
Apoptosis-prone phenotype of human colon carcinoma cells with a high level amplification of the...
10
Apoptosis-prone phenotype of human colon carcinoma cells with a high level amplification of the c-myc gene Maddalena Donzelli 1 , Rosa Bernardi 1 , Claudia Negri 1 , Ennio Prosperi 2 , Laura Padovan 3 , Christian Lavialle 4 , Olivier Brison 4 and A Ivana Scovassi 1 1 Istituto di Genetica Biochimica ed Evoluzionistica CNR, Via Abbiategrasso 207, 27100 Pavia, Italy; 2 Centro di Studio per l’Istochimica CNR, Pavia; 3 Dipartimento di Genetica, Universita ` di Pavia, Italy; 4 Laboratoire de Ge ´ne´tique Oncologique, CNRS UMR 1599, Institut Gustave-Roussy, Villejuif, France Although apoptosis can be induced by the enforced expression of exogenously introduced c-myc genes, it is not clear whether overexpression resulting from the amplification of the resident c-myc gene in tumor cells is sucient to induce apoptosis. We have investigated the relationship between c-myc gene amplification and the propensity of tumor cells to undergo apoptosis, using the SW613-12A1 and SW613-B3 cell lines, which are representatives, respectively, of tumorigenic and non- tumorigenic clones isolated from the SW613-S human colon carcinoma cell line. Tumorigenic clones are characterized by a high level of amplification and expression of the c-myc gene, whereas cells of non- tumorigenic clones have a small number of copies and a lower level of expression of this gene. Analysis of c-myc mRNA level in cells cultured under low serum conditions indicated that the expression of the gene is tightly regulated by serum growth factors in non-tumorigenic B3 cells, whereas it is poorly regulated in tumorigenic 12A1 cells, the level of mRNAs remaining relatively high in serum-starved 12A1 cells. Under these conditions, 12A1 cells showed clear evidence of apoptosis, whereas B3 cells were completely refractory to the induction of apoptosis. Moreover, the study of cell lines derived from non- tumorigenic apoptosis-resistant clones following the introduction by transfection of exogenous c-myc gene copies showed that they have acquired an apoptosis- prone phenotype. Altogether, our results strongly suggest that deregulated c-myc expression due to high-level amplification confers an apoptosis-prone phenotype to tumor cells. The possible consequences of these observa- tions for cancer therapy are discussed. Keywords: apoptosis; human colon carcinoma cells; serum starvation; c-myc; amplification; PARP proteo- lysis Introduction A number of gene products have been implicated in the control of the balance between cell proliferation and cell death. Aberrant cell survival resulting from the inhibition of cell death would be expected to contribute to oncogenesis (Marx, 1993). Paradoxically, a number of dominant oncogenes appear to act as potent inducers of apoptosis (Harrington et al., 1994b). The involvement of these oncogenes, and, more specifically, of the c-myc proto-oncogene, in the activation and/or modulation of the apoptotic process has been extensively analysed (El-Deiry, 1997; Harrington et al., 1994b). The c-myc proto-oncogene is a member of a family of related genes implicated in the control of cell proliferation and its deregulated expression can contribute to neoplasia promotion and/or progression (Spencer and Groudine, 1991). The c-Myc oncoprotein is a short-lived, sequence-specific DNA-binding pro- tein, which acts as a transcription factor in association with its partner, Max (Amati and Land, 1994). In normal cells, c-myc expression is tightly controlled by mitogenic stimuli and appears to be necessary, and in some instances sucient, to promote cell proliferation (reviewed by Evan and Littlewood, 1993), as well as to suppress dierentiation (reviewed by Homan and Liebermann, 1994). The observation that disruption of c-Myc function by gene targeting is associated with severe growth impairment and a subsequent lethal phenotype (Davis et al., 1993) confirms the essential role of this proto-oncogene in the regulation of cell proliferation. However, under some circumstances, c- Myc can also act as a potent inducer of apoptosis. c- Myc-induced apoptosis has been reported in cells deprived of specific growth factors (Askew et al., 1991), in serum-starved cells, in cells growth-arrested by drug administration (Evan et al., 1992) and in cells cultured under hypoxic conditions (Alarcon et al., 1996; Rupnow et al., 1998). The programmed cell death process induced by c- Myc can be inhibited either by activation of an antiapoptotic signal transduction pathway triggered by specific survival factors (Harrington et al., 1994a) or by the deregulated expression of genes such as those of the bcl-2 family (Reed, 1995, 1997). Bcl-2 cooperates with c-myc to immortalize pre-B cells (Vaux et al., 1988) and its product prevents apoptotic cell death induced by c-Myc (Bissonnette et al., 1992; Fanidi et al., 1992; Wagner et al., 1993). It has been demon- strated that Bcl-2 functions upstream of the caspase-3/ CPP32 enzyme (Chinnaiyan et al., 1996), a cysteine protease which is responsible for poly(ADP-ribose) polymerase proteolysis (Lazebnik et al., 1994) and which has been recently reported to play a critical role in c-Myc-mediated apoptosis (Kangas et al., 1998). The expression of the c-myc proto-oncogene is frequently deregulated in human tumors; transloca- tions and amplifications of the gene as well as Correspondence: AI Scovassi The first two authors contributed equally to this work Received 22 May 1998; revised 23 July 1998; accepted 23 July 1998 Oncogene (1999) 18, 439 – 448 ª 1999 Stockton Press All rights reserved 0950 – 9232/99 $12.00 http://www.stockton-press.co.uk/onc
-
Upload
ifom-ieo-campus -
Category
Documents
-
view
2 -
download
0
Transcript of Apoptosis-prone phenotype of human colon carcinoma cells with a high level amplification of the...

Apoptosis-prone phenotype of human colon carcinoma cells with a high levelampli®cation of the c-myc gene
Maddalena Donzelli1, Rosa Bernardi1, Claudia Negri1, Ennio Prosperi2, Laura Padovan3,Christian Lavialle4, Olivier Brison4 and A Ivana Scovassi1
1Istituto di Genetica Biochimica ed Evoluzionistica CNR, Via Abbiategrasso 207, 27100 Pavia, Italy; 2Centro di Studio perl'Istochimica CNR, Pavia; 3Dipartimento di Genetica, UniversitaÁ di Pavia, Italy; 4Laboratoire de GeÂneÂtique Oncologique, CNRSUMR 1599, Institut Gustave-Roussy, Villejuif, France
Although apoptosis can be induced by the enforcedexpression of exogenously introduced c-myc genes, it isnot clear whether overexpression resulting from theampli®cation of the resident c-myc gene in tumor cells issu�cient to induce apoptosis. We have investigated therelationship between c-myc gene ampli®cation and thepropensity of tumor cells to undergo apoptosis, using theSW613-12A1 and SW613-B3 cell lines, which arerepresentatives, respectively, of tumorigenic and non-tumorigenic clones isolated from the SW613-S humancolon carcinoma cell line. Tumorigenic clones arecharacterized by a high level of ampli®cation andexpression of the c-myc gene, whereas cells of non-tumorigenic clones have a small number of copies and alower level of expression of this gene. Analysis of c-mycmRNA level in cells cultured under low serum conditionsindicated that the expression of the gene is tightlyregulated by serum growth factors in non-tumorigenic B3cells, whereas it is poorly regulated in tumorigenic 12A1cells, the level of mRNAs remaining relatively high inserum-starved 12A1 cells. Under these conditions, 12A1cells showed clear evidence of apoptosis, whereas B3 cellswere completely refractory to the induction of apoptosis.Moreover, the study of cell lines derived from non-tumorigenic apoptosis-resistant clones following theintroduction by transfection of exogenous c-myc genecopies showed that they have acquired an apoptosis-prone phenotype. Altogether, our results strongly suggestthat deregulated c-myc expression due to high-levelampli®cation confers an apoptosis-prone phenotype totumor cells. The possible consequences of these observa-tions for cancer therapy are discussed.
Keywords: apoptosis; human colon carcinoma cells;serum starvation; c-myc; ampli®cation; PARP proteo-lysis
Introduction
A number of gene products have been implicated in thecontrol of the balance between cell proliferation andcell death. Aberrant cell survival resulting from theinhibition of cell death would be expected to contributeto oncogenesis (Marx, 1993). Paradoxically, a number
of dominant oncogenes appear to act as potentinducers of apoptosis (Harrington et al., 1994b). Theinvolvement of these oncogenes, and, more speci®cally,of the c-myc proto-oncogene, in the activation and/ormodulation of the apoptotic process has beenextensively analysed (El-Deiry, 1997; Harrington etal., 1994b). The c-myc proto-oncogene is a member ofa family of related genes implicated in the control ofcell proliferation and its deregulated expression cancontribute to neoplasia promotion and/or progression(Spencer and Groudine, 1991). The c-Myc oncoproteinis a short-lived, sequence-speci®c DNA-binding pro-tein, which acts as a transcription factor in associationwith its partner, Max (Amati and Land, 1994). Innormal cells, c-myc expression is tightly controlled bymitogenic stimuli and appears to be necessary, and insome instances su�cient, to promote cell proliferation(reviewed by Evan and Littlewood, 1993), as well as tosuppress di�erentiation (reviewed by Ho�man andLiebermann, 1994). The observation that disruptionof c-Myc function by gene targeting is associated withsevere growth impairment and a subsequent lethalphenotype (Davis et al., 1993) con®rms the essentialrole of this proto-oncogene in the regulation of cellproliferation. However, under some circumstances, c-Myc can also act as a potent inducer of apoptosis. c-Myc-induced apoptosis has been reported in cellsdeprived of speci®c growth factors (Askew et al.,1991), in serum-starved cells, in cells growth-arrestedby drug administration (Evan et al., 1992) and in cellscultured under hypoxic conditions (Alarcon et al.,1996; Rupnow et al., 1998).The programmed cell death process induced by c-
Myc can be inhibited either by activation of anantiapoptotic signal transduction pathway triggeredby speci®c survival factors (Harrington et al., 1994a) orby the deregulated expression of genes such as those ofthe bcl-2 family (Reed, 1995, 1997). Bcl-2 cooperateswith c-myc to immortalize pre-B cells (Vaux et al.,1988) and its product prevents apoptotic cell deathinduced by c-Myc (Bissonnette et al., 1992; Fanidi etal., 1992; Wagner et al., 1993). It has been demon-strated that Bcl-2 functions upstream of the caspase-3/CPP32 enzyme (Chinnaiyan et al., 1996), a cysteineprotease which is responsible for poly(ADP-ribose)polymerase proteolysis (Lazebnik et al., 1994) andwhich has been recently reported to play a critical rolein c-Myc-mediated apoptosis (Kangas et al., 1998).The expression of the c-myc proto-oncogene is
frequently deregulated in human tumors; transloca-tions and ampli®cations of the gene as well as
Correspondence: AI ScovassiThe ®rst two authors contributed equally to this workReceived 22 May 1998; revised 23 July 1998; accepted 23 July 1998
Oncogene (1999) 18, 439 ± 448ã 1999 Stockton Press All rights reserved 0950 ± 9232/99 $12.00
http://www.stockton-press.co.uk/onc

increased half-life and overexpression of the oncopro-tein have been observed in many tumors (Bishop, 1995;Brison, 1993; Marcu et al., 1992). Although it is wellestablished that apoptosis can be induced by theenforced expression of exogenously introduced c-mycgenes in several experimental systems, it is still notclear whether overexpression of the resident c-myc geneowing to its ampli®cation in tumor cells is su�cient toinduce apoptosis (Packham and Cleveland, 1995). Toinvestigate the hypothesis that deregulated c-mycexpression resulting from ampli®cation of the genecould promote apoptosis, we examined the e�ect ofgrowth-restrictive conditions on the rate and the extentof cell death in cell lines characterized by di�erent c-myc ampli®cation levels. We performed our analysis ontumorigenic and non-tumorigenic clones isolated fromthe SW613-S human colon carcinoma cell line. Cells ofthe tumorigenic clones have a high level of amplifica-tion of the c-myc gene, whereas cells of the non-tumorigenic clones have a small number of copies ofthis gene (Lavialle et al., 1988, 1990, 1989). We reporthere that cells of tumorigenic and non-tumorigenicclones respond di�erently to growth-restrictive condi-tions resulting from serum starvation, the former beingapoptosis-prone and the latter apoptosis-resistant.Moreover, we showed that introduction of multiplecopies of a plasmid bearing a complete human c-mycgene into the cells of apoptosis-resistant, non-tumori-genic clones confers on them the apoptosis-pronephenotype. Altogether, these results strongly supportthe idea that a high expression level of the c-myc gene,resulting from gene ampli®cation, contributes to theapoptosis-prone phenotype of these colon carcinomacells.
Results
Human colon carcinoma cells with a high levelampli®cation of the c-myc gene are susceptible toapoptotic cell death induced by serum deprivation
To investigate the hypothesis that c-myc overexpressionresulting from the ampli®cation of the gene couldpromote apoptosis, we used cells of tumorigenic andnon-tumorigenic clones derived from the SW613-Shuman colon carcinoma cell line. It was previouslyshown (Lavialle et al., 1988, 1990, 1989; Modjtahedi etal., 1985) that the SW613-S cell line is mainlycomposed of two cell types: (i) cells with a high levelof ampli®cation and expression of the c-myc gene andtumorigenic in nude mice; (ii) cells which harbor asmall number of copies of this gene, resulting in alower expression level, and which are non-tumorigenicin mice. Several clones representative of each of thesetwo cell types have been isolated (i.e. tumorigenicclones SW613-12A1, -3, -4A and non-tumorigenicclones SW613-B3, -2G1, -4G).Since it has been demonstrated that enforced
expression of exogenously introduced c-myc genessensitizes cells to apoptosis induced by growth-factorwithdrawal (Evan et al., 1992; Hueber et al., 1997), weexamined the e�ect of serum-deprivation on di�erenttumorigenic and non-tumorigenic clones. As represen-tatives of tumorigenic and non-tumorigenic clones, theSW613-12A1 and SW613-B3 cell lines have been
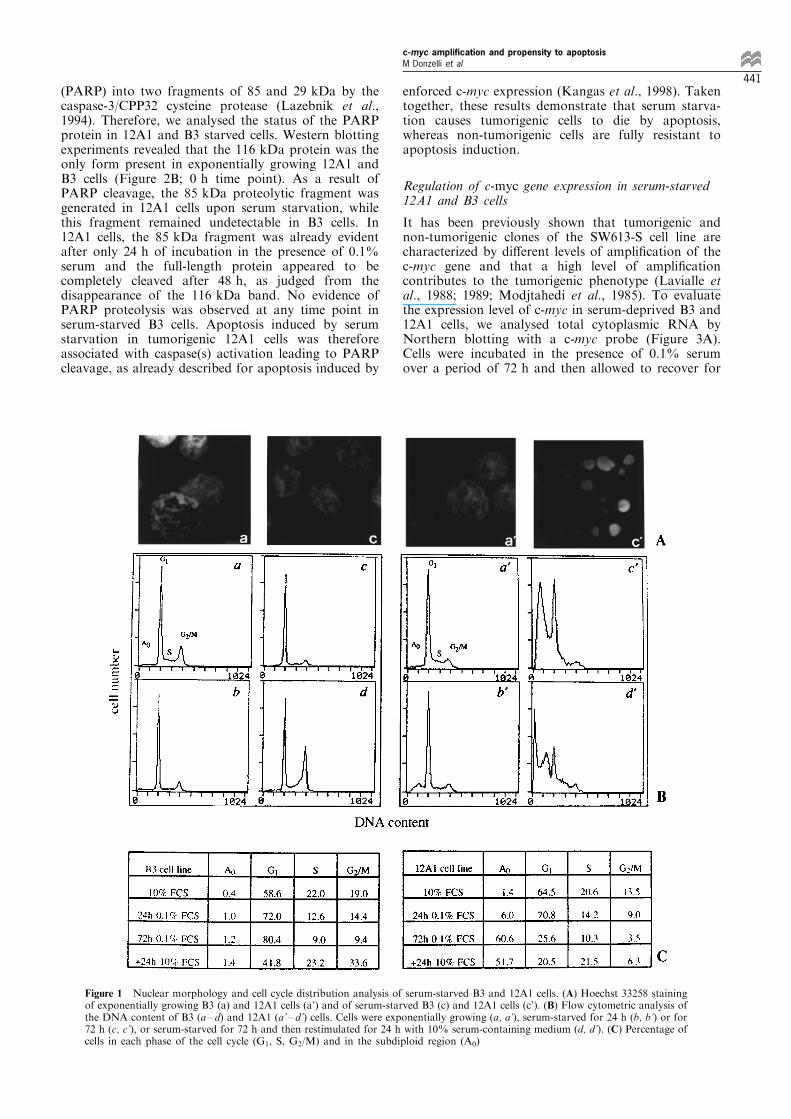
analysed. Nuclei of cells incubated for 72 h in amedium containing 0.1% serum were stained withHoechst 33258 and observed by ¯uorescence micro-scopy. As illustrated in Figure 1A, serum-starved 12A1cells showed nuclear morphological changes typical ofapoptotis such as chromatin condensation andfragmentation (c'), whereas B3 cells exhibited anormal nuclear morphology (c), under the sameexperimental conditions. The fraction of cells withcondensed or fragmented nuclei was about 60% forclone 12A1 versus 1.5% for clone B3 after 72 h ofserum deprivation.In parallel with the ¯uorescence microscopic
observations, ¯ow cytometric analysis was carriedout, using DNA content as a single parameter. Theresults are shown in Figure 1B and the quantitativeanalysis of cell cycle distribution is summarized inFigure 1C. 12A1 and B3 cells have comparable growthrates (doubling times 27 and 24 h, respectively; datanot shown) and similar cell cycle distributions whengrown in the presence of 10% serum (compare a' to a).After 24 h of serum depletion, a marked reduction inthe fraction of cells in S phase and an accumulation ofcells in the G1 phase were observed for both cell lines.Remarkably, a subdiploid cell population (A0) ap-peared in 12A1 cells incubated for 24 h in the presenceof 0.1% serum, whereas such a subpopulation was notdetectable in B3 cells, under the same conditions(Figure 1B, panels b' and b). The presence of thispopulation is indicative of cell death and is oftenconsidered as a marker of apoptosis (Darzynkiewicz etal., 1992). Prolonged incubation (72 h) in the presenceof 0.1% serum resulted in a signi®cant increase in thefraction of the subdiploid cell population in 12A1, butnot in B3 cells (compare c' to c). By that time, itrepresented about 60% of the whole 12A1 cellpopulation (Figure 1C), thus con®rming the dataobtained by microscopic observation. Both cell lineswere also tested for their capability to respond toserum addition following growth-factor withdrawal.Cell cycle distribution was analysed for cells ®rststarved during 72 h and then stimulated to grow bythe addition of 10% serum (d' and d). Cells of both celllines resumed DNA synthesis, as evidenced by anincrease in the fraction of S and G2/M phasepopulations. However, most of the 12A1 cells fromthe A0 population could not reenter the cell cycle,con®rming the occurrence of cell death.As chromatin fragmentation by internucleosomal
DNA cleavage is considered a hallmark of apoptosis,the appearance of a DNA ladder was investigated byagarose gel electrophoresis of genomic DNA extractedfrom serum-starved 12A1 and B3 cells (Figure 2A). Anincubation period of 24 h in 0.1% serum-containingmedium was su�cient to induce internucleosomalDNA degradation in 12A1 cells. Nucleosomal DNAfragments further accumulated in these cells uponprolonged incubation under low serum conditions. Inmarked contrast, B3 cells did not show any evidence ofDNA fragmentation, even after 72 h of incubation in0.1% serum-containing medium. As expected, supply-ing the medium with 10% serum did not result in thereversion of the degradation process in apoptotic 12A1cells.Another marker of apoptosis is the proteolytic
cleavage of the 116 kDa poly(ADP-ribose) polymerase
c-myc amplification and propensity to apoptosisM Donzelli et al
440
(PARP) into two fragments of 85 and 29 kDa by thecaspase-3/CPP32 cysteine protease (Lazebnik et al.,1994). Therefore, we analysed the status of the PARPprotein in 12A1 and B3 starved cells. Western blottingexperiments revealed that the 116 kDa protein was theonly form present in exponentially growing 12A1 andB3 cells (Figure 2B; 0 h time point). As a result ofPARP cleavage, the 85 kDa proteolytic fragment wasgenerated in 12A1 cells upon serum starvation, whilethis fragment remained undetectable in B3 cells. In12A1 cells, the 85 kDa fragment was already evidentafter only 24 h of incubation in the presence of 0.1%serum and the full-length protein appeared to becompletely cleaved after 48 h, as judged from thedisappearance of the 116 kDa band. No evidence ofPARP proteolysis was observed at any time point inserum-starved B3 cells. Apoptosis induced by serumstarvation in tumorigenic 12A1 cells was thereforeassociated with caspase(s) activation leading to PARPcleavage, as already described for apoptosis induced by
enforced c-myc expression (Kangas et al., 1998). Takentogether, these results demonstrate that serum starva-tion causes tumorigenic cells to die by apoptosis,whereas non-tumorigenic cells are fully resistant toapoptosis induction.
Regulation of c-myc gene expression in serum-starved12A1 and B3 cells
It has been previously shown that tumorigenic andnon-tumorigenic clones of the SW613-S cell line arecharacterized by di�erent levels of ampli®cation of thec-myc gene and that a high level of ampli®cationcontributes to the tumorigenic phenotype (Lavialle etal., 1988; 1989; Modjtahedi et al., 1985). To evaluatethe expression level of c-myc in serum-deprived B3 and12A1 cells, we analysed total cytoplasmic RNA byNorthern blotting with a c-myc probe (Figure 3A).Cells were incubated in the presence of 0.1% serumover a period of 72 h and then allowed to recover for
Figure 1 Nuclear morphology and cell cycle distribution analysis of serum-starved B3 and 12A1 cells. (A) Hoechst 33258 stainingof exponentially growing B3 (a) and 12A1 cells (a') and of serum-starved B3 (c) and 12A1 cells (c'). (B) Flow cytometric analysis ofthe DNA content of B3 (a ± d) and 12A1 (a' ± d') cells. Cells were exponentially growing (a, a'), serum-starved for 24 h (b, b') or for72 h (c, c'), or serum-starved for 72 h and then restimulated for 24 h with 10% serum-containing medium (d, d'). (C) Percentage ofcells in each phase of the cell cycle (G1, S, G2/M) and in the subdiploid region (A0)
c-myc amplification and propensity to apoptosisM Donzelli et al
441
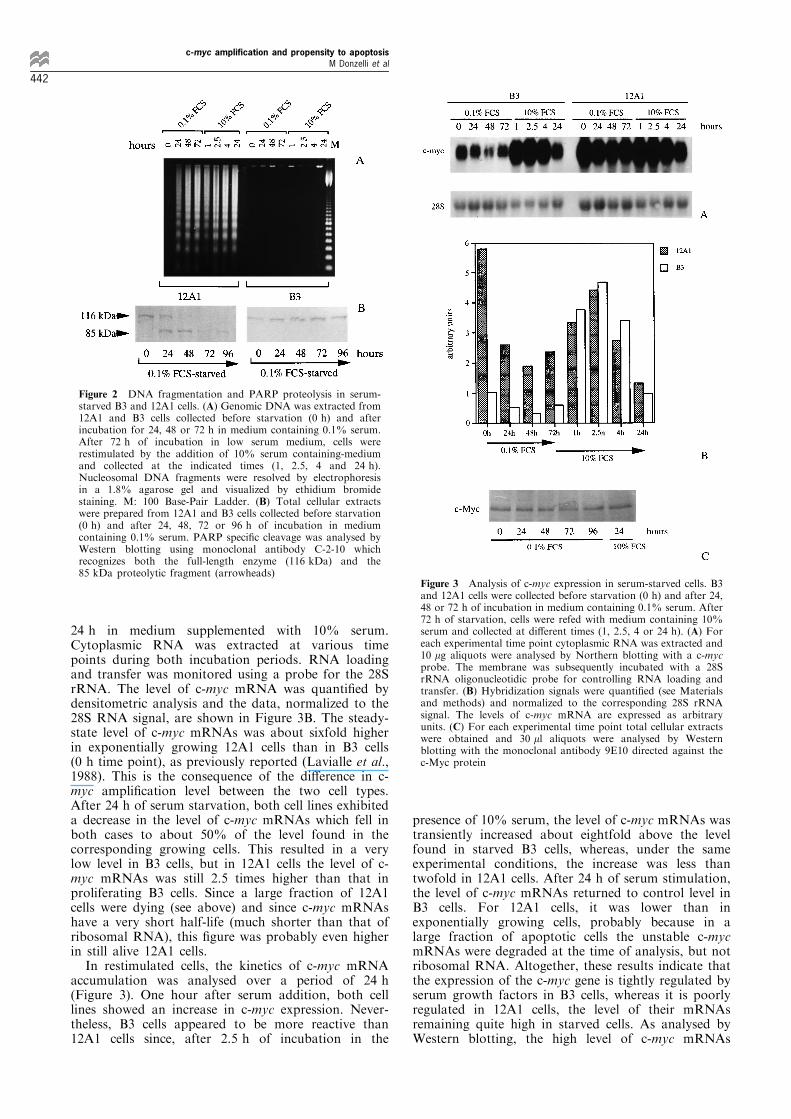
24 h in medium supplemented with 10% serum.Cytoplasmic RNA was extracted at various timepoints during both incubation periods. RNA loadingand transfer was monitored using a probe for the 28SrRNA. The level of c-myc mRNA was quanti®ed bydensitometric analysis and the data, normalized to the28S RNA signal, are shown in Figure 3B. The steady-state level of c-myc mRNAs was about sixfold higherin exponentially growing 12A1 cells than in B3 cells(0 h time point), as previously reported (Lavialle et al.,1988). This is the consequence of the di�erence in c-myc ampli®cation level between the two cell types.After 24 h of serum starvation, both cell lines exhibiteda decrease in the level of c-myc mRNAs which fell inboth cases to about 50% of the level found in thecorresponding growing cells. This resulted in a verylow level in B3 cells, but in 12A1 cells the level of c-myc mRNAs was still 2.5 times higher than that inproliferating B3 cells. Since a large fraction of 12A1cells were dying (see above) and since c-myc mRNAshave a very short half-life (much shorter than that ofribosomal RNA), this ®gure was probably even higherin still alive 12A1 cells.In restimulated cells, the kinetics of c-myc mRNA
accumulation was analysed over a period of 24 h(Figure 3). One hour after serum addition, both celllines showed an increase in c-myc expression. Never-theless, B3 cells appeared to be more reactive than12A1 cells since, after 2.5 h of incubation in the
presence of 10% serum, the level of c-myc mRNAs wastransiently increased about eightfold above the levelfound in starved B3 cells, whereas, under the sameexperimental conditions, the increase was less thantwofold in 12A1 cells. After 24 h of serum stimulation,the level of c-myc mRNAs returned to control level inB3 cells. For 12A1 cells, it was lower than inexponentially growing cells, probably because in alarge fraction of apoptotic cells the unstable c-mycmRNAs were degraded at the time of analysis, but notribosomal RNA. Altogether, these results indicate thatthe expression of the c-myc gene is tightly regulated byserum growth factors in B3 cells, whereas it is poorlyregulated in 12A1 cells, the level of their mRNAsremaining quite high in starved cells. As analysed byWestern blotting, the high level of c-myc mRNAs
Figure 2 DNA fragmentation and PARP proteolysis in serum-starved B3 and 12A1 cells. (A) Genomic DNA was extracted from12A1 and B3 cells collected before starvation (0 h) and afterincubation for 24, 48 or 72 h in medium containing 0.1% serum.After 72 h of incubation in low serum medium, cells wererestimulated by the addition of 10% serum containing-mediumand collected at the indicated times (1, 2.5, 4 and 24 h).Nucleosomal DNA fragments were resolved by electrophoresisin a 1.8% agarose gel and visualized by ethidium bromidestaining. M: 100 Base-Pair Ladder. (B) Total cellular extractswere prepared from 12A1 and B3 cells collected before starvation(0 h) and after 24, 48, 72 or 96 h of incubation in mediumcontaining 0.1% serum. PARP speci®c cleavage was analysed byWestern blotting using monoclonal antibody C-2-10 whichrecognizes both the full-length enzyme (116 kDa) and the85 kDa proteolytic fragment (arrowheads)
Figure 3 Analysis of c-myc expression in serum-starved cells. B3and 12A1 cells were collected before starvation (0 h) and after 24,48 or 72 h of incubation in medium containing 0.1% serum. After72 h of starvation, cells were refed with medium containing 10%serum and collected at di�erent times (1, 2.5, 4 or 24 h). (A) Foreach experimental time point cytoplasmic RNA was extracted and10 mg aliquots were analysed by Northern blotting with a c-mycprobe. The membrane was subsequently incubated with a 28SrRNA oligonucleotidic probe for controlling RNA loading andtransfer. (B) Hybridization signals were quanti®ed (see Materialsand methods) and normalized to the corresponding 28S rRNAsignal. The levels of c-myc mRNA are expressed as arbitraryunits. (C) For each experimental time point total cellular extractswere obtained and 30 ml aliquots were analysed by Westernblotting with the monoclonal antibody 9E10 directed against thec-Myc protein
c-myc amplification and propensity to apoptosisM Donzelli et al
442
observed in 12A1 cells resulted in a high protein levelwhich was not modulated during serum-starvation(Figure 3C). On the contrary, c-Myc was undetectableeven in exponentially growing B3 cells (data notshown). The high level of c-myc expression resultingfrom this poor regulation might contribute to theapoptosis-prone phenotype of cells from tumorigenicclones.
Transfection with an exogenous c-myc gene confers anapoptosis-prone phenotype on cells of non-tumorigenicclones
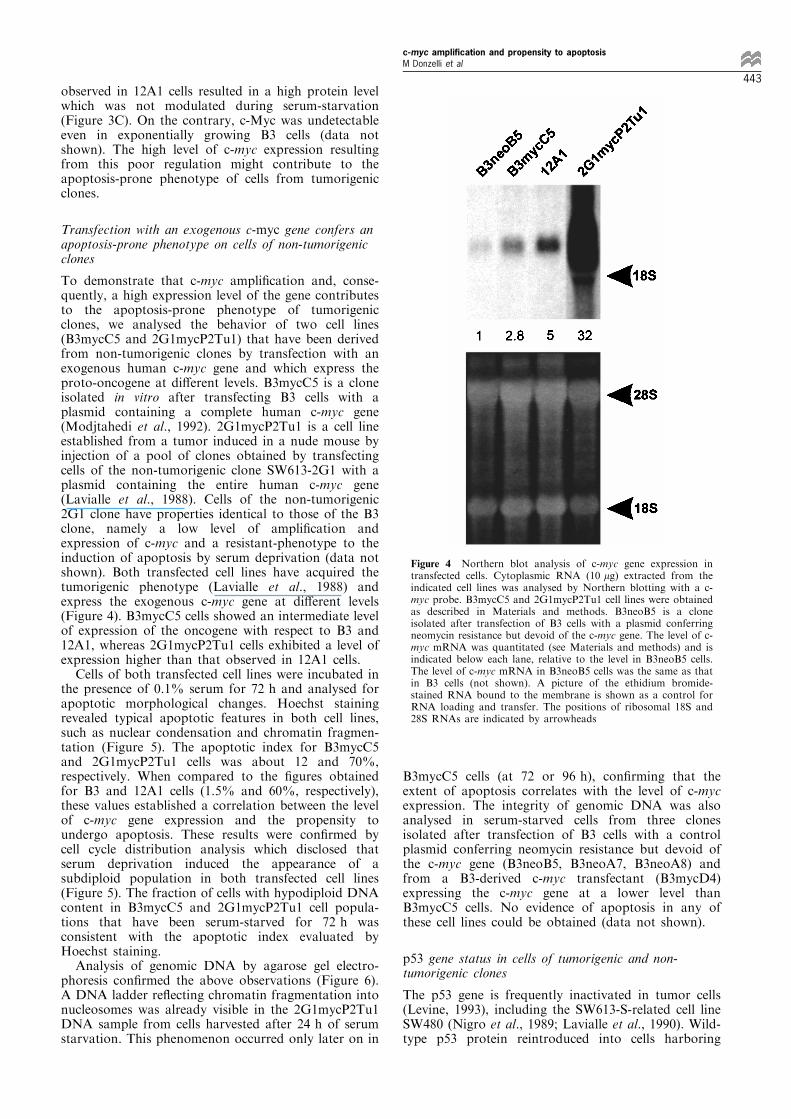
To demonstrate that c-myc ampli®cation and, conse-quently, a high expression level of the gene contributesto the apoptosis-prone phenotype of tumorigenicclones, we analysed the behavior of two cell lines(B3mycC5 and 2G1mycP2Tu1) that have been derivedfrom non-tumorigenic clones by transfection with anexogenous human c-myc gene and which express theproto-oncogene at di�erent levels. B3mycC5 is a cloneisolated in vitro after transfecting B3 cells with aplasmid containing a complete human c-myc gene(Modjtahedi et al., 1992). 2G1mycP2Tu1 is a cell lineestablished from a tumor induced in a nude mouse byinjection of a pool of clones obtained by transfectingcells of the non-tumorigenic clone SW613-2G1 with aplasmid containing the entire human c-myc gene(Lavialle et al., 1988). Cells of the non-tumorigenic2G1 clone have properties identical to those of the B3clone, namely a low level of ampli®cation andexpression of c-myc and a resistant-phenotype to theinduction of apoptosis by serum deprivation (data notshown). Both transfected cell lines have acquired thetumorigenic phenotype (Lavialle et al., 1988) andexpress the exogenous c-myc gene at di�erent levels(Figure 4). B3mycC5 cells showed an intermediate levelof expression of the oncogene with respect to B3 and12A1, whereas 2G1mycP2Tu1 cells exhibited a level ofexpression higher than that observed in 12A1 cells.Cells of both transfected cell lines were incubated in
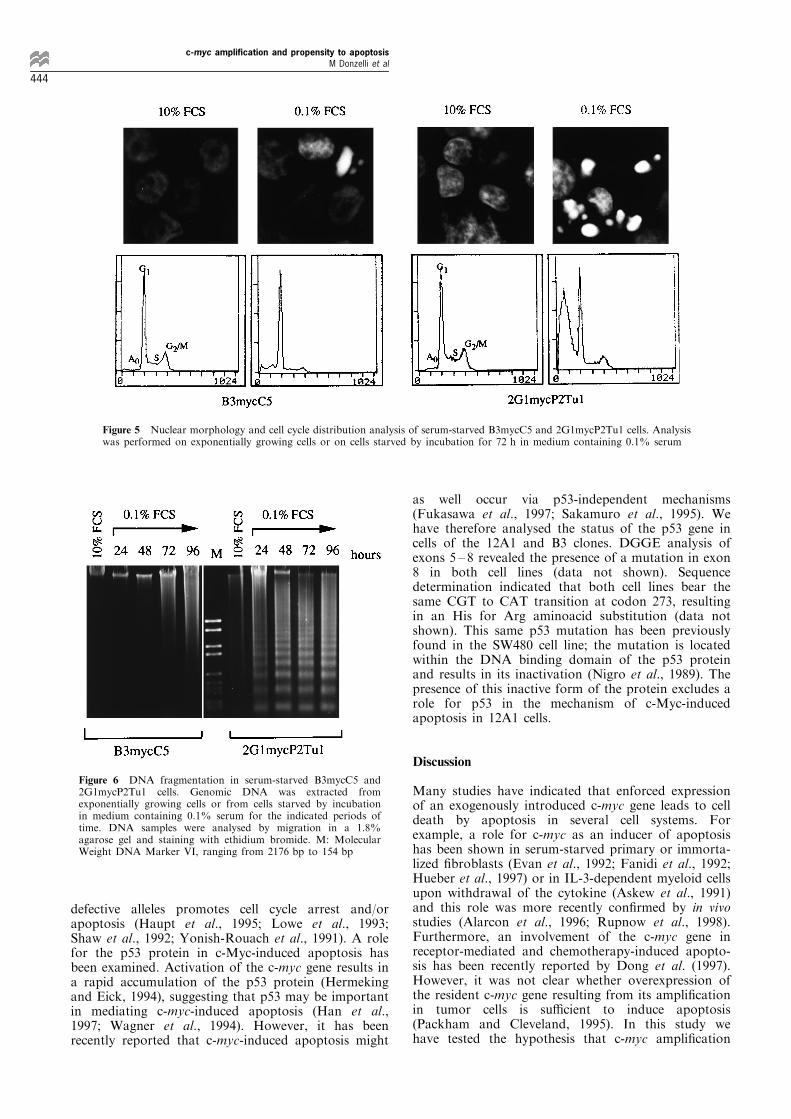
the presence of 0.1% serum for 72 h and analysed forapoptotic morphological changes. Hoechst stainingrevealed typical apoptotic features in both cell lines,such as nuclear condensation and chromatin fragmen-tation (Figure 5). The apoptotic index for B3mycC5and 2G1mycP2Tu1 cells was about 12 and 70%,respectively. When compared to the ®gures obtainedfor B3 and 12A1 cells (1.5% and 60%, respectively),these values established a correlation between the levelof c-myc gene expression and the propensity toundergo apoptosis. These results were con®rmed bycell cycle distribution analysis which disclosed thatserum deprivation induced the appearance of asubdiploid population in both transfected cell lines(Figure 5). The fraction of cells with hypodiploid DNAcontent in B3mycC5 and 2G1mycP2Tu1 cell popula-tions that have been serum-starved for 72 h wasconsistent with the apoptotic index evaluated byHoechst staining.Analysis of genomic DNA by agarose gel electro-
phoresis con®rmed the above observations (Figure 6).A DNA ladder re¯ecting chromatin fragmentation intonucleosomes was already visible in the 2G1mycP2Tu1DNA sample from cells harvested after 24 h of serumstarvation. This phenomenon occurred only later on in
B3mycC5 cells (at 72 or 96 h), con®rming that theextent of apoptosis correlates with the level of c-mycexpression. The integrity of genomic DNA was alsoanalysed in serum-starved cells from three clonesisolated after transfection of B3 cells with a controlplasmid conferring neomycin resistance but devoid ofthe c-myc gene (B3neoB5, B3neoA7, B3neoA8) andfrom a B3-derived c-myc transfectant (B3mycD4)expressing the c-myc gene at a lower level thanB3mycC5 cells. No evidence of apoptosis in any ofthese cell lines could be obtained (data not shown).
p53 gene status in cells of tumorigenic and non-tumorigenic clones
The p53 gene is frequently inactivated in tumor cells(Levine, 1993), including the SW613-S-related cell lineSW480 (Nigro et al., 1989; Lavialle et al., 1990). Wild-type p53 protein reintroduced into cells harboring
Figure 4 Northern blot analysis of c-myc gene expression intransfected cells. Cytoplasmic RNA (10 mg) extracted from theindicated cell lines was analysed by Northern blotting with a c-myc probe. B3mycC5 and 2G1mycP2Tu1 cell lines were obtainedas described in Materials and methods. B3neoB5 is a cloneisolated after transfection of B3 cells with a plasmid conferringneomycin resistance but devoid of the c-myc gene. The level of c-myc mRNA was quantitated (see Materials and methods) and isindicated below each lane, relative to the level in B3neoB5 cells.The level of c-myc mRNA in B3neoB5 cells was the same as thatin B3 cells (not shown). A picture of the ethidium bromide-stained RNA bound to the membrane is shown as a control forRNA loading and transfer. The positions of ribosomal 18S and28S RNAs are indicated by arrowheads
c-myc amplification and propensity to apoptosisM Donzelli et al
443
defective alleles promotes cell cycle arrest and/orapoptosis (Haupt et al., 1995; Lowe et al., 1993;Shaw et al., 1992; Yonish-Rouach et al., 1991). A rolefor the p53 protein in c-Myc-induced apoptosis hasbeen examined. Activation of the c-myc gene results ina rapid accumulation of the p53 protein (Hermekingand Eick, 1994), suggesting that p53 may be importantin mediating c-myc-induced apoptosis (Han et al.,1997; Wagner et al., 1994). However, it has beenrecently reported that c-myc-induced apoptosis might
as well occur via p53-independent mechanisms(Fukasawa et al., 1997; Sakamuro et al., 1995). Wehave therefore analysed the status of the p53 gene incells of the 12A1 and B3 clones. DGGE analysis ofexons 5 ± 8 revealed the presence of a mutation in exon8 in both cell lines (data not shown). Sequencedetermination indicated that both cell lines bear thesame CGT to CAT transition at codon 273, resultingin an His for Arg aminoacid substitution (data notshown). This same p53 mutation has been previouslyfound in the SW480 cell line; the mutation is locatedwithin the DNA binding domain of the p53 proteinand results in its inactivation (Nigro et al., 1989). Thepresence of this inactive form of the protein excludes arole for p53 in the mechanism of c-Myc-inducedapoptosis in 12A1 cells.
Discussion
Many studies have indicated that enforced expressionof an exogenously introduced c-myc gene leads to celldeath by apoptosis in several cell systems. Forexample, a role for c-myc as an inducer of apoptosishas been shown in serum-starved primary or immorta-lized ®broblasts (Evan et al., 1992; Fanidi et al., 1992;Hueber et al., 1997) or in IL-3-dependent myeloid cellsupon withdrawal of the cytokine (Askew et al., 1991)and this role was more recently con®rmed by in vivostudies (Alarcon et al., 1996; Rupnow et al., 1998).Furthermore, an involvement of the c-myc gene inreceptor-mediated and chemotherapy-induced apopto-sis has been recently reported by Dong et al. (1997).However, it was not clear whether overexpression ofthe resident c-myc gene resulting from its ampli®cationin tumor cells is su�cient to induce apoptosis(Packham and Cleveland, 1995). In this study wehave tested the hypothesis that c-myc ampli®cation
Figure 5 Nuclear morphology and cell cycle distribution analysis of serum-starved B3mycC5 and 2G1mycP2Tu1 cells. Analysiswas performed on exponentially growing cells or on cells starved by incubation for 72 h in medium containing 0.1% serum
Figure 6 DNA fragmentation in serum-starved B3mycC5 and2G1mycP2Tu1 cells. Genomic DNA was extracted fromexponentially growing cells or from cells starved by incubationin medium containing 0.1% serum for the indicated periods oftime. DNA samples were analysed by migration in a 1.8%agarose gel and staining with ethidium bromide. M: MolecularWeight DNA Marker VI, ranging from 2176 bp to 154 bp
c-myc amplification and propensity to apoptosisM Donzelli et al
444

could sensitize tumor cells to apoptosis. We usedtumorigenic and non-tumorigenic clones derived fromthe SW613-S human colon carcinoma cell line. Theseclones are characterized by di�erent levels of c-mycampli®cation and expression. Cells of tumorigenicclones overexpress the c-myc gene as a consequenceof its ampli®cation to a high level, whereas cells ofnon-tumorigenic clones have a low level of amplifica-tion and expression of the proto-oncogene.In this work we have shown that cells of the SW613-
12A1 clone, a representative of tumorigenic clones, aresusceptible to cell death under low serum cultureconditions. In contrast, cells of the SW613-B3 clone, arepresentative of non-tumorigenic clones, are resistantto serum deprivation. Dying 12A1 cells showedchromatin condensation, internucleosomal DNA de-gradation and proteolysis of the PARP protein,whereas these symptoms were never observed in B3cells. These observations suggested that expression ofc-myc to a high level as a result of gene ampli®cationmight contribute to the apoptosis-prone phenotype of12A1 cells. We have shown that in B3 cells c-mycexpression was down-regulated in response to serumdeprivation and that subsequent addition of serum tostarved cells led, within 2.5 h, to a transient eightfoldincrease in the level of c-myc mRNAs. These resultsindicate that in these cells c-myc expression is tightlyregulated by serum growth factors, as alreadydescribed for many other cell types (Evan andLittlewood, 1993; Spencer and Groudine, 1991). Inserum-starved 12A1 cells, the level of c-myc mRNAsdecreased, but the contribution of a downregulation ofthe expression was uncertain since a large fraction ofthe cells was committed to die. Notwithstanding, thelevel of c-myc mRNAs in starved 12A1 cells remainedhigher than in exponentially growing B3 cells. Inaddition, c-myc expression in 12A1 cells was stimulatednot more than twofold after serum addition. Alto-gether, these results suggest that, because of the highcopy number of the gene, tumorigenic cells are unableto e�ciently down-regulate the expression of c-myc inresponse to serum deprivation in order to bring thesteady-state level of the mRNA below a criticalthreshold value, that would allow them to escapeapoptotic cell death.To support this hypothesis, we investigated whether
serum starvation could induce apoptosis in cells ofstable cell lines (B3mycC5 and 2G1mycP2Tu1)obtained after transfection of two non-tumorigenicclones (SW613-B3 and SW613-2G1) with a c-myc-containing plasmid. We have shown that, in both cases,the cells have acquired an apoptosis-prone phenotypeand that, interestingly, the extent of apoptosis was incorrelation with the level of c-myc expression. High-level c-myc expression in 2G1mycP2Tu1 cells corre-lated with a massive induction of apoptosis uponserum deprivation and even the moderate increase in c-myc expression in B3mycC5 cells was apparentlysu�cient to trigger apoptosis in serum-starved cells.The tumorigenicity and the propensity to apoptosis
of SW613-S cells which overexpress the resident c-mycgene due to its ampli®cation are apparently incontradiction. This paradoxical dual role of c-myc asan inducer of both neoplastic tranformation andapoptosis can be explained by the `two signal' model®rst proposed by Bissonnette et al. (1992) and further
elaborated by Evan and Littlewood (1993). Accordingto this model, cells with deregulated c-myc genes areprimed for both proliferation and apoptosis, as anormal obligate function of the proto-oncogene. Inorder to escape apoptosis and to proliferate, the cellsrequire a second signal which can be triggered byspeci®c survival factors (Harrington et al., 1994a,b) orby the expression of anti-apoptotic genes such as bcl-2(Bissonnette et al., 1992; Fanidi et al., 1992; Reed,1997; Wagner et al., 1993). If the `two signal' modelapplies to SW613-S tumorigenic cells, one has toassume that a survival factor, essential for these cells, islacking when they are serum-starved. Harrington et al.(1994a) demonstrated that some cytokines, includingthe IGFs and PDGF, may act as survival factors andblock c-myc-induced apoptosis. Previous work ontumorigenic and non-tumorigenic SW613-S-derivedclones proved that 12A1 cells overexpress the genesof several growth factors, including PDGF-A and IGF-2, as compared to B3 cells (Galdemard et al., 1995;Lamonerie et al., 1995; Lavialle et al., 1989;Modjtahedi et al., 1992). In addition, it was shownthat tumorigenic 12A1 cells can proliferate in achemically de®ned, serum-free medium containingonly transferrin as a proteinaceous component,whereas non-tumorigenic B3 cells cannot. Moreover,we have previously reported that IGF-2 acts throughan autocrine pathway that is essential for the growth oftumorigenic cells under these serum-free cultureconditions (Lamonerie et al., 1995). Since tumorigeniccells cannot grow in de®ned medium devoid oftransferrin and since none of the growth factorsoverproduced by tumorigenic cells is apparentlysu�cient by itself to suppress c-myc-induced apoptosisin the absence of transferrin, we conclude thattransferrin is a likely candidate as a survival factorfor these cells.A role for the p53 protein in c-Myc-induced
apoptosis has been suggested (Han et al., 1997;Hermeking and Eick, 1994; Sakamuro et al., 1995;Wagner et al., 1994). Our analysis of the p53 gene statusin 12A1 and B3 cells indicated that a point mutation atcodon 273 is present in both cell lines. The SW480 cellline, which has been derived from the same patienttumor as SW613-S (Lavialle et al., 1990), bears the samemutation. The presence of this mutated form of the p53protein, which is not functional (Nigro et al., 1989),rules out a role for p53 in the mechanism of c-myc-induced apoptosis in tumorigenic SW613-S cells. Inkeeping with these observations, it has been previouslyshown that loss of wild-type p53 in colorectal tumor celllines does not a�ect sodium butyrate- or radiation-induced apoptosis (Bracey et al., 1995; Hague et al.,1993). Altogether these observations lead us to considerthat the response of colon cancer cells to growth-restrictive conditions might involve a c-myc-dependent,but p53-independent pathway. This would be atvariance with the situation described in colon adenoma(premalignant) cells in which the apoptotic response toserum starvation is apparently both c-myc- and p53-independent (Hague et al., 1997).The molecular mechanisms by which c-Myc
protein(s) triggers apoptosis are still poorly under-stood (Packham and Cleveland, 1995; Hueber et al.,1997). Recent evidences have suggested that c-Myc-induced apoptosis requires cell-surface interaction
c-myc amplification and propensity to apoptosisM Donzelli et al
445

between the CD95/Fas receptor and its ligand (Hueberet al., 1997). Preliminary results obtained by ¯owcytometric analysis have indicated that neither CD95nor its ligand can be detected at the surface of 12A1cells (unpublished data). This observation suggests thatc-Myc-induced apoptosis in cells of tumorigenicSW613-S clones does not involve the CD95 pathway.Our ®nding of an apoptosis-prone phenotype for
SW613-S cells with a high level of ampli®cation of c-myc is apparently discordant with the observation thatc-myc ampli®cation occurs quite frequently in humancarcinoma cells (Brison, 1993) and should thus conferon them a growth advantage. A high level ofampli®cation also confers on SW613-S cells atumorigenic phenotype in nude mice, i.e. a growthadvantage in vivo. It is therefore possible that apoptosisproneness is a tolerable `price to pay' by the cell tobene®t from this growth advantage, or that, in vivo,other survival factors may exert their anti-apoptoticactivity in case of serum factor shortage.It is known that colon carcinomas are very resistant
to drugs commonly used in chemotherapy (Endicottand Ling, 1989) and that a high level of ampli®cationof c-myc is rare in these tumors (Erisman et al., 1985).Actually, a recent report indicates that a fraction ofcolon carcinomas is characterized by c-myc geneampli®cation (mainly at a low level) and that thismay predispose patients to a better response totreatment (Augenlicht et al., 1997). One may wonderwhether the propensity of tumor cells with a high levelof ampli®cation of c-myc to engage in an apoptoticresponse could play a role in the induction of apoptosisby antitumoral drugs. Would the rare class of colontumors with a high level ampli®cation of c-myc beparadoxically more sensitive to chemotherapeutictreatment?
Materials and methods
Cell lines
The origin of the SW613-S cell line, which was derivedfrom a human colon carcinoma, has been previouslydescribed (Lavialle et al., 1988, 1990, 1989; Modjtahedi etal., 1985). SW613-12A1, SW613-B3 and SW613-2G1 are,respectively, a tumorigenic and two non-tumorigenic clonesderived from the parental SW613-S cell line (Lavialle et al.,1988). As previously described (Modjtahedi et al., 1992),B3mycC5 and B3mycD4 are clones derived from the non-tumorigenic SW613-B3 cell line after transfection with aplasmid containing both the neomycin-resistance gene andthe complete human c-myc gene (pSVTKneomyc1).B3neoB5, B3neoA7 and B3neoA8 are clones isolated aftertransfection of B3 cells with a plasmid conferring neomycinresistance but devoid of the c-myc gene. 2G1mycP2Tu1was established from a tumor induced in a nude mouse byinjection of a pool of neomycin-resistant colonies obtainedafter transfection of the non-tumorigenic SW613-2G1 cellswith the pSVdhfrmyc7 and pSVTKneob plasmids (Lavialleet al., 1988). The establishment procedure did not involvean in vitro cloning step.
Cell culture
Cells were maintained in DMEM (Gibco ± BRL, UK)supplemented with 10% fetal calf serum (Hyclone, NL),4 mM glutamine, 2 mM sodium pyruvate, 100 U/mlpenicillin and 0.1 mg/ml streptomycin (all from GIBCO).
Cells were grown at 378C in a humidi®ed atmospherecontaining 5% CO2. Cells were trypsinized when subcon-¯uent and seeded in 100-mm Petri dishes at a concentrationof 2.56105 cells/ml in medium containing 10% serum.Cells were grown in complete medium for up to 48 h,washed twice with DMEM and then cultured in thepresence of 0.1% serum for up to 96 h. For restimulationexperiments, cells deprived of serum as just described for72 h, were refed with medium containing 10% serum andfurther incubated for up to 24 h. For each experimentaltime point, cells were collected by trypsinization andanalysed.
Evaluation of apoptotic index
Cells were resuspended at a concentration of 106/ml in PBScontaining 10% serum and 105 cells were cytocentrifugedon a microscope slide at 500 r.p.m. for 3 min. Cells were®xed for 10 min in ice-cold 70% ethanol and washedseveral times with ice-cold PBS. DNA was then stained for10 min at room temperature with 0.1 mg/ml Hoechst 33258(Sigma, USA). Normal and apoptotic cells were countedunder ¯uorescence microscopic observation using a LeitzOrthoplan microscope equipped with a 506 objective. Theapoptotic index was expressed as the percentage of cellswith condensed or fragmented nuclei. For each sample, 500cells were counted. Pictures were obtained using a KodakTmax (400 ASA) b/w ®lm.
Cell cycle analysis
For cell cycle distribution analysis, 106 cells wereresuspended in 1 ml of cold 0.9% NaCl and then ®xedby adding 2 ml of cold ethanol. DNA was stained withpropidium iodide as previously reported (Guano et al.,1994). Ten thousands cells were analysed at each timepoint. Measurements were carried out with a Coulter EpicsXL cytometer (Coulter Co, USA). Quantitative analysis ofcell cycle distribution was performed using the softwareprovided with the instrument.
Analysis of DNA fragmentation
Cells were rinsed twice in cold PBS containing 5 mM
EDTA. Genomic DNA was extracted from 56106 cells asdescribed (Negri et al., 1995). Brie¯y, cells were resus-pended twice in a lysis bu�er containing 1% Nonidet-P40,20 mM EDTA and 50 mM Tris-HCl pH 8.0. The recoveredsupernatants were combined and incubated with 1% SDSand 0.5 mg/ml RNase A at 568C for 2 h and thereaftertreated with 1 mg/ml proteinase K at 378C for 4 h. TheDNA was precipitated by the addition of 1/10 volume of7.5 M ammonium acetate and two volumes of ethanol andanalysed by agarose gel electrophoresis. DNA wasvisualized by staining with 1 mg/ml ethidium bromide.One-hundred Base-Pair Ladder from Pharmacia (Sweden)and Molecular Weight Marker VI from BoehringerMannheim (Germany) were used as size markers.
Western blot analysis
Cells were washed twice with ice-cold PBS and resuspendedat the concentration of 56106/ml in a bu�er containing62.5 mM Tris-HCl pH 6.8, 4 M urea, 10% glycerol, 2%SDS, 5% b-mercaptoethanol and 0.003% bromophenolblue, as described by Shah et al. (1995). Cells were thendisrupted by sonication on ice, twice for 30 s (60 W). Equalvolumes (corresponding to 30 ml) of each sample wereincubated for 15 min at 658C before loading on SDS ±polyacrylamide gel. Samples were electrophoresed in a7.5% (for PARP detection) or in a 12% (for c-Mycdetection) SDS ± PAGE minigel and transferred onto a
c-myc amplification and propensity to apoptosisM Donzelli et al
446

nitrocellulose ®lter (Biorad, USA) for 3 h at 48C under aconstant voltage of 120 V (Towbin et al., 1979). Beforeincubation with antibodies, the membrane was saturatedovernight with PTN (PBS containing 0.2% Tween-20 and10% newborn calf serum). To detect PARP protein themembrane was incubated for 3 h with the monoclonalantibody C-2-10 (diluted 1 : 10 000 in PTN), whichrecognizes an epitope located between the zinc ®ngersand the automodi®cation domains, at the carboxyl-end ofthe DNA-binding domain of PARP (Lamarre et al., 1988).The antibody was kindly provided by Dr G Poirier. c-Mycdetection was carried out using the 9E10 clone from SantaCruz (USA) and the incubation was performed asdescribed above. To remove unreacted antibody themembrane was washed several times with PBS containing0.2% Tween-20. The membrane was then incubated for 2 hin the presence of anti-mouse IgG conjugated to alkalinephosphatase. Visualization of immunoreactive polypeptideswas performed using a BCIP/NBT colour developmentsubstrate (all products from Promega, USA).
Northern blot analysis
For cytoplasmic RNA isolation, about 26107 cells werescraped in growth medium, pelleted by centrifugation,washed twice with PBS and lysed with Nonidet-P40, asdescribed (Modjtahedi et al., 1985). Nuclei were sedimen-ted by low speed centrifugation, the supernatant extractedwith phenol/chloroform and the RNA precipitated underethanol. Aliquots (10 mg) of RNA were denatured byheating for 10 min at 688C in the presence of 2.2 M
formaldehyde and 50% formamide and immediatelyloaded onto a 1% agarose gel containing 2.2 M formalde-hyde (Sambrook et al., 1989). Electrophoresis wasperformed at 35 V for 14 h at room temperature. Aftertwo 15 min treatments with a solution containing 50 mM
NaOH and 10 mM NaCl, the gel was equilibrated with150 mM ammonium acetate (pH 7.5), stained with 1 mg/mlethidium bromide, destained, and the RNA was transferredonto a nylon membrane (Hybond-N, Amersham, UK),essentially as described by Dautry et al. (1988). Hybridiza-tion was carried out as previously descrived (Galdemard etal., 1995). The c-myc probe was prepared from a ClaI ±EcoRI fragment puri®ed from the pMYC-ECl plasmid(Modjtahedi et al., 1985) and labelled by the randompriming method in the presence of [a-32P]-dCTP (3000 Ci/mmol, Amersham), using the Megaprime DNA LabellingSystem (Amersham). An oligonucleotide complementary to
the human 28S rRNA (nucleotides 4011 ± 4036) was end-labeled with the T4 polynucleotide kinase in the presenceof [g-32P]-ATP (6000 Ci/mmol, Amersham) and hybridiza-tion was carried out according to Barbu and Dautry(1989). To detect the hybridization signal, membranes wereexposed to a Fuji Bio-imager plate and densitometricanalysis was performed using the MacBas 2.2 Fujiprogram. The relative c-myc mRNA content was quanti-®ed after correction for the small variations in the amountof membrane-bound RNA, as determined by hybridizationwith the 28S rRNA probe.
PCR±DGGE analysis and sequencing of p53 gene exons
DNA was extracted from B3 and 12A1 cells and regionscorresponding to exons 5 ± 8 of the p53 gene were ampli®edwith speci®c GC-clamped ampliprimers. PCR productswere analysed by DGGE as previously reported by Renaultet al. (1993). Genomic DNAs from samples showingvariant DGGE patterns were ampli®ed using the appro-priate primers without GC-clamp and the PCR productswere sequenced by the dideoxy procedure, essentially asdescribed by Pellegata et al. (1994).
AbbreviationsBCIP: 5-bromochloro-3-indolyl phosphate; DGGE: dena-turing gradient gel electrophoresis; DMEM: Dulbecco'smodi®ed Eagle medium; NBT: p-nitrotetrazolium blue;PARP: poly(ADP-ribose) polymerase; PBS: phosphatebu�ered saline; SDS ± PAGE: sodium dodecyl sulphate-polyacrylamide gel electrophoresis.
AcknowledgementsWe are greatly indebted to Dr G Poirier (Universite Laval,Que bec, Canada) for the monoclonal antibody C-2-10 andto Prof G Ranzani (IGM, UniversitaÁ di Pavia, Italy) forhelpful discussions on p53 experiments. M Donzelli wassupported by a fellowship from AIRC (AssociazioneItaliana per la Ricerca sul Cancro). C Negri and LPadovan are Telethon and Anna Villa Rusconi fellows,respectively. This work was supported in part by grantsfrom the Association pour la Recherche sur le Cancer andfrom the Ligue Nationale contre le Cancer.
References
Alarcon RM, Rupnow BA, Graeber TG, Knox SJ andGiaccia AJ. (1996). Cancer Res., 56, 4315 ± 4319.
Amati B and Land H. (1994). Curr. Opin. Genet. Dev., 4,102 ± 108.
Askew DS, Ashmun RA, Simmons BC and Cleveland JL.(1991). Oncogene, 6, 1915 ± 1922.
Augenlicht LH, Wadler S, Corner G, Richards C, Ryan L,Multani AS, Pathak S, Benson A, Haller D and HeerdtBG. (1997). Cancer Res., 57, 1769 ± 1775.
Barbu V and Dautry F. (1989). Nucleic. Acids Res., 17, 7115.Bishop JM. (1995). Genes & Dev., 9, 1309 ± 1315.Bissonnette RP, Echeverri F, Mahboubi A and Green DR.(1992). Nature, 359, 552 ± 554.
Bracey TS, Miller JC, Preece A and Paraskeva C. (1995).Oncogene, 10, 2391 ± 2396.
Brison O. (1993). Biochim. Biophys. Acta, 1155, 25 ± 41.Chinnaiyan AM, Orth K, O'Rourke K, Duan H, Poirier GGand Dixit VM. (1996). J. Biol. Chem., 271, 4573 ± 4576.
Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, HotzMA, Lassota P and Traganos F. (1992). Cytometry, 13,795 ± 808.
Dautry F, Weil D, Yu J and Dautry-Varsat A. (1988). J. Biol.Chem., 263, 17615 ± 17620.
Davis AC, Wims M, Spotts GD, Hann SR and Bradley A.(1993). Genes & Dev., 7, 671 ± 682.
Dong J, Naito M and Tsuruo T. (1997). Oncogene, 15, 639 ±647.
El-Deiry WS. (1997). Curr. Opin. Oncol., 9, 79 ± 87.Endicott JA and Ling V. (1989). Annu. Rev. Biochem., 38,137 ± 171.
Erisman MD, Rothberg PG, Diehl RE, Morse CC,Spandorfer JM and Astrin SM. (1985). Mol. Cell. Biol.,5, 1969 ± 1976.
Evan GI and Littlewood TD. (1993). Curr. Opin. Genet.Dev., 3, 44 ± 49.
c-myc amplification and propensity to apoptosisM Donzelli et al
447

Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H,Brooks M, Waters CM, Penn LZ and Hancock DC.(1992). Cell, 69, 119 ± 128.
Fanidi A, Harrington EA and Evan GI. (1992). Nature, 359,554 ± 556.
Fukasawa K, Wiener F, Vande Woude GF and Mai S.(1997). Oncogene, 15, 1295 ± 1302.
Galdemard C, Brison O and Lavialle C. (1995). Oncogene,10, 2331 ± 2342.
Guano F, Bernardi R, Donzelli M, Prosperi E, AstaldiRicotti G and Scovassi AI. (1994). Cell Death Di�., 1,101 ± 107.
Hague A, Hicks DJ, Bracey TS and Paraskeva C. (1997). Br.J. Cancer, 75, 960 ± 968.
Hague A, Manning AM, Hanlon KA, Huschtscha LI, HartD and Paraskeva C. (1993). Int. J. Cancer, 55, 498 ± 505.
Han J, Dionne CA, Kedersha NL and Goldmacher VS.(1997). Cancer Res., 57, 176 ± 182.
Harrington EA, Bennett MR, Fanidi A and Evan GI.(1994a). EMBO J., 13, 3286 ± 3295.
Harrington EA, Fanidi A and Evan GI. (1994b). Curr. Opin.Genet. Dev., 4, 120 ± 129.
Haupt Y, Rowan S and Oren M. (1995). Oncogene, 10,1563 ± 1571.
Hermeking H and Eick D. (1994). Science, 265, 2091 ± 2093.Ho�man B and Liebermann DA. (1994). Oncogene, 9, 1807 ±1812.
Hueber A-O, ZoÈ rnig M, Lyon D, Suda T, Nagata S and EvanGI. (1997). Science, 278, 1305 ± 1309.
Kangas A, Nicholson DW and HoÈ lttaÈ E. (1998). Oncogene,16, 387 ± 398.
Lamarre D, Talbot B, de Murcia G, Laplante C, Leduc Y,Mazen A and Poirier GG. (1988). Biochim. Biophys. Acta,950, 147 ± 160.
Lamonerie T, Lavialle C, Haddada H and Brison O. (1995).Int. J. Cancer, 61, 587 ± 592.
Lavialle C, Modjtahedi N, Cassingena R and Brison O.(1988). Oncogene, 3, 335 ± 339.
Lavialle C, Modjtahedi N, Cassingena R and Brison O.(1990). Oncogene, 5, 245.
Lavialle C, Modjtahedi N, Lamonerie T, Frebourg T,Landin R-M, Fossar N, Lhomond G, Cassingena R andBrison O. (1989). Anticancer Res., 9, 1265 ± 1280.
Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG andEarnshaw WC. (1994). Nature, 371, 346 ± 347.
Levine AJ. (1993). Annu. Rev. Biochem., 62, 623 ± 651.Lowe SW, Schmitt EM, Smith SW, Osborne BA and Jacks T.(1993). Nature, 362, 847 ± 849.
Marcu KB, Bossone SA and Patel AJ. (1992). Annu. Rev.Biochem., 61, 809 ± 860.
Marx J. (1993). Science, 259, 760 ± 761.Modjtahedi N, Haddada H, Lamonerie T, Lazar E, LavialleC and Brison O. (1992). Int. J. Cancer, 52, 483 ± 490.
Modjtahedi N, Lavialle C, Poupon M-F, Landin R-M,Cassingena R, Monier R and Brison O. (1985). CancerRes., 45, 4372 ± 4379.
Negri C, Bernardi R, Donzelli M and Scovassi AI. (1995).Biochimie, 77, 893 ± 899.
Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R,Cleary K, Bigner SH, Davidson N, Baylin S, Devilee P,Glover T, Collins FS, Weston A, Modali R, Harris CCand Vogelstein B. (1989). Nature, 342, 705 ± 708.
Packham G and Cleveland JL. (1995). Biochim. Biophys.Acta, 1242, 11 ± 28.
Pellegata NS, Sessa F, Renault B, Bonato M, Leone BE,Solcia E and Ranzani GN. (1994). Cancer Res., 54, 1556 ±1560.
Reed JC. (1995). Curr. Opin. Oncol., 7, 541Ð 546.Reed JC. (1997). Nature, 387, 773 ± 776.Renault B, van den Broek M, Fodde R, Wijnen J, PellegataNS, Amadori D, Khan PM and Ranzani GN. (1993).Cancer Res., 53, 2614 ± 2617.
Rupnow BA, Alarcon RM, Giaccia AJ and Knox SJ. (1998).Cell Death Di�., 5, 141 ± 147.
Sakamuro D, Eviner V, Elliott KJ, Showe L, White E andPrendergast GC. (1995). Oncogene, 11, 2411 ± 2418.
Sambrook J, Fritsch EF and Maniatis T. (1989). Cold SpringHarbor Laboratory Press: New York.
Shah GM, Poirier D, Duchaine C, Brochu G, Desnoyers S,Lagueux J, Verreault A, Ho¯ack J-C, Kirkland JB andPoirier GG. (1995). Anal. Biochem., 227, 1 ± 13.
Shaw P, Bovey R, Tardy S, Sahli R, Sordat B and Costa J.(1992). Proc. Natl. Acad. Sci. USA, 89, 4495 ± 4499.
Spencer CA and Groudine M. (1991). Adv. Cancer Res., 56,1 ± 48.
Towbin H, Staehelin T and Gordon J. (1979). Proc. Natl.Acad. Sci. USA, 76, 4350 ± 4354.
Vaux DL, Cory S and Adams J. (1988). Nature, 335, 440 ±442.
Wagner AJ, Kokontis JM and Hay N. (1994). Genes & Dev.,8, 2817 ± 2830.
Wagner AJ, Small MB and Hay N. (1993). Mol. Cell. Biol.,13, 2432 ± 2440.
Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi Aand Oren M. (1991). Nature, 352, 345 ± 347.
c-myc amplification and propensity to apoptosisM Donzelli et al
448




















