
Unrestricted modification search reveals lysine methylation as major modification induced by tissue...
12
Proteomics 2015, 00, 1–12 1 DOI 10.1002/pmic.201400454 RESEARCH ARTICLE Unrestricted modification search reveals lysine methylation as major modification induced by tissue formalin fixation and paraffin embedding Ying Zhang 1,2,3∗ , Markus Muller 2∗, ∗∗ , Bo Xu 1,4 , Yutaka Yoshida 1 , Oliver Horlacher 2 , Frederic Nikitin 2 , Samuel Garessus 2 , Sameh Magdeldin 1,5 , Naohiko Kinoshita 1 , Hidehiko Fujinaka 1,6 , Eishin Yaoita 1 , Miki Hasegawa 7 , Frederique Lisacek 2 and Tadashi Yamamoto 1,3 1 Department of Structural Pathology, Institute of Nephrology, Graduate School of Medical and Dental Sciences, Niigata University, Niigata, Japan 2 SIB-Swiss Institute of Bioinformatics, Geneva, Switzerland 3 Biofluid Biomarker Center (BB-C), Institute for Research Collaboration and Promotion, Niigata University, Niigata, Japan 4 Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, Zurich, Switzerland 5 Department of Physiology, Faculty of Veterinary Medicine, Suez Canal University, Ismailia, Egypt 6 Institute of Clinical Research, Niigata National Hospital, Kashiwazaki, Japan 7 Division of Digestive & General Surgery, Niigata University, Niigata, Japan Received: September 25, 2014 Revised: January 22, 2015 Accepted: March 25, 2015 Formalin-fixed paraffin-embedded (FFPE) tissue is considered as an appropriate alternative to frozen/fresh tissue for proteomic analysis. Here we study formalin-induced alternations on a proteome-wide level. We compared LC-MS/MS data of FFPE and frozen human kidney tissues by two methods. First, clustering analysis revealed that the biological variation is higher than the variation introduced by the two sample processing techniques and clusters formed in accordance with the biological tissue origin and not with the sample preservation method. Second, we combined open modification search and spectral counting to find modifications that are more abundant in FFPE samples compared to frozen samples. This analysis revealed lysine methylation (+14 Da) as the most frequent modification induced by FFPE preservation. We also detected a slight increase in methylene (+12 Da) and methylol (+30 Da) adducts as well as a putative modification of +58 Da, but they contribute less to the overall modification count. Subsequent SEQUEST analysis and X!Tandem searches of different datasets confirmed these trends. However, the modifications due to FFPE sample processing are a minor disturbance affecting 2–6% of all peptide-spectrum matches and the peptides lists identified in FFPE and frozen tissues are still highly similar. Keywords: Bioinformatics / FFPE tissue / Lysine methylation / Open modification search / Spectral library Additional supporting information may be found in the online version of this article at the publisher’s web-site Correspondence: Dr. Ying Zhang, Biofluid Biomarker Center (BB-C), Institute for Research Collaboration and Promotion, Niigata University, 8050, Ikarashi 2-no-cho, Nishi-ku, Niigata, Japan 950-2181. E-mail: [email protected] Fax: +81 25 2626925 Abbreviations: CC, cellular component; FFPE, formalin-fixed paraffin-embedded; LMD, laser microdissection; MF, molecular 1 Introduction Formalin fixation and paraffin embedding (FFPE) remains a standard technique for preserving tissue specimens for function; MSH, mass shift histogram; OMS, open modification search; SpecLib, spectral library; TPP, trans-proteomic pipeline ∗ Both authors contributed equally to this work. ∗∗ Additional corresponding author: Dr. Markus Muller, E-mail: [email protected] Colour Online: See the article online to view Fig. 1 in color. C 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
Transcript of Unrestricted modification search reveals lysine methylation as major modification induced by tissue...

Proteomics 2015, 00, 1–12 1DOI 10.1002/pmic.201400454
RESEARCH ARTICLE
Unrestricted modification search reveals lysine
methylation as major modification induced by tissue
formalin fixation and paraffin embedding
Ying Zhang1,2,3∗, Markus Muller2∗,∗∗, Bo Xu1,4, Yutaka Yoshida1, Oliver Horlacher2,Frederic Nikitin2, Samuel Garessus2, Sameh Magdeldin1,5, Naohiko Kinoshita1,Hidehiko Fujinaka1,6, Eishin Yaoita1, Miki Hasegawa7, Frederique Lisacek2
and Tadashi Yamamoto1,3
1 Department of Structural Pathology, Institute of Nephrology, Graduate School of Medical and Dental Sciences,Niigata University, Niigata, Japan
2 SIB-Swiss Institute of Bioinformatics, Geneva, Switzerland3 Biofluid Biomarker Center (BB-C), Institute for Research Collaboration and Promotion, Niigata University, Niigata,
Japan4 Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, Zurich, Switzerland5 Department of Physiology, Faculty of Veterinary Medicine, Suez Canal University, Ismailia, Egypt6 Institute of Clinical Research, Niigata National Hospital, Kashiwazaki, Japan7 Division of Digestive & General Surgery, Niigata University, Niigata, Japan
Received: September 25, 2014Revised: January 22, 2015Accepted: March 25, 2015
Formalin-fixed paraffin-embedded (FFPE) tissue is considered as an appropriate alternativeto frozen/fresh tissue for proteomic analysis. Here we study formalin-induced alternationson a proteome-wide level. We compared LC-MS/MS data of FFPE and frozen human kidneytissues by two methods. First, clustering analysis revealed that the biological variation is higherthan the variation introduced by the two sample processing techniques and clusters formedin accordance with the biological tissue origin and not with the sample preservation method.Second, we combined open modification search and spectral counting to find modificationsthat are more abundant in FFPE samples compared to frozen samples. This analysis revealedlysine methylation (+14 Da) as the most frequent modification induced by FFPE preservation.We also detected a slight increase in methylene (+12 Da) and methylol (+30 Da) adducts as wellas a putative modification of +58 Da, but they contribute less to the overall modification count.Subsequent SEQUEST analysis and X!Tandem searches of different datasets confirmed thesetrends. However, the modifications due to FFPE sample processing are a minor disturbanceaffecting 2–6% of all peptide-spectrum matches and the peptides lists identified in FFPE andfrozen tissues are still highly similar.
Keywords:
Bioinformatics / FFPE tissue / Lysine methylation / Open modification search / Spectrallibrary
� Additional supporting information may be found in the online version of this article atthe publisher’s web-site
Correspondence: Dr. Ying Zhang, Biofluid Biomarker Center(BB-C), Institute for Research Collaboration and Promotion,Niigata University, 8050, Ikarashi 2-no-cho, Nishi-ku, Niigata,Japan 950-2181.E-mail: [email protected]: +81 25 2626925
Abbreviations: CC, cellular component; FFPE, formalin-fixedparaffin-embedded; LMD, laser microdissection; MF, molecular
1 Introduction
Formalin fixation and paraffin embedding (FFPE) remainsa standard technique for preserving tissue specimens for
function; MSH, mass shift histogram; OMS, open modificationsearch; SpecLib, spectral library; TPP, trans-proteomic pipeline∗Both authors contributed equally to this work.∗∗Additional corresponding author: Dr. Markus Muller,E-mail: [email protected] Online: See the article online to view Fig. 1 in color.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

2 Y. Zhang et al. Proteomics 2015, 00, 1–12
pathological examination and the study of tissue morphology.Recent shotgun proteomic analyses have indicated highequivalence of MS/MS spectral counts and protein groupidentifications between paired FFPE and frozen tissues,which provides strong support for clinical attempts to ex-plore disease biomarkers and to understand pathological pro-cesses [1]. On the other hand, it was previously reported thatformalin-induced modifications reduce protein immunore-activity and protein extraction efficiency, and they may leadto misidentification of proteins during proteomic analysis[2]. Thus, an increasing amount of effort is being made todecipher chemical modifications and the subsequent proteincross-linking occurring in formalin fixation procedures.
Three types of chemical modifications after treatment ofproteins with formalin have been identified: (1) methylol(-CH2OH) adducts with mass shift of +30 Da, (2) Schiffbases (-N = CH2) with mass shift of +12 Da, (3) methy-lene bridges (-CH2-) with a mass shift of +12 Da withinthe same or between different protein molecules [3–6]. Us-ing model peptides, formalin was shown to react with theamino group of N-terminal amino acid residues and theside chains of various amino acids including lysine, argi-nine, histidine, tryptophan, and cysteine, where their sus-ceptibility is mainly dependent on formalin reaction time[3,4]. Similar investigations on model proteins demonstratedthat N-terminal amino group and lysine side chain accountfor the vast majority of formalin-induced modifications,and tertiary structure and solvent accessible surface areaof proteins played a major role in regulating the extent offormalin-induced modifications [5, 6]. Even with these ad-vances, chemical modifications occurring in FFPE tissue arestill not clearly elucidated especially in complex biologicalenvironment.
Meantime, the development of optimized protein extrac-tion strategies for FFPE tissues is in progress, aimed to breakthe cross-linking, retrieve antigen immunoreactivity, and im-prove its feasibility for proteomic analysis. Optical spectro-scopic studies indicated that formalin treatment does notappear to significantly alter the protein’s secondary or ter-tiary structure [7]. Mild heating of the formalin-fixed RNaseA solutions at 65�C in pH 4 buffer resulted in partial re-versal of protein cross-links, which lead to partial restora-tion of immunoreactivity [8]. However, SDS-PAGE analysison proteins extracted from FFPE tissue surrogates revealedthat the reversal of the intermolecular cross-links requiredhigh temperatures or elevated hydrostatic pressure at mod-erate temperatures [9, 10], which suggests protein formalinchemical adducts undergo further reactions in subsequentdehydration and paraffin-embedding processes that are notobserved in aqueous solution. The study on RNase A revealedthat ethanol dehydration induced protein conformation rear-rangement, leading to protein aggregation through the for-mation of hydrophobic �-sheets [11]. Substantial energy wasrequired to reverse the cross-links within these sheets and
regenerate protein monomers free of formalin modifications.In order to improve the applicability of FFPE tissues to pro-teomic studies, Wisniewski et al. recently described a stream-lined filter-aided sample preparation workflow that allowsefficient protein recovery from FFPE tissues, and compat-ibility to endoproteinase digestion and proteomic analysis[12, 13]. Furthermore, commercially available reagents, suchas LiquidTissueTM kits and RapiGestTM buffers, have been de-veloped and applied to protein extraction from FFPE samples[14, 15].
Finding unknown or unexpected protein modifications in-duced by FFPE tissue preservation in proteomic LC-MS/MSdata requires a different approach than standard peptide se-quence searches, where a small number of fixed or variablemodifications need to be configured in advance. Open modi-fication searches (OMS) extract the modifications present inthe sample directly from the MS/MS data without the needfor prior configuration [16]. This approach relies on the align-ment of modified query spectra to unmodified database spec-tra, where some fragment peaks are shifted in accordancewith the mass of the modification(s) [17]. Due to the drasticincrease in peptide-spectrum match scenarios, OMS usuallyis performed after a reduction of the search space in orderto avoid too many false positives and to limit search times.Either a small set of proteins likely to be present in the sam-ple [16] or spectral libraries of previously identified peptides[18] can be searched. As a search result summary, OMS pro-vides a mass shift histogram (MSH), which counts how manyhigh-quality spectrum alignments for a certain mass shift aredetected, and reveals the most common mass shifts presentin the sample.
In this study, we investigate the changes induced by FFPEprocessing compared to frozen tissue processing. We ana-lyzed the LC-MS/MS data of human glomerulus and kidneycortex tissues, which were subjected to both frozen and FFPEpreservation. The results of MS/MS spectrum clustering anal-ysis clearly show that LC-MS/MS runs form clusters in accor-dance with their biological origin indicating that the overallbiological variation is stronger than the variation due to thesample preservation method. In order to get an idea whichmodifications are induced by FFPE processing, we searchedfor modifications occurring specifically in FFPE tissues us-ing an OMS approach based on the QuickMod tool [18]. Wefound a small set of chemical modifications significantly over-represented in FFPE tissue, where lysine methylation wasthe most significant one. A subsequent standard MS/MS se-quence database search using SEQUEST/TPP [19, 20] con-firmed the enrichment of these modifications in the sameFFPE dataset, and the same trends were also found in pub-lically available datasets. However, the changes introducedby these modifications are minor compared to the biologicalvariation indicating that FFPE kidney tissue is suitable forlarge-scale LC-MS/MS proteomic analysis and comparable tofrozen tissue.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2015, 00, 1–12 3
Table 1. Human kidney FFPE and frozen samples used in this study
FFPE Frozen
Patient Number Glomerulus (G) Cortex (C) Glomerulus (G) Cortex (C)
NX020627 NX020627_FFPE_G.1a) NX020627_FFPE_C.1 NX020627_Froz_G.1 NX020627_Froz_C.1NX020627_FFPE_G.2a) NX020627_FFPE_C.2 NX020627_Froz_G.2 NX020627_Froz_C.2
NX020902 NX020902_FFPE_G.1 NX020902_FFPE_C.1 NX020902_Froz_G.1 NX020902_Froz_C.1NX020902_FFPE_G.2 NX020902_FFPE_C.2 NX020902_Froz_G.2 NX020902_Froz_C.2
NX020930 NX020930_FFPE_G.1 NX020930_FFPE_C.1 NX020930_Froz_G.1 NX020930_Froz_C.1NX020930_FFPE_G.2 NX020930_FFPE_C.2 NX020930_Froz_G.2 NX020930_Froz_C.2
NX111209 NX111209_FFPE_G1.1b) NX111209_Froz_G1.1NX111209_FFPE_G2.1 NX111209_Froz_G2.1NX111209_FFPE_G2.2 NX111209_Froz_G2.2
a) Each peptide sample extracted from kidney tissue, for example, NX020627_FFPE_G, was measured in duplicate by LC-MS/MS, producingtwo data files, NX020627_FFPE_G.1 and NX020627_FFPE_G.2.b) The sample NX111209_FFPE_G1 was measured by LC-MS/MS. The data of the second run, NX111209_FFPE_G1.2, were removed dueto unsuccessful MS/MS data conversion for Sorcerer-SEQUEST search. Its frozen counterpart, NX111209_Froz_G1.2, was accordinglyexcluded.
2 Materials and methods
2.1 FFPE and frozen human kidney tissues
This study was approved by the Ethics Committees of Ni-igata University Faculty of Medicine. Human kidney tissueswere obtained from nephrectomy due to renal cell carcinoma.Renal cortical tissue with normal morphology and most dis-tant from the tumors was excised and either fixed with 10%neutral-buffered formalin (approximately 4% formaldehyde)for 24 h or snap frozen at −80�C until use. Formalin-fixedtissues were routinely dehydrated with ethanol and xylene se-ries and embedded in paraffin. Both FFPE and frozen samplecohorts contain eight samples, five glomerular samples, andthree renal cortical samples. Each sample was measured induplicate by LC-MS/MS, producing two data files. The corre-sponding sample information is given in Table 1.
2.2 Laser microdissection (LMD), protein digestion,
and peptide purification
FFPE tissue specimens were cut into 10-�m-thick sections,mounted on membrane slides (MMI AG, Switzerland) andkept at room temperature until use. FFPE sections were de-paraffinized in xylene, rehydrated in graded ethanol, andrinsed with MilliQ. Frozen specimens were cut prior touse. Both FFPE and frozen slides were autoclaved at 110�Cfor 10 min before LMD. For each glomerulus sample, 50glomerular cross sections (�1 mm2) were collected on anadhesive tube-cap (IsolationCaps, MMI AG, Switzerland) us-ing an LMD system (CellCut Plus, MMI, Eching, Germany)equipped with Nikon’s inverted microscope (Eclipse TE2000-S, Tokyo, Japan). Similarly, cortical tissue with around 1 mm2
area was captured for each cortex sample. The tissue sectionswere directly trypsin-digested on tube-caps at 37�C overnight.This method is developed especially for peptide extractionfrom FFPE tissues (paper in preparation). After digestion,
the peptide mixture was spun down and 1 �L of 50% tri-fluoacetic acid (TFA) was added to quench the trypsin activ-ity. Peptides were eluted and purified using StageTips C18(Thermo Scientific) according to the manual instructions. Fi-nally, the peptide eluate was dehydrated in a speedvac andstored at −30�C until LC-MS/MS analysis.
2.3 LC-MS/MS analysis and peptide/protein
identification
Peptide mixtures were solubilized in the sample solution (2%ACN, 0.1% formic acid) and measured in duplicate by LTQ-Obitrap XL (Thermo Scientific) combined with nanoscaleC18 reversed phase liquid chromatography (DiNa-A, KYAtechnologies, Japan). The peptides were separated on a C18separation column (75 �m × 100 mm, particle size of3 �m, pore size of 120 A), and eluted with a 95-min mo-bile phase gradient at the flow rate of 300 nL/min. MS surveyscan (m/z 350–1600, resolution 60 000) was acquired in theObitrap and the five most intensive precursor ions were frag-mented in linear ion trap. The dynamic exclusion time wasset to 60 s. A total of 30 raw data files for MS/MS runs (15for FFPE (212 353 spectra) and 15 for frozen (217 608 spec-tra)) were used for this analysis and converted into mzXMLfiles (Table 1), and searched with Sorcerer-SEQUEST Version4.2.0 (Sage-N Research, Inc., USA) [19] against a concatenatedprotein database containing the human protein database ofUniProtKB/Swiss-Prot (Release 2012_11), as well as the re-versed sequences of all proteins and common contaminants.The database search parameters were set as follows: full-tryptic specificity and up to two missed cleavages were al-lowed; no fixed modifications were set; oxidation on Met,His, Trp were applied as variable modifications, unless spec-ified elsewhere; mass tolerance of the precursor ion was setat 50 ppm. Statistical analysis was performed using Peptide-Prophet of Trans-Proteomic Pipeline (TPP, Version 4.5.2),which uses various SEQUEST scores and other parameters
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

4 Y. Zhang et al. Proteomics 2015, 00, 1–12
to calculate a probability score for each identified peptide[20]. PeptideProphet probability score > 0.90 was used as thethreshold resulting in an overall peptide false discovery rate of2% on the spectrum level (74 542 peptide-spectrum matchesfor FFPE and 79 948 for frozen sample). The peptides abovethreshold were then used for protein identification.
2.4 Gene Ontology functional annotation and
clustering analysis
With the common proteins filtered out, the uniquely iden-tified proteins in FFPE and frozen tissues were catego-rized based on GO hierarchy vocabulary of cellular compo-nent and molecular function via DAVID knowledge platform(Database for Annotation and Integrated Discovery) (version6.7), hosted by NIAID/NIH (National Institute of Allergy andInfectious Diseases/National Institutes of Health) [21, 22].
LC-MS/MS runs were clustered according to peptide andspectral similarity. In order to calculate the peptide similar-ity, all peptide sequences with their modification annotations(PeptideProphet probability > 0.90) were extracted from thepepXML result files, and stored in lists separately for eachLC-MS/MS run. The similarity between two peptide lists wasthen calculated as two times the size of the intersection be-tween the two lists divided by the total number of entries inboth lists yielding a value between 0 (no overlap) and 1 (equallists). The spectral similarity between two runs was obtainedby calculating the average cosine similarity [23] between allinter-run spectrum pairs, where a spectrum from the firstrun pairs a spectrum from the second run if they have thesame charge and a precursor m/z value within a tolerance of0.01 Da (spectral similarity also yields values between 0 and1). Both peptide and spectrum similarities were calculated foreach pair of LC-MS/MS runs and the results were stored insimilarity matrices, which were then processed using the hi-erarchical clustering function hclust (agglomeration methodset to “average”) in the R statistical environment [24].
2.5 Creation of spectral library and QuickMod open
modification search
We built an MS/MS spectral library dubbed SpecLib usingconfidently identified peptides (PeptideProphet probability >
0.90) from the frozen samples in the SpectraST sptxt format[25, 26]. SpecLib was built using the in-house Liberator tool(version 2.0), which calculates consensus spectra in case apeptide precursor ion was identified several times. MS/MSfragment peaks were merged into a consensus peak if theirm/z values were within 0.3 units and if the peak was presentin at least 20% of the spectra. Decoy spectra were createdusing the Deliberator tool (version 2.0) to calculate the falsediscovery rate (FDR) [27]. Deliberator implements a variationof the peptide shuffling and peak repositioning algorithm[28], which creates realistic but randomized spectra. It cre-ates a decoy spectrum for each original library spectrum and
appends the decoy entries to the spectrum library file. BothLiberator and Deliberator tools are based on the MzJava classlibrary (mzjava.expasy.org, paper in preparation).
SpecLib serves as the search space for our OMS searches.The relatively small size of SpecLib (11 400 spectra withoutdecoy) compared to the total number of human tryptic pep-tides made it possible to perform the OMS searches withinreasonable time (< 1 week), whereas searching all humantryptic peptide sequences would have taken much longer. Thechoice to only include peptide spectra from the frozen samplewas one of convenience to keep the library as small as possi-ble. In the QuickMod OMS search, we were more interestedin detecting the most prominent modification types than ina complete list of peptides on which these modifications oc-cur. The subsequent SEQUEST/TPP searches then looked forspecific modifications on the entire proteome. For the total setof methylated peptide spectra found by SEQUEST/TPP, 77%had an unmodified counterpart in SpecLib indicating thatSpecLib is a valid surrogate for the entire kidney proteomefor this purpose.
All MS/MS spectra from both FFPE and frozen sam-ples were then searched by QuickMod tool (version 1.03,http://javaprotlib.sourceforge.net/packages/tools/index.html)against SpecLib. The aim of these searches is to extractthe most prominent modifications present in the samples.QuickMod aligns each query spectrum to all candidatespectra in the spectral library with the same charge andwithin the precursor mass tolerance of ±100 Da under theassumption that the precursor mass shift is due to a singleamino acid modification. The QuickMod positioning scorecounts the number of peaks contradicting a modificationon this position minus the number of peaks supporting it[18]. Then it assigns the modification to the position in thepeptide sequence with the most negative score. In this study,we only considered alignments where the absolute differencebetween the best and worst positioning scores is larger than5. Negative mass shifts were included to capture potentiallosses of peptide masses induced by sample preparation.Other QuickMod search parameters were as follows: queryprecursor ions with charges of +2 to +3 were selected; foreach FFPE query spectrum only the highest scoring libraryspectrum was considered; the mass tolerance for fragmentions was set to 0.4 Da. Query and library spectra were ranktransformed prior to searching and alignments were scoredusing the cosine or normalized dot product score [23]. TheFDR was calculated separately for modified and unmodifiedspectra and for each precursor charge using the decoyspectra. Score thresholds were set to a value that correspondsto an FDR of 1%. Most alignments passing these thresholdsare of good quality and require high-quality spectra.
2.6 Data integration and statistical analysis
The exported QuickMod OMS results were integrated using Rscripts. First, spectra previously identified by SEQUEST/TPP
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2015, 00, 1–12 5
were removed from the QuickMod results. The next stepconsisted in estimating the variation in the modification spec-trum counts in order to assign significance to the differencein these counts. The MSHs MSH j
F F P E and MSHkFr oz were
calculated for every FFPE LC-MS/MS run j ∈ Runs F F P E andfrozen sample LC-MS/MS run k ∈ Runs Fr oz, where the pre-cursor mass shifts between –100 and +100 Da were dividedinto 0.1 Da bins. For each mass shift bin i, the spectrumalignments of run j with a precursor mass difference fallinginto bin i were counted and MSH j
x [i ]; x ∈ {FFPE, Froz} wasassigned to this count. In order to compare mass shifts be-tween frozen and FFPE results, we calculated the total MSHsMSHtot
x by summing up all MSHs of the respective runs(Eq. (1)).
MSHtotx [i ] =
∑j∈Runsx
MSH jx [i ] (1)
Then we estimate the variance of the total MSH counts�2(MSHtot
x [i ]) (Eq. (2)) by sampling runs 100 times from theFFPE and frozen runs separately with replacement yielding100 sets of runs Runs k
x ; k = 1...100 . Supporting InformationFig. 1A and B shows MSHtot
x [i ] and �2(MSHtotx [i ]) for both
FFPE and frozen samples.
MSHtot,kx [i ] =
∑
j∈Runs kx
MSH jx [i ]
MSHtotx [i ] = 1
100
100∑k=1
MSHtot,kx [i ] (2)
�2(MSHtot
x [i ]) = 1
100
100∑k=1
(MSHtot,k
x [i ] − MSHtotx [i ]
)2
Finally, we calculate the ZScore for the count differencesbetween FFPE and frozen samples using Eq. (3). The resultsare shown in Supporting Information Fig. 1C and D. Gener-ally it seems that there are more modification counts in theFFPE compared to the frozen sample and we tried to removethis bias: prior to ZScore calculation MSHtot
x are normalizedin order to make the total spectral counts in the FFPE andfrozen sample equal and the standard deviations are adjustedaccordingly.
ZScor e(MSHtot
FPPE [i ] − MSHtotFr oz [i ]
)
= MSHtotF F P E [i ] − MSHtot
Fr oz [i ]√�2
(MSHtot
F F P E [i ]) + �2
(MSHtot
Fr oz [i ]) (3)
All mass shifts with a positive ZScore are more abundantin the FFPE sample compared to the frozen sample. Onlymass shifts which are confirmed by more than two uniquepeptides in SpecLib and more than five spectrum alignmentswere considered for the final ZScore calculation.
A Gaussian distribution was robustly fitted to the ZSores(Supporting Information Fig. 1E) and used to calculate thequantiles or p-values. The mass shifts were considered sig-nificantly enriched in FFPE samples when the ZScore wasmore than 1.37 (quantile of 0.9).
In order to inspect which amino acids tend to carry a givenmodification mass, we calculated the propensity of an aminoacid to be situated at or in the vicinity of the QuickMod modi-fication positions for this mass shift (Supporting InformationSection 1 and Supporting Information Eq. 1).
For the QuickMod result validation with SEQUEST/TPP,we considered the mass shifts significantly enriched in FFPEtissue together with the amino acids they predominantlyare attached to. Then we performed a variable modificationsearch for each of these mass shifts (+12 Da on Ser, Thr,Tyr, Cys, Lys; +14 Da on Lys; +30 Da on Lys; +58 Da onLys) separately using SEQUEST/TPP at a PeptideProphetprobability threshold of 0.9. In order to avoid potential falsepositives, we allowed only one modification per peptide. Sim-ilar to the QuickMod search, we removed all spectra alreadyidentified in the first SEQUEST/TPP search before searchingfor these modifications. Finally, we verified these modifica-tions using different publically available datasets obtainedfrom frozen and FFPE human tissues. MS/MS data filesfrom three different studies [29–31] (see Supporting Infor-mation Section 2 for more details) measured on Orbitrapinstruments and under similar sample preparation condi-tions (trypsin digestion, cysteine carbamidomethylated) weredownloaded from PeptideAtlas (www.peptideatlas.org) andProteomicsDB (www.proteomicsdb.org) and analyzed withX!Tandem [32] (Cys-carbamidomethylation fixed, +16 Da onMet; +12 Da on Ser, Thr, Tyr, Lys; +14 Da on Lys; +30 Da on
Figure 1. Overview of the data processing steps. Cortex (Cx) andglomerulus (Gl) regions are marked in the kidney cross sections.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

6 Y. Zhang et al. Proteomics 2015, 00, 1–12
Figure 2. Integrated peptide and protein identifications from all the human kidney FFPE and frozen (Froz) samples. (A) Venn diagrams showthat 65% of overall identified peptides (left panel) and 67% of overall proteins (right panel) are common to the two groups. (B) Clusteringanalysis shows that identified peptides from FFPE (�) and frozen (�) samples are quite similar at the level of peptide sequence while theidentified peptides from human glomerulus (�) and cortex (*) are cleared separated into two clusters. (C) GO Cellular Component (CC) and(D) molecular function annotation shows no significant difference in biological functions of uniquely identified proteins in FFPE and frozensamples.
Lys; and +58 Da on Lys variable, at most one variable modifi-cation per peptide, UniProt/SwissProt human database withreversed sequences, FDR 1%). An overview over the entiredata processing steps is presented in Fig. 1.
3 Results and discussion
3.1 High equivalence between FFPE and frozen
human kidney proteomes
The peptide and protein identifications obtained from FFPEand frozen samples exhibit fair consistency in both the num-bers and contents (Fig. 2A). A total of 5822 peptides and1395 proteins are found to be common to FFPE and frozencohorts with overlap of 65 and 67%, respectively. This obser-vation agrees with the variation in protein identities detected
previously on human pre-eclamptic placental and glioblas-toma tissues using FFPE and frozen samples [33, 34]. More-over, clustering analysis based on peptide sequences indi-cates that peptides identified in FFPE and frozen samples aresimilar with two main clusters forming for glomerulus andcortex (Fig. 2B). The similarity is also shown in the clusteringanalysis at MS/MS spectral level (Supporting InformationFig. 2). A high level of consistency is seen in GO annota-tion for subcellular localization and molecular function ofuniquely identified proteins in the two cohorts (Fig. 2C andD). The physicochemical analysis (theoretical pI and MW)of the uniquely identified peptides and proteins shows nodiscernible bias in FFPE and frozen cohorts (Supporting In-formation Fig. 3A and B). Therefore, our data indicates thatFFPE tissue provides comparable proteomic identifications tofrozen tissue as well as reliable biological information. Thisresult is in agreement with the report by Sprung et al. [1] but incontrast to some previous reports, where FFPE tissues were
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
Proteomics 2015, 00, 1–12 7
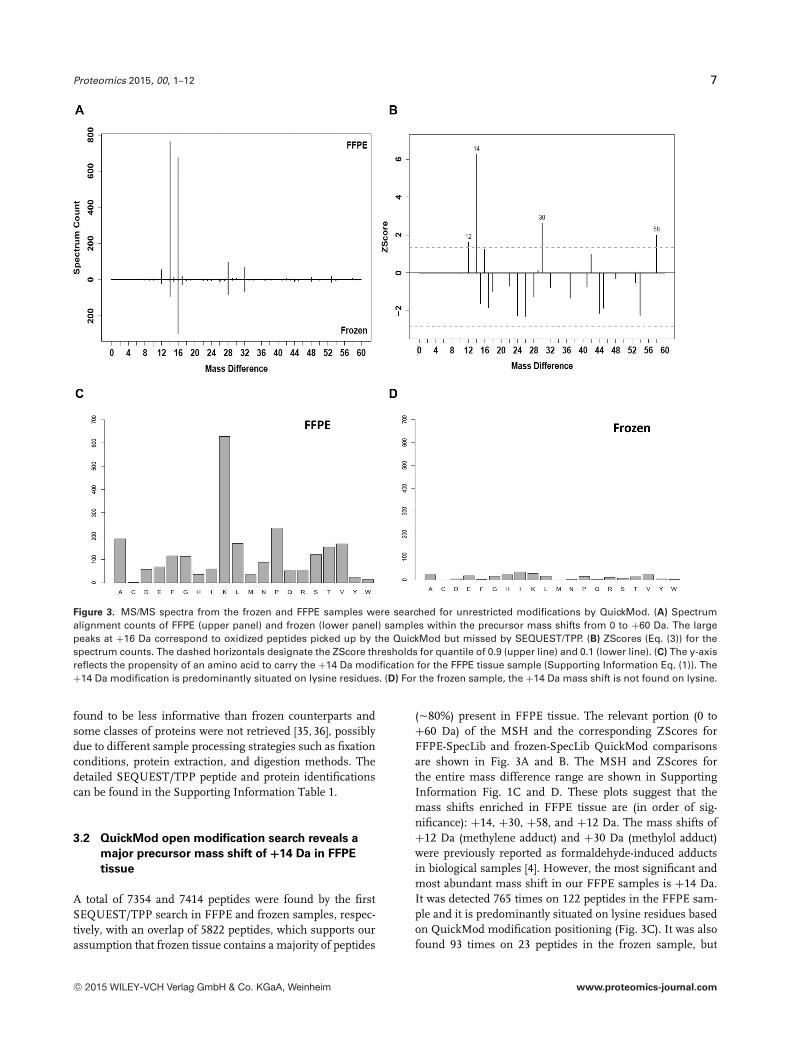
Figure 3. MS/MS spectra from the frozen and FFPE samples were searched for unrestricted modifications by QuickMod. (A) Spectrumalignment counts of FFPE (upper panel) and frozen (lower panel) samples within the precursor mass shifts from 0 to +60 Da. The largepeaks at +16 Da correspond to oxidized peptides picked up by the QuickMod but missed by SEQUEST/TPP. (B) ZScores (Eq. (3)) for thespectrum counts. The dashed horizontals designate the ZScore thresholds for quantile of 0.9 (upper line) and 0.1 (lower line). (C) The y-axisreflects the propensity of an amino acid to carry the +14 Da modification for the FFPE tissue sample (Supporting Information Eq. (1)). The+14 Da modification is predominantly situated on lysine residues. (D) For the frozen sample, the +14 Da mass shift is not found on lysine.
found to be less informative than frozen counterparts andsome classes of proteins were not retrieved [35, 36], possiblydue to different sample processing strategies such as fixationconditions, protein extraction, and digestion methods. Thedetailed SEQUEST/TPP peptide and protein identificationscan be found in the Supporting Information Table 1.
3.2 QuickMod open modification search reveals a
major precursor mass shift of +14 Da in FFPE
tissue
A total of 7354 and 7414 peptides were found by the firstSEQUEST/TPP search in FFPE and frozen samples, respec-tively, with an overlap of 5822 peptides, which supports ourassumption that frozen tissue contains a majority of peptides
(�80%) present in FFPE tissue. The relevant portion (0 to+60 Da) of the MSH and the corresponding ZScores forFFPE-SpecLib and frozen-SpecLib QuickMod comparisonsare shown in Fig. 3A and B. The MSH and ZScores forthe entire mass difference range are shown in SupportingInformation Fig. 1C and D. These plots suggest that themass shifts enriched in FFPE tissue are (in order of sig-nificance): +14, +30, +58, and +12 Da. The mass shifts of+12 Da (methylene adduct) and +30 Da (methylol adduct)were previously reported as formaldehyde-induced adductsin biological samples [4]. However, the most significant andmost abundant mass shift in our FFPE samples is +14 Da.It was detected 765 times on 122 peptides in the FFPE sam-ple and it is predominantly situated on lysine residues basedon QuickMod modification positioning (Fig. 3C). It was alsofound 93 times on 23 peptides in the frozen sample, but
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

8 Y. Zhang et al. Proteomics 2015, 00, 1–12
Ta
ble
2.
Su
mm
ary
of
Qu
ickM
od
and
SE
QU
ES
T/T
PP
resu
lts
Sea
rch
Too
lQ
uic
kMo
dS
EQ
UE
ST
/TP
P
Sam
ple
FFP
EFr
oze
nFF
PE
Fro
zen
Item
Sp
ectr
um
alig
nm
ents
a)Pe
pti
des
Pro
tein
sS
pec
tru
mal
ign
men
tsa)
Pep
tid
esPr
ote
ins
Sp
ectr
um
mat
ches
a)Pe
pti
des
Pro
tein
sS
pec
tru
mm
atch
esa)
Pep
tid
esPr
ote
ins
+12
Da
54|3
817
|12
1422
|27|
27
230|
161
121|
101
111
209|
8889
|69
82+1
4D
a76
5|69
612
2|11
163
93|1
723
|12
2013
55|1
286
238|
232
120
57|1
521
|15
18+1
6D
a22
22|2
95b
)27
4|12
0b)
147
1772
|109
b)
206|
52b
)10
356
29|9
4977
4|42
338
342
82|1
9947
8|12
724
1+3
0D
a7|
75|
55
0|0
0|0
011
4|98
21|1
920
27|1
214
|12
14+5
8D
a7|
73|
33
1|1
1|1
113
2|11
611
7|10
711
388
|77
81|7
179
Fiel
ds
sho
w:t
ota
lnu
mb
ero
fit
ems
insa
mp
le|n
um
ber
of
item
su
niq
ue
toth
esa
mp
le.
a)U
niq
ue
nu
mb
ero
fsp
ectr
um
alig
nm
ents
/mat
ches
equ
als
nu
mb
ero
fsp
ectr
am
atch
ing
wit
ha
giv
enm
ass
shift
toa
pep
tid
en
ot
fou
nd
inth
eo
ther
sam
ple
.b
)In
clu
din
gox
idiz
edsp
ectr
afo
un
db
yS
EQ
UE
ST
fro
mQ
uic
kMo
dse
arch
.
the positioning on lysine and arginine is rare (seven pep-tides) (Fig. 3D). Information on modified peptides and spec-tra detected by QuickMod is given in Supporting Informa-tion Table 2 and sptxt files in Supporting Information Data.A mass shift of +14 Da can also be produced by amino acidsubstitution (in fact given the frequency of amino acids andtheir substitutions it is the most frequent substitution massshift). However, the substitution effect should be indepen-dent of sample preparation and is not able to explain the dif-ference between frozen and FFPE samples. Subsequent SE-QUEST/TPP searching confirmed a strongly increased num-ber of methylations in the FFPE sample present on about2% of the total peptide-spectrum matches (Table 2, see Sup-porting Information Section 3 for a discussion of the differ-ences between SEQUEST/TPP and QuickMod results). Themethylated peptides on lysine detected by SEQUEST/TPP areshown in Supporting Information Table 3. Both QuickModOMS and SEQUEST/TPP search indicate that methylatedpeptides on lysine were mostly detected in more than twobiological samples and technical replicates. The methylatedproteins have a similar abundance profile to the entire set ofproteins detected in the SEQUEST/TPP search with a slightdrift toward higher abundances (Supporting Information Fig.4A). Supporting Information Fig. 4B shows methylated lysineresidues of some histone proteins based on SEQUEST/TPPsearch. It reveals that the majority of residues is not anno-tated in UniProt and is detected only in FFPE samples in-dicating that they are in fact linked to FFPE preservation.We further tested these results by searching with X!Tandemdifferent publically available MS/MS data [29–31] obtainedfrom FFPE and frozen human tissues. These results indicatethat the rate of methylation is clearly higher in FFPE com-pared to frozen samples (Fig. 4), where the effect seems tobe weaker in the PASS00375 sample indicating possible de-pendence on sample processing protocols. Despite the factthat SEQUEST/TPP and X!Tandem searches were both runat an FDR of 1%, the X!Tandem results display an increasedamount of FFPE methylation (�6% compared to 2%), whichcould be explained by differences in FDR calculation and bythe more restrictive handling of modifications in the TPP.However, the increase in methylation in FFPE compared tofrozen samples remains. Eighty two percent of the QuickModpeptide alignments in the frozen sample with mass shift of+14 Da could also be found in the FFPE sample. These com-mon +14 Da alignments are mostly of biological origin, anda discussion of some of them can be found in the SupportingInformation text section 4.
Biological protein methylation on histone lysine and argi-nine has been extensively explored [37]. It plays a key role ingenome stability, chromatin remodeling, and gene expres-sion. On the other hand, chemical methylation on lysineresidues can be carried out in the presence of formaldehydeand this reductive reaction can alter protein crystallizationproperties for the study of its structure and function [38]. Thischemical reaction, in which formaldehyde provides alkyl moi-ety and sodium cyanoborohydride acts as the reducing agent,
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2015, 00, 1–12 9
Figure 4. Percentage of methylated PSMs for publically available frozen and FFPE tissue LC-MS/MS datasets. FASP refers to the filter-aided sample preparation protocol, DT to the direct tissue trypsinization protocol and ISD to the protein extraction followed by in-solutiondigestion protocols described in [29]. IGD refers to 2D-DIGE followed by in-gel digestion described in [30].
proceeds rapidly to the dimethylated product [39]. In our data,lysine monomethylation is significantly found in FFPE tissuecompared to frozen tissue and the underlying mechanism forthis chemical process is required to be fully elucidated.
The +30 Da mass shift was only detected in FFPE Quick-Mod results, but it was quite rare (seven spectral alignments,five peptide sequences) and the QuickMod positioning sitesare not very conclusive. Some of the +30 Da adducts are sit-uated on lysine, others near the N-term of the peptides. Thelatter ones may originate in the reaction of leftover formalinwith the peptide N-term after digestion. The SEQUEST/TPPresults support the enrichment of methylol in the FFPE sam-ple (Table 2), which was confirmed by the X!Tandem search(Supporting Information Fig. 4C). Similarly, the mass shift+58 Da was detected only seven times in the FPPE and oncein the frozen tissue sample. It was predominantly situatedon lysine. The origin of this mass shift remains elusive andconfirmation by SEQUEST/TPP and X!Tandem search is lesspronounced compared to methylol (Table 2 and SupportingInformation Fig. 4D).
The mass shift +12 Da was detected in 54 spectrum align-ments in FFPE samples and in 22 spectrum alignments infrozen samples (Table 2). Twenty of the frozen sample align-ments matched a peptide that could also be found in theFFPE sample, whereas 38 of the FFPE spectral alignmentswere unique to this sample. In these alignments, the methy-lene adducts were predominantly attached to tyrosine, ser-ine, threonine, lysine, and peptide N-terminus. Two hundredand thirty methylene adduct spectra were found by the SE-QUEST/TPP search in the FFPE and 209 in frozen sample,and in contrast to the QuickMod results, there seems to beno strong enrichment here. However, the number of unique
alignments in the FFPE results is significantly higher than inthe frozen sample results indicating that some of them aredue to formalin fixation. The spurious +12 Da alignments de-tected in both samples might be due to the presence of similarpeptides with a mass difference of +12 Da and seem to occurquite frequently. In the SEQUEST/TPP FFPE results, 40% ofthe methylene was on serine, 39% on threonine, 17% on ty-rosine, and 4% on lysine. The X!Tandem search on the otherhand showed quite a strong increase of methylene adducts inFFPE samples (Supporting Information Fig. 4E). A discus-sion on differences in oxidation between FFPE and frozentissues can be found in Supporting Information Section 5.The modified peptides by QuickMod search with position-ing scores for all significant mass shifts are available in theSupporting Information Table 2.
Next, we investigated whether these modifications en-hance the number of missed cleavages after trypsin diges-tion. The majority (62%) of peptides with lysine methylationfound by QuickMod had the methyl group positioned at theC-terminal lysine indicating that trypsin still cleaves despitethe modified lysine. For example, modification site localiza-tion for peptide DYVSQFEGSALGK (APOA1_HUMAN) isclearly determined on the C-terminal lysine residue by themass shifts (+14 or +7 Da) on y2 to y12 fragment ionsin a row (Fig. 5A). Spectral alignment of another peptideTNEKVELQELNDR (DESM_HUMAN) shows a methylatedlysine which was not cleaved by trypsin both in the nonmethy-lated and methylated form (Fig. 5B). However, both Quick-Mod and SEQUEST/TPP searches showed an increased rateof missed cleavages in peptides with formalin-induced mod-ifications compared to peptides without such modifications(Table 3). Since formalin-induced modifications are rare, the
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

10 Y. Zhang et al. Proteomics 2015, 00, 1–12
Figure 5. Spectra of nonmodified peptides from the spectral library (top) aligned with the spectra of modified variants from the FFPE sam-ples (bottom), carrying a lysine modification (+ 14 Da). (A) y2 to y12-ions in the spectrum of peptide DYVSQFEGSALGK from apolipoproteinA-I (APOA1_HUMAN), annotated with an * in the FFPE spectrum, are shifted with the m/z of methylation relative to the corresponding non-modified fragment ions. (B) b6 to b9-ions and y11 to y12-ions in the spectrum of peptide TNEKVELQELNDR from desmin (DESM_HUMAN),annotated with an * in the FFPE spectrum, are shifted with the m/z of methylation relative to the corresponding nonmodified fragmentions.
Table 3. Rate of missed cleaved peptides
Mass shift QuickMod FFPE SEQUEST/TPP FFPE
#peptides withmissed cleavage/#peptidesa)
#peptides withmissed cleavage/#peptidesa)
+12 Da 0.666 0.404+14 Da 0.380 0.549+16 Da 0.108 0.086+30 Da 0.500 0.351+58 Da 0.715 0.364Unmodified
peptides- 0.089
All peptides - 0.107
a) Peptides not containing lysine were discarded.
global rate measured on all peptides irrespective of their mod-ification status was only slightly higher than nonmodifiedpeptides (0.107 versus 0.089), and it was almost the same asthe rate in the frozen sample (0.106).
4 Concluding remarks
Different from previous studies on formalin-induced modi-fications using model peptides and proteins, our data revealthat peptide adducts with a mass shift of +14 Da correspond-ing to lysine methylation are major modified products in
FFPE tissue. Our study provides a novel trial to investigatemodifications occurring in FFPE tissues by a combined pro-teomic and bioinformatic approach, which is based on OMSand differential modification count analysis. This approachis capable to detect unexpected modifications in an unbiasedmanner. As tissues are subjected to a long sample process-ing including formalin fixation, ethanol dehydration, xyleneclearing, and paraffin embedding, proteins treated with for-malin in the complex biological background could undergoreactions that are different from those observed in model pep-tides and proteins. Our data show that lysine methylation isthe major modification induced by FFPE tissue processing.Even though it only occurs on a small proportion of the totalMS/MS spectra (2–6%), it might significantly skew the re-sults when studying biological methylations, especially whencomparing FFPE to frozen samples. Despite the presence ofthe +12 Da, +14 Da, +30 Da, and +58 Da modifications,the high equivalence of the identified peptides between theFFPE and frozen tissue proteomes and the previously re-ported equivalence in protein spectral counts [1] indicates thatformalin fixation of tissue does not otherwise negatively influ-ence LC-MS/MS analysis. If these modifications are includedin MS/MS sequence searches as variable modifications, theywill add different PSMs and a few proteins. However, theywill also significantly increase the search space and thereforesearch time. The larger search space may require stricter scorethresholds to keep the FDR at 1% leading to reduced sensitiv-ity. Therefore we cannot generally recommend searching forall modifications and the optimal search settings for different
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2015, 00, 1–12 11
sample preparation protocols and different objectives need tobe evaluated in detail in further studies.
This work was supported by Grant-in-Aid for Publication ofScientific Research Results (228071) from MEXT (Ministry ofEducation, Culture, Sports, Science, and Technology) in Japanand Grant-in-aid for Young Scientists B (15K19448) from JapanSociety for the Promotion of Science.
The authors have declared no conflict of interest.
5 References
[1] Sprung, R. W. Jr., Brock, J. W., Tanksley, J. P., Li, M. et al.,Equivalence of protein inventories obtained from formalin-fixed paraffin-embedded and frozen tissue in multidimen-sional liquid chromatography-tandem mass spectrometryshotgun proteomic analysis. Mol. Cell Proteomics 2009, 8,1988–1998.
[2] Crockett, D. K., Lin, Z., Vaughn, C. P., Lim, M. S., Elenitoba-Johnson, K. S., Identification of proteins from formalin-fixedparaffin-embedded cells by LC-MS/MS. Lab. Invest. 2005, 85,1405–1415.
[3] Metz, B., Kersten, G. F., Hoogerhout, P., Brugghe, H. F. et al.,Identification of formaldehyde-induced modifications in pro-teins: reactions with model peptides. J. Biol. Chem. 2004,279, 6235–6243.
[4] Toews, J., Rogalski, J. C., Clark, T. J., Kast, J., Mass spectro-metric identification of formaldehyde-induced peptide modi-fications under in vivo protein cross-linking conditions. Anal.Chim. Acta 2008, 618, 168–183.
[5] Metz, B., Kersten, G. F., Baar,t, G. J., deJong, A. et al., Identi-fication of formaldehyde-induced modifications in proteins:reactions with insulin. Bioconjug. Chem. 2006, 17, 815–822.
[6] Toews, J., Rogalski, J. C., Kast, J., Accessibility governs therelative reactivity of basic residues in formaldehyde-inducedprotein modifications. Anal. Chim. Acta 2010, 676, 60–67.
[7] Rait, V. K., O’Leary, T. J., Mason, J. T., Modeling formalinfixation and antigen retrieval with bovine pancreatic ribonu-clease A: I—structural and functional alterations. Lab. Invest.2004, 84, 292–299.
[8] Rait, V. K., Xu, L., O’Leary, T. J., Mason, J. T., Modeling for-malin fixation and antigen retrieval with bovine pancreaticRNase A: II. Interrelationship of crosslinking, immunoreac-tivity, and heat treatment. Lab. Invest. 2004, 84, 300–306.
[9] Fowler, C. B., Cunningham, R. E., O’Leary, T. J., Mason, J.T., ‘Tissue surrogates’ as a model for archival formalin-fixedparaffin-embedded tissues. Lab. Invest. 2007, 87, 836–846.
[10] Fowler, C. B., Cunningham, R. E., Waybright, T. J., Blonder,J., et al., Elevated hydrostatic pressure promotes protein re-covery from formalin-fixed, paraffinembedded tissue surro-gates. Lab. Invest. 2008, 88, 185–195.
[11] Fowler, C. B., O’Leary, T. J., Mason, J. T., Modeling forma-lin fixation and histological processing with ribonuclease A:effects of ethanol dehydration on reversal of formaldehydecross-links. Lab. Invest. 2008, 88, 785–791.
[12] Wisniewski, J. R., Zougman, A., Nagaraj, N., Mann, M., Uni-versal sample preparation method for proteome analysis.Nat. Methods 2009, 6, 359–362.
[13] Wisniewski, J. R., Ostasiewicz, P., Mann, M., High recoveryFASP applied to the proteomic analysis of microdissectedformalin fixed paraffin embedded cancer tissues retrievesknown colon cancer markers. J. Proteome Res. 2011, 10,3040–3049.
[14] Bateman, N. W., Sun, M., Bhargava, R., Hood, B. L. et al.,Differential proteomic analysis of late-stage and recurrentbreast cancer from formalin-fixed paraffin-embedded tis-sues. J. Proteome Res. 2011, 10, 1323–1332.
[15] Nirmalan, N. J., Hughes, C., Peng, J., McKenna, T. et al., Initialdevelopment and validation of a novel extraction methodfor quantitative mining of the formalin-fixed, paraffin-embedded tissue proteome for biomarker investigations. J.Proteome Res. 2011, 10, 896–906.
[16] Ahrne, E., Muller, M., Lisacek, F., Unrestricted identificationof modified proteins using MS/MS. Proteomics 2010, 10,671–686.
[17] Pevzner, P. A., Dancık, V., Tang, C. L., Mutation-tolerant pro-tein identification by mass spectrometry. J. Comput. Biol.2000, 7, 777–787.
[18] Ahrne, E., Nikitin, Lisacek, F., Muller, F., QuickMod, M., Atool for open modification spectrum library searches. J. Pro-teome Res. 2011, 10, 2913–2921.
[19] Eng, J. K., McCormack, A. L., Yates, J. R., III. An approach tocorrelate tandem mass spectral data of peptides with aminoacid sequences in a protein database. J. Am. Soc. Mass Spec-trom. 1994, 5, 976–989.
[20] Keller, A., Nesvizhskii, A. I., Kolker, E., Aebersold, R., Em-pirical statistical model to estimate the accuracy of peptideidentifications made by MS/MS and database search. Anal.Chem. 2002, 74, 5383−5392.
[21] Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D. et al.,Gene ontology: tool for the unification of biology. The GeneOntology Consortium. Nat. Genet. 2000, 5, 25–29.
[22] Huang, da W., Sherman, B. T., Lempicki, R. A., Systematic andintegrative analysis of large gene lists using DAVID bioinfor-matics resources. Nat. Protoc. 2009, 4, 44–57.
[23] Stein, S. E., Scott, D. R., Optimization and testing of massspectral library search algorithms for compound identifica-tion. J. Am. Soc. Mass Spectrom. 1994, 5, 859–866.
[24] R Development Core Team, R: A Language and Environmentfor Statistical Computing, R Foundation for Statistical Com-puting, Austria 2011.
[25] Lam, H., Aebersold, R., Building and searching tandem mass(MS/MS) spectral libraries for peptide identification in pro-teomics. Methods 2011, 54, 424–431.
[26] Lam, H., Deutsch, E. W., Eddes, J. S., Eng, J. K. et al., Develop-ment and validation of a spectral library searching methodfor peptide identification from MS/MS. Proteomics 2007, 7,655–667.
[27] Ahrne, E., Ohta, Y., Nikitin, F., Scherl, A. et al., An improvedmethod for the construction of decoy peptide MS/MS spec-tra suitable for the accurate estimation of false discoveryrates. Proteomics 2011, 11, 4085–4095.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

12 Y. Zhang et al. Proteomics 2015, 00, 1–12
[28] Lam, H., Deutsch, E. W., Aebersold, R., Artificial decoy spec-tral libraries for false discovery rate estimation in spectrallibrary searching in proteomics. J. Proteome Res. 2010, 9,605–610.
[29] Tanca, A., Abbondio, C., Pisanu, S., Pagnozzi, D. et al., Criti-cal comparison of sample preparation strategies for shotgunproteomic analysis of formalin-fixed, paraffin-embeddedsamples: insights from liver tissue. Clin. Proteomics 2014,11, 28–38.
[30] Tanca, A., Pisanu, S., Biosa, G., Pagnozzi, D. et al., Applica-tion of 2D-DIGE to formalin-fixed diseased tissue samplesfrom hospital repositories: results from four case studies.Proteomics Clin. Appl. 2013, 7, 252–263.
[31] Wilhelm, M., Schlegl, J., Hahne, H., Gholami, A. M. et al.,Mass-spectrometry-based draft of the human proteome, Na-ture 2014, 509, 582–587.
[32] Craig, R., Beavis, R. C., A method for reducing the time re-quired to match protein sequences with tandem mass spec-tra. Rapid. Commun. Mass Spectrom. 2003, 17, 2310–2316.
[33] Guzel, C., Ursem, N. T., Dekker, L. J., Derkx, P. et al., Multiplereaction monitoring assay for pre-eclampsia related calcy-clin peptides in formalin fixed paraffin embedded placenta.J. Proteome Res. 2011, 10, 3274–3282.
[34] Guo, T., Wang, W., Rudnick, P. A., Song, T. et al., Proteomeanalysis of microdissected formalinfixed and paraffin-embedded tissue specimens. J. Histochem. Cytochem. 2007,55, 763–772.
[35] Tanca, A., Pagnozzi, D., Burrai, G. P., Polinas, M. et al., Compa-rability of differential proteomics data generated from pairedarchival fresh-frozen and formalin-fixed samples by GeLC-MS/MS and spectral counting. J. Proteomics 2012, 77, 561–576.
[36] Fowler, C. B., Cunningham, R. E., O’Leary, T. J. et al., ‘Tissuesurrogates’ as a model for archival formalin-fixed paraffin-embedded tissues. Lab. Invest. 2007, 87, 836–846.
[37] Roidl, D., Hacker, C., Histone methylation during neural de-velopment. Cell Tissue Res. 2014, May 13. [Epub ahead ofprint].
[38] Rypniewski, W. R., Holden, H. M., Rayment, I., Struc-tural consequences of reductive methylation of lysineresidues in hen egg white lysozyme: an X-ray anal-ysis at 1.8-A resolution. Biochemistry 1993, 32, 9851–9858.
[39] Rayment, I., Reductive alkylation of lysine residues to al-ter crystallization properties of proteins. Methods Enzymol.1997, 276, 171–179.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com




















