Antibody therapy to human L1CAM in a transgenic mouse model blocks local tumor growth but induces...
14
Antibody therapy to human L1CAM in a transgenic mouse model blocks local tumor growth but induces EMT Kai Doberstein 1 , Patrick N. Harter 2 , Uwe Haberkorn 3 , Niko P. Bretz 1 , Bernd Arnold 4 , Rafael Carretero 4 , Gerhard Moldenhauer 1 , Michel Mittelbronn 2 and Peter Altevogt 1 1 Translational Immunology, D015, Tumor Immunology Programme German Cancer Research Center (DKFZ), Heidelberg, Germany 2 Edinger Institute (Neurological Institute), Goethe University Frankfurt, Frankfurt, Germany 3 Department of Nuclear Medicine, University Hospital Heidelberg, Heidelberg, Germany 4 Molecular Immunology, D050, Tumor Immunology Programme German Cancer Research Center (DKFZ), Heidelberg, Germany L1 cell adhesion molecule (L1CAM) is overexpressed in many human cancers, confers bad prognosis and augments cell motil- ity, invasion and metastasis. Results from xenograft mouse models suggested that L1CAM antibodies might be promising tools for cancer therapy. Here, we generated human L1CAM-transgenic mice to study therapeutic efficacy and putative side effects in a model system. We established three transgenic lines (M2, M3 and F4) expressing the human L1CAM transgene in brain, kidney and colon with decreasing intensity (M2, M3 > F4). The expression pattern was similar to that of L1CAM in humans. No interference of the transgene with the expression of endogenous L1CAM was observed. Immunohistochemical analysis revealed correct expression of the transgene in mouse cortex and collective duct of the kidney. Injection of 125 I- labeled L1CAM antibodies resulted in specific enrichment in the kidney but not in the brain. The injection of the therapeutic anti-human L1CAM mAb L1-9.3/2a into transgenic mice even at high doses did not cause behavioral changes or other side effects. Similar results were obtained using a mouse specific L1CAM mAb in normal mice. Tumor therapy experiments were performed using syngeneic mouse tumor cells (RET melanoma and Panc02 pancreatic adenocarcinoma) transduced with human L1CAM. MAb L1-9.3/2a efficiently and specifically attenuated local tumor growth in both model systems without apparent side effects. The therapeutic effect was dependent on immune effector mechanisms. Analysis of Panc02-huL1CAM tumors after therapy showed elevated levels of EGF and evidence of immune-induced epithelial-mesenchymal transition. The results sug- gest that our transgenic mice are valuable tools to study L1CAM-based antibody therapy. The L1 cell adhesion molecule (L1CAM) is a 200–220 kDa transmembrane glycoprotein of the immunoglobulin (Ig) superfamily composed of six Ig-like domains and five fibro- nectin Type III repeats followed by a transmembrane region and a highly conserved cytoplasmic tail. 1 L1CAM can inter- act with itself (homophilic) and with heterophilic ligands such as integrins, CD24, neurocan, neuropilin-1 (NRP-1) and other members of the neural cell adhesion family. Cumulative evidence indicates that L1CAM (CD171) is ectopically expressed at high levels in a variety of cancers, including pancreatic, colorectal, ovarian and endometrial cancers, mela- noma and neuroblastoma. 2–6 Furthermore, high levels of L1CAM were found associated with increased grade of malig- nancy, 5,6 epithelial-mesenchymal transition (EMT), 6 poor patient prognosis 5,7,8 and worse response to chemotherapy. 4,7 These findings prompted extensive research towards develop- ment and preclinical testing of L1CAM specific mAbs for selective targeting of cancers. In line with the role of this molecule in cell motility, invasion and tumorigenesis, 9–11 L1CAM-specific mAbs were shown to suppress tumor cell outgrowth and metastasis in xenograft models. 12–14 The in vivo action of these mAbs was shown to depend on a combi- nation of immunologic and non-immunologic mechanisms, similar to what has been reported for mAbs targeting HER-2 (Herceptin) and CD20 (Rituxan). 13 Taken together, these findings point at the potential of anti-L1CAM mAbs as anti- cancer drugs. The use of mouse xenograft models is limited as only the therapeutic efficacy but not the potentially side effects of can- cer therapy can be evaluated. One way to overcome these problems is the development of antibodies binding with Key words: L1CAM, immunotherapy, transgenic mice, EMT Abbreviations: ADCC: antibody-dependent cellular cytotoxicity; EMT: epithelial-mesenchymal transition; huL1CAM: human L1 cell adhesion molecule; IHC: immunohistochemistry; mAb: mono- clonal antibody; mL1CAM: mouse L1 cell adhesion molecule; qRT-PCR: quantitative real-time PCR; WB: Western blot Additional Supporting Information may be found in the online version of this article. Grant sponsors: DKFZ (to G.M. and P.A.), the Deutsche Krebshilfe (Schwerpunktprogramm Invasion & Metastasis; to P.A.), the Wilhelm Sander Stiftung (Project 2010.094.1; to P.A.) DOI: 10.1002/ijc.29222 History: Received 24 June 2014; Accepted 9 Sep 2014; Online 18 Sep 2014 Correspondence to: Peter Altevogt, D015, Tumor Immunology Programme, German Cancer Research Center, Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany, Tel.: 16221-423714, Fax: 16221-423791, E-mail: [email protected] Tumor Immunology Int. J. Cancer: 00, 00–00 (2014) V C 2014 UICC International Journal of Cancer IJC
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Antibody therapy to human L1CAM in a transgenic mouse model blocks local tumor growth but induces...

Antibody therapy to human L1CAM in a transgenic mousemodel blocks local tumor growth but induces EMT
Kai Doberstein1, Patrick N. Harter2, Uwe Haberkorn3, Niko P. Bretz1, Bernd Arnold4, Rafael Carretero4,
Gerhard Moldenhauer1, Michel Mittelbronn2 and Peter Altevogt1
1 Translational Immunology, D015, Tumor Immunology Programme German Cancer Research Center (DKFZ), Heidelberg, Germany2 Edinger Institute (Neurological Institute), Goethe University Frankfurt, Frankfurt, Germany3 Department of Nuclear Medicine, University Hospital Heidelberg, Heidelberg, Germany4 Molecular Immunology, D050, Tumor Immunology Programme German Cancer Research Center (DKFZ), Heidelberg, Germany
L1 cell adhesion molecule (L1CAM) is overexpressed in many human cancers, confers bad prognosis and augments cell motil-
ity, invasion and metastasis. Results from xenograft mouse models suggested that L1CAM antibodies might be promising
tools for cancer therapy. Here, we generated human L1CAM-transgenic mice to study therapeutic efficacy and putative side
effects in a model system. We established three transgenic lines (M2, M3 and F4) expressing the human L1CAM transgene in
brain, kidney and colon with decreasing intensity (M2, M3 > F4). The expression pattern was similar to that of L1CAM in
humans. No interference of the transgene with the expression of endogenous L1CAM was observed. Immunohistochemical
analysis revealed correct expression of the transgene in mouse cortex and collective duct of the kidney. Injection of 125I-
labeled L1CAM antibodies resulted in specific enrichment in the kidney but not in the brain. The injection of the therapeutic
anti-human L1CAM mAb L1-9.3/2a into transgenic mice even at high doses did not cause behavioral changes or other side
effects. Similar results were obtained using a mouse specific L1CAM mAb in normal mice. Tumor therapy experiments were
performed using syngeneic mouse tumor cells (RET melanoma and Panc02 pancreatic adenocarcinoma) transduced with human
L1CAM. MAb L1-9.3/2a efficiently and specifically attenuated local tumor growth in both model systems without apparent side
effects. The therapeutic effect was dependent on immune effector mechanisms. Analysis of Panc02-huL1CAM tumors after
therapy showed elevated levels of EGF and evidence of immune-induced epithelial-mesenchymal transition. The results sug-
gest that our transgenic mice are valuable tools to study L1CAM-based antibody therapy.
The L1 cell adhesion molecule (L1CAM) is a 200–220 kDatransmembrane glycoprotein of the immunoglobulin (Ig)superfamily composed of six Ig-like domains and five fibro-nectin Type III repeats followed by a transmembrane regionand a highly conserved cytoplasmic tail.1 L1CAM can inter-act with itself (homophilic) and with heterophilic ligands
such as integrins, CD24, neurocan, neuropilin-1 (NRP-1) andother members of the neural cell adhesion family. Cumulativeevidence indicates that L1CAM (CD171) is ectopicallyexpressed at high levels in a variety of cancers, includingpancreatic, colorectal, ovarian and endometrial cancers, mela-noma and neuroblastoma.2–6 Furthermore, high levels ofL1CAM were found associated with increased grade of malig-nancy,5,6 epithelial-mesenchymal transition (EMT),6 poorpatient prognosis5,7,8 and worse response to chemotherapy.4,7
These findings prompted extensive research towards develop-ment and preclinical testing of L1CAM specific mAbs forselective targeting of cancers. In line with the role of thismolecule in cell motility, invasion and tumorigenesis,9–11
L1CAM-specific mAbs were shown to suppress tumor celloutgrowth and metastasis in xenograft models.12–14 The invivo action of these mAbs was shown to depend on a combi-nation of immunologic and non-immunologic mechanisms,similar to what has been reported for mAbs targeting HER-2(Herceptin) and CD20 (Rituxan).13 Taken together, thesefindings point at the potential of anti-L1CAM mAbs as anti-cancer drugs.
The use of mouse xenograft models is limited as only thetherapeutic efficacy but not the potentially side effects of can-cer therapy can be evaluated. One way to overcome theseproblems is the development of antibodies binding with
Key words: L1CAM, immunotherapy, transgenic mice, EMT
Abbreviations: ADCC: antibody-dependent cellular cytotoxicity;
EMT: epithelial-mesenchymal transition; huL1CAM: human L1
cell adhesion molecule; IHC: immunohistochemistry; mAb: mono-
clonal antibody; mL1CAM: mouse L1 cell adhesion molecule;
qRT-PCR: quantitative real-time PCR; WB: Western blot
Additional Supporting Information may be found in the online
version of this article.
Grant sponsors: DKFZ (to G.M. and P.A.), the Deutsche Krebshilfe
(Schwerpunktprogramm Invasion & Metastasis; to P.A.), the
Wilhelm Sander Stiftung (Project 2010.094.1; to P.A.)
DOI: 10.1002/ijc.29222
History: Received 24 June 2014; Accepted 9 Sep 2014; Online 18
Sep 2014
Correspondence to: Peter Altevogt, D015, Tumor Immunology
Programme, German Cancer Research Center, Im Neuenheimer Feld
280, D-69120 Heidelberg, Germany, Tel.: 16221-423714, Fax:
16221-423791, E-mail: [email protected]
Tum
orIm
mun
olog
y
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC
International Journal of Cancer
IJC

similar affinity to both the human and the murine forms ofthe target antigen.15 However, antibodies with these proper-ties may not be optimal for therapy. A further problem couldbe that the target antigen in the mouse is not expressed in asimilar cell type as in humans. Thus, these approaches toaddress putative side effects are limited in one or the otherway.
Another projection is the use of transgenic mice express-ing the human target antigen L1CAM, which has not beenperformed before. We set out to study the putative site effectsof mAb L1CAM targeted therapy by establishing humanL1CAM transgenic mice. We observed that the huL1CAMtransgene is expressed in similar cell types as seen in humansand does not alter expression of the endogenous mL1CAM.Using this transgenic model and syngeneic mouse tumorsexpressing the human L1CAM target antigen we show thatthe L1CAM mAb-based therapy of tumors is efficient andshows little site effects. The therapeutic affect was in partdependent on immune mediated effector mechanisms. Thesedata support the notion that L1CAM could be a valuablenew target antigen for tumor therapy warranting clinical test-ing. The transgenic mouse system described here will be ofgreat help for the preclinical evaluation.
Material and MethodsAntibodies
The mAbs to the ectodomain of L1CAM (L1-9.3, L1-9.3/2a, L1-11A and L1-14.10) were previously described.13 MAb to Vimen-tin (D21H3), E-Cadherin (24E10), phospho-EGFR (D7A5) andcleaved Caspase-3 (5A1E) were from Cell Signaling. The Gran-zyme B specific mAb was from Santa Cruz (sc-56117).
Immunohistochemical staining and evaluation
of expression
IHC staining was performed with mAb to the ectodomain ofL1CAM as previously described.2 Briefly, 3-mm-thick histo-logical paraffin sections were cut and mounted on SuperfrostPlus slides. HE stainings were performed according to routineprotocols. IHC staining was performed using M.O.M Kit(Dianova, Hamburg, Germany). Slides were heated in asteamer in citrate buffer (pH 6.0) for antigen retrieval.Endogenous peroxidase activity was blocked by a 15 mintreatment with 3% H2O2 in methanol and incubated withprimary antibodies for 60 min. Immunoperoxidase stainingwas accomplished using the sensitive detection kit with DAB
(DCS Diagnostics, Hamburg, Germany). Counterstaining wasperformed using hematoxylin prior to coverslipping andviewing by light microscopy. Omission of the primary anti-body was used as a negative control. Sections were reviewedby two histopathologists (PNH and MM) who were blindedto the outcome.
Cell lines
Ret melanoma cells were obtained from Prof. Viktor Umansky,(University of Mannheim)16 and the murine pancreatic adeno-carcinoma Panc0217 was obtained from Prof. E. Ryschich(University of Heidelberg). Cells were cultivated in RPMI 1640(PAA Laboratories, Pasching, Austria) medium supplementedwith 10% FBS, at 37�C, 5% CO2 and 100% humidity. Cellswere transduced with recombinant retrovirus encoding humanL1CAM or control virus as described before.18
Animal studies
Animal experiments were carried in accordance with GermanCoordinating Committee on Cancer Research guidelines andwith approval of the Regierungspr€asidium Karlsruhe (G156/09).
In vivo imaging
For antibody imaging studies, 200 ml of a solution of the125I-labeled L1-9.3 mAb (6.076 1.29 MBq/mouse; range4.72–8.03 MBq) was injected into the tail vein of normal ortransgenic mice. Scintigraphic images were taken for 10 minusing a gamma camera (g imager, Biospace, France). Theaccumulation of the radioactive tracer was monitored bystatic planar images at 2 hr, 4 hr, 24 hr, 48 hr, 72 hr, 96 hr,and 168 hr after injection.
For in vivo tumor cell imaging D-Luciferin (Biosynth,Switzerland) (0.15 g/g mice), dissolved in PBS, was injectedi.p. Imaging was performed using an IVIS Lumina device(Caliper). Mice were anesthetized with 1.5% isofluranethrough an O2 flow modulator. Ten minutes after injectionimage was acquired (60s). Photon flux was measured at base-line by drawing a rectangular region of interest (ROI). TheROI was duplicated each time the mouse was imaged.
Quantitative RT-PCR
Total RNA was isolated using the Quiagen RNeasy mini kit(Qiagen Hilden, Germany). Reverse transcription into cDNAwas performed using RevertAid First Strand cDNA SynthesisKits (Fementas, St. Leon-Rot, Germany). For qPCR, the
What’s new?
L1CAM is overexpressed in many human cancers, and is associated with poor prognosis and a reduced response to chemo-
therapy. In this study, the authors generated both mouse tumors and transgenic mice that express human L1CAM (huL1CAM)
in a pattern similar to that seen in humans. They found that monoclonal antibodies (mAb) directed against huL1CAM reduced
the growth of the tumors in vivo, with few side effects. These data support L1CAM as a potential therapeutic target. The
transgenic-mouse system described in this study may also be useful for preclinical evaluation of anti-L1CAM
immunotherapies.
Tum
orIm
mun
olog
y
2 L1CAM and tumor therapy
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

cDNA was purified on Microspin G-50 columns (GE Health-care, M€unchen, Germany) and quantified by NanoDrop spec-trophotometer (ND-1000, Kisker-Biotechnology, Steinfurt,Germany). Primers for qPCR were designed with the DNAStar Program and were produced by MWG Eurofines (Ebers-berg, Germany). b-Actin was used as an internal standard.The PCR reaction was performed with the SYBRgreen mas-termix (Applied Biosystems, Darmstadt, Germany). Thesequence of primers used is available on request. The mouseEMT array was performed using the Qiagen RT2 profilerPCR array (Cat.: 330231, PAMM-090ZA) as described in themanufacturer protocol.
FACS analysis
The staining of cells with mAbs and PE-conjugated second-ary antibodies has been previously described.19 Additionally,the primary anti-mouse antibodies and the procedure usedfor the analysis of mouse organs and tissues have been pub-lished before.20 Stained cells were analyzed with a FACSCanto II using FlowJo software (Becton & Dickinson, Heidel-berg, Germany).
Cell motility assay
This assay was described previously.21
Western blot analysis
SDS-PAGE under non-reducing conditions and transfer ofproteins to an Immobilon membrane using semi-dry blottinghas been described.19 After blocking with 5% skim milk inTBS, the blots were developed with the respective primaryantibody followed by peroxidase conjugated secondary anti-body and ECL (Perkin Elmer, Rodgau, Germany) detection.The human tissue blot was obtained from BioChain(W1234404, Biocat (Heidelberg, Germany).
Cytokine and angiogenesis protein arrays
Relative levels of cytokines/chemokines or angiogenic factorsin tumor lysates were evaluated using mouse antibody arraykits (Proteome ProfilerTM, mouse cytokine array panel A[catalog number ARY006] and mouse angiogenesis array kit[catalog number ARY015], R&D Systems, Abingdon, UK)according to the manufacturer’s recommendations. Proteinswere extracted from tumors using the following lysis buffer:250 mM NaCl, 50 mM HEPES, 0.5% NP-40, 10% glycerol,2 mM EDTA, 10 mM NaF, 1 mM Na-orthovanadate, 1 mMPMSF, 10 mg/ml of each leupeptin and aprotinin. Tumor tis-sue samples were homogenized in the lysis buffer using aFastPrep-24 homogenizer (MP Biomedicals, Eschwege, Ger-many). Pixel density in each spot of the array was quantifiedusing ImageJ 1.44o software.
Statistical analysis
p-Values were calculated by an unpaired t-test and 95% con-fidential intervals (CI) were calculated with the Graph Prismprogram.
ResultsCharacterization of human L1CAM transgenic mice
We generated transgenic mice using the complete genomichuman L1CAM sequence isolated from a BAC clone. Weincluded a large portion 30- and 50-sequences of the gene andinjected the excised DNA-fragment of �180,000 bp into fertil-ized eggs. Founder mice were typed using a huL1CAM specificPCR and three lines (M2, M3 and F4) were established by back-crossing with normal C57Bl/6 mice. We reasoned that the regu-latory sequences included in the DNA fragment, might result ina similar organ specific expression in mice as in humans. To testthis hypothesis, we analyzed the transgenic lines for organ-specific expression of huL1CAM by WB and RT-PCR.
L1CAM is most strongly expressed in the cerebral cortex, thecollecting duct cells of the kidney and in peripheral nerves of thecolon.22–25 We could confirm this by analyzing the lysates of humanorgans by WB (Fig. 1a). In addition, we detected L1CAM in theintestine and the rectum most likely due to the high innervation.Furthermore, also pancreatic tissue displayed high L1CAM levels(see Discussion section). In accordance with published human data,we also detected strongest expression of endogenous mL1CAM inmurine brain, kidney and colon (Figs. 1b and 1c, upper panel). Sim-ilarly, transgenic mice expressed huL1CAM most prominently inbrain followed by kidney and colon (Figs. 1b and 1c).
Interestingly, in addition to full-length huL1CAM of 220kDa (L1-220), we detected fragments of 200 kDa (L1-200)and 80 kDa (L1-80) (Fig. 1b), suggesting a conserved process-ing and cleavage of huL1CAM by proteases as describedbefore.26 Besides the organs known to express huL1CAM,low amounts were detected in the spleen, pancreas and heartof transgenic mice (Figs. 1b and 1c).
When comparing the transgenic lines among each otherfor protein and mRNA expression, M2 and M3 linesappeared to express higher amounts of huL1CAM comparedto F4 line (Figs. 1d and 1e). Of note, the mRNA expressionof mL1CAM was not altered in transgenic mice when com-pared to non-transgenic animals (Fig. 1f).
IHC analysis of human L1CAM in mouse kidney and brain
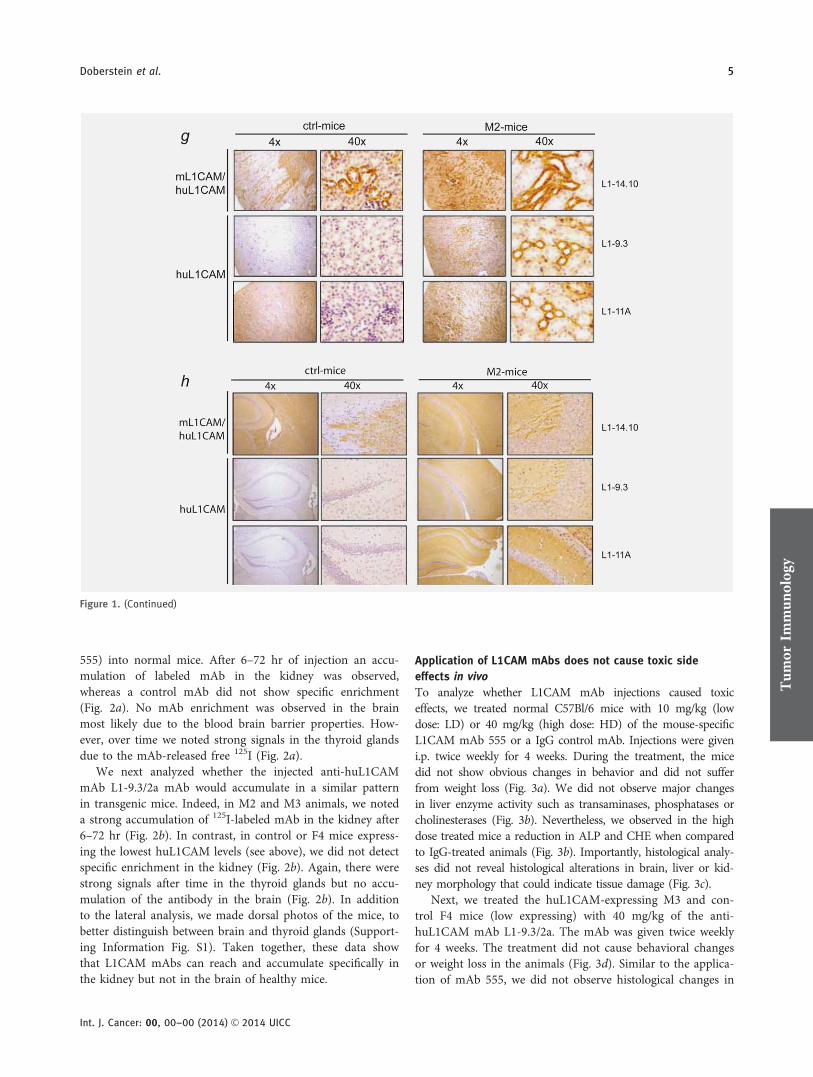
In humans, the expression of L1CAM in the kidney isrestricted to collecting duct cells.27,28 Therefore, we deter-mined whether the transgene was expressed in respectivecells. Using a mAb reactive with both huL1CAM andmL1CAM (L1-14.10), we found expression in the collectingduct cells of the kidney in both control and transgenic mice(Fig. 1f). In contrast, using the huL1CAM specific mAbs (L1-9.3 and L1-11A), we noticed that only the collecting ductcells from transgenic mice were positive (Fig. 1g).
Next, we studied the expression of huL1CAM in mousebrain. Using mAb L1-14.10, we observed strong L1CAMexpression in the hippocampus, which is the major part ofthe archicortex, of control and transgenic animals (Fig. 1g).In contrast, with huL1CAM-specific mAbs (L1-9.3 andL1-11A), staining in murine hippocampus was only detectedin the transgenic animals (Fig. 1h).
Tum
orIm
mun
olog
y
Doberstein et al. 3
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

In summary, our data indicate that the expression patternof huL1CAM is largely preserved in transgenic mice and,therefore, the M2 and M3 lines showing highest expressionare suitable to study L1CAM antibody therapy.
Radiolabeled anti-L1CAM mAbs localize to mouse kidney
We analyzed whether mAbs to L1CAM could reach the kid-ney and bind specifically to L1CAM expressing cells. There-fore, we first injected 125I-labeled anti-mL1CAM mAb (L1-
Figure 1. Expression of human L1CAM in transgenic mice. (a) WB analysis of human organ lysates with a huL1CAM specific antibody. A b-
actin antibody was used as a loading control. (b) WB analysis of mL1CAM in a control animal (ctrl) and huL1CAM expression in M2, M3
and F4 transgenic mice. A b-actin antibody was used as a loading control. (c) Relative qPCR analysis of mL1CAM and huL1CAM expression
in relation to b-actin in organs of a control animal (ctrl) or M2, M3 and F4 transgenic animals. (d) Comparison of huL1CAM expression
between M2, M3 and F4 in brain, kidney and colon as detected by WB analysis. A b-actin-specific antibody was used as a loading control.
Comparison of huL1CAM (e) and mL1CAM (f) expression between different mouse lines (ctrl, M2, M3 and F4) in brain, kidney and colon as
detected by RT-PCR. b-Actin expression was used as internal control. Expression of huL1CAM and m L1CAM in kidney (g) and brain (h) was
analyzed by IHC in control and transgenic mice. For the detection of huL1CAM, the mAbs L1-9.3 or L1-11A and for mL1CAM/huL1CAM mAb
L1-14.10 antibody were used.
Tum
orIm
mun
olog
y
4 L1CAM and tumor therapy
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC
555) into normal mice. After 6–72 hr of injection an accu-mulation of labeled mAb in the kidney was observed,whereas a control mAb did not show specific enrichment(Fig. 2a). No mAb enrichment was observed in the brainmost likely due to the blood brain barrier properties. How-ever, over time we noted strong signals in the thyroid glandsdue to the mAb-released free 125I (Fig. 2a).
We next analyzed whether the injected anti-huL1CAMmAb L1-9.3/2a mAb would accumulate in a similar patternin transgenic mice. Indeed, in M2 and M3 animals, we noteda strong accumulation of 125I-labeled mAb in the kidney after6–72 hr (Fig. 2b). In contrast, in control or F4 mice express-ing the lowest huL1CAM levels (see above), we did not detectspecific enrichment in the kidney (Fig. 2b). Again, there werestrong signals after time in the thyroid glands but no accu-mulation of the antibody in the brain (Fig. 2b). In additionto the lateral analysis, we made dorsal photos of the mice, tobetter distinguish between brain and thyroid glands (Support-ing Information Fig. S1). Taken together, these data showthat L1CAM mAbs can reach and accumulate specifically inthe kidney but not in the brain of healthy mice.
Application of L1CAM mAbs does not cause toxic side
effects in vivo
To analyze whether L1CAM mAb injections caused toxiceffects, we treated normal C57Bl/6 mice with 10 mg/kg (lowdose: LD) or 40 mg/kg (high dose: HD) of the mouse-specificL1CAM mAb 555 or a IgG control mAb. Injections were giveni.p. twice weekly for 4 weeks. During the treatment, the micedid not show obvious changes in behavior and did not sufferfrom weight loss (Fig. 3a). We did not observe major changesin liver enzyme activity such as transaminases, phosphatases orcholinesterases (Fig. 3b). Nevertheless, we observed in the highdose treated mice a reduction in ALP and CHE when comparedto IgG-treated animals (Fig. 3b). Importantly, histological analy-ses did not reveal histological alterations in brain, liver or kid-ney morphology that could indicate tissue damage (Fig. 3c).
Next, we treated the huL1CAM-expressing M3 and con-trol F4 mice (low expressing) with 40 mg/kg of the anti-huL1CAM mAb L1-9.3/2a. The mAb was given twice weeklyfor 4 weeks. The treatment did not cause behavioral changesor weight loss in the animals (Fig. 3d). Similar to the applica-tion of mAb 555, we did not observe histological changes in
Figure 1. (Continued)
Tum
orIm
mun
olog
y
Doberstein et al. 5
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

the brain, liver or kidney (Fig. 3e). When analyzing differentliver enzymes, aspartate-transaminase, cholinesterase (CHE)and alanine-transaminase showed no major differenceswhereas alkaline phosphatase levels were elevated in bothtransgenic animals after mAb L1-9.3/2a treatment (Fig. 3f).We further analyzed blood albumin and total blood proteindisplaying no major differences (Fig. 3g). In urine of M3mice treated with L1-9.3/2a, we observed elevated creatinineand protein levels compared to animals treated with controlmAb, yet the ratio between protein and creatinine was nearlyequal (Fig. 3h). Taken together, these data indicated that theprolonged treatment with anti-L1CAM mAbs did not lead tomajor side effects in mice.
Tumor therapy in transgenic mice
To set up a therapeutic model for tumor therapy in trans-genic animals, we used the syngeneic mouse melanoma cell
line RET and the pancreatic carcinoma cell line Panc02.16,17
Both cell lines were retrovirally transduced with huL1CAMand expressed high levels as detected by FACS or Westernblot analysis compared to control cells transduced with acontrol vector (Supporting Information Figs. S2A and S2B).
The analysis of cell proliferation did not reveal differencesbetween Panc02-huL1CAM cells or vector control transducedcells (Panc02-ctrl) (Supporting Information Fig. S2C). In con-trast, RET cells expressing human L1CAM (RET-huL1CAM)proliferated significantly faster than RET control (RET-ctrl)cells (Supporting Information Fig. S2D). In cell motility assays,we noticed only minor effects in RET and Panc02 cells over-expressing huL1CAM (Supporting Information Fig. S2E).
To test the therapeutic potential of mAb L1-9.3/2a, wefirst injected s.c. Panc02-huL1CAM (5 3 105 cells) into theflanks of M3 mice. Panc02-ctrl cells were injected into theopposite flank. The mAb L1-9.3/2a or the isotype control was
Figure 2. Injection of radiolabeled anti-L1CAM mAbs into mice. (a) Lateral radioimages of control mice after intravenous administration of
anti-mouse L1CAM antibody (L1-555) or control antibody (IgG) coupled to coupled to 125I. A time course analysis was performed from 2 to
72 hr and pictures were taken dorsally. Orange arrowhead symbols indicate thyroid glands; red arrows indicate accumulation in the kidney.
(b) Radioimages of control (ctrl), M2, M3 and F4 mice after intravenous administration of anti-huL1CAM antibody (L1-9.3/2a) coupled to125I. Time course analysis was performed from 2 to 72 hr and pictures were taken dorsally.
Tum
orIm
mun
olog
y
6 L1CAM and tumor therapy
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

Figure 3. Administration of anti-L1CAM mAbs is non-toxic in mice. (a) Mice were treated twice weekly with PBS, low dose (LD, 10 mg/kg) or
high dose (HD, 40 mg/kg) of control antibody (IgG) or anti-mouse L1CAM antibody (L1-555). Weight of animals was measured after 3 and 5
weeks. (b) Pooled blood plasma from five animals in (a) was analyzed for aspartate transaminase (AST), alanine transaminase (ALT), alka-
line phosphatase (ALP) and cholinesterase (CHE) by routine clinical analysis after 5 weeks. (c) H&E staining of brain, kidney and liver sec-
tions of mice from (a) treated with high dose L1-555 or control antibody (IgG2a). (d) Mice were treated twice weekly (40 mg/kg) with a
control antibody (IgG2a) or the anti-human L1CAM antibody (L1-9.3/2a). The weight of mice was measured after 3 and 5 weeks. (e) H&E
staining of brain, kidney and liver sections of F4 and M3 mice treated with control (IgG2a) or L1-9.3/2a mAb. (f) Blood plasma of mice
from (d) was analyzed for the indicated enzymes after 5 weeks. (g) Albumin and total blood protein content were analyzed from blood
plasma. (h) After 5 weeks, urine was collected for 24 hr from M3 mice from (d) and analyzed for creatinine and protein.
Tum
orIm
mun
olog
y
Doberstein et al. 7
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

given twice weekly for 4 weeks, starting 3 days after tumorinjection (Fig. 4a). Ten days after injection, tumor incidenceand size were first measured. For Panc02-huL1CAM tumors,we observed a reduced tumor incidence (87–67%) and tumorsize was 3.8-fold smaller in mice treated with L1-9.3/2a ascompared to control tumors (Fig. 4b).
Animals were sacrificed after 4 weeks of treatment andtumor tissue was analyzed. In non-treated animals, the tumorsize was similar irrespective of huL1CAM expression (Fig.
4c). In contrast, in mAb L1-9.3/2a-treated animals, thePanc02-huL1CAM tumors were significantly reduced in sizecompared to isotype control-treated animals (0.73–0.37 g/tumor) (Fig. 4d). Importantly, tumors not expressing huL1-CAM did not respond to mAb L1-9.3/2a treatment (Fig. 4e).
As a second model system, we used RET-huL1CAM andthe respective RET-ctrl cells. As described above, mice weretreated twice weekly with mAb L1-9.3/2a or control mAb(Fig. 4f). The experiment was terminated after 3 weeks and
Figure 4. Treatment with anti-human L1CAM mAb L1-9.3/2 is effective in syngeneic cancer models. (a) 5 3 105 Panc02 cells were in
injected s.c. into flanks of transgenic animals. Antibody treatment (20 mg/kg) was performed twice weekly for 4 weeks with either control
mAb or L1-9.3/2a mAb (n 5 15 for each group). Mice were sacrificed 35 days after tumor injection. (b) Ten days after tumor injection, tumor
size was measured by luciferase imaging. Left: fluorescence (ROI) in tumors treated with control mAb (IgG2a) or anti-huL1CAM mAb (L1-
9.3/2a) (p 5 0.04; 95% CI 5 0.26–7.42). Right: tumor incidence. Mice were sacrificed on day 35 and tumors were analyzed. (c) Comparison
of tumor growth between Panc02-ctrl and Panc02-huL1CAM. Left: Tumor weight in grams (p 5 0.84; 95% CI 5 20.42 to 0.35). Right: a rep-
resentative H&E section is shown. (d) Comparison of Panc02-huL1CAM tumors treated with control mAb (IgG2a) or L1-9.3/2a mAb. Left:
Tumor weight in grams (p 5 0.02; 95% CI 5 0.0085–0.62). Right: a representative H&E section is shown. (e) Comparison of Panc02-ctrl
tumors treated with control mAb (IgG2a) or L1-9.3/2a mAb. Left: Tumor weight in grams (p 5 0.87; 95% CI 5 20.40 to 0.33). Right: a repre-
sentative H&E section is shown. (f) 5 3 105 RET cells were injected s.c. into the flanks of transgenic animals. Antibody treatment (20 mg/
kg) was given twice weekly for 3 weeks with either a control mAb (IgG2a) or the L1-9.3/2a mAb (n 5 10 for each group). Mice were sacri-
ficed 22 days after tumor injection. (g) Comparison of tumor growth between RET-ctrl and RET-huL1CAM. Left: tumor weight in grams
(p 5 0.06; 95% CI 5 20.014 to 0.86). Middle: tumor incidence. Right: a representative H&E section is shown. (h) Comparison of RET-
huL1CAM tumors treated with control antibody (IgG2a) or L1-9.3/2a antibody. Left: Tumor weight in grams (p 5 0.09; 95% CI 5 20.039 to
0.48). Middle: tumor incidence. Right: a representative H&E section is shown. (i) Comparison of RET-ctrl tumors treated with control anti-
body (IgG2a) or L1-9.3/2a mAb. Left: tumor weight in grams (p 5 0.5; 95% CI 5 20.41 to 0.84). Middle: tumor incidence. Right: a represen-
tative H&E section is shown.
Tum
orIm
mun
olog
y
8 L1CAM and tumor therapy
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

mice were sacrificed. In non-treated animals RET-huL1CAMtumors showed an average of 0.26 g/tumor and were smallerthan the RET-vector tumors (0.68 g/tumor). Also the tumorincidence was lower in RET-huL1CAM tumors (90–60%)(Fig. 4g).
When comparing RET-huL1CAM tumors treated with L1-9.3/2a to tumors treated with control mAb, a strong reduc-tion in tumor size was observed (0.26–0.04 g/tumor) (Fig.4h). Additionally, tumor incidence was much lower in L1-9.3/2a-treated animals (60–20%) (Fig. 4h). In contrast, RET-ctrl tumors showed only little difference in tumor size (0.68–0.47g/tumor) and no differences in tumor incidence (both
90%) after mAb treatment (Fig. 4i). Taken together, thesedata demonstrated that the L1-9.3/2a mAb treatment effi-ciently attenuates tumor growth in two syngeneic mousetumor models.
Analysis of effector mechanisms
Previous studies in the SKOV3ip xenograft model had shownthat therapeutic effects by L1-9.3/2a could be inhibited byclodronate-mediated depletion of phagocytic cells suggestingthe involvement of ADCC.13 This is in line with the fact thatthe IgG2a isotype is the preferred isotype for ADCC in themouse.29 To investigate the role of ADCC in our syngeneic
Figure 4. (Continued)
Tum
orIm
mun
olog
y
Doberstein et al. 9
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

Panc02-huL1CAM system, we injected clodronate twiceweekly at the site of the primary tumor in the absence orpresence of mAb L1-9.3/2a therapy (i.p. injected). Clodronate
treatment enhanced the growth of both Panc02-ctrl andPanc02-L1CAM and eliminated the therapeutic effect of mAbL1-9.3/2a (Figs. 5a and 5b).
Figure 5.
Tum
orIm
mun
olog
y
10 L1CAM and tumor therapy
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

FACS analysis of tumor infiltrating leukocytes showed nostriking differences in the percentage of dendritic cells, eosin-ophils, neutrophils and macrophages (not shown). Also thetotal number of leukocytes was only marginally increased.However, the percentage of CD4 or CD8 T lymphocytes andNK cells was significantly increased (Figs. 5c and 5d).
To further proof a role for NK cells, we prepared tumortissue lysates and compared the five largest Panc02-huL1CAM tumors treated with L1-9.3/2a or control antibodyfor NK-cell markers by WB analysis. Interestingly, in three offive L1-9.3/2a tumors, we found an increase in Granzyme-Bexpression, which is a marker for active NK cells (Figs. 5eand 5f). Furthermore, samples expressing higher amounts ofGranzyme-B also showed an increase in cleaved Caspase-3,indicating an induction of apoptosis in treated tumors (Figs.5e and 5f). Taken together, these results suggested theinvolvement of immune effector mechanisms in the anti-tumor effect of mAb L1-9.3/2a.
Antibody therapy of Panc02-L1CAM tumors induces EMT
Tumor cell lysates were examined for cytokines expressionusing protein arrays (Fig. 6a). Importantly, in Panc02-huL1CAM tumors treated with mAb L1-9.3/2a, we observedsignificantly elevated levels of EGF as compared to Panc02-control tumors (Figs. 6a and 6b). As EGF is a potent inducerof EMT,30 we analyzed mRNAs extracted from tumors forEMT markers using specific mRNA arrays (Fig. 6c). Indeed,we observed a strong decrease of E-Cadherin and up-regulation of Vimentin in L1CAM-positive tumors treatedwith mAb L1-9.3/2a (Fig. 6c). These findings were confirmedby WB analysis of tumor cell lysates (Fig. 6d). We furtheranalyzed by qPCR the expression of the E-Cadherin repress-ors Snail, Slug and Twist (Fig. 6e). We found an induction ofSnail and Twist gene expression that was not significant butmight cause the down-regulation of E-Cadherin in the tumorcells.
EGF is known to signal via EGFR that in its active state isauto-phosphorylated at Tyr1068. To find out whether EGF inthe tumor tissue led to activation of EGFR, we stained tumorsections by IHC. We observed strong staining with the
phospho-EGFR-specific antibody that was only detected inmAb L1-9.3/2a-treated but not in control tumors (Fig. 6f).Nevertheless, we could not observe this effects when Panc02-huL1CAM tumors were treated with increasing doses of mabL1-9.3/2a in vitro, indicating that the tumor microenviron-ment might be essential for this phenomenon (data notshown). These results suggest that prolonged treatment withtherapeutic antibodies in the Panc02-huL1CAM model leadsto the induction of EMT.
DiscussionL1CAM is a biomarker for ovarian, endometrial carcinomasand pancreatic adenocarcinoma with the potential to serve asa therapeutic target.2,12,13,31–33 In this study, we have set up atransgenic mouse model system to allow the study of thera-peutic efficacy and putative side effects of an L1CAM-antibody-based tumor therapy. We found that (i) the humanL1CAM transgene is expressed in mouse tissues with highsimilarity to human tissues; (ii) injections of mAbs directedagainst mouse or human L1CAM do not cause toxic sideeffects; (iii) in syngeneic tumor models antibodies to humanL1CAM specifically attenuate tumor growth; (iv) in Panc02-huL1CAM tumors the therapy with mAb to L1CAM leads tothe induction of EMT. These unexpected findings suggestedthat mAb therapy can support both positive (curative) aswell as potentially harmful (EMT induction) effects withinthe tumor microenvironment.
The prime goal of our study was to develop mouse lineswith a similar organ expression pattern of human L1CAM asin humans. As determined by WB analysis of human tissues,L1CAM expression is highest in brain and kidney and in theintestine most likely due to strong neuronal innervation (seeFig. 1). The expression of L1CAM in pancreatic tissue asseen on tissue blot (see Fig. 1) could not be confirmed byIHC on normal pancreatic tissues in our hands and not byothers.22,34 It is not excluded that the material on the blotwas derived from an individual suffering from chronic pan-creatitis that is positive for L1CAM.34,35 The tissues collectedfrom our transgenic mouse lines recapitulated the expressionpattern in humans with high fidelity. It should be noted that
Figure 5. Anti-L1CAM treatment induce T-/NK cell recruitment into tumors. (a) Average tumor growth in volume (mm3) over time (days) was
measured in M3 mice carrying Panc02-huL1CAM tumors. 5 3 105 Panc02 cells were in injected s.c. into flanks. Mice were treated i.p. with
control mAb (IgG2a) or L1-9.3/2a mAb twice weekly. Additionally, clodronate liposomes (Clo) or control liposomes (ctrl) were injected s.c.
close to the tumor, twice weekly. Clodronate-treated mice were sacrificed after day 25 because of large tumor size, whereas the control
liposome-treated mice were sacrificed on day 34. (b) Tumor volume (mm3) at day 25 and tumor mass (mg) at day 34 of mice as described
in (a) (day 25, IgG: p 5 0.2; 95% CI 5 231 to 87; day 25, L1-9.3/2a: p 5 0.03; 95% CI 5 295 to 7.15; day 34: p 5 0.05; 95% CI 5 237.3
to 2,569). (c) Analysis of tumor infiltrating immune cells by FACS using appropriate gating. Representative FACS blots of a Panc02-huL1CAM
tumor that were treated with control mAb (IgG2a) or L1-9.3/2a. Left: PI versus CD45 for leukocytes; middle: NK1.1 versus CD3 for NK-cells,
NKT-cells and T-lymphocytes; right: CD8 versus CD4 for cytotoxic T-cells and T-helper cells. Given numbers are the percentages of gated
cells to the counted tumor cells. The gating strategy is shown in detail in Supporting Information Figure S3. (d) Analysis of tumor infiltrating
immune cells shown in percent cells in tumor (c). (n 5 3 for each group). Shown are leukocytes (p 5 0.5; 95% CI 5 218.66 to 10.71), T-
helper cells (p 5 0.03; 95% CI 5 23.99 to 20.36), Cytotoxic T-Cells (p 5 0.2; 95% CI 5 22.66 to 0.76) and NK cells (p 5 0.03; 95%
CI 5 20.38 to 20.03). (e) WB analysis of control IgG2a, L1-9.3/2a-treated tumors and Panc02-huL1CAM cell lysate. Analysis was performed
with specific mAbs for cleaved caspase 3 (c-Caspase 3), Granzyme B and as a loading control huL1CAM. (f) Densitometric analysis of c-
Caspase 3 (p 5 0.4; 95% CI 5 21.25 to 0.51) or Granzyme B (p 5 0.2; 95% CI 5 228 to 6.94) of blot shown in (e).
Tum
orIm
mun
olog
y
Doberstein et al. 11
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

the pancreas was always negative for huL1CAM and this wasalso confirmed on the level of the mRNA. The mRNAexpression pattern was coherent to the obtained protein
expression data. In addition, immunohistochemical stainingof organs expressing highest levels of L1CAM revealed thatthe transgene was expressed in the correct type of cells.
Figure 6.
Tum
orIm
mun
olog
y
12 L1CAM and tumor therapy
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

Although we have not yet examined all transgenic tissues byIHC in detail, it seems that the transgenic mouse linesreflected the expression pattern in humans quite well.
To examine whether the transgenic protein was accessiblevia the blood stream during the course of an antibody ther-apy, we injected 125I-labeled anti-L1CAM mAbs into wild-type or transgenic mice. In both settings, we observed specificenrichment of the labeled mAb in the kidney but not in thebrain. It is quite likely that an intact blood–brain barrier pre-vented the entry of antibodies to the brain.
It is known that the application of pertussis-toxoid can inpart open the blood brain barrier and allow the entry ofimmune cells.36 When using a similar protocol to augment entryof labeled antibody we failed to detect any enhanced signals inthe brain (data not shown). Thus, at presence, we cannot ruleout the possibility that in the situation of a leaky blood–brainbarrier anti-L1CAM antibody may get access to the brain.
To assess the consequences of systemic and long-lastingL1CAM mAb application we injected high doses of the thera-peutic mAb L1-9.3/2a into transgenic M3 mice for several weeks.Looking at parameters such as behavior, weight, blood enzymemarkers and histochemical appearance of the kidney, we did notdetect striking changes induced by mAb injection. This is in linewith previous findings showing that prolonged and high-doseapplication of L1-555 mAb into wild-type C57Bl/6 mice did notshow adverse effects either.13 In our study, we observed slightlyincreased urine creatinine and protein values. However, we didnot observe any damage of the glomeruli. The significance ofthese findings needs to be further investigated.
In contrast, in the setting with syngeneic tumor cellsexpressing huL1CAM, the therapeutic mAb was very welleffective. In both model systems, the administration of mAb-L1-9.3/2a significantly reduced the tumor incidence and sub-cutaneous tumor growth. We provide evidence that innateand adaptive immune mechanisms are involved as the num-ber of tumor-infiltrated T cells and NK cells were signifi-cantly augmented in groups receiving the therapeutic mAbcompared to isotype controls. These findings are reminiscentto previous work on anti-Her2/neu mAb therapy strategies inmice.37 In our experiments, ADCC was most likely importantbased on the IgG2a isotype of the therapeutic mAb and our
previous experience in the SKOV3ip xenograft model wherethe same mAb was applied.13
It is quite important to note that tumor cells are moresensitive to the targeting with anti-L1CAM mAbs comparedto normal tissue. The binding of antibodies to L1CAM ontumor cells leads to rapid internalization and degradation38,39
that can proceed in a clathrin-dependent and independentmanner.40 Similar studies have not been done for normal epi-thelial cells expressing L1CAM. If indeed the rate or themode of internalization of L1CAM in tumor cells is distinctfrom that in normal cells this could open a therapeutic win-dow for the anti-tumor therapy based on L1CAM.
In breast cancer, Her2 mAb therapy has greatly improvedthe survival rate of patients. But despite the initial success oftherapy about 70% of treated patients will experience relapse.41
This may be related to phenotypic changes that tumor cellsundergo during therapy. Interestingly, the analysis of Panc02-huL1CAM tumors revealed some unexpected findings uponanti-L1CAM treatment. Tumors that were treated with mAb L1-9.3/2a expressed higher levels of EGF. From the control experi-ments it became clear that the expression of huL1CAM as wellas the therapeutic antibody was required for this effect. We alsonoticed a down-regulation of E-cadherin and an up-regulationof vimentin in the tumor tissues that are typical signs ofEMT.30,42 It is quite known that EMT promotes metastasis andtumor progression and can be triggered by EGFR signaling.43
We found evidence for active EGFR in the tumor tissues sug-gesting that the EGF-EGFR axis was involved in the inductionof EMT. It is currently unclear whether EGF was secreted fromtumor cells or from infiltrating immune cells. Interestingly, arecent study by Zhang et al. has shown that endothelial cell-derived EGF induces EMT and acquisition of stem-like proper-ties in head and neck tumor cells.44 There is increasing evidencethat the tumor microenvironment has an important influenceon metastatic behavior of tumor cells.45 Collectively, our resultsclearly support the notion that long-lasting mAb therapies canhave an adverse effect by promoting tumor progression.
Finally, a clear limitation of this work is that our mousemodel system represents only an approximation to the clini-cal situation of the cancer patient. At present, we do notknow whether our findings can be extrapolated to clinical
Figure 6. Anti-L1CAM treatment induces EMT in tumor cells in vivo. (a) Pooled lysates (n 5 9) of Panc02-huL1CAM and Panc02-ctrl tumors
treated either with control IgG or L1-9.3/2a were analyzed with an angiogenesis antibody array. The signal for EGF is boxed. (b) Combined
densitometric analysis of the angiogenesis array shown in (a) and the cytokine array (not shown) is represented in a logarithmic scale of
L1-9.3/2a-treated tumors against control IgG-treated tumors. Left: showing Panc02-huL1CAM tumors. Right: showing Panc02-ctrl tumors.
(c) Pooled RNAs (n 5 9) from Panc02-huL1CAM tumors were analyzed with a qPCR using an EMT array. The results are displayed in a
logarithmic scale of L1-9.3/2a-treated tumors versus IgG-treated tumors. Highlighted are the expression values of Vimentin (Vim), EGFR and
E-Cadherin (Cdh1). (d) Left: Western blot analysis of five control IgG, 5 L1-9.3/2a-treated tumors and a Panc02/huL1CAM cell line lysate for
control. Blot was analyzed with specific antibodies for the expression of Vimentin (Vim), E-Cadherin (E-Cad) and as a loading control human
b-actin. Middle and right: relative densitometric analysis of Vimentin (p 5 0.002; 95% CI 5 20.55 to 20.17) and E-Cadherin (p 5 0.02;
95% CI 5 2.54 3 1027 to 2.71 3 1026) expression in relation to the b-actin loading control. (e) Relative qPCR analysis normalized to
b-actin of Snail (p 5 0.16; 95% CI 5 213.7 to 2.73), Slug (p 5 0.43; 95% CI 5 20.63 to 1.36), Twist (p 5 0.38; 95% CI 5 212.66 to 5.40)
and E-Cadherin (p 5 0.05; 95% CI 5 22.36 3 1023 to 0.57) of Panc02/huL1CAM tumors treated with IgG/2a or L1-9.2/2a. n 5 5 for
each group. (f) Detection of p-EGFR (Tyr1068) in Panc02-huL1AM tumors after treatment with IgG or L1-9.3/2a. A representative picture is
shown.
Tum
orIm
mun
olog
y
Doberstein et al. 13
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC

application. Our model system will also not make additionalpreclinical toxicology testing in primates dispensable. How-ever, we provided good evidence that our L1CAM transgenicmice could be helpful for the development, testing and opti-mization of L1CAM-mAb based therapies.
AcknowledgementsThe authors thank Natalie Erbe-Hofmann and Cornelia Zachskorn for skill-ful technical assistance and G€unther K€ublbeck for compentent help with theestablishment of huL1CAM transgenic mice. The authors are particularlythankful to their colleague Rienk Offringa for discussions and ideas and toNico van Rooijen for clodronate liposomes.
References
1. Moos M, Tacke R, Scherer H, et al. Neural adhe-sion molecule L1 as a member of the immuno-globulin superfamily with binding domainssimilar to fibronectin. Nature 1988;334:701–3.
2. Fogel M, Gutwein P, Mechtersheimer S, et al. L1expression as a predictor of progression and sur-vival in patients with uterine and ovarian carci-nomas. Lancet 2003;362:869–75.
3. Kaifi JT, Reichelt U, Quaas A, et al. L1 is associ-ated with micrometastatic spread and poor out-come in colorectal cancer. Mod Pathol 2007;20:1183–90.
4. Sebens Muerkoster S, Werbing V, Sipos B, et al.Drug-induced expression of the cellular adhesionmolecule L1CAM confers anti-apoptotic protec-tion and chemoresistance in pancreatic ductaladenocarcinoma cells. Oncogene 2007;26:2759–68.
5. Ben QW, Wang JC, Liu J, et al. Positive expres-sion of L1-CAM is associated with perineuralinvasion and poor outcome in pancreatic ductaladenocarcinoma. Ann Surg Oncol 2010;17:2213–21.
6. Huszar M, Pfeifer M, Schirmer U, et al. Up-regu-lation of L1CAM is linked to loss of hormonereceptors and E-cadherin in aggressive subtypesof endometrial carcinomas. J Pathol 2010;220:551–61.
7. Bondong S, Kiefel H, Hielscher T, et al. Prognos-tic significance of L1CAM in ovarian cancer andits role in constitutive NF-kappaB activation. AnnOncol 2012;23:1795–802.
8. Boo YJ, Park JM, Kim J, et al. L1 expression as amarker for poor prognosis, tumor progression,and short survival in patients with colorectal can-cer. Ann Surg Oncol 2007;14:1703–11.
9. Mechtersheimer S, Gutwein P, Agmon LN, et al.Ectodomain shedding of L1 adhesion moleculepromotes cell migration by autocrine binding tointegrins. J Cell Biol 2001;155:661–74.
10. Gavert N, Conacci-Sorrell M, Gast D, et al. L1, anovel target of beta-catenin signaling, transformscells and is expressed at the invasive front ofcolon cancers. J Cell Biol 2005;168:633–42.
11. Meier F, Busch S, Gast D, et al. The adhesionmolecule L1 (CD171) promotes melanoma pro-gression. Int J Cancer 2006;119:549–55.
12. Arlt MJ, Novak-Hofer I, Gast D, et al. Efficientinhibition of intra-peritoneal tumor growth anddissemination of human ovarian carcinoma cellsin nude mice by anti-L1-cell adhesion moleculemonoclonal antibody treatment. Cancer Res 2006;66:936–43.
13. Wolterink S, Moldenhauer G, Fogel M, et al.Therapeutic antibodies to human L1CAM: func-tional characterization and application in amouse model for ovarian carcinoma. Cancer Res2010;70:2504–15.
14. Fischer E, Grunberg J, Cohrs S, et al. L1-CAM-targeted antibody therapy and (177) Lu-Radioimmunotherapy of disseminated ovariancancer. Int J Cancer 2012;130:2715–21.
15. Farady CJ, Sellers BD, Jacobson MP, et al.Improving the species cross-reactivity of an anti-body using computational design. Bioorg MedChem Lett 2009;19:3744–7.
16. Kato M, Takahashi M, Akhand AA, et al. Trans-genic mouse model for skin malignant melanoma.Oncogene 1998;17:1885–8.
17. Corbett TH, Roberts BJ, Leopold WR, et al.Induction and chemotherapeutic response of twotransplantable ductal adenocarcinomas of thepancreas in C57BL/6 mice. Cancer Res 1984;44:717–26.
18. Kiefel H, Bondong S, Erbe-Hoffmann N, et al.L1CAM-integrin interaction induces constitutiveNF-kappaB activation in pancreatic adenocarci-noma cells by enhancing IL-1beta expression.Oncogene 2010;29:4766–78.
19. Gutwein P, Oleszewski M, Mechtersheimer S,et al. Role of Src kinases in the ADAM-mediatedrelease of L1 adhesion molecule from humantumor cells. J Biol Chem 2000;275:15490–7.
20. Bretz NP, Salnikov AV, Doberstein K, et al.CD24 expression alters leukocyte trafficking tothe colon. Immunol Lett 2014;161:140–8.
21. Gast D, Riedle S, Kiefel H, et al. The RGD integ-rin binding site in human L1-CAM is importantfor nuclear signaling. Exp Cell Res 2008;314:2411–8.
22. Huszar M, Moldenhauer G, Gschwend V, et al.Expression profile analysis in multiple humantumors identifies L1 (CD171) as a molecularmarker for differential diagnosis and targetedtherapy. Hum Pathol 2006;37:1000–8.
23. Nolte C, Moos M, Schachner M. Immunolocali-zation of the neural cell adhesion molecule L1 inepithelia of rodents. Cell Tissue Res 1999;298:261–73.
24. Martini R, Schachner M. Immunoelectron micro-scopic localization of neural cell adhesion mole-cules (L1, N-CAM, and myelin-associatedglycoprotein) in regenerating adult mouse sciaticnerve. J Cell Biol 1988;106:1735–46.
25. Rathjen FG, Schachner M. Immunocytologicaland biochemical characterization of a new neuro-nal cell surface component (L1 antigen) which isinvolved in cell adhesion. EMBO J 1984;3:1–10.
26. Schafer MK, Altevogt P. L1CAM malfunction inthe nervous system and human carcinomas. CellMol Life Sci 2011;67:2425–37.
27. Helbert MJ, Dauwe SE, De Broe ME. Flow cyto-metric immunodissection of the human distaltubule and cortical collecting duct system. KidneyInt 2001;59:554–64.
28. Doberstein K, Wieland A, Lee SB et al. L1-CAMexpression in ccRCC correlates with shorterpatients survival times and confers chemoresist-ance in renal cell carcinoma cells. Carcinogenesis2011;32:262–70.
29. Nimmerjahn F, Ravetch JV. Divergent immuno-globulin g subclass activity through selective Fcreceptor binding. Science 2005;310:1510–2.
30. Thiery JP, Sleeman JP. Complex networks orches-trate epithelial-mesenchymal transitions. Nat RevMol Cell Biol 2006;7:131–42.
31. Schafer H, Dieckmann C, Korniienko O, et al.Combined treatment of L1CAM antibodies andcytostatic drugs improve the therapeutic responseof pancreatic and ovarian carcinoma. Cancer Lett2012;319:66–82.
32. Schafer H, Geismann C, Heneweer C, et al. Myo-fibroblast-induced tumorigenicity of pancreaticductal epithelial cells is L1CAM dependent. Car-cinogenesis 2012;33:84–93.
33. Zeimet AG, Reimer D, Huszar M, et al. L1CAMin early-stage type I endometrial cancer: resultsof a large multicenter evaluation. J Natl CancerInst 2013;105:1142–50.
34. Bergmann F, Wandschneider F, Sipos B, et al. Ele-vated L1CAM expression in precursor lesions andprimary and metastastic tissues of pancreatic ductaladenocarcinoma. Oncol Rep 2010;24:909–15.
35. Geismann C, Morscheck M, Koch D, et al. Up-regulation of L1CAM in pancreatic duct cells istransforming growth factor beta1- and slug-dependent: role in malignant transformation ofpancreatic cancer. Cancer Res 2009;69:4517–26.
36. Kugler S, Bocker K, Heusipp G, et al. Pertussistoxin transiently affects barrier integrity, organelleorganization and transmigration of monocytes ina human brain microvascular endothelial cell bar-rier model. Cell Microbiol 2007;9:619–32.
37. Park S, Jiang Z, Mortenson ED, et al. The thera-peutic effect of anti-HER2/neu antibody dependson both innate and adaptive immunity. CancerCell 2010;18:160–70.
38. Novak-Hofer I, Amstutz HP, Macke HR, et al.Cellular processing of copper-67-labeled mono-clonal antibody chCE7 by human neuroblastomacells. Cancer Res 1995;55:46–50.
39. Novak-Hofer I. The L1 cell adhesion molecule asa target for radioimmunotherapy. Cancer BiotherRadiopharm 2007;22:175–84.
40. Long KE, Asou H, Snider MD, et al. The role ofendocytosis in regulating L1-mediated adhesion.J Biol Chem 2001;276:1285–90.
41. Diessner J, Bruttel V, Stein RG, et al. Targetingof preexisting and induced breast cancer stemcells with trastuzumab and trastuzumab emtan-sine (T-DM1). Cell Death Dis 2014;5:e1149.
42. Gavert N, Ben-Ze’ev A. Epithelial-mesenchymaltransition and the invasive potential of tumors.Trends Mol Med 2008;14:199–209.
43. Misra A, Pandey C, Sze SK, et al. Hypoxia activatedEGFR signaling induces epithelial to mesenchymaltransition (EMT). PLoS One 2012;7:e49766.
44. Zhang Z, Dong Z, Lauxen IS, et al. Endothelialcell-secreted EGF induces epithelial tomesenchymal transition and endows head andneck cancer cells with stem-like phenotype. Can-cer Res 2014;74:2869–81.
45. Joyce JA, Pollard JW. Microenvironmental regula-tion of metastasis. Nat Rev Cancer 2009;9:239–52.
Tum
orIm
mun
olog
y
14 L1CAM and tumor therapy
Int. J. Cancer: 00, 00–00 (2014) VC 2014 UICC