
Angiotensin II type 1 and bradykinin B2 receptors expressed in early stage epithelial cells derived...
10
Angiotensin II Type 1 and Bradykinin B2 Receptors Expressed in Early Stage Epithelial Cells Derived From Human Embryonic Stem Cells ZHENHUA HUANG, 1 JUN YU, 1 PAUL TOSELLI, 1 JAG BHAWAN, 2 VASANTHI SUDIREDDY, 2 LINDA TAYLOR, 1 AND PETER POLGAR 1 * 1 Department of Biochemistry, Boston University School of Medicine, Boston, Massachusetts 2 Department of Dermatology, Boston University School of Medicine, Boston, Massachusetts When human embryonic stem (hES) cells were placed into suspension culture followed by culture on BD matrigel TM coated plates in the presence of medium conditioned by NIH-3T3 cells, they differentiated into cells of which more than 95% stained positive for keratin 8 by day 14, demonstrating that the hES cells had committed to an epithelial lineage. Approximately 50% of the keratin 8 staining cells became positive for cytokeratin 14 after 26 days. Binding experiments supported by real time PCR showed that the expression of bradykinin B2 (BKB2) and angiotensin II type 1 (AT1) receptors accompanied this differentiation. Neither receptor was expressed in the pluripotent H9 stem cells. However, transduction of the hES cells with lentivirus containing BKB2 or AT1R cDNA resulted in ligand binding and ERK1/2 activation but not in Gai or Gaq coupled signaling. In the differentiated cells, both BKB2R and AT1R were expressed constitutively and effected typical Gai and Gaq coupled signaling characterized by the release of arachidonate, generation of inositol phosphates, and Ca 2þ mobilization. These signals were abolished by the receptor antagonists, losartan, and HOE 140. Angiotensin II and bradykinin also stimulated the phosphorylation of ERK1/2, JNK1/2, and p70S6 in the differentiated cells. Our results demonstrate that human embryonic stem cells can be differentiated effectively into the epithelial lineage and that when differentiated express functional, signaling AT1 and BKB2 receptors. J. Cell. Physiol. 211: 816–825, 2007. ß 2007 Wiley-Liss, Inc. Human embryonic stem (hES) cells are immortal and pluripotent. These cells under appropriate conditions remain undifferentiated and retain a normal karyotype. Both mouse and human ES cells can be differentiated in vitro into various cell types (Thomson et al., 1998; Amit et al., 2000; Itskovitz-Eldor et al., 2000; Reubinoff et al., 2000; Schuldiner et al., 2000; Watt and Hogan, 2000). When injected into a host blastocyst, ES cells contribute to a variety of cell types (Bradley et al., 1984; Nagy et al., 1993). This potential defines the pluripotency of these cells. Theoretically, these cells can produce more restricted progenitor cells. In fact, protocols for directed differentiation into a wide variety of cell types are now being established. In addition to mice, pluripotent, embryonic stem cells have been isolated from a number of vertebrates, including chicken (Pain et al., 1996), rabbit (Graves and Moreadith, 1993), pig (Wheeler, 1994), cow (Cherny et al., 1994), non-human primates (Thomson et al., 1995), and most recently, humans (Shamblott et al., 1998; Thomson et al., 1998). An unlimited supply of cells open to a number of genetic manipulations and possessing unlimited differentiation capacity is an invaluable advantage of the ES cells as tools for gene therapy and transplantation therapy. The ability to genetically manipulate hES cells is gradually resulting in the purification of specific cell types. Human ES cells have potential as an in vitro source of cells for cellular manipulation toward various pathologies including cardiovascular and pulmonary diseases. Differentiation toward a specific lineage has been achieved by various procedures such as transfection of ES cells with transcription factors; exposure of hES cells to selected growth factors; or coculture of hES cells with cell types capable of lineage induction (Pera and Trounson, 2004; Trounson, 2004). hES cells may be induced to form the lineage of interest by a combination of growth factors and/or their antagonists (Loebel et al., 2003). Another culture system for derivation of differentiated hESCs involves the formation of embryoid bodies (EBs)(Itskovitz-Eldor et al., 2000). In vitro, the aggregation of hES cells into EBs allows for the spontaneous differentiation of hES into progeny representing various lineages (Levenberg et al., 2002). In this report, we develop an effective strategy to differentiate hES cells in vitro along the epithelial lineage as substantiated by the expression of cytokeratin 8 and 14 and the appearance of desmosomes among the differentiated cells under the EM. We also observe the accompanying changes in the expression of two G-protein coupled receptors, bradykinin B2 (BKB2) and angiotensin type 1 (AT1). We find that both the receptors are expressed and evoke Gai and Gaq coupled signaling and mediate phosphorylation of the kinases, ERK1/2, JNK1/2, and p70S6 in the differentiated hES cells, while no expression of these receptors is observed in undifferentiated hES cells. Materials and Methods Materials Antibodies were purchased from the following vendors: phospho-ERK, ERK, phospho-JNK, JNK, phospho-p70 S6 kinase, p70 S6 kinase were from Cell Signaling Technologies (Beverly, MA); SSEA-4, TRA-1-60, Contract grant sponsor: NIH; Contract grant number: HL25776. *Correspondence to: Dr. Peter Polgar, Department of Biochemistry, Boston University School of Medicine, 80 East Concord Street, Boston, MA 02118. E-mail: [email protected] Received 25 August 2006; Accepted 21 November 2006 DOI: 10.1002/jcp.20985 ORIGINAL ARTICLE 816 Journal of Journal of Cellular Physiology Cellular Physiology ß 2007 WILEY-LISS, INC.
Transcript of Angiotensin II type 1 and bradykinin B2 receptors expressed in early stage epithelial cells derived...

ORIGINAL ARTICLE 816J o u r n a l o fJ o u r n a l o f
CellularPhysiologyCellularPhysiology
Angiotensin II Type 1 andBradykinin B2 ReceptorsExpressed in Early StageEpithelial Cells Derived FromHuman Embryonic Stem Cells
ZHENHUA HUANG,1 JUN YU,1 PAUL TOSELLI,1 JAG BHAWAN,2 VASANTHI SUDIREDDY,2LINDA TAYLOR,1 AND PETER POLGAR1*1Department of Biochemistry, Boston University School of Medicine, Boston, Massachusetts2Department of Dermatology, Boston University School of Medicine, Boston, Massachusetts
When human embryonic stem (hES) cells were placed into suspension culture followed by culture on BD matrigelTM coated plates in thepresence of medium conditioned by NIH-3T3 cells, they differentiated into cells of which more than 95% stained positive for keratin 8 byday 14, demonstrating that the hES cells had committed to an epithelial lineage. Approximately 50% of the keratin 8 staining cells becamepositive for cytokeratin 14 after 26 days. Binding experiments supported by real time PCR showed that the expression of bradykinin B2(BKB2) and angiotensin II type 1 (AT1) receptors accompanied this differentiation. Neither receptor was expressed in the pluripotent H9stem cells. However, transduction of the hES cells with lentivirus containing BKB2 or AT1R cDNA resulted in ligand binding and ERK1/2activation but not in Gai or Gaq coupled signaling. In the differentiated cells, both BKB2R and AT1R were expressed constitutively andeffected typical Gai and Gaq coupled signaling characterized by the release of arachidonate, generation of inositol phosphates, and Ca2þ
mobilization. These signals were abolished by the receptor antagonists, losartan, and HOE 140. Angiotensin II and bradykinin alsostimulated the phosphorylation of ERK1/2, JNK1/2, and p70S6 in the differentiated cells. Our results demonstrate that human embryonicstem cells can be differentiated effectively into the epithelial lineage and that when differentiated express functional, signaling AT1 andBKB2 receptors.
J. Cell. Physiol. 211: 816–825, 2007. � 2007 Wiley-Liss, Inc.
Contract grant sponsor: NIH;Contract grant number: HL25776.
*Correspondence to: Dr. Peter Polgar, Department of Biochemistry,Boston University School of Medicine, 80 East Concord Street, Boston,MA 02118. E-mail: [email protected]
Received 25 August 2006; Accepted 21 November 2006
DOI: 10.1002/jcp.20985
Human embryonic stem (hES) cells are immortal andpluripotent. These cells under appropriate conditions remainundifferentiated and retain a normal karyotype. Both mouseand human ES cells can be differentiated in vitro into various celltypes (Thomson et al., 1998; Amit et al., 2000; Itskovitz-Eldoret al., 2000; Reubinoff et al., 2000; Schuldiner et al., 2000; Wattand Hogan, 2000). When injected into a host blastocyst, ES cellscontribute to a variety of cell types (Bradley et al., 1984; Nagyet al., 1993). This potential defines the pluripotency of thesecells. Theoretically, these cells can produce more restrictedprogenitor cells. In fact, protocols for directed differentiationinto a wide variety of cell types are now being established. Inaddition to mice, pluripotent, embryonic stem cells have beenisolated from a number of vertebrates, including chicken (Painet al., 1996), rabbit (Graves and Moreadith, 1993), pig(Wheeler, 1994), cow (Cherny et al., 1994), non-humanprimates (Thomson et al., 1995), and most recently, humans(Shamblott et al., 1998; Thomson et al., 1998). An unlimitedsupply of cells open to a number of genetic manipulations andpossessing unlimited differentiation capacity is an invaluableadvantage of the ES cells as tools for gene therapy andtransplantation therapy. The ability to genetically manipulatehES cells is gradually resulting in the purification of specific celltypes. Human ES cells have potential as an in vitro source of cellsfor cellular manipulation toward various pathologies includingcardiovascular and pulmonary diseases.Differentiation toward a specific lineage has been achieved byvarious procedures such as transfection of ES cells withtranscription factors; exposure of hES cells to selected growthfactors; or coculture of hES cells with cell types capable oflineage induction (Pera and Trounson, 2004; Trounson, 2004).hES cells may be induced to form the lineage of interest by acombination of growth factors and/or their antagonists (Loebelet al., 2003). Another culture system for derivation ofdifferentiated hESCs involves the formation of embryoid bodies
� 2 0 0 7 W I L E Y - L I S S , I N C .
(EBs)(Itskovitz-Eldor et al., 2000). In vitro, the aggregation ofhES cells into EBs allows for the spontaneous differentiation ofhES into progeny representing various lineages (Levenberget al., 2002).In this report, we develop an effective strategy to differentiatehES cells in vitro along the epithelial lineage as substantiated bythe expression of cytokeratin 8 and 14 and the appearance ofdesmosomes among the differentiated cells under the EM. Wealso observe the accompanying changes in the expression oftwo G-protein coupled receptors, bradykinin B2 (BKB2) andangiotensin type 1 (AT1). We find that both the receptors areexpressed and evoke Gai and Gaq coupled signaling andmediate phosphorylation of the kinases, ERK1/2, JNK1/2, andp70S6 in the differentiated hES cells, while no expression ofthese receptors is observed in undifferentiated hES cells.
Materials and MethodsMaterials
Antibodies were purchased from the following vendors: phospho-ERK,ERK, phospho-JNK, JNK, phospho-p70 S6 kinase, p70 S6 kinase werefrom Cell Signaling Technologies (Beverly, MA); SSEA-4, TRA-1-60,

H U M A N E M B R Y O N I C S T E M C E L L E P I T H E L I A L L I N E A G E 817
and cytokeratin 14 were from Chemicon (Temecula, CA); andcytokeratin 8 was from Dako (Carpinteria, CA). Angiotensin II,Bradykinin, Hoe 140, and PD123,319 were obtained from Sigma(St Louis, MO). Losartan was from Cayman Chemical Company (AnnArbor, MI) and hLIF from Chemicon (Temecula, CA). KO DMEM, KOserum replacement, MEM nonessential amino acids, b-mercaptoethanol, penicillin/ streptomycin, 0.05% trypsin-EDTA,Gluta-MAXTM, and hrbFGF were from Invitrogen (Carlsbad, CA).Plasmanate was from National Hospital Specialties (Hackensack, NJ).Basement Membrane MatrigelTM Matrix, Growth Factor Reduced(GFR) was from Fisher Scientific (Springfield NJ). The humanembryonic stem cell line, H9, was obtained from WiCell (Madison, WI).The RNeasy kit and plasmid purification kits were from Qiagen(Valencia, CA). Lipofectamine 2000 and Superscript II were fromInvitrogen. 3H-AngII (52.5 Ci/mmol) and 3H-BK (114 Ci/mmol) werepurchased from Perkin Elmer Life Sciences (Boston, MA).
Cell culture
Undifferentiated hES cells (H9) were maintained on irradiated mouseembryo fibroblast (MEF) feeder layers in KO-DMEM, 10% KnockoutSerum Replacement, 10% plasmanate, 2 mM Gluta-MAXTM, 0.1 mMMEM non-essential amino acids, 50 units/ml penicillin, 50 mg/mlstreptomycin, 12 ng/mL hLIF, 0.1 mMb-mercaptoethanol, and 10 ng/mlhrbFGF as described by Cowan et al. (2004). Cells were passaged bydetachment with 0.05% trypsin-EDTA every 4 days and replated onfresh feeder layers (Cowan et al., 2004, 2005; Dravid et al., 2006;Mitalipova and Palmarini, 2006). Unless otherwise stated, detachmentof the hES cells was carried out by this procedure. All data from theundifferentiated hES cells were obtained from feeder-layer freecultures. The feeder layer free hES cells were cultured following aprocedure described by Xu and coworkers (2001) which maintainsthese cells in their undifferentiated state for extended time. In thisprocedure, the feeder layer free hES cells are cultured in conditionedmedium (CM) obtained from MEF cultures at a 24-h cycle. To obtainfeeder-free hES cultures, the irradiated (non-dividing) MEFs wereseparated from the hES cells by differential adhesion. The hES cellscultured on the MEFs were detached and resuspended in 10–12 ml ofthe conditioned medium and plated on 10-cm plates. After 2 h in a378C, 5% CO2 incubator, the MEFs adhered to the plate, while most ofthe hES cells remained suspended. The hES cells in suspension wereseeded onto 0.1% gelatin coated tissue culture plates. The feeder-layerfree hES cells remained undifferentiated for at least 1 month. Theretention of the undifferentiated state was further confirmed bySSEA-4 and TRA-1-60 staining as reported by (Xu et al., 2001).The MEF, IMR90, and HEK-293T cells were cultured at 378C with5% CO2 in 90% Dulbecco’s Modified Eagle’s medium (DMEM), 10%fetal bovine serum (FBS), 2 mM L-glutamine, 50 units/ml penicillin, and50 mg/ml streptomycin.
Plasmid constructs, virus production, and transduction of cells
pCD/NL-BH�DDD helper plasmid, pLTR-G Env plasmid, and self-inactivating (SIN) pNL-EGFP/CMV-WPREDU3 vectors were kindlyprovided by Jakob Reiser (Gene Therapy Program, Department ofMedicine, Louisiana State University Health Sciences Center NewOrleans, New Orleans, LA). The woodchuck hepatitis viruspost-transcriptional regulatory element (WPRE) that increasestransgene expression by stabilizing mRNAs in cis (Donello et al., 1998;Ramezani et al., 2000) is present in EGFP/CMV-WPREDU3 whichcontains the enhanced green fluorescent protein (EGFP) gene. Tocreate pNL-AT1R/CMV-WPREDU3 (pNL-AT1R) or pNL-BKB2R/CMV-WPREDU3 (pNL-BKB2R), the NheI (blunt)/Xhol EGFP fragmentof pNL-EGFP/CMV-WPREDU3 was removed and substituted with anXbal(blunt)/Xhol fragment containing AT1R or BKB2R sequences frompcDNA3.1(�) AT1R or pcDNA3.1(�) BKB2R. Virus particles wereproduced in 293 T cells by transient co-transfection of a three-plasmidexpression system using Lipofectamine 2000 as described by (Plutaet al., 2005). Briefly, 293 T cells were plated in 6-well plates (6� 105
cells/well) and, 24 h later, 5 mg of vector plasmid DNA, 3.5 mg of pCD/NL-BH�DDD helper plasmid DNA and 1.75 mg of pLTR-G Env plasmidDNA were added. Approximately 48 h later, virus-containingsupernatant was harvested, filtered through a 0.45 mM filter unit,aliquoted, and frozen at �808C. For transductions, feeder layer freehES cells (3 days after seeding at 5� 104 cells/well) in 6-well plateswere infected in a total volume of 2 ml supernatant and 8 mg/mlpolybrene. Analyses were carried out 3 days after infection.
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
Differentiation of hES cells to epithelial lineage
Feeder layer free hES cells were placed in non-adherent 6-well dishes inthe presence of either (a) differentiation medium, F12/DMEM (1:2)with 15% fetal bovine serum (FBS), 2 mM Gluta-MAXTM, 0.1 mM MEMnon-essential amino acids, 50 units/ml penicillin, and 50 mg/mlstreptomycin or (b) conditioned differentiation medium:differentiation medium conditioned by exposure to non-dividing(X-irradiated) NIH-3T3 cells at 75% confluency. The 3T3 cells wererendered non-diving in order to maintain a constant cell numberthroughout the conditioning and to avoid excessive consumption ofculture medium nutrients by highly dividing 3T3 cells. The medium wascollected from the 3T3 cell cultures every 24 h. The EBs were exposedto fresh conditioned differentiation medium daily by allowing thesuspended cells to settle in a conical tube. After 6 days in suspension,the EBs were plated on gelatin- or BM matrix-coated culture dishes asindicated, and maintained in the presence of either differentiationmedium or conditioned differentiation medium. The cell outgrowthsfrom the EBs were followed by light microscopy. Reference to thenumber of days of differentiation includes the 6 days in which the EBswere maintained in suspension. For example, the 7th day ofdifferentiation refers to the EBs in suspension for 6 days and thencultured in the attached state for one day.
Immunohistochemistry
For keratin 8 detection, the hES cells and the differentiated cells werecultured on gelatin coated 24-well plates. The cells were fixed withformalin for 24 h. The cells were washed twice with TBS (Tris-bufferedsaline) followed by blocking with ‘‘peroxidase block’’ (Dako,Carpinteria, CA) for 5 min. After three washes with TBS, the cells werestained with keratin 8 monoclonal antibody at a dilution of 1:20 for30 min. The cells were then washed twice with TBS and incubated inHRP (Horseradish peroxidase) labeled secondary antibody (Dako) for30 min. After two washes with TBS, the cells were stained withchromogen substrate AEC (3-amino, 9 ethyl-carbazole) (Dako). Thestained cells were visualized with a light microscope. For keratin 14detection, the hES cells and the differentiated cells (obtained withdifferent treatments) were detached and cultured in gelatin-coated 24-well plates for 3 days, and then fixed with 3% paraformaldehyde in PBSfor 30 min at room temperature followed by two washes with PBS. Thecells were then permeabilized with 0.2% triton for 5 min followed bytwo washes with PBS. The cultures were then blocked with 10%normal goat serum blocking solution (Zymed1, Invitrogen, Carlsbad,CA) for 10 min followed by two washes with 0.2% gelatin in PBS(PBS-gel). The cultures were incubated with antibody to keratin 14 for45 min diluted in 5% PBS-gel followed by four 5-min washes withPBS-gel. The cells were then incubated sequentially with secondantibody (goat anti-mouse IgG, Oregon Green 488 from MolecularProbesTM, Invitrogen, Carlsbad, CA), diluted in PBS-gel for 45 minfollowed by four 5-min washes with PBS-gel. The permeabilized cellswere mounted with permanent mounting medium (from VectorLaboratories, Inc., Burlingame, CA).
Transmission electron microscopy (TEM)
The WT hES cells and differentiated cells on 6-well plates were washedtwice with phosphate buffered saline. The cells were then fixed with4.3% glutaraldehyde (Polysciences; Warrington, PA) in 0.03 M sodiumbarbital–sodium acetate buffer (pH 7.4) containing 0.07 M potassiumchloride for 1 h. The samples were then rinsed with phosphate bufferedsaline three times for 15 min each, and post-fixed in a solution of 1%osmium tetroxide (Ted Pella; Redding, CA) in 0.03 M sodium barbital–sodium acetate buffer (pH 7.4), 0.07 M potassium chloride, for 1–2 h atroom temperature. This was followed by dehydration in a graded seriesof ethanol solutions starting with 70% ethanol, embedded in a 1:1mixture of Araldite 502 and dodecenyl succinic anhydride at 608C.After polymerization of the Araldite mixture, sections were cut on anLKB Ultratome V. Ribbons of sections showing gray, silver, or slightlygold interference colors were picked up on uncoated 200 mesh AtheneThin Bar copper grids. Electron micrographs were obtained fromsections stained with uranyl acetate followed by lead citrate.
Calcium mobilization
Mobilization of Ca2þ was determined as reported previously withsome modifications (Prado et al., 1997). The WT hES cells, infected hEScells, and 14-day differentiated cells were detached and washed two

818 H U A N G E T A L .
times in physiological buffer solution (140 mmol/L NaCl, 5 mmol/L KCl,1 mmol/L MgCl2, 10 mmol/L glucose, 0.9 mmol/L CaCl2, 15 mmol/LHEPES, 0.1% BSA). The cells were resuspended at 2� 107 cells/ml andincubated with Fura-2/AM for 30 min (2 mmol/L final concentration).After 30 min, the cell suspension was diluted five times withphysiological buffer solution and incubated for another 15 min. Cellswere pelleted and resuspended at 1� 106 cells/ml. Ca2þ mobilizationexperiments were performed using a Hitachi 2500 FluorescenceSpectrophotometer.
Arachidonic acid release
The hES cells and 14-day differentiated cells in 24-well plates werepre-labeled with [3H]-arachidonate (0.2mCi/well) for 18 h as describedpreviously (Prado et al., 1997). Cells were washed with MEM containing2 mg/ml bovine serum albumin, and incubated with stimulant for 20 minat 378C. Medium was removed and centrifuged at 800g. Radioactivity inthe supernatant was determined in a b-counter.
Phosphoinositide turnover
For the measurement of phosphoinositide (PI) turnover, cells in12-well plates were incubated with 1 mCi/ml of myo-[3H]-inositol in1 ml of growth medium for 18 h and the levels of inositol phosphatewere determined as previously described (Prado et al., 1997). Briefly,10 min prior to ligand stimulation, cells were exposed to DMEMcontaining 20 mmol/L LiCl2 and 20 mmol/L HEPES, pH 7.4. Cells werethen exposed to 100 nmol/L ligand for 30 min at 378C. Incubation wasterminated by the addition of 0.5 ml of 10 mmol/L ice-cold formic acid.Cells were scraped and the formic acid soluble material isolated bycentrifugation and neutralized with 10 ml 5 mmol/L sodiumtetraborate. Total [3H]IPs were extracted using a Dowex AG 1-X8formate resin in an anion exchange column and eluted with 2 mol/Lammonium formate, pH 5.0, as described. Following the addition of4 ml of Ecolite scintillation fluid (ICN Biomedical, Inc., Aurora, OH),samples were counted for radioactivity in a b-counter.
Western blot analysis
Cells cultured in 6-well plates were either unstimulated or incubatedwith desired concentrations of BK or AngII for indicated time intervalsat 378C. Cells were harvested in RIPA buffer, 150 mM NaCl, 1.0% IgepalCA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0,complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis,IN) as described previously (Huang et al., 2006). The insoluble celldebris was removed by centrifugation at 48C for 20 min at 12,000g. Theprotein concentration of the supernatant fraction was determined bythe Bio-Rad assay. The boiled samples were separated on 12% SDS–PAGE and transferred to nitrocellulose. The nonspecific binding siteson the membrane were blocked by treatment with TBS containing 5%w/v nonfat dry milk and 0.25% Tween 20 for 2 h. The membrane wasincubated overnight at 48C with a specific indicated primary antibodyand developed with HRP-conjugated secondary antibody by enhancedchemiluminescence (ECL).
Receptor binding
Receptor binding determinations were carried out as describedpreviously with some modification (Yu et al., 2005). Briefly, cellspropagated in feeder layer free cultures (absence of MEFs) wereincubated in receptor binding buffer (50 mM Tris-HC1, 10 mM HEPES,pH 7.4, 120 mM NaCl, 4 mM KCl, 1 mM CaCl2, 5 mM MgCl2 10 mg/mlbacitracin, 0.25% BSA and 2 mg/ml glucose) containing specifiedconcentrations of [3H]AngII or [3H]BK in the absence (total binding) orpresence (nonspecific binding) of 1 mM BK or AngII for 2 h, at 48C. Thecells were washed three times with ice cold binding buffer and thensolubilized with 0.2% sodium dodecyl sulfate. Radioactivity wasdetermined in a scintillation counter. The Kd and Bmax weredetermined using Sigma Plot 8.
Real-time PCR
Total RNAs isolated from hES cells or differentiated cells using theRNeasy kit (Qiagen, Inc.) were subjected to RT-PCR as follows: 1mg oftotal RNA was reverse transcribed to cDNA using random hexamerpriming with Superscript II. The cDNA was diluted 50-fold and then2ml of this solution was used in a 50-ml PCR reaction. The total dilutionwas 1,250-fold. The expression levels of indicated mRNA were
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
analyzed using an ABI 7700 system (Applied Biosystems, Inc., FosterCity, CA). The results were normalized with GAPDH. The sequencesof the primers used were as follows: AT1R forward primer, 50-GCATTA TGT GGA CTG AAC CG-30; reverse primer 50-GAG CAG CCGTCA TCT GTC TA-30. BKB2R forward primer 50-TCT TAA CGGGAC CTT TGC-30 ; reverse primer 50-CCA GTC GAA GTT GTTGGA-30; GAPDH forward: 50-AGC CAC ATC GCT CAG ACA CC-30,reverse: 50-GTA CTC AGC GGC CAG CAT CG-30.
ResultsFeeder layer free hES cultures
Human embryonic stem cells are cultured on feeder layers,generally consisting of mouse embryonic fibroblasts (MEF). Thisprocedure allows for the continuous growth of the hES cells inan undifferentiated state. However, to study receptor action,receptor expression, and signaling properties of hES cellsaccurately, free of contaminating cells, which may also beexpressing the same receptors with similar signal cascades, theMEF have to be removed. Differential adhesion between MEFand hES cells was used to separate the two cell types. Greaterthan 95% of the detached MEF and a small number of hES cellsadhered to tissue culture plates after 2 h in the incubator, whilemost of the detached hES cells remained in suspension. The useof this simple and quick procedure resulted in purity of hES cellcultures of greater than 95%. After removal of the MEF, the H9cells were seeded onto 0.1% gelatin-coated plates andmaintained in the presence of conditioned medium derivedfrom MEF cultures. The hES cells in the feeder layer freecultures remain in clusters and retain their typical morphologyof round shape, compactness, and a high ratio of nucleus tocytoplasm (Fig. 1A). Additionally, the cells retain the molecularmarkers of undifferentiated hES cells including SSEA-4 andTRA-1-60 even after 1 month (Fig. 1B). All subsequentbiochemical data from the hES cells were obtained withfeeder-layer free cultures containing MEF-conditioned mediumas the culture medium.
Differentiation of hES cells into epithelial lineage
To induce epithelial cell development, the undifferentiated hEScells were detached and cultured in suspension on ultra lowattachment plates (Fisher Scientific Co., Pittsburg, PA) in thepresence of differentiation medium or differentiation mediumconditioned by exposure to NIH-3T3 cells for 24 h(conditioned differentiation medium). This promoted theformation of floating EBs. After 6 days in suspension, theembryoid bodies (EBs) were plated on either gelatin orBasement Membrane MatrigelTM (BM)-coated tissue cultureplates (6th day of differentiation). As shown inFigure 2A, approximately 80% of the EBs in the differentiationmedium on the gelatin-coated plates attached, and only a few ofthe attached clusters displayed outward cell migration after 1day (7th day of differentiation). All of the EBs in the conditioneddifferentiation medium, seeded onto gelatin-coated plates andthe EBs in differentiation medium or conditioned differentiationmedium seeded onto BM matrix-coated plates attached atapproximately 100% and displayed outward cell migration. TheEBs incubated on gelatin in differentiation medium displayed thepoorest adherence. Figure 2A shows that these cultures alsodisplayed the poorest cell migration. Whereas the EBs culturedin conditioned differentiation medium and seeded onto the BMmatrix were the quickest to adhere and, as illustrated inFigure 2A, were the quickest to migrate. After three days ofattachment (on the 9th day of differentiation), all treatmentsresulted in some outgrowth from the EBs. The migrating cellson either gelatin or BM matrix-coated plates, in the presence ofconditioned differentiation medium, but not differentiationmedium, formed epithelial-like sheets (Fig. 2A). The cultures inthe presence of differentiation medium showed both sheet-like
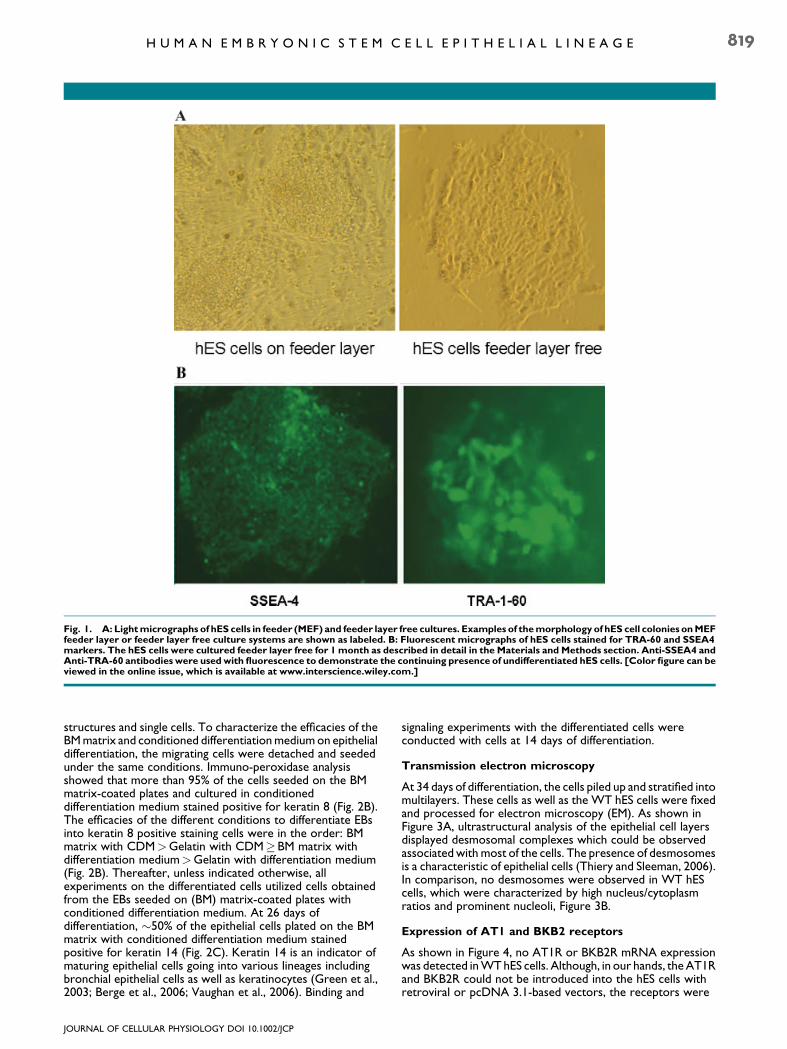
Fig. 1. A: Lightmicrographs of hES cells in feeder (MEF) and feeder layer free cultures. Examples of themorphology of hES cell colonies onMEFfeeder layer or feeder layer free culture systems are shown as labeled. B: Fluorescent micrographs of hES cells stained for TRA-60 and SSEA4markers. The hES cells were cultured feeder layer free for 1month as described in detail in the Materials and Methods section. Anti-SSEA4 andAnti-TRA-60 antibodies were usedwith fluorescence to demonstrate the continuing presence of undifferentiated hES cells. [Color figure can beviewed in the online issue, which is available at www.interscience.wiley.com.]
H U M A N E M B R Y O N I C S T E M C E L L E P I T H E L I A L L I N E A G E 819
structures and single cells. To characterize the efficacies of theBM matrix and conditioned differentiation medium on epithelialdifferentiation, the migrating cells were detached and seededunder the same conditions. Immuno-peroxidase analysisshowed that more than 95% of the cells seeded on the BMmatrix-coated plates and cultured in conditioneddifferentiation medium stained positive for keratin 8 (Fig. 2B).The efficacies of the different conditions to differentiate EBsinto keratin 8 positive staining cells were in the order: BMmatrix with CDM>Gelatin with CDM�BM matrix withdifferentiation medium>Gelatin with differentiation medium(Fig. 2B). Thereafter, unless indicated otherwise, allexperiments on the differentiated cells utilized cells obtainedfrom the EBs seeded on (BM) matrix-coated plates withconditioned differentiation medium. At 26 days ofdifferentiation, �50% of the epithelial cells plated on the BMmatrix with conditioned differentiation medium stainedpositive for keratin 14 (Fig. 2C). Keratin 14 is an indicator ofmaturing epithelial cells going into various lineages includingbronchial epithelial cells as well as keratinocytes (Green et al.,2003; Berge et al., 2006; Vaughan et al., 2006). Binding and
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
signaling experiments with the differentiated cells wereconducted with cells at 14 days of differentiation.
Transmission electron microscopy
At 34 days of differentiation, the cells piled up and stratified intomultilayers. These cells as well as the WT hES cells were fixedand processed for electron microscopy (EM). As shown inFigure 3A, ultrastructural analysis of the epithelial cell layersdisplayed desmosomal complexes which could be observedassociated with most of the cells. The presence of desmosomesis a characteristic of epithelial cells (Thiery and Sleeman, 2006).In comparison, no desmosomes were observed in WT hEScells, which were characterized by high nucleus/cytoplasmratios and prominent nucleoli, Figure 3B.
Expression of AT1 and BKB2 receptors
As shown in Figure 4, no AT1R or BKB2R mRNA expressionwas detected in WT hES cells. Although, in our hands, the AT1Rand BKB2R could not be introduced into the hES cells withretroviral or pcDNA 3.1-based vectors, the receptors were
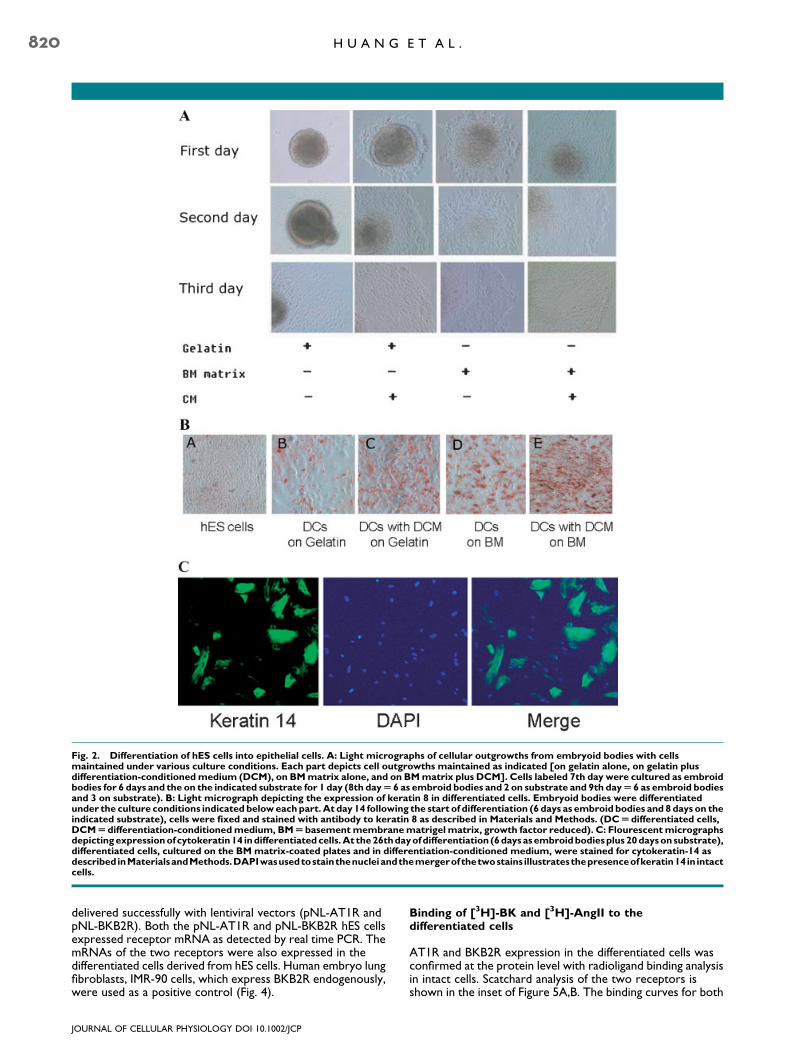
Fig. 2. Differentiation of hES cells into epithelial cells. A: Light micrographs of cellular outgrowths from embryoid bodies with cellsmaintained under various culture conditions. Each part depicts cell outgrowths maintained as indicated [on gelatin alone, on gelatin plusdifferentiation-conditionedmedium (DCM), on BMmatrix alone, and on BMmatrix plus DCM]. Cells labeled 7th day were cultured as embroidbodies for 6 days and the on the indicated substrate for 1 day (8th dayU 6 as embroid bodies and 2 on substrate and 9th dayU6 as embroid bodiesand 3 on substrate). B: Light micrograph depicting the expression of keratin 8 in differentiated cells. Embryoid bodies were differentiatedunder the culture conditions indicated beloweachpart.At day 14 following the start of differentiation (6 days as embroid bodies and 8days on theindicated substrate), cells were fixed and stained with antibody to keratin 8 as described in Materials and Methods. (DCUdifferentiated cells,DCMUdifferentiation-conditionedmedium, BMUbasementmembranematrigel matrix, growth factor reduced). C: Flourescentmicrographsdepictingexpressionofcytokeratin14 indifferentiatedcells.At the26thdayofdifferentiation (6daysasembroidbodiesplus20daysonsubstrate),differentiated cells, cultured on the BM matrix-coated plates and in differentiation-conditioned medium, were stained for cytokeratin-14 asdescribed inMaterialsandMethods.DAPIwasusedtostainthenucleiandthemergerofthetwostains illustratesthepresenceofkeratin14in intactcells.
820 H U A N G E T A L .
delivered successfully with lentiviral vectors (pNL-AT1R andpNL-BKB2R). Both the pNL-AT1R and pNL-BKB2R hES cellsexpressed receptor mRNA as detected by real time PCR. ThemRNAs of the two receptors were also expressed in thedifferentiated cells derived from hES cells. Human embryo lungfibroblasts, IMR-90 cells, which express BKB2R endogenously,were used as a positive control (Fig. 4).
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
Binding of [3H]-BK and [3H]-AngII to thedifferentiated cells
AT1R and BKB2R expression in the differentiated cells wasconfirmed at the protein level with radioligand binding analysisin intact cells. Scatchard analysis of the two receptors isshown in the inset of Figure 5A,B. The binding curves for both
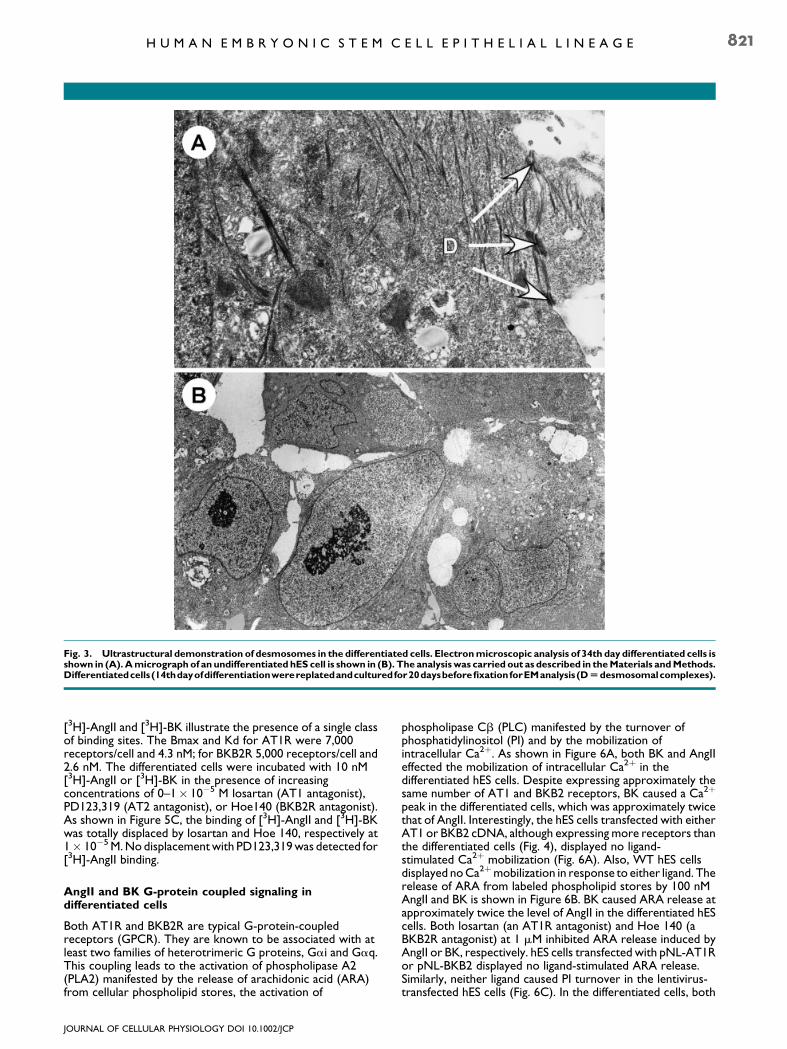
Fig. 3. Ultrastructural demonstration of desmosomes in the differentiated cells. Electronmicroscopic analysis of 34th day differentiated cells isshown in (A).Amicrograph of an undifferentiated hES cell is shown in (B). The analysis was carried out as described in theMaterials andMethods.Differentiatedcells(14thdayofdifferentiationwerereplatedandculturedfor20daysbeforefixationforEManalysis(DUdesmosomalcomplexes).
H U M A N E M B R Y O N I C S T E M C E L L E P I T H E L I A L L I N E A G E 821
[3H]-AngII and [3H]-BK illustrate the presence of a single classof binding sites. The Bmax and Kd for AT1R were 7,000receptors/cell and 4.3 nM; for BKB2R 5,000 receptors/cell and2.6 nM. The differentiated cells were incubated with 10 nM[3H]-AngII or [3H]-BK in the presence of increasingconcentrations of 0–1� 10�5 M losartan (AT1 antagonist),PD123,319 (AT2 antagonist), or Hoe140 (BKB2R antagonist).As shown in Figure 5C, the binding of [3H]-AngII and [3H]-BKwas totally displaced by losartan and Hoe 140, respectively at1� 10�5 M. No displacement with PD123,319 was detected for[3H]-AngII binding.
AngII and BK G-protein coupled signaling indifferentiated cells
Both AT1R and BKB2R are typical G-protein-coupledreceptors (GPCR). They are known to be associated with atleast two families of heterotrimeric G proteins, Gai and Gaq.This coupling leads to the activation of phospholipase A2(PLA2) manifested by the release of arachidonic acid (ARA)from cellular phospholipid stores, the activation of
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
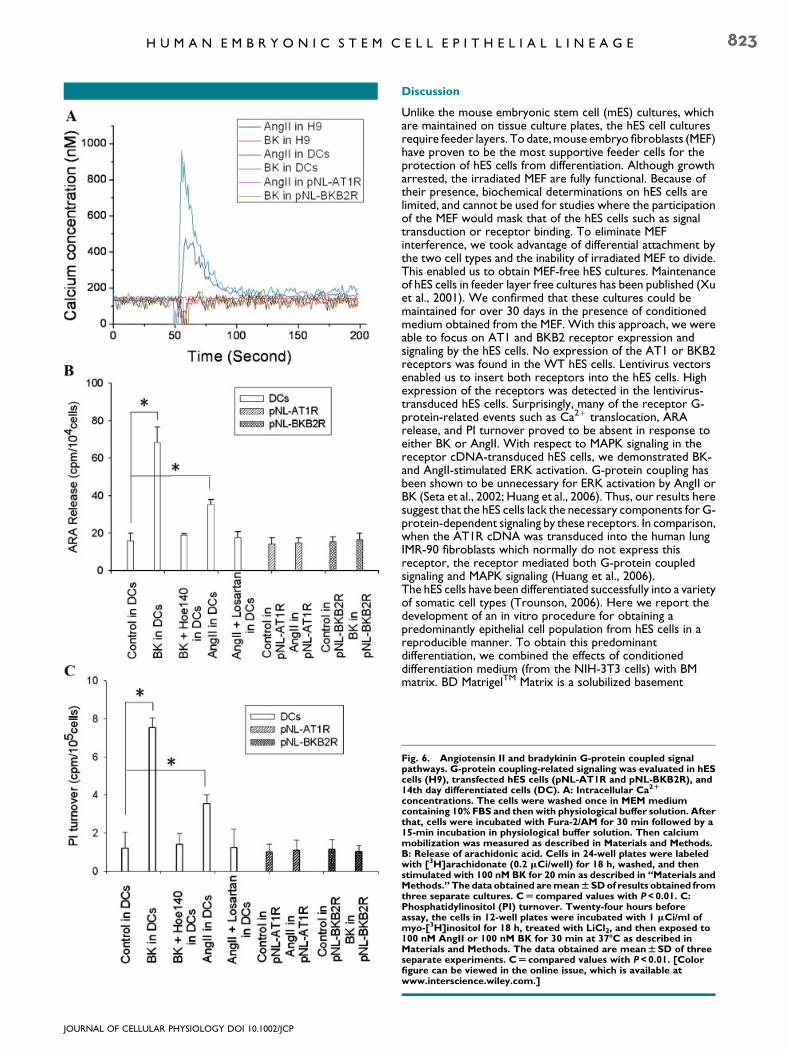
phospholipase Cb (PLC) manifested by the turnover ofphosphatidylinositol (PI) and by the mobilization ofintracellular Ca2þ. As shown in Figure 6A, both BK and AngIIeffected the mobilization of intracellular Ca2þ in thedifferentiated hES cells. Despite expressing approximately thesame number of AT1 and BKB2 receptors, BK caused a Ca2þ
peak in the differentiated cells, which was approximately twicethat of AngII. Interestingly, the hES cells transfected with eitherAT1 or BKB2 cDNA, although expressing more receptors thanthe differentiated cells (Fig. 4), displayed no ligand-stimulated Ca2þ mobilization (Fig. 6A). Also, WT hES cellsdisplayed no Ca2þmobilization in response to either ligand. Therelease of ARA from labeled phospholipid stores by 100 nMAngII and BK is shown in Figure 6B. BK caused ARA release atapproximately twice the level of AngII in the differentiated hEScells. Both losartan (an AT1R antagonist) and Hoe 140 (aBKB2R antagonist) at 1 mM inhibited ARA release induced byAngII or BK, respectively. hES cells transfected with pNL-AT1Ror pNL-BKB2 displayed no ligand-stimulated ARA release.Similarly, neither ligand caused PI turnover in the lentivirus-transfected hES cells (Fig. 6C). In the differentiated cells, both
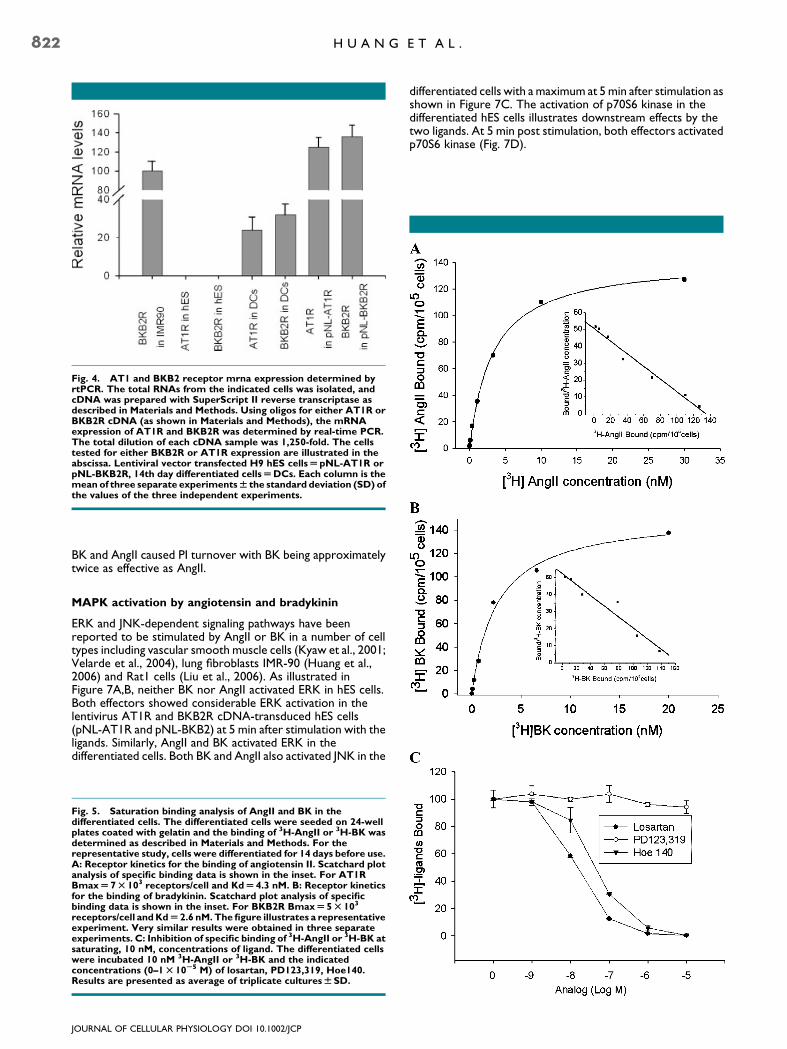
Fig. 4. AT1 and BKB2 receptor mrna expression determined byrtPCR. The total RNAs from the indicated cells was isolated, andcDNA was prepared with SuperScript II reverse transcriptase asdescribed in Materials and Methods. Using oligos for either AT1R orBKB2R cDNA (as shown in Materials and Methods), the mRNAexpression of AT1R and BKB2R was determined by real-time PCR.The total dilution of each cDNA sample was 1,250-fold. The cellstested for either BKB2R or AT1R expression are illustrated in theabscissa. Lentiviral vector transfected H9 hES cellsUpNL-AT1R orpNL-BKB2R, 14th day differentiated cellsUDCs. Each column is themean of three separate experimentsW the standard deviation (SD) ofthe values of the three independent experiments.
822 H U A N G E T A L .
BK and AngII caused PI turnover with BK being approximatelytwice as effective as AngII.
MAPK activation by angiotensin and bradykinin
ERK and JNK-dependent signaling pathways have beenreported to be stimulated by AngII or BK in a number of celltypes including vascular smooth muscle cells (Kyaw et al., 2001;Velarde et al., 2004), lung fibroblasts IMR-90 (Huang et al.,2006) and Rat1 cells (Liu et al., 2006). As illustrated inFigure 7A,B, neither BK nor AngII activated ERK in hES cells.Both effectors showed considerable ERK activation in thelentivirus AT1R and BKB2R cDNA-transduced hES cells(pNL-AT1R and pNL-BKB2) at 5 min after stimulation with theligands. Similarly, AngII and BK activated ERK in thedifferentiated cells. Both BK and AngII also activated JNK in the
Fig. 5. Saturation binding analysis of AngII and BK in thedifferentiated cells. The differentiated cells were seeded on 24-wellplates coated with gelatin and the binding of 3H-AngII or 3H-BK wasdetermined as described in Materials and Methods. For therepresentative study, cells were differentiated for 14 days before use.A: Receptor kinetics for the binding of angiotensin II. Scatchard plotanalysis of specific binding data is shown in the inset. For AT1RBmaxU 7T 103 receptors/cell and KdU 4.3 nM. B: Receptor kineticsfor the binding of bradykinin. Scatchard plot analysis of specificbinding data is shown in the inset. For BKB2R BmaxU 5T 103
receptors/cell andKdU 2.6 nM.Thefigure illustrates a representativeexperiment. Very similar results were obtained in three separateexperiments. C: Inhibition of specific binding of 3H-AngII or 3H-BK atsaturating, 10 nM, concentrations of ligand. The differentiated cellswere incubated 10 nM 3H-AngII or 3H-BK and the indicatedconcentrations (0–1T 10S5 M) of losartan, PD123,319, Hoe140.Results are presented as average of triplicate culturesWSD.
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
differentiated cells with a maximum at 5 min after stimulation asshown in Figure 7C. The activation of p70S6 kinase in thedifferentiated hES cells illustrates downstream effects by thetwo ligands. At 5 min post stimulation, both effectors activatedp70S6 kinase (Fig. 7D).
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
H U M A N E M B R Y O N I C S T E M C E L L E P I T H E L I A L L I N E A G E 823
Discussion
Unlike the mouse embryonic stem cell (mES) cultures, whichare maintained on tissue culture plates, the hES cell culturesrequire feeder layers. To date, mouse embryo fibroblasts (MEF)have proven to be the most supportive feeder cells for theprotection of hES cells from differentiation. Although growtharrested, the irradiated MEF are fully functional. Because oftheir presence, biochemical determinations on hES cells arelimited, and cannot be used for studies where the participationof the MEF would mask that of the hES cells such as signaltransduction or receptor binding. To eliminate MEFinterference, we took advantage of differential attachment bythe two cell types and the inability of irradiated MEF to divide.This enabled us to obtain MEF-free hES cultures. Maintenanceof hES cells in feeder layer free cultures has been published (Xuet al., 2001). We confirmed that these cultures could bemaintained for over 30 days in the presence of conditionedmedium obtained from the MEF. With this approach, we wereable to focus on AT1 and BKB2 receptor expression andsignaling by the hES cells. No expression of the AT1 or BKB2receptors was found in the WT hES cells. Lentivirus vectorsenabled us to insert both receptors into the hES cells. Highexpression of the receptors was detected in the lentivirus-transduced hES cells. Surprisingly, many of the receptor G-protein-related events such as Ca2þ translocation, ARArelease, and PI turnover proved to be absent in response toeither BK or AngII. With respect to MAPK signaling in thereceptor cDNA-transduced hES cells, we demonstrated BK-and AngII-stimulated ERK activation. G-protein coupling hasbeen shown to be unnecessary for ERK activation by AngII orBK (Seta et al., 2002; Huang et al., 2006). Thus, our results heresuggest that the hES cells lack the necessary components for G-protein-dependent signaling by these receptors. In comparison,when the AT1R cDNA was transduced into the human lungIMR-90 fibroblasts which normally do not express thisreceptor, the receptor mediated both G-protein coupledsignaling and MAPK signaling (Huang et al., 2006).The hES cells have been differentiated successfully into a varietyof somatic cell types (Trounson, 2006). Here we report thedevelopment of an in vitro procedure for obtaining apredominantly epithelial cell population from hES cells in areproducible manner. To obtain this predominantdifferentiation, we combined the effects of conditioneddifferentiation medium (from the NIH-3T3 cells) with BMmatrix. BD MatrigelTM Matrix is a solubilized basement
Fig. 6. Angiotensin II and bradykinin G-protein coupled signalpathways. G-protein coupling-related signaling was evaluated in hEScells (H9), transfected hES cells (pNL-AT1R and pNL-BKB2R), and14th day differentiated cells (DC). A: Intracellular Ca2R
concentrations. The cells were washed once in MEM mediumcontaining 10% FBS and then with physiological buffer solution. Afterthat, cells were incubated with Fura-2/AM for 30 min followed by a15-min incubation in physiological buffer solution. Then calciummobilization was measured as described in Materials and Methods.B: Release of arachidonic acid. Cells in 24-well plates were labeledwith [3H]arachidonate (0.2 mCi/well) for 18 h, washed, and thenstimulated with 100 nM BK for 20 min as described in ‘‘Materials andMethods.’’ The data obtained aremeanWSDof results obtained fromthree separate cultures. CU compared values with P<0.01. C:Phosphatidylinositol (PI) turnover. Twenty-four hours beforeassay, the cells in 12-well plates were incubated with 1 mCi/ml ofmyo-[3H]inositol for 18 h, treated with LiCl2, and then exposed to100 nM AngII or 100 nM BK for 30 min at 37-C as described inMaterials and Methods. The data obtained are meanWSD of threeseparate experiments. CU compared values with P<0.01. [Colorfigure can be viewed in the online issue, which is available atwww.interscience.wiley.com.]
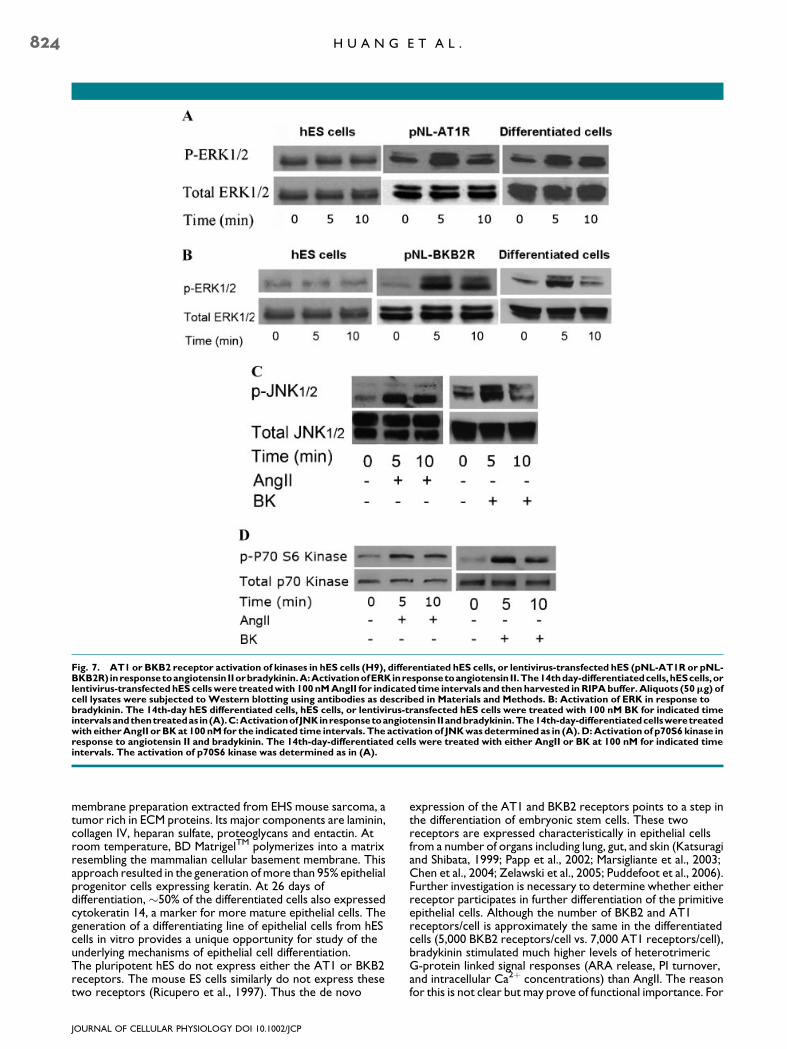
Fig. 7. AT1 or BKB2 receptor activation of kinases in hES cells (H9), differentiated hES cells, or lentivirus-transfected hES (pNL-AT1R or pNL-BKB2R) inresponsetoangiotensin IIorbradykinin.A:ActivationofERKinresponsetoangiotensin II.The14thday-differentiatedcells,hEScells,orlentivirus-transfectedhES cellswere treatedwith 100nMAngII for indicated time intervals and thenharvested inRIPAbuffer.Aliquots (50mg) ofcell lysates were subjected to Western blotting using antibodies as described in Materials and Methods. B: Activation of ERK in response tobradykinin. The 14th-day hES differentiated cells, hES cells, or lentivirus-transfected hES cells were treated with 100 nM BK for indicated timeintervalsandthentreatedas in(A).C:ActivationofJNKinresponsetoangiotensinIIandbradykinin.The14th-day-differentiatedcellsweretreatedwith eitherAngII orBKat 100nM for the indicated time intervals. The activation of JNKwasdetermined as in (A).D:Activationof p70S6 kinase inresponse to angiotensin II and bradykinin. The 14th-day-differentiated cells were treated with either AngII or BK at 100 nM for indicated timeintervals. The activation of p70S6 kinase was determined as in (A).
824 H U A N G E T A L .
membrane preparation extracted from EHS mouse sarcoma, atumor rich in ECM proteins. Its major components are laminin,collagen IV, heparan sulfate, proteoglycans and entactin. Atroom temperature, BD MatrigelTM polymerizes into a matrixresembling the mammalian cellular basement membrane. Thisapproach resulted in the generation of more than 95% epithelialprogenitor cells expressing keratin. At 26 days ofdifferentiation, �50% of the differentiated cells also expressedcytokeratin 14, a marker for more mature epithelial cells. Thegeneration of a differentiating line of epithelial cells from hEScells in vitro provides a unique opportunity for study of theunderlying mechanisms of epithelial cell differentiation.The pluripotent hES do not express either the AT1 or BKB2receptors. The mouse ES cells similarly do not express thesetwo receptors (Ricupero et al., 1997). Thus the de novo
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
expression of the AT1 and BKB2 receptors points to a step inthe differentiation of embryonic stem cells. These tworeceptors are expressed characteristically in epithelial cellsfrom a number of organs including lung, gut, and skin (Katsuragiand Shibata, 1999; Papp et al., 2002; Marsigliante et al., 2003;Chen et al., 2004; Zelawski et al., 2005; Puddefoot et al., 2006).Further investigation is necessary to determine whether eitherreceptor participates in further differentiation of the primitiveepithelial cells. Although the number of BKB2 and AT1receptors/cell is approximately the same in the differentiatedcells (5,000 BKB2 receptors/cell vs. 7,000 AT1 receptors/cell),bradykinin stimulated much higher levels of heterotrimericG-protein linked signal responses (ARA release, PI turnover,and intracellular Ca2þ concentrations) than AngII. The reasonfor this is not clear but may prove of functional importance. For

H U M A N E M B R Y O N I C S T E M C E L L E P I T H E L I A L L I N E A G E 825
example, it may be important in determining further lineage ofthese cells.
Literature Cited
Amit M, Carpenter MK, Inokuma MS, Chiu C-P, Harris CP, Waknitz MA, Itskovitz-Eldor J,Thomson JA. 2000. Clonally derived human embryonic stem cell lines maintainpluripotency and proliferative potential for prolonged periods of culture. Dev Biol 227:271.
Berge U, Kristensen P, Rattan SI. 2006. Kinetin-induced differentiation of normal humankeratinocytes undergoing aging in vitro. Ann N Y Acad Sci 1067:332–336.
Bradley A, Evans M, Kaufman MH, Robertson E. 1984. Formation of germ-line chimaeras fromembryo-derived teratocarcinoma cell lines. Nature 309:255–256.
Chen BC, Yu CC, Lei HC, Chang MS, Hsu MJ, Huang CL, Chen MC, Sheu JR, Chen TF, ChenTL, Inoue H, Lin CH. 2004. Bradykinin B2 receptor mediates NF-kappaB activation andcyclooxygenase-2 expression via the Ras/Raf-1/ERK pathway in human airway epithelialcells. J Immunol 173:5219–5228.
Cherny RA, Stokes TM, Merei J, Lom L, Brandon MR, Williams RL. 1994. Strategies forthe isolation and characterization of bovine embryonic stem cells. Reprod Fertil Dev6:569–575.
Cowan CA, Klimanskaya I, McMahon J, Atienza J, Witmyer J, Zucker JP, Wang S, Morton CC,McMahon AP, Powers D, Melton DA. 2004. Derivation of embryonic stem-cell lines fromhuman blastocysts. N Engl J Med 350:1353–1356.
Cowan CA, Atienza J, Melton DA, Eggan K. 2005. Nuclear reprogramming of somatic cellsafter fusion with human embryonic stem cells. Science 309:1369–1373.
Donello JE, Loeb JE, Hope TJ. 1998. Woodchuck hepatitis virus contains a tripartiteposttranscriptional regulatory element. J Virol 72:5085–5092.
Dravid G, Hammond H, Cheng L. 2006. Culture of humanembryonic stem cells on human andmouse feeder cells. Methods Mol Biol 331:91–104.
Graves KH, Moreadith RW. 1993. Derivation and characterization of putative pluripotentialembryonic stem cells from preimplantation rabbit embryos. Mol Reprod Dev 36:424–433.
Green H, Easley K, Iuchi S. 2003. Marker succession during the development of keratinocytesfrom cultured human embryonic stem cells. Proc Natl Acad Sci USA 100:15625–15630.
Huang Z, Taylor L, Liu B, Yu J, Polgar P. 2006. Modulation by bradykinin of angiotensin type 1receptor-evoked RhoA activation of connective tissue growth factor expression in humanlung fibroblasts. Am J Physiol Lung Cell Mol Physiol 290:L1291–L1299.
Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, Soreq H, BenvenistyN. 2000. Differentiation of human embryonic stem cells into embryoid bodiescompromising the three embryonic germ layers. Mol Med 6:88–95.
Katsuragi T, Shibata K. 1999. Modulations by stimulation of angiotensin II type 1 receptor ofgastrointestinal functions. Nippon Rinsho 57:1054–1059.
Kyaw M, Yoshizumi M, Tsuchiya K, Kirima K, Tamaki T. 2001. Antioxidants inhibit JNK andp38 MAPK activation but not ERK 1/2 activation by angiotensin II in rat aortic smoothmuscle cells. Hypertens Res 24:251–261.
Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R. 2002. Endothelial cells derivedfrom human embryonic stem cells. PNAS 99:4391–4396.
Liu B, Yu J, Taylor L, Zhou X, Polgar P. 2006. Microarray and phosphokinase screeningsleading to studies on ERK and JNK regulation of connective tissue growth factorexpression by angiotensin II 1a and bradykinin B2 receptors in Rat1 fibroblasts. J CellBiochem 97:1104–1120.
Loebel DAF, Watson CM, De Young RA, Tam PPL. 2003. Lineage choice and differentiation inmouse embryos and embryonic stem cells. Dev Biol 264:1.
Marsigliante S, Muscella A, Elia MG, Greco S, Storelli C. 2003. Angiotensin II AT1 receptorstimulates Naþ-Kþ ATPase activity through a pathway involving PKC-zeta in rat thyroidcells. J Physiol (Lond) 546:461–470.
Mitalipova M, Palmarini G. 2006. Isolation and characterization of human embryonic stemcells. Methods Mol Biol 331:55–76.
Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. 1993. Derivation of completelycell culture-derived mice from early-passage embryonic stem cells. PNAS 90:8424–8428.
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
Pain B, Clark ME, Shen M, Nakazawa H, Sakurai M, Samarut J, Etches RJ. 1996. Long-term invitro culture and characterisation of avian embryonic stem cells with multiplemorphogenetic potentialities. Development 122:2339–2348.
Papp M, Li X, Zhuang J, Wang R, Uhal BD. 2002. Angiotensin receptor subtype AT1 mediatesalveolar epithelial cell apoptosis in response to ANG II. Am J Physiol Lung Cell Mol Physiol282:L713–L718.
Pera MF, Trounson AO. 2004. Human embryonic stem cells: Prospects for development.Development 131:5515–5525.
Pluta K, Luce MJ, Bao L, Agha-Mohammadi S, Reiser J. 2005. Tight control of transgeneexpression by lentivirus vectors containing second-generation tetracycline-responsivepromoters. J Gene Med 7:803–817.
Prado GN, Taylor L, Polgar P. 1997. Effects of intracellular tyrosine residue mutation andcarboxyl terminus truncation on signal transduction and internalization of the ratbradykinin B2 receptor. J Biol Chem 272:14638–14642.
Puddefoot JR, Udeozo UKI, Barker S, Vinson GP. 2006. The role of angiotensin II in theregulation of breast cancer cell adhesion and invasion. Endocr Relat Cancer13:895–903.
Ramezani A, Hawley TS, Hawley RG. 2000. Lentiviral vectors for enhanced gene expressionin human hematopoietic cells. Mol Ther 2:458–469.
Reubinoff BE, Pera MF, Fong C-Y, Trounson A, Bongso A. 2000. Embryonic stem cell linesfrom human blastocysts: Somatic differentiation in vitro. Nat Biotech 18:399.
Ricupero DA, Polgar P, Taylor L, Sowell MO, Gao Y, Bradwin G, Mortensen RM. 1997.Enhanced bradykinin-stimulated phospholipase C activity in murine embryonic stem cellslacking the G-protein alphaq-subunit. Biochem J 327:803–809.
Schuldiner M, Yanuka O, Itskovitz-Eldor J, Melton DA, Benvenisty N. 2000. From the cover:Effects of eight growth factors on the differentiation of cells derived from humanembryonic stem cells. PNAS 97:11307–11312.
Seta K, Nanamori M, Modrall JG, Neubig RR, Sadoshima J. 2002. AT1 receptor mutant lackingheterotrimeric G protein coupling activates the Src-Ras-ERK pathway without nucleartranslocation of ERKs. J Biol Chem 277:9268–9277.
Shamblott MJ, Axelman J, Wang S, Bugg EM, Littlefield JW, Donovan PJ, Blumenthal PD,Huggins GR, Gearhart JD. 1998. Derivation of pluripotent stem cells from cultured humanprimordial germ cells. PNAS 95:13726–13731.
Thiery JP, Sleeman JP. 2006. Complex networks orchestrate epithelial-mesenchymaltransitions. Nat Rev Mol Cell Biol 7:131.
Thomson JA, Kalishman J, Golos TG, Durning M, Harris CP, Becker RA, Hearn JP. 1995.Isolation of a primate embryonic stem cell line. PNAS 92:7844–7848.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM.1998. Embryonic stem cell lines derived from human blastocysts. Science 282:1145–1147.
Trounson A. 2004. Derivation characteristics and perspectives for mammalian pluripotentialstem cells. Reprod Fertility Dev 17:135–141.
Trounson A. 2006. The production and directed differentiation of human embryonic stemcells. Endocr Rev 27:208–219.
Vaughan MB, Ramirez RD, Wright WE, Minna JD, Shay JW. 2006. A three-dimensionalmodel of differentiation of immortalized human bronchial epithelial cells. Differentiation74:141–148.
Velarde V, de la Cerda PM, Duarte C, Arancibia F, Abbott E, Gonzalez A, Moreno F, Jaffa AA.2004. Role of reactive oxygen species in bradykinin-induced proliferation of vascularsmooth muscle cells. Biol Res 37:419–430.
Watt FM, Hogan BLM. 2000. Out of Eden: Stem cells and their niches. Science287:1427–1430.
Wheeler MB. 1994. Development and validation of swine embryonic stem cells: A review.Reprod Fertil Dev 6:563–568.
Xu C, Inokuma MS, Denham J, Golds K, Kundu P, Gold JD, Carpenter MK. 2001. Feeder-freegrowth of undifferentiated human embryonic stem cells. Nat Biotechnol 19:971–974.
Yu J, Polgar P, Lubinsky D, Gupta M, Wang L, Mierke D, Taylor L. 2005. Coulombic andhydrophobic interactions in the first intracellular loop are vital for bradykinin B2 receptorligand binding and consequent signal transduction. Biochemistry 44:5295–5306.
Zelawski W, Witalinska-Labuzek J, Stadnicki A. 2005. A role of kallikrein-kinin system in gutdiseases. Wiad Lek 58:331–334.




















