donne non avevano le
mascherine nelle fabbriche).
Prevalenza di circa 7/100.000, che sale a 12/100.000 considerando che qui in Italia i pazienti seguiti in centri ad alto livello
assistenziale.
Età di esordio. 55-75 anni, pur essendoci pazienti anche dai 18 anni.
Patogenesi. Il 90% dei casi sono casi sporadici, ma anche tra questi il 20% presenta delle mutazioni ricorrenti; questo può
essere per la bassa penetranza (che non si manifesta in tutte le generazioni) o per nuove mutazioni.

68
In generale, si tratta di una MALATTIA MULTIFATTORIALE dove concorrono:
1. Genetica: Il fatto di essere portatori di uno di questi geni non implica il fatto che si manifesterà la malattia. Esistono
casi in cui sono presenti due mutazioni diverse contemporaneamente. In ogni regione diversa si trovano
prevalenze di una o di un’altra mutazione (es. in Piemonte prevale la c9orf72, in Sardegna la TARDBP). Mutazioni:
• SOD1: superossido dislocasi (primo identificato, poi ne sono stati scoperti altri).
• TARDBP o TDP-43
• FUS
• C9orf72→ responsabile anche di demenza fronto-temporale e forma congiunta di SLA-demenza.
• NEK1
2. Età (60 anni circa)
3. Ambiente:
a. Pesticidi
b. Piombo e metalli pesanti
c. Fumo di sigaretta
Si sono studite le popolazioni in cui sono presenti dei grappoli di pazienti; nell’isola di Guam la prevalenza era di
400/100.000→ si sono studiati i contaminanti ambientali e si è scoperta una tossina prodotta da un cianobatterio
nella terra che veniva assorbito dalla piante e che quindi poi arrivava all’uomo tramite l’alimentazione.
Reduci della guerra del Golfo: prima di partire per la guerra erano stati analizzati prima di partire con tanto di
cartella medica, e poi al ritorno erano sottoposti a follow up→ incidenza di SLA aumentata di 10 volte.
Anche nei calciatori si è scoperto esserci un aumento di incidenza di 7/100.000 sotto i 59 anni. Analizzando poi
quelli che avevano giocano a livello agonistico per più di 5 anni, si saliva a 15 casi. Su un terreno di predisposizione
genetica, gli eccessivi traumi a livello cervicale causati dai colpi di testa sembrano avere un ruolo nel favorire la
malattia (allo stesso modo per cui i pugili hanno un rischio maggiore di ammalarsi di Morbo di Parkinson).
Fisiopatologia. Vari meccanismi di degenerazione:
• Grazie alle mutazioni geniche sono state scoperte molte proteine responsabili della propagazione e della
trasmissione del danno.
• Cellule gliali hanno un ruolo fondamentale anche nella trasmissione neuronale e sono il target principale di alcune
malattie→ reazione gliale importante con astrogliosi e attivazione della microglia. Si pensa che possano essere
addirittura loro le prime cellule ad ammalarsi.
• Difetto del trasporto assonale
• Danno del mitocondrio
C’è una sorta di infezione tra una cellula all’altra, cioè un passaggio simil-prionico tra cellule. Il danno avviene focalmente
in un punto, e poi si propagai n senso caudale o craniale.
Da parte di TBP-43 c’è una diffusione a livello cerebrale dalla corteccia fronto-parietale estendendosi fino al lobo temporale
(e quindi motori) o in senso inverso, a seconda se si manifestino prima i segni frontali o motori→ dimostrazione in vivo che
TBP-43 gioca un ruolo di trasmissione della malattia.

69
Si arriva a fare diagnosi quando ormai c’è la disconnessione neurone-muscolo in almeno il 50% dei neuroni, infatti nelle
prime fasi ci sono dei meccanismi di compenso per cui i motoneuroni sani recuperano quello che i neuroni malati non sanno
fare. Quando compare il disturbo di forza per la prima volta ormai sono persi circa il 50%.
QUADRO CLINICO
Sindrome del II motoneurone. Sono sintomi da sofferenza del secondo motoneurone:
➢ Ipostenia: i movimenti fini si perdono, diventa difficile mettere la chiave nella serratura e aprire la bottiglia.
➢ Ipotrofia e ipotonia
➢ Fascicolazioni: scariche spontanee che determinano il movimento involontario di poche fibre muscolari, clinicamente
visibili; a volte interessano più fibre muscolari determinando la contrazione vera e propria (miochimie). Le fascicolazioni
si vedono anche alla lingua, che appare ipotrofica.
➢ Crampi: contrazione spontanea dolorosa di più fibre muscolari. si distinguono in crampi a riposo e da sforzo.
➢ Sintomi bulbari: rappresentano il sintomo di esordio in circa un terzo dei casi di SLA, soprattutto nelle donne in età
menopausale. In rari casi la malattia non progredisce oltre la regione bulbare, delineando il quadro di paralisi bulbare
progressiva.
o Disartria
o Disfagia
o Scialorrea
➢ Molto più rare sono le forme che esordiscono come insufficienza respiratoria acuta; sono la condizione più frequente
di insufficienza ventilatoria senza altre cause che arriva in PS→ vengono intubati e tracheotomizzati. Spesso sono legate
a forme selettive sui motoneuroni del diaframma, quindi sono difficili da individuare.
Sindrome del I motoneurone. Sono sintomi da sofferenza del secondo motoneurone:
➢ Iperreflessia: diminuiscono i controlli dai centri superiori sui motoneuroni delle corna ventrali.
➢ Segno di Babinski
➢ Segno di Hoffman-Tinel (corrispettivo di Babinski ma agli arti superiori)
➢ Spasticità
➢ Riflesso masseterino
➢ Riso e pianto spastico: avvengono in moto incongruo e incontrollato.
Disfunzione cognitiva. Non è raro trovare la concomitanza di SLA e demenza fronto-temporale, sono il continuum di
una stessa malattia, infatti hanno le stesse mutazioni. Solitamente prima viene fatta diagnosi di demenza FT poi di SLA o
prima di SLA e poi manifestano demenza→ ESTREMA ETEROGENEITÀ FENOTIPICA.
40% circa di SLA ha un disturbo frontale.
È dovuta alla trasmissione del danno dai lobi temporali ai lobi frontali.
Classificazione dei disordini cognitivi:
• Alterazioni comportamentali precoci, insidiose e progressive.
• Precoce compromissione funzioni esecutive.
• Disturbi del linguaggio (denominazione).
• Relativa conservazione delle funzioni strumentali di percezione, abilità spaziali, abilità prassiche.
• Relativa conservazione della memoria.
→ non riguarda decadimento di memoria ma su capacità di shifting
dell’attenzione, di linguaggio, …. Bisogna cercare di capire se la difficoltà di
espressione e di linguaggio è dovuta solo al deficit motorio o se c’è anche un
decadimento fronto-temporale.
I pazienti con disturbi cognitivi hanno una sopravvivenza più bassa e una
prognosi peggiore: sia perché è una malattia più estesa sia perché non essendo
cognitivamente integro è difficile da gestire anche l’aspetto sintomatico→ è
poco compliante alle terapie.

70
ESAME STRUMENTALE
EMG. Il più importante per valutare il danno del II motoneurone è l’EMG: attraverso un ago nel muscolo si registra
l’attività. Permette soprattutto nella SLA di trovare pattern caratteristici e identificare un danno laddove clinicamente anche
non è visibile→ un muscolo apparentemente normale può apparire già compromesso tramite l’EMG.
Quando si inserisce l’ago nel muscolo, le fascicolazioni che clinicamente non sono ancora visibili, generano delle scariche
continue sul tracciato, tipo a mitragliatrice. Sono causati dai tanti tentativi di rigenerazione che scaricano in maniera
disordinata.
La muscolatura paraspinale è importante perché è quella in cui per prima si vedono fascicolazioni, infatti è la prima
muscolatura innervata dal II motoneurone; spesso qui si vedono fascicolazioni prima ancora che su altri muscoli più
periferici.
Elettroneuronografia. Permette di analizzare tutto il nervo e la sua conduzione dello stimolo→ soprattutto nella fase
iniziale, la periferia è integra, non ci sono difetti di mielina che rallentano lo stimolo. Permette di fare DD con alcune forme
di neuropatia periferica, in cui la clinica può simulare quella della SLA.
RM. Permette soprattutto di individuare l’atrofia della via piramidale discendente→ i segni del I motoneurone infatti non
sono così visibili clinicamente rispetto a quelli del II, quindi può essere utile questo esame strumentale. Permette di
individuare il fascio e vedere quindi la perdita di fibre. Possibili segni:
• Corteccia: ridotta intensità di segnale in T2 a livello della corteccia motoria (aspecifico)
• Tratto cortico-spinale: incremento di segnale in T2 (secondario alla degenerazione walleriana).
Altra DD importante è con la mielopatia spondilogena cervicale, che causa compressioni sul midollo cervicale; con la RM si
riesce a distinguere.
Stimolazione magnetica transcranica. Registra la conduzione dello stimolo
dopo uno stimolo magnetico sulla corteccia parietale che evoca una risposta motoria
in un muscolo periferico; permette inoltre di discernere le vie, quindi di individuare se
il danno è nella via centrale o periferica: infatti se a livello centrale la conduzione è
conservata ma poi rallenta dal livello cervicale alla periferia, significa che il danno è a
carico del II motoneurone e non del I.
Esami biochimici. Sono marcatori indiretti aspecifici.
- CK può essere alterata perché il muscolo sta soffrendo
- Si abbassa la Crea
SINDROMI SLA-SIMILI • Forme paraneoplastiche: da tumore al polmone e all’ovaio. Sono più subdoli perché possono manifestarsi con una
sindrome neurologica prima ancora che sia individuato il tumore. In un soggetto fumatore con sintomi neurologici si fa
RX e TC.
• Tumori ematologici, soprattutto linfomi e mielomi.
• Infezioni: bisogna escludere prima di tutto la malattia di Lyme.
DIAGNOSI DI SLA CRITERI DI EL ESCORIAL:
A. Presenza di:
A1 degenerazione del II motoneurone con evidenza clinica, neurofisiologica o neuropatologica
A2 evidenza clinica di degenerazione del I motoneurone
A3 progressiva diffusione dei sintomi o segni nella regione o in altre regioni con evidenza anamnestica o clinica
B. Assenza di:
B1 evidenze neurofisiologiche o patologiche di altro processo patologico interessante il II o I motoneurone
B2 evidenze neuroradiologiche di altro processo patologico che possa spiegare i segni clinici e neurofisiologici
→ punta sul fatto che deve esserci compromissione di I e II motoneurone insieme e che deve essere progressiva.

71
DECORSO CLINICO ✓ Sempre progressivo: non è mai contemplata la stabilizzazione, al massimo rare fasi di stabilizzazione.
✓ Diversa velocità: la progressione cambia da soggetto a soggetto, non ha la stessa rapidità in tutti.
✓ Nessuna remissione
✓ Nessuna insorgenza acuta
✓ Aspettativa media di vita di 3-5 anni (oltre i 10 anni nel 10% dei soggetti)
Gli unici due fattori prognostici abbastanza attendibili, anche se non con specificità del 100%, sono:
- Età: i più giovani hanno un decorso più lento, sia perché hanno più neuroni giovani (ci sono più neuroni da perdere),
sia perché sono più reattivi a eventuali problematiche respiratorie ecc.
- Localizzazione: quelli con esordio bulbare vanno incontro più facilmente a complicanze respiratorie, cardiache,
alimentari, ….
VARIANTI CLINICHE DI SLA Oltre agli estremi (in cui abbiamo coinvolgimento solo di I o solo di II motoneurone, ma che non si tratta di SLA e vedremo
dopo), ci sono molte varianti cliniche, con infinite sfumature.
FORMA SPINALE: È la forma classica, si ha coinvolgimento soprattutto della periferia e solo tardivamente coinvolgimento
bulbare (sopravvivenza più prolungata).
PARALISI BULBARE PROGRESSIVA: colpisce solo il bulbo senza compromissione spinale. È la peggiore perché causa
velocemente disartria, dispnea, fascicolazioni linguali, caduta del capo. Entra in DD con miastenia gravis, SM, tumori bulbari.
SINDROME DI VULPIAN: colpisce solamente gli arti superiori (diplegia brachiale ingravescente), continua a degenerare
senza compromettere il resto del corpo per molto molto tempo; alla lunga tuttavia viene coinvolto anche il respiro (perché
colpisce muscolatura accessoria respiratoria). È una sindrome che colpisce solamente gli uomini.
FORMA PSEUDOPOLINEUROPATICA: simula una polineuropatia periferica, ma il realtà è una forma di SLA che colpisce
prima solo il II motoneurone ma successivamente anche il I.
FORMA MONOMERICA: colpisce solo un arto.
FORMA POST-POLIO: non è una SLA che viene in un malato di Polio. È una forma di rivilurentazione del virus in quel
territorio, peggiorando la poliomelite.
→ tutte queste forme, dopo anni possono evolvere e dare una SLA classica.
TERAPIA DELLA SLA I farmaci in grado id modificare il decorso di malattia non esistono.
L’unico farmaco utilizzabile è il riluzolo, migliora leggermente la sopravvivenza ma non risolve→ è un inibitore
glutammatergico, con effetto di ridurre il danno eccitotossico che porta alla morte neuronale.
L’unico trattamento in atto al momento è la miglior gestione multidisciplinare possibile per cercare di gestire e prevenire
le complicanze.

72
MALATTIE DEL I MOTONEURONE Forme in cui si ha compromissione solo del I motoneurone. Si può fare diangosi solo dopo 4 anni in cui non sono comparsi
insorti del II motoneurone (perché potrebbe essere una forma di SLA che però si manifesta gradatamente).
Comprendono:
- Sclerosi Laterale Primaria.
- SLA emiplegica, colpisce il I motoneurone solo di un emisoma.
- Paraparesi spastiche infettive: tra cui da HIV.
- PARAPARESI SPASTICHE EREDITARIE: forme di degenerazione del I motoneurone spesso confinata solo agli arti
inferiori. È mutato il gene della spastina, di cui esistono tante mutazioni diverse: la più frequente è la SP64. A volte
si associano anche danni del cervelletto.
MALATTIE DEL II MOTONEURONE MALATTIA DI KENNEDY Atrofia Muscolare Bulbo-Spinale (BSMA). È una mutazione recessiva del cromosoma X del recettore degli androgeni: si
associano a:
- Interessamento del II motoneurone con tantissime fascicolazioni e miochimie della muscolatura bulbare e del
cingolo scapolare.
- Spesso è associata ginecomastia, infertilità, diabete (per compromissione endocrina).
- Spesso hanno anche un tremore strutturale degli arti superiori.
Per la diangosi è importante la consulenza genetica.
ATROFIA MUSCOLARE SPINALE (SMA)
SMA I. Forma neonatale, a trasmissione autosomica ereditaria recessiva del gene SMN1 (survival motoneuron). SMN2 è
associato alle forme ad esordio più tardivo.
Colpisce solo motoneuroni alfa (II), ed è la causa più frequente di morte neonatale per insufficienza respiratoria; morivano
durante il parto o sotto i due anni→ venivano intubati e attaccati al ventilatore fin dalla nascita.
SMA II. Esordio intorno a 5-6 anni, a volte raggiungo la posizione assisa ma vi è una degenerazione rapida di muscolatura
del tronco e respiratoria.
SMA III e IV. Sono forme più tardive ancora, nell’età adulta. Colpiscono entrambi i cingoli (superiore e inferiore) e la IV
raramente causa danno respiratorio.
Terapia. Oligonucleoitidi anti-senso, la prognosi è migliorata nettamente. Raggiungo la forma assisa e riescono a
respirare. Sono in studio di fase 3 i farmaci genici, che permettono un risoluzione definitiva della malattia.

73
SCLEROSI MULTIPLA È una malattia che colpisce i giovani; da qualche anno si è riuscita a controllare bene e ci sono state molte conquiste
terapeutiche e farmacologiche. Sta crescendo anche in ambito pediatrico e ha un picco di prevalenza intorno ai 30 anni.
È una malattia che inizialmente veniva definita come puramente infiammatoria, in cui viene danneggiata la mielina formata
da oligodendrociti (malattia infiammatoria demielinizzante). Poi si è scoperto che in realtà era una malattia
autoimmunitaria; si è infatti scoperto che anche il neurone con il suo assone poteva essere colpito da questa malattia:
questo perché il danneggiamento della mielina espone l’assone al SI (che funge da antigene perché in condizioni normali
non visibile al SI). Sembra che ci siano anche del meccanismi non così chiaramene correlabili a un processo autoimmune,
quindi viene ultimamente definita anche degenerativa (per indicare questo processo di danneggiamento anche neuronale,
che la avvicina anche alla SLA).
→ MALATTIA DEGENERAZIONE INFIAMMATORIA-AUTOIMMUNE DEL SNC.
Patogenesi. I meccanismi autoimmuni in atto nella malattia sono:
A. Mimetismo molecolare: omologia di strutture self con superficie di microrganismi, per cui il SI compie un errore
di riconoscimento.
B. Diffusione dell’epitopo: la parte riconosciuta dal TCR delle cellule T viene diffusa anche in relazione al danno
tissutale cronico.
PERMEABILITÀ DELLA BEE: nella SM ci sono meccanismi autoimmuni sia mediati da cellule T effettrici (con produzione di
citochine) ma anche meccanismi B-mediati (con produzione di anticorpi): sono meccanismi effettori nel SNC, ma sono in
comunicazione con l’immunità periferica tramite la BEE. È una malattia un cui ci sono manifestazioni autoimmuni centrali
ma anche la periferia partecipa alla malattia. Infatti alcun farmaci usati oggigiorno agiscono a livello linfonodale
perifericamente.
Fasi. - Prevalenza di infiammazione, con sintomi subacuti. Gli esami del liquor e neuroimmagini evidenziano una risposta
infiammatoria.
- Componente degenerativa, in cui i meccanismi sono diversi e le possibilità di intervenire sono meno.

74
L’immunità è più facile da attaccare rispetto al sistema degenerativo perché, come detto, essa è collegata con la periferia:
agendo quindi sul sistema immunitario periferico (mandano in apoptosi le cellule e deperimento la risposta immunitaria) si
può riuscire ad agire anche centralmente. Inducendo la morte di cellule T si può consentire il recupero: è un sistema a
chiamata, che si accende e si spegne, e questo facilita nella terapia nel momento in cui è acceso.
EPIDEMIOLOGIA Non è così frequente: ha una prevalenza di 70 casi su 100.000, ma in realtà in Piemonte sono superiori al 100 su 100.000:
probabilmente per la migliore capacità diagnostica o forse perché l’autoimmunità sta aumentando come frequenza.
Dopo i traumi, è la malattia neurologica più invalidante.
Rapporto donne:uomini di 3:2, intorno ai 20-40 anni, con rari casi pediatrici e casi più tardivi (gaussiana dai 10 ai 60 anni).
Fattori di rischio. • Etnia: quella bianca è più colpita.
• Latitudine: paesi più vicini ai poli hanno una frequenza maggiore→ dovuto al clima e agli agenti microbiologici. Si
è visto che soggetti che migrano dopo i 15 anni conservano il rischio dell’area dove ha passato l’infanzia, mentre
soggetti che migrano prima dei 15 anni assumono il rischio dell’area in cui migrano→ sembra essere più rilevante
il periodo dell’infanzia.
• Familiarità: tra gemelli omozigoti si arriva al 30% di concordanza, ma se questi sono cresciuti in luoghi diversi il
rischio di concordanza è pressoché 0.
→ la malattia quindi si manifesta per la concomitanza di fattori genetici e ambientali, singolarmente non bastano.
QUADRO CLINICO Sclerosi= cicatrice, riparazione, tessuto connettivo che sostituisce tessuto sano→ aree di danno che vengono riparate.
Multipla=può interessare molteplici zone. Teoricamente può dare qualsiasi manifestazione neurologica: perdita di forza, di
sensibilità, disturbi memoria, della vista, della coordinazione, …. Alcuni sono più frequenti quindi ci sono sindromi che la
rendono più identificabile; in alcuni casi però viene riscontrata incidentalmente; ci sono delle zone che sono più eloquenti
(e quindi danno sintomi) mentre altre sono “mute”.
→ non sempre è sintomatica, e quando si fanno esami solitamente si ritrovano più lesioni di quelle che ci si aspettava dai
sintomi.
Sintomi più frequenti. - INTERESSAMENTO DEI NERI CRANICI:

75
o Riduzione dell’acuità visiva: relativa all’infiammazione del nervo ottico→ NEURITE OTTICA
RETROBULBARE, soprattutto in giovani donne.
o DIPLOPIA: paresi degli oculomotori per lesione a livello dei nuclei bulbari di II, IV e VI.
o OFTALMOPLEGIA INTERNUCLEARE: paralisi oculare da un lato e nistagmo nel controlaterale, perché
viene compromessa l’integrità del circuito oculo-vestibolare tramite il fascio longitudinale mediale→
consente di regolare il tono dei muscoli estensori del tronco durante il cammino, controlla l’equilibrio in
base a informazioni visive. Se danneggiato si ha sia un problema di visione nitida che di nistagmo.
o NEVRALGIA TRIGEMINALE
o PARESI FACCIALE: con una ipostenia completa del volto.
- INTERESSAMENTO DELLE VIE SENSITIVE:
o Parestesie, ipoestesia, disestesie
o Segno di Lhermitte: segno di scossa elettrica dovuto alla degenerazione dei cordoni posteriori del
midollo→ una volta questo segno semeiologico era patognomonico di SM quando ancora non c’era la
neuroimaging.
o Dolore neurogeno
- INTERESSAMENTO MOTORIO CORTICO-SPINALE:
o Emisindrome motoria
o Sindrome midollare: può dare una sindrome in fase acuta proprio come se ci fosse stato un trauma, se a
livello cervicale si chiamerà tetraparesi, se a livello toracico ci sarà paraparesi. Ci può essere anche una
associazione tra la sensibilità termico-dolorifica e quella propriocettiva. Come si studia?
▪ Il diapason è molto preciso anche su deficit di grado lieve (modo migliore).
▪ Prova di posizione del dito (a occhi chiusi)
▪ Analisi della deambulazione: andatura atassica sensitiva che si distingue da quella cerebellare
perché chiudendo gli occhi peggiora (se invece fosse cerebellare rimarrebbe invariata).
o Spasticità: muscolatura diventa ipertonica per mantenere la stazione eretta→ ipertono di muscoli anti-
gravitari (estensori di arti inferiori -stampella naturale- e flessori di arti superiore). Ipertono è la resistenza
che in muscolo offre al movimento passivo e può essere:
▪ Ipertono da rigidità: è una rigidità costante, come piegare un tubo di piombo.
▪ Ipertono da spasticità: coltello a serramanico, è velocità- dipendente (presente in SM).
▪ Ipertono da plasticità: segno della troclea, dovuto a contrazione contemporanea di muscoli
gravitari e antigravitari per danno di sistema extrapiramidale.
- INTERESSAMENTO MOTORIO CEREBELLARE:
o Triade di Charcot:
▪ Nistagmo
▪ Disastria (scandisce le parole)
▪ Tremore cinetico (che peggiora quando raggiunge l’obiettivo, è un tremore intenzionale e
aggravato dalla mira).
- INTERESSAMENTO SFINTERICO: urgenza minzionale alternata a fenomeni di incontinenza sia urinario che fecale o
ritenzione→ tutti i quadri di disturbo urologico, per danneggiamento di fibre di SNA.
- INTERESSAMENTO COGNITIVO/COMPORTAMENTALE, quasi mai presente all’esordio ma in fase molto avanzata.
Decorso clinico. Esistono 2 principali andamenti all’esordio:
1. Andamento a recidive e remissioni (non dire remissioni e recidive
all’esame!!): primo episodio seguito da un intervallo di tempo indefinito
libero da malattia, seguito da un secondo episodio e così via. 50% circa.
Ci sono vari tipi di gravità: quelle che accumulano poche disabilità o quelle
che accumulano sempre più disabilità (che poi diventerà progressiva).
2. Primariamente progressive, in cui si ha un peggioramento continuo senza
remissioni (10%) fin dall’esordio di malattia.
3. Secondariamente progressive: dopo circa 25 anni dall’esordio (alcuni prima),
tutte le forme recidivanti-remittenti evolvono e diventano progressive.
4. Recidivanti progressive: altre forme non molto ben definite, hanno
andamento primariamente progressivo ma con attacchi acuti.

76
DIAGNOSTICA STRUMENTALE
RM. Evidenzia la presenza di lesioni ovalari sottocorticali, cotonose intorno a ventricoli laterali, nervo ottico, cervelletto,
midollo, sottocorticali.
Esame liquor cerebrospinale. Ricercava la presenza di bande oligoclonali→ evidenziava la presenza di una
attivazione immunitaria specifica, che al di fuori del SNC (nel plasma) non si ritrova. In realtà ora esistono metodi più veloci
quantitativi che hanno sostituito l’elettroforesi→ calcolo del rapporto delle catene K (K index).
Potenziali evocati. Poco usati ormai, è una tecnica che dimostra che una determinata via è stata colpita, ma non
individua la causa (individua solo il rallentamento della conduzione). A volte servono perché una neurite ottica ormai
passata da anni non si individua alle RM, quindi viene individuata con i potenziali.
Criteri di McDonald. Più attacchi sono presenti, più la diagnosi è certa
anche senza accertamenti di imaging.
Se invece ha avuto un solo attacco o nessuno, è importante evidenziare la
disseminazione spazio-tempo: si va a ricercare con RM o liquor se ci sono
lesioni in posti diversi con tempistiche diverse→ lesioni ipointense in T1 è
vecchia, mentre una lesione iperintensa in T2 è nuova.
Il mezzo di contrasto aiuta a distinguere una lesione vecchia o nuova, non è
che permette di vedere lesioni che prima non si vedevano; la presa di
contrasto indica una immunità aggressiva attraverso la barriera, per cui il
contrasto riesce a passare→ le lesioni si vedono anche senza contrasto ma
il MdC permette di distinguer l’aggressività biologica.
PROGNOSI Fattori prognostici positivi:
✓ Esordio con NORB
✓ Esordio con sintomi sensitivi
✓ Sesso femminile
✓ Poche ricadute, a distanza elevata tra loro.
✓ Alta capacità riparativa (propria del soggetto)
Fattori prognostici negativi:
Sintomi motori
Età avanzata
Sesso maschile
Distanza ravvicinata e alta frequenza di ricadute
Bassa capacità riparativa (propria del soggetto):
alcune volta è presente una sola lesione che però
difficilmente viene riparata. Non c’entra con il
tipo di malattia, ma dipende dalla capacità propria
di ogni soggetto.
Sono indicazioni generali, che aiutano a decidere se usare farmaci più efficaci ma ance più pericolosi.
Scala EDSS. Scala più usata di disabilità; è dato dalla somma di:
- da 0 a 4 sono la somma dei deficit all’esame neurologico
- Punteggio per la camminata.
→ Ha molti limiti questa scala sia perché è estremamente variabile sia inter che intra-operatore, ma è quelle più diffusa.
In caso di riscontro occasionale alla RM di placche in assenza di sintomi, si tratta o no? Se si riscontrano più lesioni in stadi
di evoluzione diversi in T1 e T2 (cioè che sono nate in momenti diversi), si tratta e si fa anche esame del liquor, perché si è
visto che è meglio trattare. Invece se si tratta di un esordio monofasico (cioè anche più lesioni ma insorte insieme), conviene
spettare a trattare e fare esame del liquor.
Gravidanza. Durante i mesi di gravidanza le ricadute sono più rare, ma sono più frequenti nel puerperio: fino ai 6-7
mesi dopo il parto non si hanno ricadute, ma tutte le ricadute che sarebbero dovute avvenire in quel periodo si manifestano
tutte dopo, come se la gravidanza congelasse la malattia. Dal punto di vista prognostico a lungo termine non cambia nulla
avere figli, perché la progressione totale sul periodo è uguale: quello che è difficile è la gestione della terapia: non tutti i
farmaci si possono usare perché alcuni passano barriera placentare, e soprattutto sui farmaci nuovi non ci sono ancora dati
a sufficienza. Ci sono due farmaci disponibili: INFbeta e glatiramer acetato, che possono essere usati in gravidanza.

77
Vaccinazioni. È meglio farle che non farle. Non si possono fare quelli con agenti vivi inattivati che si possono riattivare,
ma sono pochi i vaccini così ormai. Con vaccinazione anti-epatite B non ci sono rischi.
Stress e traumi. Non sembrano incidere più di tanto; forse lo stress può innescare la ricaduta ma non è chiaro.
TERAPIA Bisogna trovare il giusto abbinamento farmaco-paziente. Si potrebbero usare marcatori immunologici per strutturare le
terapie personalizzate, ma si stanno ancora studiando; per ora si procede ancora in maniera empirica.
TERAPIA DELLA RICADUTA Per la terapia in acuto, si fanno alte dosi di metilprednisolone in vena per ricadute gravi o deltacortene (prednisone) per
bocca se non sono ricadute gravi. È utile ad accelerare il recupero del paziente ma non incide sul danno strutturale: non
riduce l’entità del danno ma togliendo un po’ di infiammazione permette di recuperare prima.
Farmaci di prima linea. Sono immunomodulanti e possono essere orali o iniettivi. Si usano per forme con bassa
attività biologica.
FARMACI ORALI DI PRIMA LINEA: si adattano bene al paziente che inizia ad avere la malattia ma non in forma aggressiva
(nelle forme più frequenti). Sono:
• Teriflunomide: blocca la sintesi di DNA di linfociti B e T, quindi blocca la loro replicazione.
• Dimetilfumarato: citoprotettivo e antinfiammatorio.
→ meccanismo d’azione non è specifico, riducono l’attività dei linfociti Th1 e Th17 e attenuano la componete autoreattiva.
FARMACI INIETTIVI DI PRIMA LINEA: da metà anni ’90. • IFN-beta (3-4 tipi diversi): riduce migrazione in SNC, inibisce proliferazione linfociti T e riduce produzione
citochine pro-infiammatorie.
• Glatiramer acetato (Copolimero 1): si legano all’MHC spiazzando l’antigene mielinico (proteina basica), azione
soppressiva anti-Th2.
Un po’ più fastidiosi.
Farmaci orali di seconda linea. Vengono usati nelle forme aggressive che non rispondono agi immunomodulanti;
hanno un effetto più marcato in tempi brevi. - Fingolimod: impedisce l’uscita di linfociti dagli organi linfoidi per raggiungere il target.
- Cladribina: effetto su cellule T e B di tipo soppressivo, resetta il sistema immune; è una terapia induttiva.
I tre anticorpi monoclonali, i più efficaci e nuovi:
- Natalizumab: Ab che lavora sul sistema delle integrine riducendo l’ingresso di linfociti in SNC.
- Alentuzimab: meccanismo meno specifico di soppressione, causa deplezione di linfociti B, T e NK tramite
attivazione del complemento.
- Ocrelizumab: agisce sul CD20.
Come scegliere quali usare? In un paziente con bassa attività biologia, si cerca solo di tenere sotto controllo la malattia quindi bastano farmaci di prima
linea. In un paziente con attività biologica media, è più difficile scegliere. Si può scegliere una combinazione in base alle
necessità, all’imaging→ nelle forme intermedie si vede più variabilità, cioè che ogni medico darebbe una terapia differente.
In un paziente con attività biologica elevata, si va subito sull’anticorpo monoclonale.
→ Si è visto che se fallisce una delle linee, è difficile riuscire poi a inserirne un’altra; infatti alcuni centri puntano sull’usare
fin da subito la terapia più radicale che resetta tutto subito, ma che rende il paziente più vulnerabile.
L’unica vera terapia per la rigenerazione nervosa è l’ATTIVITÀ FISICA: bisogna sfruttare al 100% la capacità fisica e mentale
dell’individuo per permettere il più possibile una rigenerazione. Per quanto riguarda la rigenerazione, si è tanto paralto di
cellule stminali, ma attualmente non c’è ancora nulla di disponibile, l’unica cosa che si po' sfruttare è la capacità di
rigeneraizone propria del pazinete.
Sono farmaci in generale che riducono l’attività di malattia a lungo termine ma difficilmente riescono ad arrestarla
completamente. L’obbiettivo di nuovi farmaci è proprio questo, di bloccare l’attività di malattia→ i nuovi farmaci in trial

78
infatti vengono classificati secondo il grado di “NEDA” (Non Evidence Of Disease Activity), cioè in base alla loro capacità di
bloccare la malattia. Il problema è comunque la componente degenerativa: magari si ferma l’infiammazione ma la
degenerazione continua, quindi non si riesce a bloccare del tutto la progressione del danno.
→ moltissimi progressi ma ancora molto da studiare perché ci sono pazienti che hanno ancora tante disabilità.
NEUROMIELITE OTTICA Questa forma di malattie infiammatoria non è parente della SM ma è un’altra malattia.
• Ha un andamento acuto o subacuto
• con un danno necrotico di midollo e nervi ottici contemporaneamente o non.
• con un precedente di infezione o di malattia autoimmune sistemica
• che risponde a trattamento immunosoppressivo.
Esistono diverse varianti nel mondo:
- Forma classica: in cui convivono neurite ottica bilaterale e mielite; si tratta di una mielite lunga→ interessa diversi
metameri, è più estesa rispetto alla SM. La forma classica è monofasica. Non ci devono essere altre lesioni.
- Forme più polimorfe: ci possono essere anche altre lesioni, es. pavimento del quarto ventricolo (area postrema) e
altre aree encefaliche; inoltre, la mielite e la lesione del nervo ottico sono molto distanziate tra loro.
Entra in DD con:
- SM
- Encefalomielite acuta disseminata
- LES
- Sjogren
Diagnosi. Bande oligoclonali nel liquor non sono molto presenti. Al contrario si ritrovano auto-anticorpi anti-acquaporina
4: è un canale che controlla l’ingresso di acqua nel SN. È stato ritrovato anche un anticorpo anti-MOG: è una proteina della
melina e dà forme di neuromielite ottica.
Criteri di diagnosi:
- Storia di neurite ottica
- Storia di mielite acuta
- Due criteri tra:
o Positività a autoanticorpi (acquaporina o MOG)
o Lesioni midollari di almeno tre metameri.
o RM non indicativa di SM
Epidemiologia. È variabile a seconda della latitudine: in Giappone è molto frequente la NMO (ma non la SM), così come
in Inghilterra.
Terapia. Si usa sempre il cortisone in fase acuta, ma si può anche usare plasmaferesi per togliere anticorpi; sono utili
anche le immunoglobuline in vena.

79
TUMORI CEREBRALI
Distinguiamo in PRIMITIVI e SECONDARI. La diagnostica differenziale spesso è diversa, perché le metastasi possono
comparire ancora prima del primitivo (tumore occulto).
I tumori primitivi si localizzano soprattutto a livello sovratentoriale. Il cervelletto è raramente coinvolto nei tumori cerebrali
primitivi nell’adulto, mentre è coinvolto frequentemente dà metastasi. Per le lesioni emisferiche invece il quadro è più
complesso.
A parità di gravità biologica del tumore, gioca un ruolo importante la sede: una lesione emisferica destra è meno grave in
termini di deficit funzionali rispetto al sinistro, dove c’è l’area del linguaggio→ la stessa lesione dal punto di vista biologico
e dimensionale si gestisce con difficoltà diverse.
Inoltre nel cervello ci sono aree più eloquenti e altre mute; un tumore può essere piccolo in un’area eloquente e già dare
segni di sé, oppure crescere lentamente in un’area muta e dare segni di se quando ormai è molto grande.
I tumori cerebrali sono tra le prime cause di morte oncologica dopo pancreas e polmone. L’istotipo più frequente infatti è
anche quello più grave, il glioblastoma.
Le metastasi cerebrali invece sono 10 volte più frequenti e sono la causa di ¼ dei pazienti oncologici; tuttavia per i tumori
secondari cerebrali si hanno più armi, infatti sono maggiormente radiosensibili rispetto i glioblastomi e con farmaci che
passano la BEE si possono eradicare.
Il problema dei glioblastomi è che non sono eradicabili, e anche quando raramente lo sono, essendo diffusi, hanno già
invaso anche il tessuto circostante quindi ha un criterio di radicabilità molto discutibile (nel SNC non si possono tenere
margini di resezione ampi).
Dopo l’ictus, sono la seconda causa di morte per cause cerebrali.
Malignità. Dipende da:
• istologia
• localizzazione→ forse è il fattore più importante.
• dimensione
Epidemiologia. 5 nuovi casi su 100.000 persone, abbastanza stabile. Ovviamente sono aumentati i casi in over 65, ma
non è preoccupante. Sesso maschile è il più colpito, tranne nel meningioma che colpisce prevalentemente donne.
Nei bambini sono tumori abbastanza frequenti, soprattutto in fossa cranica posteriore (medulloblastomi, astrocitomi,
ependimomi); hanno una presentazione clinica differente rispetto all’adulto.
Negli adulti, sono più frequenti astrocitoma anaplastico e glioblastoma. Nella donna, il meninginoma è più frequente ma si
può scollare e resecare il più possibile in modo da evitare recidiva,
CLASSIFICAZIONE PER ETÀ 0-20 ANNI: fossa posteriore. Il meningioma cresce verso l’esterno e anche dal punto di vita biologico è meno grave, ma se
cresce troppo arriva a causare compressione anche sul parenchima cerebrale; se viene rimosso bene la prognosi è buona
(solo un 10% è maligno).

80
FATTORI DI RISCHIO Per tumori primitivi:
- Radiazioni ionizzanti: esplosioni nucleari, vecchie diagnostiche invasive usate in gravidanza.
- Campi elettromagnetici: è difficile condurre studi epidemiologici; non è chiaro l’effetto che hanno nell’adulto, nel
bambino non ci sono studi soddisfacenti ma sembra che dipenda dall’intensità del campo.
- Esposizione professionale: formaldeide, industria petrolchimica.
Le metastasi invece hanno gli stessi FdR del tumore principale→ in ordine di frequenza: polmone, mammella, melanoma,
genitourinari, gastroenterici, ….
TUMORI PRIMITIVI GLIOMI Più frequenti.
Il problema di questi tumori è che con il tempo cambiano e si
sviluppano aree eterogenee dal punto di vista cellulare; se quindi
viene biopsiato.
Astrocitoma. Tipico dell’età giovanile-adulta
Localizzazione prevalentemente emisferica, a lento sviluppo. Nel 50-
70% dei casi indifferenziazione verso forma anaplastica.
Oligodendroglioma. È importante avere il dato TC, perché può
mettere in evidenza calcificazioni. Anche nel meningioma è utile la TC perché si vede bene la base di attacco delle meningi
all’osso. Per tutti gli altri tumori invece si fa solo RM.
Glioblastoma. Emisferico soprattutto, cresce velocemente e ha prognosi rapidamente peggiorativa; ha una levata
attività biologica. Si sviluppa controlateralmente a farfalla lungo il corpo calloso: può avere aspetti di angiogenesi, edema,
effetto massa, aree emorragiche, aree cistiche→ replicazione cellulare e movimento biologico molto marcato.
MENINGIOMI Conflitto con il parenchima cerebrale; ha sedi tipiche ed è solitamente
multifocale.
La sede parasagittale è peculiare perchè può dare paraparesi perché
comprime area degli arti inferiori.
SEGNI E SINTOMI
Sintomi generali. Il principale è ipertensione endocranica.
Sintomi focali. La clinica è subdola, on come quella di un ictus; è graduale e progressiva. Un tumore genererà due
possibili quadri:
❖ IRRITATIVI: il tessuto tumorale può danneggiare il circuito elettrico della zona di parenchima, soprattutto se a
livello del lobo temporale→ lobo più elettricamente carico e quindi più epilettogeno.
❖ DEFICITARIO: Con questa definizione si intende la perdita di una o più funzioni cerebrali. I sintomi deficitari sono
più frequenti nei tumori maligni; infatti durante la veloce crescita, in questi tumori, avvengono fenomeni di necrosi
❖ con conseguente distruzione di regioni cerebrali.
Sintomi secondo la sede. Le manifestazioni di un tumore cerebrale dipendono soprattutto dalla sua localizzazione e
dalle dimensioni della massa: poiché ogni zona è responsabile di una funzione specifica, infatti, sarà quella stessa funzione a essere più o meno compromessa, con una grande varietà di sintomi.
LOBO FRONTALE:

81
• Alterazioni cognitive
• Alterazione della personalità
• Deficit di attenzione
• Ricomparsa di automatismi motori (suzione, grasping)→ segni di liberazione frontale quando si ha danneggiamento
frontale; sono normali ne bambino
• Deficit motori (emiparesi, emiplegia)
• Deficit del linguaggio (afasia di Broca non fluente)
• Crisi epilettiche
• Anosmia (se tumore alla base del lobo frontale)
TUMORI DEL LOBO TEMPORALE:
• Alterazioni del comportamento (delirio) e disturbo mnesico
• Alterazione del campo visivo (radiazioni ottiche): quadropsia superiore laterale omonima. In realtà per edema e
crescita veloce genera emianopsia.
• Disturbo del linguaggio (afasia di Wernicke, fluente con contenuto incomprensibile).
• Crisi epilettiche (crisi uncinate, psicomotoria, stati onirici).
TUMORE DEL LOBO PARIETALE:
• Aprassia ideomotoria
• Agnosia
• Disturbo sensitivo (per coinvolgimento area sensitiva primaria)
• Disturbo campimetrico (quadrantopsie)
• Crisi epilettiche (sensitive) meno frequenti.
TUMORE DEL LOBO OCCIPITALE: è un lobo moonofunzionale quindi è più facile.
• Disturbo del campo visivo (quadrantopsie)
• Agnosia visiva
• Allucinazioni visive
TUMORE DEL TRONCO ENCEFALICO→ Sindrome alterna di Wallenberg.
• Disfunzione dei nervi cranici
• Paralisi di sguardo verticale
• Nistagmo
• Atassia vestibolo cerebellare di tronco e arti.
• Deficit motori (ipotono), deficit di stop.
• Idrocefalo ostruttivo per compressione dell’acquedotto del Silvio o del quarto ventricolo
Cefalea. È il sintomo più frequente di esordio; bisogna sempre preoccuparsi quando si manifesta una cefalea in un
soggetto che non ne ha mai avute. Può essere intermittente, ma spesso si cronicizza perché il tumore cresce.
Si localizza principalmente bilaterale frontale. Intensità variabile. È principalmente tensiva, in rari casi emicrania.
Possibile associazione con nausea e vomito per via di ipertensione endocranica.
Epilessia. Sintomo molto frequente delle neoplasie cerebrali, con incidenza all’esordio del 20-40% e in progressione
malattia 20-45%.
L’epilessia tumorale rappresenta il 3,6-6% di tutte le epilessie
Netta preponderanza delle crisi focali (75%) rispetto alle generalizzate; non raro lo Stato Epilettico convulsivo o non
convulsivo (10-14%).
Solitamente sono farmaco-resistenti
Crisi più frequenti in tumori vicini alla corteccia, sono più epilettogene le lesioni a bassa malignità e a lenta crescita rispetto
al glioblastoma.
Ipertensione endocranica. Consiste generalmente in un aumento della pressione all’interno della scatola cranica, e
può essere dovuto generalmente a tre fattori nei tumori cerebrali:
1) Aumento di volume del tumore all’interno della scatola cranica, con conseguente compressione delle
strutture adiacenti

82
2) Blocco della circolazione del liquido cerebrospinale
3) Edema (nei tumori cerebrali si tratta di edema vasogenico)
Sintomi:
❖ Cefalea, prevalente mattutina
❖ Nausea e vomito a getto
❖ Papilla da stasi
❖ Rallentamento psicomotorio
DIAGNOSI Cosa principale è l’anamnesi: sintomi ad insorgenza lenta.
Imaging. Si sta abbandonando sempre di più l’uso di raggi X, mentre sta aumentando l’uso di RM e ECO.
➢ TC: oggi riveste un ruolo di secondo piano nella diagnosi. Utile in alcune situazioni:
o Escludere sanguinamenti
o Ricerca calcificazioni (es oligodendrogliomi)
o Unitamente alla RM nel planning operatorio
o Utile per la pianificazione di biopsia: sono caschi che si avvitano sulla testa del paziente con un archetto
su cui si muove l’ago→ la TC permette di fare la centratura quado il paziente ha già su il caschetto.
➢ RM: è lo strumento più sensibile in:
o Diagnosi differenziale preoperatoria.
o Precisa localizzazione anatomica in vista del trattamento
o Valutazione della progressione di malattia
o Valutazione della risposta al trattamento, soprattutto alla radio che causa necrosi e quindi bisogna
differenziarla da una recidiva di tumore.
o Valutazione degli effetti collaterali legati al trattamento
o Identificazione dell’istotipo: si sta cercando sempre di più di abbandonare la biopsia pre-operatoria ma di
arrivare alla diangosi istologica tramite imaging. Ci si basa soprattutto sull’enhancement al mezzo di
contrasto, che passa solo quando si ha una permeabilità di barriera (perché l’angiogenesi indotta dal
tumore non è quella “corretta”, quindi non si ha la formazione della giusta BEE):
▪ Negli astrocitomi la presenza di enhancement implica solitamente un elevato grado di malignità,
anche quando la biopsia dimostra un grado II (follow up ravvicinato!). Negli oligodendrogliomi
questo non avviene.
▪ L’assenza di enhancement non implica necessariamente un basso grado, poiché circa 1/3 dei
gliomi non enhancing sono di alto grado (50% dopo i 45 anni).
→ non per forza quindi presa di contrasto= maligno.
Nuove tecniche. Oggi sono utilizzati diversi nuovi approcci nell’imaging dei tumori cerebrali; sebbene il valore di queste
tecniche non sia ancora completamente definito, esse trovano ampio spazio sia nei trial clinici sia nella routine. Le tecniche
più importanti sono:
1) SPETTROSCOPIA (Proton MR): tecnica basata sulle proprietà magnetica dei nuclei di alcuni atomi/isotopi, per
ricavare informazioni sulla frequenza di risonanza dei nuclei attivi; si quantificano n-acetilaspartato, colina, creatina
+ fosfocreatina, glutammato e glutammina, GABA, mio-inositolo e acido lattico.
2) IMAGING DINAMICO VASCOLARE: Sono tecniche che misurano la vascolarizzazione del tumore in vivo, incluse la
densità e la permeabilità dei capillari (Perfusion MR)→ correla con la angiogenesi e quindi con aggressività e
evoluzione. Permette la DD con necrosi post-radioterapia: se si è in dubbio, si fa RM perfusion e se risulta
vascolarizzazione si tratta di recidiva di tumore (perché necrosi non genera angiogenesi).
3) DIFFUSION IMAGING: questa tecnica è sensibile al movimento browniano delle molecole d’acqua all’interno delle
cellule tumorali. I tumori con elevata densità cellulare presentano ridotta diffusione, mentre tumori a scarsa
cellularità o la presenza di necrosi presentano valori di diffusione normali o aumentati. Può essere importante nella
diagnosi differenziale fra tumori primitivi e metastatici (DWI ridotta intorno ai tumori primitivi e aumentata
nell’edema associato alle metastasi).
4) RM FUNZIONALE: si fanno compiere gesti e azioni. Ha il limite che quando si apre la scatola cranica, il cervello si
sposta quindi il confronto con l’imaging non è così fedele.

83
5) RM INTRAOPERATORIA: risolve il problema della non affidabilità di imaging pre-operatoria.
MARCATORI MOLECOLARI MGMT: proteina di riparazione che rimuove l’alchilazione del DNA determinata da agenti alchilanti; pazienti che hanno la
metilazione del promotore, hanno questa proteina silenziata, mentre pazienti non metilati esprimono questa proteina e
quindi hanno un profilo di farmaco-resistenza→ valore prognostico e predittivo della risposta a radio e chemio con
Temozolomide (in realtà il farmaco si dà a tutti perché la risposta c’è un poì in tutti tutti).
LOH di 1p e 19q: generalmente presente negli oligodendrogliomi, e si ritrova in una buona % di pazienti (60-90%). La perdita
combinata correla con un aumento della sopravvivenza e con una migliore risposta ad alcune chemioterapie.
Mutazioni IDH1 e 2: se presenti, sono associate a miglior sopravvivenza.
FUS: focus ultra Sound: usata per tremori o per causare un momentaneo danno di barriera per permettere il passaggio di
farmaci, infatti uno dei problemi maggior in chemioterapia è far arrivare il farmaco al tumore, superando la BEE.
Esistono poi alcune mutazioni che sono in grado di distinguere se un glioblastoma è de novo o secondario:
Glioblastoma “de novo”
• Lesione primaria
• Overespressione di EGFR
• Età avanzata
• > radioresistenza
• > recidive dopo terapia
• > angiogenesi, edema, invasione
Glioblastoma secondario
• Lesione secondaria
• Mutazione di TP53
• Età giovanile
• < radioresistenza
• < recidive dopo terapia
• < angiogenesi, edema, invasione
→ prognosi migliore.
TRATTAMENTO L’attuale atteggiamento terapeutico prevede in generale:
• Gliomi basso grado: chirurgia + wait and see o RT
• Gliomi anaplastici: chirurgia + RT e CT
• Glioblastomi: chirurgia + RT e CT
• Gliomi recurrent: eventuale chirurgia + CT seconda linea e/o locoregionale
Intervento chirurgico. se si può si opera!! Nei gliomi resta indispensabile per la diagnosi istologica, ma spesso è
insufficiente. La chirurgia è controindicata se:
a. Condizioni del paziente compromesse, cioè un grado basso della Karnofsky Prtfomrance Status (KPS): scala che
valuta la condizione generale.
b. Età sopra gli 80 anni.
c. Sede molto eloquente
Chemioterapia. La chemioterapia sistemica prevede l’utilizzo di farmaci in mono terapia o in associazione (schema
PCV, temozolamide ecc). La TEMOZOLAMIDE è sicuramente il chemioterapico che più ha modificato l’atteggiamento del

84
neurologo e dell’oncologo nei confronti dei pazienti affetti da glioma. Nel glioblastoma attualmente è in uso lo schema
STUPP che prevede l’uso di radioterapia e chemioterapia concomitanti (aumento della sopravvivenza di qualche mese).
Discussa è la chemioterapia loco-regionale: lo scopo della CT loco-regionale è quindi quello di:
- Ottenere nella cavità residua all’intervento chirurgico concentrazioni di chemioterapico elevate e per periodi lunghi
- Superare la BEE e dare meno effetti collaterali sistemici
- Solitamente il tumore recidiva entro i 2 cm dal tumore iniziale, quindi potrebbe bastare la loco-regionale.
- La CT sistemica ha importanti limitazioni, quali il difficile superamento della BEE ed i suoi effetti sistemici.
→ in realtà, non sembra esserci un impatto così rivoluzionario.
Optune di Novocure. Utilizzo di campi elettrici superficiali (tramite un casco) che sembra aver dato ottimi risultati
sulla capacità di ridurre la crescita dei glioblastomi→ molto scomodo e costoso ma sembra essere efficace, anche se allunga
la sopravvivenza di solo qualche mese.

85
INFEZIONI DEL SNC
MENINGITI
Malattie infiammatorie delle membrane che avvolgono l’encefalo, soprattutto della pia madre e dell’aracnoide (leptomeningi) e del liquido cefalo-rachidiano→ esame principale è l’esame biochimico di liquor.
SINDROME MENINGEA La meningite si manifesta come un insieme di segni di irritazione; si basano su manifestazioni dovute a una contrattura secondaria al dolore causato dallo stiramento dell’infiammazione→ trasudato e essudato.
1. RIGIDITÀ NUCALE: segno più fedele di irritazione meningea, dall’infanzia in poi. Si fa giacere supino e si sposta il capo nelle varie direzioni; il collo dovrebbe opporre una resistenza non troppo elevata, ma se si riscontra una difficoltà eccessiva (associata a dolore se il paziente è vigile) è segno di irritazione meningea. Nei casi più gravi, capo e collo formano un blocco unico. Non è sempre specifico: anche in caso di cefalea si può riscontrare una certa rigidità. Nel neonato questo segno non è molto apprezzabile, non si sa perché, quindi fondamentale è la puntura lombare. → N.B.: la rigidità nucale può anche essere l’unico segno!
o Segno di Lasegue: flessione contro resistenza dell’arto inferiore esteso, con flessione del piede. si evoca dolore in qualsiasi caso di dolore lombare o irritazione radicolare, poco specifico.
o Segno di Kernig o Segno di Brudzinski o Addome a barca o Decubito a cane di fucile: giace su un lato con cosce flesse sull’addome e gambe flesse sulle cosce.
2. SEGNI DI IPERTENSIONE ENDOCRANICA: a. Cefalea b. Vomito cerebrale c. Bradicardia d. Papilla da stasi: non solo l’oculista dovrebbe essere dotato di
oftalmoscopio: è una lente graduata con una luce che puntata nella pupilla permette di vedere il fondo della retina (fondo rosso) con i vasi che dipartono dalla papilla del nervo ottico (di colore biancastro): è il punto in cui le fibre mieliniche del nervo stesso si raggruppano in fascio e fuoriescono dal polo posteriore. Vicino alla papilla c’è la macula, la zona con massima densità di coni, in cui la vista è massima. In caso di ipertensione, il liquor tende a infiltrarsi tra le guaine del nervo ottico e rendere il margine della papilla meno netto o addirittura rialzarlo rispetto al piano di fondo. Al tempo stesso le vene appaiono congeste. → PAPILLA DAI MARGINI SFUMATI O RIALZATI E VENE CONGESTE.
3. SEGNI DI IRRITAZIONE DEL SNC: confusione mentale, foto-fonofobia, vomito, crisi epilettiche etc… 4. SEGNI DI DEFICIT NEUROLOGICI: sonnolenza, sopore, stupore, coma. Ritenzione urinaria, paralisi dei nervi cranici,
emiparesi. Alterazione del respiro tipo Cheynes-Stokes. 5. IPERPIRESSIA (90% dei casi)
RACHICENTESI L’iter diagnostico parte sempre dalla rachicentesi. Si fa in un punto più basso rispetto al midollo, cioè tra L4 e L5; per individuare il processo spinoso di L4 si fa riferimento ad una linea immaginaria che unisce i punti più alti delle due creste iliache. Ovviamente serve una adeguata sterilità, per evitare di inoculare batteri. Si fa anestesia con lidocaina (una volta gas freddo). Si inclina leggermente l’ago in modo che segua la stessa inclinazione del processo spinoso. L’ago in questo modo procede all’interno di tessuti molli fino ad arrivare al legamento giallo; superato quest’ultimo, lago penetrerà più facilmente e si troverà all’interno del canale spinale.

86
Cosa mi permette di capire che sono nel canale midollare? Togliendo il mandrino dell’ago, si vedrà fluire liquor, descritto come acqua di rocca.
Cosa si controlla? Aspetto: Se il liquido è rosato (misto a sangue), è possibile che sia stato preso un plesso venoso: si fa la prova delle 3 provette→ si riempiono 3 provette e si confrontano: se l’ultima è più trasparente della prima allora si era preso un plesso, se sono uguali allora significa che il liquor è ematico. Pressione: apprezzata a vista, ad esempio se fuoriesce a zampillo significa che la pressione elevata. In condizioni normali dovrebbe sgocciolare. Esistono dei manometri che si raccordano con l’ago e permettono di misurarla in mmH2O o cmH2O. la pressione cambia se il soggetto è supino o seduto (nei casi in cui è più difficile eseguirlo, può essere fatto da seduto perché è più
semplice, e in questo caso la pressione è più elevata. Pressione nella norma: 60-200 mmH2O.
Cellularità: Globuli bianchi fino a 5/mm3, con PMN assenti.
Glicorrachia: tenere presente che corrisponde al 55-60% della glicemia, quindi in un soggetto diabetico scompensato sarà
più alta. È importante misurarla perché i batteri consumano glucosio, mentre i virus no.
Proteinorrachia: sotto i 45 mg/dl.
Esami colturali e colorazione Gram nel sospetto di batteri. Antigeni batterici ed indagini molecolari PCR nel sospetto di virus.
CLASSIFICAZIONE DI MENINGITI ACUTE A SECONDA DEL LIQUOR La DD può essere fatta analizzando 3 fattori:
1) ETÀ:
• Neonato: listeria o enterobatteri
• Adulti: pneumococco e meningococco 2) LIQUOR:
• Torbido, cioè con pus→ eziologia batterica. o Globuli bianchi elevati (5000-10000). o Proteine: aumentata fino a 20 volte. o Glicorrachia: quasi azzerata.
• Limpido, cioè con essudato→ eziologia virale. o Globuli bianchi lievemente elevati o Proteine lievemente aumentate o Glicorrachia normale
Esistono meningiti non purulente ma batteriche, dove il liquor è come quello delle meningiti virali: l’unica cosa che cambia è che la glicorrachia è bassa→ meningite tubercolare.
3) FATTORI PREDISPONENTI:
• Fratture craniche, soprattutto della base cranica (rinoliquorrea)
• Neurochirurgia

87
• Gravidanza
• Immunosoppressione (alcolismo, neoplasie, diabete, setticemia, infezioni vie urinarie, trapiantati)
• HIV
PRINCIPALI AGENTI EZIOLOGICI
Neisseria meningitidis (meningococco) Grave perché tende a dare setticema e necrosi di midollare e corticale del surrene→ SINDROME DI WATERHOUSE-FRIEDRICKSON. Causa una meningite della volta cranica (più raramente della base). - Diplococco Gram negativo - Causa più comune bambini e giovani adulti - Mortalità ~10% - Trasmissione per via aerea o contatto stretto con soggetti
infetti (5-10% portatori asintomatici nel nasofaringe) - Sierogruppi: A e C epidemie, B sporadico.
- Sviluppo rapido in minuti/ore.
Streptococcus pneumoniae (pneumococco) - Cocco Gram positivo, catenella - Causa più comune negli adulti - Mortalità ~20% - Trasmissione per via aerea, portatori sani nel nasofaringe - Fattori predisponenti: otite media, sinusite, fratture base
cranica, soluzioni di continuità dura madre, immunodepressione, alcolismo, diabete.
- Volta cranica
Listeria moncitogenes - β-emolitico intracellulare - Mortalità ~15% - Trasmissione: cibi contaminati - Fattori predisponenti: gravidanza, neoplasie, età > 50,
immunodepressione
- Varie manifestazioni: meningite, meningoencefalite, romboencefalite (del ponte), neuropatie nervi cranici
Haemophilus influenzae - Coccobacillo Gram-negativo - Mortalità ~5% - Oggi disponibile vaccinazione per tipo B (in precedenza
prima 1° causa meningite infantile) - Trasmissione: via aerea
- Fattori predisponenti: otite media, traumi cranici, neurochirurgia
- Posizione basale
PATOGENESI
TERAPIA
Meningiti batteriche. Nel momento in cui si individua che si tratta di meningite bisogna SUBITO iniziare terapia:

88
1. ANTIBIOTICOTERAPIA EMPIRICA a. Verso gli agenti patogeni più verosimilmente responsabili (età, immunocompetenza, provenienza,
modalità di esordio) b. Ad ampio spettro:
i. Neonati-lattanti: ampicillina (corrispettivo endovenoso di amoxicillina). ii. Bambini-adulti: cefalosporine 3° gen (uniche a passare la BEE!).
c. Via endovenosa d. Elevato dosaggio (per raggiungere liquor)
2. ANTIBIOTICOTERAPIA MIRATA
Meningiti virali. • Controllo delle funzioni vitali • Controllo eventuali disturbi metabolici • Valutazione eventuale interessamento encefalitico • Terapia di supporto (ossigenoterapia, antiedema) • Decorso favorevole in 7-15 gg. • ACICLOVIR endovena
COMPLICANZE
Precoci. • Ipertensione endocranica • Crisi epilettiche focali e generalizzate (33% dei casi) • Empiema subdurale • Shock e coagulazione intravascolare disseminata (meningite meningococcica) • Ischemia arteriosa o infarto venoso cerebrale (per vasculite o infiltraz. parete)
Tardive. • Idrocefalo ostruttivo (per coaguli di fibrina nei forami di coniugazione) • Ascessi intracerebrali • Emorragie cerebrali: spesso in anamnesi si ritrova una meningite durante l’infanzia.
MENINGITI CRONICHE
Meningite tubercolare. Colpisce sia immunocompetenti che immunocompromessi. È un processi infettivo subdolo: spesso causa una cachessia (con perdita di peso, anemia), spesso interessa i nervi cranici (segno più evidente è lo strabismo perché coinvolge primariamente VI, III e IV nc); infatti per motivi non chiari ha una predilezione per la base del cranio→ meningite della base cranica. Con i flussi migratori degli ultimi anni è una infezione che è ritornata ad aumentare, spesso molti casi sono misconosciuti. DIAGNOSI:
- Esame liquor: liquor limpido, glicorrachia ridotta, pleiocitosi linfocitaria, iperproteinorrachia). - Colorazione di Ziehl-Nielsen per bacilli alcool-acido resistenti - Esame colturale liquorale per conferma diagnostica - PCR su liquor - 50% dei casi segni RX torace di pregressa TBC

89
Neurosifilide. La sua incidenza è tornata ad aumentare. Agente eziologico: Treponema Pallidum. Contagio: via venerea, transplacentare, accidentale. Storia naturale della sifilide non trattata:
• Incubazione
• Sifilide primaria (ulcera indolente, sifiloma)
• Sifilide secondaria (sintomatologia simil-influenzale, manifestazioni cutannee diffuse e linfoadenopatia)
• Sifilide latente (asintomatica, test sierologici +)
• Sifilide terziaria: tre possibili evoluzioni: o Tardiva benigna o Cardiovascolare o Neurosifilide
MANIFESTAZIONI: molte diverse, neurologiche e psichiatriche:
1) Meningite precoce 2) Neurosifilide asintomatica 3) Meningite sifilitica acuta 4) Sifilide terziaria benigna 5) Forme atipiche (in corso di HIV)
6) SIFILIDE MENINGOVASCOLARE: la più frequente Segni di sofferenza focale dell’encefalo o del midollo spinale
(come paralisi dei nervi cranici) associata a episodi vasculitici (o endoarteritici)→ possono determinare fenomeni ischemici. È una forma che può comparire dopo 5-10 anni dal contagio. Prodromi aspecifici con cefalea, vertigini, deficit mnesici, labilità emotiva Esordio improvviso: afasia, emiparesi, crisi focali Gli infarti spinali: grave paraparesi, turbe sfinteriche, anestesia sottolesionale TD. Diagnosi: esame liquor, VDRL + (tes sierologico non treponemico), TC e RM possono evidenziare lesioni infartuali. Angiografia: focali restringimenti delle piccole e grandi arterie
7) SIFILIDE PARENCHIMATOSA: o PARALISI PROGRESSIVA: invasione diretta dell’encefalo da parte del Treponema Pallidum. Demenza a
rapida evoluzione (atrofia frontale e temporale), aspetti psichiatrici da turbe dell’umore fino a delirio e allucinazioni, vari segni neurologici→ pupilla di Argyll-Robertson: riflesso fotomotore diretto conservato in accomodazione, assente per stimolazione luminosa diretta (manca il riflesso fotomotore alla stimolazione luminosa ma non quello alla convergenza, quindi si mantiene il riflesso di accomodazione alla convergenza). Il deficit è causato da una lesione a livello della zona pretettale del mesencefalo, nel nucleo di Edinger-Westphal. Si associano anche: disartria, crisi epilettiche, segni piramidali ed extrapiramidali, tremore della lingua e delle estremità.
o TABE DORSALE: atassia sensitiva, disestesie dolorose, crisi tabetiche. Insorge entro 10 anni dall’infezione primaria. È una meningoradicolite, da localizzazione del Treponema a livello dei gangli sensitivi delle radici dorsali. Conseguente degenerazione dei cordoni posteriori del midollo e delle fibre nervose di grosso calibro deputate alla sensibilità epicritica.
Neuroborreliosi Borrelia Burgdorferi: spirocheta Gram-negativa. Spesso entra in diagnosi differenziale con la sindrome di Guillen-Barré.
8) Stadio 1: eritema migrante 9) Stadio 2: entro tre mesi, diffusione per via ematica
o interessamento cardiaco o interessamento SNC
▪ meningite ▪ neuriti craniali ▪ meningoradicoliti linfocitarie
10) Stadio 3: meningite cronica, mieloradicolopatia progressiva, encefalomielite, encefalopatia subacuta

90
ENCEFALITI Processo infiammatorio del tessuto cerebrale sostenuto per lo più da una causa infettiva, di origine virale, batterica, fungina, parassitaria. Frequente è il coinvolgimento delle meningi. La definizione di encefalite non implica necessariamente la presenza dell’agente infettivo nell’encefalo, dal momento che alcune forme possono essere scatenate da un meccanismo (auto) immunitario.
Meccanismo patogenetico. Distinguiamo in:
11) Encefaliti primarie: diretta invasione del SNC da agente patogeno. Porta di ingresso: mucosa tratto GE, app. respiratorio, cute Crescita locale del virus in tessuto non neurale Invasione dell’encefalo:
o Diffusione ematogena o Via neurale (es. migrazione centripeta: rabbia, herpes) o Per contiguità (orecchio medio, mastoidi, seni paranasali)
12) Encefaliti secondarie: causa post-infettiva o post-vaccinica, paraneoplastica. il danno è mediato da reazione immunitaria innescata dall’infezione sistemica.
Distinguiamo anche in:
❖ ENCEFALITE ACUTA: sintomi aspecifici: febbre, astenia, mialgie, malessere, associate ad alterazioni di laboratorio. Sintomi neurologici da:
o Sofferenza cerebrale diffusa: Dist. coscienza, crisi, ipertensione endocranica, segni di irritazione meningea.
o Sofferenza cerebrale focale: ▪ Irritativi ▪ Deficitari ▪ Disturbi psichici/della sfera cognitiva
❖ ENCEFALITE CRONICA: Progressivo deterioramento mentale, accompagnato da crisi epilettiche e da deficit neurologici focali. Solitamente i segni di malattia infettiva acuta sono assenti.
Iter diagnostico. 1) anamnesi - esame obiettivo 2) esami di laboratorio, studi sierologici 3) Esame del liquor (standard + coltura + titolazione anticorpale + PCR)→ iperproteinorrachia, pleiocitosi modesta
(10-200/mm3); se pleiocitosi elevata, c’è una concomitante meningite. 4) EEG: si riscontrano quadri aspecifici, come un rallentamento diffuso (onde lente teta e delta) o quadri
patognomonici, come le PLEDs dell’encefalite erpetica. 5) Neuroimmagini (TC, RM)→ soprattutto RM individua processi infiammatori e necrotici.
(faremo solo quelle assolutamente da sapere)
ENCEFALITE ERPETICA La principale encefalite virale acuta dalle nostre parti è quella erpetica da Herpes Simplex, si trasmette per via respiratoria e raggiunge l’encefalo per contiguità attraverso i filuzzi olfattivi. È un virus che rimane silente e in alcuni casi si attiva, determinando encefalite. La flogosi coinvolge il LOBO TEMPORALE (basale e mesiale), solitamente è asimmetrico.

91
Base anatomo-patologica. È la necrosi emorragica asimmetrica dei lobi temporali (possibile domanda d’esame).
Quadro clinico. Ha un esordio insidioso.
• Febbre, cefalea, astenia • Alterazioni psichiche: agitazione psicomotoria, cambiamento caratteriale, allucinazioni, deliri. • Deficit neurologici focali: crisi epilettica focale del lobo temporale. • Segni neurologici da sconfinamento oltre il lobo temporale, come emiparesi o afasia.
Diagnosi. ESAME DEL LIQUOR: si riscontrerà una elevata cellularità: pleiocitosi linfocitaria e PMN (anche granulociti!!), proteinorrachia elevata. PCR: esame fondamentale per fare diagnosi precoce!!! RM: le alterazioni sono precoci, soprattutto a carico di uno dei due lobi temporali, soprattutto la parte mesiale (aree iperintense in T2). Con il tempo invaderà anche l’altro lato. È l’esame migliore, in quanto alla TC si potrebbe vedere qualcosa solo dopo tanto tempo (infatti essendo una necrosi emorragica, dopo un po’ si vedranno gli stravasi ematici). EEG aiuta parecchio: onde lento-aguzze ad alto voltaggio, quasi periodiche, che si trovano nei canali pari (lato destro)→ si parla di scariche periodiche epilettiformi lateralizzate (PLEDs). Non sono quadri patognomonici, ma sono altamente suggestivi! Se PLEDs, allucinazioni, febbre, delirio e segni di irritazione meningea→ si procede con rachicentesi per fare definitivamente rachicentesi.
Terapia. Aciclovir 10 mg/Kg per via endovenosa tre volte al giorno (per 14-21 giorni).
ENCEFALITI SUBACUTE/CRONICHE
Leucoencefalopatia multifocale progressiva (PML) Tipica di immunodepressi, ma si è aggiunta negli ultimi anni una causa iatrogena: l’uso di anticorpo monoclonali per sclerosi multipla→ sono farmaci che riducono la risposta infiammatoria a livello del SNC, quindi possono causare la riattivazione del virus JC (polioma virus), che è un virus ubiquitario ma latente. Infetta gli oligodendrociti e causa demielinizzazione del SNC. CLINICA:
- Esordio insidioso con modificazioni della personalità e decadimento cognitivo, nell’arco di alcune settimane.
- Segni neurologici focali (emiparesi, disturbi del visus, deficit campimetrici, cecità corticale, afasia, atassia)
NEUROPATOLOGIA: Multiple lesioni demielinizzanti con tendenza alla confluenza (emisferi, cervelletto, tronco). In fase iniziale coinvolge la giunzione fra sostanza bianca e grigia, nelle regioni posteriori dell’encefalo (margini non così netti tra sostanza bianca e grigia).
ASCESSO CEREBRALE Processo infettivo focale del parenchima cerebrale, dovuto a diffusione ematogena a distanza (da endocardite batterica) o per continuità (es. da oto-mastoidite acuta, frattura della base con fistola meningea con raccolta saccata di pus). NB: Non sempre in caso di ascesso c’è un evidente quadro infettivo e segni liquorali di infezione→ la diagnostica differenziale può essere incerta con tumori cistici. Si può prendere in considerazione una biopsia con i neurochirurghi; infatti non sempre RM o PET riescono a stabilire una chiara differenza. La RM teoricamente dovrebbe essere in grado di discriminare in modo preciso tra ascessi e processi patologici di altra natura, ma nella realtà non sempre è così. L’esame del liquor on è indicato per via del basso valore diagnostico.
PATOLOGIE SNC DA HIV Avere il concetto che alcuni agenti infettivi posso anche colpire il SNC. Complicanze PRIMARIE: encefalopatia HIV-correlata, mielopatia vacuolare, neuropatia e miopatia HIV-correlate.

92
Complicanze secondarie. ❖ Infezioni opportunistiche più frequenti:
- Riattivazione di CMV, tre possibili quadri: encefalite, retinite emorragico essudativa, radicolopatia lombosacrale.
- Riattivazione di Toxoplasma (focolai di encefalite necrotica non purulenta). - PML (riattivazione JCV) - Meningite criptococcica
❖ Linfoma cerebrale primitivo
MIELITI Processi infiammatori a livello del midollo spinale. Se coinvolto anche encefalo si parla di encefalomielite
Patogenesi. Distinguiamo in:
• MIELITE DA CAUSA INFETTIVA DIRETTA: o Da virus: HZV, HSV, EBV, CMV, enterovirus, virus della rabbia. o Da batteri, miceti, protozoi e parassiti pluricellulari
• MIELITI DA CAUSA IMMUNITARIA o A decorso monofasico: post-infettive e post-vaccinali o Recidivanti: sclerosi multipla, lupica.
• Mieliti trasverse acute infettive
• Poliomielite acuta e sindrome post-polio
(faremo una lezione sulla presentazione clinica di patologie del midollo spinale) Spesso si manifestano con sindrome midollare trasversa:
• Dolore al collo o alla schiena • Parestesie, perdita di sensibilità con livello sensitivo • Ipostenia (para o tetraparesi) • Disturbi sfinterici
DIAGNOSI: RMN midollo con contrasto da eseguire d’urgenza. Distingue le forme compressive/non compressive. DD con forme immunomediate (demielinizzanti, parainfettive, malattia infiammatorie sistemiche).
ENCEFALOMIELITE ACUTA DISSEMINATA (ADEM) Può essere post-infettiva o post-vaccinica. Ha genesi disimmune, per mimetismo molecolare. Entra in DD con SM. È un episodio demielinizzante, monofasico, associato ad una vaccinazione o ad una infezione virale sistemica→ non progressivo, non ha un decorso cronico o recidivante (a differenza di SM).
MALATTIE DA PRIONI
Patogenesi. (non importante) Un prione è un isomero conformazionale di una glicoproteina normalmente espressa. La
sua modalità di infezione è data da una particolare catena proteica alfa e beta ripiegata in maniera scorretta, che induce altre proteine ad assumere la stessa conformazione anomala. Queste proteine sono poi in grado a loro volta di infettare le proteine adiacenti. Esistono forme sporadiche, genetiche o iatrogene.
MALATTIA DI CREUTZFELDT-JAKOB (MCJ) Chiedono praticamente solo questa. Si tratta di una demenza rapidamente progressiva, nell’ordine di settimane o mesi, a cui si accompagnano segni neurologici, tantissimi diversi:
1) MIOCLONO: segno principe delle Creutzfeldt-Jakob, di solito bilaterale simmetrico, coinvolge arti superiori e capo. 2) Segni piramidali, paresi, paralisi, Babinski, riflessi accentuati. 3) Segni simil-parkinsoniani: rigidità, acinesia. 4) Segni cerebellari:
o Atassia della marcia

93
o Disturbi vestibolari 5) Turbe visive o oculomotorie:
o Nistagmo o Alterazioni motilità oculare o Disturbi visivi corticali (agnosie visive e allucinazioni visive)
6) Mutismo acinetico: stato di vigilanza con mutismo→ sorta di coma. A volte questi segni avvengono prima del decadimento cognitivo quindi possono confondere le idee.
Base anatomo-patologica. I prioni causano una necrosi massiva di aree corticali (vacuolizzazione intraneuronale),
chiamata spongiosi.
Diagnosi. EEG: importantissimo per fare diagnosi perché riscontreremo un EEG periodico→ scariche aguzze periodiche che coinvolgono tutto lo scalpo (nella fase iniziale magari si ritrova solo un rallentamento). In sincronia con queste onde aguzze ci può essere uno scatto mioclonico. LIQUOR: si cerca la proteina 14.3.3: è un prodotto di degradazione del tessuto nervoso. RM: dà informazioni già in fase precoce: iperintensità di segnale di putamen/caudato in T2. Altro segno importante sono le alterazioni nastriformi di segnale in corteccia, chiamate cortical ribboning: nel dovuto contesto è molto suggestivo di malattia prionica. Nel morbo della mucca pazza si evidenzia invece una degenerazione del pulvinar con maggiori sintomi psichiatrici associati.
Terapia. Nessuna specifica
Profilassi. Norme igieniche, regime speciale di disinfezione.

94
CEFALEE
È importante distinguere:
• CEFALEA: mal di testa, qualsiasi dolore che interessa il capo (non indica una specifica patologia, ma solo un sintomo) • CEFALEA PRIMARIA: il mal di testa è la patologia (non riconosce alcuna causa organica o funzionale dannosa o
potenzialmente tale). • CEFALEA SECONDARIA: il mal di testa è il sintomo di una patologia sottostante→ fondamentale ricercare quale sia.
CLASSIFICAZIONE Si fa riferimento alla classificazione delle cefalee primarie del 2013.
1. Parte prima: cefalee primarie. La diagnosi si fa con l’anamnesi, analizzando i sintomi.
2. Parte seconda: cefalee secondarie, divise per cause ben determinate. La diagnosi si basa sulla ricerca delle cause.
3. Parte terza: nevralgie o dolori facciali considerati primari.
Diagnosi in base alle caratteristiche fenotipiche della cefalea presentata nell’ultimo anno in accordo con i criteri diagnostici elencati. Ciascun tipo di cefalea deve essere separatamente diagnosticata e codificata. Quando sono presenti più diagnosi, devono essere tutti classificati; la prima diagnosi deve essere quella più frequente, devono essere elencate in ordine di importanza per il paziente. In caso in cui la cefalea sia insorta in seguito al trauma cranico, viene classificata come secondaria; allo stesso modo, se una cefalea precedente viene peggiorata e modificata in seguito a un trauma cranico, deve comunque essere catalogata come secondaria. Quando un mal di testa precedente peggiora in concomitanza a una causa (trauma o sinusite) bisogna classificarla come
secondaria. La conferma certa di cefalea secondaria si ha solo quando togliendo la causa principale la cefalee regredisce.
La cefalea non può essere classificata come primaria quando alla visita neurologica ci sono segni obiettivi che testimoniano
la sofferenza del sistema nervoso (guardare sempre anche il fondo dell’occhio!)→ L’OBIETTIVITÀ NEUROLOGICA DEVE
ESSERE NELLA NORMA.
1. EMICRANIA Epidemiologia. Interessa soprattutto il sesso femminile, più probabile entro i 50 anni, se insorge dopo bisogna
guardarla con molto sospetto. Gli attacchi avvengono circa 1-5 volte al mese.
Attacco emicranico. • PRODROMI: sono disturbi aspecifici: frequenti sbadigli, modificazioni dell’umore, torpore, irritabilità, desiderio di
specifici alimenti, difficoltà a concentrarsi, rigidità al collo, visione annebbiata, pallore. Non devono essere confusi con l’aura emicranica, che invece sono disturbi focali molto specifici.
• CEFALEA: il dolore interessa la regione fronto-temporale o occipito-nucale, frequentemente si concentra a livello dell’occhio o della tempia, ma può essere diffuso a tutto il cranio.
• COMPORTAMENTO DURANTE L’ATTACCO: la cefalea viene esacerbata dai movimenti e dagli sforzi fisici, il paziente è quindi costretto a sospendere le sue normali attività, mettersi a riposo, al buio lontano dai rumori. Fotofobia, fonofobia e disturbi per i rumori.
Fattori scatenanti. 7) Fattori alimentari: vino, cioccolato, formaggi stagionati, ristorante cinese (per l’alta presenza di glutammati).
Anche alimentazione disordinata e ipoglicemia. Si può dire che una cefalea sia attribuibile a un alimento quando mangiandolo viene sempre mal di testa, se viene due volte sì e cinque no allora non serve toglierlo dall’alimentazione.
8) Fattori ormonali: molto importanti nella donna; alcune cefalee sono classificate come CEFALEE CATAMENIALI: si manifestano nei due giorni prima delle mestruazioni o nei tre giorni successivi. In questi casi si può avviare una terapia solo progestinica (che blocca il ciclo) o estrogel. Se oltre che in queste fasi si manifesta anche in altri momenti del ciclo, si parla solo di cefalea correlata al ciclo.

95
9) Fattori emotivi: stress fisico o psicologico, ma anche dal rilassamento dopo lo stress (emicrania da week-end o da vacanza).
10) Altri fattori: cambiamenti atmosferici, farmaci, stimoli visivi e uditivi. → esistono quindi dei fattori che sono eliminabili e altri no, se si possono togliere lo si fa.
1.1. EMICRANIA SENZ’AURA Criteri diagnostici:
A. Almeno 5 attacchi che rispettino i criteri successivi. B. Cefalea della durata di 4-72 ore (non trattata o trattata successivamente). C. D. Cefalea che rispetti i seguenti criteri fenotipici:
C. Caratteristiche del dolore (almeno 2) - Localizzazione unilaterale - Dolore di tipo pulsante - Intensità moderata o grave - Aggravamento indotto da attività fisiche
D. Sintomi associati (almeno 1): - Nausea - Vomito - Fotofobia - Fonofobia
1.2. EMICRANIA CON AURA Criteri diagnostici:
A. Almeno 2 attacchi che rispettino i criteri B-C B. Uno o più dei seguenti disturbi d’aura completamente reversibili:
1. Visivi 2. Sensoriali 3. Del disturbo dell’eloquio o del linguaggio 4. Motori 5. Del tronco-encefalo 6. Retinici
C. Almeno due delle seguenti caratteristiche: * 1. Almeno uno dei sintomi dell’aura si sviluppa gradualmente in più di 5 min. e/o differenti sintomi dell’aura si presentano in successione 2. Ciascun sintomo d’aura dura 5-60 min 3. Almeno un sintomo è unilaterale 4. L’aura è accompagnata o seguita, entro 60 min, dalla cefalea
D. La cefalea non attribuibile ad altri disordini della classificazione e devono essere esclusi attacchi ischemici transitori.
* Per parlare di emicrania con aura ci vogliono solo 2 attacchi e non più 5, in cui ci sono uno o più disturbi d’aura. I sintomi si devono manifestare gradatamente, non possono insorgere all’improvviso: es. comincia a sentire formicolio al braccio, o comunica ad avere la vista offuscata. Prima di tutti solitamente insorge il sintomo visivo, poi quello somato-sensoriale o afasico e solitamente ci si ferma qui. L’aura deve essere completamente reversibile: se non lo è, non è aura. La reversibilità avviene in un’ora circa se si tratta di un solo sintomo, mentre se si manifestano più sintomi uno dopo l’altro può durare anche 3 ore. Cosa importante nelle cefalee con aura è sempre escludere che si tratti di attacchi ischemici transitori; si distinguono per:
- Insorgenza improvvisa dei sintomi di aura e non graduali (es. emianopsia istantanea senza prima una fase di solo annebbiamento della vista).
- Assenza di fenomeni positivi: solitamente chi ha un ictus non ha scotomi (sintomi positivi), ma solo emianopsia (negativo).
1.2.1. Emicrania con aura tipica
Due possibili sottotipi: - Aura tipica con cefalea→ episodi emicranici seguiti entra in DD con ictus, ischemia ed emorragia, quindi è difficile. - Aura tipica senza cefalea→ fenomeni di aura sine emicrania.
AURA TIPICA: sintomi neurologici focali che permettono di individuare la zona interessata dal fenomeno, sofferenze reversibili di alcune zone del SNC che danno sintomi particolari:
- Aura visiva con offuscamento della vista e con visione di scotomi, lampi luminosi come stelline o a palizzata (linee zigzaganti), emianopsia laterale omonima (coinvolge lobo occipitale).
- Aura somestesica: parestesie, formicolio peribuccale e all’arto superiore (raramente all’arto inferiore). - Aura afasica: se colpisce lobo temporale dell’emisfero dominante; non è più in grado di parlare.
Criteri di aura tipica:

96
A. Almeno 2 attacchi che rispettino i criteri B e C. B. Aura consistente in disturbi visivi, sensitivi o del linguaggio, completamente reversibili, non disturbi motori,
del tronco-encefalo o retinici. C. e D. uguali a sopra a 1.2.
NB: non sono presenti disturbi motori, o del tronco-encefalo o retinici!! Nel momento in cui si manifestano, si passa all’aura atipica.
1.2.2. Emicrania con aura del tronco encefalico Qui ci sono sia sintomi dell’aura tipica a cui si aggiungono anche sintomi del tronco!
- Disturbi visivi: diplopia. - Disturbi della veglia: sopore e stato simil-comatoso. - Disturbi dell’udito: vertigini, tinnito, acufeni.
Criteri diagnostici:
A. Almeno due attacchi che rispettino i criteri B – D B. Aura consistente in disturbi visivi, sensitivi o del linguaggio, completamente reversibili, non disturbi motori, o
retinici. C. Almeno uno dei seguenti sintomi di interessamento del tronco encefalo:
1. disartria 2. vertigine 3. tinnito 4. ipoacusia 5. diplopia 6. atassia 7. alterazione del livello di coscienza
D. e E. come 1.2.
1.2.3. Emicrania emiplegica familiare. Rarissima. I sintomi sono quelli di aura tipica a cui si aggiungono sintomi motori. Hanno una durata molto più lunga, più di
72 ore, quindi risulta più difficile distinguerli da un ictus.
Si suddivide in 3 sottotipi a seconda della patogenesi:
1) Alterazione del cromosoma 19 sul canale del calcio.
2) alterazione del cromosoma 1 sulla pompa sodio-potassio.
3) Alterazione del cromosoma 2 sulla pompa del sodio.
Criteri diagnostici:
A. Almeno due attacchi che rispettino i criteri B-C
B. Aura caratterizzata da entrambe i seguenti:
1. Disturbi motori (emiparesi o emiplegia) completamente reversibili
2. Disturbi visivi, sensitivi, dell’eloquio o del linguaggio completamente reversibili
C. Almeno due dei seguenti 4 caratteristiche:
1. Almeno uno dei sintomi dell’aura si sviluppa gradualmente in >= 5 min e/o differenti sintomi dell’aura
si presentano in successione.
2. Ogni sintomo non motorio dell’aura dura 5 - 60 min e i sintomi motori durano più di 72 ore
3. Almeno un sintomo dell’aura è unilaterale
4. l’aura è accompagnata o seguita, entro 60 min, da cefalea
D. Cefalea non attribuibile ad altri disordini della classificazione e devono essere esclusi attacchi ischemici transitori.
Aura retinica. È un disturbo periferico legato a un ipoafflusso retinico e di solito sono monoculari completamente
reversibili: non vede più da un occhio, è diverso dall’emianopsia laterale omonima.
1.3. EMICRANIA CRONICA S basa solo sul criterio del numero di giorni al mese: se ha più di 15 giorni al mese per almeno 3 mesi consecutivi di mal di testa, con caratteristiche tensive, ma almeno 8 giorni al mese attacchi che emicranici. Criteri diagnostici:

97
A. Cefalea (tension type o migraine like) della durata durata >= 15 gg al mese per più di tre mesi e che rispecchia i criteri B e C
B. Si presenta in pazienti che hanno avuto almeno 5 attacchi di che rispettano i criteri di emicrania con aura o senz’aura C. Per >= 8 gg/mese per > di 3 mesi devono essere rispettati ciascuno dei seguenti criteri:
1. criteri C e D per 1.1. di emicrania senz’aura 2. criteri B e C per 1.2 di emicrania con aura 3. storia di emicrania all’ esordio responsiva a triptani o ergot derivati
D. Non rispetta altri criteri diagnostici della classificazione
1.4. COMPLICANZE DI EMICRANIA Stato di male emicranico: È un criterio solo temporale, se dura più di 72 ore non si tratta più di attacco emicranico. Aura persistente senza infarto: Solitamente l’aura è reversibile, in rari casi però può persistere e durare anche settimane; si distingue da un infarto solo grazie all’imaging perché sarà negativo. Infarto emicranico: In alcuni casi in un’aura persistente, l’imaging rivela una lesione congrua: le immagini neuroradiologiche indicano che c’è stato un insulto cerebrale nelle zone congrue con i sintomi. Epilessia indotta dall’emicrania: Rarissima; in corso di attacco emicranico entro un’ora dall’attacco può manifestare sintomi sovrapponibili con una crisi epilettica.
1.5. EMICRANIA PROBABILE In un paziente con quasi tutti i criteri di fare diagnosi di emicrania ma ne manca uno, si può comunque fare diagnosi ma di emicrania probabile.
1.6. SINDROMI PERIODICHE ASSOCIATE A EMICRANIA Si chiede sempre ai pazienti adulti se hanno sofferto di coliche addominali immotivate da bambini, o da vomito ciclico, o vertigini torcicollo parossistico→ sono considerati equivalenti emicranici, cioè episodi che presagiscono una possibile emicrania da adulti, quindi si ricercano sempre in anamnesi! Vomito ciclico: attacchi ricorrenti episodici, generalmente stereotipati nel singolo paziente, di vomito e nausea intensa. Gli attacchi si associano a pallore e letargia. Vi è completa risoluzione tra gli attacchi (durata 1 h 5 gg). Emicrania addominale: disordine idiopatico ricorrente prevalentemente pediatrico e caratterizzato da dolore addominale che si manifesta in attacchi della durata di 1-72 ore con risoluzione tra gli attacchi. Il dolore è di intensità da moderata a severa e associato a sintomi vasomotori, nausea, vomito, pallore. Vertigine benigna parossistica dell’infanzia: questo disordine probabilmente eterogeneo è caratterizzato da attacchi brevi, ricorrenti ed episodici di vertigine che si manifestano senza preavviso e che si risolvono spontaneamente in bambini altrimenti sani; almeno un sintomo associato tra ny, atassia, vomito, pallore.
PATOGENESI DELL’EMICRANIA L’evento per cui un paziente tranquillo senza fattori scatenanti determini l’innesco, non si sa. Si pensa ci sia un tipo di “cervello emicranico”, cioè più suscettibile di altri a sviluppare emicrania, ma non si sa da dove parta.
Ipotesi vascolare. Prima ipotesi formulata, che parte da una vasocostrizione iniziale che genera ipossia e sofferenza
del parenchima→ aura. A questa ipossia segue una vasodilatazione reattiva→ genera la cefalea (teoria di Moschoviz).
Ipotesi neurogena. L’evento scatenante non è l’ipossia ma è una depolarizzazione dei neuroni he parte dalle corteccia
occipitale che si propaga a cerchi concentrici anteriormente alla velocità di 2-3 mm/s, seguita da una depolarizzazione di

98
fibre nervose corticali e poi perivascolari→ determina una prima fase di iperemia della corteccia cerebrale a ci segue una iperpolarizzazione sostenuta dei neuroni che concomita con una oligoemia (ipoperfusione)→ responsabile dell’aura emicranica. È un modello sperimentale, non direttamente riconoscibile nell’uomo.
Ipotesi trigemino-vascolare. Mette insieme ipotesi neurogena e vascolare.
Dal Ganglio di Gasser partono le tre branche trigeminali, di cui la prima è coinvolta nell’emicrania (innerva occhio e fronte); una sua branca efferente entra in contatto con le arterie meningee e seni venosi, che irrorano il cervello. Di per sé l’encefalo non ha fibre dolorifiche, sono localizzate solo a livello meningeo. Le fibre afferenti del trigemino arrivano alla parte caudale-bulbare del nucleo del trigemino, da cui parte il secondo neurone della via che si dirige al nucleo ventro-postero-laterale del talamo e poi in corteccia somato-sensoriale e frontale.
Il CGRP (Calcitonin Gene Related Peptide) è fondamentale per la trasmissione del dolore, sia a livello centrale che
periferico. È un potente vasodilatatore ed è espresso vicino ai recettori 1B e 1D della serotonina. Viene liberato dalle terminazioni del trigemino sia a livello periferico vasale sia a livello del nucleo bulbare. È responsabile di due cose: ❖ Tramite la branca efferente, in seguito all’onda di depolarizzazione, è responsabile di VASODILATAZIONE E
INFIAMMAZIONE NEUROGENA a livello dei vasi meningei. ❖ Tramite la branca afferente, è responsabile dell’AMPLIFICAZIONE DELLA SENSAZIONE NOCICETTIVA. → bloccare il CGRP è fondamentale per bloccare il meccanismo di innesco dell’emicrania. Infatti esistono dei farmaci nuovi che agiscono proprio a questo livello.
→ ipotesi unificante che permette di spiegare anche l’ipotesi vascolare che neurogena, infatti l’onda di depolarizzazione corticale (CSD) responsabile dell’innesco dell’aura sarebbe in grado di attivare il sistema trigemino-vascolare, inducendo una stimolazione dei nocicettori durali attraverso proteine della matrice extracellulare e conseguente attivazione dei nocicettori trigeminali con la cascata dei fenomeni biologici appena descritti (tramite CGRP).
Emicrania e serotonina. Un’altra sostanza implicata nella trasmissione del dolore è la SEROTONINA: nei pazienti emicranici prima, durante e dopo
l’attacco si rilevavano livelli di serotonina variabili: infatti risultavano aumentati 24 ore prima, dimezzati nel corso dell’attacco e poi normalizzati alla risoluzione dell’attacco. Inoltre durante l’attacco aumentava anche la concentrazione di serotonina urinaria. La somministrazione endovenosa di serotonina era in grado di risolvere l’attacco. La serotonina ha 7 recettori ma quelli più importante sono il 1B e 1D: sono presenti nel SNC e hanno permesso di studiare i farmaci antiemicranici selettivi, cioè i triptani→ fornaci agonisti serotoninergici che agiscono sul 5-HT1B sui vasi e 5-HT1D su terminazioni trigeminali perivascolari. → ora si utilizzano farmaci selettivi. Ha una predisposizione familiare, ma non è direttamente determinata. Solo in patologie ereditarie come il CADASIL (di cui la cefalea è il sintomo principale) si parla di trasmissione genetica (autosomica dominante).
2. CEFALEA DI TIPO TENSIVO Più frequente di emicrania: la prevalenza varia nei diversi studi dal 30 al 78 % della popolazione. Inizialmente era considera psicogena, ulteriori studi hanno individuato basi neurobiologiche:

99
• Si considerava la presenza di alterazioni a livello della muscolatura pericranica utilizzando il sistema manuale (dolorabilità della muscolatura pericranica alla palpazione -muscoli frontali, temporali, massetere, pterigoidei, sternocleidomastoidei, splenio e trapezio)→ sovrapposizione di cefalea con contrattura della muscolatura pericranica molto frequente.
• L‘esatto meccanismo della cefalea tensiva non è conosciuto; possibili meccanismi di dolore periferico e/o centrale. → non è possibile individuare un farmaco specifico, sia per la profilassi sia per la cura sintomatica. Distinguiamo in:
2.1. EPISODICA SPORADICA INFREQUENTE: almeno 10 episodi con frequenza inferiore a 1 gg/mese in media (meno di 12 gg/anno) che rispecchia i criteri detti sopra. 2.2. EPISODICA SPORADICA FREQUENTE: Almeno 10 episodi con frequenza trc 1 e 15 gg/mese per almeno tre mesi (da 12 a 180 g/anno). 2.3. CRONICA: cefalea presente per più di 15 gg/mese per più di 3 mesi (più di 180 gg /anno) e che soddisfi i criteri detti sopra. 2.4. PROBABILE: se manca un criterio si fa diagnosi di probabile cefalea tensiva.
→ i sintomi sono sempre gli stessi, cambia solo la frequenza. → Ogni categoria si divide a sua volta in due, a seconda di se abbia o no associata la dolorabilità e contrazione dei muscoli paravertebrali e cervicali.
A. Occorrono più episodi rispetto all’emicrania ci vogliono almeno 10 episodi. Sono infrequenti gli attacchi, devono essere meno di una volta al mese.
B. Devono rispettare le caratteristiche di: - Durata: maggiore dell’emicrania, infatti va dai 30 minuti ai 7 giorni. - Bilaterale - Gravativa-costrittiva (non pulsante) - Intensità di media o lieve intensità, non elevata come in emicrania. - Non aggravata da attività fisica.
C. Inoltre ci devono essere: - Nausea o vomito assenti.
D. Solo fotofobia o solo fonofobia isolate, non insieme.
PATOGENESI Molto confusa. Il fattore scatenante consiste in stress estrinseci e una predisposizione alla suscettibilità allo stress→ parte una attivazione aspecifica di nocicettori di tessuti profondi, con una ipereccitabilità dei gamma-motoneuroni: questo determina una contrattura dei fusi con conseguente aumento di afferente delle radici spinali e trigeminali. Si ha poi una secondaria attivazione di strutture sovraspinali deputate alla percezione del dolore. → meccanismo sia periferico che centrale. Quando il processo si autolimita per la presenza di stimoli che inibiscono il dolore, la cefalea tensiva allora rimane episodica; quando invece il processo non si autolimita (o per contrazione di muscolatura o per iperattiva zone gamma) la cefalea diventa cronica.
3. CEFALEA A GRAPPOLO E ALTRE AUTONOMICO-TRIGEMINALI
3.1. CEFALEA A GRAPPOLO Prevalenza: circa 0,1%, più presente nel sesso maschile (rapporto 5:1). Insorge tra i 20 e i 40 anni. Spesso viene confusa con nevralgia del trigemino. Chiamata anche cefalea del suicido per la sua forza e dolore. Sono crisi concentrate a cluster (a grappolo appunto) in un periodo, in media 2 mesi (fase attiva della malattia); si verificano in modo ripetuto, da 1 a 8 al giorno in periodi specifici dell’anno, come primavera e autunno (nel caso episodico). Criteri clinici:
A. Almeno 5 attacchi. B. Tutte le seguenti caratteristiche:
o Dolore di intensità severa o Unilaterale (sede orbitaria, sovraobitaria e/o raramente temporale). o Durata da 15 min a 3 ore (senza trattamento)

100
C. Almeno uno dei seguenti segni ispilaterali: o Iniezione congiuntivale e/o lacrimazione o Ostruzione nasale e/o rinorrea o Edema palpebrale o Sudorazione facciale o Miosi e/o ptosi palpebrale o Irrequietezza ed agitazione
D. La frequenza degli attacchi va da uno ogni due gg ad 8 attacchi al giorno E. Si verifica una delle seguenti condizioni:
o Sono esclusi i disturbi elencati nei gruppi 5-12 o I disturbi dei gruppi 5 –12 sono esclusi da indagini strumentali o Sono presenti disturbi dei gruppi 5-12 ma senza relazione temporale con cefalea
CEFALEA DA ABUSO DI SINTOMATICI Delle cefalee secondarie vediamo solo la 8, da abuso di sintomatici. Molti pazienti prendono tutti i giorni un sintomatico per il mal di testa.
CEFALEA DA ABUSO DI TRIPTANI Esistono criteri per porre diagnosi:
➢ Più di 15 giorni (quindi cronica) ➢ Assunzione concomitante di triptani in qualsiasi formulazione superiore a 10 giorni al mese per almeno 3 mesi
(superiori a 15 giorni nel caso di analgesici). → a questo punto è una probabile cefalea da abuso di sintomatici. Per porre diagnosi certa ci deve essere una regressione del numero di attacchi entro due mesi dalla sospensione di sintomatici. Si cerca di convincere il paziente a ridurre il più possibile l’uso del sintomatico; se la cefalea si riduce dopo la sospensione dell’abuso, si può fare con certezza la diagnosi.
Quali esami fare? EEG: si faceva una volta, ora non più. RM con angio- RM: unico esame che ormai ha senso fare per escludere cause vascolari, anche se le linee guida non lo prevedono a meno che: cambi il tipo di cefalea o si manifesti sempre nella stessa zona→ l’emicrania si manifesta sempre dallo stesso lato, ma se si manifesta nello stesso preciso punto dell’emiencefalo, allora c’è da sospettare ci sia una causa organica.
TERAPIA EMICRANIA Come ci si comporta davanti a un paziente con mal di testa? L’approccio cambia se si tratta di una cefalea occasionale o
ricorrente:
• Il trattamento dell’attacco occasionale è sintomatico→ si punta alla regressione (se possibile completa).
• Nella ricorrente, bisogna capire se è secondaria (e in tal caso si cura la causa) o primaria: in tal caso bisogna
eliminare i fattori scatenanti. Se non ha fattori scatenanti, esistono dei farmaci che vengono dati come terapia
profilattica→ si cerca quindi di ridurre il più possibile la frequenza degli attacchi.
Linee guida nel 2011.
CRITERI GENERALI • quando vengono evidenziati fattori favorenti o scatenanti, occorre eliminarli ove possibile.
• terapia sintomatica al bisogno in caso di attacchi occasionali o quando la frequenza critica è bassa
• terapia profilattica a lungo termine
• la profilassi deve essere preceduta da un periodo di osservazione di tre mesi solo con farmaci sintomatici, per
individuare le caratteristiche del mal di testa e capire di cosa di tratta.
OBIETTIVI 1. Scomparsa del dolore

101
2. Scomparsa dei sintomi d’accompagnamento→ esistono dei farmaci come i FANS che agiscono bene sul dolore ma
non tanto su sintomi di accompagnamento.
3. Rapida restituzione del paziente alla normale capacità funzionale.
4. Senza effetti indesiderati né recidive
5. Opportuno dire al paziente che non sempre è possibile ottenere pienamente questo obiettivo.
TERAPIA SINTOMATICA DELL’EMICRANIA
Criteri generali. • Da effettuarsi solo in crisi occasionali in cui è stata esclusa la sintomaticità
• quando la frequenza critica è di due attacchi al mese e/o cefalea presente per non più di 4 gg al mese. Due attacchi
dalla durata di pi di 4 gionri, prevedono un terpaia profilattica, non sintomatica!
• Il farmaco ritenuto appropriato deve essere assunto al minor dosaggio utile alla risoluzione della crisi ed il più
precocemente possibile.
• Consigliati prodotti contenenti un unico principio attivo, almeno in prima battuta.
Che differenza c’è dal punto di vista clinico tra FANS e triptani?
I FANS sono sintomatici solo sul dolore in generale quindi vanno
bene per qualsiasi cefalea. I triptani e gli ergotaminici invece
sono anti-emicranici, possono essere usati solo nell’emicrania o
nella cefalea a grappolo.
Triptani. Il farmaco di prima scelta per la terapia sintomatica
dell’attacco di emicrania in pazienti con crisi di grado 2 o 3 (cioè intensissimo). Il SUMATRIPTAN è il capostipite. Sono agonisti dei recettori serotoninergici, soprattutto sul 5-HT1. Ha una struttura simile alla serotonina e ha un’azione vasocostrittrice quasi solo a livello carotideo→ molto selettivo a differenza degli ergotaminici. MECCANISMO D’AZIONE: agiscono a livello dei vasi sui recettori agonisti 5-HT1B riducendo la vasocostrizione, ma anche sulle terminazioni nervose del trigemino determinando un’inibizione del rilascio di neuropeptidi responsabili di infiammazione neurogena ma riducono anche la sensibilizzazione centrale perché i 5-HT1B sono presenti nel nucleo caudale del trigemino (da cui partono afferenze verso il talamo e corteccia)→ meccanismo d’azione sia neurogeno che vascolare. CONTROINDICAZIONI: sono soprattutto a livello vascolare: bisogna sempre indagare in anamnesi una cardiopatia ischemica o un pregresso infarto, pregressi eventi ischemici cerebrali, ipertensione non controllata. In alcun forme di emicrania non si può dare, come in quella emiplegica, basilare o oftalmoplegica. È controindicato anche in caso di assunzione ai SSRI, tuttavia molti pazienti sono in cura con questi. Limiti di età: sotto i 18 e sopra i 65 non si possono dare, ad eccezione del sumatriptan spray nasale 10 mg, che è indicato anche in pazienti dai 16 ai 18 anni. EFFETTI COLLATERALI: in alcuni casi dà un senso di costrizione a livello stenocardico; si pensava fosse dato da una vasocostrizione a livello cardiaco ma dopo studi condotti con ECG dopo dolore stenocardico sono risultati normali. Si pensa sia legato ad alterazioni della motilità esofagea. Altri:
• Reazioni locali nella sede di somministrazione sotto cute.
• Astenia, mialgie, sonnolenza
• Sensazione di caldo e freddo alla testa ed agli arti
• Parestesie, vampate di calore al volto ed in regione toracica
• Vertigini, instabilità posturale
• Dolore e sensazione di rigidità nucale
• Acatisia, crisi distoniche, euforia
• Raramente e senza sicuro rapporto causale: angina instabile, ima, arresto cardiaco, ictus ischemico.

102
INTERAZIONI FARMACOLOGICHE: derivati dell’ergot: bisogna arrendere 24 ore dopo assunzione prima di somministrare triptano. Dopo triptano bisogna aspettare 6 ore prima di prendere derivato di ergot. SSRI: possono dare sindrome serotoninergica, fino al coma. IMAO: sospese 2 settimane prima, sono le più pericolose. FORME FARMACEUTICHE: Il sumatripatan è efficace in tutti i pazienti, la cosa che cambia è la velocità di azione.
La formazione iniettiva è quella di prima scelta nel caso di attacchi acuti devastanti, come quelli della cefalea a grappolo. DATI DI EFFICACIA: A differenza dei FANS, ha effetti sia sul dolore che sui sintomi di accompagnamento (nausea, vomito, fotofobia, …). La formulazione sottocute è quella che risponde meglio, e sono più efficaci quanto più è precoce la somministrazione: devono prenderlo appena sentono il mal di testa o in fase d’aura; se invece è già iniziata l’emicrania è meglio prendere un fans perché il triptano ha meno effetto. Se dopo l’assunzione il dolore si riduce da 3 a 1 o 2 ma non passa totalmente, si può assumere un secondo triptano dopo 2 ore, solo se però c’è stato un miglioramento. Non si possono assumere più di due triptani al giorno, qualunque formulazione sia. Se dopo 2 ore dall’assunzione di triptano il dolore non si è modificato, c’è indicazione per un farmaco di salvataggio, che è un FANS. L’abuso del sintomatico, che consiste in più di 15 somministrazioni in un mese, cronicizza l’emicrania, quindi bisogna provvede con altri farmaci.
FANS/analgesici. Sono farmaci da impiegarsi soprattutto per il trattamento di crisi di intensità lieve o moderata, e
delle crisi di intensità forte quando triptani ed ergotaminici sono controindicati o sono risultati inefficaci. I più usati sono:
DATI DI EFFICACIA: hanno scarsa efficacia nel trattare i sintomi di accompagnamento e hanno numerosi effetti collaterali. L’associazione di vari FANs con metaclopramide o domperidone (anti-emetici) non ne migliora effetto anti -emicranico Associazione di fans con caffeina non ne aumenta efficacia. Non testata efficacia su aura emicranica CONTROINDICAZIONI:
- Ipersensibilità ai prodotti, diatesi emorragica o patologie dell’emocoagulazione - ulcera gastrica e duodenale - insuff epatica o renale gravi: nemmeno il paracetamolo si può dare perché epatolesivo. - ibuprofene, naprossene, acido tolfenamico, piroxicam, diclofenac e ketorolac controindicati in scompenso
cardiaco congestizio - paracetamolo controindicato in deficit di glucoso 6 fosfato deidrogenasi ( dà anemia emolitica) - molti fans non si usano al di sotto dei 14 anni e devono essere dati con cautela ad anziani - sindrome di Reye da
ASA in bambini. EFFETTI AVVERSI:
• Gastralgia, pirosi gastrica, nausea, vomito, raramente ulcera gastrica e duodenale-_> possono aumentare nausea e vomito anziché ridurli.
• rare allergie con talora anafilassi • sonnolenza, astenia, alterazione della crasi ematica • paracetamolo: rari neutropenia, trombocitopenia, pancitopenia, necrosi epatica e renale per dosi massive

103
• indometacina: disturbi della visione, soprattutto nei trattamenti prolungati e alterazioni della crasi ematica (anemia, agranulocitosi, porpora trombocitopenica), sensazione di confusione, stordimento, vertigini, ronzii, edemi, iperglicemia, glicosuria.
INTERAZIONI FARMACOLOGICHE: Cautela se associati ad anticoagulanti e a steroidi per aumento del rischio di sanguinamento. Evitare assunzione di alcool Incrementano concentrazione ematica di digossina, barbiturici e litio Riducono effetto di aldosterone, diuretici risparmiatori di potassio ed anti-iperttensivi
Ergotaminici. Derivati della segale cornuta. Sono vasocostrittori dotati di attività antiemicranica riconducibile alla
vasocostrizione delle arterie craniche dilatate nel corso dell’attacco. Sono farmaci alternativi da impiegarsi per il
trattamento di crisi di grado 2 o 3 in pazienti a bassa frequenza di crisi (e per massimo 5 giorni) e per il trattamento di crisi
resistenti a triptani ed agli analgesici.
Differenza tra ergotaminici e triptani? Gli ergotaminici sono molto meno selettivi, quindi hanno un potente effetto
vasocostrittore anche a livello periferico, a differenza dei triptani che sono molto più selettivi.
Si siano veramente poco.
CONTROINDICAZIONI: sono le stesse dei triptani perché il meccanismo è lo stesso.
EFFETTI COLLATERALI:
- Nausea, vomito
- rari dolori addominali, diarrea, crampi muscolari, parestesie distali
- ergotismo in somministrazione cronica con acrocianosi→ uso cornico di ergotaminici può causare ulcere cutanee
fino alla vera e propria necrosi con amputazione su base di acrocianosi.
- necrosi ulcerose distali
- neuropatie ischemiche
- fibrosi pericardica, pleurica o retroperitoneale.
INTERAZIONI FARMACOLOGICHE: Mai dare nelle sei ore successive a somministrazione di triptano
Maggior rischio di vasocostrizione periferica in pazienti che usano beta-bloccanti→ sempre stare attenti a beta-bloccanti
perché l’ergotaminico dà un aumento degli effetti collaterali del beta-bloccante ma anche della vasocostrizione periferica.
Eritromicina, josamicina ed altri macrolidi rallentano il metabolismo di derivati di ergot, aumentandone i loro livelli
plasmatici
→ in generale non somministrare in concomitanza ad altri farmaci vasocostrittori.
FORMULAZIONI:
Quando associare anti emetico? Quando sui sintomi nausea e vomito i farmaci usati come anti-dolorifici hanno scarso
effetto→ con i triptani non serve.
Che farmaco associare? Solitamente metoclopramide o domperidone
Nella cefalea di tipo tensivo I TRIPTANI NON SI USANO! Rimangono solo i FANS ed eventualmente anche dei miorilassanti
(che però sono più usati nella profilassi che nell’attacco acuto).
TERAPIA DI PROFILASSI Serve a ridurre la frequenza degli attacchi.
IDENTIFICARE I FATTORI SCATENANTI: sono quelli in grado di scatenare attacco entro breve tempo dalla loro esposizione
anche se non sono sempre in grado di indurlo, necessitando a volte di cofattori

104
• stress (64 - 90%), non facilmente eliminabile.
• alcool (20 -52 %)
• cibi (formaggio, cioccolato, agrumi, cibi grassi, fritti) (10 -45%)
• digiuno prolungato (40%)
• Mestruazioni (24 - 64 %), infatti nella classificazione esiste l’emicrania catameniale.
• riduzione od eccesso di sonno
• variazioni metereologiche (7 - 43 %), soprattutto vento ed esposizione al gelo.
• luci fioche o molto forti e psichedeliche
Quando iniziare la profilassi farmacologica? quando sono presenti più di due crisi al mese parzialmente o totalmente
disabilitanti della durata complessiva di almeno 4 gg.
Quanto trattare? Per almeno tre mesi anche se è consigliabile rivedere il paziente che giunge per la prima volta
all’attenzione del medico dopo i primi due mesi di terapia
Come iniziare la profilassi? Iniziare a basse dosi possibilmente con un singolo farmaco da aumentare lentamente sino ad
ottenere gli effetti terapeutici in assenza di effetti collaterali. Raccomandare attenta compilazione del diario delle cefalee,
è l’unico modo per valutare l’efficacia della profilassi→ il trattamento è considerato efficace quando si ha una RIDUZIONE
DEL 50% DEGLI ATTACCHI.
Considerazioni prima della terapia profilattica. Essendo terapie profilattiche a lungo termine, prima di dare
questi farmaci, bisogna considerare altre patologie concomitanti, come ipertensione, cardiopatie, problemi di FC,
sovrappeso (alcuni farmaci fanno aumentare la fame).
Chiedere se il paziente assume già alcune di queste terapie per motivi cardiologici; si può chiedere eventualmente di
chiedere di andare dal cardiologo e farsi cambiare la terapia con qualche farmaco che possa essere utile anche per
l’emicrania.
Che farmaci usare?
➢ Beta bloccanti
➢ Antidepressivi triciclici
➢ Antiepilettici
➢ Calcio antagonisti
➢ Antagonisti 5-HT2
Calcio antagonisti. Meccanismo d’azione: il meccanismo preciso per cui i ca antagonisti funzionino sul mal di testa non si sanno con precisione,
si possono supporre. Due meccanismi ipotizzati:
▪ Modulazione della neurotrasmissione ed azione sul tono vascolare
▪ Inducono vasodilatazione ed esercitano azione citoprotettiva riducendo l’ingresso di calcio all’interno della cellula
con conseguente riduzione del danno ipossico.
L’effetto è graduale dopo settimane di trattamento, l’azione di prevenzione si vede dopo 3 mesi.
Flunarizina, l’unico farmaco che ha in scheda tecnica l‘indicazione di profilassi. Cinnarizina e Verapamile non hanno
l’indicazione in scheda tecnica ma è comune indicato per profilassi, bisogna però far firmare il consenso informato.
FLUNARIZINA
Farmaco di prima scelta in presenza di
insonnia.
Schema terapeutico consigliato:
- 5 mg alla sera a cicli di 20 gg (e 10
di pausa) al mese per tre mesi
Oppure
- 5 gg alla settimana per tre mesi
La compressa da 10 mg è meno usata
perché ha molti effetti collaterali.
Le sospensioni servono per ridurre gli
effetti collaterali piramidali.
CINNARIZINA
Farmaco di prima scelta in presenza
di ansia e/ o insonnia.
Schema terapeutico consigliato:
- 75 - 150 mg/die alla sera a cicli di
20 gg al mese per tre mesi
Oppure
- 5 gg alla settimana per 3 mesi
Precauzioni: evitare in età senile,
obesità, depressione anche familiare
od anamnestica ed in parkinsonismo.
VERAPAMILE
Farmaco di prima scelta in cefalea a
grappolo (dosaggi molto elevati in
cefalea a grappolo, più bassi in
emicrania).
Farmaco da preferire in presenza di
depressione, ipertensione, tachicardia,
in pazienti con pregresso stroke.
Schema terapeutico consigliato:
- 160 -320 mg/die per tre mesi

105
Precauzioni: evitare in età senile,
obesità, depressione anche familiare
od anamnestica ed in parkinsonismo.
Precauzioni: evitare associazione con
betabloccanti, in caso di ipotensione,
insufficienza cardiaca congestizia o
aritmie, blocco atrio-ventricolare, in
associazione con IMAO.
Beta-bloccanti. Agiscono a diversi livelli: sistema RAS, broncocostrizione (no in asmatici), ….
Meccanismo d’azione: non noto, probabilmente agiscono su sistema monoaminergici centrali incluso quello
serotoninergico.
Farmaco di prima scelta in presenza di ipertensione e tachicardia. Beta-bloccanti più usati:
CONTROINDICAZIONI: evitare in caso di depressione anche solo anamnestica, asma bronchiale, bradicardia, in anziani,
diabetici, scompenso cardiaco, malattie vascolari periferiche - tipo Reynaud, gravidanza.
Non indicati in emicrania con aura e se vi è uso cronico di ergot derivati→ NO ASSOCIAZIONE CON ERGOTAMINICI.
Non controindicazioni ad associazione con flunarizina o cinnarizina.
PRECAUZIONI:
- Inizio (e sospensione) graduale in circa 2 settimane per evitare effetto rimbalzo-
- Monitoraggio costante di PA ed FC durante la terapia
- Possibili astenia, vertigini
- La non risposta ad un beta -bloccante non esclude altri tentativi usando altro farmaco di stessa classe
Antidepressivi triciclici. Il più usato è l’AMITRIPTILINA (laroxil); è anche l’unico ad avere azione di profilassi nella
cefalea tensiva. Ha anche un’azione sedativa quindi è utile per i casi di insonnia, depressione, ansia (dato che la
somministrazione è serale).
MECCANISMO D’AZIONE: non è correlato al meccanismo antidepressivo, infatti si usano dosaggi molto più bassi del
dosaggio sa antidepressivo. Determina un ‘inibizione del reuptake di noradrenalina e serotonina, inducendo down
regulation dei recettori beta adrenergici e serotoninergici a livello centrale.
Si usa la formulazione in gocce perché permette di aumentare gradualmente la posologia ed evitare gli effetti collaterali. Si
inizia con 3 gocce (6 mg) e poi si arriva a 12-15 gocce.
CONTROINDICAZIONI: ha azione anticolinergica quindi è da usare con cautela in pazienti con glaucoma, ipertrofia
prostatica, tendenza all’ostruzione intestinale.
EFFETTI COLLATERALI: incremento ponderale e sonnolenza, che può anche essere ricercato.
Antiepilettici. Uno solo ha indicazione in scheda tecnica ed è il TOPIRAMATO.
EFFETTI INDESIDERATI: aumento del tono dell’occhio fino a glaucoma, calcolosi renale, disturbi cognitivi, sedazione, diarrea,
perdita di peso, vertigini, depressione→ è una delle principali cause per cui si sospende la terapia (preferiscono il mal di
testa alla depressione).
Altri effetti sono: acroparestesie, sedazione, calo ponderale→ può essere farmaco di scelta per una paziente obesa non
depressa.
Dosaggio: 100 mg al giorno; si inizia con 25 mg e si aumenta di 25 ogni settimana, fino ad arrivare a 50 mg due volte al
giorno.
ACIDO VALPROICO: funziona molto bene, il problema è che la maggior parte dei pazienti sono donne in età fertile ed è
controindicato; in un uomo va benissimo. Si fa un dosaggio costante: serve solo per vedere se è sovradosato, invece se è
sottodosato (sotto al range terapeutico) non deve essere aumentato se comunque è efficace.
EFFETTI COLLATERALI: tremori, alopecia.
LAMOTRIGINA: ha una indicazione molto precisa: prevenzione di attacchi di emicrania con aura ad elevata frequenza. Si
inizia con dosaggi basi e si aumenta con molta molta cautela→ rischio di sindrome di Stevens-Johnson.

106
SNRI. Sono farmaci duali, cioè sono inibitori del reutpake di serotonina e noradrenalina. Hanno dei trial che evidenziano
la loro efficacia nella terapia preventiva di emicrania, soprattutto VENLAFAXINA e DULOXETINA. Non sono di prima scelta
ma si è visto che in caso di non risposta ad altri tipi di profilassi erano molto efficaci.
Profilassi dell’emicrania catameniale. Sono “mini-profilassi” limitate in caso di emicrania catameniale:
- Dididroergotamina a lento rilascio (5 mg x 2/die)
- Naprossene sodico (550 mg x 2/ die per os o rettale)
Si danno 4-5 giorni prima dell’inizio del ciclo e per tutta la durata del ciclo.
Primo controllo dopo terapia profilattica. Quando il paziente si presenta 3 mesi dopo dall’inizio della profilassi
con il diario:
- Se gli attacchi si sono ridotti del 50%, si fa continuare la terapia in atto per altri 2-3 mesi.
- Se invece la terapia non è stata efficace, si cambia il farmaco, tenendo sempre in considerazione tutte le
controindicazioni.
- Dopo aver utilizzato vari farmaci in monoterapia per un periodo di tempo di almeno tre mesi senza ottenere una
riduzione delle crisi di almeno il 50%, si può tentare l’associazione di più farmaci.
Bisogna avvisare il paziente che non è detto si riesca rimuovere tutte le cefalee; ci si aspetta che rimarranno sempre delle
crisi residue, che si trattano con i farmaci della fase acuta (tenendo in considerazione le interazioni tra farmaci).
Una volta trattato il mal di testa secondo le linee guida ma il paziente continua ad avere mal di testa, si prende in
considerazione l’abuso farmacologico: si tenta di trattare il paziente con l’abuso e disassuefazione e si cercano altre cause
del mal di testa.
NUOVE TERAPIE PROFILATTICHE: mAb Dopo il 2011, sono uscite altre due terapie profilattiche per l’emicrania, che sono innovative. Hanno ricevuto l’approvazione
per l’uso nel Dicembre 2018 ma tuttora sono poco utilizzate e sono a pagamento.
Secondo l’ipotesi trigemino-vascolare, il mediatore più importante nella trasmissione del dolore e di vasodilatazione è il
CGRP: viene prodotto sia a livello di terminazioni nervose (centrali e periferiche) che dei vasi. È responsabile anche
dell’infiammazione neurogena. La branca afferente del trigemino rilascia CGRP durante l’attacco emicranico nel nucleo
caudale del trigemino, che ha l’effetto di amplificazione del meccanismo dell’infiammazione e della trasmissione
dell’impulso doloroso.
Un farmaco che blocca il legame del CGRP è un farmaco che ha una azione elettiva sull’emicrania→ è stato studiato
partendo da un meccanismo patogenetico.
Sono i farmaci migliori disponibili per la cura dell’emicrania e si dividono in due categorie:
❖ Anticorpi monoclonali anti- CGRP:
o Galcanezumab: è stato approvato nel 2018 ed è stato studiato per la prevenzione di emicrania episodica;
somministrazione sottocutanea. È in grado di ridurre in modo significativo il numero di giorni di emicrania
in un mese. Gli eventi avversi sono minimi: eritema e dolore nella sede dell’iniezione o dolore addominale.
o Fremanezumab: sottocutaneo, ha dimostrato una netta riduzione dell’uso di farmaci sintomatici.
o Eptinezumab: meno maneggevole, non ancora in commercio perché viene somministrato endovena in
regime ospedaliero.
❖ Anticorpi monoclonali anti-CGRPR: Erenumab: eventi avversi sono rino-faringiti, astenia e cefalea.

107
ALTRE PROCEDURE 1) TOSSINA BOTULINICA: Meccanismo d’azione non è finora chiaro. Si sa che inibisce il rilascio di acetilcolina a livello
della giunzione neuromuscolare, e previene la contrattura muscolare ma probabilmente questo meccanismo non
è sufficiente a spiegare le proprietà anti-nocicettive osservate in diversi trials clinici e preclinici.
È stata ipotizzata una inibizione al rilascio di NT correlati alla nocicezione, quali la sostanza P, il CGRP e il
glutammato. Ultimamente è stato proposto un nuovo meccanismo anti- nocicettivo della tossina botulinica: esso
consisterebbe nell’inibizione della sensibilizzazione periferica delle fibre trigeminali, che a loro volta potrebbero
ridurre la sensibilizzazione centrale a livello del nucleo trigeminale caudale.
Procedura: inoculazione di tossina botulinica di tipo A (Botox) in specifiche aree dei muscoli della testa e del collo
(cervicali, temporali, frontali).
TERAPIA CEFALEA A CLUSTER TERAPIA SINTOMATICA DEL CLUSTER
Obiettivi. - Trattare attacco appena si manifesta
- Determinare nel paziente la risoluzione o significativa attenuazione di dolore e fenomeni vegetativi ad esso correlati
- Ottenere tale risultato nel più breve tempo possibile (entro 15 minuti da assunzione di farmaco) e limitare effetti
collaterali.
Farmaci. ➢ SUMATRIPTAN SOTTOCUTE 6 mg, farmaco di prima scelta (max due volte al di almeno a due ore di distanza) con
scomparsa contestuale di fenomeni vegetativi mentre miosi, ptosi palpebrale ed iniezione congiuntivale
scompaiono dopo.
➢ SUMATRIPTAN SPRAY NASALE: dose di 20 mg, meno efficace con meno effetti collaterali; dose max 2
somministrazioni die con intervallo di almeno due ore.
➢ OSSIGENO TERAPIA: O2 al 100% a 6-7 lt/min per 15 min. Indicato come seconda scelta o in caso sia controindicato
sumatriptan o in caso di attacchi giornalieri frequenti in attesa che funzioni la profilassi.
→ Spesso consente di fare DD: se la sintomatologia regredisce con l’ossigenoterapia, allora è sicuramente una
cefalea a grappolo. Spesso viene confusa con nevralgia del trigemino che in genere non ha comparsa stagionale e
non regredisce con O2.
TERAPIA PROFILATTICA DEL CLUSTER
Obiettivi. - Indurre rapida scomparsa degli attacchi con conseguente conclusione del grappolo.
- Riduzione frequenza, intensità e durata degli attacchi.
- Ritenuto efficace solo nel cluster cronico (in forme ricorrenti o cicliche dubbio che cluster si esaurisca da solo).
Importante è iniziare precocemente e continuare ancora per 10-14 giorni dopo la scomparsa delle crisi. La terapia poi viene
ripresa quando ricompare u nuovo cluster.
Farmaci. ➢ VERAPAMILE: farmaco di prima scelta nel trattamento del cluster episodico e cronico. È possibile associazione con
litio in casi severi. Non interazioni con sumatriptan, corticosteroidi.
➢ PREDNISONE: farmaco di seconda scelta nel trattamento del cluster episodico (rapida azione); in cluster cronico
da rapida riduzione delle crisi quando altri farmaci non sono ancora efficaci. Usato per indurre remissione in casi
più gravi con attacchi ad alta frequenza ed intensità, a grappolo già iniziato. Possibile recrudescenza cefalea
quando prednisone < 25 mg: in tal caso possibile associare a prednisone farmaci di profilassi di prima scelta.
➢ LITIO: nel trattamento del cluster episodico e cronico (dosaggio: 300 mg x 3/die fino a max di 1200 mg/die).
Monitoraggio della litiemia tra 0,4 -1,2 mEq/l, controllo funzionalità tiroidea e renale.
o Effetti collaterali: tremori, diarrea, confusione mentale.
o Cautela se associato con: calcio antagonisti, SSRI, diuretici tiazidici, indometacina, diclofenac.

108
PARKINSONISMI
Oggi, Parkinson e malattie affini vengono raggruppati sotto il nome di disordini del movimento, mentre una volta venivano chiamate malattie extra-piramidali o dei gangli della base.
CLASSIFICAZIONE Parkinson è un nome sindromico: indica cioè una costellazione di segni e sintomi dovuta a diverse cause ma con qualcosa in comune. I parkinsonismi si dividono in:
• PATOLOGIE DEGENERATIVE: che comprendono: o PARKINSON IDIOPATICO: è quello più frequente, è la malattia di Parkinson così
come la conosciamo. o PARKINSONISMI ATIPICI o PLUS: si aggiungono altri segni e sintomi relativi al
coinvolgimento degenerativo di più sistemi neuronali (es. cervelletto, sistema autonomico, sistema piramidale, …).
Cosa è più grave, il parkinsonismo atipico o idiopatico? Quello atipico è certamente più sfavorevole sia come decorso che come qualità di vita; quello idiopatico infatti si lascia curare e consente anni di vita quasi normale.
• CAUSE SECONDARIE o Farmaci: neurolettici tipici. o Veleni e tossine: CO, manganese, idrocarburi. o PARKINSONISMO VASCOLARE: Cerebropatia vascolare cronica (lacune multiple dei gangli della base) o Idrocefalo normoteso (NPH) (ne parleremo) o POST-TRAUMATICO: traumi ripetuti cronici (pugili). o Infezioni: l’epidemia di influenza asiatica poteva causare encefalite (chiamata letargica) con
parkinsonismo. o PARKINSONISMO TUMORALE: tumori della linea mediana.
. Percentuali:
- Idiopatica: 85% - Neurolettici: 7% - Vascolare: 3% - Degenerativi atipici: 5%
PARKINSON IDIOPATICO EPIDEMIOLOGIA Prevalenza di 200 per 100.000 abitanti (per difetto). L’insorgenza tende ad aumentate con l’età, la media è intorno ai 60 anni ma può manifestarsi anche prima dei 20 anni→ si parla di Parkinson giovanile se prima di 20 anni e precoce se prima di 40 anni. Le forme geneticamente determinate si manifestano principalmente come giovanili e la malattia è molto più problematica se esordisce come giovanile perché è da gestire per un lungo periodo: il Parkinson curato non comporta un’abbreviamento del corso della vita, quindi sono da gestire per tutta la vita. L’unico problema della terapia del Parkinson è perde efficacia con il passare del tempo.

109
PATOGENESI È stata descritta nel 1877 per la prima volta, ma il primo caso anatomo-patologico è stato descritto solo nel 1919.
La chiave neuropatologia del Parkinson è la DEGENERAZIONE DELLA PARS COMPACTA DELLA SOSTANZA NERA DEL MESENCEFALO: qui ci sono i neuroni pigmentati di neuromelanina (pigmento che deriva dal catabolismo di catecolammine -ammine simpaticomimetiche), sono quindi neuroni dopaminergici→ perdita di proiezioni dopaminergiche che dalla sostanza nera raggiungono il corpo striato o corpo lenticolare. Fisiologicamente, con l’età si perde molta dopamina nella sostanza nera (tant’è che l’anziano ha un’andatura molto simile a quella del parkinsoniano), tuttavia finché ne resta il 50% non si manifestano segni di malattia, è necessario perderne più della metà per manifestarsi. Nei neuroni degenerati si trovano anche corpi inclusi, chiamati CORPI DI LEWY: sono accumuli patologici di proteine ubiquitinate. Avviene qualche errore per cui determinate parti proteiche non vengono smaltite correttamente, si tratta soprattutto dell’alfa-sinnucleina; una volta era considerato un fatto patognomonico, ma successivamente si è visto che non si formano solo in sostanza nera ma anche in tutto l’encefalo, non solo nelle forme idiopatiche ma anche in alte forme, chiamate nel complesso sinnucleinopatie. Cosa succede nel resto dei gangli della base se vengono meno le fibre nigro-striatali? Il circuito è molto complesso e ha un significato soprattutto motorio ma non solo (anche cognitivo e affettivo). Dalla corteccia partono fibre motorie che arrivano al putamen e ai gangli della base, poi attraverso il talamo torna in corteccia. A questo punto l’output elaborato viene inviato al midollo. Lo squilibri più evidente, e che interessa di più, è il determinarsi di una iperattività anomala del globus pallidus interno e del nucleo subtalamico di Luys; questa iperattività ha un ruolo fondamentale perché turba tutta la circuitazione tra nuclei della base e corteccia, in un modo molto complesso, in modo tale da causare segni e sintomi tipici (non è importante sapere cosa accade perché troppo complesso). È importane focalizzare l’attenzione su queste strutture perché sono i possibili bersagli di terapia chirurgica e farmacologica (microcoagulazione, DPS -deep brain stimulation). Una lesione chirurgica o un blocco elettrico del nucleo subtalamico o del pallido interno gioveranno quindi ai segni/sintomi della MP.
SEGNI CARDINALI Le funzioni dei gangli della base non sono solo motrici, ma hanno valenza cognitiva e affettiva, e anche sul sistema vegetativo. I segni cardinali del Parkinson idiopatico sono motori:
1. ESORDIO ASIMMETRICO: rimane per parecchio tempo, quindi si parla di emiparkinson: è un dato molto
caratteristico. Con il passare del tempo si bilateralizza.
2. TREMORE: esistono diversi tipi di tremori:
a. A RIPOSO: è la forma più comune di Parkinson, scompare quando si chiede di muoversi o di assumere determinate posture. Frequenza di 4-6 Hz.
b. POSTURALE: è un tremore scatenato da determinate posture; esistono anche forme dove coesistono più tipi di tremori: es. tremore a riposo, ma se si chiede di assumere posizione di Mingazzini il tremore persiste. (a riposo + posturale).
c. DA AZIONE: in una proporzione ancora minore di pazienti. Si manifesta quando si chiede di compiere dei movimenti fini come fare pollice-indice con la mano (gesto anche per valutare bradicardia).
d. MANDIBOLARE: a volte oscilla la mandibola. Inizia con una lieve intensità, ma poi col tempo peggiora; come tutti i tremori, peggiora notevolmente con lo stato emotivo (fretta, ansia, agitazione aumentano il tremore).

110
3. RIGIDITÀ: ipertonia che coinvolge tutti i muscoli dell’organismo (sia flessori che estensori) ma predomina nei
flessori. L’atteggiamento del paziente infatti è prevalentemente flesso; alla palpazione del bicipite, appare una certa resistenza, se poi si tenta di estendere il bicipite sul braccio si nota che cede a scatti al movimento passivo→ segno della troclea. Si può aumentare la sensibilità della prova facendo muovere gli arti controlaterali: si chiede di flettere ed estendere continuamente l’avambraccio dell’altro lato, e si osserva un aumento di rigidità controlaterale.
4. BRADICINESIA: si tratta di una lentezza all’inizio e nel corso dei movimenti, in ogni tipo di movimento (sia
volontari che automatici). Spesso si saggia con movimenti di fine manualità che risultano più impacciati, meno ampi, più lenti e più rapidamente esauribili per cui, con la ripetizione, diventano quasi impercettibili. Bradicinesia implica difficoltà nei passaggi posturali, quali ad esempio scendere dall’automobile o girarsi nel letto o vestirsi. Spesso arrivano dal neurologo perché lamentano eccessiva lentezza o difficoltà nel fare alcuni movimenti o viene notato da qualche parente. Conseguenza di bradicinesia:
a. Impoverimento di movimenti automatici, ipomimia o amimia. b. Ridotta frequenza dell’ammiccamento spontaneo (il fisiologico sbattere le palpebre) che causa fissità
dello sguardo. c. Micrografia: lamenta un cambio di scrittura e una
diminuzione della dimensione della scrittura (progressione da grande a piccolo sulla stessa riga). Non vale solo per la scrittura, ma anche nel fare ghirigori.
d. Diminuzione delle sincinesie pendolari (fisiologico pendolamento dell’arto superiore durante la marcia), asimmetrico nelle fasi iniziali: uno pendola normalmente l’altro è più fermo.
5. DISTURBI DELL’EQUILIBRIO E ALTERAZIONE DELLA POSTURA: più tardivi, talvolta non si presentano affatto;
sintomi meno favorevoli. Causa una riduzione dei riflessi di raddrizzamento: il soggetto non è più in grado di correggere spontaneamente eventuali squilibri. Si ricerca verificando la capacità di correggere una spinta all’indietro a occhi chiusi (Pull test).
6. ANDATURA: Interessata fin dai primi anni di malattia. Passo piccolo e strascicato. Assume una posizione durante il cammino definita “festinazione”: il paziente piega il busto in avanti e tende ad accelerare il passo come se inseguisse il proprio baricentro, fa dei passettini piccoli e veloci come se stesse cadendo in avanti (non è vero che si mettono a correre!).
7. FREEZING: negli stadi avanzati della malattia (talvolta come effetto collaterale del trattamento con levodopa) possono verificarsi episodi di blocco motorio improvviso. Può colpire vari aspetti del movimento, ma più spesso la deambulazione. I piedi del paziente sembrano incollati al pavimento. Di solito si verifica nelle strettoie o sulle soglie, più raramente, anche negli spazi aperti. Questa difficoltà può essere superata adottando alcuni accorgimenti quali alzare le ginocchia come per marciare, oppure considerando le linee del pavimento come ostacoli da superare (es. un bastone) o anche con un ritmo verbale o musicale come quello che si utilizza durante la marcia militare→ il vedere un riferimento che limita lo spazio davanti al piede può vincere il freezing, probabilmente per interazione con il sistema visivo (ma è un meccanismo difficile da capire).
→ Sono alla base della diagnosi di malattia di Parkinson idiopatico; oltre a verificare questi insomma, bisogna anche verificare che ci sia una risposta alla terapia dopamino-stimolante. Veniva chiamato TEST ALLA LEVODOPA, se è positivo si avvalora la diagnosi di Parkinson idiopatico→ permette di fare DD con parkinsonismi atipici, che sono di gran lunga più negativi. Un altro test è il TEST ALL’APOMORFINA (un dopamino-agonista diretta).
SINTOMI ATIPICI Sono sintomi che depongono per parkinsonismi atipici:
- Scarsa risposta al test alla levodopa - Cadute precoci (entro 3 anni esordio) - Freezing precoce - Allucinazioni non terapia-correlate→ fanno sempre pensare a un parkinsonismo atipico, addirittura a una
demenza a corpi di Lewy. - Decadimento intellettivo precoce: oggi non è più vero che ci sono così tanti deficit cognitivi, ma se ci sono allora
non si tratta di Parkinson idiopatici. - Paralisi sguardo verso il basso: paralisi sovranucleare progressiva (la vedremo). - Grave disautonomia precoce non terapia-relata - Movimenti involontari anomali atipici e precoci - Cause accertate di parkinsonismo secondario

111
CRITERI DIAGNOSTICI PER MALATTIA DI PARKINSON Per fare diagnosi definita: serve una biopsia, possibile solo dopo il decesso. Per fare diagnosi probabile: almeno 3 segni cardinali, assenza di segni atipici, documentata e persistente risposta a levodopa e apomorfina. Per fare diagnosi possibile: almeno 2 segni cardinali, assenza di segni atipici, buon risposta a levodopa o apomorfina. La causa di errore nella diagnosi più frequente sono i parkinsonismi atipici. Su 100 casi autoptici, 76 sono stati dichiarati idiopatici.
TREMORE ESSENZIALE NON È UN PARKINSONISMO, ma è un’altra frequente causa di tremore e spesso viene scambiata erroneamente per Parkinson dai non esperti. Forte tendenza eredofamiliare, familiarità autosomica dominante.
o È un tremore soprattutto cinetico, cioè intenzionale, ma in una minor percentuale di casi può anche essere posturale o a riposo (esattamente il contrario del Parkinson): fanno fatica soprattutto a compiere gesti fini, e il tremore aumenta più si avvicini nano al bersaglio. Si associano a tremori del capo, che causano uno scuotimento continuo della testa tipo “no”; a volte si associa a tremore della voce.
o È bilaterale, non è più asimmetrico. o Non risponde alla levodopa, ma è invece un tremore alcol-sensibile.
Sembra essere un fattore di rischio aggiuntivo a sviluppare una forma di Parkinson in pazienti con tremore essenziale, ma il prof non lo ha mai riscontrato.
Terapia. Terapia standard sono i betabloccanti. Se non funzionano si dà il primidone, un pro-barbiturico.
PARKINSONISMI SECONDARI PARKINSONISMO VASCOLARE Tra i parkinsonismi secondari, cioè non degenerativi, c’è primo fra tutti quello da farmaci seguito da quello vascolare. La base anatomo-patologica sono le lacune multiple dei gangli della base→ dovuto a cerebropatia ischemica cronica. I sintomi quindi sono un misto di parkinsonismo e di malattia cerebrovascolare:
• Tremore è molto raro • Rigidità • Bradicinesia • Interessamento preferenziale per arti inferiori e turbe della deambulazione molto pronunciate→ si parla di LOWER
BODY PARKINSONISM. Può esordire infatti con cadute precoci, deambulazione ondulante-uniforme. • Segni focali dati da cerebropatia ischemica:
o Emiparesi pura o Emianestesia pura o Decadimento cognitivi precoce
Prognosi. Hanno una progressione variabile, con assai modesta risposta alla levodopa. L’imaging è positivo, nel Parkinson
idiopatico non ci si aspettano queste lacune.
PARKINSONISMI ATIPICI/PLUS ATROFIA MULTISISTEMICA (MSA) Il termine plus indica che c’è un’aggiunta di segni e sintomi relativi alla degenerazione di altri sistemi neurali oltre a quelli coinvolti nel Parkinson idiopatico; a seconda della degenerazione “aggiuntiva” si avranno quadri clinici diversi con nomi diversi:
❖ Associazione a degenerazione del sistema autonomico: VARIANTE AUTONOMICA DI MSA (o insufficienza autonomica progressiva o malattia di Shy-Grainer). Si associa a:
o Disturbi genito-urinari, incontinenza sfinterica o Turbe della sfera sessuale o Ipotensione ortostatica e sincopi o Anidrosi
❖ Associazione a degenerazione del cervelletto: MSA CEREBELLARE. Si associa a turbe cerebellari: o Atassia cerebellare (per gli arti si fa la prova indice-naso) o Nistagmo (flocculo e nodulo -archicerebello- si collegano con i nuclei vestibolari).

112
o Dismetria, tremore d’azione. ❖ Associazione a degenerazione del sistema cortico-spinale: MSA PIRAMIDALE o DEGENERAZIONE STRIO-NIGRICA:
avremo una sindrome del I motoneurone con segni piramidali: o Riflessi accentuati o Paresi o Segni di Babinski
Altri segni di MSA (non tutti da conoscere, lui vuole solo i più significativi): 1. Distonia oro-facciale: una distonia dei muscoli mimici della faccia che simula un sorriso sardonico, beffardo. 2. Distonia del tronco con lateroflessione: si ha una deviazione laterale della colonna, chiamata anche SINDROME
DI PISA. 3. Turbe comportamentali del sonno REM (danno pugni e calci). 4. Apnee notturne e stridor: è un rumore emesso dalla glottide che rimane semichiusa durante l’espirazione.
PARALISI SOPRANUCLEARE PROGRESSIVA (PSP) Chiamata anche Steele-Richardson-Olszewski. Si fa riferimento a una paralisi verticale dello sguardo: non riesce a dirigere lo sguardo sia in senso orizzontale che verticale. Anche nella malattia di Parkinson idiopatico può esseri una difficoltà nello sguardo verso l’alto (sindrome di Parinaud); qui non si ha nemmeno lo sguardo verso il basso e poi si perde anche la capacità di muoverlo in orizzontale. Se si chiede di muovere gli occhi, un pochino verso destra e sinistra riesce, ma verso alto e basso no; dopodiché si sfruttano i riflessi oculo-cefalici: si muove il capo fissando un punto e la motilità sembrano conservati. → Il termine sopranucleare allude proprio a una disfunzione del controllo che i centri corticali hanno sui nuclei degli oculomotori: il comando motorio volontario non è attivo perché la corteccia non parla con i nuclei oculomotori ma i nuclei stessi funzionano perché il circuito elementare riflesso che include gli oculomotori è conservato.
DEGENERAZIONE CORTICO-BASALE Al quadro di Parkinsonismo si aggiungono segni legati a una degenerazione asimmetrica della corteccia sensorimotoria. Oggi si parla più di sindrome della degenerazione cortico-basale.
➢ Sindrome parkinsoniana asimmetrica. ➢ Segni corticali:
o Problematiche di sensibilità coordinate con coinvolgimento frontale: astereognosia. ➢ Disordini del movimento:
o Distonia o Mioclonia→ si può dimostrare pizzicando la falange per vedere se genera una mioclono riflesso. o Fenomeno dell’arto alieno, perché l’arto vive di vita propria, non risponde più ai comandi. In certi casi è
così inutile che si preferisce amputarlo. → È l’unico del parkinsonismi degenerativi che manifesta una asimmetria, come il Parkinson idiopatico.
DEMENZA A CORPI DI LEWY È sempre una sinnucleinopatia: si distingue dalle altre demenze per:
- Rapidamente progressiva - Allucinazioni visive - Decorso fluttuante, ondulate. Variazioni del comportamento nel tempo, non giustificate. - Risposta paradossa ai neurolettici: in una persona allucinata con iniziale allucinazioni e turbe del comportamento,
se si iniziano i neurolettici si ha un peggioramento (perché i neurolettici sono antagonisti dopaminergici)→ tipico di demenza a corpi di Lewy.
Ci sono delle caratteristiche in comune con la encefalopatia di Creutzfeldt-Jakob (era stata scambiata per tale ma poi all’indagine istologica è stato confermato esse Lewy)→ simili per insorgenza repentina e per EEG (in entrambi periodico).
IMAGING DI PARKINSONISMI SECONDARI
TC. Alcuni sono individuabili anche con una semplice TC, come:
- Lacune del parkinsonismo vascolare. - In una forma particolare, chiamata malattia di Fahr, c’è un accumulo patologico di calcio nei nuclei della base. - Tumori della linea mediana, sono chiaramente visibili a TC. - Parkinsonismo in un quadro di idrocefalo normoteso.

113
RM. Unica metodica che in alcuni casi permette di notare piccole anomalie che possono contraddistinguere i parkinsonismi
plus. - MSA tipo C: Aree ipointense a livello pontino, con aspetto a croce (Hot cross bun). Atrofia di cervelletto e ponte a
forma di pinguino o colibrì. - PSP: atrofia tetto mesencefalo, iperintensità grigio periacqueduttale. - Degenerazione perirolandica in degenerazione cortico-basale
→ permettono di fare DD tra Parkinson idiopatico e secondario, ma è un concetto molto specialistico.
DAT-SCAN. SPECT (Scintigrafia a emissione di fotone singolo)
con un particolare radio-tracciante, con cui si possono evidenziare i sistemi di trasporto della dopamina nei gangli della base (DAT: dopamine transporter). Evidenzierà anomalie sia nei parkinsonismi che nel Parkinson idiopatico. Ottimo per distinguere il tremore essenziale e il Parkinson, infatti nel primo caso è assolutamente normale perché non ha nulla a che vedere con la degenerazione dopaminergica. Questo esame è molto comune e molto spesso viene prescritto. Alcuni tipi particolari di SPECT permettono di distinguere anche tra parkinsonismi primitivi e secondari.
Scintigrafia miocardica. Il radiotracciante MIBG si lega al sistema simpatico cardiaco post-gangliare e ha la
particolarità di evidenziare una deplezione abbastanza selettiva in corso di malattia di Parkinson. La scinti quindi risulterà patologica in Parkinson, mentre nei parkinsonismi sarà negativa→ al contrario di quanto può sembrare, dato che nei secondari c’è degenerazione autonomica: questa però è una degenerazione pre-gangliare, che non si può vedere in nessun modo.
→ nei primari è alterata la componente simpatica post-gangliare (evidenziabile con scinti), nei secondari è alterata la componente simpatica pre-gangliare (non visibile con nessuna scinti).
PERDITA DELL’OLFATTO: è un sintomo precoce di malattia di Parkinson, è una particolarità. È un deficit quantificabile con determinati test→iposmia idiopatica.
VALUTAZIONE NEUROPSICOLOGICA Anche nella malattia di Parkinson vengono meno delle funzioni cognitive, soprattutto quelle esecutive: prese di decisione, scelta di atti motori, capacità di inibire iniziative motorie non necessarie. Tuttavia un soggetto con Parkinson nei primi anni di malattia non sarà decaduto cognitivamente. Esistono delle scale di valutazione, come il Minimental State Test. È importante per la diagnosi di demenze.

114
TERAPIA PARKINSON È una delle prime e non moltissime malattie neurologiche degenerative con una terapia. Sono terapie sintomatiche, e non patogenetiche: non contrastano la degenerazione, sono solo terapie di rimpiazzo o di correzione di disfunzioni neurofisiologiche (DBS o chirurgia funzionale).
LEVODOPA Inizialmente si dava pura, in grandissime quantità perché passa difficilmente la BEE e perché viene rapidamente trasformata (circa il 70%), ora invece viene associata a inibitori di carbossilasi periferici, come la carbidopa: impediscono che la levodopa sia degradata nel circolo periferico e raggiunga il SNC in quantità maggiore e qui si trasformi in dopamina e rimpiazzi la dopamina mancante.
Preparazioni più comuni:
• Levodopa + Benserazide = Madopar • Levodopa + Carbidopa = Sinemet • Levodopa + Carbidopa+ Entacapone = Stalevo • Duo-DOPA: consiste in una stomia duodenale per somministrare dopa direttamente nel duodeno in modo da
saltare la tappa gastrica, dove con l’avanzare degli anni l’assorbimento diventa incostante e imprevedibile.
EFFETTI COLLATERALI Nausea e vomito
Disturbi gastrointestinali
Ipotensione ortostatica →si usano anti-dopaminergici che non interferiscono con trasmissione a livello dei gangli della base: Domperidone (Peridon, Motilium) da utilizzare per qualche tempo fino a quando il disturbo non si risolve. No metoclopramide, perché agisce sui gangli ella base con effetto parkinsonizzante. SINDROME DA TRATTAMENTO A LUNGO TERMINE: sono complicazioni più tardive, dopo 4-5 anni o anche mai, la situazione ottimale cambia e la terapia non ha più un effetto ottimale:
Fluttuazioni motorie: nel corso della giornata ci sono dei momenti in cui la motricità non è più controllata. Compaiono:
o Discinesie: bucco-linguali (incessante movimento di masticazione o della lingua, non voluti), coreiformi (mimano le coree, a volte fino a essere violenti -prendono il nome di ballismo).
o Distonie o Freezing o cadute
Possono essere dovute a diversi fenomeni: ➢ Deterioramento da fine dose (wearing off): questo inizia solitamente con la sensazione che lontano
dall’assunzione del farmaco si fa fatica a muoversi. È dose-correlato, prevedibile, quindi si risolve aumentando la dose o aumentando il numero di dosi nel corso della giornata. Solitamente questo effetto avviene dopo pranzo, perché c’è una competizione della dopa con gli amminoacidi del pasto, quindi importante è spiegare di assumere il farmaco almeno mezz’ora prima dei pasti. Se non dovesse migliorare nemmeno con questi accorgimenti, si potrebbe pensare di modificare la dieta.
➢ Meccanismo ON/OFF: sono variazioni imprevedibili e immediate della motricità, non dose-correlato: passa da una fase di immobilità a una di ipercinesie; non si può agire in nessun modo. In fase OFF (cioè quando viene meno l’effetto di dopa) ci possono essere fenomeni distonici, spesso anche dolorosi (classica è la distonia dolorosa del piede).

115
Disturbi di tipo psicotico, più frequenti nei pazienti anziani e con i dosaggi più elevati: confusione, allucinazioni notturne, deliri, agitazione psicomotoria→ questo perché i farmaci agiscono su tutti i sistemi dopaminergici, anche quelli mesolimbici.
Sono dovute alla terapia a pungo termine con DOPA (ma qualcuno sostiene che possano essere anche altri farmaci)→ per questo motivo si tende al risparmio della DOPA: si cerca di darne il meno possibile e il più tardi possibile e si ricorre ad altri farmaci (ovviamente considerando l’età del paziente, se anziano non mi interessa). Mantenimento del ruolo sociale: se il paziente ha un lavoro che prevede una certa motricità, se non lo curo gli impedirei il mantenimento del ruolo sociale, quindi in questo caso anche se è giovane do la dopa.
DOPAMINO-AGONISTI Possono essere dati se non si vuole iniziare subito la terapia con la DOPA; non hanno tuttavia lo stesso effetto quindi magari si fa un’associazione con basse dosi di DOPA. Di diverse generazioni:
❖ BROMOCRIPTINA e alcaloidi dell’ergot, di vecchia generazione → effetto collaterale raro: fibrosi idiopatica retroperitoneale, talvolta polmonare e delle valvole cardiache. Sono molto gravi, quindi le linee guida li suggeriscono di II linea.
❖ PRAMIPEXOLO e ROPIRINOLO: più nuovi, che non condividono gli effetti collaterali dell’ergot. Si parte con dosi basse e si sale fino alla dose efficace con titolazione, altrimenti causano: nausea, ipotensioni, colpi di sonno. EFFETTI COLLATERALI: soprattutto durante dose piena o in associazione con atri farmaci: manifestazioni psicotiche, agitazione psicmotoria, allucinazioni notturne. Soprattutto i dopamino-agonisti sono stati imputati come scatenanti la tendenza al gioco d’azzardo: le dipendenze sono associate ai circuiti dopaminergici.
INIBITORI DI MAO-B I MAO-A non si usano praticamente più come antidepressivi. Tra i MAO-B si usano SELEGILINA e RASAGILINA, a rinforzo della DOPA o in associazione con altri farmaci. Si pensava che fossero in grado di cambiare il decorso della malattia per via dell’effetto neuroprotettivo: tuttavia questa teoria non è mai stata confermata, tuttavia si danno comunque. L’utilità dei due è sovrapponibile e l’effetto più importante è quello di aumentare la quantità di dopamina disponibile nell’encefalo in quanto ne riduce la degradazione. Esistono in formulazione di cerotto.
INIBITORI DI COMT ENTACAPONE agisce a livello periferico soprattutto, mentre il TALCAPONE ha azione anche a livello centrale; quest’ultimo era stato ritirato dal commercio perché imputato di causare gravi epatopatie necrotiche, ma poi è stato riabilitato. Stalevo: è un farmaco che contiene la combinazione di entacapone, levoDOPA e inibitori di carbossilasi.
ANTICOLINERGICI Sono nati prima della DOPA, erano delle preparazioni officinali a base di Belladonna. Si dice siano particolarmente utili sui tremori. Se il paziente è giovane con una sintomatologia molto pronunciata, si può associare anche questa categoria di farmaci. Effetti collaterali:
- Annebbiamento visus - Stipsi - Xerostomia (secchezza delle fauci) - Difficoltà minzionali (controindicati nella grande ipertrofia prostatica) - Aggravamento glaucoma - Confusione e turbe mnesiche→ accusa di peggiorare lo stato cognitivo, quindi sono stati declassati. - Riducendo la motilità gastrointestinale possono interferire con l’assorbimento della levodopa.
Spesso si associano ai neurolettici in pazienti psichiatrici per ridurre gli effetti anti-dopaminergici (anche questo non si fa più ora).
AMANTADINA È un anti-influenzale, che ha un ruolo anche nella gestione del Parkinson complicato:
- In pazienti con discinesie ha discreto successo

116
- Ha effetto nuche sul freezing: è controverso perché può allungare l’intervallo QT (come neurolettici tipici), quindi è difficoltoso il suo uso.
TERAPIA NEUROCHIRURGICA Tecniche stereotassiche. Non ci sono solo farmaci, ma anche dei risvolti di neurochirurgia funzionale: mira a correggere delle disfunzioni neurofisiologiche. Qualche decina di anni fa si era iniziato a coagulare un certo nucleo del talamo (TALAMOTOMIA) e portava a un ottimo controllo del tremore controlaterale→ stiamo parlando di un epoca dove la Levodopa non c’era, quindi era l’unico modo per controllare il tremore. Il limite di questo intervento era la sua transitorietà: dopo un certo periodo di tempo, il tremore ricompariva. Si sono quindi cercare altre strade, altri bersagli; i principali sono l’intervento stereotassico di microablazione del nucleo pallido o del nucleo subtalamico (PALLIDOTOMIA o SUBTALAMOTOMIA)→ viene posizionato un casco con delle coordinate, che consentono di dirigere dalla superfice verso la profondità una sonda o elettrodo, in un determinato punto. Una volta a guidare la stereotassi c’era un atlante con le coordinate, mentre ora ci si basa sul neuroimaging: sistema coordinato e integrato computerizzato per raggiungere precisamente il punto prescelto→ neuronavigazione.
Neurostimolazione. Si è visto che, prima di lesionare il punto prescelto, una sua stimolazione elettrica ad alta frequenza produceva un effetto analogo, cioè spegneva la sua attività. Dall’1987 questo meccanismo è stato usato sul corpo di Luys. Si posizionano le sonde e si verifica se si è raggiunto il punto registrando l’attività elettrica: infatti il nucleo subtalamico si comporta in modo particolare elettricamente (interpretato da un neurofisiologo che affianca il neurochirurgo). La sonda è allo stesso tempo registrante e stimolante, quindi una volta registrata l’attività elettrica e verificato che sia il punto giusto si procede stimolando. Sistema di neurostimolazione DBS (Deep Brain Stimulator):
- cavo metallico sottilissimo e flessibile - elettrodo di un numero di poli variabile da 1 a 4 i e attivabili separatamente come poli negativi o positivi - sistema di fissazione cranio e del collo - generatore di impulsi posizionato sottocute nella regione anteriore e superiore del torace, regolabile dall’ esterno
La DBS si usa anche per altri scopi, come la cefalea a grappolo. Ci sono delle limitazioni e effetti collaterali, per cui non si può usare su tutti i parkinsoniani:
• Sopra i 70 anni si riduce l’efficacia della stimolazione. • Emorragie intracerebrali di piccole-medie dimensioni • Rischio più frequente: malposizionamento dell'elettrodo stimolatore, con scarsi benefici terapeutici, che può
portare ad un reintervento. • Aggrava la depressione e sintomi cognitivi (demenza), quasi tutti i parkinsoniani sono depressi, quindi vuol dire
escludere una grossa fetta di pazienti.
TERAPIA DEI SINTOMI SECONDARI Depressione→ antidepressivi, come l’AMITRIPTILINA (Laroxil): viene usato nelle fasi iniziali della malattia di Parkinson perché è sia antidepressivo che anticolinergico. Allucinosi→ si curerebbero con neurolettici, che però aggraverebbero il Parkinson, quindi ci si affida a neurolettici di II generazione.
→ il pilastro della terapia è la DOPA: il problema è capire come gestirla, quando darne, e supplire alla sua inefficacia con altri farmaci, fino ad arrivare a volte a 4-5 farmaci.

117
NEUROPATIE Qualsiasi patologia del nervo periferico indipendentemente dalla causa. Si parla di neuropatie comprendendo qualsiasi malattia che coinvolge anche solo una delle strutture che formano il SNP.
Anatomia del nervo. I nervi sono strutture anatomiche del sistema nervoso
periferico formate da fascicoli di assoni (provenienti da un gruppo di neuroni) che trasportano informazioni da o verso il sistema nervoso centrale. Il nervo contiene, inoltre, vasi sanguigni utili al rifornimento di ossigeno e nutrienti. Nel nervo sono presenti guaine di tessuto connettivo che si fanno via via più piccole, ricoprendo prima l'intero nervo poi fasci e fascetti di assoni:
• Endonervio: all’intero dei fascicoli • Perinervio: delimita i fascicoli • Epinervio: circonda e racchiude tutto.
Le fibre nervose possono essere efferenti ossia motorie (quando trasmettono impulsi dal sistema nervoso centrale alla periferia) oppure afferenti ovvero sensitive (quando trasmettono gli stimoli sensoriali dagli organi periferici al sistema nervoso centrale). I nervi possono contenere fibre efferenti, afferenti o entrambe (in caso di nervi misti).
Struttura del SNP. • Nervi cranici (tranne I e II) • Radici spinali • Gangli dorsali • Plessi
o Cervicale (C1-C4) o Brachiale (C5-T1) o Lombosacrale (T12-S5)
• Nervi periferici
ITER DIAGNOSTICO GENERALE 1) Anamnesi 2) EO neurologico 3) ENG e EMG (estensione dell’EO)→ esame principe nelle neuropatie (non i potenziali evocati, perché qui ci interessa
solo la periferia!). a. Degenerazione assonale/demielinizzazione b. Sede c. Estensione d. Sofferenza subclinica
4) Esami biochimici: a. Ematochimici (routine e specifici)→ sempre dosare vitamine perché ci sono neuropatie da ipovitaminosi. Si
dosano anche alcuni anticorpi specifici (sia in sangue che in liquor). b. Esame liquor: nella sede di origine del nervo, cioè nell’impianto delle radici sul midollo→ un danno di un nervo
periferico causerà alterazioni anche nel liquor a livello centrale. 5) Biopsia di nervo (nervo surale): solo in centri specializzati, richiede una particolare competenza.
a. Neuropatia multifocale asimmetrica b. (vasculite, sarcoidosi, amiloidosi, lebbra)
6) Biopsia di cute: a volte si possono dimostrare alterazioni peculiari delle piccole fibre nervose→ non esiste infatti un esame neurofisiologico per studiare le fibre nervose più piccole, quindi ci si affida alla biopsia.
Esame principe nelle neuropatie: EMG e ENG. Esame principe nella SM: PE.
CLASSIFICAZIONE TOPOGRAFICA Si basa sul distretto interessato: Radicolopatia→ causato più comunemente è una compressione per via di un trauma. Ganglionopatia→ causato da Herpes Zoster.

118
Plessopatie. Neuropatie: patologie dei tronchi nervosi, che si distinguono in: - MONONEUROPATIE (singolo tronco nervoso), ad eziologia soprattutto meccanica. Es. Sindrome del Tunnel carpale. - MULTINEVRITI: sono solitamente multifocali, più nervi non contigui e asimmetrici. Molto spesso associate a vasculiti
del SN: LES, AR, PAN. - POLINEUROPATIE: coinvolge la maggior parte dei nervi in modo bilaterale→ deficit bilaterale, simmetrico, sincrono. - POLIRADICOLONEUROPATIE
GANGLIONITE DA HERPES ZOSTER Infiammazione isolata che segue il decorso di un dermatomo o di una branca del trigemino (o più di una); ha una manifestazione papulare. Il problema, oltre al dolore in fase acuta che risente favorevolmente di terapie, può ripresentarsi a distanza con una SINDROME DOLOROSA di tipo NEVRALGICO: è un aggettivo che si usa con parsimonia e indica fenomeni ben precisi→ dolore che compare a crisi/accessi di durata variabile, da pochi secondi a qualche minuto, con un dolore intensissimo simile a scossa elettrica o urente simile a una ustione, a volte scatenato da particolari manovre come la compressione di punti di emergenza dei nervi dai loro forami. →Nevralgia post-Herpetica è una condizione molto invalidante; l’antivirale ormai non ha più nessun effetto e servono altri farmaci.
Farmaci di prima scelta in una nevralgia o dolore neuropatico: antiepilettici (bloccano scariche!)→ CARBAMAZEPINA e PREGABALIN (Lyrica), lamotrigina, valproato, difenilidantoina. (DOMANDA D’ESAME FREQUENTE).
MONONEUROPATIE
Sindrome del tunnel carpale. La storia clinica inizia con parestesie, fastidio, dolore prevalentemente notturni, riferiti alla mano o in risalita verso avambraccio e braccio. Co più precisione, si può individuare che le più colpite sono le prime 3 dita e metà del 4 dito. A lungo andare, queste parestesie e fastidi diventano importanti e causano numerosi risvegli nella notte. Col tempo, i muscoli dell’eminenza tenar si ipertrofizzano→ segno dello scalino: segno di depressione sul lato esterno dell’eminenza tenar. Il paziente riferisce l’incapacità di svolgere movimenti fini e una ipoestesia. CAUSE SCATENANTI: di tutto e di più: professionale, tenosinovite, tumefazione dei tessuti molli (ipotiroidismo, acromegalia, amiloidosi, gravidanza), diabete. DIAGNOSI: con ENG (studio di velocità sensitiva di nervi digitali con elettrodi ad anello). TERAPIE: - Infiltrazioni con corticosteroidi - Intervento chirurgico che crea spazio al nervo nel canale trasverso del carpo. Le pareti del canale carpale aumentano di volume perché si sclerotizzano e il nervo viene compresso.
Sindrome di intrappolamento dell’ulnare al gomito. Le pareti del canale cubitale si ispessiscono e il nervo ulnare al suo interno ne risulta compresso. CAUSE: - Aver subito una frattura del gomito precedente con immobilizzazione. - Soggetti che si servono di stampelle per deambulare, che preme proprio sul gomito. CLINICA: causerà deficit di sensibilità delle ultime due dita e di tutta la superficie dorsale e laterale dell’avambraccio. Se si chiede al paziente di tenere un foglio tra primo e secondo dito, si verifica il segno di Froment: invece che tenere entrambe le dita estese e tenere il foglio solo con la forza dell’adduttore del pollice, compensa con la flessione della falange del pollice.
Lesione del nervo peroneo comune. Il nervo peroneo comune si stacca dallo sciatico e arriva al poplite; qui si divide nei suoi rami terminali, di cui uno è il tibiale anteriore: si sposa dal poplite posteriormente alla superficie anteriore della gamba, girando intorno alla testa del perone→ qualsiasi trauma che coinvolge la regione peronea può dare un disturbo del peroneo comune con parestesie formicolanti e debolezza della dorsi flessione del piede, con conseguente andatura steppante (steppage). Può succedere in soggetti che rimangono troppo a lungo con le gambe accavallate e per una loro eccessiva suscettibilità possono presentare un danno più o meno perdurante al peroneo.

119
Paralisi periferica del nervo facciale. • Angolo della bocca è ptosico.
• Solco naso-labiale appiattito.
• Deficit di muscoli periorbitari, con ptosi della rima inferiore→ possibile lagoftalmo perché non avviene il corretto drenaggio delle lacrime (per il corretto drenaggio serve che occhio e palpebra inferiore siano aderenti). Questo può portare a una infezione congiuntivale per via del fatto che la congiuntiva rimane esposta all’ambiente esterno. Segno di Bell: all’ordine di chiudere gli occhi, entrambi i bulbi oculari si rovesciano verso indietro, uno dei due rimane aperto e si vede il bianco.
• NERVO INTERMEDIO: è un nervo sensitivo di pertinenza del trigemino, che viaggia attaccato al facciale, quindi è lesionato sempre insieme al facciale. È responsabile della sensibilità dei 2/3 inferiori della lingua→ quindi la paralisi di Bell è caratterizzata anche dalla mancanza di sensibilità linguale.
• Si accompagna anche alla distorsione dei suoni (cacofonia): il facciale infatti va ad innervare il muscolo stapedio: è responsabile della corretta trasduzione meccanica dei suoni.
EZIOLOGIA: È causata da una irritazione del nervo periferico. Il complesso faciale intermediario scorre in un canalicolo osseo: in caso di infiammazione, si ha edema, che causa aumento del volume del nervo in un canale osseo inespansibile, quindi viene compresso. Se il sito di infiammazione è il segmento iniziale del nervo (cioè dalla faccia ventrale del ponte), si avrà paralisi competa: - Paralisi di muscoli mimici - Cacofonia - Ipogeusia dei 2/3 inferiori Se il segmento interessato è più distale (es. dopo l’origine dello stapedio), non si avranno sintomi acustici. Se il segmento interessato è ancora più distale, dopo il distacco dell’intermediario, non si avranno sintomi linguali. DD con una forma di Herpes zoster che colpisce il ganglio genicolato: fornisce il contingente sensitivo trigeminale gustativo. Una infiammazione del nervo per causa erpetica darà una paralisi del faciale non più idiopatica, ma ci aspettiamo anche un’eruzione erpetica nel meato acustico esterno. In questo caso quindi un ORL individua la presenza di papule erpetiche, e se presenti la terapia è a base di aciclovir, con supplementi vitaminici. Si fanno anche esami del sangue: glicemia (il diabete è predisponente). La guarigione può non essere completa, può rimanere un emispasmo facciale dal lato ex-paretico: sono delle più o meno continui spasmi (ammiccamento o stiramento della rima orale, simili a mioclonie). In questo caso si induce una paralisi iatrogena con tossina botulinica. Differenza con la paralisi centrale facciale: la paralisi periferica coinvolge sia il ramo superiore che inferiore, mentre la paralisi centrale coinvolge solo il territorio del ramo inferiore (bocca). C’è la conservazione della motricità del facciale superiore legata al fatto che la regione superiore è servita da entrambi gli emisferi, quindi riceve anche fibre controlaterali.
MULTINEVRITI Interessamento multifocale, simultaneo o successivo, di più nervi non contigui (DD con polineuropatie). Distinguiamo in forme: - Assonali: spesso si ritrovano nel contesto di malattie sistemiche come le vasculiti, o diabete, infezioni, …
Es: paziente in degenza in urologia per orchiectomia (edema e dolore testicolare, dovuto a infarto epididimo-testicolare), ha iniziato a sviluppare uno steppage→ si associavano anche segni di sofferenza di altri nervi periferici. Si trattava di una PAN (che infatti dà infarti) associata a multinevriti.
- Demielinizzanti: causate soprattutto da malattie immuno-mediate che colpiscono selettivamente la mielina.

120
POLINEUROPATIE Compromissione bilaterale, simmetrica e sincrona dei nervi. Tantissime cause: - Genetica: molte forme sono geneticamente determinate. - Tossico-metaboliche - Infettive - Carenziali Si contraddistingue per una prima fase con sintomi sensitivi positivi (parestesie formicolanti o dolorose o urenti e deficit motori) e solo in una seconda fase subentra una fase deficitaria:
• Deficit sensitivo con tipica distribuzione a «calza» e a «guanto»
• Andamento disto-prossimale
• Riduzione fino a scomparsa di ROT→ DD con un danno centrale (SLA).
• Ipotrofia muscolare secondaria Ricordiamo che la maggior parte dei nervi sono misti, con componente sia sensitiva che motoria. A eccezione di:
- Nervi motori puri: ipoglosso, accessorio spinale. - Nervi sensitivi puri: nervo surale.
→ quindi bisogna sempre pensare a deficit sensitivi, motori e autonomici. Dal punto di vista temporale si distinguono in: acute, subacute o croniche. Si possono distinguere anche in base alla componente nervosa interessata:
➢ POLINEUROPATIE MIELINICHE: è colpita prevalentemente la parte mielinica, acquisite (solitamente sono immuno-mediate) o ereditarie.
➢ POLINEUROPATIE ASSONALI: sono molto più frequenti. Sono lunghezza-dipendenti: la patologia si manifesta sempre più in senso distale, infatti le patologie sono legate a problemi di trasporto i materiali lungo gli assoni. Es: polineuropatie da farmaci anti-neoplastici. Sono farmaci che agiscono sul sistema tubulare.
Se coinvolgono anche le radici si parla di POLIRADICOLONEUROPATIA.
POLINEUROPATIE ACUTE 1. POLIRADICOLONEUROPATIA INFIAMMATORIA ACUTA: è assolutamente da conoscere da chiunque.
a. Sindrome di Guillain Barré: è un quadro acuto di poliradicoloneuropatia infiammatoria mielinica acquisita. b. Sindrome di Miller Fisher
2. PORFIRIE ACUTE 3. NEUROPATIE TOSSINFETTIVE (botulismo, difterite) 4. NEUROPATIE PARANEOPLASTICHE
SINDROME DI GUILLEN-BARRÈ Chiamata anche PARALISI ASCENDENTE PROGRESSIVA.
Quadro clinico. • Esordio con parestesie o deficit di forza agli arti inferiori • Estensione verso l’alto di parestesie e deficit di forza, fino a coinvolgere tronco, arti superiori e a volta anche il
distretto cranico. • Infezione gastro-intestinale in anamnesi qualche settimane prima: è dovuta al Campylobacter, cioè la diarrea del
viaggiatore. Solo in una bassa percentuale di casi, ci può essere una cross-reazione tra epitopi batterici ed epitopi di guaina mielinica per mimetismo molecolare. Anche altre infezioni (anche virali) possono causare questa complicanza (CMV, EBV, Mycoplasmi). Anche una vaccinazione.

121
EO neurologico. ➢ Disturbi motori:
o Ipostenia dei nervi periferici interessati, con magari steppage se interessa il peroniero. o ROT indeboliti o assenti. o Paralisi facciale bilaterale (diplegia facciale): è una condizione molto rara, causata da poche malattie. o Plegia di nervi oculomotori. o Paralisi di muscolatura respiratoria→ nelle forme più gravi, costituisce una minaccia alla vita, quindi non può
essere tenuto in un ospedale senza rianimazione! ➢ Turbe di sensibilità modeste→ il disturbo di sensibilità è soggettivo, si sentono parestesie ma non ci sono dei veri deficit
obiettivabili. ➢ Segni di disfunzione autonomica: aritmie cardiache, ipotensione. ➢ Dolore
→ è una PATOLOGIA PREVALENTEMENTE MOTORIA!!
→ esiste un tasso di mortalità non indifferente.
Diagnosi. è clinica. Si possono fase altri esami:
- ENG: si positivizza dopo alcuni giorni, e questo è dovuto al fatto che è una poli-radicolo-neuropatia: nelle fasi iniziali infatti il danno è molto prossimale e investe soprattutto le radici. Bisogna quindi attende alcuni giorni per poter dimostrare il danno neuropatico.
- ESAME DEL LIQUOR: anche questo esame ci mette fino a una settimana per positivizzarsi e mostrare un aumento della proteinorrachia (normale: 45 mg/dL) senza un aumento della cellularità (es. nelle meningiti batteriche aumenta la proteinorrachia ma anche la cellularità)→DISSOCIAZIONE ALBUMINO-CITOLOGICA.
Varianti. In realtà quando si parla di Guillen Barrè si intendono una serie di molte varianti. In alcune forme, il danno non
è più mielinico ma assonale (non sono da sapere tutti i vari nomi). Da ricordare: sindrome di Miller-Fisher: variante di Guillen-Barrè caratterizzata da un inevitabile paresi dei muscoli oculomotori→ oftalmoparesi unita a una atassia predominante.
Sono stati individuati degli autoanticorpi circolanti IgG, diretti contro i gangliosidi (lipidi di membrana particolari con componente glucidica); si ritrovano solo nelle varianti: a una determinata variante corrisponde la positività verso un determinato sotto tipo di IgG mentre nella forma tipica non sono stati identificati anticorpi specifici!
Trattamento. a seconda della gravità si può avere da un paziente con solo formicolio ai piedi a uno prossimo alla
rianimazione. 1) Assistenza: sarà calibrata sul grado di evoluzione, ma in linea generale ci deve sempre essere lì vicino una
rianimazione. 2) Ig ENDOVENA: trattamento di base nelle polineuropatie acute. 3) PLASMAFERESI: non è indicata in tutti i casi, è più invasiva, non sempre è ben tollerata (fenomeni vasomotori). 4) STEROIDE: un volta usato per tutte le forme acute, ora invece non più. È indicata soprattutto nelle forme associate
a dolore. Altri trattamenti collaterali:
• Per prevenzione TVP: calze compressive (non più così indicate ultimamente) ed eparina a basso peso molecolare.

122
• Ventilazione meccanica se necessaria.
NEUROPATIE TOSSINFETTIVE
Neuropatia da botulismo. Causano una polineuropatia con alla base un difetto neurotrasmettitoriale: blocco del rilascio di acetilcolina a livello del terminazioni nervose nella giunzione neuromuscolare nei muscoli striati e sistema nervoso autonomo (blocca fusione delle vescicole). Può sembrare un paziente curarizzato, è interessato anche il sistema autonomico. Clinica: Sintomi neurologici compaiono dopo qualche ora o giorno dall’ingestione: paralisi flaccida, blocco oculomozione, midriasi areattiva, disturbi fonazione e deglutizione, deficit muscoli estensori del collo e della radice degli arti, paralisi vescicale ed intestinale. Intensa secchezza delle fauci.
Neuropatia difterica. Tossina difterica con azione selettiva sulle cellule di Schwann. Clinica (dopo tre settimane): paralisi del velo palatino e dell’accomodazione, poi coinvolgimento degli arti, atassia, instabilità posturale e areflessia.
NEUROPATIE PARANEOPLASTICHE Possono interessare anche il SNC. Questi effetti paraneoplastici sono immuno-mediati. Si verifica una cross-reazione tra antigeni tumorali e antigeni nervosi: sono chiamati AUTOANTICORPI ONCO-NEURALI (anti-Hu o anti-ANA1) rivolti contro antigeni all’interno di cellule neurali. Neuropatia sensitiva di Denny Brown: ha una particolare trofia per i cordoni posteriori, quindi sarà soprattutto sensitiva: disestesie, parestesie, dolori, atassia sensitiva→ per danno di fibre propriocettive (sono le fibre mieliniche più grandi). (Domanda esame frequente!)
POLINEUROPATIE CRONICHE • Poliradicoloneuropatia Infiammatoria Demielinizzante Cronica (CIDP), • Neuropatie carenziali • Neuropatia diabetica • Neuropatie tossiche • Neuropatie amiloidi • Neuropatie da malattia ematologica
CIDP Analogo cronico della sindrome di Guillen Barrè, è una forma immuno-mediata che si esprime con un andamento temporale diverso. Eziologia. Solitamente è idiopatica, ma esistono casi in cui è associata a una malattia sistemica come HIV, linfoma, melanoma, MGUS (gammopatia monoclonale).
Decorso. - Progressivo può esserci il caso i cui un paziente non guarisce da Guillen Barrè ed evolve in forma cronica. Può essere
una evoluzione lentamente progressiva o a gradini. - Recidivante-remittente, con ricadute e miglioramenti. La connotazione mielinica è più importante.
Terapia. • Gli steroidi funzionano e sono indicati come primo approccio, a differenza della GB→ gold standard: prednisone 1
mg/kg al giorno. • Le Ig endovena devono essere riservate a eventuali fasi di acuzie; alcuni pazienti fanno Ig a cicli ripetuti per molto
tempo. Questo ha dei costi non indifferenti.
NEUROPATIE ASSONALI Sono per lo più croniche e spesso vengono chieste all’esame.

123
Neuropatia alcolica. I meccanismi di danno sono vari: ricordiamo ipovitaminosi B1. Clinica:
- Affaticamento nella deambulazione, crampi notturni. - Disestesie a calza, atassia sensitiva. - Steppage bilaterali - ROT achillei assenti.
Neuropatia diabetica. Più del 50% dei diabetici hanno varie forme di neuropatie. Non tutte hanno uguale importanza.
❖ Le più frequenti sono le polineuropatie simmetriche: sensitiva acuta (esacerbata da crisi iperglicemiche), sensitivo-motoria cronica (la più frequente, si manifestano con parestesie a gambe e piedi, crampi, ipostenia e ipotrofia muscolare, atassia sensitiva), autonomica. Mononeuropatia craniale, del III paio di nervi cranici→ coinvolgimento selettivo della muscolatura oculare estrinseca con risparmio di quella intrinseca. Clinicamente si manifesta con ptosi palpebrale, impossibilità ad aprire l’occhio, strabismo divergente ma senza miosi→ oftalmoplegia. Entra in DD con aneurisma del sifone carotideo nel seno cavernoso: causa una condizione analoga ma in modo iperacuto, può causare compressione del III nervo, accompagnata da dolore e da un soffio sistolico al fonendoscopio poggiato sul globo oculare.
❖ Neuropatie asimmetriche focali e multifocali: o Tronculari o Da intrappolamento o Troacolombare o Amiotrofia diabetica di Garland: ipostenia e ipotrofia agli arti inferiori prossimali.
Eziologia: sembra avere sia una causa vascolare (anterazioni endoteliali dei vasa nervorum) sia una causa metabolica (l’eccesso di glucosio induce a livello dei nervi una reazione che porta alla produzione di ROS). Terapia: FANS, antiepilettici, antidepressivi.
NEUROPATIE EREDITARIE Hanno un meccanismo patogenetico diverso. Non dobbiamo entrare troppo nel dettaglio dei geni. Il danno può essere di tipo demielinizzante, assonale o misto. Distinguiamo in forme: motorie pure, sensitive pure, sensitivo-motorie.
Forme sensitivo-motorie. Tra queste troviamo la NEUROPATIA DI CHARCOT-MARIE-TOOTH, chiamata anche atrofia
muscolare peroneale (HSMN). Clinica:
- Ipostenia distale, ipo-atrofia peroneale - Deficit sensibilità superficiale e profonda (ipopallestesia, atassia) - ROT ridotti/assenti - Ipotrofia inizialmente agli arti inferiori, poi mani e avambracci
Fasi precoci: Ipotrofia ed ipostenia dei muscoli loggia anteriore gamba e i muscoli intrinseci del piede:
- Steppage progressivo - Deformità scheletriche del piede (piede cavo, dita a martello) - Amiotrofia (gambe con aspetto di bottiglia rovesciata)
Una forma (neuropatia tomaculare) comporta una patologica suscettibilità alla compressione, condivide lo stesso substrato della CIMP: dopo aver tenuto le gambe accavallate o aver premuto contro il lettino della sala chirurgica, può manifestare uno steppage→ una pressione di poco conto dà effetti gravi visibili. In questo caso bisogna studiare il paziente per una neuropatia ereditaria.

124
PATOLOGIE MIDOLLO SPINALE
Abbiamo già trattato le lesioni di origine traumatica. Cause non traumatiche più frequenti:
• Neoplasie • Vascolari • Infiammatorie • Degenerative • Altre
È possibile distinguerle in: ❖ MIELOPATIE NON COMPRESSIVE:
o Mielite acuta o Localizzazione midollare di SM
❖ MIELOPATIE COMPRESSIVE: o Neoplasie o Mielopatie spondilogena
Quali sono le conseguenze cliniche di una lesione del midollo? Dipende dal livello della leisone e dalla porzione di sezione interessata. Per una lesione completa a livello D2 avremo: paraplegia arti inferiori, anestesia, turbe sfinteriche e autonomiche.
ANATOMIA DEL MIDOLLO Caratteristiche:
• È più breve rispetto al rachide: finisce tra L1-L2, dopo di che c’è il cono. • Ha due rigonfiamenti, uno cervicale e uno lombare. Questo perché ci
sono più neuroni motori destinati agli arti. • Discrepanza tra numero di vertebre cervicali e segmenti midollari
cervicali (e relative radici): 7 vertebre cervicali con 8 segmenti e radici. La prima esce tra atlante e osso occipitale. Tra C2 e C3 esce C3, mentre tra L1 e L2 esce L1!
Sezione trasversale. Vedo sostanza bianca con all’interno sostanza grigia
formata tra corno anteriore, posteriore e a volte laterale. In mezzo c’è la commissura che unisce la parte destra a sinistra e al centro c’è il canalicolo dentro cui scorre liquor. Dove ci sono i rigonfiamenti, le dimensioni della sostanza grigia sono maggiori: c’è un impacchettamento di neuroni. DERMATOMERI (o dermatomi): non è vero che gli ultimi dermatomi corrispondano al piede, infatti i dermatomi più caudali servono la zona perineale→ questo per eredità filogenetica, da quando eravamo quadrupedi. Gli ultimi segmenti quindi riguardano perineo, vescica e organi genitali. Organizzazione somato-topica di sostanza grigia: nel corno anteriore, la rappresentazione dei muscoli è fatta in modo che muscoli distali vengano rappresentati nel bordo anteriore, mentre i muscoli più prossimali vengano rappresentati nel bordo più posteriore. Esistono delle tabelle che indicano con precisione per ogni singolo muscolo quale innervazione riceve (da che segmento). Muscolo deltoide: innervato dal C5-C6, nervo circonflesso o ascellare. Cordone laterale:
- Discendente: fasci piramidali o cortico-spinali. Non tutto il fascio corticospinale è crociato e decorre interamente nel cordone laterale: in realtà una parte non crociata decorre nel cordone anteriore e un’altra componente non crociata decorre nel cordone laterale→ non c’è solo una componente del fascio cortico-spinale!
- Ascendente: spino-talamico. Cordone posteriore: sensibilità tattile protopatica. L’organizzazione somatotopica c’è anche nei fasci ascendenti e discendenti: le fibre che arrivano dall’arto inferiore si trovano a livello del bordo esterno del midollo.

125
→ In generale, possiamo dire che:
- Se la lesione coinvolge sostanza grigia, avremo una SINDROME SEGMENTARIA: es. plegia di tutti i muscoli di C5-C6 e anestesia completa nel dermatomo C5-C6→ non di tutto il midollo sottostante. Si manifesta come paralisi flaccida, amiotrofia, fascicolazioni, assenza di ROT.
- Se la lesione coinvolge i cordoni, avremo delle SINDROMI CORDONALI, quindi anche effetti a distanza dalla lesione stessa.
In realtà, quasi sempre si tratta di un mix tra queste due condizioni, a meno che non si tratti di una malattia neurodegenerativa che colpisca selettivamente solo una di queste due componenti (es. SLA). → Una lesione di midollo spinale determinerà disturbi motori sia segmentali che cordonali; bisogna quindi vedere quale delle due cose prevale e cosa si apprezza. Nella SLA spinale, ci sarà coesistenza di questi due elementi: sindrome da I motoneurone, ipotrofia muscolare, fascicolazioni., … → anche per i disturbi sensitivi si avrà una ipo-anestesia di solo un dermatomo o di tutti i livelli sottostanti a seconda se il danno è segmentale o cordonale.
Livello sensitivo. La linea di confine tra la zona integra di sensibilità e la zona patologica si definisce LIVELLO SENSITIVO; sono disposti in senso rostro-caudali (e non prossimali o distali!): una volta individuato dove si trova il livello sensitivo (es. linea ascellare media), tramite delle mappe si risale al segmento midollare interessato. Eccezione al concetto di livello: una compressione estrinseca su C5-C6 (che viene da fuori) si fa sentire sui cordoni e non su sostanza grigia; questa determina dei disturbi di sensibilità molto distali→ infatti le fibre destinate ai segmenti più distali e all’arto inferiore sono disposte esternamente nei cordoni. Es. una persona con mielopatia spondilogena che comprime dal di fuori su C6, si presenta con formicolio ai piedi: è infatti una compressione incompleta che dall’esterno coinvolge le fibre verso i distretti più distali→ danno cordonale.
MIELOPATIA CERVICALE SPONDILOGENA La sciatalgia all’arto inferiore è paragonabile alla brachialgia all’arto superiore: dovuta alla compressione del midollo da parte di ernie discali o vertebre deformate dal processo osteoartrosico. RM: le discopatie appaiono come dei salsicciotti che comprimono. In T2 non si vede più il bianco del liquor tra midollo e vertebre.
Clinica. Inizia a carico di arti inferiori, in sede distale:
- Turbe di sensibilità - Turbe di deambulazione, aggravate da ipostenia distale→ andatura para-pareto-spastica o para-pareto-atassica.
Anche la sostanza grigia soffre in questo caso, quindi avremo anche ipotrofia dei muscoli degli arti superiori, fascicolazioni, perdita dei riflessi relativi al segmento, mentre ci aspettiamo che al di sotto di questo livello i riflessi son aumentati. → riflessi ridotti a livello di arti superiori e aumentati a livello di arti inferiori. Se si escludesse la parte sensiva (se non ci fossero segnali di ipoestesia o anestesia) ma solo segni motori, potrebbe essere confusa con una SLA! Quindi bisogna analizzare bene la componente sensitiva anche se sembra normale. Segni autonomici in una lesione midollare:
• turbe del controllo sfinterico→ prima ritenzione poi incontinenza.
• ipotensione ortostatica
• sindrome di Claude-Bernard-Horner→ per quest’ultima serve una lesione più rostrale, circa C8-T1.
Lesione midollare acuta, il disturbo sfinterico sarà di incontinenza o di ritenzione? In fase acuta c’è RITENZIONE.
Distinguiamo le sindromi midollari in: sindromi da sezione completa trasversa e da sezione incompleta.
SEZIONE TRASVERSA COMPLETA Può essere causata da malattie come:
- Infarti midollari - Tumori intramidollari

126
- Ematomielia: è l’equivalente di emorragia intracerebrale interessa tutta la sostanza grigia. MAV= malformazione artero-venosa.
- Mielite acuta: processo infamatorio acuto che può avere numerose eziologia.
Quadro clinico. ➢ A seconda del livello avremo paraplegia o tetraplegia a seconda del livello della lesione (se sopra o sotto il
rigonfiamento cervicale)→In acuto, spesso la para o tetraplegia sono FLACCIDE: ci vuole un po’ di tempo prima che si verifichi una iperreflessia, perché prima si ha la fase di shock spinale.
➢ Anestesia per tutte le forme ➢ Turbe autonomiche.
La gravità ovviamente dipende dal livello: se una lesione trasversa completa è a livello cervicale alto, è il caso peggiore perché qui c’è anche la sede del nervo frenico (C4)→ paralisi del diaframma con blocco respiratorio. Più si sale di livello, più si aggiungono sintomi clinici. Man mano che si sale, si aggiungono nuovi fenomeni clinici:
- Lesione di segmenti sacrali (cono): anestesia perineale a sella, turbe genitourinarie. Il cono midollare è al confine tra L1-L2.
- Segmenti con nuclei motori dei muscoli degli arti inferiori: paralisi di muscoli posteriori della coscia (lesioni più basse) e anteriori della coscia (lesioni più alte).
- Lesione di rigonfiamento cervicale: tetraplegia.
MIELITE ACUTA TRASVERSA Mielite è una delle cause più frequenti di lesione midollare trasversa completa. È un termine generico che indica una infiammazione acuta che può essere di origine:
- Infettiva - Immuno-mediata: il LES può dare una mielite longitudinale, cioè estesa per molti segmenti.
Il segmento preferito da questi processi è quello toracico o cervico-toracico.
Indagini strumentali. Se si sospetta una mielite, si impone l’esame del liquor (proteine, cellule, PMN); si cercano
anche le bande oligoclonali e l’indice K. RM: da C6 in giù si vede un “biancore” che non dovrebbe esserci; oppure può essere un danno più focale. → una volta individuato che si tratta di una mielite, si va a ricercare la causa con indagini più specifiche. La diangosi di mielite è una diagnosi importante, non sono mai patologie benigne e poco preoccupanti.
Neuromielite ottica. C’è un processo demielinizzante acuto con una preferenza per midollo spinale e nervo ottico. La
placca della SM è limitata longitudinalmente a meno di 3 segmenti, mentre in caso di neuromielite servono lesione di più di 3 segmenti→ per poter parlare di neuromielite servono lesioni più estese della SM. La SM non è una mielite, mentre la neuromielite ottica viene chiamata così nonostante sia parente di SM perché coinvolge più di 2-3 segmenti.
TUMORI INTRAMIDOLLARI Un tumore intramidollare (es. meningioma) si può presentare con una sindrome trasversa completa, anche se non è frequente; causa più frequentemente compressione con lesione incompleta. Il quadro non è improvviso come quello di una mielite perché il tumore si accresce con tempi più lunghi, ma a volte succede. I segni son sempre uguali, a meno che non predomini anche il dolore. Alla RM è evidente la presenza id un meningioma ad esempio, non serve fare anche ricerca id bande policlonali per fare DD con mielite.
SINDROME DELLA CAUSA EQUINA Non è propriamente una sindrome midollare, ma è più radicolare. Si intende una lesione da L2 a S5. Ha numerose cause (es. canale lombosacrale stretto). È una presentazione cinica dove sono interessate molte radici, perché decorrono tutte in modo parallelo e adiacenti: - Disturbi genito-urinari e intestinali. - Anestesia a sella - Paralisi distale degli arti inferiori, di solito asimmetrica→ il processo patologico infatti si sviluppa prima da una parte
poi pian piano interesserà in ugual modo entrambi i lati.

127
Sono più colpiti i muscoli distali, infatti la maggior parte delle radici che formano la cauda si riferiscono a muscoli distali. Si distingue dalla sindrome del cono midollare (interessamento S3-S5) perché quest’ultima ha un maggior interessamento sfinterico e sintomi simmetrici..
SINDROME DA COMPRESSIONE MIDOLLARE O sindrome del canale stretto (o stenosi midollare): stenosi del canale lombo-sacrale che da segni e sintomi: - Quadri di sciatica da compressione di una singola o alcune radici. - Claudicatio midollare: l’esercizio esalta dolori preesistenti agli arti inferiori, crampi, contratture, ipostenia e anestesia,
parestesie→ viene riferito come gambe dolenti dopo qualche centinaio di metri di cammino; in presenza di un ECO-doppler normale (che esclude claudicatio vascolare), si pensa alla claudicatio midollare.
→ sono patologie molto comuni, sono tutte parenti.
SEZIONE TRASVERSA INCOMPLETA SINDROME DA EMISEZIONE MIDOLLARE È lesionato il cordone laterale, quindi si avrà: - Paralisi piramidale omolaterale. - Anestesia propriocettiva-vibratoria profonda omoletalerale (perché si incrocia più in alto). - Anestesia termico-dolorifica controlaterale perché si incrocia nel midollo.
SINDROME CORDONALE ANTERIORE Detta anche sindrome dell’arteria spinale anteriore. - Paralisi piramidale bilaterale - Anestesia termico-dolorifica bilaterale - Risparmiata la sensibilità profonda, perché i cordoni posteriori non sono coinvolti. La causa è infarto del midollo nel territorio dell’ARTERIA SPINALE ANTERIORE: lungo tutto il decorso del midollo si trovano una arteria spinale anteriore e due spinali posteriori. La gran parte del midollo (più di 2/3) è di pertinenza dell’arteria anteriore. Sono serviti da rami arteriosi che derivano dall’aorta con una organizzazione segmentale: dove ci sono le coste, i rami che riforniscono i rami spinali provengono dalle arterie intercostali, a livello cervicale il discorso è più complicato perché deriva da arterei cervicali ma anche da altri→ non è da sapere, ricordarsi solo che deriva quasi tutta da aorta o da grossi vasi del collo. → questo spiega come mai spesso insorge questo quadro in seguito a un grande aneurisma dell’aorta: a volte il paziente può presentarsi con paraparesi, perdita del controllo sfinterico, assenza di polsi arti interiori, dolore addominale, …→ manovre sull’aorta hanno caudato una diminuzione de flusso dei rami che riforniscono il circolo spinale. Spesso un quadro improvviso di paraparesi e discontrollo sfinterico può suggerire un aneurisma dissecante, che ha privato il circolo spinale dell’apporto di sangue e ha determinato un infarto midollare. Un’altra causa può essere la poliomielite.
INFARTO MIDOLLARE Si accompagna quasi sempre al DOLORE, come in molte sindromi midollari acute. La presentazione è sempre la stessa: paraperesi, debiti sensibilità, ….
SINDROME CORDONALE POSTERIORE Causa atassia sensitiva. Cause:
• Lue o tabe dorsale

128
• Malattie dei cassoni • Avitaminosi B12: soggetti con problemi gastrointestinali (gastrici croniche), gastroresecati, dieta vegana. Causa
una SCLEROSI COMBINATA: non degenerano solo quelli posteriori ma anche quelli laterali; soggetti con una vera ipovitaminosi non avranno quindi solo sintomi sensitivi ma anche piramidali.
SIRINGOMIELIA O sindrome centro-midollare. È un lesione nel mezzo del midollo, un allargamento del canale centrale. È la zona dove decussano le fibre della sensibilità termico-dolorifica. Determina la presenza di:
• Banda dove la sensibilità termico-dolorifica è diminuita o assente: è definito deficit a mantellina sulle spalle che si espande ad arti superiori.
• Sensibilità profonda è indenne. • Se la lesione si espande ancora interessa tutta sostanza grigia quindi può dare problemi di ipostenia e amiotrofia
degli arti superiori. Il distretto preferito è il passaggio cervico-toracico, quindi le mani. RM: appare un bianco intenso all’interno del midollo in T2, che è il liquor. Molto spesso si associata alla MALFORMAZIONE DI ARNOLD-CHIARI: è la discesa delle strutture dell’encefalo verso il canale spinale: le tonsille cerebellari erniano oltre il forame occipitale. Ci sono molti gradi diversi, molti dei quali sono assolutamente benigni; solo le varianti più gravi, con una importante discesa lungo il canale spinale, sono patologiche e si associano a siringomielia.
TERAPIA Una sindrome midollare acuta va saputa sospettare da chiunque; se è acuta bisogna pensare ci sia una causa traumatica, un ascesso, un tumore, un ematomielia, cioè tutte condizioni che richiedono un correzione chirurgica. Il problema è che si aspetta tropo tempo, non sarà più necessario intervento chirurgico perché il midollo è estremamente snesibile alla compressione e va in necrosi rapidamente. In una sindrome midollare acuta siamo tenuti a insistere per avere una DIAGNOSI PRECOCE: → RM entro 4 ore!! → Se non c’è una compressione alla RM, si fa puntura lombare e si cerca processo infiammatorio. → Se non ci sono segni di infiammazione, si pensa all’ischemia.

129
DISTURBI DEL MOVIMENTO
Esistono diverse tipologia di movimenti alterati; non tutti riguardano la neurologia, come ad esempio l‘encefalopatia epatica: a causa di un problema epatico, si osserva un tipo particolare di mioclono che viene definito flapping tremor ma non è un tremore. Ricordarsi che due cose fondamentali in neurologia sono anamnesi ed esame obiettivo: la maggior parte dei disturbi del movimento si diagnosticano con la clinica, magari poi la neuroimaging serve per distinguer le cause. Distinguiamo in: ➢ SINDROMI IPOCINETICHE: tra cui sindromi parkinsoniane→ movimento meno ampio, difficoltoso, impacciato. ➢ SINDROMI IPERCINETICHE: ci sono movimenti in più, che possono essere ipercinesi più ritmiche e regolari o irregolari,
imprevedibili e variabili. ➢ SINDROMI ATASSICHE: possono essere considerate ipocinetiche per certi versi, ma in realtà spesso si trovano tremore
(soprattutto interzonale) che è una ipercinesia, quindi non possono essere comprese nelle due categorie precedenti. A seconda delle cause, possiamo distinguere in:
• Sindromi del sistema piramidale • Disordini dei gangli della base • Disordini cerebellari.
SINDROMI IPERCINETICHE
MOVIMENTI INVOLONTARI Sono movimenti involontari, cioè non controllabili dalla volontà; ci sono disturbi del movimento con TIC che sono gli unici a metà strada tra movimento volontario e involontario→ il soggetto con tic non vuole farli ma li po' parzialmente contenere e limitare con una spesa energetica ed emotiva elevata. Quindi non è una classificazione così definita quella i dire che non è controllabile dalla volontà. Per definizione sono:
❖ INVOLONTARI ❖ VARIABILI CON LE EMOZIONI→ con sollecitazioni emotive e cognitive durante la visita si può sfruttare questa
“facilità di induzione” per slatentizzarla e farlo emerge davanti al medico. ❖ SOPPRESSI NEL SONNO
Definizione topografica. Quando si definisce un disturbo bisogna sempre definire anche la sede con i termini:
• Focale: una sola regione. • Segmentale: interessanti più regioni contigue. Es. regione capo-collo nei tremori essenziali. • Generalizzata: estesi in molte regioni corporee.
Es. Tremori sono soprattutto focali o segmentali, raramente sono generalizzati (a meno che non ci sia un problema psichico). Le distonie (alterazioni di tono posturale) possono essere molto lievi e focali (crampo dello scrivano o distonia del musicista). Un atteggiamento distonico invece è una lieve distonia focale che può essere notata dal clinico ma che non dà problemi dal punto di vista clinico; molte volte ha più senso no trattarle piuttosto che continuare ad aggiungere farmaci→ da notar è che molti disturbi del movimento sono conseguenza di farmaci come amiodarone, antiemetici.
TREMORE È il più comune di tutti; molte persone hanno tremore ma molte se lo tengono, infatti è compatibile in genere con una qualità di vita normale e perché i farmaci in grado di ridurre il dolore deprimono l’attività di altri neuroni; non esistono prodotti per tremori non parkinsoniani specifici, di solito si usano sedativi→ ne vale la pena di avere un paziente sedato solo per un tremore? O è meglio con un po’ di tremore ma lucido? Il tremore ha due componenti: bisogna immaginare un asse di equilibrio e immaginare il movimento che va un po’ sopra e un po’ sotto→ ha due componenti, una agonista e una antagonista. Questo è la differenza con il mioclono, che invece ha solo una componente che però poi ha un ritorno. Sono molto simili, ci vuole un occhio esperto per vedere le differenze. Modalità di insorgenza:
➢ TREMORE A RIPOSO: si associa al Parkinson. Comprare con segmento a riposo, solitamente negli arti distali e mandibolare/peribuccale. Scompare quando compie movimento volontario.

130
➢ TREMORE POSTURALE: quando si mantiene una posizione. Causato da attivazione adrenergica, compare nel movimento attivo di una postura e persiste o peggiora durante i movimenti di previsione. Peggiora in caso di iperattiva zone adrenergica (caffè per stare svegli, privazione di sonno) o altre condizioni come iperitoridismo.
➢ TREMORE INTENZIONALE: è quello diretto a un bersaglio, è classicamente un tremore cerebellare: tentando di raggiungere un bersaglio, aumenta il tremore e diventa ampio e si associa a frenage.
Tremore intenzionale. Caratteristico di malattie del cervelletto. Classificazione sindromica (tabella slide):
➢ Frequenza ➢ Attivato da: riposo, postura, movimento mirato.
Tremore essenziale. È molto comune perché c’è una genetica molto determinante; si localizza a:
• Arti superiori • Capo: a testa si muove in direzione verticale (Sì-sì) o orizzontale (no-no). Generalmente i parkinsoniani non hanno
tremore del capo, ma tutto può essere, a volte si tratta di un tremore distonico, cioè un tremore causato da alterazione del tono→ se il sistema di propriocezione individua che la testa è una posizione non naturale (es. distonia in rotazione) tenta di correggerla e quindi si manifesta tremore, ma si tratta di un tremore secondario alla distonia.
• Può interessare la voce. • Risparmia gli arti inferiori.
La maggior parte dei tremori essenziali viene scambiato per Parkinson ma non vengono curati.
MOVIMENTI COREICI Ipercinesie, sono discinesie che però in realtà non hanno caratteristiche della danza perché non sono movimenti armonici e simmetrici: cambia velocemente sede, ampiezza, velocità→ molto disorganizzato, disarmonico, imprevedibile, anarchici. L’imprevedibilità della corea serve per distinguerla dal TIC, che è stereotipato e sempre uguale a se stesso per un certo periodo della vita, la corea invece è irregolare e mai uguale.
COREA= IRREGOLARE, TIC= STEREOTIPATO
Esiste la stereotipia al di fuori del tic che è conservato anche negli animali. Atetosi: discinesia più distale e lento, tentacolare. Corea: è un movimento più veloce e prossimale. Entrambe possono coinvolgere nasce il volto, con smorfie; la corea è sempre più veloce. Si manifestano in corea di Huntington ma esistono altre condizioni: - Diabete scompensato con sbalzo glicemico: spesso manifestano un danno al caudato, - Ischemie - Malattie autoimmuni:
o Sindrome anti-fosfolipidi (a volte associata a LES o a altre vasculiti) e corea lupica. A volte sono causate da infarto o anche senza infarto, con un meccanismo legato ad auto-anticorpi.
o Corea minor del bambino o corea di Sydenham: dovuta a una reazione anticorpale post-streptococcica→ per questo motivo si è ipotizzato che gli anticorpi anti-fosfolipidi e del LES possano causare danno direttamente anche senza infarto.
BALLISMO Discinesia esplosiva in cui vengono letteralmente lanciati gli arti o muove il tronco in modo esplosivo e di impatto, può farsi male; è caudata da una lesione del nucleo subtalamico di Luys. Solitamente la causa è emorragica, difficilmente ischemica. È molto rara. Solitamente è un emiballismo, difficilmente è bilaterale.
DISTONIA È una contrazione lenta e sostenuta che determina un movimento torsionale, all’inizio intermittente poi consolidato in postura abnorme. Se è un monito non consolidato, che continua a scomparire può essere in realtà una discinesia. Definizione più inclusiva: è un disturbo del movimento eterogeneo, caratterizzato da spasmi muscolari involontari che inducono posture abnormi della postura del corpo interessata.

131
FENOMENO DELLA CO-CONTRAZIONE: deve esserci sia contrazione dell’agonista che dell’antagonista; se uno dei due prevale sull’altro, ci si alterna nel movimento e si arriva ad avere un disturbo che ricorda una atetosi (che è sempre una discinesia). Spesso si associa al tremore distonico, perché esiste un sistema di correzione della postura. Esistono distonie compito-specifiche: distonia del musicista (quando utilizza l’indice per suonare parte automaticamente anche il movimento del pollice), crampo dello scrivano. Bisogna cercare il gesto antagonista: speso i pazienti compiono della azioni che correggono la posizione abnorme: es. appoggiarsi la mano sulla guancia in caso di distonia del capo può inviare afrezze sensitive che correggono la postura abnorme. Distonie generalizzate essere anche causate da somministrazione acuta di aloperidolo; spesso in pazienti schizofrenici con terapia a ita di neurolettici si sviluppa una distonia generalizzata.
Classificazione per eziologia. - PRIMITIVA: è una malattia vera e propria e non solo un segno.
o Torcicollo (distonia cervicale): è una distonia segmentale tipica di donne tra 55-65 anni, ha acquisito importanza perché viene impiegata la tossina botulinica.
o Blefarospasmo: spasmi di orbicolare dell’occhio, idiopatica post-innervazione patologica da paralisi facciale (anche solo dopo la paralisi di Bell, insieme alla lacrime di coccodrillo).
o Distonia oro-mandibolare: se associata a blefarospasmo si parla di sindrome di Meig. o Distonia laringea: Le gestisce l’otorino con ossina botulinica in corde vocali.
▪ In adduzione (quando le corde vocali sono ravvicinate), con un voce strangolata. ▪ In abduzione, con voce sussurrata.
- SECONDARIA: è un segno di un danno cerebrale o altro. In Parkinson avanzato con fenomeni di off, si manifestano distonie che sono secondarie al Parkinson (non si tratta di due malattie sovrapposte); lo stesso si manifesta nelle malattie da accumulo di metallo. Anche un ictus in una determinata zona frontale può interrompere circuiti e dare distonia.
- DYSTONIA-PLUS: nel corso di altre malati neurodegenerative. - PAROSSISTICHE: nel corso degli attacchi è normale ma poi si manifestano ne distonie tra un attacco e l’altro. - EREDO-DEGENERATIVA:
o DYT1: Più frequente negli ebrei askenaziti. Rispondono alla levodopa, esordiscono nell’età infantile e sono distonie focali con associate un Parkinson.
o Distonia mioclonica: poco comuni, sono un’associazione di distonia e mioclono, sono associate a mutazione dell’Y-sarcoglicano.
o Malattia di Wilson: può esordire anche con disturbi del movimento, di solito di tipo distonico. o NBIA1: accumulo di ferro nei nuclei della base. Segno RM dell’occhio di tigre.
TIC È un argomento di neurologia infantile: compaiono in età pediatrica poi tendono a scomparire e ricomparire in età adulta ma in forma “benigna”: ne costituiscono una caratteristica gestuale dell’individuo, si manifestano come persone veloci, “elettriche”. Possono essere semplici o complessi, motori o vocali. Sono preceduti da una sensazione di coazione (necessità) e sono peggiorati da disagio emotivo.
Sindrome di Gilles de la Torrette. È la presenza di tic ma anche un deficit di attenzione con iperattività (ADHD) e
un disturbo ossessivo-compulsivo (DOC); è una via di mezzo tra neurologia e psichiatria. Il tic vocale è caratterizzato da coprolalia, cioè dire parole socialmente sconvenienti. Storia naturale: un bambino in età prescolare inizia a avere ADHD (bisogno di muoversi, non sta mai fermo), poi compaiono in onro a 6 anni i tic motori, poi vocali, ai 10 anni c’è l’esacerbazione della malattia e poi intorno ai 18 anni si ha la remissione. Per la diangosi, devono essere presenti più volte al giorno per almeno un anno e devono esserci anche tic vocali. Terapia: neurolettici per la componente DOC e antidepressivi serotoninergici.
DIAGNOSI Oltre ad anamnesi ed esame obiettivo, bisogna considerare che tutte queste patologia possono essere sia primitive che secondarie, quindi una TC o RM si fa a tutti per escludere la presenza di qualche patologi organica. Quello che cambia è la tempistica: se si sospetta una lesione vascolare (perché non c’è familiarità) si fa imaging urgente, altrimenti si può fare senza fretta.

132
ATASSIE EREDITARIE
Sono malattie da triplette, e per questo sono accomunate alle coree. Il difetto è dato da una espansione di trinucleotidi. Son un gruppo eterogenee di malattie con:
1) una base genetica e un fenotipo enormemente variabile 2) Atassia progressiva 3) Coinvolgimento di cervelletto e sue connessioni
Classificazione:
- Atassie progressive a esordio precoce, AR. Un sottotipo ne costituisce il 90%, le altre osno poco ripevanti. - Atassie progressive ad esordio tardivo, AD. Sono le ATASSIE SPINOCEREBELLARI (SCA), ce ne sono 40 sottotipi.
SCA: AUTOSOMICHE DOMINANTI
Epidemiologia. Rendono conto di 3 casi su 100.000, quindi sono molto rare. Sono
più frequenti nelle isole. La SCA-3 ha una prevalenza maggiore di tutte, soprattutto in Brasile e altre colonie portoghesi. Variabilità di età: enorme, perché dipende da come si espande o restringe la sequenza, da casi in età pediatrica a oltre 60 anni. Età media: terza decade. ¼ di SCA in Italia sono SCA-1 e ¼ sono le SCA-2. Circa il 30% di mutazioni di SCA sono conosciute.
Genetica. Le ripetizioni di triplette si possono espandere o ritrarre nel corso di
trasmissione, ma più frequentemente si espandono, soprattutto se la trasmissione è paterna (FENOMENO DELL’ANTICIPAZIONE), e di solito si associano a forme giovanili. Le poliglutammine (perché sono formate da CAG) fanno sì che la proteina diventa tossica sia perché non riesce a essere digerita (proteasoma) sia perché genera un danno immunologico (lisosomiale).
Neuropatologia. Macroscopicamente: atrofia (corteccia e nuclei cerebellari, nuclei della base, ponte, corteccia
cerebrale, sostanza nera, nervi cranici e midollo spinale) Microscopicamente: degenerazione neuronale e gliosi.
CLINICA - Atassia della stazione eretta e della marcia. Gambe larghe, difficoltà a partire, frenare e cambiare direzione. - Dismetria e tremore cinetico agli arti: esame classico è far mettere il tappo alla penna. - Ipotonia, riduzione tono muscolare. - Disturbi dell’oculomozione (imprecisione) e nistagmo. SEGNO DEL RIMBALZO: si chiede al paziente di fare un movimento e di fare resistenza (flettere avambraccio sul braccio), dopo di che si lascia la presa e il paziente non avere una fase di stop pulita, tende a farsi rimbalzare il braccio addosso→ impossibilità a frenare in maniera pulita il movimento. degenerazione può coinvolgere anche retina, nervi ottici, gangli della base, corteccia cerebrale, midollo spinale, nervi cranici e tronco cerebrale, con conseguenti sintomi. Vecchia dizione di ADCA di Harding (atassia cerebellare autosomica dominante): prevedeva, a seconda della sintomatologia prevalente, la distinzione in:
- ADCA tipo 1 (prevalenza sintomi extracerebellari) - ADCA tipo 2 (atassia cerebellare e retinite pigmentosa) - ADCA tipo 3 (rilevanza di sintomi cerebellari)
Con questi cluster si restringeva il numero di esami da fare; oggi invece questa classificazione è stata superata dalla classificazione genetica. Se il paziente ha:

133
→ deficit neurologici, si fanno esami per SCA-1, SCA-2, SCA-3. → Atassia cerebrale e ritinite pigmentosa: si fanno esami per SCA-7. → Sintomi cerebellari prevalenti: SCA-6, SCA-12.
SEGNI CLINICI DISTINTIVI SCA1: fenotipo è altamente variabile con iniziale sindrome cerebellare, e successivamente deficit dell’oculomozione neuropatia periferica e segni piramidali. Difficilmente distinguibile clinicamente dalle altre forme di SCA. SCA2: Sono più pronunciati il tremore, l’iporeflessia e in rari casi vi è una forma pura di parkinsonismo familiare, senza segni cerebellari. SCA-3: sindrome di Machado-Joseph. Si fenotipo è altamente variabile e sono state descritte alcune forme pure di atassia, di parkinsonismo, di paraplegia spastica e di neuropatia. Sono comuni i disturbi del sonno con restless leg syndrome, sensibile ai dopamino-agonisti. Fenotipo vario con atassia, di parkinsonismo, di paraplegia spastica e di neuropatia.
DIAGNOSI Non è mai dettata dalla clinica, perché esseno così aspecifiche non esiste un prototipo unico per queste forme, l’overlap tra le varie forme è molto frequente. L’analisi genetica è molto importante e deve essere indirizzata dalla presenza dei segni clinici:
RM: può essere normale nel primo anno dopo l’esordio. Neurofisiologia: soprattutto in presenza di neuropatia periferica. Meno utili sono i PE.
TERAPIA Non c’è nulla, se non sperimentazioni con staminali o eventualmente silencing dell’RNA: si sta tentando di rendere la poliglutammina non dannosa, mettendo una sequenza complementare che la blocca.

134
ATASSIE AUTOSOMICHE RECESSIVE Le atassie autosomiche recessive sono un gruppo eterogeneo di malattie neurodegenerative rare, caratterizzate da esordio precoce di progressiva degenerazione cerebellare e dei tratti spino cerebellari, associata a vari segni e sintomi neurologici, oftalmologici e sistemici. Rispetto alle eredo-atassie autosomiche dominanti, questo gruppo di atassie è ad oggi meno caratterizzato e soltanto alcune di esse sono inquadrate dal punto di vista genetico e molecolare. La patogenesi sembra essere associata a “perdita di funzione” di specifiche proteine cellulari coinvolte nei processi di omeostasi metabolica, nel controllo del ciclo cellulare o nei processi di riparazione del DNA
ATASSIA DI FRIEDERICH Causata da una mutazione della proteina FRATASSINA (da Friederich Ataxia). Normalmente la malattia si manifesta quando si superano le 42 triplette (da 66 a 1700); più grave è l’espansione, più precoce è l’esordio. Ruolo di fratassina: ha un ruolo nel coinvolgimento mitocondriale, quindi genera un danno energetico→ cellule se soffrono di più sono cellule miocardiche e cellule dei neuroni sensitivi dei gangli delle radici dorsali.
Esordio. Pubertà, meno di 25 anni.
Quadro clinico. - Progressiva atassia degli arti e della marcia - Riflessi assenti agli arti inferiori e segno di Babinski
→ simile alla SCA-1. Aiutano: scoliosi, piede cavo, diabete. Genetica: neuropatologia conferma sofferenza di gangli e radi ci dorsali.
Diagnosi. RMN cerebrale e midollare: metodica di scelta, che mostra tipicamente atrofia del midollo cervicale con minima o assente atrofia del cervelletto Indagini cardiologiche: l’ECG mostra nel 65% inversione di onda T e l’ecocardiogramma rivela ipertrofia ventricolare concentrica simmetrica Neurofisiologia: Velocità di conduzione normali o lievemente ridotte, potenziali d’azione neurosensoriali assenti nel 90%, SEP, BAEP e PEV spesso anormali.
Terapia. Non funziona.

135
MALATTIA DI HUNTINGTON
È una malattia da espansione di triplette sul cromosoma 4, si forma una proteina più grande malfunzionante. 20 casi a Novara (circa 5 su 100.000).
PRESENTAZIONE CLINICA Tre possibili esordi:
1) MOTORIA: discinesie coreiche isolate. 2) SINTOMI PSICHIATRICI: DOC, depressione, disfunzioni affettive, le discinesie vengono solo dopo. 3) DECADIMENTO COGNITIVO: più frequente quando la corea compare nell’anziani (corea senile): è una corea a
basso numero di triplette, che in caso di deplezione neuronale fisiologia per l’età può diventare sintomatica in età molto avanzata.
Tre sintomi classici:
• DEAMBULAZIONE IRREGOLARE: passi lungi, brevi, lateralmente. Marcia anarchica, senza ritmo e criterio. • IPOTONIA MUSCOLARE • IMPERSISTENZA MOTORIA: incapacità si mantenere una postura al lungo (es. tenere lingua porta per 30 secondo,
Mingazzini per 1 minuto). Una condizione simile può essere causata anche da neurolettici, ed è chiamata acatisia, che però è più una incapacità di stare fermi. Sedia a dondolo: stereotipia tipica del soggetto psichiatrico
• DEPRESSIONE • OCD • Tasso di suicidio più alto: sono malattie ereditarie e portano a morte entro 15 -17 anni dopo la diagnosi, quindi
spesso i pazienti sono portati al suicida perché sanno qual è la loro sorte. Inoltre poi ci sono disfunzioni biochimiche tipiche della depressione che possono portare a questo gesto (attenzione quando si somministrano antidepressivi perché può essere la mesa in atto).
Il neurone è una cellula molto sensibile, che ha 3 meccanismi di difesa:
- Proteasi: libere o all’intero di sistemi organizzati (proteasoma) - Lisosomi enzimi litici racchiusi in un lisosoma. - Sistema immunitario: il neurone sta bene se il SI lo protegge da danni causati da agenti infettivi, neoplastici ma
anche da metaboliti o proteine anomale. I detriti infatti sono eliminati da fagociti, che fanno parte di SI.
TERAPIA Neurolettici:
- Clozapina: da monitorare perché da granulocitopenia. - Quetapiana: effetto moderato.
Depletore dopamina: Tetrabenazina: è n farmaco reseprino-simile, toglie la dopamina dal vallo sinaptico. Antidepressivi: servono per OCD e depressione.

136
MIOPATIE
Ci concentriamo sulle miopatie primitive.
SINTOMI E SEGNI CLINICI Sospettiamo una malattia primitiva del muscolo in presenza di:
1. IPOSTENIA: debolezza muscolare è prossimale, tende a interessare cingoli, collo e tronco (es. riferita fatica a pettinarsi, lavarsi); più rara è invece una distribuzione distale. Non è sempre presente ma è il sintomo più comune. Si richiede che la forza dei muscoli sia documentata con la scala MRC. È simmetrico.
Esempi di manifestazioni di ipostenia:
- Cingolo scapolare: si manifesta con la scapola alata: appoggiarsi al muro e spingerlo con le braccia tese, il bordo mediale della scapole si solleva dal dorso. È dato da una ipostenia del gran dentato (o dentato anteriore). La si può vedere nei pallavolisti che fanno schiacciate molto forti per lesione del nervo toracico lungo. Un altro muscolo interessato nella soppressione della scapola è il trapezio, quindi un danno del nervo accessorio.
- Cingolo pelvico: difficoltà nel salire le scale e deambulazione anserina: eccessivo basculamento del bacino durante la camminata dato da una debolezza del gluteo medio. Segno di Gowers: difficoltà di nell’alzarsi da terra; prima il paziente assume una posizione di quadrupedia con mani e braccia estese, poi avvicina le mani ai piedi e si fa leva con le mani sulle cosce, arrampicandosi su se stesso. È un segno che si fa spesso se si sospetta una miipatia.
- Quadricipite: iperestensione del ginocchio durante la marcia (genu recurvatum) come se il quadricipite non avesse forza a sufficienza per limitare l’escursione della gamba.
Più raramente è un andamento asimmetrico. Raramente può anche coinvolgere la muscolatura più distale→ distrofia miotonica: debolezza dei muscoli anteriori della gamba si manifesta con steppage. A volte la debolezza coinvolge i muscoli del capo e collo:
o M. viso (distrofia facio-scapolo-omerale, distrofia miotonica) o M. extraoculari: ptosi palpebrale con oftalmoplegie. Causata da alcune miopatie mitocondriali. o M. masticatori (distrofia miotonica) o M. faringolaringei (distrofia oculofaringea, distrofia miotonica) o M. flessori del collo (distrofia miotonica) o Dropped-head: caduta del capo.
Possono interessare anche muscoli respiratori. Alcune miotonie congenite si manifestano come un floppy baby: è un neonato flaccido, ipotonico. Quali altri quadri possono causare un floppy infant? SMA1 e miastenia gravis congenita.
2. ESAURIBILITÀ MUSCOLARE: la debolezza, se anche non è tanto grave a riposo, aumenta con l’esercizio. 3. MIALGIE E CRAMPI: i muscoli fanno male quando c’è una necrosi massiva di fibre muscolari, con edema e
liberazione di mediatori algogeni→ prototipo è la polimiosite. Alcune miopatie sono dovute a difetti enzimatici nella catena del consumo del glicogeno (glicogenosi o mitocondriopatie) è facile avere dolori e ipostenia da sforzo.

137
4. ALTERATO TROFISMO MUSCOLARE: le dimensioni del muscolo sono ridotte, atrofia o ipertrofia. Spesso si ha un fenomeno di pseudoipetrofia: incremento volume dei muscoli dovuto a un’abbondante sostituzione connettivale del tessuto che ne provoca anche una retrazione→ cosce assottigliate e polpacci di dimensioni sproporzionate. Ipetrofia: si osserva nelle fasi precoci delle miopatie distrofiche, come compenso dei muscoli sinergici risparmiati dal processo patologico o in alcune canalopatie→ difetto nel transito ionico attraverso la membrana.
5. MIOTONIE: Difficoltà a decontrarre i muscoli dopo contrazione forzata. Il paziente è impacciato nei movimenti distali perché non riesce a rilasciarli. Percuotendo con un martelletto si stimola una miotonia meccanica.
6. RETRAZIONI, CONTRATTURE MUSCOLARI, DEFORMITÀ ARTICOLARI: Sostituzione del tessuto muscolare da parte di tessuto connettivo, dotato di minore elasticità. Debolezza dei muscoli paravertebrali e posture anomale, favoriscono la comparsa di deformità a carico della colonna vertebrale (cifoscoliosi, iperlordosi, etc.). spesso una soluzione è la tenotomia perché la retrazione tendinea può rendere difficile la deambulazione.
7. COINVOLGIMENTO DI ALTRI DISTRETTI MUSCOLARI: possono essere interessati anche altri organi e apparati. Se si parla di difetti metabolici ed enzimatici, sono generali in tutto l’organismo, non solo muscolari. spesso nel contesto delle miopatie si trovano difetti del muscolo cardiaco.
EMG: uno degli strumenti principali nella diagnosi. BIOPSIA: importante è la parte ultrastrutturale e immunoistochimica (IIC), che permettono di marcare determinati anticorpi diretti contro particolari antigeni→ permettono di marcare determinati elementi della biopsia. non solo esami di routine. Distinguiamo le miopatie in: ➢ DESTRUENTI: perdita integrità della fibrocellula muscolare che subisce un danno necrotico associato a fenomeni di
rigenerazione secondaria. Ne fanno parte le distrofie musoclari. ➢ NON DESTRUENTI: Difetti enzimatici o da disfunzioni di organuli o membrane cellulari che determinano un’alterazione
della funzione muscolare senza distruzione delle cellule. Ne fanno parte le distrofie miotoniche, le miopatie mitocondriali, le canalopatie. E le miopatie secondarie.
DISTROFIE MUSCOLARI Sono un gruppo di malattie che risalgono a una alterazione di geni di proteine connesse con la membrana cellulare del muscolo, ossia con il sarcolemma. DISTROFINA: è la principale proteina coinvolta, è un componente di un insieme estremamene complesso di strutture proteiche, che garantiscono l’integrità funzionale e strutturale della fibra muscolare.
DISTROFIA DI DUCHENNE Patogenesi: manca la distrofina per via di una mutazione frameshift. È una malattia ereditaria ma non è congenita: infatti non si manifesta alla nascita, insorge circa a 3 anni. i bambini quindi imparano a camminare dopo di che si ha un regresso nell’attività motoria: poco per volta si accentuano i segni fondamentali visti prima (ipostenia dei cingoli e tronco, meno quella distale). Può avere qualche problema di tipo cognitivo. Interessamento cardiaco è molto frequente: cardiomiopatia dilatativa→ può darsi che un paziente sopravviva più dell’atteso e se la cavi bene dal punto si vista della motricità ma prima o poi muore per cardiomiopatia.
DISTROFIA DI BECKER La produzione di distrofina c’è ma è ridotta e anomala, l’esordio è più tardivo (adolescenti e giovani adulti), sopravvivono fino a 30-40 anni. L’interessamento cardiaco è meno frequente di Duchenne ma è più grave.
DIAGNOSI Il pattern enzimatico comprende:
• Aumento di CPK
• Aumento di transaminasi.
• Mioglobinuria: le fibre muscolari contengono grandi quantità di mioglobina, in caso di danno muscolare vengono liberate ed espulse con le urine.
Altre cause di mioglobinuria: immobilità, schiacciamento.

138
Importante è anche la biopsia muscolare, in cui si può ricercare la proteina mutata.
TRATTAMENTO Agire sulle retrazioni tendinee con fisioterapia. Vengono somministrati steroidi; non si sa bene con quale meccanismo, ma aumentano la sopravvivenza in queste distrofie muscolari. Importante è ricordare la terapia genica.
DISTROFIA DI EMERY-DREIFUSS Due varianti: X-linked e AD: mutazione di emerina e laminina→ meccanismo simile a Duchenne e Becker ma il bersaglio è leggermente diverso. Si manifesta con distribuzione diversa dell’ipostenia muscolare: è anche un po’ distale, rimane possibilità della grave cardiopatia.
DISTROFIE MIOTONICHE Hanno un meccanismo diverso, sono malattie da espansione di triplette. Importante è che non sono patologie solo del muscolo, ma coinvolgono anche altri distretti. Due forme: DM1 o malattie di Steinert. DM2. Cambia la distribuzione distrettuale di ipostenia e di miotonia; la 2 è più prossimale→ si hciama anche PROMM (Proximal Miotomy).
DM1 (domanda frequente!) Incidenza: 15/100.000. Maggiore è l’espansione, maggiore è la gravità di malattia e più precoce è l’esordio.
Clinica. Ipostenia e ipotrofia più distali che prossimale: loggia antero-laterale della gamba, volto, linguali. Una delle particolarità è che hanno una precoce calvizie frontale; unita all’ipostenia dei muscoli mimici, conferisce un aspetto funereo, lugubre. Altri coinvolgimenti sistemici:
• Cataratta
• Difetti del sistema endocrino: diabete, ipotiroidismo, ipogonadismo, problemi cognitivi.
• Problemi cardiaci con disturbi del ritmo.
Terapia. È possibile cercare di diminuire un po’ la miotonia: antiepilettici (carbamazepina) e antiaritmico (mexiletina, si
può tentare di controllare la miotonia).
MIOPATIE DA ACCUMULO METABOLICO Utile è ricordare le glicogenosi:
• Si manifestano con ipostenia ingravescente.
• Intolleranza allo sforzo, esauribilità muscolare. La glicogenosi di tipo II si accompagna a un fenomeno chiamato second-wing (wind??)→ pausa dallo sforzo. Glicoenosi: malattia di Pompe e malattia di Mc Ardle.
MIOPATIE MITOCONDRIALI I mitocondri si ereditano per via materna. Tutte le miopatie mitocondriali alla biopsia si presentano con ragged red fibers. Sono tante, dobbiamo ricordarne due:

139
- MELAS (Encefalomiopatia Mitocondriale, Acidosi Lattica, Stroke): l’abbiamo citata nell’ictus→ si manifesta con STROKE GIOVANILE che non rispettano un preciso territorio arterioso, mentre c’è una prevalenza per le regioni parieto-occipitali.
- MERFF (Epilessia Mioclonica con ragged red fibers)→ le fibre rosse muscolari con la colorazioni di Gomori si presentano frastagliate. Si presenta con una EPILESSIA MIOCLONICA PROGRESSIVA, aggrava notevolmente l’aspettativa di vita del paziente.
MIOPATIE ACQUISITE Tossiche o infiammatorie.
MIOPATIE TOSSICHE - Statine: causano un aumento di CPK e transaminasi. - Alcol: può dare miopatie sia un acuto (ingestione massiva episodica) che in cronico. - Amiodarone - Steroidi: CK si alza poco.
MIOPATIE INFIAMMATORIE Necrosi muscolare con ipostenia prossimale. La CK si alza moltissimo. DM e PM si differenziano perché la DM interessa anche la pelle; molto spesso è paraneoplastica, è sottostante un tumore (noto o no)→ ovaio, polone, stomaco.
Quale ha la prognosi peggiore? La DM perché è quasi sempre specchio di un tumore!
Ci sono manifestazioni al di là del muscolo (indipendentemente dl tumore): - polmoniti interstiziali - febbre, perdita di peso.
EMG: oltre che facile interferenza, anche fibrillazioni→ date da reinnervazione. IBM→ non è sensibile a steroidi.

140
MIASTENIA GRAVIS
Malattia della placca motrice: viene interessata da un processo autoimmunitario, tale per cui si producono autoanticorpi anti-recettore acetilcolina; viene quindi alterata la normale capacità di trasmissione. Bisogna ledere metà dell’apparato recettoriale prima di avere un riscontro clinico.
Patogenesi. Non si da di preciso come si sviluppino questi auto-anticorpi, ma una possibile causa sembra essere il timo.
Il maggior parte dei Pazienti è positiva agli anticorpi anti-recettore, ma una certa percentuale di pazienti si è dimostrata essere siero-negativi→ si sono scoperti altri anticorpi responsabili della MG, come gli anti-MUSK.
→ se anti-AchR negativi, si fanno anti-MUSK e anche altri (anti-contattina).
Clinica. Il sintomo principale è l’esauribilità e facile faticabilità, che predilige il distretto oculare→ muscoli oculomotori.
Ptosi palpebrale, strabismo, diplopia→ peggiorano durante la sera.
Diagnosi All’EO:
• Test dell’esauribilità: si fa masticar una cicca per qualche minuto, dopo un po’ lamenta una difficoltà a masticare. Oppure si fa fissare il dito dell’esaminatore mantenendo lo sguardo verso l’alto, e dopo un po’ subentra la ptosi.
• Test del ghiaccio: se si trova una paziente con occhio ptosico, si prende del ghiaccio e lo si mette a contatto; dopo pochi minuti si ha un miglioramento della sintomatologia.
Esami strumentali:
- Ricerca di Ab: antirecettore, anti-musk, anti-contattina. - Stimolazione sovramassimale ripetitiva del nervo motore (si parla di EMG per intendere in generale questo tipo di
esame, anche se in realtà è una tecnica elettroneurografico)→ stimolazione ripetuta del nervo ulnare (un treno di stimoli, alla frequenza di 3 Hz) e con un elettrodo di superficie sul muscolo (NO AGO) si dimostra una caduta delle onde M (o CMAP).
Un altro test possibile è somministrando anti-colinesterasici: prostigmina (in acuto) o piridostigmina (in qualche giorno)→ permette di fare diagnosi ex juvantibus in seguito a miglioramento della sintomatologia. Di fronte a una sintomatologia solo oculare, si danno anti-acetilcolinesterasici periferici: come piridostigmina (la rivastigmina invece è centrale). Se invece la sintomatologia è generalizzata, allora si dovrà applicare terapia immunosoppressiva, perché potrà essere colto da insufficienza respiratoria→ terapie cortisoniche: una volta instaurate vano portate avanti a dose piena per molto tempo, anche anni. Timectomia

141
POTENZIALI EVOCATI
Abbiamo parlato di velocità di conduzione dei nervi; qui il concetto è analogo ma lo si applica anche al SNC→ si segue la propagazione degli impulsi lungo le vie sensitive o sensoriali. I più noti potenziali evocati sono: visivi, acustici e somestesici.
POTENZIALI EVOCATI MOTORI Esistono anche i potenziali evocati motori, dove la questione è al contrari: si stimola la corteccia e si registrano le risposte dai muscoli. Si usano Stimolazioni Magnetici Transcranici (TMS): è una bobina molto forte che genera un campo magnetico rapidamente variabile, che induce correnti sulla corteccia motoria (che funge da conduttore)→ così facendo si stimola la corteccia motoria. È indolore. In base a dove si mette la bobina, si muoveranno di più gli arti di destra o di sinistra. Si stimola la corteccia e si registra il tempo di latenza dall’onda M. Dopo di che si stimolano le radici cervicali (per arto superiore) o lombari (per arto inferiore) e anche qui si misura il tempo di latenza dall’onda M. →Poi si fa la differenza tra le due e si ottiene il TEMPO DI CONDUZIONE MOTORIA CENTRALE, cioè il tempo che lo stimolo ci ha messo ad andare dalla corteccia al livello midollare interessato, cioè lungo il sistema piramidale (I motoneurone).
Applicazioni. MONITORAGGIO NEUROFISIOLOGICO INTRAOPERATORIO: i neurochirurghi vogliono che sia registrata in continuazione l’attività neurofisiologica delle strutture limitrofe per accertarsi di non danneggiarle (ad esempio la via piramidale per evitare di rendere un paziente emiplegico). PARALISI PSICOGENE: esistono molti fenomeni neurologici che non hanno una base organica ma emotiva (afasia, alessia, paralisi). Spesso ci sono casi piuttosto evidenti (es. paralisi ma con assenza di Babinski o assenza di iperreflessia), ma ci sono casi più sfumati (perché a volte sono addestrati bene) e quindi a volte è necessario un esame neurofisiologico che non mostra nessun danno delle vie motorie→ quello che manca è lo stimolo volontario da parte del soggetto. A volte anche solo il fatto di vedere i propri arti muoversi, li fa convincere che non ci sia nessuna patologia e quindi poi riescono a camminare→ ha anche un effetto curativo. SLA: infatti permette di studiare selettivamente il I motoneurone. A volte non sono così chiari i sintomi (Babinski non sempre c’è, ecc).
POTENZIALI EVOCATI VISIVI (PEV) Molto usati nella SM e nella neurite ottica retrobulbare. Nella routine, si stimola la retina facendo fissare al paziente con un occhio alla volta il centro di uno schermo (possibilmente con tubo catodico) con una scacchiera che continua ad alternare alla frequenza di 1 Hz→ stimolazione a pattern reversal. Hanno determinato che è il metodo più efficace per ottenere una risposta simile nella popolazione (bassa variabilità interindividuale). Così facendo si stimola la macula (sito della visione distinta, quini la visione centrale che è quella implicata anche nelle neuriti ottiche). Si registino poi i potenziali evocati dallo scalpo che sovrasta la corteccia visiva. Si ottiene un’onda con un picco positivo dopo 100 ms dallo stimolo→ per questo si chiama P100. Se quest’onda è ritardata, c’è qualcosa di patologico.
Tre possibili pattern (con registrazione a 3 canali per ciascun occhio):

142
- Assenza monoculare: Stimolando l’occhio destro si ottiene la P100, ma stimolando l’occhio sinistra non si riscontra nessuna P100→ evidente segno di patologia (o ci vede male da un occhio -ma bisogna mettere gli occhiali- o non stava guardando il bersaglio).
- Ritardo monoculare - Ritardo binoculare
Si usa sia per fare diagnosi di SM sia per fare follow-up: a intervalli fissi si fanno questi potenziali per vedere il miglioramento.
POTENZIALI EVOCATI ACUSTICI DEL TRONCO (BAEP) Quelli corticali non si usano molto in neurologia perché sono tropo variabili; si usano di più quelli del tronco (BAEP): hanno una latenza più breve, di pochi ms. si ottengono 5 onde sequenziali (non una sola come nei VEP). In base a quale onda manca, si individua il livello della lesione.
Applicazioni. Neurinoma dell’acustico: tumore istologicamente benigno ma che cresce in vicinanza del nervo acustico, causando ipoacusia unilaterale. Quando si va a togliere questo tumore, il chirurgo tenta di mantenere o la stessa sensibilità sonora o addirittura migliora→maggior utilità quindi è il MONITORAGGIO INTRAOPERATORIO per evitare di arrecare ulteriori danni. DIAGNOSTICA DEL COMA: di fronte a un paziente in coma, bisogna capire se si sveglierà o no e una risposta può arrivare proprio dai BAEP→ se le onde finali (III, IV e V) sono perse perché non passa più nulla dal nervo al tronco (rimangono solo le onde proprie dl nervo acustico, I e II), questo si associa a morte cerebrale o stato vegetativo persistente.
POTENZIALI EVOCATI SOMATOSENSORIALI (PES) Si stimolano le fibre sensitive di un nervo e si segue l’attività elettrica che ne risulta, nel SNP e nel SNC in base a dove si posizionano gli elettrodi si possono registrare onde del passaggio del potenziale d’azione che noi abbiamo suscitato. Si può fare per l’arto superiore o inferiore: es. si stimola in nervo ulnare e si misura la latenza del potenziale nel punto di Erb a livello, del plesso brachiale→ disposizione strategica degli elettrodi per ottenere più informazioni possibili circa il passaggio lungo il nervo. L’onda più celebre è l’onda N20: stimolando il nervo mediano al polso, dopo 20 ms si suscita nella corteccia somatosensoriale un’onda elettrica.
Applicazioni. MONITORAGGIO INRAOPERATORIO DIAGNOSI DI COMI ANOSSO-ISCHEMICI o TRAUMATICI

143
AVERAGING Come distinguere un piccolo PE da un rumore di fondo? Spesso i PE (o anche segnali con altri strumenti neurofisiologici- EEG, EMG, …) sono talmente piccoli che deve esserci un sistema che tira fuori questi segnali e li risalti; il metodo è quello di fare la media (AVERAGING): si registra molte volte lo stimolo nello stesso istante. Questo ingigantisce gli effetti, perché cadranno nello stesso istante di tempo. Invece la sommazione di onde casuali darà un grafico piatto→ Principio fisico chiamato ESTRAZIONE DEL SEGNALE DAL RUMORE. Più si aumenta il numero di stimoli, più si riduce l’errore di fondo e spiccano le onde dei PE. Più piccolo è il segnale, più numerosi devono essere i campioni necessari: nei PEV ne bastano un centinaio perché sono grossi, per i BAEP ne servono circa 2000. AVERAGING: significa fare la media, si fa una sommatoria media tra tutte le onde.
Copyright © 2022 FDOKUMEN




















