
Antinociceptive effects of N-acetylaspartylglutamate (NAAG) peptidase inhibitors ZJ-11, ZJ-17 and...
12
Antinociceptive effects of N-acetylaspartylglutamate (NAAG) peptidase inhibitors ZJ-11, ZJ-17 and ZJ-43 in the rat formalin test and in the rat neuropathic pain model Tatsuo Yamamoto, 1 Serabi Hirasawa, 1 Barbara Wroblewska, 2 Ewa Grajkowska, 4 Jia Zhou, 3 Alan Kozikowski, 3 Jarda Wroblewski 4 and Joseph H. Neale 2 1 Department of Anaesthesiology, Graduate School of Medicine, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba-shi, Chiba 260– 8670, Japan 2 Department of Biology, Georgetown University, Washington, DC, 20007, USA 3 Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at Chicago, Chicago, Illinois 60612, USA 4 Department of Pharmacology, Georgetown University, Washington, DC, 20007, USA Keywords: Fos, group II metabotropic glutamate receptors, inflammatory pain, LY-341495, mechanical allodynia, NAAG peptidase, NAAG, neuropathic pain Abstract The peptide neurotransmitter N-acetylaspartylglutamate (NAAG) acts as an agonist at group II metabotropic glutamate receptors (mGluRs). NAAG is inactivated by extracellular peptidase activity yielding glutamate and N-acetylaspartate. We recently developed a series of potent NAAG peptidase inhibitors, including ZJ-11, ZJ-17 and ZJ-43. In the present study, we examined the effects of intrathecally administered ZJ-11 and ZJ-17 and intravenously administered ZJ-11 and ZJ-43 in the rat formalin test (an inflammatory pain model) and in the rat partial sciatic nerve ligation model (a neuropathic pain model). Intrathecal injection of ZJ-11 or ZJ-17 or intravenous injection of ZJ-11 or ZJ-43 suppressed both phases of the agitation behaviour induced by paw formalin injection. Intrathecal and intravenous injection of ZJ-11 suppressed the expression of Fos-like immunoreactivity, induced by paw formalin injection, in laminae I–II in segments L4–L5 of the spinal cord, suggesting an action on sensory spinal transmission. Partial sciatic nerve ligation induced significant mechanical allodynia 7 days after the nerve injury. Intrathecal injection of ZJ-11 or ZJ-17 or intravenous administration of ZJ-11 or ZJ-43 attenuated the level of mechanical allodynia induced by this nerve ligation. These effects of intrathecally or intravenously administered ZJ compounds in both the formalin test and the partial sciatic nerve ligation model were completely antagonized by pretreatment with LY-341495, a highly selective group II mGluR antagonist. Thus, elevation of extracellular NAAG, induced by the inhibition of NAAG peptidase, activates group II mGluRs and produces an analgesic effect in neuropathic and inflammatory and pain models. In contrast, peptidase inhibition did not affect the threshold for withdrawal from a noxious mechanical stimulus or from an acute thermal stimulus in the hotplate test. Introduction N-acetylaspartylglutamate (NAAG) is the most prevalent and widely distributed peptide transmitter in the mammalian nervous system (reviewed by Neale et al., 2000). This peptide is present in highest concentration in the spinal cord (Fuhrman et al., 1994), has been identified in synaptic vesicles (Williamson & Neale, 1988; Renno et al., 1997), and has been localized in spinal sensory neurons (Cangro et al., 1987), in ascending and descending spinal tracts and in spinal interneurons (Moffett et al., 1993, 1994). NAAG is released from spinal cord cells by depolarizing stimuli (Zollinger et al., 1994). It selectively activates the type 3 metabotropic glutamate receptor (mGluR3), with 10-fold less activity at mGluR2 (Wroblewska et al., 1997; Schweitzer et al., 2000). Group II mGluRs, primarily mGluR3, are expressed by spinal and sensory neurons (Carlton et al., 2001; Ohishi et al., 1993; Boxall et al., 1998; Berthele et al., 1999; Azkue et al., 2000). Group II mGluR agonists markedly reduce monosynaptic excitation and primary afferent synaptic transmission in the spinal cord (Ishida et al., 1993; Gerber et al., 2000) and are antinociceptive in both inflammatory and neurogenic thermal hyperalgesia (Dolan & Nolan, 2000; Neugebauer, 2002; Sharpe et al., 2002). Additionally, activation of presynaptic mGluR3 by NAAG suppresses synaptic transmission (Zhao et al., 2001; Xi et al., 2002; Garrido Sanabria et al., 2004). Following release from neurons, NAAG is inactivated by at least two extracellular peptidases to produce glutamate and N-acetylaspar- tate (Riveros & Orrego, 1984; Robinson et al., 1987; Serval et al., 1990; Slusher et al., 1990; Bzdega et al., 1997; Luthi-Carter et al., 1998; Bacich et al., 2002; Bzdega et al., 2004). Inhibition of NAAG peptidase activity in the nervous system results in increased extracel- lular levels of NAAG and decreased glutamate levels under conditions of elevated neuronal activity (Slusher et al., 1999). We recently found that intrathecal injection of 2-(phosphonomethyl)pen- tanedioic acid (2-PMPA), an inhibitor of NAAG peptidase, failed to reduce the threshold for perception of acute painful thermal stimulus but did produce an analgesic effect in both phases of the formalin model and in the carrageenan model of inflammatory pain (Yamamoto et al., 2001a,b). In an elegant series of experiments, Carpenter et al. (2003) Correspondence: Dr Tatsuo Yamamoto, as above. E-mail: [email protected] Received 12 February 2004, revised 10 May 2004, accepted 14 May 2004 European Journal of Neuroscience, Vol. 20, pp. 483–494, 2004 ª Federation of European Neuroscience Societies doi:10.1111/j.1460-9568.2004.03504.x
Transcript of Antinociceptive effects of N-acetylaspartylglutamate (NAAG) peptidase inhibitors ZJ-11, ZJ-17 and...

Antinociceptive effects of N-acetylaspartylglutamate(NAAG) peptidase inhibitors ZJ-11, ZJ-17 and ZJ-43 in therat formalin test and in the rat neuropathic pain model
Tatsuo Yamamoto,1 Serabi Hirasawa,1 Barbara Wroblewska,2 Ewa Grajkowska,4 Jia Zhou,3 Alan Kozikowski,3
Jarda Wroblewski4 and Joseph H. Neale21Department of Anaesthesiology, Graduate School of Medicine, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba-shi, Chiba 260–8670, Japan2Department of Biology, Georgetown University, Washington, DC, 20007, USA3Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at Chicago, Chicago, Illinois 60612, USA4Department of Pharmacology, Georgetown University, Washington, DC, 20007, USA
Keywords: Fos, group II metabotropic glutamate receptors, inflammatory pain, LY-341495, mechanical allodynia, NAAG peptidase,NAAG, neuropathic pain
Abstract
The peptide neurotransmitter N-acetylaspartylglutamate (NAAG) acts as an agonist at group II metabotropic glutamate receptors(mGluRs). NAAG is inactivated by extracellular peptidase activity yielding glutamate and N-acetylaspartate. We recently developed aseries of potent NAAG peptidase inhibitors, including ZJ-11, ZJ-17 and ZJ-43. In the present study, we examined the effects ofintrathecally administered ZJ-11 and ZJ-17 and intravenously administered ZJ-11 and ZJ-43 in the rat formalin test (an inflammatorypain model) and in the rat partial sciatic nerve ligation model (a neuropathic pain model). Intrathecal injection of ZJ-11 or ZJ-17 orintravenous injection of ZJ-11 or ZJ-43 suppressed both phases of the agitation behaviour induced by paw formalin injection.Intrathecal and intravenous injection of ZJ-11 suppressed the expression of Fos-like immunoreactivity, induced by paw formalininjection, in laminae I–II in segments L4–L5 of the spinal cord, suggesting an action on sensory spinal transmission. Partial sciaticnerve ligation induced significant mechanical allodynia 7 days after the nerve injury. Intrathecal injection of ZJ-11 or ZJ-17 orintravenous administration of ZJ-11 or ZJ-43 attenuated the level of mechanical allodynia induced by this nerve ligation. These effectsof intrathecally or intravenously administered ZJ compounds in both the formalin test and the partial sciatic nerve ligation model werecompletely antagonized by pretreatment with LY-341495, a highly selective group II mGluR antagonist. Thus, elevation ofextracellular NAAG, induced by the inhibition of NAAG peptidase, activates group II mGluRs and produces an analgesic effect inneuropathic and inflammatory and pain models. In contrast, peptidase inhibition did not affect the threshold for withdrawal from anoxious mechanical stimulus or from an acute thermal stimulus in the hotplate test.
Introduction
N-acetylaspartylglutamate (NAAG) is the most prevalent and widely
distributed peptide transmitter in the mammalian nervous system
(reviewed by Neale et al., 2000). This peptide is present in highest
concentration in the spinal cord (Fuhrman et al., 1994), has been
identified in synaptic vesicles (Williamson &Neale, 1988; Renno et al.,
1997), and has been localized in spinal sensory neurons (Cangro et al.,
1987), in ascending and descending spinal tracts and in spinal
interneurons (Moffett et al., 1993, 1994). NAAG is released from
spinal cord cells by depolarizing stimuli (Zollinger et al., 1994). It
selectively activates the type 3 metabotropic glutamate receptor
(mGluR3), with 10-fold less activity at mGluR2 (Wroblewska et al.,
1997; Schweitzeret al., 2000).Group IImGluRs, primarilymGluR3, are
expressed by spinal and sensory neurons (Carlton et al., 2001; Ohishi
et al., 1993;Boxall et al., 1998;Berthele et al., 1999;Azkue et al., 2000).
Group II mGluR agonists markedly reducemonosynaptic excitation and
primary afferent synaptic transmission in the spinal cord (Ishida et al.,
1993; Gerber et al., 2000) and are antinociceptive in both inflammatory
and neurogenic thermal hyperalgesia (Dolan & Nolan, 2000;
Neugebauer, 2002; Sharpe et al., 2002). Additionally, activation of
presynaptic mGluR3 byNAAG suppresses synaptic transmission (Zhao
et al., 2001; Xi et al., 2002; Garrido Sanabria et al., 2004).
Following release from neurons, NAAG is inactivated by at least
two extracellular peptidases to produce glutamate and N-acetylaspar-
tate (Riveros & Orrego, 1984; Robinson et al., 1987; Serval et al.,
1990; Slusher et al., 1990; Bzdega et al., 1997; Luthi-Carter et al.,
1998; Bacich et al., 2002; Bzdega et al., 2004). Inhibition of NAAG
peptidase activity in the nervous system results in increased extracel-
lular levels of NAAG and decreased glutamate levels under conditions
of elevated neuronal activity (Slusher et al., 1999).
We recently found that intrathecal injection of 2-(phosphonomethyl)pen-
tanedioic acid (2-PMPA), an inhibitor of NAAG peptidase, failed to
reduce the threshold for perception of acute painful thermal stimulus but
did produce an analgesic effect in both phases of the formalin model and
in the carrageenan model of inflammatory pain (Yamamoto et al.,
2001a,b). In an elegant series of experiments, Carpenter et al. (2003)
Correspondence: Dr Tatsuo Yamamoto, as above.
E-mail: [email protected]
Received 12 February 2004, revised 10 May 2004, accepted 14 May 2004
European Journal of Neuroscience, Vol. 20, pp. 483–494, 2004 ª Federation of European Neuroscience Societies
doi:10.1111/j.1460-9568.2004.03504.x

assayed the effects of 2-PMPA in the isolated spinal cords of
anaesthetized rats and found that NAAG peptidase inhibition decreased
noxious evoked activity but not low threshold Ab fibre responses in
normal animals. These data suggested that NAAG peptidase plays an
important role in spinal nociceptive transmission during inflammation
and that decreasing the rate of inactivation of NAAG following synaptic
release and ⁄ or decreasing the extracellular glutamate level in the spinal
cord produces an analgesic effect during inflammation. However, the
precise mechanism through which inhibition of NAAG peptidase
produced an analgesic effect was not resolved.
In order to independently test the role of NAAG in nervous system
functions, we recently developed a series of novel urea-based NAAG
peptidase inhibitors (Nan et al., 2000; Kozikowski et al., 2001;
Kozikowski et al., 2004), including ZJ-11, ZJ-17 and ZJ-43 (Fig. 1).
In the present study, we tested the potential of these compounds to
interact with cloned mGluRs, their efficacy as inhibitors of glutamate
carboxypeptidase II (GCPII) and their efficacy in reducing pain
perception during inflammatory and neuropathic pain states. Specif-
ically, the effects of intrathecal administration of ZJ-11 and ZJ-17 and
intravenous administration of ZJ-11 and ZJ-43 were tested in the
formalin test, an inflammatory pain model, and the partial sciatic nerve
ligation model, a neuropathic pain model. The involvement of group II
mGluRs in peptidase-mediated analgesia also was tested using the
group II-selective antagonist LY341495.
Expression of the immediate–early protooncogene c-fos as assayed
by detection of Fos-like immunoreactivity (Fos-LI) has been widely
used to identify populations of neurons that are activated by noxious
stimuli (Hunt et al., 1987) and to concomitantly examine the ability of
drugs such as morphine to suppress activation of these pathways in
pain models (Hammond et al., 1998; Yamamoto et al., 2000). In the
present study, we tested the hypothesis that ZJ-11 would suppress
expression of Fos-LI in the spinal sensory pain pathway that is
activated by formalin injection in the rat footpad.
Materials and methods
The following investigations were performed according to a protocol
approved by the Institutional Animal Care Committee of Chiba
University, Chiba, Japan. Immediately following the completion of
formalin tests and partial sciatic nerve injury tests, the rats were killed
with an overdose of barbiturate.
Interactions with recominantly expressed metabotropicglutamate receptors
ZJ-11, ZJ-17 andZJ-34 (10 lm) were assayed for their ability to serve asagonists or antagonists on membranes from cell lines that expressed
group I (mGluR1a andmGluR5), group II (mGluR2 andmGluR3 [ZJ-43
only and at 100 lL) and group III (mGluR4 and mGluR6) metabotropic
glutamate receptors. Changes in the signal transduction responses
(increase in phosphoinositide hydrolysis or decrease in cyclic AMP)
were measured after the exposure to the ZJ compounds as has been
described previously (Wroblewska et al., 1997; Nan et al., 2000).
NAAG peptidase assays
Membranes from cell lines that express human and rat GCPII (Bzdega
et al., 1997; Nan et al., 2000) were assayed for their ability to
hydrolyse NAAG in vitro. Transfected cells were harvested in 50 mm
Tris–HCl buffer, pH 7.5, frozen at )80 �C, thawed and sonicated.
Membranes were sedimented in a refrigerated microcentrifuge for
15 min, washed once and resuspended in the same buffer. Protein
concentrations were determined by the bicinchoninic acid method
(Pierce, Rockford, IL, USA). NAAG peptidase activity was deter-
mined using 4 lm NAAG as a substrate. The GPCPII activity was
assayed in a two-step process. In the first step, aliquots of membranes
(4 lg in 100 lL) were incubated for 2 h at 37 �C in 50 mm Tris-HCl
(pH 7.4) in the presence of 4 lm NAAG and the given inhibitor, to
allow for the accumulation of glutamate produced in the GPCII
reaction. In the second step, the amount of accumulated glutamate was
measured using the Amplex Red Glutamic Acid Assay Kit (Molecular
Probes), and the fluorescence was read with a Dynex fluorescent plate
reader using excitation at 530 nm and emission at 590 nm. For all
inhibitors, dose–response curves were obtained and were used to
calculate IC50 values by nonlinear regression using the Sigma-Plot
software. In each series of experiments, a concentration curve of
NAAG was used to determine the Km. The final Ki values for each
Fig. 1. Chemical structures of ZJ-11, ZJ-17 and ZJ-43.
484 T. Yamamoto et al.
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494

inhibitor were calculated from the respective IC50 and Km values using
the Cheng and Prusoff equation.
Chemistry methods: preparation of ZJ-11, ZJ-17 and ZJ-43
ZJ-11, ZJ-17 and ZJ-43 were prepared following methods previously
described (Nan et al., 2000; Kozikowski et al., 2001; Kozikowski
et al., 2004; Olszewski et al., 2004). In general, these asymmetrical
ureas were prepared by the reaction of the isocyanate generated in situ
from the tosylate salt of glutamic acid dibenzyl ester and triphosgene–
Et3N with the second appropriately protected amino acid benzyl ester
component. The benzyl ester intermediates were then converted to the
free acids by catalytic hydrogenation to deliver the desired ureas.
Intrathecal catheters
Chronic intrathecal catheters were inserted, during halothane anaes-
thesia, by passing a PE-10 catheter through an incision in the atlanta–
occipital membrane to a position 8 cm caudal to the cisterna at the
level of lumbar enlargement (Yaksh & Rudy, 1976; Yamamoto et al.,
2001b).
Formalin test
The formalin test was excuted according to the protocol in Yamamoto
et al. (2001b). Briefly, rats were lightly anaesthetized with halothane
and the dorsal surface of the right hindpaw was injectd with 50 lL of
5% formalin, the animal was placed in a plexiglass box and the
spontaneous flinching of the paw as observed. For the intrathecal (IT)
dose–response study, ZJ-11 or ZJ-17 was administered intrathecally
10 min before the formalin injection. IT catheters were inserted 7 days
before the formalin injection. For the intravenous (IV) dose–response
study, ZJ-11 or ZJ-43 was administered intravenously 10 min before
the formalin injection. To verify that the analgesic effect of ZJ
compounds is mediated by the activation of mGluR3, 1 mg ⁄ kg of
LY-341495, a Group II mGluR antagonist, was administered intra-
peritoneally 20 min before either the intrathecal administration or
intravenous administration of ZJ compounds and formalin was
injected 10 min after the administration of ZJ compounds. To examine
the effect of intraperitoneal injection of 1 mg ⁄ kg of LY-341495 itself,
LY-341495 was administered intraperitoneally 20 min before either
the intrathecal administration or the intravenous administration of
saline and formalin was injected 10 min after the saline administra-
tion.
Partial sciatic nerve injury model
Anaesthesia was induced by inhalation of 5% halothane, maintained at
a concentration of 2–3%, as needed. After a local incision, the biceps
femoralis of right leg was bluntly dissected at mid-thigh to expose the
sciatic nerve. The right sciatic nerve was carefully mobilized with care
being taken to avoid undue stretching. After the mobilization of the
sciatic nerve, an 8–0 silicon-treated silk suture was inserted into the
right sciatic nerve just proximal to the sciatic trifurcation with a 3 ⁄ 8curved, reversed-cutting mini-needle, and tightly ligated so that the
dorsal 1 ⁄ 3–1 ⁄ 2 of the nerve thickness was trapped in the ligature
(Seltzer et al., 1990). The incision was closed layer to layer, with 3–0
silk suture, and the rats were allowed to recover from the anaesthetics.
All animals postoperatively displayed normal feeding and drinking
behaviour.
Mechanical thresholds were measured using von Frey filaments
with logarithmically incremental stiffness (0.41, 0.70, 1.20, 2.00,
3.63, 5.50, 8.50 and 15.1 g; Stoelting, Wood Dale, IL, USA) to
calculate the 50% probability thresholds for mechanical paw
withdrawal, as previously described (Chaplan et al., 1994). For the
measurement of mechanical threshold in the nerve injury model, rats
were placed in a plastic cage with a wire mesh bottom. Beginning
with the 2.00-g probe, filaments were applied to the plantar surface
of a right (nerve injured) hind paw for 6–8 s, in a stepwise
ascending or descending order following negative or positive
withdrawal responses, respectively, until six consecutive responses
were noted. Stimuli were presented at intervals of several seconds,
allowing for apparent resolution of any behavioural responses to
previous stimuli. Fifty per cent probability thresholds were calcu-
lated according to the method of Dixson (1980). In cases where
continuous positive or negative responses were observed to the
exhaustion of the stimulus set, values of 15.00 and 0.25 g,
respectively, were assigned.
A preliminary study performed revealed that the maximum level of
mechanical allodynia occurred 7 days after the nerve injury and lasted
for > 2 weeks after the nerve injury. Thus, we administered drugs
7 days after the partial sciatic nerve injury. Before and 7 days after the
nerve injury, the mechanical threshold of the right hind paw (nerve-
injured paw) was measured as control data. For the IT dose–response
study, ZJ-11 or ZJ-17 was administered intrathecally, and the right
hind paw was tested at 5, 15, 30, 45 and 60 min after the drug
administration. IT catheters were inserted 3 days before the nerve
injury. For the IV dose–response study, ZJ-11 or ZJ-43 was
administered intravenously and the right hind paw was tested at 5,
15, 30, 45 and 60 min after the drug administration. To verify that the
analgesic effect of ZJ compounds is mediated by the activation of
mGluR3, 1 mg ⁄ kg of LY-341495 was administered intraperitoneally
30 min before the administration of ZJ compounds. To examine the
effect of intraperitoneal injection of 1 mg ⁄ kg of LY-341495 itself,
LY-341495 was administered intraperitoneally 30 min before either
the intrathecal administration or the intravenous administration of
saline and the right hind paw was tested at 5, 15, 30, 45 and 60 min
after the saline administration.
For the measurement of the effect of NAAG peptidase inhibition
on the response to noxious mechanical stimulus (mechanical
threshold), rats were placed in a plastic cage with a wire mesh
bottom and allowed to habituate for a period of � 5 min. After
acclimation, pain-related behaviours were induced with a von Frey
monofilament (46.5 g) which was applied to the plantar surface of
the right and left hind paw every 2 s for 40 s (10 stimuli to each
hind paw). Before and 5, 15, 30, 45 and 60 min after the drug
administration (ZJ-11, ZJ-43 and saline for IV study and ZJ-11 and
saline for IT study), the number of paw withdrawal responses was
measured.
The hotplate test was carried out to assess the effect of NAAG
peptidase inhibitors on the thermal nociceptive threshold. Rats were
placed on 52.5 �C hotplate. The response latency to either a hindpaw
lick or jump was recorded. In the absence of a response, the animals
were removed from the hotplate at 60 s to avoid tissue injury, and a
60 s latency was assigned as the response.
Two baseline measurements were made before the drug adminis-
tration. For the IT study, ZJ-11 or saline was administered intrathecally
and the hotplate latency was measured at 5, 15, 30, 45 and 60 min
after the drug injection. For the IV study, ZJ-11, ZJ-43 or saline was
administered intravenously and the hotplate latency was measured at
5, 15, 30, 45 and 60 min after the drug injection.
NAAG peptidase inhibitor and analgesia 485
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494

Behavioural analysis
The general behaviour of each rat was carefully observed and tested.
Motor functions were evaluated by the performance of two specific
behavioural tasks. (i) The placing ⁄ stepping reflex: this response was
evoked by drawing the dorsum of either hindpaw over the edge of a
table top. In normal animals, this stimulus elicits an upward lifting of
the paw onto the surface of the table, called stepping. Animals with
any degree of hind limb flaccidity will demonstrate an altered or
absent reflex. (ii) The righting reflex: an animal placed horizontally
with its back on the table will normally show an immediate
coordinated twisting of the body around its longitudinal axis to regain
its normal position on its feet. Animals displaying ataxic behaviour
will show a decreased ability to right themselves. To quantify the
evaluation of motor functions, both tasks were scored on a scale of
0–2 in which 0 ¼ absence of function and 2 ¼ normal motor
functions. Animals that were able to perform the motor tasks but
did so more slowly than normal animals were assigned a score of 1.
Drugs
The intrathecally administered drugs were delivered in a total volume
of 10 lL followed by 10 lL of saline to flush the catheter. The
intravenously administered drugs were delivered in a total volume of
1 mL. For the intravenous administration, drugs were administered via
the tail vein. The agents used in this study were ZJ-11 (molecular
weight 322.3; Fig. 1), ZJ-17 (molecular weight 354.3; Fig. 1), and
ZJ-43 (molecular weight 304.3; Fig. 1). LY-341495, a very highly
selective group II metabotropic glutamate receptor antagonist (Kings-
ton et al., 1998).
Assay of Fos-like immunoreactivity
Fos-LI was assayed and quantified using methods previously
described (Yamamoto et al., 2000). For the IT study, a most-
effective dose (1000 lg) of ZJ-11 or saline was administered
intrathecally 10 min before the formalin injection, and the expression
of Fos-LI was examined 2 h after the formalin injection. For the IV
study, a most-effective dose (100 mg ⁄ kg) of ZJ-11 or saline was
administered intravenously 10 min before the formalin injection, and
the expression of Fos-LI was examined 2 h after the formalin
injection.
Statistical analyses
Formalin test
The time-response data are presented as the mean flinches (±SEM)
per min for the periods of 1–2 min and 5–6 min and then for
1-min periods at 5-min intervals up to 60 min. For the dose–
response analysis, data from phase 1 (0–6 min) and phase 2
(10–60 min) observations were considered separately. In each case,
the cumulative instances of formalin-evoked flinches during the
phase 1 and phase 2 were calculated for each rat. These individual
rat data were then used to construct phase 1 and phase 2 dose–
response curves. To compare the dose–response curves between
phase 1 and phase 2, the percentage of the phase 1 and phase 2
vehicle responses (% vehicle response) was calculated. To evaluate
the dose-dependence, one-way anova was used. For multiple
comparison, Dunnett’s test was used. To compare the dose–
response curve of phase 1 with that of phase 2 of each drug, two-
way anova was used. To evaluate the effect of LY-341495 on the
analgesic effect of ZJ compounds, the unpaired t-test (two-tailed)
was used.
Partial sciatic nerve ligation model
In the present study, we estimated the level of mechanical allodynia by
50% probability threshold for mechanical paw withdrawal of the right
(nerve injured) hind paw. To determine whether partial sciatic nerve
ligation induced significant mechanical allodynia, we compared the
preinjury mechanical threshold with predrug (7 days after nerve
injury) mechanical threshold with an unpaired t-test (two-tailed). To
analyse the effect of drugs on the mechanical threshold, the maximum
50% probability threshold was used. The maximum 50% probability
threshold was defined as the maximum value of the 50% probability
threshold during the entire time course. To analyse the dose-
dependency, one-way anova with Dunnett’s test was used. To
evaluate the effect of LY-341495 on the antiallodynic effect of ZJ
compounds, the unpaired t-test (two-tailed) was used.
Noxious mechanical stimulus model
To compare the base-line data (before the drug administration)
between groups, one-way analysis of variance (one way anova; IV
study) or t-test (IT study) was used. The time–response data are
presented as the mean number of paw withdrawal responses (± SEM)
induced by the 20 applications (10 stimuli to each hind paw) of a von
Frey filament. To analyse the effect of ZJ compounds on the
mechanical threshold, the minimum and maximum number of paw
withdrawal responses during the entire time course was used. To
analyse the drug effects, one-way anova (IV study) or t-test (IT study)
was used.
Thermal stimulus test
To compare the baseline hotplate latencies between groups, one-way
anova (IV study) or t-test (IT study) was used. To analyse the effects
of ZJ compounds on the hotplate test, the per cent maximum possible
effect (%MPE) was calculated, where %MPE ¼ ([postdrug maximum
response latency – predrug response latency] ⁄ [cut-off time (60 s) –
predrug response latency]) · 100. The postdrug maximum response
latency was defined as the single longest response latency during the
first 30 min after the intrathecal drug injection. To analyse the drug
effects, one-way anova (IV study) or t-test (IT study) was used.
Immunohistochemical study
For comparison of the number of Fos-LI neurons between 1000 lg of
ZJ-11 IT-treated group and saline IT-treated group or between
100 mg ⁄ kg of ZJ-11 IV-treated group and saline IV-treated group,
the unpaired t-test (two-tailed) was used.
Wherever appropriate, results are expressed as mean ± SEM.
Critical values that reached a P < 0.05 level of significance were
considered statistically significant.
Results
ZJ compounds
For inhibition of cloned human GCPII, ZJ-11, ZJ-17 and ZJ-43 had Ki
values of 1.4, 3.0 and 0.8 nm, respectively, under assay conditions in
which 2-PMPA had a Ki of 1.4 nm. For the recombinantly expressed
rat GCPII, the Ki values were 6, 1.5 and 3 nm.
486 T. Yamamoto et al.
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494

ZJ-17, ZJ-11 and ZJ-43 failed to significantly interact at 10 lm as
agonists or antagonists with recombinantly expressed group I
metabotropic receptors (mGluR1 and 5), group II (mGluR2 and
mGluR3) or group III (mGluR4, 6 and 8) as shown in Table 1.
Behavioural analysis
After the IT injection of ZJ-11, ZJ-17 or saline and the IV injection of
ZJ-11, ZJ-43 or saline, all animals scored 2 (normal motor function) in
the placing ⁄ stepping reflex and righting reflex tests at doses applied in
this study. Similarly, systemic ZJ-43 had no significant effect on open
field locomotor behaviour in the same strain of rats (Olszewski et al.,
2004).
Formalin test
In the saline-treated rats (both IT and IV injection), a subcutaneous
injection of formalin resulted in a highly reliable biphasic display of
Fig. 2. Effects of intrathecal injection of 1000 lg of NAAG peptidase inhibitors ZJ-11, 1000 lg of ZJ-17 and saline, and effects of intravenous injection of100 mg ⁄ kg of ZJ-11, 100 mg ⁄ kg of ZJ-43 and saline on the time course of the flinches observed after the formalin injection into the dorsal surface of the ratright hindpaw. Drugs were administered 10 min before the formalin injection. The effects of pretreatment with 1 mg ⁄ kg of the group II mGluR antagonistLY341495 on the effect of the ZJ compounds were also plotted. Effects of 1 mg ⁄ kg of LY341495 + saline (intrathecal or intravenous) also are presented.LY341495 was injected intraperitoneally 20 min before either ZJ compound injection or saline injection. The number of flinches ⁄min is plotted vs. time after theformalin injection. Each point represents the mean response and SEM (n ¼ 5 or 6).
Table 1. Agonist and antagonist activity of ZJ compounds at recombinantly expressed mGluRs
Agonist activity(percentage of agonist activity)
Antagonist activity(percentage of inhibition)
ZJ-11 ZJ-17 ZJ-43 ZJ-11 ZJ-17 ZJ-43
mGluR1 0.4 ± 0.8 0.7 ± 0.3 )2.8 ± 2.2 1.8 ± 2.5 2.8 ± 6.1 3.3 ± 3.9mGluR5 4.2 ± 4.0 1.4 ± 1.5 )0.6 ± 0.3 4.0 ± 5.4 2.8 ± 7.3 5.1 ± 2.4mGluR2 4.0 ± 2.6 )1.8 ± 4.5 )3.0 ± 5.1 3.3 ± 2.4 )0.5 ± 2.0 4.0 ± 3.1mGluR3 NT NT 8.0 ± 4.2 NT NT 3.6 ± 2.0mGluR4 5.6 ± 5.7 )0.9 ± 2.9 3.2 ± 4.9 0.5 ± 4.5 )3.4 ± 2.1 5.8 ± 7.8mGluR6 3.9 ± 6.6 )4.8 ± 7.3 )0.1 ± 3.4 )4.2 ± 8.3 )4.8 ± 9.4 1.2 ± 4.8mGluR8 2.2 ± 5.9 4.9 ± 3.8 )2.5 ± 7.9 2.4 ± 4.7 2.0 ± 5.5 )2.5 ± 3.6
Activity assays were performed on membranes of cell lines expressing the individual mGluRs. All ZJ compounds were used at 10 lm concentrations (exceptmGluR3, at 100 lm). Agonist activity is expressed as a percentage of maximal activity induced by the agonist. Antagonist activity is expressed as the percentage ofinhibition obtained in the presence of the agonist. In inhibition experiments the following concentrations of agonists were used: 30 lm Glu for mGluR1, 10 lm Glufor mGluR5, 2 lm Glu for mGluR2, 100 lm for mGluR3, 1 lm AP4 for mGluR4, 0.5 lm AP4 for mGluR6 and 0.75 lm AP4 for mGluR8. Results are expressed asmeans ± SEM from 6–12 experiments. ZJ-11 and ZJ-17 were not tested (NT) against mGluR3.
NAAG peptidase inhibitor and analgesia 487
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494

flinching of the injected paw (Fig. 2), and this behaviour was similar to
that previously reported (Wheeler-Aceto et al., 1990; Yamamoto et al.,
2001b).
IT administration of either ZJ-11 or ZJ-17 decreased the cumulative
instances of formalin-evoked flinches of phase 1 and phase 2 (Fig. 2)
and this effect was dose-dependent (Fig. 3: ZJ-11, phase 1 P < 0.05,
phase 2 P < 0.001; ZJ-17, phase 1 P < 0.05, phase 2 P < 0.001; one-
way anova). While there was a significant effect of ZJ-11 and ZJ-17
on phase 2 at 100 lg and the phase 1 response was not significantly
affected by this dose of the peptidase inhibitor, the difference between
the dose–response curves of the phase 1 and phase 2 responses in
either IT ZJ-11 or ZJ-17 group did not reach statistical significance
(Fig. 3: P > 0.2, two-way anova). Pre-treatment with the group II
antagonist, LY-341495, blocked the effect of IT injection of 1000 lgof either ZJ-11 or ZJ-17 (Fig. 2: ZJ-11, phase 1 P < 0.01; phase 2
P < 0.001; ZJ-17, phase 1 P < 0.001, phase 2 P < 0.001, one-way
anova). Intraperitoneal injection of 1 mg ⁄ kg of LY-341495 + IT
saline injection had no effect on the cumulative instances of formalin-
evoked flinches of phase 1 and phase 2 as compared with the IT
saline-treated rats (Fig. 2: phase 1 P > 0.5, phase 2 P > 0.8, t-test).
IV administration of either ZJ-11 or ZJ-43 (Fig. 2) decreased the
cumulative instances of formalin-evoked flinches of phase 1 and
phase 2 in a dose-dependent manner (Fig. 3: ZJ-11, phase 1 P < 0.05,
phase 2 P < 0.005; ZJ-43, phase 1 P < 0.005, phase 2 P < 0.001;
one-way anova). There was no significant difference between the
dose–response curve of the phase 1 response and that of phase 2 in
either the IV ZJ-11 group or the IV ZJ-43 group (Fig. 3: P > 0.2, two-
way anova). Pre-treatment with LY-341495 significantly antagonized
the effect of IV administration of 100 mg ⁄ kg of ZJ-11 or ZJ-43
(Fig. 2: ZJ-11, phase 1 P < 0.05, phase 2 P < 0.005; ZJ-43, phase 1
P < 0.05, phase 2 P < 0.001; one-way anova). Intraperitoneal
injection of 1 mg ⁄ kg of LY-341495 + IV saline injection had no
effect on the cumulative instances of formalin-evoked flinches in
phase 1 or phase 2 compared with the IV saline-treated rats (Fig. 2:
phase 1 P > 0.5, phase 2 P > 0.8; t-test).
Partial sciatic nerve ligation model study
In the study of peptidase inhibitors administered by IT injection, the
level of 50% probability threshold of the injured paw 7 days after the
nerve injury was significantly less than the preinjury level (preinjury,
14.7 ± 1.2 g; 7 days after the nerve injury, 2.4 ± 1.4 g; n ¼ 53,
P < 0.001, t-test). This indicated that partial sciatic nerve injury
induced significant mechanical allodynia 7 days after the nerve
injury. There were no differences in the predrug (7 days after the
nerve injury, Fig. 4) level of 50% probability threshold between any
groups.
IT injection of either 100 or 1000 lg of ZJ-17 or IT injection of
1000 lg of ZJ-11 significantly increased the level of maximum 50%
probability threshold as compared with the saline-treated rats (Figs 4
and 5: ZJ-11, P < 0.001; ZJ-17, P < 0.001, one-way anova). Pre-
treatment with LY-341495 significantly antagonized the effect of IT
injection of either 1000 lg of ZJ-11 or 1000 lg of ZJ-17 (Fig. 4:
Fig. 3. Dose–response curves for intrathecal injection of ZJ-11 and ZJ-17 and intravenous administration of ZJ-11 and ZJ-43. The cumulative instances offormalin-evoked flinches in rats, expressed as a percentage of vehicle (saline)-evoked flinches during phase 1 and phase 2 of the formalin test are presented.Drugs were administered intrathecally 10 min before the formalin injection. Each point represents the mean and SEM of five or six rats. The abscissa showsthe log dose (lg in the intrathecal study and mg ⁄ kg in the intravenous study) and the ordinate shows the percentage of vehicle (saline) control flinches duringphase 1 and phase 2. *P < 0.05 as compared with responses at 10 lg of ZJ-11, ZJ-17 or ZJ-43.
488 T. Yamamoto et al.
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494

ZJ-11, P < 0.001; ZJ-17, P < 0.001, one-way anova). Intraperitoneal
injection of 1 mg ⁄ kg of LY-341495 + IT saline injection had no effect
on the level of maximum 50% probability threshold as compared with
the IT saline-treated rats (Fig. 4: P > 0.7, t-test).
In the study of peptidase inhibitors administered by IV injection, the
level of 50% probability threshold of the injured paw 7 days after the
nerve injury again was significantly less than preinjury level of 50%
probability threshold (preinjury, 15.0 ± 0 g; 7 days after the nerve
injury, 1.7 ± 0.5 g; n ¼ 50, P < 0.001, t-test). There were no
differences in the predrug level of 50% probability threshold among
these groups.
IV injection of either 30 or 100 mg ⁄ kg of ZJ-11 or IV injection of
100 mg ⁄ kg of ZJ-43 significantly increased the level of maximum
50% probability threshold as compared with the saline-treated rats
(Figs 4 and 5: ZJ-11, P < 0.001; ZJ-43, P < 0.001; one-way anova).
Pre-treatment with LY-341495 completely blocked the effect of IV
injection of either 100 mg ⁄ kg of ZJ-11 or 100 mg ⁄ kg of ZJ-43 (Fig. 4:ZJ-11, P < 0.001; ZJ-43, P < 0.001; one-way anova). Intraperitoneal
injection of 1 mg ⁄ kg of LY-341495 + IV saline administration had no
effect on the level of maximum 50% probability threshold as compared
with the IV saline-treated rats (Fig. 4: P > 0.7, t-test).
Mechanical and thermal threshold tests
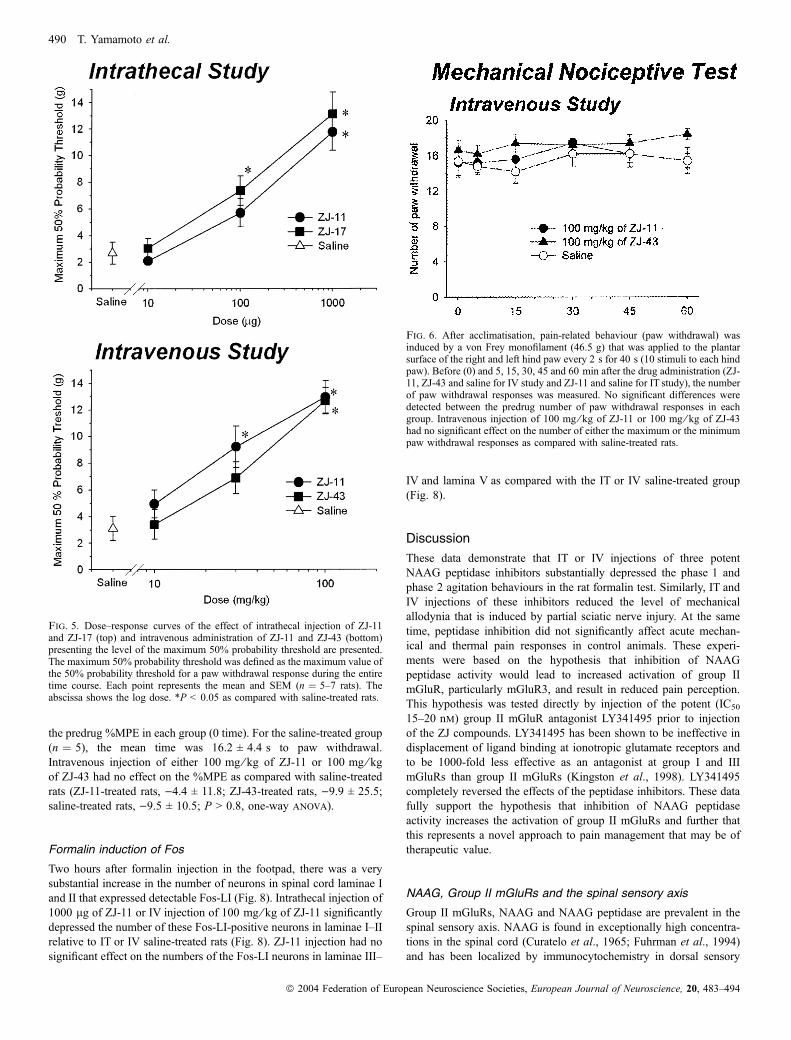
No difference was apparent between predrug number of paw
withdrawal responses to the mechanical stimuli in each group
following intrathecal administration (rats treated with 1000 lg of
ZJ-11, n ¼ 5, 13.2 ± 3.7; saline-treated rats, n ¼ 5, 13.4 ± 2.7;
P > 0.9, t-test). Intrathecal injection of 1000 lg of ZJ-11 had no
effect on the number of either the maximum or the minimum paw
withdrawal responses as compared with saline-treated rats (maximum
paw withdrawal responses: ZJ-11-treated rats, 15.6 ± 2.1; saline-
treated rats, 15.0 ± 2.0, P > 0.6; minimum paw withdrawal responses:
ZJ-11-treated rats, 13.0 ± 2.1; saline-treated rats, 12.0 ± 2.8; P > 0.5,
t-test).
Following intravenous injection, no difference was apparent
between predrug number of paw withdrawal responses (Fig. 6, time 0)
in each group. In the saline-treated group (n ¼ 5), the mean value was
15.4 ± 3.6 paw withdrawals. Intravenous injection of either
100 mg ⁄ kg of ZJ-11 or 100 mg ⁄ kg of ZJ-43 had no effect on the
number of either the maximum or the minimum paw withdrawal
responses as compared with saline-treated rats (maximum paw
withdrawal responses: ZJ-11-treated rats, 18.4 ± 1.5; ZJ-43-treated
rats, 18.8 ± 0.8; saline-treated rats, 17.0 ± 1.9; P > 0.1; minimum
paw withdrawal responses: ZJ-11-treated rats, 14.0 ± 3.2; ZJ-43-
treated rats, 15.6 ± 1.8; saline-treated rats, 13.4 ± 2.3; P > 0.3, one-
way anova).
In the hotplate assay following intrathecal injection of peptidase
inhibitor, no difference was apparent between the predrug % MPE in
each group (rats treated with 1000 lg of ZJ-11, n ¼ 5, 19.2 ± 4.7 s;
saline-treated rats, n ¼ 5, 14.7 ± 4.4 s; P > 0.1, t-test). Intrathecal
injection of 1000 lg of ZJ-11 had no effect on the %MPE as
compared with saline-treated rats (ZJ-11-treated rats, 13.4 ± 19.3;
saline-treated rats, 11.5 ± 15.2; P > 0.8, t-test).
Following intravenous injection of peptidase inhibitors in this
thermal stimulus assay (Fig. 7), no difference was apparent between
Fig. 4. Effects of intrathecal injection of 1000 lg of ZJ-11, 1000 lg of ZJ-17 and saline, and effects of intravenous injection of 100 mg ⁄ kg of ZJ-11, 100 mg ⁄ kgof ZJ-43 and saline on the time courses of the mechanical threshold in the partial sciatic nerve ligation model are presented. Drugs were administered 7 days after thenerve lesion. The effects of pretreatment with 1 mg ⁄ kg of LY341495 on the effect of either intrathecal or intravenous injection of ZJ compounds also are presented.LY341495 was injected intraperitoneally 30 min before the either ZJ compound injection or saline injection. Ordinate, 50% probability threshold of the nerve-injuredpaw (g); abscissa, time after drug administration (min). Each line represents the group mean and SEM of five to seven rats.
NAAG peptidase inhibitor and analgesia 489
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494
the predrug %MPE in each group (0 time). For the saline-treated group
(n ¼ 5), the mean time was 16.2 ± 4.4 s to paw withdrawal.
Intravenous injection of either 100 mg ⁄ kg of ZJ-11 or 100 mg ⁄ kgof ZJ-43 had no effect on the %MPE as compared with saline-treated
rats (ZJ-11-treated rats, )4.4 ± 11.8; ZJ-43-treated rats, )9.9 ± 25.5;
saline-treated rats, )9.5 ± 10.5; P > 0.8, one-way anova).
Formalin induction of Fos
Two hours after formalin injection in the footpad, there was a very
substantial increase in the number of neurons in spinal cord laminae I
and II that expressed detectable Fos-LI (Fig. 8). Intrathecal injection of
1000 lg of ZJ-11 or IV injection of 100 mg ⁄ kg of ZJ-11 significantly
depressed the number of these Fos-LI-positive neurons in laminae I–II
relative to IT or IV saline-treated rats (Fig. 8). ZJ-11 injection had no
significant effect on the numbers of the Fos-LI neurons in laminae III–
IV and lamina V as compared with the IT or IV saline-treated group
(Fig. 8).
Discussion
These data demonstrate that IT or IV injections of three potent
NAAG peptidase inhibitors substantially depressed the phase 1 and
phase 2 agitation behaviours in the rat formalin test. Similarly, IT and
IV injections of these inhibitors reduced the level of mechanical
allodynia that is induced by partial sciatic nerve injury. At the same
time, peptidase inhibition did not significantly affect acute mechan-
ical and thermal pain responses in control animals. These experi-
ments were based on the hypothesis that inhibition of NAAG
peptidase activity would lead to increased activation of group II
mGluR, particularly mGluR3, and result in reduced pain perception.
This hypothesis was tested directly by injection of the potent (IC50
15–20 nm) group II mGluR antagonist LY341495 prior to injection
of the ZJ compounds. LY341495 has been shown to be ineffective in
displacement of ligand binding at ionotropic glutamate receptors and
to be 1000-fold less effective as an antagonist at group I and III
mGluRs than group II mGluRs (Kingston et al., 1998). LY341495
completely reversed the effects of the peptidase inhibitors. These data
fully support the hypothesis that inhibition of NAAG peptidase
activity increases the activation of group II mGluRs and further that
this represents a novel approach to pain management that may be of
therapeutic value.
NAAG, Group II mGluRs and the spinal sensory axis
Group II mGluRs, NAAG and NAAG peptidase are prevalent in the
spinal sensory axis. NAAG is found in exceptionally high concentra-
tions in the spinal cord (Curatelo et al., 1965; Fuhrman et al., 1994)
and has been localized by immunocytochemistry in dorsal sensory
Fig. 5. Dose–response curves of the effect of intrathecal injection of ZJ-11and ZJ-17 (top) and intravenous administration of ZJ-11 and ZJ-43 (bottom)presenting the level of the maximum 50% probability threshold are presented.The maximum 50% probability threshold was defined as the maximum value ofthe 50% probability threshold for a paw withdrawal response during the entiretime course. Each point represents the mean and SEM (n ¼ 5–7 rats). Theabscissa shows the log dose. *P < 0.05 as compared with saline-treated rats.
Fig. 6. After acclimatisation, pain-related behaviour (paw withdrawal) wasinduced by a von Frey monofilament (46.5 g) that was applied to the plantarsurface of the right and left hind paw every 2 s for 40 s (10 stimuli to each hindpaw). Before (0) and 5, 15, 30, 45 and 60 min after the drug administration (ZJ-11, ZJ-43 and saline for IV study and ZJ-11 and saline for IT study), the numberof paw withdrawal responses was measured. No significant differences weredetected between the predrug number of paw withdrawal responses in eachgroup. Intravenous injection of 100 mg ⁄ kg of ZJ-11 or 100 mg ⁄ kg of ZJ-43had no significant effect on the number of either the maximum or the minimumpaw withdrawal responses as compared with saline-treated rats.
490 T. Yamamoto et al.
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494

neurons, spinal interneurons, motoneurons and ascending and des-
cending spinal tract axons (Cangro et al., 1987; Moffett et al., 1993).
NAAG immunoreactivity is highest in rat spinal layers III–X and
lower in layers I–II with the exception of large-caliber dorsal root
fibers that pass through layers I–II and terminate in layers III–X
(Moffett et al., 1993, 1994). NAAG is released from spinal cord
neurons in a calcium-dependent manner following depolarization
(Zollinger et al., 1994). Substantial levels of NAAG peptidase activity
have been identified in spinal cord (Fuhrman et al., 1994; Bacich
et al., 2001) and dense NAAG peptidase immunoreactivity has been
reported in dorsal horn laminae I and II, while mild staining has been
detected in lamina X (Slusher et al., 1992). Group II mGluR,
predominantly mGluR3, message is expressed in spinal cord (Ohishi
et al., 1993; Jia et al., 1999). Carlton et al. (2001) also localized these
receptors in both the myelinated and unmyelinated central axons and
peripheral neurites of dorsal root ganglion cells. The highest density of
group II mGluRs is found in lamina IIi with lesser amounts of
immunoreactivity in laminae III and IV. Transection studies suggested
that most, but not all, of these receptors are localized on primary
afferent terminals (Carlton et al., 2001). These data provide a
substantial framework within which to interpret the influence of
NAAG peptidase inhibition on pain perception at the spinal sensory
level.
Site of analgesic action of NAAG peptidase inhibition
Neither the IT nor IV data permit a definitive conclusion as to the site
of action of the peptidase inhibitors with respect to analgesia.
However, the efficacy of IT and IV injection of ZJ-11 in reducing the
formalin-induced increase in Fos-LI in laminae I-II, but not in laminae
III-V, is highly supportive of the hypothesis that systemically applied
ZJ-11 suppresses the effect of nociceptive input from the primary
afferents. Intrathecal morphine administration has a similar effect in
the rat formalin test and on the reduction of inflammation-induced Fos
expression (Yamamoto et al., 2000). The observation that the
responses in both phases of the formalin test were decreased by
peptidase inhibition might suggest actions on primary afferents and
spinal cord neurons. Three reports are consistent with a role for
peripheral neurons in NAAG peptidase-induced analgesia. NAAG
peptidase inhibition by 2-PMPA attenuated afferent discharge in the
partial sciatic nerve ligation model (Chen et al., 2002). Injection of
group II agonists into the plantar surface of the mouse hindpaw did not
alter mechanical thresholds, but did moderate the mechanical thresh-
old increase induced by PGE2 or carrageenan (Yang & Gereau, 2003).
The conclusion that the sensory spinal synapse is an important site of
action of NAAG peptidase inhibitors in reducing pain perception is
further supported by the recent observation that 2-PMPA decreased
noxious evoked activity but not low threshold Ab fibre responses in
Fig. 8. Effect of intrathecal injection of 1000 lg of ZJ-11 and the effect ofintravenous injection of 100 mg ⁄ kg of ZJ-11 on the number of Fos-LI neuronsin different laminae of spinal segments L4 and L5 ipsilateral to the site offormalin injection. Drugs were administered 10 min before the formalininjection and the expression of Fos-LI was examined in tissue fixed 2 h afterthe formalin injection. Either intrathecal injection or intravenous injection ofZJ-11 decreased the number of Fos-LI neurons in laminae I–II, but not inlaminae III–V as compared with the saline-treated rats. Each bar represents thegroup mean and SEM of five rats.
Fig. 7. The hotplate test was carried out to assess the effect of NAAG pept-idase inhibitors on the thermal nociceptive threshold. Rats were placed on a52.5 �C hotplate. The response latency to either a hindpaw lick or jump wasrecorded. Two baseline measurements were made before the drug administra-tion. ZJ-11, ZJ-43 or saline was administered intravenously and the hotplatelatency was measured prior to (0) and at 5, 15, 30, 45 and 60 min after the druginjection. No significant differences were apparent between the predrug %MPEin each group. Intravenous injection of 100 mg ⁄ kg of ZJ-11 or 100 mg ⁄ kg ofZJ-43 had no significant effect on the %MPE as compared with saline-treatedrats.
NAAG peptidase inhibitor and analgesia 491
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494

normal animals (Carpenter et al., 2003). Strikingly, following
carrageenan-induced inflammation, 10-fold lower amounts of 2-PMPA
decreased the evoked activity. These reports together with the data
presented here suggest a significant role for sensory neurons in the
analgesia provide by NAAG peptidase inhibitors. The greater efficacy
of NAAG at mGluR3 and the low level of expression of mGluR2 in
the rat spinal cord (Ohishi et al., 1993) additionally emphasize the
potential role of mGluR3 in this process.
Group II mGluRs and pain perception
IT injection of a group II mGluR agonist significantly increased
mechanical withdrawal threshold in an inflammatory pain model in
adult female sheep (Dolan & Nolan, 2000). Similarly, IP injection of a
selective group II mGluR agonist, LY379268, reduced both
inflammatory and neurogenic thermal hyperalgesia in rats but did
not affect withdrawal latencies to mechanical or thermal stimulation
(Sharpe et al., 2002). Using group II- and group III-selective mGluR
agonists, Gerber et al. (2000) demonstrated that activation of receptors
in either group suppressed synaptic transmission via presynaptic
receptors on primary afferents in rat spinal cord slices. Similarly, a
group II agonist inhibited the C-fibre evoked response of dorsal horn
neurons in the rat carrageenan test (Stanfa & Dickenson, 1998). While
the mechanism by which group II mGluR activation contributes to
analgesia is unknown, these data suggest that inhibition of glutamate
release from primary afferent terminals may be involved. Consistent
with this, group II mGluR agonist inhibition of primary afferent
trasnsmission in spinal cord has been reported (Ishida et al., 1993).
Acting at presynaptic mGluR3 receptors, NAAG inhibits transmitter
release from cortical neurons in culture (Zhao et al., 2001) and
hippocampal neurons (Anwyl, 1999). More relevant to the actions of
the ZJ compounds, NAAG peptidase inhibition in hippocampal slices
reduces glutamate release via a presynaptic mechanism (Garrido
Sanabria et al., 2004).
The absence of an effect of LY341495 alone on pain perception
suggests that in these models the rate of NAAG release in the presence
of normal NAAG peptidase activity is not sufficient to provide
sustained activation of relevant group II mGluRs to overcome
consequences of the painful stimulus. As is the case for many peptide
cotransmitters, the rate of release of NAAG relative to the release of
small amine transmitters from the same cells is not known. It is clear,
however, that NAAG is rapidly hydrolysed by extracellular peptidase
activity in the nervous system (Robinson et al., 1987; Serval et al.,
1990; Slusher et al., 1990; Bzdega et al., 1997; Luthi-Carter et al.,
1998; Bacich et al., 2002; Bzdega et al., 2004). It may be, for
example, that the highest release of NAAG occurs outside the laminae
in which mGluR receptor activation is required to achieve analgesia.
In the absence of peptidase inhibition, hydrolysis would preclude the
extended diffusion required to reach these receptors. The data from
this study would suggest that inhibition of inactivation of NAAG
permits the extracellular peptide levels to rise to or to maintain an
elevated steady state for a longer period and to activate group II
mGluRs.
Specificity of ZJ actions
The three peptidase inhibitors used in this study were selected from a
group of more than 100 NAAG analogues that we have synthesized.
ZJ-11, -17 and -43 are among a group with considerable potency as
inhibitors of cloned human, rat and mouse NAAG peptidases.
Confidence in the conclusion that ZJ compounds are achieving
analgesia by elevating NAAG levels derives from several sources.
First, these actions are similar to those obtained by a structurally
different peptidase inhibitor, 2-PMPA. We have previously
demonstrated that this compound was antinociceptive in the formalin
pain model and in mechanical allodynia induced by carrageenan,
while also lacking an effect in the hotplate test (Yamamoto et al.,
2001a,b). Second, similar results are obtained from three different
potent peptidase inhibitors in our urea-based series of compounds. For
reasons of economy, we did not test all three compounds by both
routes of administration. These compounds do not significantly
interact with metabotropic glutamate receptors. ZJ-11 and ZJ-43
(100 lm) previously were found to be without significant activity in
competitive binding assays for a series of neurotransmitter receptors,
including NMDA receptors (Olszewski et al., 2004). Additionally,
ZJ-43 has been found to substantially reduce some of the schizophre-
nia-like behaviours induced by phencyclidine administration and this
action is similarly blocked by the group II antagonist LY341495
(Olszewski et al., 2004).
The data obtained using the group II mGluR antagonist, LY341495,
in these experiments militate against a direct role for the NMDA
receptor in the analgesic effects of this series of peptidase inhibitors.
The question of potential NMDA involvement arises because IT
injection of an NMDA receptor antagonist produced analgesia in the
formalin pain model (Yamamoto & Yaksh, 1992). However, ZJ-43
(100 lm) is without agonist or antagonist activity at NMDA receptor-
induced conductances or antagonist activity on glutamate-mediated
spontaneous end plate currents in cerebellar granule neurons
(Olszewski et al., 2004). Additionally, based on indirect data, some
have speculated that NAAG may act as a partial agonist at this
receptor without directly testing this concept (Puttfarcken et al., 1993;
Greene, 2001). We recently tested this hypothesis directly and found
no evidence of a partial agonist action of NAAG at NMDA receptors
or on NMDA-mediated spontaneous postsynaptic currents (Losi et al.,
2004).
Efficacy of ZJ compounds
The ZJ compounds are potent inhibitors of GCPIII. In preliminary
assays, ZJ-11, ZJ-17 and ZJ-43 also were found to inhibit the NAAG
peptidase activity of recombinantly expressed mouse GCPIII with Ki
values of � 25 nm. Nonetheless, relatively high concentrations of
these inhibitors were required for efficacy in pain assays following
both IT and IV administration. This is not surprising in that these
compounds are hydrophilic and highly charged, qualities that are
likely to limit diffusion into the spinal cord and passage via the blood–
brain barrier. In the intrathecal injection experiments, relatively high
amounts of these compounds were required to achieve effects in either
pain model. Indeed, we found that intrathecal injection of 100 lg of
2-PMPA attenuated the level of the agitation behaviour in the rat
formalin test (Yamamoto et al., 2001b) while10-fold higher amounts
of intrathecal ZJ-11 and ZJ-17 than 2-PMPA were required to obtain
maximal analgesia in this model. The difficulty with this mode of
administration as a means of assessing biological activity of a
compound relates to the requirement that the test compound be
sufficiently lipid soluble to penetrate the substantial layer of myeli-
nated tracks that surround the grey matter of the cord if it is to act
there. Given the amounts of ZJ-11 and ZJ-17 that were administered
intrathecally, it is not possible to determine whether the site of action
of the drug was in the spinal cord or whether a significant quantity of
peptidase inhibitor diffused within the intrathecal space to the brain or
sensory ganglia.
492 T. Yamamoto et al.
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494

Taken together, these data suggest that NAAG peptidase activity
represents a significant target in the development of analgesic
compounds that function by enhancing normal processes of synaptic
transmission. Additionally important from a therapeutic perspective,
NAAG peptidase inhibition does not interfere directly with the ability
to detect and respond to acute mechanical and thermal stimuli under
normal conditions.
Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research (B) ofJapan 12470315 (T.Y.) and by NIH grants NS38080 and NS42672.
Abbreviations
2-PMPA, 2-(phosphonomethyl)pentanedioic acid; Fos-LI, Fos-like immunore-activity ⁄ immunoreactive; GCPII, glutamate carboxypeptidase II; IT, intrathe-cal; IV, intravenous; mGluR, metabotropic glutamate receptor; %MPE, per centmaximum possible effect; NAAG, N-acetylaspartylglutamate.
References
Anwyl, R. (1999) Metabotropic glutamate receptors: electrophysiologicalproperties and role in plasticity. Brain Res. Rev., 29, 83–120.
Azkue, J.J., Mateos, J.M., Elezgarai, I., Benitez, R., Osorio, A., Diez, J.,Bilbao, A., Bidaurrazaga, A. & Grandes, P. (2000) The metabotropicglutamate receptor subtype mGluR 2 ⁄ 3 is located at extrasynaptic loci in ratspinal dorsal horn synapses. Neurosci. Lett., 287, 236–238.
Bacich, D.J., Pinto, J.T., Tong, W.P. & Heston, W.D. (2001) Cloning,expression, genomic localization, and enzymatic activities of the mousehomolog of prostate-specific membrane antigen ⁄NAALADase ⁄ folatehydrolase. Mamm. Genome., 12, 117–123.
Bacich, D.J., Ramadan, E., O’Keefe, D.S., Bukhari, N., Wegorzewska, I.,Ojeifo, O., Olszewski, R., Wrenn, C.C., Bzdega, T., Wroblewska, B.,Heston, W.D.W. & Neale, J.H. (2002) Deletion of the glutamatecarboxypeptidase II gene I mice reveals a second enzyme activity thathydrolyzes N-acetylaspartylglutamate. J. Neurochem., 83, 20–29.
Berthele, A., Platzer, S., Laurie, D.J., Weis, S., Sommer, B., Zieglgansberger, W.,Conrad,B.&Tolle, T.R. (1999)Expressionofmetabotropic glutamate receptorsubtype mRNA (mGluR1–8) in human cerebellum. Neuroreport, 10, 3861–3867.
Boxall, S.J., Berthele, A., Laurie, D.J., Sommer, B., Zieglgansberger, W.,Urban, L. & Tolle, T.R. (1998) Enhanced expression of metabotropicglutamate receptor 3 messenger RNA in the rat spinal cord during ultravioletirradiation induced peripheral inflammation. Neuroscience, 82, 591–602.
Bzdega, T., Crowe, S.L., Ramadan, E.R., Sciarretta, K.H., Olszewski, R.,Ojeifo, O.A., Rafalski, V.A., Wroblewska, B. & Neale, J.H. (2004) Thecloning and characterization of a second brain enzyme with NAAG peptidaseactivity. J. Neurochem., 89, 627–635.
Bzdega, T., Turi, T., Wroblewska, B., She, D., Chung, H.S., Kim, H. & Neale,J.H. (1997) Molecular cloning of a peptidase against N-acetylaspartylglu-tamate from a rat hippocampal cDNA library. J. Neurochem., 69, 2270–2277.
Cangro, C.B., Namboodiri, M.A.A., Sklar, L.A., Corigliano-Murphy, A. &Neale, J.H. (1987) Immunohistochemistry and biosynthesis of N-acetylas-partylglutamate in spinal sensory ganglia. J. Neurochem., 49, 1579–1588.
Carlton, S.M., Hargett, G.L. & Coggeshall, R.E. (2001) Localization ofmetabotropic glutamate receptors 2 ⁄ 3 on primary afferent axons in the rat.Neuroscience, 105, 957–969.
Carpenter, K.J., Sen, S., Matthews, E.A., Flatters, S.L., Wozniak, K.M.,Slusher, B.S. & Dickenson, A.H. (2003) Effects of GCP-II inhibition onresponses of dorsal horn neurones after inflammation and neuropathy: anelectrophysiological study in the rat. Neuropeptides, 37, 298–306.
Chaplan, S.R., Bach, F.W., Pogrel, J.W., Chung, J.M. & Yaksh, T.L. (1994)Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci.Meth., 53, 55–63.
Chen, S.-R., Wozniak, K.M., Slusher, B.S. & Pan, H.L. (2002) Effect of2-(phosphono-methyl)-pentanedioic acid on allodynia and afferent ectopicdischarges in rat model of neuropathic pain. J. Pharmacol. Exp. Ther., 300,662–667.
Curatelo, A., D’Archangelo, P., Lino, A. & Brancati, A. (1965) Distribution ofN-acetyl-aspartic and N-acetyl-aspartyl-glutamic acids in nervous tissue.J. Neurochem., 12, 339–342.
Dixson, W.J. (1980) Efficient analysis of experimental observations. Annu. Rev.Pharmacol. Toxicol., 20, 441–462.
Dolan, S. & Nolan, A.M. (2000) Behavioral evidence supporting a differentrole for group I and II metabotropic glutamate receptors in spinal nociceptivetransmission. Neuropharmacology, 39, 1132–1138.
Fuhrman, S., Neale, J.H., Cassidy, M. & Palkovits, M. (1994) The regionaldistribution of N-acetylaspartylglutamate (NAAG) and peptidase activityagainst NAAG in the rat nervous system. J. Neurochem., 62, 275–281.
Garrido Sanabria, E.R.,Wozniak, K.M., Slusher, B.S. &Keller, A. (2004) GCPII(NAALADase) inhibition suppresses mossy fiber-CA3 synaptic neurotrans-mission by a presynaptic mechanism. J. Neurophysiol., 91, 182–193.
Gerber, G., Zhong, J., Youn, D. & Randic, M. (2000) Group II and group IIImetabotropic glutamate receptor agonists depress synaptic transmission inthe rat spinal cord dorsal horn. Neuroscience, 100, 393–406.
Greene, R. (2001) Circuit analysis of NMDAR hypofunction in the hippo-campus, in vitro, and psychosis of schizophrenia.Hippocampus, 11, 569–577.
Hammond, D.L., Wang, H., Nakashima, N. & Basbaum, A.I. (1998)Differential effects of intrathecally administered delta and mu opioidreceptor agonist on formalin-evoked nociception and on the expression ofFos-like immunoreacrivity in the spinal cord of the rat. J. Pharmacol. Exp.Ther., 284, 378–387.
Hunt, S.P., Pini, A. & Evan, G. (1987) Induction of c-fos-like protein in spinalcord neurons following sensory stimulation. Nature, 328, 632–634.
Ishida, M., Saitoh, T., Shimamoto, K., Ohfune, Y. & Shinozaki, H. (1993) Anovel metabotropic glutamate receptor agonist: marked depression ofmonosynaptic excitation in the newborn rat isolated spinal cord. Br. J.Pharmacol., 109, 1169–1177.
Jia, H., Rustioni, A. & Valtschanoff, J.G. (1999) Metabotropic glutamatereceptors in superficial laminea of the rat dorsal horn. J. Comp. Neurol., 410,627–642.
Kingston, A.E., Ornstein, P.L., Wright, R.A., Johnson, B.G., Mayne, N.G.,Burnet, J.P., Belagaje, R., Wu, S. & Schoepp, D.D. (1998) LY341495 is ananomolar potent and selective antagonist of group II metabotropicglutamate receptor. Neuropharmacology, 37, 1–12.
Kozikowski, A.P., Nan, F., Conti, P., Zhang, J., Ramadan, E., Bzdega, T.,Wroblewska, B., Neale, J.H., Pshenichkin, S. & Wroblewski, J.T. (2001)Design of remarkably simple, yet potent urea-based inhibitors of glutamatecarboxypeptidase II (NAALADase). J. Med. Chem., 44, 298–301.
Kozikowski, A.P., Zhang, J., Nan, F., Petukhov, P., Grajkowska, E., Wroblewski,J.T.,Yamamoto,T.,Bzdega,T.,Wroblewska,B.&Neale, J.H. (2004)Synthesisof urea-based inhibitors as active site probes of glutamate carboxypeptidase II:efficacy as analgesic agents. J. Med. Chem., 47, 1729–1737.
Losi, G., Vicini, S. & Neale, J. (2004) NAAG fails to antagonize synaptic andextrasynaptic NMDA receptors in cerebellar granule neurons. Neurophar-macology, 46, 490–496.
Luthi-Carter, R., Berger, U.V., Barczak, A.K., Enna, M. & Coyle, J.T. (1998)Isolation and expression of a rat brain cDNA encoding glutamatecarboxypeptidase II. Proc. Natl Acad. Sci. USA, 95, 3215–3220.
Moffett, J.R., Namboodiri, M.A.A. & Neale, J.H. (1993) Enhancedcarbodiimide immunohistochemistry: Application to the comparativedistributions of N-acetylaspartylglutamate and N-acetylaspartate immunor-eactivities in the rat brain. J. Histochem. Cytochem., 41, 559–570.
Moffett, J.R., Palkovits, M., Namboodiri, M.A.A. & Neale, J.H. (1994)Comparative distribution of N-acetylaspartylglutamate and GAD-67 in thecerebellum and precerebellar nuclei of the rat utilizing enhanced carbodii-mide fixation and immunocytochemistry. J. Comp. Neurol., 346, 2–22.
Nan, F., Bzdega, T., Pshenichkin, S., Wroblewski, J.T., Wroblewska, B., Neale,J.H. & Kozikowski, A.P. (2000) Dual function glutamate-related ligands:discovery of a novel, potent inhibitor of glutamate carboxypeptidase IIpossessing mGluR3 agonist activity. J. Med. Chem., 43, 772–774.
Neale, J.H., Bzdega, B. & Wroblewska, B. (2000) N-acetylaspartylglutamate:The most abundant peptide neurotransmitter in the mammalian centralnervous system. J. Neurochem., 75, 443–452.
Neugebauer, V. (2002) Metabotropic glutamate receptors – important modu-lators of nociception and pain behavior. Pain, 98, 1–8.
Ohishi, H., Shigemoto, R., Nakanishi, S. & Mizuno, N. (1993) Distribution ofthe messenger RNA for a metabotropic glutamate receptor, mGluR2, in thecentral nervous system of the rat. Neuroscience, 53, 1009–1018.
Olszewski, R., Buhkari, N., Zhou, J., Kozikowski, A., Wroblewski, J.,Shamimi-Noori, S., Wroblewska, B., Bzdega, T., Barton, F., Vicini, S. &Neale, J.H. (2004) NAAG peptidase inhibition reduces locomotor activity
NAAG peptidase inhibitor and analgesia 493
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494

and some stereopypes in the PCP model of schizophrenia via group IImGluR. J. Neurochem., 89, 876–885.
Puttfarcken, P.S., Handen, J.S., Montgomery, D.T., Coyle, J.T. & Werling, L.L.(1993) N-acetyl-aspartylglutamate modulation of N-methyl-D-aspartate-stimulated [3H]norepinephrine release from rat hippocampal slices.J. Pharmacol. Exp. Ther., 266, 796–803.
Renno, W.M., Lee, J.H. & Beitz, A.J. (1997) Light and electron microscopicimmunohistochemical localization of N-acetylaspartylglutamate (NAAG) inthe olivocerebellar pathway of the rat. Synapse, 26, 140–154.
Riveros, N. & Orrego, F. (1984) A study of possible excitatory effects ofN-acetylaspartylglutamate in different in vivo and in vitro brain preparations.Brain Res., 299, 393–395.
Robinson, M.B., Blakely, R.D., Couto, R. & Coyle, J.T. (1987) Hydrolysis ofthe brain dipeptide N-acetyl-L-aspartyl-L-glutamate. Identification andcharacterization of a novel N-acetylated alpha- linked acidic dipeptidaseactivity from rat brain. J. Biol. Chem., 262, 14498–14506.
Schweitzer, C., Kratzeisen, C., Adam, G., Lundstrom, K., Malherbe, P.,Ohresser, S., Stadler, H., Wichmann, J., Woltering, T. & Mutel, V. (2000)Characterization of [(3)H]-LY354740 binding to rat mGlu2 and mGlu3receptors expressed in CHO cells using semliki forest virus vectors.Neuropharmacology, 39, 1700–1706.
Seltzer, Z., Dubner, R. & Shir, Y. (1990) A novel behavioral model ofneuropathic pain disorders produced in rats by partial sciatic nerve injury.Pain, 43, 205–218.
Serval, V., Barbeito, L., Pittaluga, A., Cheramy, A., Lavielle, S. & Glowinski, J.(1990) Competitive inhibition of N-acetylated-alpha-linked acidic dipepti-dase activity by N-acetyl-L-aspartyl-beta-linked 1-glutamate. J. Neurochem.,55, 39–46.
Sharpe, E.F., Kingston, A.E., Lodge, D., Monn, J.A. & Headley, P.M. (2002)Systemic pre-treatment with a group II mGlu agonist, LY379268, reduceshyperalgesia in vivo. Br. J. Pharmacol., 135, 1255–1262.
Slusher, B.S., Robinson, M.B., Tsai, G., Simmons, M.L., Richards, S.S. &Coyle, J.T. (1990) Rat brain N-acetylated alpha-linked acidic dipeptidaseactivity. Purification and immunologic characterization. J. Biol. Chem., 265,21297–21301.
Slusher, B.S., Tsai, G., Yoo, G. & Coyle, J.T. (1992) Immunocytochemicallocalization of the N-acetyl-aspartyl-glutamate (NAAG) hydrolyzing enzymeN-acetylated alpha-linked acidic dipeptidase (NAALADase). J. Comp.Neurol., 315, 217–229.
Slusher, B.S., Vornov, J.J., Thomas,A.G., Hurn, P.D., Harukuni, I., Bhardwaj,A.,Traystman, R.J., Robinson, M.B., Britton, P., May Lu, X.-C., Tortella, F.C.,Wozniak, K.M., Yudkoff, M., Potter, B.M. & Jackson, P.F. (1999) Selective
inhibition of NAALADase, which converts NAAG to glutamate, reducesischemic brain injury. Nature Med., 5, 1396–1402.
Stanfa, L.C. & Dickenson, A.H. (1998) Inflammation alters the effects of mGlureceptor agonist on spinal nociceptive neurons. Eur. J. Pharmacol., 347,165–172.
Wheeler-Aceto, H., Porreca, F. & Cowan, A. (1990) The rat formalin test:comparison of noxious agents. Pain, 40, 229–238.
Williamson, L.C. & Neale, J.H. (1988) Calcium-dependent release ofN-acetylaspartylglutamate from retinal neurons upon depolarization. BrainRes., 475, 151–155.
Wroblewska, B., Wroblewski, J.T., Pshenichkin, S., Surin, A., Sullivan, S.E. &Neale, J.H. (1997) N-acetylaspartylglutamate selectively activates mGluR3receptors in transfected cells. J. Neurochem., 69, 174–181.
Xi, Z.-X., Baker, D.A., Shen, H., Carson, D.S. & Kalivas, P.W. (2002) Group IImetabotropic glutamate receptors modulate extracellular glutamate in thenucleus accumbens. J. Pharmacol. Exp. Ther., 300, 162–171.
Yaksh, T.L. & Rudy, T.A. (1976) Chronic catheterization of the spinalsubarachnoid space. Physiol. Behav., 17, 1031–1036.
Yamamoto, T. & Yaksh, T.L. (1992) Comparison of the antinociceptive effectsof pre- and post-treatment with intrathecal morphine and MK801, an NMDAantagonist, on the formalin test in the rat. Anesthesiology, 77, 757–763.
Yamamoto, T., Nozaki-Taguchi, N. & Sakashita, T. (2001a) SpinalN-acetylated-alpha-linked acidic dipeptidase (NAALADase) inhibitionattenuates mechanical allodynia induced by paw carrageenan injection inthe rat. Brain Res., 909, 138–144.
Yamamoto, T., Nozaki-Taguchi, N., Sakashita, Y. & Inagaki, T. (2001b)Inhibition of spinal N-acetylated-alpha-linked acidic dipeptidase produces anantinociceptive effect in the rat formalin test. Neuroscience, 102, 473–479.
Yamamoto, T., Ohtori, S. & Chiba, T. (2000) Inhibitory effect of intrathecallyadministered nociceptin on the expression of Fos-like immunoreactivity inthe rat formalin test. Neurosci. Lett., 284, 155–158.
Yang, D. & Gereau, R.W. (2003) Peripheral group II metabotropic glutamatereceptors mediate endogenous anti-allodynia in inflammation. Pain, 106,411–417.
Zhao, J., Cappiello, M., Ramadan, E., Wroblewska, B., Bzdega, T. & Neale,J.H. (2001) NAAG inhibits [3H]-GABA release from cortical neuronsvia the type 3 metabotropic glutamate receptor. Eur. J. Neurosci., 13, 340–346.
Zollinger, M., Brauchli-Theotokis, J., Gutteck-Amsler, U., Do, K.Q., Streit, P.& Cuenod, M. (1994) Release of N-acetylaspartylglutamate from slices of ratcerebellum, striatum, and spinal cord, and the effect of climbing fiberdeprivation. J. Neurochem., 63, 1133–1142.
494 T. Yamamoto et al.
ª 2004 Federation of European Neuroscience Societies, European Journal of Neuroscience, 20, 483–494




















