
Regioselective Reactions on a Chiral Substrate Controlled by the Configuration of a Chiral Catalyst
12
DOI: 10.1002/adsc.200900822 Regioselective Reactions on a Chiral Substrate Controlled by the Configuration of a Chiral Catalyst Raju Ranjith Kumar a and Henri B. Kagan a, * a Laboratoire de Catalyse MolȖculaire, Institut de Chimie MolȖculaire et des MatȖriaux d’Orsay (UMR CNRS 8182), BȦtiment 420, UniversitȖ Paris-Sud, 91405 Orsay, France Fax: (+ 33)-1-6915-4680; e-mail: [email protected] Received: December 26, 2009; Published online: January 27, 2010 Abstract: A racemic mixture may be partially trans- formed in the presence of a chiral catalyst by kinetic resolution and formation of products with new struc- tural features. If the starting material is fully con- sumed the products may still be enantiomerically en- riched. The situation is summarized in the Introduc- tion. A brief discussion on the regioselective trans- formations occurring on a racemic mixture under the influence of a chiral catalyst is presented in Section 2. Often stereo-differences occur, each enantiomer of the starting material resulting in a different prod- uct. It allows one to predict what the behaviour of some enantiopure substrates should be in presence of each of the enantiomers of a chiral catalyst. Many examples are presented in Section 3. The chiral sub- strates under consideration have two different react- ing sites, usually of the same nature (OH, C = C, allyl- ic positions, C À H for carbene insertion, epoxide frag- ment, etc.). In some cases the absolute configuration of the catalyst allows an excellent control of the re- gioselectivity. This approach is promising for the se- lective transformation of chiral molecules. 1 Introduction 2 Regioselective Transformation on a Racemic Mixture 3 Chiral Catalysts on Chiral Substrates 3.1 Sharpless Epoxidation 3.2 Allylic Substitution 3.3 Epoxide Opening 3.4 C À H Insertions 3.5 Nitroso Diels–Alder Reaction 3.6 Intramolecular Hydroacylation on a Triple Bond 3.7 Alcoholysis of Substituted Succinic Anhydrides 3.8 Miscellaneous 4 Conclusions Note Added in Proof Keywords: asymmetric catalysis; match-mismatch; regiodivergence; regioselectivity; site selection; ste- roids 1 Introduction Non-enzymatic asymmetric catalysis has developed very much in the last thirty years and become a pow- erful method to create a chiral product, [1–5] or to dif- ferentiate between the two enantiomers of a racemic mixture. [6–9] The classical enantioselective synthesis [10] involves an achiral substrate often equipped with a C = O, C = N or C = C double bond. An enantioface is selectively attacked with the help of the catalyst. An- other type of enantioselective transformation is relat- ed to the preferential selection of one site (topos) [5] as described in Scheme 1. Asymmetric catalysts are also able to differentiate between two enantiomers of a racemic mixture by inducing a faster transformation of one of them (kinetic resolution). Processes in Scheme 1 involve substrates either devoid of stereo- genic units or which are of meso-type structures Scheme 1. Some examples of site selection on achiral compounds. Adv. Synth. Catal. 2010, 352, 231 – 242 # 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 231 REVIEWS
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Regioselective Reactions on a Chiral Substrate Controlled by the Configuration of a Chiral Catalyst

DOI: 10.1002/adsc.200900822
Regioselective Reactions on a Chiral Substrate Controlled by theConfiguration of a Chiral Catalyst
Raju Ranjith Kumara and Henri B. Kagana,*a Laboratoire de Catalyse Mol�culaire, Institut de Chimie Mol�culaire et des Mat�riaux d’Orsay (UMR CNRS 8182),
B�timent 420, Universit� Paris-Sud, 91405 Orsay, FranceFax: (+33)-1-6915-4680; e-mail: [email protected]
Received: December 26, 2009; Published online: January 27, 2010
Abstract: A racemic mixture may be partially trans-formed in the presence of a chiral catalyst by kineticresolution and formation of products with new struc-tural features. If the starting material is fully con-sumed the products may still be enantiomerically en-riched. The situation is summarized in the Introduc-tion. A brief discussion on the regioselective trans-formations occurring on a racemic mixture under theinfluence of a chiral catalyst is presented in Section2. Often stereo-differences occur, each enantiomerof the starting material resulting in a different prod-uct. It allows one to predict what the behaviour ofsome enantiopure substrates should be in presenceof each of the enantiomers of a chiral catalyst. Manyexamples are presented in Section 3. The chiral sub-strates under consideration have two different react-ing sites, usually of the same nature (OH, C=C, allyl-ic positions, C�H for carbene insertion, epoxide frag-ment, etc.). In some cases the absolute configurationof the catalyst allows an excellent control of the re-
gioselectivity. This approach is promising for the se-lective transformation of chiral molecules.
1 Introduction2 Regioselective Transformation on a Racemic
Mixture3 Chiral Catalysts on Chiral Substrates3.1 Sharpless Epoxidation3.2 Allylic Substitution3.3 Epoxide Opening3.4 C�H Insertions3.5 Nitroso Diels–Alder Reaction3.6 Intramolecular Hydroacylation on a Triple Bond3.7 Alcoholysis of Substituted Succinic Anhydrides3.8 Miscellaneous4 ConclusionsNote Added in Proof
Keywords: asymmetric catalysis; match-mismatch;regiodivergence; regioselectivity; site selection; ste-roids
1 Introduction
Non-enzymatic asymmetric catalysis has developedvery much in the last thirty years and become a pow-erful method to create a chiral product,[1–5] or to dif-ferentiate between the two enantiomers of a racemicmixture.[6–9] The classical enantioselective synthesis[10]
involves an achiral substrate often equipped with aC=O, C=N or C=C double bond. An enantioface is
selectively attacked with the help of the catalyst. An-other type of enantioselective transformation is relat-ed to the preferential selection of one site (topos)[5] asdescribed in Scheme 1. Asymmetric catalysts are alsoable to differentiate between two enantiomers of aracemic mixture by inducing a faster transformationof one of them (kinetic resolution). Processes inScheme 1 involve substrates either devoid of stereo-genic units or which are of meso-type structures
Scheme 1. Some examples of site selection on achiral compounds.
Adv. Synth. Catal. 2010, 352, 231 – 242 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 231
REVIEWS

where pairs of stereogenic centers are related by aplane or center of symmetry.
In a kinetic resolution the reaction may involve theformation of a stereogenic unit from a C=X or C=Cdouble bond, giving a mixture of diastereomeric prod-ucts and of enantioenriched starting material.
For a full transformation of the racemic substratethe products will consist of a mixture of enantioen-riched diastereomers. Horeau and Guett� discussedthis case in the asymmetric reduction of racemic cam-phor, they established that the mole ratio of the dia-stereomeric alcohols (dr) is equal to the inverse ratioof their corresponding enantiomeric excesses (ees).[12]
We subsequently considered the case of the partialtransformation of racemates with a kinetic resolutionor not of the remaining starting material. The systemstudied was the incomplete hydrogenation of a racemicdehydropeptide catalyzed by a chiral rhodium com-plex.[13] We established Eq. (1) which relates the rela-tive amounts and ees of the various components, it is ageneralization of the above Horeau relation.[12] In thisequation the two diastereomers (ee1 and ee2) are pres-ent in the respective amounts n1 and n2, in addition thestarting material is recovered in amount n3 with ee3.
It is possible to deduce qualitatively or quantita-tively the diastereoselectivity of the reaction on eachenantiomer of the starting material for a given enan-tiomer of the catalyst.
An asymmetric catalyst can also transform a race-mic mixture by a site-selection process instead of aface differentiation, giving a mixture of regioisomersinstead of a mixture of diastereomers. This less fre-quent situation affords information on the behaviourof each enantiomer of the racemic substrate. Eq. (1)applies with n1, n2 and n3 being the relative amountsof the recovered starting material and the two regioi-someric products. In the present article we intend toshortly review this area on some selected examplesbefore considering the regioselective control on achiral substrate under the influence of a chiral cata-lyst.
2 Regioselective Transformation on aRacemic Mixture
The first example of a non-enzymatic Baeyer–Villigeroxidation of a racemic ketone was described in mid-1990s.[14,15] Bolm et al. used a copper complex (R)-4 to
Henri B. Kagan was born inFrance in 1930. He graduatedfrom the Sorbonne and EcoleNationale Sup�rieure deChimie de Paris in 1954. Heprepared his Ph.D. under thesupervision of Dr. J. Jacques.He joined Prof. A. Horeau atthe Coll�ge of France inParis in 1962 as a research as-sociate. In 1965 he workedwith Prof. T. Mabry at theUniversity of Texas, Austin. He joined the Univer-sit� Paris-Sud, Orsay in 1968. He is emeritus Profes-sor of Universit� Paris-Sud since 1999. He is amember of the French Academy of Sciences. H. B.Kagan developed investigations in various areas,such as asymmetric synthesis, asymmetric catalysis,lanthanide reagents (for example, diiodosamarium).His awards include the Prelog Medal, the August-Wilhelm-von-Hofmann Medal, the Chirality Medal,the Nagoya Medal of Organic Chemistry, the Tetra-hedron Prize, the 2001 Wolf Prize for Chemistry(shared with K. B. Sharpless and R. Noyori), the2002 Grand Prix de la Fondation de la Maison de laChimie (shared with H. Yamamoto), the RyojiNoyori Prize in 2002. He was the recipient of theBower Award of Franklin Institute in 2005 and ofthe Horst-Pracejus Prize in 2007.
Raju Ranjith Kumar wasborn in Udhagamandalam(India) in 1977. He obtainedhis M.Sc. (2001) in Chemistryfrom Government Arts Col-lege (Bharathiar University),Udhagamandalam and Ph.D.(2008) from Madurai Kamar-aj University, India under thesupervision of Prof. S. Peru-mal. His Ph.D. research in-cluded synthesis, structuralelucidation and antimycobacterial evaluation ofnovel organic heterocycles obtained via 1,3-dipolarcycloaddition, cyclocondensation and one-pot multi-step reactions. Since September 2008, he has beenworking as a post-doctoral research fellow underProf. Henri B. Kagan at Universit� Paris-Sud. Hispresent research project involves the investigation ofthe influence of absolute configuration of chiral cat-alysts on the reactivity and selectivity of chiral sub-strates.
232 asc.wiley-vch.de � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Synth. Catal. 2010, 352, 231 – 242
REVIEWS Raju Ranjith Kumar and Henri B. Kagan
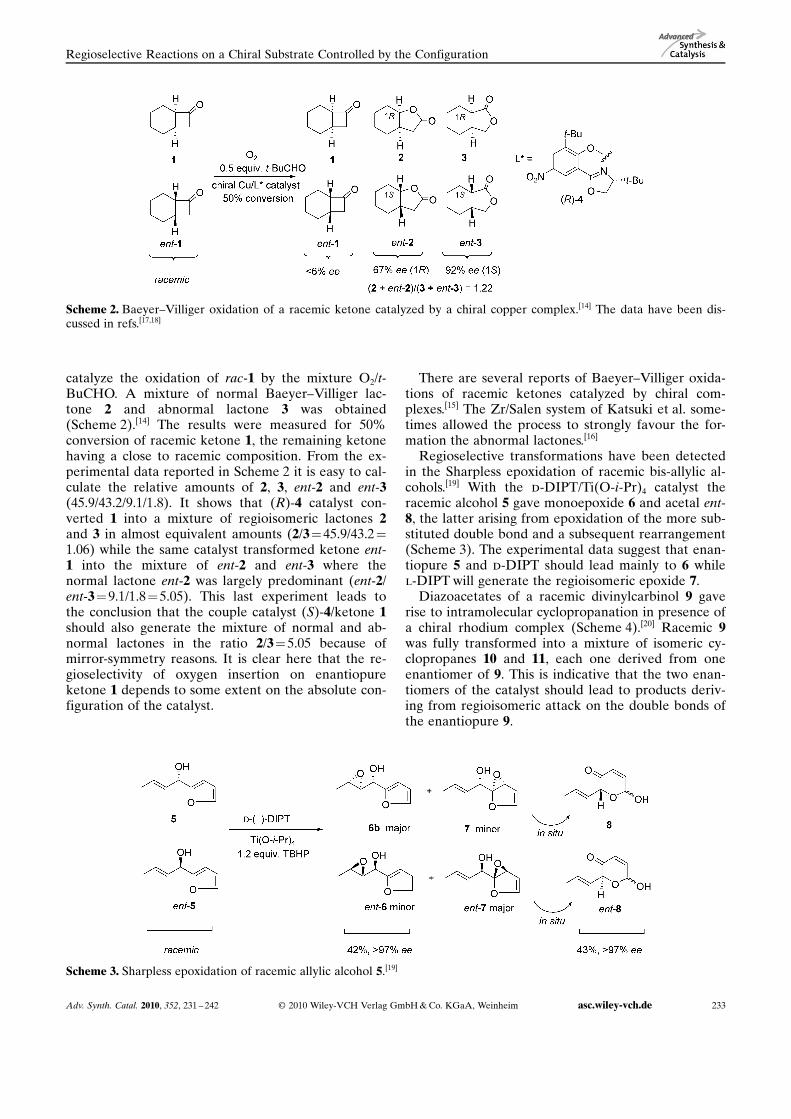
catalyze the oxidation of rac-1 by the mixture O2/t-BuCHO. A mixture of normal Baeyer–Villiger lac-tone 2 and abnormal lactone 3 was obtained(Scheme 2).[14] The results were measured for 50%conversion of racemic ketone 1, the remaining ketonehaving a close to racemic composition. From the ex-perimental data reported in Scheme 2 it is easy to cal-culate the relative amounts of 2, 3, ent-2 and ent-3(45.9/43.2/9.1/1.8). It shows that (R)-4 catalyst con-verted 1 into a mixture of regioisomeric lactones 2and 3 in almost equivalent amounts (2/3= 45.9/43.2 =1.06) while the same catalyst transformed ketone ent-1 into the mixture of ent-2 and ent-3 where thenormal lactone ent-2 was largely predominant (ent-2/ent-3=9.1/1.8= 5.05). This last experiment leads tothe conclusion that the couple catalyst (S)-4/ketone 1should also generate the mixture of normal and ab-normal lactones in the ratio 2/3= 5.05 because ofmirror-symmetry reasons. It is clear here that the re-gioselectivity of oxygen insertion on enantiopureketone 1 depends to some extent on the absolute con-figuration of the catalyst.
There are several reports of Baeyer–Villiger oxida-tions of racemic ketones catalyzed by chiral com-plexes.[15] The Zr/Salen system of Katsuki et al. some-times allowed the process to strongly favour the for-mation the abnormal lactones.[16]
Regioselective transformations have been detectedin the Sharpless epoxidation of racemic bis-allylic al-cohols.[19] With the d-DIPT/TiACHTUNGTRENNUNG(O-i-Pr)4 catalyst theracemic alcohol 5 gave monoepoxide 6 and acetal ent-8, the latter arising from epoxidation of the more sub-stituted double bond and a subsequent rearrangement(Scheme 3). The experimental data suggest that enan-tiopure 5 and d-DIPT should lead mainly to 6 whilel-DIPT will generate the regioisomeric epoxide 7.
Diazoacetates of a racemic divinylcarbinol 9 gaverise to intramolecular cyclopropanation in presence ofa chiral rhodium complex (Scheme 4).[20] Racemic 9was fully transformed into a mixture of isomeric cy-clopropanes 10 and 11, each one derived from oneenantiomer of 9. This is indicative that the two enan-tiomers of the catalyst should lead to products deriv-ing from regioisomeric attack on the double bonds ofthe enantiopure 9.
Scheme 2. Baeyer–Villiger oxidation of a racemic ketone catalyzed by a chiral copper complex.[14] The data have been dis-cussed in refs.[17,18]
Scheme 3. Sharpless epoxidation of racemic allylic alcohol 5.[19]
Adv. Synth. Catal. 2010, 352, 231 – 242 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim asc.wiley-vch.de 233
Regioselective Reactions on a Chiral Substrate Controlled by the Configuration

Diazoacetates of racemic cyclohexenol 12 in pres-ence of the Rh2[(S)-MEOX]4 catalyst were trans-formed into a mixture of lactone 13 and cyclohexen-ACHTUNGTRENNUNGone 14 (Scheme 5).[21] Compound 14 is derived froman intermediate b-lactone produced by a carbene in-sertion on the CH of the asymmetric center of (R)-12followed by a fragmentation releasing ketene.
Ethylzirconation and ethylalumination of thedouble bond of racemic 2-phenyl-3,4-dihydrofuranhave been realized by the use of a chiral zirconiumcomplex.[22,23] The full conversion of the racemic sub-strate gave a mixture of two products, each one with>95–99% ee. The data indicate that the two enantio-mers of the substrate have reacted differently withthe organometallic reagent, with a regioselective con-trol in the attack of the double bond.
3 Chiral Catalysts on Chiral Substrates
The use of chiral reagents or catalysts to create a newstereogenic center on a chiral substrate is verycommon. The concepts behind these reactions have
been elaborated by Masamune et al. , they are usefulin stereocontrolled syntheses.[24] The authors proposedthat the inherent steric controls of a chiral susbtrateand a chiral catalyst for a given transformation coop-erate (match pair)[25] or are antagonist when the enan-tiomeric catalyst is involved (mismatch pair). Thematch/mismatch concept is very apparent in Scheme 3where the pair 5/d-DIPT lead to 6 while the pair ent-5/d-DIPT gives the isomeric epoxide ent-7. The exam-ples of Scheme 3 and Scheme 4 clearly indicate thatthe site selectivity on a chiral substrate can be con-trolled by the absolute configuration of the catalyst.This is a case of regiodivergent reactions under cata-lyst control.[7,17,18]
3.1 Sharpless Epoxidation
The transformations of Scheme 3 have been con-firmed by a reaction carried out on enantiopure 5. d-DIPT gave epoxide 6 (from epoxidation on the a site)while l-DIPT exclusively led to attack on b site withformation of ent-6 (Scheme 6).
Scheme 4. Intramolecular cyclopropanation of a racemic diene catalyzed by a chiral rhodium complex.[20]
Scheme 5. Regiodivergent reaction on a racemic mixture: intramolecular cyclopropanation (a) on (S)-12 and C�H insertion(b) followed by ketene extrusion on (R)-12.
234 asc.wiley-vch.de � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Synth. Catal. 2010, 352, 231 – 242
REVIEWS Raju Ranjith Kumar and Henri B. Kagan

3.2 Allylic Substitution
The allylic substitution in the presence of an organo-metallic catalyst is a complex process. It involves theformation of an intermediate p-allylic complex whichmay be able to interconvert into a stereoisomeric p-allylic system according to the conditions and the cat-alyst. Usually palladium catalysts favour the linearproduct, Mo and W catalysts give predominantly thebranched product.[26] In the course of a detailed mech-anistic study involving chiral molybdenum catalystsReider et al. found that enantiopure 15 (Scheme 7)gave a different product distribution according to theconfiguration of ligand 18.[26] Some results are tabulat-ed in Scheme 7. The match situation (R)-15/ACHTUNGTRENNUNG(R,R)-18generated a large amount of the branched product inexcellent ee while in the mismatch pair (R)-15/ACHTUNGTRENNUNG(S,S)-18 the regioselectivities and ees are lower. In this lastcase the rate of equilibration between p-allyl com-plexes was competitive with the subsequent malonateattack.
Kinetic resolution in palladium-catalyzed allylicsubstitution on racemic compounds is well document-ed. What is less common is the possibility to get re-gioisomeric products deriving from different enantio-mers of the starting material (regiodivergent kineticresolution).[27–32] As expected, the chemical transfor-mations performed on the enantiopure allylic sub-strates show a very strong catalyst control on the re-gioselectivity. It will be exemplified in Scheme 8,Scheme 9 and Scheme 10.
The reaction of dimethyl malonate on some enan-tioenriched allylic acetates allowed Pfaltz et al. to
produce at will regioisomeric products in high ees (>99% ee).[28] For example, 19 (94% ee) gave 20 or 21with (S) or (R) catalyst 22, respectively (Scheme 8).
The substitution reaction of dimethyl malonateanion on a steroidal allylic acetate 23 (Scheme 9) hasbeen studied by Shimizu et al.[29] The palladium cata-lysts involving (S) or (R)-binap promoted different re-actions. With (S)-binap the main product was derivedby the substitution reaction at the C-3 position, withalmost exclusive retention of configuration. Theminor product was the diene 25. With (R)-binap (mis-
Scheme 6. Sharpless epoxidation of enantiopure allylic alco-hol 5.[19] Scheme 7. Regioselective allylic substitution of (R)-15.
Scheme 8. Reaction of the anion of dimethyl malonate onallylic acetates catalyzed by a chiral Pd complex.
Adv. Synth. Catal. 2010, 352, 231 – 242 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim asc.wiley-vch.de 235
Regioselective Reactions on a Chiral Substrate Controlled by the Configuration

match combination) the product distribution is differ-ent: diene 25 is predominant while the minor substitu-tion product 24 was obtained as a mixture of epimersat C-3. These results were confirmed by similar reac-tions performed in the kinetic resolution of rac-26with an (S)-binap-palladium catalyst.
Cook et al. investigated the formation of vicinal di-amines from racemic 5-vinyloxazolidinones 27 andphtalimide in presence of a chiral palladium com-plex.[30] They also briefly studied the use of enantio-pure substrate 27 (Scheme 10). They found that thetwo enantiomers of binap led to a different ratio ofregioisomeric diamides 28 and 29, although there isnot a reversal of the regioselectivity with the mis-match pair.
Fiaud et al. have studied the catalytic nucleophilicsubstitution of allylic acetate (R)-30 by potassium di-methyl malonate (Scheme 11).[31] The catalyst precur-sor was an equimolar amount of Pd ACHTUNGTRENNUNG(dba)2 and a chiralbidentate ligand L*. Due to the harsh conditions(120 8C, DMSO) the initial product 31a or 32a waspartially transformed into the monoesters 31b or 32b,respectively, with full conversion of the starting mate-rial 30. (E)-Configuration has been proposed for 32aand 32b. The authors found that (R)-30 and (S)-30gave different regioselectivities for a chiral ligand of agiven configuration (see data in Scheme 11). For ex-ample (S)-i-PrPhox provided predominantly products
Scheme 9. Substitution versus elimination in some Pd-binap-catalyzed reactions of steroidal allylic esters.
Scheme 10. Chiral Pd-binap-catalyzed synthesis of diamides.
Scheme 11. Catalytic nucleophilic substitution of allylic ace-tate (R)-30.
236 asc.wiley-vch.de � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Synth. Catal. 2010, 352, 231 – 242
REVIEWS Raju Ranjith Kumar and Henri B. Kagan

32 derived from an SN’-like process on (R)-30 (66:34)while (S)-30 gave mainly the formal SN products(87:13). One can predict that (R)-i-PrPhox on (R)-30must necessarily generate (S)-31b with an overall SN/SN’ ratio of 87:13 as mixture of (R)-31a and (R)-31b.It means that the regioselectivity of the nucleophilicsubstitution on enantiopure (R)-30 is to some extentcontrolled by the absolute configuration of i-PrPhox.
Allylic substitution on some oxabicyclic systems 33has been widely studied, especially by Lautenset al.[32] Chiral rhodium complexes in methanol pro-vided a regioselective cleavage according to the con-figuration of the diphosphine ligand 36 in the complex(Scheme 12). The isomeric alcohols formerly derivedfrom the cleavage (a or b types) of the bridgedoxygen, with endo attack of methanol on one or theother side of the double bond. Alcohol 34 is com-pletely free of its isomer 35 and vice versa. These ex-periments have been confirmed by regiodivergent ki-netic resolutions of racemic mixture.[33]
3.3 Epoxide Opening
Gans�uer et al. described in 2007 the regiodivergentreductive opening of chiral epoxides 37 catalyzed bytitanium complexes 40 or ent-41 (Scheme 13).[37] Man-ganese metal and cyclohexadiene are the reducingsystem. Catalyst 40 afforded alcohol 38 (71%, 99%ee) as a major product while the minor alcohol 39(13%) was obtained with 8% ee. With ent-40 it was al-cohol 39 which predominated (76%, 94% ee) against
the regioisomer 38 (10%, 50% ee). The authors pro-vided many additional examples of regioselectiveopening of enantioenriched epoxides which stronglydepend of chiral catalyst 40 or ent- 40.
3.4 C�H Insertions
Doyle et al. brillantly developed in 1996 the con-trolled intramolecular C�H insertion reaction of di-ACHTUNGTRENNUNGazoacetates.[38] The authors used the chiral dirho-ACHTUNGTRENNUNGdium(II) carboxamidate catalysts 42–45 shown inScheme 14. Enantiopure (1S,2R)-cis-cyclohexanol 46was converted into its diazoacetate 47. Various sitesare potentially available for the insertion reaction asshown in Scheme 15. Sites (a)–(d) may lead to lac-tones 48–51. Diazoacetates 47 in presence of catalyst42 gave in excellent yield almost exclusively lactone
Scheme 12. Regioselective methanolysis of an oxabicyclicalkene under catalyst control.
Scheme 13. Regioselective reduction of a chiral epoxide.
Scheme 14. Some chiral dirhodium(II) carboximidate com-plexes.
Adv. Synth. Catal. 2010, 352, 231 – 242 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim asc.wiley-vch.de 237
Regioselective Reactions on a Chiral Substrate Controlled by the Configuration

48, while catalyst ent-42 produced mainly lactone 49.Catalyst 43 gave 49 (82%) and some 50 (7%,Scheme 15).
The pair of enantiomeric catalysts 42 and ent-42achieved a full control of regioselectivity of the car-bene insertion into C�H (a) versus C�H (b). The cat-alyst ent-43 gave an excellent control in the diastereo-selective insertion involving C�H (b) versus C�H (c).Catalyst 44 afforded mainly b-lactone 51 (56%) and amixture of 48 and 49, while ent-44 generated almostexclusively 49.[38]
A similar study has been done with the decomposi-tion of menthyl diazoacetate 52 (Scheme 16). Anachiral rhodium catalyst gave an almost equivalentamount of lactones 53 and 54 (substrate control). Cat-alyst 42 slightly modified the product distribution; but43 was able to almost fully give lactone 53 (matchpair). The mismatch situation with ent-43 catalyst af-forded a mixture of lactones 53, 54 and 55.
The regio- and diastereoselectivities of the insertionreaction of cyclohexyl or menthyl diazoacetates werediscusssed taking into account the structure of the cat-alysts and the conformations of the substrates. Atrend for the insertion into an equatorial C�H bondwas noticed. The results obtained with the cyclohexylsystems were extended to some steroidal diazoace-tates.[39] The experiments were done on various 3-hy-droxy steroids, in Scheme 17 are indicated the product
distributions observed for the decomposition of thediazoacetate of 3b-hydroxy-(5a)-androstane 56. In thepresence of an achiral catalyst Rh2ACHTUNGTRENNUNG(OAc)4 the mixtureof lactones 57–59 was obtained. Catalyst 43 gave b-
Scheme 15. Lactone formation by catalyzed decompositionof diazoester 47.
Scheme 16. Competitive intramolecular C�H insertions inthe decomposition of menthyl diazoacetate 52.
Scheme 17. Competitive carbene insertions in some C�Hbond of the (5a)-androstane series.
238 asc.wiley-vch.de � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Synth. Catal. 2010, 352, 231 – 242
REVIEWS Raju Ranjith Kumar and Henri B. Kagan

lactone 58 as the main product whereas ent-43 gener-ated a different distribution of lactones with 57 as themain component formed by the insertion to the equa-torial C�H bond at the 2-position. Clearly the struc-ture and the absolute configuration of the rhodiumcatalysts play a key role in the regioselectivity anddiastereoselectivity of the insertion reaction.
3.5 Nitroso Diels–Alder Reaction
The nitroso Diels–Alder reaction between an arylni-troso compound and a racemic diene can give rise toa regiodivergent reaction, which was studied byStuder et al.[40] The reaction was catalyzed by a Cu(I)/chiral phosphine complex. The authors also studiedthe effect of various chiral diphosphines in the cyclo-addition on the chiral diene 60. The best results wereobtained with walphos 63 (Scheme 18). An almostperfect control of the regioselectivity was observed,together with the exclusive formation of the anti-ad-ducts.
3.6 Intramolecular Hydroacylation on a Triple Bond
Tanaka and Fu discovered that the parallel kineticresolution of a racemic 4-alkynal 64 catalyzed by aRh(I)/Tol-binap complex generated a 1:1 mixture ofenantioenriched cyclobutanone 65 and cyclopentan-ACHTUNGTRENNUNGone 66.[41] As a consequence they treated alkynal (R)-64 with (R)- or (S)-tolyl-binap rhdodium catalyst andobtained an excess of cyclobutanone (R)-65 or cyclo-pentanone (R)-66, each one in 99% ee (Scheme 19).As outlined by the authors the appropriate choice of
the appropriate enantiomer of Tol-binap dictates if acyclopentanone or a cyclobutanone is formed. Thistype of catalyst selectivity can be categorized as a cat-alyst control in the regioselective addition of an inter-mediate acylrhodium hydride on the triple bond(Scheme 19).
3.7 Alcoholysis of Substituted Succinic Anhydrides
Regiodivergent kinetic resolution of racemic mono-substituted succinic anhydrides was established duringalcoholysis catalyzed by a modified Cinchona alkaloid70.[42] This was a good indication that different regio-selectivity should be achieved by each of the enantio-mers of the catalyst. Since the enantiomer of the cata-lyst 70 was not available the reaction was done oneach enantiomer of the anhydride (Scheme 20). Cata-lyst 70 [(DHQD)2AQN] is based on the dihydroquini-dine fragment. It is well documented that dihydroqui-nine, which is a diastereomer of dihydroquinidine,often leads in asymmetric synthesis to enantiomericproducts.[43] Consequently, the authors used(DHQ)2AQN as a catalyst for alcoholysis of enantio-pure (S)-2-methyl succinic anhydride (S)-67, they re-covered the two monoesters (S)-68 and (S)-69 in aratio 6:94. This is the opposite regioselectivity to thatobtained with (DHQD)2AQN 70 (see Scheme 20).
Methanolysis of racemic lactone 71 mediated byquinidine gave quantitatively the mixture of mono-methyl esters 72 and 73 (Scheme 21).[44] The stereo-chemical analysis done by Bolm et al. indicated that71 and ent-71 generated regioisomeric products 72and 73 (99% ee versus 91% ee, respectively). It meansScheme 18. Regioselective nitroso Diels–Alder reaction.
Scheme 19. Regioselective intramolecular hydroacylation.
Adv. Synth. Catal. 2010, 352, 231 – 242 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim asc.wiley-vch.de 239
Regioselective Reactions on a Chiral Substrate Controlled by the Configuration

that one expects quite high regioselectivity in themethanolysis of 71 with formation of 72 if quinidine isthe catalyst, while quinine (a mimic of ent-quinidine)should mainly produce 73, in a process where thechiral base can be taken in catalytic amount as estab-lished in similar cases.[45]
3.8 Miscellaneous
Diols lead to various kinds of transformation in thepresence of a chiral catalyst. Most of the reactionshave been performed on a racemic mixture and maygive rise to regiodivergent kinetic resolutions. Anearly report noticed that dehydrogenation of chiralunsymmetrical a,w-diols catalyzed by a chiral com-plex should give rise to the formation of lactones withdifferent regioselectivity (Scheme 22).[46] Some experi-ments were carried out with a ruthenium/diop catalyston (R)-74. A small difference of regioselectivity hasbeen evidenced between the two enantiomeric cata-lysts.
Several racemic 1,2-diols have been monoacylatedin presence of a chiral catalyst.[47–50] It can be deducedfrom the product distribution of the kinetic resolutionthat some regioselectivities driven by the configura-tion of the catalyst are expected for a reaction on achiral diol.[49,50] An interesting study compared themonoacetylation of erythromycin A in the presenceof catalytic amounts of N-methylimidazole or a smallpeptide analogue. A very large site selectivity was no-ticed, however there were no comparisons betweenthe two enantiomers of the catalytic peptide.[49]
Catalytic silylation of racemic 1,2-diols in the pres-ence of an organocatalyst (a peptide derivative) canbe highly site and enantioselective.[50] It gives somehope that, in such a type of reaction performed on achiral diol, the regioselectivity could be closely linkedto the absolute configuration of the catalyst.
There are various ways to screen enantioselectivecatalyst candidates by high throughput methods.[51]
The fast screening of new chiral catalysts for selectivereactions on achiral substrates with two enantiotopicsites (such as in Scheme 1, for example) has been pio-neered by Reetz et al.[52] These authors used thetransformation of pseudo-meso substrates 77 orpseudo-prochiral substrates 78 into chiral products(Scheme 23). For example, enantiopure pseudo-mesodiester 79 was monohydrolyzed in the presence of alipase to generate predominantly monoalcohol 80
Scheme 20. Regioselectivity in catalytic alcoholysis of 2-methyl succinic anhydride.
Scheme 21. Regiodivergent methanolysis of a cyclic racemicanhydride.
Scheme 22. Formation of lactones from chiral diols.
240 asc.wiley-vch.de � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Synth. Catal. 2010, 352, 231 – 242
REVIEWS Raju Ranjith Kumar and Henri B. Kagan

with loss of deuterium. The regioselectivity can bethen easily measured by mass spectroscopy on smallamounts because of the deuterium labelling. The abil-ity of the catalyst to desymmetrize the meso-diacetateof cis-cyclopentene 3,5-diol is then known.
4 Conclusions
The control of regioselective transformations of chiralcompounds is an important problem which oftenneeds several steps. In this review it was shown thatthe use of a chiral catalyst with the suitable absoluteconfiguration is a straightforward approach to get thedesired reaction on one or the other site. Much usefulinformation has been afforded when a racemic mix-ture gives rise to a regiodivergent kinetic resolution.
Usually the regioselectivity involves two sites of thesame kind, for example, two double bonds(Scheme 6), two carbonyl groups in an anhydride(Scheme 20) or two ester functionalities (Scheme 23).The regioselectivity can be observed in the reactionof a dissymmetric functional group as in the ringopening of some epoxides (Scheme 13) or in theDiels–Alder addition on a nitroso compound(Scheme 18). More unusual is the chiral catalyst con-trol between two different kinds of reactive centerslocated at different sites of the chiral substrate as thecarbene insertion into a C�H bond versus a C=C cy-clopropanation (see Scheme 5 for the correspondingdivergent kinetic resolution scenario).
An unselective catalytic reaction because of a dualreactivity (for example, epoxidation and allylic oxida-tion)[53] should also be improved by the chiral catalystcontrol. It will be interesting to see to what extentthis methodology will apply to polyhydroxylated com-pounds, for example, to trigger their regioselective ox-idation or acylation. In conclusion, one can predictthat the spectacular advances in enantioselective cat-
alysis will indirectly contribute to a great extent to setup new approaches concerning selective transforma-tions of chiral compounds under the chiral catalystcontrol.
Note Added in Proof
A highly regiodivergent ring opening of chiral aziri-dines by trimethylsilyl azide has been recently pub-lished.[55] This reaction is catalyzed by a chiral Y com-plex and provides 1,2-diamine derivatives.
Acknowledgements
We thank Universit� Paris-Sud and CNRS for financial sup-port. One of us (R.R.K.) acknowledges Institut des Substan-ces Naturelles du CNRS (Gif-sur-Yvette) for its financial sup-port.
References
[1] Asymmetric Organic Reactions, Vol. 5, Catalysis, (Ed.:J. D. Morrison), Academic Press, New York, 1985.
[2] Asymmetric Catalysis in Organic Synthesis, (Ed.: R.Noyori), Wiley, New York, 1994.
[3] Catalytic Asymmetric Synthesis, (Ed.: I. Ojima), 2nd
edn., Wiley-VCH, New York, 2001.[4] Comprehensive Asymmetric Catalysis, Vols. I–III, (Eds.:
E. N. Jacobsen, A. Pfaltz, H. Yamamoto), Springer,Heidelberg, New York, 1999.
[5] Stereo-Differentiating Reactions, (Eds.: Y. Izumi, A.Tai), Kodansha, Tokyo and Academic Press, New York,1977.
[6] H. B. Kagan, J. C. Fiaud, in: Topics in Stereochemistry,(Eds.: N. L. Allinger, E. L. Eliel), Wiley, New York,1988 ; Vol. 18, pp 249 – 330.
[7] E. Vedejs, M. Jure, Angew. Chem. 2005, 117, 4040 –4069; Angew. Chem. Int. Ed. 2005, 44, 3974 – 4001.
[8] A. H. Hoveyda, M. T. Didiuk, Curr. Org. Chem. 1998,2, 489 – 526.
[9] J. M. Keith, J. F. Larrow, E. N. Jacobsen, Adv. Synth.Catal. 2001, 343, 5 – 26.
[10] The expression enantioselective synthesis is widely usedand does not always refer to a key step involving anasymmetric synthesis.[11] For example, multi-step totalsyntheses of natural products are commonly qualifiedas enantioselective because the final product has beensynthesized as a single enantiomer. The expression“enantioselectivity” will be used in the present articlewhen meaning that an achiral starting material is trans-formed in one step into an enantioenriched product.Often a given reaction involves several consecutivesteps and one is stereodeterminating. We will not usethe word “selection” in a mechanistic discussion butonly when comparing the starting material to the prod-uct (see some examples in Scheme 1).
Scheme 23. Enantiopure pseudo-meso or pseudo-prochiralsubstrates for evaluation of chiral catalysts.
Adv. Synth. Catal. 2010, 352, 231 – 242 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim asc.wiley-vch.de 241
Regioselective Reactions on a Chiral Substrate Controlled by the Configuration

[11] For the misuse of stereochemical words see: a) K.Mislow, Chirality 2002, 14, 126 – 134; b) E. L. Eliel,S. H. Wilen, L. N. Mander, Stereochemistry of OrganicCompounds, Wiley, New York, 1994.
[12] J. P. Guett�, A. Horeau, Bull. Soc. Chim. Fr. 1967,1747 – 1752.
[13] S. El Baba, J. C. Poulin, H. B. Kagan, Tetrahedron 1984,40, 4275 – 4284.
[14] a) C. Bolm, G. Schlingloff, K. Weickhard, Angew.Chem. 1994, 106, 1944 – 1946; Angew. Chem. Int. Ed.Engl. 1994, 33, 1848 – 1849; b) C. Bolm, G. Schlingloff,J. Chem. Soc. Chem. Commun. 1995, 1247 – 1248.
[15] a) A. Gusso, C. Baccin, F. Pinna, G. Strukul, Organo-metallics 1994, 13, 3442 – 3451; b) G. Strukul, Angew.Chem. 1998, 110, 1256 – 1267; Angew. Chem. Int. Ed.1998, 37, 1199 – 1209.
[16] A. Watanabe, T. Uchida, K. Ito, T. Katsuki, Tetrahe-dron Lett. 2002, 43, 4481 – 4485.
[17] H. B. Kagan, Croat. Chem. Acta 1996, 69, 669 – 680.[18] H. B. Kagan, Tetrahedron 2001, 57, 2449 – 2459.[19] T. Honda, N. Sano, K. Kanai, Heterocycles 1995, 41,
425 – 429.[20] S. F. Martin, M. R. Spaller, S. Liras, B. Hartmann, J.
Am. Chem. Soc. 1994, 116, 4493 – 4494.[21] M. P. Doyle, A. B. Dyatkin, A. V. Kalinin, D. A.
Ruppar, J. Am. Chem. Soc. 1995, 117, 11021 – 11022.[22] M. S. Visser, A. H. Hoveyda, Tetrahedron 1995, 51,
4383 – 4394.[23] G. Dawson, C. A. Durrant, G. G. Kirk, R. J. Whitby,
R. V. H. Jones, M. C. H. Standen, Tetrahedron Lett.1997, 38, 2335 – 2338.
[24] S. Masamune, W. Choy, J. S. Peterson, L. R. Sita,Angew. Chem. 1985, 97, 1 – 31; Angew. Chem. Int. Ed.Engl. 1985, 24, 1 – 30.
[25] A. Horeau, H. B. Kagan, J. P. Vigneron, Bull. Soc.Chim. Fr. 1968, 3795 – 3797.
[26] D. L. Hughes, M. Palucki, N. Yasuda, R. A. Reamer,P. J. Reider, J. Org. Chem, 2002, 67, 2762 – 2768, andreferences cited therein.
[27] T. Hayashi, A. Yamamoto, Y. Ito, J. Chem. Soc. Chem.Commun. 1986, 1090 – 1092.
[28] O. Loiseleur, M. C. Elliott, P. von Matt, A. Pfaltz, Helv.Chim. Acta 2000, 83, 2287 – 2294.
[29] H. Daimon, R. Ogawa, S. Itagaki, I. Shimizu, Chem.Lett. 2004, 33, 1222 – 1223.
[30] G. R. Cook, S. Sankaranarayanan, Org. Lett. 2001, 3,3531 – 3533.
[31] O. Jacquet, J. Y. Legros, M. Coliboeuf, J.-C. Fiaud, Tet-rahedron 2008, 64, 6530 – 6536.
[32] R. Webster, C. Bçig, M. Lautens, J. Am. Chem. Soc.2009, 131, 444 – 445.
[33] For examples of regiodivergent kinetic resolutions ofallylic epoxides see refs.[34–36]
[34] F. Bertozzi, P. Crotti, F. Macchia, M. Pineschi, B. L.Feringa, Angew. Chem. 2001, 113, 956 – 958; Angew.Chem. Int. Ed. 2001, 40, 930 – 932.
[35] M. Pineschi, F. Del Moro, F. Crotti, V. Di Bussolo, F.Macchia, J. Org. Chem. 2004, 69, 2099 – 2105.
[36] a) R. Millet, A. Alexakis, Synlett 2007, 435 – 438; b) R.Millet, A. Alexakis, Synlett 2008, 1797 – 1800.
[37] A. Gans�uer, C.-A. Fan, F. Keller, J. Keil, J. Am.Chem. Soc. 2007, 129, 3484 – 3485.
[38] M. P. Doyle, A. V. Kalinin, D. G. Ene, J. Am. Chem.Soc. 1996, 118, 8837 – 8846.
[39] M. P. Doyle, S. B. Davies, E. J. May, J. Org. Chem.2001, 66, 8112 – 8119.
[40] C. K. Jana, A. Studer, Angew. Chem. 2007, 119, 6662 –6664; Angew. Chem. Int. Ed. 2007, 46, 6542 – 6544.
[41] K. Tanaka, G. C. Fu, J. Am. Chem. Soc. 2002, 125,8078 – 8079.
[42] Y. Chen, L. Deng, J. Am. Chem. Soc. 2001, 123, 11302 –11303.
[43] S. Zhang, C. Girard, H. B. Kagan, Tetrahedron: Asym-metry 1995, 6, 2637 – 2640.
[44] C. Bolm, I. Schiffers, C. L. Dinter, L. Defr�re, A. Ger-lach, C. Raabe, Synthesis 2001, 1719 – 1730.
[45] C. Bolm, I. Schiffers, C. L. Dinter, A. Gerlach, J. Org.Chem. 2000, 65, 6984 – 6991.
[46] Y. Ishii, K. Osakada, T. Ikariya, M. Saburi, S. Yoshika-wa, J. Org. Chem. 1986, 51, 2034 – 2039.
[47] I. Iwasaki, T. Maki, W. Nakashima, O. Onomura, Y.Matsumura, Org. Lett. 1999, 1, 969 – 972.
[48] C. Mazet, S. Roseblade, V. Kçhler, A. Pfaltz, Org. Lett.2006, 8, 1879 – 1882.
[49] C. A. Lewis, S. J. Miller, Angew. Chem. 2006, 118,5744 – 5747; Angew. Chem. Int. Ed. 2006, 45, 5616 –5619.
[50] Y. Zhao, A. W. Mitra, A. H. Hoveyda, M. L. Snapper,Angew. Chem. 2007, 119, 8623 – 8626; Angew. Chem.Int. Ed. 2007, 46, 8471 – 8474.
[51] a) M. Tsukamoto, H. B. Kagan, Adv. Synth. Catal. 2002,344, 453 – 463; b) M. T. Reetz, Angew. Chem. 2002, 114,1391 – 1394; Angew. Chem. Int. Ed. 2002, 41, 1335 –1338.
[52] M. T. Reetz, M. H. Becker, H. W. Klein, D. Stçckigt,Angew. Chem. 1999, 111, 1872 – 1875; Angew. Chem.Int. Ed. 1999, 38, 1758 – 1761.
[53] The oxidation of enantiomeric aryl-substituted allylicalcohols by iodosyl benzene catalyzed by a chiral Fe-ACHTUNGTRENNUNG(III) porphyrin complex has been described.[54] There isa competition between the C=C epoxidation and the al-lylic oxidation into enone. The chemoselectivity isslightly different according to the absolute configura-tion of the substrate.
[54] W. Adam, S. Prikhodovski, K. J. Roschmann; C. R.Saha-Mçller, Tetrahedron: Asymmetry 2001, 12, 2677 –2681.
[55] B. Wu, J. R. Parquette, T. V. RajanBabu, Science 2009,326, 1662.
242 asc.wiley-vch.de � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Adv. Synth. Catal. 2010, 352, 231 – 242
REVIEWS Raju Ranjith Kumar and Henri B. Kagan




















