Chiral building blocks for amphotericin B
10
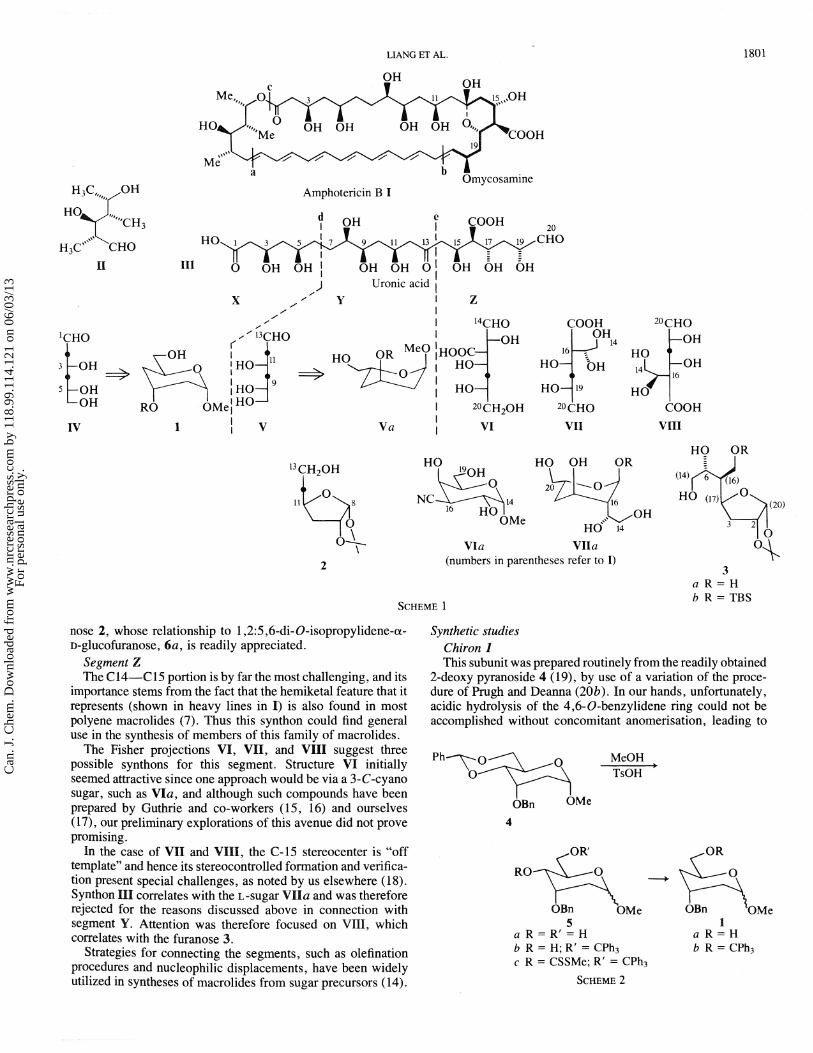
Ghiral building blocks for amphotericin B DAVID LIANC,~ HENRY W. PAULS, AND BERT FRASER-RE ID^ Paul M. Gross Chemical Laborato~y, Duke University, Durham, NC 27706, U.S.A. MICHAEL GEORGES The Guelph-Waterloo Centre for Graduate Work in Chemistry, Universih of Waterloo, Waterloo, Ont., Canada N2L 36L AND AZEEZ M. MUBARAK AND SLAWOMIR JAROSZ Department of Chemistry, University of Maryland, College Park, MD 20742, U.S.A. Received November 4, 1985 DAVD LIANG, HENRY W. PAULS, BERT FRASER-REID, MICHAEL GEORGES, AZEEZ M. MUBARAK, and SLAWOMIR JAROSZ. Can. J. Chem. 64, 1800 (1986). A general plan is outlined for disconnection of the polyene macrolides, which is aimed at achieving their syntheses, as well as facilitating the determination of their absolute configurations. The plan is exemplified by amphotericin B, I, the only family member of known absolute configuration. The major chiral component is a 20-carbon chain, 111, which upon further retrosynthetic disconnection leads to three subunits, two of which correspond to the dideoxy hexoses IV and V. Although these are formally mirror images, they are best represented as the 3,4-dideoxyhexopyranoside, 1, and the 3,5-dideoxyfuranose, 2. Syntheses of 1 and 2 from readily available starting materials are described. DAVID LIANG, HENRY W. PAULS, BERT FRASER-REID, MICHAEL GEORGES, AZEEZ M. MUBARAK et SLAWOMIR JAROSZ. Can. J. Chem. 64, 1800 (1986). On presente un plan gkneral de dkmembrement des macrolides poly6niques qui a pour but d'en rkaliser la synthkse ainsi que de faciliter la determination de leurs configurations absolues. Utilisant comme exemple I'amphotCricine B, le seul membre de la famille dont la configuration soit connue, on dtmontre comment le plan peut &tre opBrationalisk. Le composant chiral principal est une chaine a 20 atomes de carbone, 111, qui conduit a trois sous-unites par un dkmembrement rktrosynthetique; deux de ces sous-unites correspondent aux dideoxy hexoses IV and V. M&me si ces sous-unites sont forrnellement des images mirroirs, leurs meilleures representations sont le dideoxy-3,4 hexopyrannoside 1 et le dikoxy-3,s furannose 2. On dkcrit les syntheses des composCs 1 et 2 2 partir de produits de depart facilement accessibles. [Traduit par la revue] Introduction The polyene macrolides comprise a group of approximately 100 members (I), some of which have been in widespread clinical use for over 30 years, mainly for the treatment of yeast and candida infections (2-4), but also as antitumor agents (5) and in prostate therapy (6). Their skeleta are characteristically divided into a conjugated polyene chain (7) (from which the family name is derived), and an array that contains secondary hydroxyl groups in (almost) regular 1,3 relationships. The latter stereochemical feature currently attracts considerable attention (8). In spite of the remarkable profile based on their clinical importance and unique structural features, the absolute stereo- chemistry is known for only one member, amphotericin B (I), which has yielded a crystalline derivative (9). However, in general, these antibiotics are amorphous, and their sensitivity to a wide variety of chemical reagents, as well as to light, renders chemical proof of structure very difficult. Synthetic activity has been remarkably light in comparison with other types of macrolides; however, the studies spearheaded by Masamune 'Taken in part from Ph.D. theses: D.L. , Duke University, 1985; H.P., Duke University, 1984; M.G., University of Waterloo, 1983. 'For preliminary communications dealing with this work, see refs. 27 and 3 1. 3~resent addresses: D.L., Chemistry Department, University of South Carolina. H.P., Syntex, Inc., Mississauga, Ont., Canada. M.C., Xerox Research Centre, Mlsslssauga, Ont . , Canada. A.M., Ceylon Institute of Scientific and Polish Academy of Sciences, Warsaw, Poland. 4Author to whom correspondcncc may be addressed. and co-workers (lo), Nicolaou and Uenishi (1 l), Brooks et al. (12), and Floyd et al. (13) deserve particular note. Our laboratory has been developing a general program on the polyene macrolides, which should facilitate proof of their structures and stereochemistry, relative and absolute, by a combination of degradative and synthetic studies. Our plans are necessarily based on the only member whose structure is known with certainty, that of amphotericin B, I, but there are several structural features that I shares with other polyene macrolides, which suggest that a general plan for degradation and (or) synthesis should be possible. Retrosynthetic analysis Logical points of disconnection of I are termini of the polyene chain (bonds a and b), and the lactone (bond c). The subunits thereby obtained are hexose 11, and the uronic acid III. The regions of the latter designated d and e do not contain any contiguous chiral centers, and hence are logical sites for further retrosynthetic disconnections, resulting in segments X, Y, and 2;. Segment X A plausible synthon for segment X is the 2,4-dideoxyhexose IV, which is conveniently represented as the methyl a-D- pyranoside, I. Segment Y Interestingly, a synthon for this segment is V, which, being the mirror image of IV, leads to the pyranoside Va. However, pursuit of this analysis would require the use of an L-sugar, which would be uneconomical for a major synthetic under- taking. A more acceptable chiron (14) is the 3,5-dideoxyfura- Can. J. Chem. Downloaded from www.nrcresearchpress.com by 118.99.114.121 on 06/03/13 For personal use only.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Chiral building blocks for amphotericin B

Ghiral building blocks for amphotericin B
DAVID LIANC,~ HENRY W. PAULS, AND BERT FRASER-RE ID^ Paul M. Gross Chemical Laborato~y, Duke University, Durham, NC 27706, U.S.A.
MICHAEL GEORGES The Guelph-Waterloo Centre for Graduate Work in Chemistry, Universih of Waterloo, Waterloo, Ont., Canada N2L 3 6 L
AND
AZEEZ M. MUBARAK AND SLAWOMIR JAROSZ Department of Chemistry, University of Maryland, College Park, MD 20742, U.S.A.
Received November 4, 1985
DAVD LIANG, HENRY W. PAULS, BERT FRASER-REID, MICHAEL GEORGES, AZEEZ M. MUBARAK, and SLAWOMIR JAROSZ. Can. J . Chem. 64, 1800 (1986).
A general plan is outlined for disconnection of the polyene macrolides, which is aimed at achieving their syntheses, as well as facilitating the determination of their absolute configurations. The plan is exemplified by amphotericin B, I , the only family member of known absolute configuration. The major chiral component is a 20-carbon chain, 111, which upon further retrosynthetic disconnection leads to three subunits, two of which correspond to the dideoxy hexoses IV and V. Although these are formally mirror images, they are best represented as the 3,4-dideoxyhexopyranoside, 1, and the 3,5-dideoxyfuranose, 2. Syntheses of 1 and 2 from readily available starting materials are described.
DAVID LIANG, HENRY W. PAULS, BERT FRASER-REID, MICHAEL GEORGES, AZEEZ M. MUBARAK et SLAWOMIR JAROSZ. Can. J . Chem. 64, 1800 (1986).
On presente un plan gkneral de dkmembrement des macrolides poly6niques qui a pour but d'en rkaliser la synthkse ainsi que de faciliter la determination de leurs configurations absolues. Utilisant comme exemple I'amphotCricine B, le seul membre de la famille dont la configuration soit connue, on dtmontre comment le plan peut &tre opBrationalisk. Le composant chiral principal est une chaine a 20 atomes de carbone, 111, qui conduit a trois sous-unites par un dkmembrement rktrosynthetique; deux de ces sous-unites correspondent aux dideoxy hexoses IV and V. M&me si ces sous-unites sont forrnellement des images mirroirs, leurs meilleures representations sont le dideoxy-3,4 hexopyrannoside 1 et le dikoxy-3,s furannose 2. On dkcrit les syntheses des composCs 1 et 2 2 partir de produits de depart facilement accessibles.
[Traduit par la revue]
Introduction The polyene macrolides comprise a group of approximately
100 members ( I ) , some of which have been in widespread clinical use for over 30 years, mainly for the treatment of yeast and candida infections (2-4), but also as antitumor agents (5) and in prostate therapy (6). Their skeleta are characteristically divided into a conjugated polyene chain (7) (from which the family name is derived), and an array that contains secondary hydroxyl groups in (almost) regular 1,3 relationships. The latter stereochemical feature currently attracts considerable attention (8).
In spite of the remarkable profile based on their clinical importance and unique structural features, the absolute stereo- chemistry is known for only one member, amphotericin B (I) , which has yielded a crystalline derivative (9). However, in general, these antibiotics are amorphous, and their sensitivity to a wide variety of chemical reagents, as well as to light, renders chemical proof of structure very difficult. Synthetic activity has been remarkably light in comparison with other types of macrolides; however, the studies spearheaded by Masamune
'Taken in part from Ph.D. theses: D.L. , Duke University, 1985; H.P., Duke University, 1984; M.G., University of Waterloo, 1983.
'For preliminary communications dealing with this work, see refs. 27 and 3 1.
3~resent addresses: D.L., Chemistry Department, University of South Carolina. H.P., Syntex, Inc., Mississauga, Ont., Canada. M.C. , Xerox Research Centre, Mlsslssauga, Ont . , Canada. A.M., Ceylon Institute of Scientific and Polish Academy of Sciences, Warsaw, Poland.
4Author to whom correspondcncc may be addressed.
and co-workers (lo), Nicolaou and Uenishi (1 l), Brooks et al. (12), and Floyd et al . (13) deserve particular note.
Our laboratory has been developing a general program on the polyene macrolides, which should facilitate proof of their structures and stereochemistry, relative and absolute, by a combination of degradative and synthetic studies. Our plans are necessarily based on the only member whose structure is known with certainty, that of amphotericin B , I, but there are several structural features that I shares with other polyene macrolides, which suggest that a general plan for degradation and (or) synthesis should be possible.
Retrosynthetic analysis Logical points of disconnection of I are termini of the polyene
chain (bonds a and b), and the lactone (bond c). The subunits thereby obtained are hexose 11, and the uronic acid III. The regions of the latter designated d and e do not contain any contiguous chiral centers, and hence are logical sites for further retrosynthetic disconnections, resulting in segments X, Y, and 2;.
Segment X A plausible synthon for segment X is the 2,4-dideoxyhexose
IV, which is conveniently represented as the methyl a-D- pyranoside, I .
Segment Y Interestingly, a synthon for this segment is V, which, being
the mirror image of IV, leads to the pyranoside V a . However, pursuit of this analysis would require the use of an L-sugar, which would be uneconomical for a major synthetic under- taking. A more acceptable chiron (14) is the 3,5-dideoxyfura-
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
118.
99.1
14.1
21 o
n 06
/03/
13Fo
r pe
rson
al u
se o
nly.
LIANG ET AL
O f C H 3 20
H ~ C ' CHO HO CHO
I1 111
I Uronic acid i X / / / Y I z
/ / I
/ I l4cwo COOH 2 0 ~ ~ ~
'CHO HO I Hoo~{OH OH
HO OH ==3
HO
I 20CH20H 20CH0 COOH
1 I I HV I V Va I VI VII VIII
VIa VIIa (numbers in parentheses refer to I)
3 a R = H b R = TBS
nose 2, whose relationship to 1,2:5,6-di- O-isopropylidene-a- D-glucofuranose, 6a, is readily appreciated.
Segment Z The C14-C15 portion is by far the most challenging, and its
importance stems from the fact that the hemiketal feature that it represents (shown in heavy lines in I) is also found in most polyene macrolides (7). Thus this synthon could find general use in the synthesis of members of this family of macrolides.
The Fisher projections VI, VII, and VIII suggest three possible synthons for this segment. Structure VP initially seemed attractive since one approach would be via a 3-C-cyano sugar, such as VIa, and although such compounds have been prepared by Guthrie and co-workers (15, 16) and ourselves (17, our preliminary explorations of this avenue did not prove promising.
In the case of VII and VIHI, the 62-15 stereocenter is "off template" and hence its stereocontrolled formation and verifica- tion present special challenges, as noted by us elsewhere (1 8). Synthon IPI correlates with the L-sugar VIHa and was therefore rejected for the reasons discussed above in connection with segment Y. Attention was therefore focused on VIII, which correlates with the furanose 3.
Strategies for connecting the segments, such as olefination procedures and nucleophilic displacements, have been widely utilized in syntheses of macrolides from sugar precursors (14).
Synthetic studies Chiron I This subunit was prepared routinely from the readily obtained
2-deoxy pyranoside 4 (19), by use of a variation of the proce- dure of Prugh and Deanna (20b). In our hands, unfortunately, acidic hydrolysis of the 4,6-0-benzylidene ring could not be accomplished without concomitant anomerisation, leading to
MeOH ph%w -Gx+ Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
118.
99.1
14.1
21 o
n 06
/03/
13Fo
r pe
rson
al u
se o
nly.

1802 CAN. .I. CHEM.
6 u Y = O H b Y = OBn c Y = OBz d Y = H
I
7 X = MeSCS
8 2 u ~ 1 = ~ 2 = H b R' = Tr; hi2 = H
9 SCHEME 3
the a,@ mixture 5a. Deoxygenation of the alcohol 5b was effected by the classical Barton-McCombie (21) procedure.
Chiron 2 The chiron V is a 3,5-dideoxy hexose and it seemed most
expedient to remove the C-3 and C-5 oxygens simultaneously. However, we were unable to effect double deoxygenation of several doubly activated derivatives 7 (e.g. X = S02R, SCimidazoyl, SCSMe) by a variety of procedures. The product in each case was a complex mixture.
The deoxygenations therefore had to be done separately, and the logical starting point was the well-known 3-deoxy derivative 6d (21, 22), prepared most conveniently from 6a by the Barton procedure (21). Selective hydrolysis of 6d afforded the diol8a (21, 22) and the tritylated derivative there of 8b was then subjected to the Barton deoxygenation followed by hydrolysis. However, the overall yield of 2 from 8 a obtained by this route was 60% and so an alternative route was examined.
A more satisfactory yield (-80%) was achieved by hydro- boration (23) of the alkene 8. Of various procedures (24) tested for the preparation of this oiefin, the reductive elimination of vicinal disulfonates, originated by Jones and Thomson (24a), proved to be the most convenient.
Chiron 3 In our first approach to the synthesis, we had hoped to make
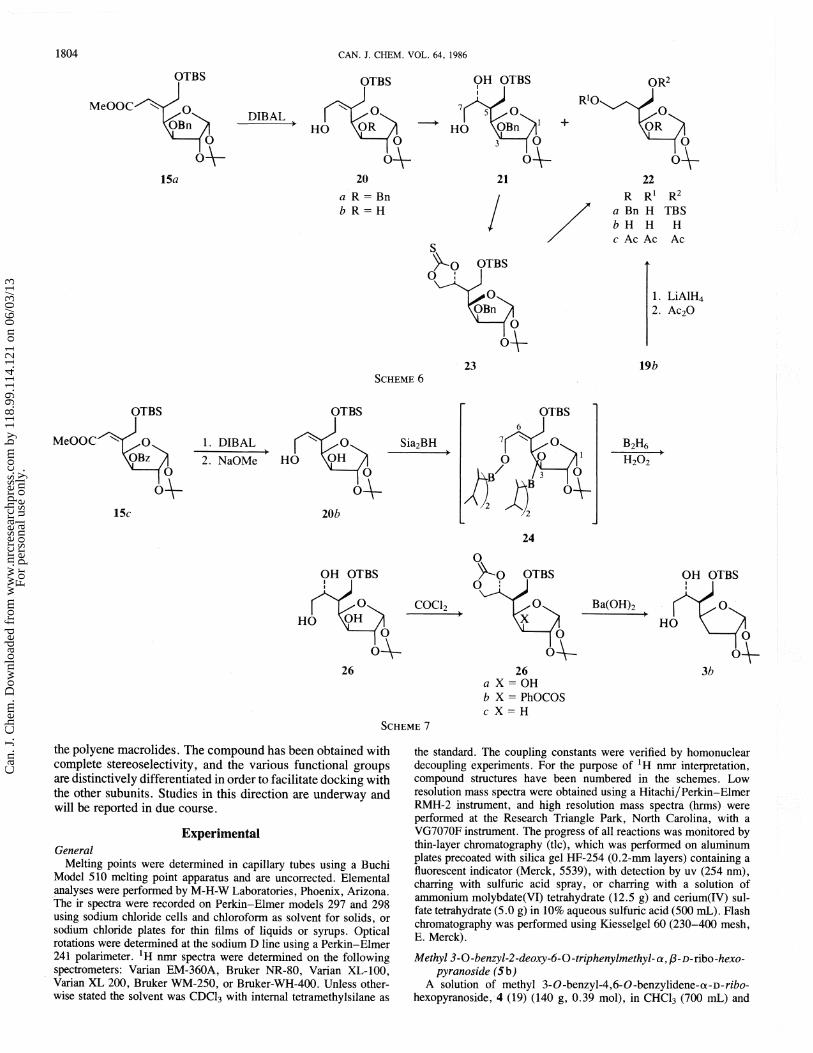
use of the alcohol 8b. Thus oxidation to 10 and a Horner- Emmons reaction afforded a 4:1 mixture ('H nmr estimate) of the geometric isomers 1 l a . Reduction with DIBAL (diisobutyl-
aluminum hydride) gave the allylic alcohols l l b , and hydro- boration then led to 12 in excellent overall yield. However, the latter was obtained as a mixture of diastereomers of undetermined composition, as indeed as to be expected in view of the fact that the olefinic precursor, I l b , was a mixture of geometric isomers.
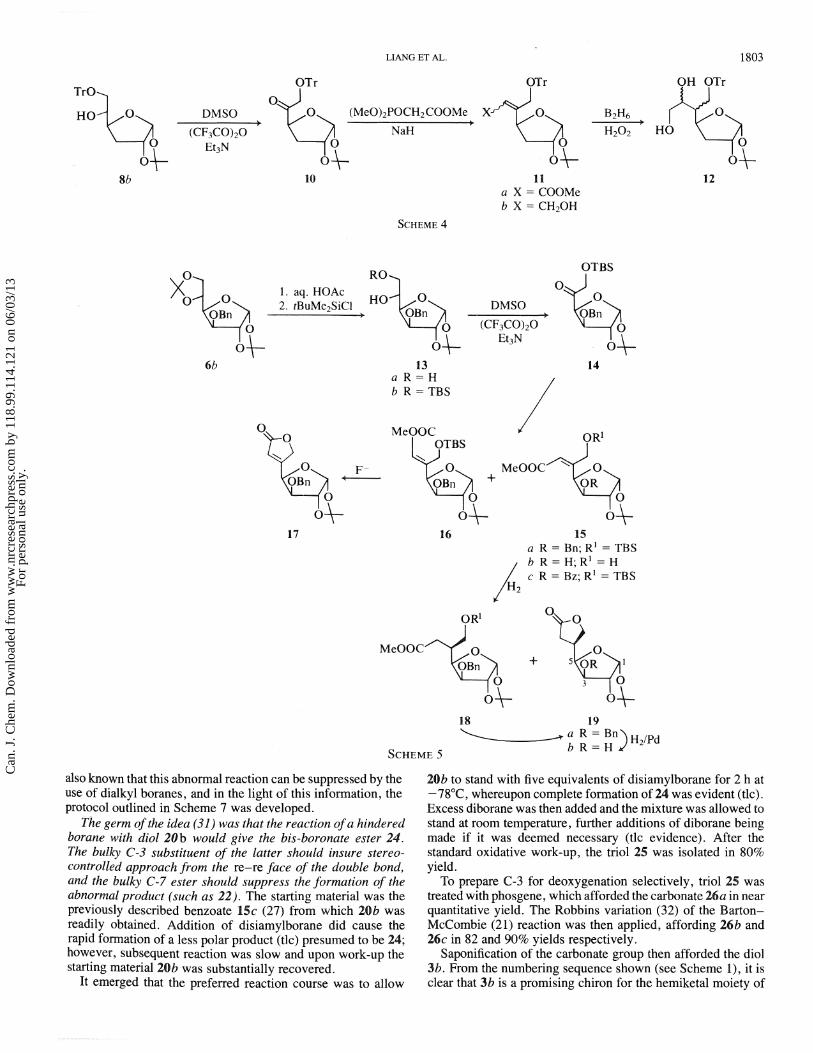
The uncertainty about the C-5 and C-6 configurations of 12 was a major problem for us. Our preliminary studies had suggested that, with C-5 alkenes such as l l b , the presence of a bulky substituent at C-3 could be used to control the facial selectivity of electrophilic additions. According to this observa- tion, the desired C-5 and C-6 configurations of 3 could both be achieved, by ensuring that addition to the C-5 alkene occurred from the re-re face of a (Z) olefin such as 15. The correspond- ing (E) olefin, 16, would give the correct C-5, but incorrect C-6, configuration.
Stereoselective formation of a (2) olefin therefore became the first requirement. The known diol 13a (25) was converted into ketone 14 by routine transformations. It was found that reaction with trimethylphosphonoacetate gave an inseparable 6: 1 mixture of 15a and 16. However, the corresponding Wittig reagent, carbomethoxymethylenetriphenylphosphorane, afforded 15a exclusively. Assignment of structure to these geometric isomers followed from the fact that treatment of each isomer with fluoride ion gave the hydroxyester 15b and the butenolide 17, respectively, based upon their ir (infrared) and 'H nrnr spectral characteristics. Compound 15b, therefore, had the desired (2) configuration, and hydrogenation over palladium gave two products that were assigned as the hydroxyester 18 and lactone 19a since, upon treatment of the mixture with p-toluenesulfonic acid for 6 h at room temperature, the more polar component was gradually transformed quantitatively into the less polar product, i.e. 18 -, 19a.
A timely publication by Redlich and Neumann (26) also reported stereocontrolling effects on the hydrogenation of C-5 alkenes and, on the basis of these observations, the C-5 configuration of the lactone was tentatively assigned as shown in 19a. This assignment was subsequently established by X-ray analysis of a transformation product (27), and it was therefore apparent that hydrogenation of the double bond of 15 was favored from the re-re face. We then hoped to extend this selectivity to hydroboration.
The allylic alcohol 20a was obtained by DIBAL reduction of 15a, and upon hydroboration a 5:3 mixture of compounds 21 and 22a was formed. The structures of these compounds were established as follows. Firstly 21 was treated with thiocarbonyl- diimidazole to give the thionocarbonate 23, which was then reduced with tri-n-butyltin hydride according to the Barton- Subramanian procedure (28) to give 22a. The latter was hydrogenated over palladium in the presence of formic acid, and the resulting trio1 22b was acetylated directly to give triacetate 22c.
Secondly, compound 19a was correlated with 22c by hydrogenolysis to give the hydroxylactone 19b, which upon reduction and acetylation gave the triacetate 22c.
These correlations indicated that the C-5 configurations in 19 and 21 were the same. Given (a) the Z configuration of 28 and (b) the mechanism of hydroboration (29), the C-6 hydroxyl configuration must therefore be as shown in 21.
However, the high proportion of the by-product, 22a, was intolerable. This abnormal course of hydroboration responsible for its fornation has been observed previously, and the mechanism that has been proposed is shown below (30). It is
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
118.
99.1
14.1
21 o
n 06
/03/
13Fo
r pe
rson
al u
se o
nly.
LIANG ET AL. 1803
DMSO
QTr OTr
(Me0)2POCH2COOMe +
NaH
RO 1. aq. HOAc 2 . ;BuMe:SiC
OTBS
16 P 5 a R = Bn; R ' = TBS
, b R = H : R 1 = H c R = Bz; R1 = TBS
also known that this abnormal reaction can be suppressed by the use of dialkyl boranes, and in the light of this information, the protocol outlined in Scheme 7 was developed.
The germ ofthe idea (31) was that the reaction o fa hindered borane with diol 2 0 b would give he bis-boronate ester 24. The bulky C-3 substituent of the latter should insure srereo- controlled approach from the re-re face of the double bond, and the bulky C-7 ester should suppress the formation of the abnormal product (such as 22). The starting material was the previously described benzoate 15c (27) from which 20b was readily obtained. Addition of disiamylborane did cause the rapid formation of a less polar product (tlc) presumed to be 24; however, subsequent reaction was slow and upon work-up the starting material 20b was substantially recovered.
It emerged that the preferred reaction course was to allow
20b to stand with five equivalents of disiarnylborane for 2 h at -78"C, whereupon complete formation of 24 was evident (tlc). Excess diborane was then added and the mixture was allowed to stand at room temperature, further additions of diborane being made if it was deemed necessary (blc evidence). After the standard oxidative work-up, the trio1 25 was isolated in 80% yield.
To prepare C-3 for deoxygenation selectively, trio1 25 was treated with phosgene, which afforded the carbonate 26a in near quantitative yield. The Robbins variation (32) of the Bxton- McCombie (21) reaction was then applied, affording 26b and 26c in 82 and 90% yields respectively.
Saponification of the carbonate group then afforded the diol 3b. From the numbering sequence shown (see Scheme I ) , it is clear that 3b is a promising chiron for the hemiketal moiety of
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
118.
99.1
14.1
21 o
n 06
/03/
13Fo
r pe
rson
al u
se o
nly.
CAN. J . CHEM. VOL. 64, 1986
OTBS OTBS OH OTBS OR2
MeOOc%O OBn ____+ DIBAL + + RIO
0%
OTBS
1. DIBAL _____t
2. NaOMe
20 21 22
a R = B n R R' R2 b R = H a Bn H TBS
b H H H c Ac Ac Ac
S
the polyene rnacrolides. The compound has been obtained with complete stereoselectivity, and the various functional groups are distinctively differentiated in order to facilitate docking with the other subunits. Studies in this direction are underway and will be reported in due course.
OTBS
a - H O
206
oi-
Experimental General
Melting points were determined in capillary tubes using a Buchi Model 510 melting point apparatus and are uncorrected. Elemental analyses were performed by M-H-W Laboratories, Phoenix, Arizona. The ir spectra were recorded on Perkin-Elmer models 297 and 298 using sodium chloride cells and chloroform as solvent for solids, or sodium chloride plates for thin films of liquids or syrups. Optical rotations were determined at the sodium D line using a Perkin-Elmer 241 polarimeter. 'H nrnr spectra were determined on the following spectrometers: Varian EM-360A, Bruker NR-80, Varian XL- 100, Varian XL 200, Bruker WM-250, or Bruker-WH-400. Unless other- wise stated the solvent was CDC13 with internal tetramethylsilane as
the standard. The coupling constants were verified by homonuclear decoupling experiments. For the purpose of 'H nrnr interpretation, compound structures have been numbered in the schemes. Low resolution mass spectra were obtained using a Hitachi/Perkin-Elmer RMH-2 instrument, and high resolution mass spectra (hrms) were performed at the Research Triangle Park, North Carolina, with a VG7070F instrument. The progress of all reactions was monitored by thin-layer chromatography (tlc), which was performed on aluminum plates precoated with silica gel HF-254 (0.2-mm layers) containing a fluorescent indicator (Merck, 5539), with detection by uv (254 nm), charring with sulfuric acid spray, or charring with a solution of ammonium molybdate(V1) tetrahydrate (1 2.5 g) and cerium(1V) sul- fate tetrahydrate (5.0 g) in 10% aqueous sulfuric acid (500 mL). Flash chromatography was performed using Kiesselgel 60 (230-400 mesh, E. Merck).
24
?H OTBS O H OTBS
- F H O
- -
- -
Methyl 3 - 0 -benzyl-2-deoxy-6-0-triphenylrnethyl- a , P-D-ribo-hexo- pyranoside (5 b)
A solution of methyl 3-0-benzyl-4,6-0-benzylidene-a-D-ribo- hexopyranoside, 4 (19) (140 g, 0.39 mol), in CHC13 (700 mL) and
B2H6 - Hz02
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
118.
99.1
14.1
21 o
n 06
/03/
13Fo
r pe
rson
al u
se o
nly.

LIANG ET AL. 1805
MeOH (700 mL) containing p-toluenesulfonic acid (5 g) was refluxed for 3 h. After cooling, the solution was neutralized with triethylamine (5 mL), concentrated in vacuo, and the oily residue was chromato- graphed on silica gel (400 g). Elution with cyclohexane - ethyl acetate (2:l) and EtOAc gave diol5a (84.9 g, 80.5%) as an oil. The material was dissolved in methylene chloride (500 mL) containing triethylamine (70 mL) and triphenylmethyl chloride (87.3 g, 0.3 1 mmol) and N, N- dimethylaminopyridine (2 g) were added. After 15 h stirring at room temperature, the reaction mixture was evaporated in vacuo, diluted with ethyl acetate, washed with aqueous saturated NH4Cl, NaCl, and NaHC0, solutions, then chromatographed on a silica gel column (1.5 kg). Elution with EtOAc-C&I12 (1:4) gave 140 g (87%) of 5b as a syrupy, inseparable mixture of anomers. [a]:' + 37.3" ( c 1.1, chloroform); v,,(film): 3510 (OH) c m ' ; 'H nmr (60 MHz) 6: 1.50- 2.60 (m, 3H, H-2a, H-2e, OH), 3.42 (s, 3H, OCH,), 3.20-4.20 (m, 5H, H-3, H-4, H-5, H-6a, H-6b), 4.55 (q, 2H, 0CH2Ph), 4.6 (m, lH, H-1), 7.00-7.75 (m, 20H, 0CH2Ph, CPh3). Anal. calcd. for c33H3405: C 77.62, H 6.71; found: C 77.38, H 6.56.
Methyl 3-0-benzyl-2-deoxy-4-O-[(methylthio)thiocarbonyl]-6-0-tri- phenylmethyl- a , P-D-ribo -hexopyranoside (5c)
To a suspension of NaH (12 g, oil free) in tetrahydrofuran (500 rnL) was added a solution of compound 5 a (128 g, 0.25 mol) in tetrahydro- furan (800 mL), and irnidazole (1.2 g). The mixture was refluxed for 3 h under argon, and then carbon disulfide (75 mL) was added gradually. After 30 min under reflux methyl iodide (75 mL) was added gradually and, after an additional 2 h, the reaction mixture was cooled and quenched with acetic acid (40 mL), diluted with ethyl acetate, and washed with 3% dilute HCl, NaHC03, and NaCl solutions, then dried over anhydrous sodium sulfate, filtered, and evaporated in vacuo. The residue was purified by flash silica gel chromatography to give 140 g (95%) of the thiocarbonate 5 c as a yellow syrup; Rf 0.54 (15% ethyl acetate - petroleum ether) for the P isomer, 0.44 for the a isomer; 'H nmr (60 MHz, CDC13) for the a isomer, 6: 1.66-2.20 (m, 2H, H-2a, H-2e), 2.30 (s, 3H, SCH,), 2.96-3.34 (m, 2H, H-6, H-6), 3.38 (s, 3H, OMe), 3.96-4.32 (m, 2H, H-3, H-5), 4.50 (s, 2H, PhCH20), 4.74(brs, lH,H-1),5.86(dd, lH, J4,5 = 9.6Hz, J3,.,=4.0Hz,H-4), 6.93-7.90 (m, 20H, PhCH20, Ph3C); for the P isomer, 6 : 1.50-2.20 (m, 2H, H-2a, H-2e), 2.34 (s, 3H, SMe), 2.90-3.22 (m, 2H, H-6, H-6), 3.42 (s, 3H, OMe), 4.06-4.35 (m, 2H, H-3, H-5), 4.46 (s, 2H, PhCH20), 4.78 (dd, lH, JlZza = 8.0 HZ, = 3.0 HZ, H-1), 5.90 (dd, lH, J4,5 = 9.0 HZ, J3,4 = 4.0 HZ, H-4), 6.90-7.70 (m, 20H, PhCH20, Ph3C). Anal. calcd. for C35H3605S2: C 69.97, H 6.04; found: C 69.42, H 6.12.
Methyl 3-0-benzyl-2,4-dideoxy- a, P-D-erythro-hexopyranoside (1 a) The crude oily xanthate 5 a (140 g) was dissolved in toluene (1 L)
containing 2,2-azobis(2-methylpropionitrile) (250 mg). The solution was added to tributyltin hydride (100 mL) in toluene (0.5 L). The mixture was boiled under argon for 24 h. Additional tributyltin hydride (50 mL) was added, and reflux was continued for 24 h. Concentration in vacuo gave a crude mixture, which was diluted with n-hexane (150 mL) and extracted with acetonitrile (100 mL X 2). The acetoni- trile layer was concentrated in vacuo, and chromatographed on a silica gel column. Elution with cyclohexane - ethyl acetate (4: 1) gave 120 g of material, which was assumed to be the tritylated derivative l b . To the crude substance was added, directly, methanol (2.5 L) containing p-toluenesulfonic acid (5 g), and the mixture was stirred at room temperature for 6 h. The solution was then neutralized with triethyl- amine (10 mL), concentrated in vacuo, and chromatographed on a silica gel column. Elution with cyclohexane - ethyl acetate (4:1, then 1: 1) gave 44 g (68%) of the anomeric mixture l a ; Rf 0.16 (30% ethyl acetate - petroleum ether); 'H nmr (60 MHz) 6: 1.20-2.38 (m, 4H, H-2, H-4), 2.80 (br s, IH, OH), 3.53 (s, 3H, OMe), 3.34-4.25 (m, 4H, M-3, H-5, H-6, H-6'), 4.55 (s, 2H, PhCH2), 4.78 (dd, lH, J1,2e = 2.0Hz, J1 , la = lOHz, H-I), 7.35 (s, 5H, Ph). Anal. calcd. for C14H2004: C 66.64, N 7.99; found: C 65.80, H 7.89.
3-Deoxy-l,2-O-isopropylidene-6-O-triphenylrnethyl- a- D-hexofura- nose (8 b)
A mixture of 3-deoxy-1 ,2-0-isopropylidene-a-D-ribo-hexofura-
nose, 8a (21,22) (10.2 g, 50 rnmol), triphenylmethyl chloride (14.6 g, 52.5 rnmol), and N, N-dimethylaminopyridine (500 mg) was dissolved dimethylformarnide (120 mL) and stirred at room temperature for 20 h. The reaction mixture was poured into ice water (100 mL) and then extracted with dichloromethane (3 x 200 mL). The combined organic extracts were washed with saturated ammonium chloride solution, water, and brine, dried (Na2S04), and concentrated in vacuo. Flash chromatography of the residue afforded 19.6 g (88%) of 8b; Rf 0.50 (30% ethyl acetate - petroleum ether); [ a ] i 3 - 15.8' (c 1 .O, CHC13); 'H nrnr (80 MHz, CDC1,) 6: 1.32, 1.52 (s, s, 6H, C(CH3)2), 1.67-2.55 (m, 3H, H-3, H-3', OH, D 2 0 exchange), 3.22 (m, IH, H-5), 3.74-4.50 (m, 3H, H-4, H-6, H-6'), 4.70 (t, 1H, J = 3.7 Hz, H-2), 5.78 (d, lH, Jl,2 = 3.7 Hz, H-1). Anal. calcd. for C28H3005: C 75.31, H 6.77; found: C 75.44, H 6.67.
3,5,6-Trideoxy-I ,2-0-isopropylidene- a-D-erythro -hex-5-enofura- nose ( 9 )
To a solution of the diol $a (78 g, 0.382 mol) in tetrahydrofuran (400 mL) and pyridine (180 mL) was added methylsulfonyl chloride (131 g, 1.146 mol). The mixture was refluxed for 1.5 h and, after cooling, was concentrated in vacuo, diluted with ethyl acetate, washed with dilute solutions of HCl, NaHC03, and NaCl, then dried (MgS04). Evaporation gave a crude oil that was chromatographed on silica gel to yield compound 8 c as a crystalline solid (46 g, 33.4%). The material was dissolved directly in butanone (1 L), sodium iodide (60 g ,0 .4 mol) was added, and the solution was boiled under reflux overnight. The reaction mixture was diluted with diethyl ether, and washed with solutions of sodium thiosulfate, NaHC03, and NaCl. After drying over MgS04, the solution was concentrated in vacuo and the residue distilled at45'C (1 Torr(l Torr = 133.3 Pa)) to give9 (15 g, 88%) as an oil; Rf0.83 (50% ethyl acetate-petroleumether); [a]: -38.7" (c 1 .O, CHC13); 'H nmr (80 MHz) 6: 1.38, 1.58 (s, s, 6H, C(CH1)2), 1.60-1.86 (m, IH, H-3), 2.22 (dd, lH, J = 4.6Hz, H-3P), 4.66 (m, lH, H-4), 4.78 (t, lH, J = 3.7Hz, H-2), 5.14, 5.26, and 5.46 (m, 2H, H-6, H-6'), 5.88 (d, 1H, J1,2 = 3.7 Hz, H-1), 5.92 (m, IH, H-5). Anal. calcd. for C9HI4o3: C 63.51, H 8.29; found: C 63.47, H 8.26.
3,5-Dideoxy-l,2-0-isopropylidene- a-D-erythro -hexofuranose (2) (a) Deoxygenation and detritylation of compound 8 b (646 mg,
1 3 . 4 ~ 0 1 ) was carried out as described above for 5b-z 5c+ l a . The product 2 (337 mg), obtained in 70% overall yield from 8b, was isolated as a colorless oil by chromatography.
(b) To a solution of disiamylborane (4.8 mmol) in tetrahydrofuran was added, dropwise, a solution of 9 (540 mg, 3.18 mmol) in tetrahydrofuran (5 mL) at O°C under argon. After stirring at room temperature for 3 h, water ( 1 mL) was added dropwise at O°C to destroy the excess of the reagent. The reaction mixture was oxidized by addition of 3 N sodium hydroxide (2 mL) in one portion and then 30% hydrogen peroxide (2 mL) dropwise, while maintaining the tempera- ture at 20-25OC. After stirring for another 3 h at room temperature, the aqueous phase was saturated with sodium chloride. The tetrahydro- furan phase was separated and the aqueous layer was extracted with ether. The combined organic layers were dried (MgS04) and concen- trated in vacuo and the excess water was removed azeotropically with toluene. Flash chromatography afforded compound 2 (537 mg, 90%) as a colorless syrup; Rf 0.20 (50% ethyl acetate - hexane); -8.0" (c 1.05, chloroform); 'H nmr (80 MHz) 6: 1.28, 1.48 (s, s, 6H, C(CH3)*), 1.60-2.35 (m, 5H, H-3, H-3', H-5, H-5', OH, D 2 0 exchange), 3.75 (m, 2H, H-6, H-6'), 4.33 (m, IN , H-4), 4.68 (t, lH, J = 3.7 Hz, H-2), 5.78 (d, 1H, J1,2 = 3.7 Hz, H-1). Anal. calcd. for C9Hl6o4: C 57.43, H 8.57; found: C 57.21, H 8.61.
3-Deoxy-I ,2-0-isopropylidene-6-0-triphenylmethy- a-D-erythro- hexofuran-5-ulose (10)
To a solution of dry dimethylsulfoxide (3.9 mL) in dichloromethane (50 mL), trifluoroacetic anhydride (5.8 mL) was added dropwise at -78°C under an argon atmosphere. After 15 min a solution of $b (6.1 g, 13.7 mmol) in dichloromethane (60 mL) was added dropwise over a period of 45 min, and after another hour triethylamine (12 rnL) was added dropwise, and the resulting solution was warmed up to
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
118.
99.1
14.1
21 o
n 06
/03/
13Fo
r pe
rson
al u
se o
nly.

1806 CAN. J. CHEM.
room temperature. The reaction solution was diluted with 200 mL of dichloromethane and then washed with saturated sodium bicarbonate, water, and brine, dried (Na2S04), then concentrated in vacuo, Flash chromatography gave 5.8 g (96%) of ketone 10 as a syrup; K f 0.42 (15% ethyl acetate - petroleum ether); [ a ] 2 - 1.33" (c 1 .O, chloro- form); v,, (CHCl;): 3000, 1730 cm-'; 'H nmr (60 MHz) 6: 1.15, 1.35 (5, S , 6H, C(CH3)2), 1.60 (dd, lH, J = 4.0 HZ, J = 13.0 Hz, H-3a), 2.20 (dd, IH, J = 4.0 Hz, H-3P), 3.90 (s, 2H, H-6, H-6'), 4.50 (t, lH, J = 4.0 Hz, H-2), 4.62 (dd, IH, J = 5.0 Hz, H-5), 5.62(d, IH, J=4.OHz,H-1) , 7.20(m, 15H,Ph,C). Anal. calcd. for C28H2805: C 75.65, H 6.35; found: C 75.59, H 6.31.
Methyl 3,5,6-trideoxy-5-C -triphenylmethyloxy-I ,2-0-isopropyli- dene- a- D-erylhro-hept-5-(E, Z)-enofuranosyluronate ( I 1 a)
To a suspension of 1.0 g (20.8 rnmol) of 50% sodium hydride dispersion (prewashed with hexane) in dry benzene (60 mL) was added trimethylphosphonoacetate (2.6 g, 14.3 mmol) at room temperature under an argon atmosphere. After stirring at 35'C for 1 h, a solution of 10 (5.8 g, 13.06 mrnol) in dry benzene (60 mL) was added to the reaction flask dropwise at room temperature. After stirring for 2 days the solution was diluted with ether and processed in the usual way. Flash chromatography gave 5 .6 g (86%) of I l a as an inseparable 4:1 mixture of geometric isomers (based upon the integration of the methoxy peaks); Rf0.58 (30% ethyl acetate - petroleum ether); [a];' -33" (c 1.0, chloroform); v,,, (CHCI,): 3000, 1710 (C=O), 1450cm-I; 'Hnmr (80MHz) 8: 1.30, 1.58 (s, s, 6H, C(CH3)2), 1.50 (m, IH, H-3), 2.53 (dd, IH, J = 5.0 Hz, J = 13.0 Hz, H-3@), 3.80 (s, 3H, 0CH3), 4.00 (AB, 2H, J = 11.0 Hz, A6 = 0.13 ppm, H-5, H-5'), 4.64 (t, lH, J = 3.7 Hz, H-2), 5.63 (d, lH, J = 3.7 Hz, H-I), 5.84(dd, lH, J = 4.4Hz, J = 5.0Hz, H-4), 6.45 (m, lH, H-6), 7.32 (m, 15H, Ph3C). Anal. calcd. for C31H3206: C 74.38, H 6.44; found: C 74.34, H 6.20.
3,5,6-Trideoxy-5-C-triphenylmethyloxy-l,2-O-isopropylidene- a- D-erythro-hept-5-(E, Z)-enofuranose ( I I b)
To a solution of I l a (3.5 g , 7 mmol) in diethyl ether, disobutylalu- minum hydride (21 mL, 1 .OM in toluene) was added dropwise at O°C under an argon atmosphere. After 4 h, 1 mL of water was added slowly at O°C, followed by 1 mL of 15% aqueous sodium hydroxide and 3 mL of water. The inorganic salts were removed by filtration and the residue was rinsed with diethyl ether. The combined filtrates were dried (MgS04) and then concentrated in vacuo. Flash chromatography afforded 2.64 g (80%) of the allylic alcohol Plb as a white amorphous solid, mp 104-106°C (from ether - petroleum ether); R f 0.29 (30% ethyl acetate - petroleum ether); [a ] i3 - 18.4" (c 1 .O, chloroform); 'H nmr (80 MHz) 6: 1.18, 1.38 (s, s, 6H, C(CH3)2), 1.66-2.20 (m, 3H, H-3, H-3', OH, D 2 0 exchange), 3.52 (br s, 2H, H-5, H-5'), 4.16 (d, 2H, J = 7.0 HZ, H-7, H-7'), 4.52 (t, IH, J = 3.7 HZ, H-2), 4.84(dd, lH, J = 4 . 4 H z , J=5 .0Hz.N-4) ,5 .58(d , IH, J = 3.7Hz, H-1), 5.94 (t, lH, J = 7.0 Hz, H-6), 7.35 (m, 15H, Ph3C). Anal. calcd. for C30H3205: C 76.25, H 6.82; found: C 76.06, H 6.61.
Hydroboration of compound 11 b To a solution of diborane (16 mL, 1.0 M in tetrahydrofuran), which
had been cooled to O°C, was added a solution of 11b (2.5 g, 5 .3 mmol) in tetrahydrofuran (20 mL) dropwise over a period of 1 h under an argon atmosphere. After 6 h, 1 mL of water was added slowly, followed by addition of 6 mL of 3 N aqueous sodium hydroxide and 6 mL of 30% hydrogen peroxide, maintaining the temperature at 20-25'C. After stirring for another 3 h at room temperature, the aqueous phase was saturated with sodium chloride. The tetrahydrofuran phase was separated and the aqueous layer was extracted with diethyl ether. The combined organic layers were dried over anhydrous magnesium sulfate and concentrated in vacuo. The excess of water was removed azeotropically with toluene, after which flash chromatography afforded 1% (2.3 g, 90%) as an amorphous solid; Rf 0.26 (50% ethyl acetate - petroleum ether); 'H nmr (60 MHz) 6: 1.30, 1.50 (s, s , 6H, C(CH3)l), 1.70-2.24 (m, 3H, H-P', H-5), 2.50-3 80 (m, 8H, N-4, H-6, H-7, H-7', H-5, H-5', 2 x OH), 4.68 (m, lH, H-2), 5.75 (brd, lH, H-1), 7.40 (m, 15H, Ph3C). Anal. calcd. for C30H3406: C 73.45, H 6.98; found: C 73.38, H 6.93.
3-O-Benql-6-O-tert-butyldirnetk~~/silyl-l,2-O-isopropylidene- a-D- glucofuranose (13 b)
To a solution of 3-0-benzyl-1 ,2-0-isopropylidene-a-D-glucofura- nose (13a) (25) (17.4 g, 56.13 mmol), N, N-dimethylaminopyridine ( 1 .O g), and trimethylarnine (23 mL) in dichloromethane (250 mL) was added tert-butyl-dimethylsilyl chloride (8.46 g, 56.13 rnmol) at room temperature under an argon atmosphere. After 20 h the reaction mixture was poured into 100 mL of ice water, extracted with dichloromethane, and processed in the usual way to give 13b (23.1 g, 97%) as a colorless syrup; Rf 0.41 (15% ethyl acetate - petroleum ether); [a]K3 -24.2' (c 1 .O, chloroform); 'H nmr (80 MHz) 6: 0.03 (s, 6H, &Me2), 0.89 (s, 9H, tBuSi), 1.30, 1.46 (s, s, 6H, C(CH3)>), 2.62 (d, lH, J = 5.5 Hz, OH), 3.68-4.20 (m, SH, H-3, H-4, H-5, H-6, H-6'), 4.57 (d, IH, J = 3.7 HZ, H-2), 4.66 (s, 2H, PhCH,), 5.89 (d, IH, J = 3.7 Hz, H-1), 7.33 (s, 5H, Ph). Anal. calcd. for C22H3606Si: C 62.23, H 8.55; found: C 62.31, H 8.64.
3 - 0 -Benzyl-6-0-tert -butyldimethylsilyl-,2- 0 -isopropylidene- a - D-
xylo-hexofuran-5-ulose (14) Compound 13b (23.1 g, 54.48 mmol) was oxidized as described
above for 8 b + 10. The product 14 (20.7 g, 90%) was a colorless syrup; Rf 0.41 (15% ethyl acetate - petroleum ether); [a];' -91.98" (c 1.87, chloroform); v,,, (CHC13): 2950, 1740 (C=O), 1460 cm-'; 'H nmr (80 MHz) 6: 0.04 (s, 6H, SiMe2), 0.90 (s, 9H, tBuSi), 1.30, 1.48 (s, s, 6H, C(CH3)2), 4.37 (d, IH, J3,4 = 3.5 Hz, H-3), 4.48 (s, 2H, H-6, H-6'1, 4.52 (d, 2H, J = 3.5 HZ, PhCH2), 4.58 (d, lH, J 3 , 4 = 3.5 HZ, H-4), 4.88 (d, lH, J = 3.7 HZ, H-2), 6.02 (d, IH, J1 ,2 = 3.7 Hz, H-1), 7.25 (s, 5H, Ph). Anal. calcd. for C22H3406Si: C 62.53, H 8.11; found: C 62.62, H 8.15.
Methyl 3-O-benzyl-5-C-(tert-bu~ldimethylsilylo~methyl)-5,6- dideoxy-1 ,2-0-isopropylidene-a-D-xylo-hept-5-(Zj-eno- jifilranosyluranote (15a)
A solution of 14 (16 g, 37.9 rnmol) and carbomethoxymethylenetri- phenylphosphorane (16 g, 50 mmol), in dry acetonitrile (50 mL) under argon, was refluxed for 2 h. Work-up in the usual way afforded P5a (15.2 g, 84%) after chromatography; Rf 0.63 (20% ethyl ether - petroleum ether); v,, (CHCI,): 2950, 1740 (C=O, unsaturated ester), 1660 cm-I ; 'H nmr (80 MHz) 6: 0.05 (s, 6H. SiMe,), 0.92 (s, 9H, tBuSi), 1.31, 1.50 (s, s , 6H, C(CH3)2), 3.62 (s, 3H, 0CH3), 4.35-4.73 (m, 6H, H-2, H-3,H15, H-5', PhCH2), 5.86 (m, lH, H-6), 5.95(d, 1H, J 1 , 2 = 3.7Hz, H-I), 6.17(m, lH, H-4), 7.30(s, 5H, Ph). Anal. calcd. for C25H3807Si: C 62.73, H 8.00; found: C 62.70, H 7.96.
5-C-Hydroxymethyl-5,6-dide0xy-I ,2-0-isopropylidene- a-D-gluco- heptofuranuronic 1 ', 7-lactone ( I 9 b)
To a solution of 15a (5.0 g, 10 mmol) in methanol (50 mL) was added ammonium iiuoride (0.75 g, 20 mmol) in water (4 mL). The reaction mixture was refluxed for 3 h, cooled, and the methanol removed in vacuo. A solution of the residue (15b) in benzene (100 mL) was hydrogenated over 5% platinum on carbon at a starting pressure of 500 psi (1 psi = 6.9 kPa). After 45 min the catalyst was removed by filtration and the filtrate was concentrated in vacuo to yield an oil that showed two components (tlc) assigned as 18 and P9a. To a solution of the oil in diethyl ether (100 I&) was added p-toluenesulfonic acid (120 mg, 0.6 mmol), and after 6 h at 23OC the reaction mlxture was quenched with triethylamine, washed with brine, dried (Na2SB,), and concentrated in vacuo to afford 3.4 g (96%) of 19a. Debenzylation by hydrogenolysis (10% Pd/C) in ethanol was carried out in the usual way, to give 19b (1.9 g, 79%) as acrystailine material, rnp 138-139°C; R f 0.44 (5% methanol - methylene chloride); [alto -47 .Y (c 0.4, chloroform); v,,, (CHC13): 3400 (hydrogen bonded OH), 2910, 1775 (lactone) cm-' ; 'H nmr (400 MHz) 6: 1.31 (s, 3H, CCH;), I .49 (s, 3H, CCH?), 1.57 (s, l H , OH), 2.70 (ddd, 2H, H-7, H-7'), 3.00 (q, J = 8.0 Hz, lH, H-5), 4.09-4.17 (m, 3H, H-3, H-6, H-6'), 4.46 (dd, J = 8.0 Hz, lH, H-4), 4.50 (d, J = 3.7 Hz, IN, H-21, 5.91 (d, J = 3.7 HZ, IH, H-I). Anal. calcd. for CllHI6O6: C 54.09, H 6.60; found: C 54.14, H 6.76.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
118.
99.1
14.1
21 o
n 06
/03/
13Fo
r pe
rson
al u
se o
nly.

ET AL. 1807
3-0-Benzyl-5-C-(tert-butyldimethylsiloxymethyl)-5,6-dideo~-1,2- 0-isopropylidene-a-D-xylo-hept-5-(Z)-enofuranose (20a)
Reduction of 1Sa (15.0 g, 31.4 mmol) with diisobutylaluminum hydride was carried out as described above for I l a + 11 b. The product 20a (1 1.6 g, 82%) was a syrup; Rf0.54 (50% ether- petroleum ether); 'H nmr (80 MHz) 6: 0.05 (s, 6H, SiMe2), 0.90 (s, 9H, tBuSi), 1.32, 1.48 (s, s, 6H, C(CH,),), 1.75 (t, lH, J = 5.4 HZ, OH), 3.92 (d, lH, J3,4 = 3.3 Hz. H-3), 4.00-4.28 (m, 4H, two H-7, two H-5'), 4.54 (d, 2H, J - 2.7 HZ, PhCH2), 4.58 (d, lH, J = 3.7 Hz, H-2), 4.92 (br d, 1H. J = 3.3 Hz, H-4), 5.94 (d and m, 2H, J1,2 = 3.7 Hz, H-1, H-6; overlapping), 7.29 (s, 5H, Ph). Anal. calcd. for C24H3806Si: C 63.97, H 8.50; found: C 63.86, H 8.57.
Hydroboration of compound 20a (formation of compounds 15 and 16a)
Hydroboration of 20a (1.72 g , 3.78 mmol) was carried out as described for I l b + 12. Flash chromatography of the product gave 21 (500 mg) and 22a (283 mg). For 21: Rf0.41 (50% ethyl acetate-petro- leum ether); [a]: -42.8' (c 0.90, chloroform); 'H nmr (250 MHz) 6: 0.03 (d, 6H, J = 10.4 Hz, %Me2), 0.80 (s, 9H, tBuSi), 1.28, 1.44 (s, S, 6H, C(CH3)2), 2.20 (m, lH, H-5), 2.70 (br s, 2H, two OH, D 2 0 exchange), 3.47 (dd, lH, J6,7a = 4.3 Hz, J,,, = 11.3 Hz, H-7a), 3.70 (d, 2H, J = 5.0 HZ, two H-5'), 3.75 (dd, lH, J6.7b = 3.0 HZ, J,,, = 11.3 HZ, H-7b), 3.84 (d, lH, J3,4 2.5 HZ, H-3), 3.98 (m, IH, H-6), 4.32 (dd, lH, J,,, = 2.5 Hz, J = 11.3 Hz, H-4), 4.54 (ABq, 2H, J = 11.3 HZ, PhCH2), 4.60 (d, lH, J 1 , 2 = 4.2 HZ, H-2), 5.90 (d, 1H, J 1 , 2 = 4.2 Hz, H-1), 7.30 (m, 5H, Ph). Anal. calcd. for C24H4007Si: C 61.51, H 8.60; found: C 61.36, H 8.83.
For 22a: Rf 0.67 (50% ethyl acetate - petroleum ether); 'H nmr (250 MHz) 6: 0.03 (d, 6H, %Me2), 0.82 (s, 9H, tBuSi), 1.30, 1.45 (s, s, 6H, C(CH3)2), 1.82 (m, 2H, two H-6), 2.16 (m, lH, H-5), 3.40 (dd, lH, J5.5,a = 5.0 HZ, Jgem = 10.0 Hz, H-5'a), 3.56 (dd, lH, J5,5,b = 2.5 HZ, J,,, = 10.0 Hz, H-5'b), 3.69 (t, 2H, J = 5.5 Hz, two H-7), 3.82 (d, lH, J3,4 = 2.5 Hz, H-3), 4.12 (dd and s, 2H, J3,4 = 2.5 Hz, J 4 , 5 = 10.0 Hz, H-4, OH, D20 exchange), 4.55 (ABq, 2H, J = 10.0 HZ, PhCH2), 4.60 (d, lH, J1,2 = 4.2 HZ, H-2), 5.88 (d, 1H, J 1 , 2 = 4.2 Hz, H-1), 7.28 (m, 5H, Ph). Anal. calcd. for C24H40O6Si: C 63.68, H 8.91; found: C 63.56, H 8.83.
5-C-Acetoxymethyl-3,7-dr-O-ace~l-5,6-dideoq~-l,2-O-isepropyli- dene- a-D-gluco-heptofuranose (22c)
(a) Eactone 19b (0.123 g, 0.543 mmol) was dissolved in dry ether (25 mL), cooled in an ice water bath, and treated with lithium aluminum hydride (0.067 g, 1.76 mmol). The reaction mixture was warmed to room temperature and stirred, with the exclusion of moisture, for 1 h. Excess lithium aluminum hydnde was destroyed with water and the solvent was removed. The residue (22b) was dissolved in dry pyridine (30 mL) and treated with excess acetic anhydride (5 mL) for 1.5 h at room temperature. Work-up in the usual way afforded 22c (0.156 g, 76%); Rf 0.40 (50% ethyl acetate - petroleum ether); -13.6"(c 1.1, CHC13); ' H n m r ( 2 5 0 ~ H z ) G : 1.28, 1.48 (twos, 2 X
3H, 02C(CH3)2), 1.74, 1.97-2.06 (two m, 2H, H-6a, H-6b), 2.01, 2.03, 2.09 (three s, 3 x 3H, 3 x OCOCH,), 2.15 (m, lH, H-5), 3.91-4.26 (m, 5H, H-4, H-7a, H-7b, H-l 'a, H-1 'b), 4.46 (d, lH, H-2, J1,2=3.7H~),5.14(d,1H,H-3,J3,4=3.2Hz),5.84(d,1H,H-l). Anal. calcd. for C17H26O9: C 54.54, H 7.00; found: C 54.36, H 6.86.
(b) A mixture of compound 21 (530 mg, 1.08 mmol) and 1 , l ' - thiocarbonyldiimidazole (228 mg, 1.3 rnmol) in 20 mL of dry tetra- hydrofuran was refluxed under an argon atmosphere for 1 h. The reaction solution was partitioned between diethyl ether (50 mL) and cold 2 N hydrochloric acid (10 mL). The organic layer was separated, washed with water and brine, dried over anhydrous sodium sulfate, filtered, and evaporated irz vacuo to give 5 18 mg (90%) of crude thionocarbonate 23. A portion of the crude thionocarbonate (388 mg, 0.72 mmol), in tri-n-butyltin hydride (424 g, 1.44 mmol), and a catalytic amount of 1,1 '-azobis(isobutyronitri1e) in 20 mL of dry toluene was added to refluxing toluene over 10 min. After refluxing 3 h the mixture was treated with 5 mL of 10% aqueous sodium hydroxide at 40°C for 12 h. Work-up with the standard processing and flash chromatography provided pure compound 22a. A portion of the
material (162 mg, 0.36 mmol), a catalytic amount of 10% palladium on activated carbon, and a few drops of 88% aqueous formic acid in 10 mL of methanol were stirred at room temperature under a hydrogen atmosphere. After 6 h the reaction mixture was filtered and evaporated in vacuo. The crude residue (22b) was acetylated using the standard procedure, then treated with acetic anhydride and pyridine, to yield compound 22c (121 mg, 90%) whose physical properties were iden- tical with those of the material described in part (a).
5-C-( tert-Buty1dimethylsiloxymethyl)-5,6-dideo- ,2-0-isopropyli- dene- a- D-xylo-hept-5-( Z)-enofuranose (20 b)
To a solution of 4.1 g (8.1 mmol) of the ester 15c (27) in 100 mL of dry toluene was added 24 mL of 1 M diisobutylaluminum hydride in toluene at P C , dropwise, over a period of 20 min under an argon atmosphere. After 3 h, tlc indicated that most of the starting material had been consumed. The reaction was quenched by addition of water at 0°C followed by 15% aqueous sodium hydroxide (1 mL) and water (3 mL). After vigorous stirring at room temperature for a few minutes, the white inorganic salts were removed by filtration, the residue was rinsed several times with ethyl acetate, and the combined filtrates were dried over anhydrous magnesium sulfate. The residue from evaporation was dissolved in dry methanol (100 mL) and then treated with a catalytic amount of sodium methoxide at room temperature. The saponification was monitored by tlc and, upon completion, the reaction mixture was poured into ice water and extracted with dichloromethane. The combined extracts were washed with cold dilute hydrochloric acid, saturated sodium bicarbonate, water, and brine, dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by flash chromatography to give 2.1 g (72%) of purediol20bas acolorless syrup; Rf0.45 (50% ethyl acetate-petroleum ether); [a]: -38.4' (c 1.0, chloroform); 'H nmr (80 MHz) 6: 0.08 (s, 6H, %Me2), 0.88 (s, 9H, tBuSi), 1.28, 1.45 (s, s, 6H, C(CH3)2)r 2.48 (brs, lH, OH), 3.80-4.46 (m, 6H, H-3, H-7, H-7', H-5, H-5', OH), 4.52 (d, lH, J = 3.7 Hz, H-2), 5.03 (br s, lH, H-4), 5.88 (m, 2H, J1,2 = 3.7 Hz, H-1, H-6). Anal. calcd. for C17H3206Si: C 56.64, H 8.95; found: C 56.73, H 8.93.
5-C-( tert-Bu~ldimethylsiloq~methyl)-5-deoxy-l,2-0-isopropylidene- L-glycero- a-D-gluco-heptofuranose (25)
To a solution of disiamylborane (38 mmol) in dry tetrahydrofuran was added a solution of diol 20b (2.65 g, 7.36 mmol) in dry tetra- hydrofuran (45 mL) at -78'C under an argon atmosphere, dropwise, over a period of 45 min. After stirring at -78OC for 2 h, the temperature was raised to -20°C, on the assumption (tlc evidence) that the formation of 24 was optional. A solution of 1 M borane in tetrahydro- furan (46 mL) was then added dropwise over a period of 45 min and the reaction mixture was stirred at room temperature for 16 h. Further portions of borane solution were added if necessary (tlc evidence). The reaction was quenched by addition of water (2 mL) to destroy the excess of borane, followed by 3 N sodium hydroxide and 30% hydrogen peroxide (30 mL of each) at O°C. After vigorous stirring at room temperature for 6 h, the mixture was saturated with sodium chloride. The tetrahydrofuran phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic extracts were dried over anhydrous magnesium sulfate, filtered, and concen- trated in vacuo. Excess water was removed azeotropically with toluene several times. The resulting residue was purified by flash chromato- graphy to afford 2.22 g (80%) of pure triol 25 as a colorless syrup; Rf 0.21 (50% ethyl acetate - petroleum ether); [a]:, +7.8" (c 0.97, chloroform); 'H nmr (250 MHz) 6: 0.06 (s, 6H, SiMe2), 0.88 (s, 9H, tBuSi), 1.28, 1.47 (s, s, 6H, C(CH3)2), 2.14 (m, 2H, H-5, OH, D 2 0 exchange), 2.92 (br s, lH, OH, D 2 0 exchange), 3.35 (br s, lH, OH, D20 exchange), 3.57-3.83 (m, 4H, H-3, H-6, H-5, H-5'), 3.86 (dd, lH, J3,4 = 2.5Hz, J = 11.3Hz, H-4), 4.13 (m, 2H, H-7, H-7'), 4.52(d,!H,J=4.2Hz,H-2),5.88(d, l H , J 1 2=4.2Hz,H-l) .Anal . calcd. for C17H3407Si: C 53.94, H 9.12; found: C 53.76, H 9.05.
5-C-(tert-Butyldimethylsiloxymethy1)-5-deoxy- ,2-0-isopropylidene- L-glycero- a-D-gluco-heptofuranose-6, 7-carbonate (26a)
To a stirred solution of triol 25 (1.14 g, 3.0 mmol) in dry dichloro- methane (70 mL) and dry pyridine (35 rnL) was added dropwise
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
118.
99.1
14.1
21 o
n 06
/03/
13Fo
r pe
rson
al u
se o
nly.

1808 CAN. J. CHEILI. VOL. 64, 1986
14.4 mL of 12.5% phosgene in toluene at O°C under an argon atmosphere over a period of 20 min. After 1 h the reaction was poured into 50 mL of ice water and then extracted with dichloromethane (2 X
50 mL). The combined organic extracts were washed with cold dilute hydrochloric acid, water, and brine, and dried (Na2S04), filtered, and concentrated in vacuo. The residue was purified by flash chromato- graphy to give26a (1.1 g, 90%) as acolorless syrup; Rf0.64 (50% ethyl acetate - petroleum ether); [a]: +10.7" (c 1.47, chloroform); v,,, (CHC13): 3450 (OH), 2950, 2900, 1880 (C=O, cyclic carbo- nate) cm-'; 'H nmr (250 MHz) 6: 0.05 (s, 6H, SiMe2), 0.85 (s, 9H, tBuSi), 1.27, 1.45 (s, s, 6H, C(CH3)2), 2.32 (m, IH, H-5), 3.17 (br s, lH, OH. D20 exchange), 3.68 (dd, 1H, J5,5,a = 7.5Hz, J,, = 10.0Hz, H-5'a), 3.90 (dd, lH, J5,53b = 2.5Hz, J,,, = lO.OHz, H-S'b), 4.04 (dd, 1H, J3,4=2.5Hz, J 4 , 5 = lO.OHz,H-4),4.18(d, l H , J = 2 . 5 H z , H-3), 4.35 (t, IH, J = 8.8 Hz, H-7a), 4.50 (d, IH, J = 4.2 Hz, H-2), 4.61 (dd, 1H, J = 7.5 HZ, J = 10.0 HZ, H-7b), 4.76 (q, lH, J5,6 = .f6,7a = 8.8 HZ, H-6), 5.82 (d, 1H, J 1 , 2 = 4.2 Hz, H- 1). Anal, calcd. for C18H3208Si: C 53.44, H 7.97; found: C 53.43, H 8.00.
5-C-( tert-Buty1dimethylsiloxymethyl)-5-deoxy ,2-0-isopropylidene- 3-0-phenoxythiocarbonyl-L-glycero- a-D-gluco-heptofuranose- 6,7-carbonate (26 b)
To a solution of the alcohol 26a (1.1 g, 2.7 mmol) and a catalytic amount of N,N-dimethylaminopyridine (-10 mg) in dry dichloro- methane (50 mL) and dry pyridine (2 mL) was added phenyl chloro- thionocarbonate (0.62 mL, 4 .5 mmol) at room temperature under an argon atmosphere. After 3 h another more phenyl chlorothionocar- bonate (0.2 mL) was added and the reaction was monitored by tlc. The reaction mixture was poured into 10 mL of ice water and extracted with 3 X 20 mL of dichloromethane. The combined organic extracts were washed with cold dilute hydrochloric acid, water, and brine, and dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography gave 26b (1.2 g, 82%) as a yellow syrup; Rf 0.50 (50% ethyl acetate - hexane); [ a 1 2 +9.4° (c 0.98, chloro- form); v,,, (CNC13): 2950, 2900, 1800 (C=O), 1580, 1480cm-'; 'H nmr (250 MHz) 6 : 0.06 (d, 6H, %Me2), 0.88 (s, 9H, tBuSi), 1.3 1, 1.50 (s, s, 6H, C(CH3),), 2.30 (ttt, lH, J = 2.9 Hz, J = 9.8 Hz, H-5),3.63(dd, lH, J=3 .3Hz,Jge ,= 10.8Hz,H-Sra),3.96(dd, lH, J = 3.3 Hz, J,,, = 10.8 Hz, H-5'b), 4.38 (t, lH , J = 8.8 Hz, H-7a), 4.49(dd, lW, J 3 , 4 = 2.9Hz, J4,5 = 9.8Hz,H-4),4.62(dd, lH, J = 7.8 Hz, J = 8.8 Hz, H-7b), 4.72 (d, l H , J = 4.2 Hz, H-2), 4.96 (q, IH, J = 7.8 Hz, H-6), 5.64 (d, lH, J3 ,4 = 2.9 Hz, H-3), 5.92 (d, lH, J1,, = 4.2 Hz, H- l) , 7.05-7.50 (m, SH, Ph). Anal. calcd. for C25H3609SSi: C 55.53, H 6.71, S 5.93; found: C 55.61. H 6.50, S 6.15.
5-C-(tert-Butyldimethylsiloxlyrnet/1yl)-3,5-dide0-1,2-O-isopro- pylidene-P-L- talo-heptofuranose-6,7-carbonate (26c)
A mixture of compound 26b (1.2 g, 2.22 mmol), tri-n-butyltin- hydride (0.9 mL, 3.33 mmol) and 2,2'-azobis(2-methylpropionitrile) (30 mg) in toluene (40 mL) was heated to reflux under an argon atmosphere. After 3 h the reaction mixture was cooled to room temperature and then concentrated in vacuo. Flash chromatography afforded compound 26c (775 mg, 90%) as a colorless syrup; Rf 0.15 (30% ethyl acetate - hexane); [ a ] i 3 + 1.9" (c 1.35, chloroform); v,,, (CHC13): 3000, 1800 (C=O, cyclic carbonate), 1500 cm-'; ' H n m r ( 2 5 0 M ~ z ) 6: 0.04 (s, 6H, SiMe2), 0.88 (s, 9H, tBuSi), 1.29, 1.46 (s, s, 6H, C(CH3)2), 1.55 (m, lH, H-3a), 1.80 (m, lH, H-5), 2.25 (dd, 1H, J 2 , 3 p = J3P,4 = 4.2Hz, J,,, = 13.0Hz, H-3a), 3.67 (dd, lH, J = 3.1 Hz, J,,, = 10.2 Hz, H-S'a), 3.91 (dd, lH, J = 3.1 HZ, J,,, = 10.2 Hz, H-5'b), 4.25 (ddd, lH, J 3 p , 4 = 4.2 HZ, J4,5 = 10.2 HZ, H-4), 4.38 (t, IN, J = 9.3 Hz, H-7a), 4.62 (t, lH, 3 = 8.3 HZ, H-7b), 4.70 (t, lH , J,,, = J 2 , 3 p = 4.2 HZ, M-2), 4.90 (q, lH, J = 8.3 Hz, H-6), 5.74 (d, IH, J 1 , 2 = 4.2 Hz, H-1). Anal. calcd. for C18H3207Si: C 55.64, I1 8.30; found: C 55.81, H 8.36.
temperature for 30 min, the reaction mixture was neutralized with a stream of carbon dioxide gas, and then extracted with 3 x 20 mL of ethyl acetate. The combined organic extracts were dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. Flash chromatography gave diol 3 b (197 mg, 98%) as a colorless syrup; Rf 0.13 (30% ethyl acetate - petroleum ether); [ a 1 2 -9.8" (C 1.25, chloroform); 'H nmr (250 MHz) 6: 0.06 (s, 6H, SiMe2), 0.88 (s, 9H, tBuSi), 1.28, 1.48 (s, s, 6H, C(CH3),), 1.57 (m, lH, H-3P), 1.82 (m, lH, H-51, 2.15 (dd, lH, J 2 , 3 p = J3P,4 = 4.2 Hz, J,,, = 13.0 HZ, H-3P), 2.70 (dd, 1H, J = 6.3 Hz, J = 8.3 Hz, OH, D 2 0 exchange), 3.21 (d, lH, J = 6.3Hz, OH, D 2 0 exchange), 3.65-3.83 (m, 4H, two H-5', H-7, H-7'), 3.93 (m, IH, H-6), 4.32 (m, lH, H-4), 4.71 (t, lH, J1,2 = J 2 . 3 ~ = 4.2Hz, H-2), 5.78 (d, lH, J 1 , 2 = 4.2Hz, H-I). Anal. calcd. for C17H3406Si: C 56.32, H 9.45; found: C 56.32, H 9.31.
Acknowledgements We are grateful to the Burroughs Wellcome Trust, Duke
University, the University of Maryland, the Natural Sciences and Engineering Research Council of Canada, and the National Science Foundation (CHE 8304283) for support of this work. The help of Drs. Naoki Mitsuo and Masao Shiozaki is acknowledged.
1. J. D. DUTCHER, M. B. YOUNG, J . H. SHERMAN, W. HIBBITS, and D. R. WALTERS. Antibiotic Annu. 886 (1956); J . D. DUTCHER, D. R. WALTERS, and D. WINTERSTEINER. J. Org. Chem. 28, 995 (1963); M. VON SALTZA, J. D. DUTCHER, J. REID, and 0 . WINTERSTEINER. J . Org. Chem. 28,999 (1963); A. C. COPE, V. AXEN, E. P. BURROWS, and J. WEINLICH. J . Am. Chem. Soc. 88, 4228 (1966); E. BOROWSKI, J. ZIEIINSKI, T. ZIMINSKI, L. FALOWSKI, P. KOLODZIEJCZYK, J. GOLIK, and E. JERECZEK. Tetrahedron Lett. 3909 (1970).
2. S. WAKSMAN, and H. LECHEVALIER. In The actinomycetes. Vol. 3. Williams and Wilkins, Baltimore. 1962.
3. G. HILDICK-SMITH, H. BLANK, and I. SARKANY. In Fungus diseases and their treatment. Little, Brown and Co., Boston. 1964.
4. S. C. KINSKY. In Antibiotics. Vol. 1. Edited by D. Gottlieb and P. D. Shaw. Springer Verlag, New York. 1967. p. 122, and references cited therein; R. W. HOLZ. In Antibiotics. Vol. 2. Edited by F. E. Hahn. Springer Verlag, New York. 1979. p. 3 13, and references cited therein.
5. I. K. ARONSON, C. H. L. RIEGER, K. SOLTANI, V. TKALCEVIC, W. C. CHAN, A. L. LORINCZ, and G. MATZ. Cancer, 43, 101 (1979); B. A. ADELMAN, A. BENTMAN, P. ROSENTHAL, B. R. SMITH, K. R. BRIDGES, and W. HOLMES. Ann. Intern. Med. 91, 323 (1979); E. Z. EZDINLI, D. D. O'SULLIVAN, L. P. WASSER, U. KIM, andL. STUTZMAN. J. Am. Med. Assoc. 262,258 (1979).
6. I. M. TERESHIN. In Polyene antibiotics-present and future. University of Tokyo Press, Japan. 1976.
7. W. ORosHNrK and A. D. MEBANE. Prog. Chem. Org. Nat. Prod. 37, 166 (1973); A. W. NORMAN, A. M. SPIELVOGEL, and R . W. WONG. Adv. Lipid Res. 14, 127 (1976).
8. T. NAKATA, S. TAKAO, M. FUKUI, T. TANAKA, and T. OISHI. Tetrahedron Lett. 24, 3873 (1983); K. NARASAKA and F. C. PAI. Tetrahedron, 40, 2233 (1984); S. KIYOOKA, H. SASAOKA, R. FUJIYAMA, and C. H. HEATHCOCK. Tetrahedron Lett. 25, 5331 (1984); P. A. BARTLETT and K. K. JERNSTEDT. J . Am. Chem. Soc. 99,4829 (1977); M. HIRAMA and M. UEI. Tetrahedron Lett. 23, 5307 (1982); B. H. LIPSHUTZ and J. A. KOZLOWSKI. J. Org. Chem. 49, 1149 (1984).
9. W. MECHLINSKI. C. P. SCHAFFNER. P. GANIS, and G. AVITA- 5-C-( tert-Butyldirnethylsiloxymethyl/-3,5-~-1,2-O-isopropyli- BILE. Tetrahedron Lett. 3873 (1970); P. GANIS, G. AVITABILE,
dene- p-L-talo-heptofuranose (3 b) W. MECHLINSKI, and C. P. SCHAFFNER. J. Am. Chem. Soc. 93, To a stirred solution of the carbonate 26c (215 mg, 0.56 mmol) in 4560 (1970).
215 mL of a water-methanol-tetrahydrofuran (1:l:l) mixture was 10. D. BOSCHELLI, J. W. ELLINGBOE, and S. MASAMUNE. Tetra- added barium hydroxide octahydrate (100mg). After stirring at room hedron Lett. 25, 3395 (1984); S. MASAMUNE, T. KAIWO, and
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
118.
99.1
14.1
21 o
n 06
/03/
13Fo
r pe
rson
al u
se o
nly.

LIANG ET AL. 1809
D. S. GARVEY. J. Am. Chem. Soc. 104, 5521 (1982); S. MASAMUNE, P. MA, H. OKUMOTO, J . W. ELLINGBOE, and Y. ITO. J. Org. Chem. 49, 2837 (1984); P. MA, V. S. MARTIN, S. MASAMUNE, K. B. SHARPLESS, and S. M. VITI. J . Org. Chem. 47, 1378 (1982).
1 1 . K. C. NICOLAOU and J. UENISHI. J . Chem. Soc. Chem. Commun. 1292 (1982).
12. D. W. BROOKS and R. P. KELLOGG. Tetrahedron Lett. 23, 4991 (1982); D. W. BROOKS and J. T. PALMER. Tetrahedron Lett. 24, 3059 (1983).
13. D. M. FLOYD and A. W. FRITZ. Tetrahedron Lett. 22, 2847 (1981); D. M. FLOYD and C. M. CIMARUSTI. Tetrahedron Lett. 20,4129 (1979).
14. S. HANESSIAN. In Total synthesis of naturalproducts: the 'chiron' approach. Pergamon Press, New York. 1982.
15. B. E. DAVIDSON and R. D. GUTHRIE. J. Chem. Soc. Perkin Trans. 1 , 658 (1972).
16. B. E. DAVIDSON, R. D. GUTHRIE, and A. T. MCPHAIL. J. Chem. Soc. Chem. Cornmun. 1273 (1968).
17. A. MUBARAK and B. FRASER-REID. J. Org. Chem. 47, 4265 (1982).
18. B. FRASER-REID, L. MAGDZINSKI, and B. MOLINO. In Current trends in organic synthesis. Edited by H. Nozaki. Pergamon, New York. 1983; B. FRASER-REID, L. MAGDZINSKI, and B. MOLINO. J. Am. Chem. Soc. 106, 731 (1984).
19. D. A. PRINS. J. Am. Chem. Soc. 70, 3955 (1948). 20. (a) P. T. HO and S. CHUNG. Carbohydr. Res. 110, 217 (1982);
(6) J. D. PRUGH and A. A. DEANA. Tetrahedron Lett. 23, 281 (1982); ( c ) Y. L. YANG and J. R. FALCK. Tetrahedron Lett. 23, 4305 (1982).
21. D. H. R. BARTON and S. W. MCCOMBIE. J. Chem. Soc. Perkin Trans. 1, 1574 (1975).
22. E. J. HEDGELEY, W. G . OVEREND, and R. A. C. RENNIE. J . Chem. Soc. 4701 (1963).
23. K. C. NICOLAOU, M. R. PAVIA, and S. P. SEITZ. J . Am. Chem. SOC. 104, 2027 (1982).
24. (a) J . K. N. JONES and J. L. THOMPSON. Can. J . Chem. 35, 955 (1957); (b) D. HORTON, J . K . THOMSON, and C. G. TINDALL, JR. Methods Carbohydr. Chem. 6 , 297 (1972).
25. R. L. WHISTLER and W. C. LAKE. Methods Carbohydr. Chem. 6, 286 (1972).
26. H. REDLICH and 13. J. NEUMANN. Chem. Ber. 114,2020 (1981). 27. M. GEORGES, T. F. TAM, and B. FRASER-REID. J. Org. Chem. 50,
5747 (1985); B. FRASER-REID, Z. BENKO, R. GUILIANO, K. M. SUN, and N. TAYLOR. J . Chem. Soc. Chem. Commun. 1029 (1984).
28. D. H. R. BARTON and R. SUBRAMANIAN. J. Chem. Soc. Perkin Trans. 1, 1718 (1977).
29. H. C. BROWN. In Boranes in organic chemistry. Cornell University Press, Ithaca, NY. 1972. p. 266.
30. H. C. BROWN and M. K. UNNI. J. Am. Chem. Soc. 90, 2902 (1968); H. C. BROWN and R. M. GALLIVAN, JR. J. Am. Chem. Soc. 90, 2906 (1968); H. C. BROWN and R. L. SHARP. J. Am. Chem. Soc. 90, 2915 (1968), and references therein; K. H. SCHULTE-ELTE and G. OHLOFF. Helv. Chim. Acta, 50, 153 (1967); D. J. PASTO and J. HICKMAN. J. Am. Chem. Soc. 90, 4445 (1968).
31. D. LIANG, H. W. PAULS, and B. FRASER-REID. J. Chem. Soc. Chem. Commun. 1122 (1984).
32. M. J. ROBBINS and J. S. WILSON. J . Am. Chem. Soc. 103, 932 (1981).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
118.
99.1
14.1
21 o
n 06
/03/
13Fo
r pe
rson
al u
se o
nly.




















