
Developmental Neurotoxicity Study of Dietary Bisphenol A in Sprague-Dawley Rats
Upload
independentCategory
view
1download
0
Journal of Chromatography A, 1216 (2009) 449–469
Contents lists available at ScienceDirect
Journal of Chromatography A
journa l homepage: www.e lsev ier .com/ locate /chroma
Review
Analytical methods for the determination of bisphenol A in food
Ana Ballesteros-Gómez, Soledad Rubio ∗, Dolores Pérez-BenditoDepartment of Analytical Chemistry, Facultad de Ciencias, Edificio Anexo Marie Curie, Campus de Rabanales, 14171 Córdoba, Spain
a r t i c l e i n f o
Article history:Available online 3 July 2008
Keywords:Bisphenol AReviewEndocrine disruptersFood analysisSample treatmentMass Spectrometry
a b s t r a c t
Food constitutes the primary route for human exposure to bisphenol A (BPA), one of the highest vol-ume chemicals produced worldwide. The estrogenic properties of BPA, its wide dispersive use and therecent extensive literature describing low-dose BPA effects in animals, have raised concerns about itspossible adverse effects on human health. A reliable health risk assessment of BPA relies basically on itsunambiguous identification and accurate quantification in food, and the aim of the present review is togive an overview of the analytical methods reported so far for the determination of BPA in these matri-ces. Emphasis is placed on the main strategies developed for sample treatment, which usually consistsof several laborious and time-consuming steps in order to achieve the required sensitivity and selectiv-ity. Separation, identification and quantitation of BPA is today reliably made with mass spectrometricmethods, namely liquid chromatography–mass spectrometry (LC–MS) and gas chromatography–mass
Chromatographyspectrometry (GC–MS), and thus main attention is devoted to these techniques, but other methods usingC
0d
LC coupled to fluorescence or electrochemical detection, as well as immunochemical methods are alsocovered. Recent and expected future developments are discussed.
© 2008 Elsevier B.V. All rights reserved.
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4502. Sources and removal of background contamination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4503. Sample treatment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 450
3.1. Solvent-based extraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4513.1.1. Solvent extraction (SE) and liquid–liquid extraction (LLE) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4513.1.2. Microwave-assisted extraction (MAE) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4523.1.3. Pressurized liquid extraction (PLE) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 455
3.2. Solid-phase extraction (SPE) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4553.2.1. Non-selective sorbents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4553.2.2. Selective sorbents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 456
3.3. Less common extraction techniques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4573.3.1. Solid-phase microextraction (SPME) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4573.3.2. Stir bar sorptive extraction (SBSE) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4583.3.3. Matrix solid-phase dispersion (MSPD) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 458
4. Separation and detection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4624.1. Liquid chromatography . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 462
4.1.1. Fluorescence detection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4624.1.2. Electrochemical detection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4624.1.3. LC–MS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 462
4.2. GC–MS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .4.3. Immunochemical methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
5. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Acknowledgment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
∗ Corresponding author. Tel.: +34 957 218643; fax: +34 957 218644.E-mail address: [email protected] (S. Rubio).
021-9673/$ – see front matter © 2008 Elsevier B.V. All rights reserved.oi:10.1016/j.chroma.2008.06.037
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 465. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 467. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 467. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 468. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 468

4 hrom
1
tB5hNe22oOawmwatfsp
shc2[sc[i(5ihii0t
TfwibwaccteguatrbuAsm[
d
inE[af
[oflhafsdrsn
2
csa4m
MtptOatai
tce1mSa[tiBpv
3
aBts
50 A. Ballesteros-Gómez et al. / J. C
. Introduction
Bisphenol A (BPA), 2,2-bis(4-hydroxyphenyl) propane, is one ofhe highest volume chemicals in the world [1,2]. Global demand forPA is predicted to grow from 3.9 million tonnes in 2006 to aboutmillion tonnes in 2010 [3]. Many countries throughout the worldave large production capacities for BPA, especially Germany, theetherlands, the USA and Japan. BPA capacity in West Europe wasstimated at 830 million tonnes in 2000 [4], it grew by 4%/year from000 to 2006 and it has been predicted to grow by 2%/year in the006–2010 period [3]. The main market for BPA is the productionf polycarbonate with the second largest outlet being epoxy resins.ther uses include flame retardants, unsaturated polyester resinsnd polyacrylate, polyetherimide and polysulphone resins [5,6]. Aide variety of food contact materials stand out among their uses,ainly derived from polycarbonates (infant feeding bottles, table-are, microwave ovenware, storage containers, returnable water
nd milk bottles and water pipes) and epoxy resins (internal pro-ective lining for food and beverage cans, coating on metal lidsor glass jars and bottles and surface-coating on drinking watertorage tanks and wine vats) [7]. The chemical structure and somehysico-chemical properties of BPA are shown in Table 1.
The extensive use of BPA-based polymers, with ester bondsubject to hydrolysis and non-polymerized monomer residues,as led to widespread environmental contamination. BPA con-entrations in the ranges 5–320 ng L−1 in river waters [8–10],0–700 ng L−1 in sewage effluents [9–11], 2–208 ng m−3 in air12–14], 0.2–199 ng g−1 in dust [12–14] and 0.1–384 ng g−1 in food-tuffs [15–17] have been reported. Its presence in food is of specialoncern since it constitutes the primary route of human exposure6,7,14]. The scientific panel on food additives, flavourings, process-ng aids and materials in contact with food of the European UnionEU) has reported estimates of potential dietary exposure of 13,.3 and 1.5 �g/kg body weight/day in 6- to 12-month-old breastfed
nfants, young children and adults, respectively [7]. The widespreaduman exposure to BPA has been highlighted by measurements
n human fluids and tissues (reviewed in [18]). Concentrationsn blood and urine were on average in the 0.3–4.4 �g L−1 and.47–9.5 �g L−1 ranges with a detection rate above 90% in most ofhe studies.
The estrogenic activity of BPA was first reported in 1993 [19].he affinity of BPA for estrogen receptors is 10,000- to 100,000-old weaker than that of estradiol, so it has been considered a veryeak environmental estrogen. However, a large number of recent
n vitro studies have shown that the effects of BPA are mediated byoth genomic and non-genomic estrogen-response mechanisms,ith the disruption of the cell function occurring at doses as low
s 1 pM (0.23 ng L−1) (reviewed in [20]). Recent reports also indi-ate the potential of BPA to disrupt thyroid hormone action [21], toause proliferation of human prostate cancer cells [22] and to blockestosterone synthesis [23] at very low part-per-trillion doses. Thisxtensive new literature concerning low-dose effects of BPA hasiven rise to controversy about the BPA limit values set by reg-latory agencies for consumer health protection and a new riskssessment has been strongly recommended [20]. Currently, theolerable daily intake (TDI) set by the EU Commission [7] and theeference dose (RfD) established by EPA [24] is 0.05 mg BPA/kgody weight/day. This value was derived by applying a 100-foldncertainty factor to the currently accepted overall Non-Observed-dverse-Effect Level (NOAEL) of 5 mg/kg. On the other hand, a
pecific migration limit (SML) for BPA from food contact plasticaterials of 600 ng g−1 was set by the EU Commission in 200425].Because of the high volume, wide dispersive use and endocrine
isrupting and toxic properties of BPA, it is a clear candidate to be
cbaai
atogr. A 1216 (2009) 449–469
ncluded in the list of substances subject to authorization in theew policy on chemicals approved by the EU, REACH (Registration,valuation, Authorisation and Restriction of Chemicals, Annex XIV)26]. So, there are needs for research (new methods) and for revisionnd optimization of existing methods in order to have reliable toolsor risk assessment and control of human exposure to BPA.
Although different authors [18,20,27] and regulatory agencies6,7] have offered reviews concerning the toxicological propertiesf BPA and the levels of this contaminant in human tissues anduids, no review dealing with the determination of BPA in foodas been reported so far. Here, we summarize the state-of-the-rt of the analytical methodologies developed, including strategiesor removal of background contamination, sample preparation andeparation and detection of BPA. Conventional methods and recentevelopments will be critically discussed in terms of simplicity,obustness, sample size, cost, consumption of organic solvents, sen-itivity and selectivity, highlighting their main drawbacks and theeed for future developments.
. Sources and removal of background contamination
BPA is inherently ubiquitous in the environment. Backgroundontamination of BPA occurs at ng L−1 levels and mainly arises fromolvents, SPE columns, glassware, plastic ware and other reagentsnd laboratory tools. In general, heat-treated glassware (4 h at00 ◦C) and solvent-washed materials are used as a precautionaryeasure to prevent background contamination [28].BPA concentrations around 0.02 �g L−1 have been found in
illi-Q water using highly sensitive methods (instrumental quan-ification limit of 5 pg) [29–31]. The contamination arose from thelastics used in the purification system and was removed by fil-ering the water through a hydrophobic membrane (Empore disk).ther authors, however, did not find BPA contamination when theynalysed different ultra-high quality waters, such as Water Pes-anal from Riedel-de Haën or those obtained from an Elgastastnd a Millipore Milli-Q system, although in these experiments thenstrumental quantification limit was around 10 ng [32].
SPE cartridges (e.g. Oasis HLB from Waters and Bond Elut Cer-ify from Varian) have been known to cause BPA contamination atoncentrations of around 0.04 �g L−1 [31]. The contamination wasffectively removed by pre-washing the cartridges which at least5 mL of methanol and was probably derived not from the sorbentaterial, but from the manufacturing process. Sample loading in
PE can introduce strong BPA contamination when glass syringesre utilized, mainly caused by the adhesive used to fix the needle30,32]. Using a peristaltic pump with vinyl tubes, the BPA con-amination became insignificant although the contamination wasmportant for two BPA derivatives (i.e. 4-tert-butylbenzoic acid,BA, and 4-tert-butylphenol, t-BP). Contamination from SPE sam-le loading can be almost completely eliminated by replacing theinyl tubes with Viton tubes from DuPont [32].
. Sample treatment
There are a wide variety of BPA-containing foods including freshnd canned solid and liquid samples (Fig. 1). Determination ofPA in these matrices often requires extensive sample prepara-ion prior to instrumental analysis. The typical steps within foodample preparation include pre-treatment, extraction, clean-up,
oncentration and sometimes derivatization, and constitute theottleneck in current food analysis (Fig. 1). The solid samplesre usually first homogenized while the liquid ones are filterednd/or centrifuged. Special treatments can be required depend-ng on the matrix composition; e.g. carbonated beverages are
A. Ballesteros-Gómez et al. / J. Chromatogr. A 1216 (2009) 449–469 451
Table 1Physico-chemical properties of bisphenol A
Chemical structure Molecular mass Water solubility (mg L−1) log Kowb pKa H donor and acceptor suma
227 89 3.25 9.7 4
for So
dtflr
fmtsoctcaTaOuaofa
3
3
tetSmcfbaaas[
s
Fss
a Calculated using Advanced Chemistry Development (ACD/Labs) Software V9.04b Logarithm of the octanol–water partition coefficient.
egassed [16,33], samples with high protein content may requireheir removal by precipitation and fish tissues are crushed andreeze-dried before homogenization. Canned foods containing bothiquid and solid portions are usually filtered and treated sepa-ately.
Solvent extraction and SPE are the most widely used techniquesor the isolation of BPA from solid and liquid samples, respectively,
ainly because of its simplicity and wide-range applicability. Otherechniques (e.g. MAE, PLE, MSPDE, SPME, SBSE, cf. Fig. 1), althoughcarcely used so far, may improve the extraction of BPA in termsf sample sizes, automation and solvent consumption. The extractsontaining BPA are commonly subject to extensive clean-up and inhis respect liquid–liquid extraction and SPE are preferred. Con-entration of BPA by solvent evaporation of the final extract isn inevitable step because of its low concentration in food (cf.able 2). In general, the combination of different techniques is usu-lly inevitable and methods become frequently matrix-dependent.nly those methods intended to study the migration of BPA fromnused commercial cans by extraction with food-simulating liquids
re simpler [34–37]. Table 3 gives some representative examplesf sample treatment procedures for the analysis of BPA-containingoods. Below, the main techniques used for extraction and clean-upre discussed in detail.t[b5
ig. 1. Analytical methodologies for the determination of bisphenol A in foodstuffs. Abburized liquid extraction; MSPS, matrix solid-phase dispersion; LLE, liquid–liquid extractorptive extraction; FL, fluorescence detection; ED, electrochemical detection.
laris.
.1. Solvent-based extraction
.1.1. Solvent extraction (SE) and liquid–liquid extraction (LLE)Solvent extraction (SE) is still the most common technique for
he isolation of BPA from solid foodstuffs (cf. Table 3). Liquid–liquidxtraction (LLE), despite being a reliable technique for the extrac-ion of BPA from liquid foods, has been used to a lesser extent thanPE. Most extraction methods have been developed for specific foodatrices such as fish [38,39], fruit and vegetables [40,41], yoghurt,
ream, butter, and pudding [42], infant formula powders [43], petoods [44] and mineral water [45], and only few of them are applica-le to a wide range of foods. In this respect, Goodson et al. proposedmethod for the extraction of BPA and isomers of bisphenol F fromvariety of canned products, including fish, fruit, vegetables, bever-ges, soup, desserts, infant formula, meat and pasta [16], which wasubsequently applied by Thomson et al. to a wide array of foodstuffs17].
The amount of sample required for the extraction of BPA fromolid an liquid food is usually in the ranges 0.5–30 g (being 5 g
he most frequent selected size [39,40,41,44,46]) and 10–50 mL15–17,33], respectively, although higher sample sizes have alsoeen proposed, e.g. 120 g for canned jalapeno peppers [47] and00 mL for mineral water [45]. Among solvents, acetonitrile isreviations: SE, solvent extraction; MAE, microwave-assisted extraction; PLE, pres-ion; SPE, solid-phase extraction; SPME, solid-phase microextraction; SBSE, stir bar

452 A. Ballesteros-Gómez et al. / J. Chromatogr. A 1216 (2009) 449–469
Table 2Levels o BPA in foods reported by several authors
Foodstuff Number of samples (number ofpositive quantifiable samples)
BPA concentration inpositive quantifiablesamples
Reference
Fruit and vegetables 14(8) 18.4–95 ng g−1a Yoshida et al. [40]10(6) 29–458 ng mL−1b Brotons et al. [48]10(10) 9–48 ng g−1 Goodson et al. [16]33(11) 12–24 ng g−1 Thomson et al. [17]10(10) 5–35 ng g−1 Braunrath et al. [78]
Milk 8(2) 7.11–15.2 ng g−1 Maragou et al. [65]10(3) 0.4–10 ng g−1 Shao et al. [114]
Milk or soy-based infant formula 5(5) 44–113 ng g−1 Kuo and Ding [43]14(14) 0.1–13.2 ng mL−1 Biles et al. [15]
Fish 8(4) 24–109 ng g−1 Thomson et al. [17]10(9) 10–43 ng g−1 Goodson et al. [16]
2(2) 2.1–43 ng g−1a Braunrath et al. [78]7(5) 0.3–1.0 ng g−1 Shao et al. [28]
Seafood 5(5) 13–213 ng g−1 Basheer et al. [51]
Meat 6(2) 29–98 ng g−1 Thomson et al. [17]2(2) 9.6–22.0 ng g −1 Braunrath et al. [78]
20(8) 0.3–7.1 ng g−1 Shao et al. [28]
Eggs 10(1) 0.5 ng g−1 Shao et al. [114]
Soup and sauces 8(4) 11–21 ng g−1 Thomson et al. [17]10(4) 8–21 ng g−1 Goodson et al. [16]
4(4) 9.6–37.6 ng g−1 Braunrath et al. [78]
Infant formula, other than milk or non-specified 13(11) 9–384 ng g−1 Goodson et al. [16]
Honey 107(17) 3–33 ng g−1 Inoue et al. [62]Pet foods 26(26) 13–206 ng g−1 Kang and Kondo [44]
Wine (vat) 10(8) 0.2–2.1 ng mL−1 Brenn-Struckhofova and Cichna-Markl [79](Bottle) 48(37) 0.2–1.6 ng mL−1 Brenn-Struckhofova and Cichna-Markl [79]
Soft-drinks 7(6) 0.1–3.4 ng mL−1 Braunrath et al. [78]M
ua[egttasdaidait
tyattrib[ac
oo(rrbl(lebc
3
pqscThsw
ineral water (PET bottles) 9(9)
a Concentrations referred to the solid portion of the container.b Concentrations referred to the liquid portion of the container.
sually preferred for extraction of solid foods [16,17,39,40,42,44,46]lthough others like acetone [41], methanol [39,47] and ethanol43] may also extract BPA efficiently. Liquid foods are extracted withthyl acetate [45], chloroform [48] or dichloromethane [33]. As aeneral rule, repeated extractions are usually necessary to ensurehe complete isolation of BPA. Typical overall solvent consump-ion, including repeated extractions and washing, is between 40nd 300 mL. Extraction times range from 10 min to 120 min usingtirring or sonication to favour equilibrium partition. Sodium anhy-rous sulphate is frequently added to the sample before [40,41] orfter extraction [16,17] in order to remove trace amounts of watern the organic layer. The importance of controlling the enzymaticegradation of BPA during extraction in samples such as fresh fruitnd vegetables has been highlighted since it leads to poor recover-es [41]. Degradation may be prevented by addition of 0.1N HCl tohe extractant solvent [41].
Because of the limited selectivity of solvent-based extractionshere is a need for extensive clean-up prior to instrumental anal-sis. Removal of lipids from the extract is essential for samples ofnimal origin (e.g. fish, meat) since they can significantly reducehe analytical performance of LC and GC. Lipidic material affectshe active surface of the stationary phase in LC and degrades theesolution power of the column. In GC/MS, lipids accumulate in the
njection port, column and ion source. Fat removal is mainly madey liquid–liquid extraction with n-heptane [16], trimethylpentane17] and n-hexane [39,40,46] or by freezing the lipids in the extractt −24 ◦C for 40 min followed by filtration [38]. The use of SPE forlean-up of the BPA-containing extracts becomes inevitable in mostvhPet
3–10 ng L−1 Toyo’oka and Oshige [45]
f sample treatment procedures [38,40–44,46]. After that, the evap-ration of elutes, reconstitution with organic solvent and filtration0.20–0.5 �m) completes the sample treatment. Although overallecoveries of these procedures are usually well above 75%, lowecoveries (<50%) due to matrix–analyte interactions were reportedy Thomson et al. for a variety of foods [17]. Kuo et al. also obtainedow recoveries (18%) and high relative standard deviation (RSD)24%) in a specific type of infant formula powders, namely a hypoal-ergenic brand, this fact being related to the presence of hydrolyzednzymes influencing the SPE [49]. Table 3 compares several solvent-ased extraction methods in terms of overall solvent consumption,oncentration factors and recoveries, among other characteristics.
.1.2. Microwave-assisted extraction (MAE)MAE is based on the application of microwave energy to the sam-
le during extraction, which is consequently agitated and heateduickly. It constitutes a good alternative to the solvent extraction ofolid and semi-solid food samples because good extraction efficien-ies can be achieved using less solvent and shorter extraction times.he application of MAE to the extraction of BPA-containing foodsas been a rather limited and restricted to fish [50,51]. Low sampleizes (typically between 0.2 and 1 g of thawed and crushed tissue)ere subject to MAE with 20 mL of dichloromethane:methanol (2:1,
/v) [50] or 10 mL (20% water content) of tetramethylammoniumydroxide (TMOH) and 1 mL n-nonane [51] for 15–20 min. SPE (Sep-ak NH2) [49] and Oasis-HLB [50] were used for clean-up of thextracts with the subsequent solvent evaporation and reconstitu-ion prior to LC/MS [50] or GC/MS [51]. Average recoveries were
A.Ballesteros-G
ómez
etal./J.Chrom
atogr.A1216
(2009)449–469
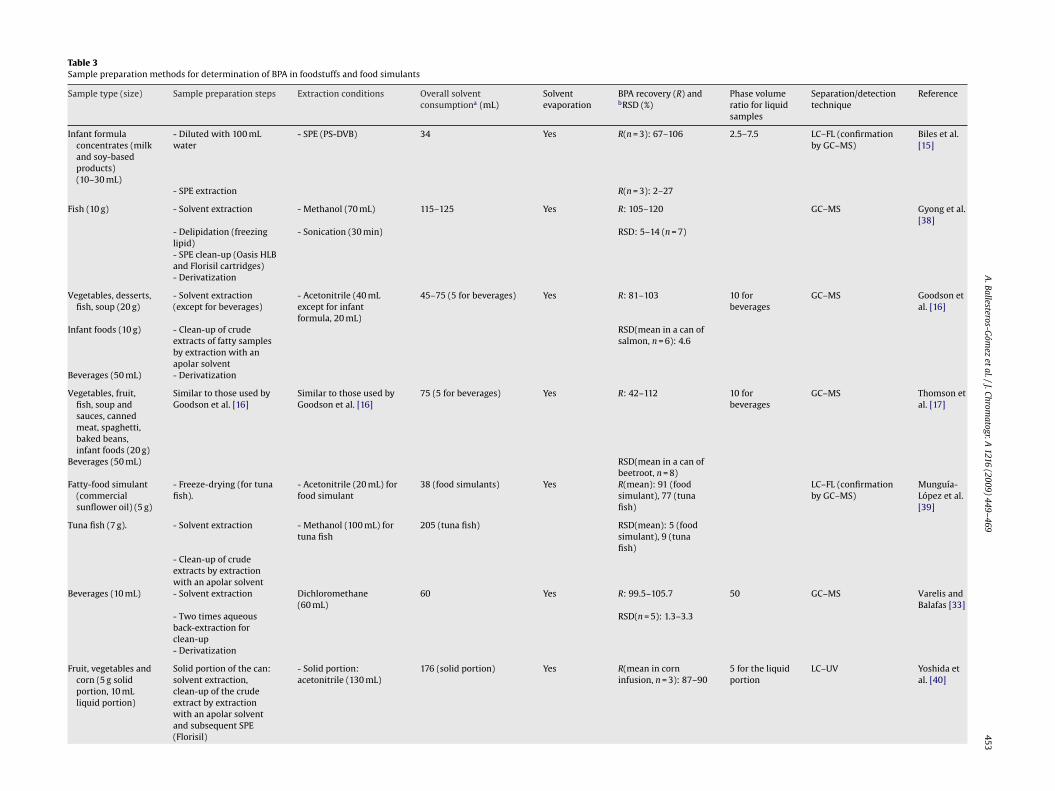
453Table 3Sample preparation methods for determination of BPA in foodstuffs and food simulants
Sample type (size) Sample preparation steps Extraction conditions Overall solventconsumptiona (mL)
Solventevaporation
BPA recovery (R) andbRSD (%)
Phase volumeratio for liquidsamples
Separation/detectiontechnique
Reference
Infant formulaconcentrates (milkand soy-basedproducts)(10–30 mL)
- Diluted with 100 mLwater
- SPE (PS-DVB) 34 Yes R(n = 3): 67–106 2.5–7.5 LC–FL (confirmationby GC–MS)
Biles et al.[15]
- SPE extraction R(n = 3): 2–27
Fish (10 g) - Solvent extraction - Methanol (70 mL) 115–125 Yes R: 105–120 GC–MS Gyong et al.[38]
- Delipidation (freezinglipid)
- Sonication (30 min) RSD: 5–14 (n = 7)
- SPE clean-up (Oasis HLBand Florisil cartridges)- Derivatization
Vegetables, desserts,fish, soup (20 g)
- Solvent extraction(except for beverages)
- Acetonitrile (40 mLexcept for infantformula, 20 mL)
45–75 (5 for beverages) Yes R: 81–103 10 forbeverages
GC–MS Goodson etal. [16]
Infant foods (10 g) - Clean-up of crudeextracts of fatty samplesby extraction with anapolar solvent
RSD(mean in a can ofsalmon, n = 6): 4.6
Beverages (50 mL) - Derivatization
Vegetables, fruit,fish, soup andsauces, cannedmeat, spaghetti,baked beans,infant foods (20 g)
Similar to those used byGoodson et al. [16]
Similar to those used byGoodson et al. [16]
75 (5 for beverages) Yes R: 42–112 10 forbeverages
GC–MS Thomson etal. [17]
Beverages (50 mL) RSD(mean in a can ofbeetroot, n = 8)
Fatty-food simulant(commercialsunflower oil) (5 g)
- Freeze-drying (for tunafish).
- Acetonitrile (20 mL) forfood simulant
38 (food simulants) Yes R(mean): 91 (foodsimulant), 77 (tunafish)
LC–FL (confirmationby GC–MS)
Munguía-López et al.[39]
Tuna fish (7 g). - Solvent extraction - Methanol (100 mL) fortuna fish
205 (tuna fish) RSD(mean): 5 (foodsimulant), 9 (tunafish)
- Clean-up of crudeextracts by extractionwith an apolar solvent
Beverages (10 mL) - Solvent extraction Dichloromethane(60 mL)
60 Yes R: 99.5–105.7 50 GC–MS Varelis andBalafas [33]
- Two times aqueousback-extraction forclean-up
RSD(n = 5): 1.3–3.3
- Derivatization
Fruit, vegetables andcorn (5 g solidportion, 10 mLliquid portion)
Solid portion of the can:solvent extraction,clean-up of the crudeextract by extractionwith an apolar solventand subsequent SPE(Florisil)
- Solid portion:acetonitrile (130 mL)
176 (solid portion) Yes R(mean in corninfusion, n = 3): 87–90
5 for the liquidportion
LC–UV Yoshida etal. [40]
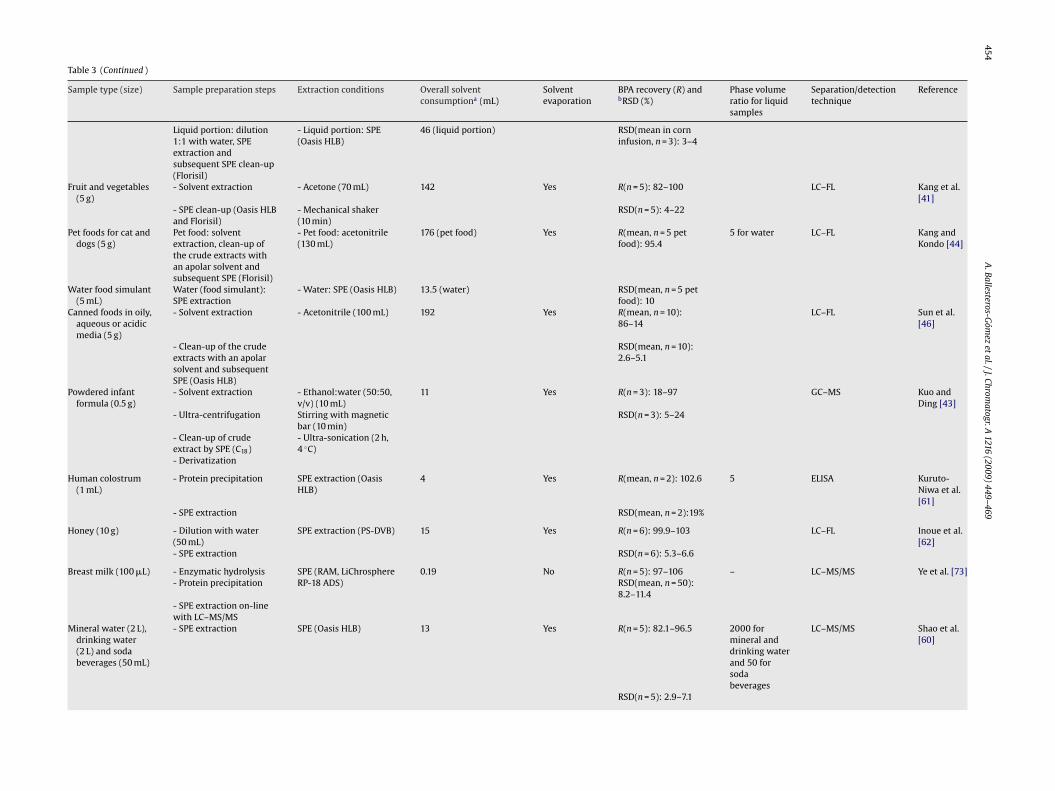
454A
.Ballesteros-Góm
ezet
al./J.Chromatogr.A
1216(2009)
449–469Table 3 (Continued )
Sample type (size) Sample preparation steps Extraction conditions Overall solventconsumptiona (mL)
Solventevaporation
BPA recovery (R) andbRSD (%)
Phase volumeratio for liquidsamples
Separation/detectiontechnique
Reference
Liquid portion: dilution1:1 with water, SPEextraction andsubsequent SPE clean-up(Florisil)
- Liquid portion: SPE(Oasis HLB)
46 (liquid portion) RSD(mean in corninfusion, n = 3): 3–4
Fruit and vegetables(5 g)
- Solvent extraction - Acetone (70 mL) 142 Yes R(n = 5): 82–100 LC–FL Kang et al.[41]
- SPE clean-up (Oasis HLBand Florisil)
- Mechanical shaker(10 min)
RSD(n = 5): 4–22
Pet foods for cat anddogs (5 g)
Pet food: solventextraction, clean-up ofthe crude extracts withan apolar solvent andsubsequent SPE (Florisil)
- Pet food: acetonitrile(130 mL)
176 (pet food) Yes R(mean, n = 5 petfood): 95.4
5 for water LC–FL Kang andKondo [44]
Water food simulant(5 mL)
Water (food simulant):SPE extraction
- Water: SPE (Oasis HLB) 13.5 (water) RSD(mean, n = 5 petfood): 10
Canned foods in oily,aqueous or acidicmedia (5 g)
- Solvent extraction - Acetonitrile (100 mL) 192 Yes R(mean, n = 10):86–14
LC–FL Sun et al.[46]
- Clean-up of the crudeextracts with an apolarsolvent and subsequentSPE (Oasis HLB)
RSD(mean, n = 10):2.6–5.1
Powdered infantformula (0.5 g)
- Solvent extraction - Ethanol:water (50:50,v/v) (10 mL)
11 Yes R(n = 3): 18–97 GC–MS Kuo andDing [43]
- Ultra-centrifugation Stirring with magneticbar (10 min)
RSD(n = 3): 5–24
- Clean-up of crudeextract by SPE (C18)
- Ultra-sonication (2 h,4 ◦C)
- Derivatization
Human colostrum(1 mL)
- Protein precipitation SPE extraction (OasisHLB)
4 Yes R(mean, n = 2): 102.6 5 ELISA Kuruto-Niwa et al.[61]
- SPE extraction RSD(mean, n = 2):19%
Honey (10 g) - Dilution with water(50 mL)
SPE extraction (PS-DVB) 15 Yes R(n = 6): 99.9–103 LC–FL Inoue et al.[62]
- SPE extraction RSD(n = 6): 5.3–6.6
Breast milk (100 �L) - Enzymatic hydrolysis SPE (RAM, LiChrosphere 0.19 No R(n = 5): 97–106 – LC–MS/MS Ye et al. [73]- Protein precipitation RP-18 ADS) RSD(mean, n = 50):
8.2–11.4- SPE extraction on-linewith LC–MS/MS
Mineral water (2 L),drinking water(2 L) and sodabeverages (50 mL)
- SPE extraction SPE (Oasis HLB) 13 Yes R(n = 5): 82.1–96.5 2000 formineral anddrinking waterand 50 forsodabeverages
LC–MS/MS Shao et al.[60]
RSD(n = 5): 2.9–7.1

A. Ballesteros-Gómez et al. / J. ChromM
ilk
(con
den
sed
and
pow
der
ed)
(1g)
-R
econ
stit
uti
onw
ith
1m
Lw
ater
(con
den
sed
)an
d∼6
.4m
Lw
ater
(pow
der
ed).
SPE
(C18
)12
Yes
R(n
=6)
:83
–106
LC–M
SM
arag
ouet
al.[
65]
-A
dd
itio
nw
ater
:met
han
ol18
mL
(8:1
,v/v
)to
des
esta
bili
zeth
eem
uls
ion
RSD
(n=
6):
2.1–
12.5
-SP
Eex
trac
tion
Min
eral
wat
er(5
00
mL)
Solv
ent
extr
acti
onEt
hyla
ceta
te30
0m
LYe
sR
(mea
n,n
=3)
:63
500
0LC
–ED
Toko
’oya
etal
.[45
]R
SD
Jala
pen
op
epp
ers
(120
g)So
lven
tex
trac
tion
-M
eth
anol
(270
mL)
275
mL
Yes
R(m
ean
,n=
3):
89(f
ood
sim
ula
nt)
,91
(jal
apen
op
epp
ers)
LC–F
L(c
onfi
rmat
ion
byG
C–M
S)M
un
guía
-Ló
pez
etal
.[4
7]-
Stir
rin
g(1
h)
and
allo
wed
tost
and
over
nig
ht
RSD
(mea
n,n
=3)
:1(f
ood
sim
ula
nt)
,6(j
alap
eno
pep
per
s)
Abb
revi
atio
ns:
SPE,
soli
d-p
has
eex
trac
tion
;PS
-DV
B,p
olys
tyre
ne
div
inyl
ben
zen
e;R
AM
,res
tric
ted
acce
ssm
ater
ial.
aO
vera
llso
lven
tco
nsu
mpt
ion
per
sam
ple
du
rin
gth
ed
iffe
ren
tsa
mp
lep
rep
arat
ion
step
s.b
Spik
edsa
mp
les,
RSD
asre
pea
tabi
lity
.
if[c
3
tpeT[fauametsodLcPwSitam
3
oaiTeofe(
3
fTttrielAt[casrpao
atogr. A 1216 (2009) 449–469 455
n the ranges 47–49% and 78–79% for spiked liver or muscle tissuerom rainbow trout (RSD 4.1–4.7% [50]) and 92–111% (RSD 3–8%51]) for a variety of seafood including prawns, crabs, cockles, whitelams, squids and fish.
.1.3. Pressurized liquid extraction (PLE)PLE, also known as accelerated solvent extraction (ASE), involves
he use of liquid solvents at elevated temperatures (40–200 ◦C) andressures (1000–2500 psi). Under these conditions, solvents havenhanced solvation power and have increased the extraction rates.he application of PLE to food analysis has been recently reviewed52,53]. Although scarcely used for the extraction of BPA-containingoods, the suitability of PLE for extracting samples of animal [28,54]nd vegetal [32] origin prior to LC/MS has been proved. The solventssed have been dichloromethane for meat (pork, meat, rabbit, ducknd chicken) [28], acetone–n-hexane (1:1, v/v) for fish liver [54] andethanol for cereals [32]. Sodium sulphate [54] and diatomaceous
arth [32] have been utilized as dispersing agents. The optimisa-ion of the extraction conditions for PLE includes the study of theolvent type, temperature, pressure, extraction time and numberf cycles and this can be performed using statistical experimentalesign procedures to minimise the number of experiments [32].ipid removal from meat is carried out by mixing the samples withelite and putting activated neutral alumina on the bottom of theLE cells [28], while its removal from fish liver is made by extractionith n-hexane after PLE [54]. Further clean-up of PLE-extracts by
PE (Oasis HLB [32], Florisil 60 [54] and aminopropyl sorbents [28])s always required to achieve selectivity. The recoveries obtained inhe above applications were from 81% to 104% (RSD 4–9%) for cere-ls [32], 53% (RSD 20%) for fish liver [54] and from 91% to 100% foreat [28].
.2. Solid-phase extraction (SPE)
SPE is by far the most used technique for both the extractionf BPA-containing liquid foods and the clean-up of crude extractsfter solvent extraction. The basis of SPE [55] and its applicationn food analysis [56–58] have been comprehensively reviewed.he selection of a suitable sorbent for BPA will mainly be gov-rned by its properties (moderately polar character and presencef hydrogen acceptor/donor groups, cf. Table 1) and the type ofood matrix. Both, non-selective and selective sorbents have shownxcellent efficiency for the isolation and clean-up of BPA from foodcf. Table 3).
.2.1. Non-selective sorbentsThe divinylbenzene/N-vinylpyrrolidone copolymer (OASIS HLB
rom Waters, 30–200 mg) has been the most used sorbent to date.he hydrophilic N-vinylpyrrolidone polymer affords good wet-ability of the sorbent and acts as a hydrogen acceptor, whilehe hydrophobic divinylbenzene polymer provides reversed-phaseetention for BPA. It offers advantages over classical silica sorbents,.e. high specific area (800 m2/g), possibility to dry out during thextraction procedure without reducing its ability to retain BPA andike other polymeric resins, stability over the entire pH range [59].mong other liquid foods, OASIS HLB has been applied to the isola-
ion of BPA from the aqueous portion of canned fruit and vegetables40], the leachate originated from the treatment of empty pet foodans with distilled water at 121 ◦C for 30 min [44], drinking waternd soda beverages [60], human colostrum [61] and highly viscous
amples, such as honey [62]. Removal of proteins and lipids wasequired for human colostrum samples prior to SPE. For this pur-ose, the samples were diluted with distilled water, treated withcetonitrile or 2-propanol, centrifuged and filtered [61]. The sizef samples was in the ranges 1–50 mL [40,44,61] and 10 g [62]
4 hrom
aw
ac(ecSsvtpoa
pw[iwttftaBbpDrrodtat
h[iaatamf4
fscSupici
3
dcid
aiis
3a[csbsi(icp
fpcCp6apleSta
3tistfem(otp
icdlttfc[taobbt
56 A. Ballesteros-Gómez et al. / J. C
lthough the breakthrough volume increased up to 2 L for drinkingater [60].
On the other hand, as OASIS HLB efficiently removes hydrophilicnd lipophilic interferences [63] and it has been applied to thelean-up of a variety of foods after solvent extraction, namely fishto eliminate the co-extracted polar lipids) [38], fruit and veg-tables (total content or solid portion) [40,41] and a variety ofanned foods [46]. A second clean-up step with a normal-phasePE sorbent (Florisil, a synthetic magnesium silicate) was neces-ary in some applications, namely in the treatment of fish, fruit andegetable samples [38,40,41]. Retention on Florisil mainly occurshrough adsorption, and clean-up has to be carried out from sam-le extracts previously evaporated and redissolved in a non-polarrganic solvent such as n-hexane. Overall recoveries in all the citedpplications were above 80%.
Chemically bonded reversed-phase silica (C18) has been pro-osed as a SPE sorbent for the isolation of BPA from mineralater and wines [64] and canned condensed and powdered milk
65]. Milk samples were diluted with water–methanol (8:1, v/v)n order to obtain extracts as clean as possible. The addition ofater reduced the viscosity of the sample, thus resulting in a bet-
er flow rate during SPE, while the addition of methanol aimed athe destabilization of the milk emulsion. It was argued that if milkat globule membranes are not disrupted, BPA might not be effec-ively and reproducibly extracted. Milk samples with a fat contentbove 3.5% were previously diluted with water. SPE recoveries forPA ranged between 57 and 85% and the internal standard [2H16]isphenol A, BPA-d16 was used for quantitation [65]. The use ofolystyrene-divinylbenzene (PS-DVB, Isolute, and hydroxylated PS-VB, Isolute ENV+) sorbents in this application did not improve
ecoveries despite they offer hydrophobic and �–� interactions foretention of BPA. Contrariwise, recoveries higher than 85% werebtained using PS-DVB sorbents for the isolation of BPA from wateriluted and acidified honey [62], water diluted, centrifuged and fil-ered infant formula and canned vegetables and fruit juices [15,66]nd untreated canned drinks [67]. Recoveries were always higherhan 85%.
Finally, multi-mode phases (Isolute multi-mode cartridges)ave also been proposed for isolation of BPA from instant coffee68]. Multi-mode phases were introduced to exploit multiple-modenteractions and were originally designed for the screening of drugsnd metabolites [69,70]. Besides exploiting the multiple inter-ctions to retain mixtures of analytes with different properties,hese sorbents have been lately used to obtain clean extracts forcompound that can be retained by multiple interactions. Isoluteulti-mode cartridges combine cationic, anionic and non-polar
unctionalities. Recoveries in coffee ranged from 85% to 89% (RSD:–7%).
In general, the use of SPE for isolation of BPA from liquidoods offers two advantages as compared with LLE, namely higherelectivity and lower solvent consumption. On the other hand, con-entrations factors, enhanced by solvent evaporation, are similar forPE and LLE. The SPE of liquid foods usually requires further clean-p and great care must be taken regarding the small particulatesresent in the sample extracts that can produce low recoveries and
rreproducibility by adsorption of analytes or clogging. A detailedomparison between different SPE and solvent extraction methodss given in Table 3.
.2.2. Selective sorbents
A variety of highly selective SPE sorbent materials have beeneveloped that are especially suitable for the determination of traceontaminants in such complex samples as foods, thus perform-ng extraction and clean-up in one step. On the other hand, theevelopment of these smart materials is time-consuming, complex
tt
im
atogr. A 1216 (2009) 449–469
nd expensive, as is the case for immunosorbents and molecularlymprinted polymers. Their application in food analysis, and specif-cally to the determination of BPA, is emerging but a rather limitedo far.
.2.2.1. Restricted access materials (RAMs). The term “restrictedccess materials” (RAMs) was introduced by Desilets et al. in 199171]. These sorbents were particularly developed for the on-linelean-up/extraction of biological samples (such as plasma anderum) in order to perform a high throughput analysis. RAMs com-ine size exclusion of proteins and other macromolecules with theimultaneous enrichment of low molecular mass analytes at thenner pore surface, which are retained by conventional mechanismshydrophobic, ionic or affinity interactions) [72]. The size exclusions achieved by a physical diffusion barrier (pore diameter) or by ahemical barrier created by a polymer at the outer surface of thearticles.
A RAM (LiChrosphere RP-18 ADS from Merck) has been usedor the simultaneous on-line SPE-LC–MS/MS analysis of BPA, otherhenolic compounds and triclocarban in breast milk [73]. The RAMonsists of a bonded reverse-phase covering the internal pore (C4,8 or C18) and a surface modified with hydrophilic groups (glyceryl-ropyl, i.e. diol moieties), which acts as a physical barrier (pore sizenm) that excludes the macromolecules. The method involves theddition of the internal standard (100 �L, 13C12-BPA) to the sam-le, the enzymatic hydrolysis of the conjugated BPA, protein and
ipid removal by the addition of 2-propanol, and centrifugation toxtract the clean supernatant (200 �L), which is subject to on-linePE-LC–MS/MS. It can also be used to measure the free BPA by omit-ing the enzymatic treatment. Recoveries for BPA were between 94nd 106% and the precision of the method was in the range 5–19%.
.2.2.2. Immunosorbents (ISs). Immunoaffinity columns (IAC) orhe so-called immunosorbents (ISs) are made by covalently bond-ng antibodies onto an appropriate support. They provide uniqueelectivity on the basis of molecular recognition, which is par-icularly suited to complex food matrices. ISs can be designedor targeting a single analyte or by exploiting cross-reactivity toxtract a whole class of structurally related compounds. Althoughost applications are for biological and environmental samples
reviewed in [74]), ISs have also been exploited for the extractionf foods and are commercially available for the analysis of naturaloxins [75,76]. The main drawbacks of this technique is the cost toroduce the antibodies and the short-life of columns.
Several methods have been reported that use sol–gelmmunoaffinity chromatography for the clean-up of liquid foods orrude extracts prior to the determination of BPA by LC–fluorescenceetection. Non-antibody-containing sol–gel precolumns were on-
ine coupled with the immunoaffinity columns in order to preventhem from the clogging caused by the small particles present inhe sample extracts. The approach was applicable to a variety ofoods including canned beverages, fruits, vegetables [77,78], fat-ontaining foodstuffs (e.g. tuna, cream, potato soup) [78] and wine79]. The ISs produced by immobilization of the antibodies intohe pores of a sol–gel generated silica matrix are simpler, cheapernd more versatile than those prepared by the covalent couplingf polyclonal antibodies to CNBr-activated Sepharose 4B [80]. Theinding capacity of the IAC columns for BPA was 280 ng and goodatch to batch reproducibility was found, being reusable at least 15imes. Substances showing high cross-reactivity (>1%) did not affect
he determination of BPA since they were efficiently separated byhe LC system.In general, food samples have to be treated to some extentn order to make them compatible with immunoaffinity chro-
atography. Dilution with water is essential since organic solvents
A. Ballesteros-Gómez et al. / J. Chrom
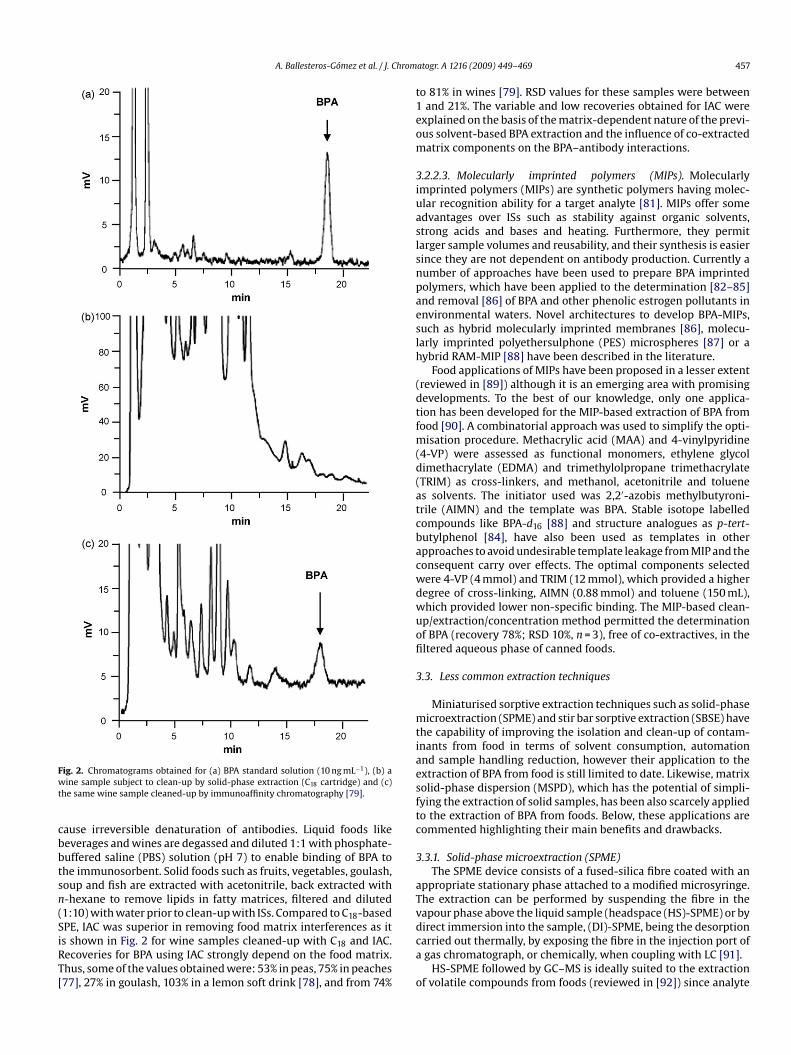
Fwt
cbbtsn(SiRT[
t1eom
3iuaslsnpaeslh
(dtfm(d(atcbacwdwuofi
3
mtiaesftc
3
aTvdirect immersion into the sample, (DI)-SPME, being the desorption
ig. 2. Chromatograms obtained for (a) BPA standard solution (10 ng mL−1), (b) aine sample subject to clean-up by solid-phase extraction (C18 cartridge) and (c)
he same wine sample cleaned-up by immunoaffinity chromatography [79].
ause irreversible denaturation of antibodies. Liquid foods likeeverages and wines are degassed and diluted 1:1 with phosphate-uffered saline (PBS) solution (pH 7) to enable binding of BPA tohe immunosorbent. Solid foods such as fruits, vegetables, goulash,oup and fish are extracted with acetonitrile, back extracted with-hexane to remove lipids in fatty matrices, filtered and diluted1:10) with water prior to clean-up with ISs. Compared to C18-basedPE, IAC was superior in removing food matrix interferences as it
s shown in Fig. 2 for wine samples cleaned-up with C18 and IAC.ecoveries for BPA using IAC strongly depend on the food matrix.hus, some of the values obtained were: 53% in peas, 75% in peaches77], 27% in goulash, 103% in a lemon soft drink [78], and from 74%ca
o
atogr. A 1216 (2009) 449–469 457
o 81% in wines [79]. RSD values for these samples were betweenand 21%. The variable and low recoveries obtained for IAC were
xplained on the basis of the matrix-dependent nature of the previ-us solvent-based BPA extraction and the influence of co-extractedatrix components on the BPA–antibody interactions.
.2.2.3. Molecularly imprinted polymers (MIPs). Molecularlymprinted polymers (MIPs) are synthetic polymers having molec-lar recognition ability for a target analyte [81]. MIPs offer somedvantages over ISs such as stability against organic solvents,trong acids and bases and heating. Furthermore, they permitarger sample volumes and reusability, and their synthesis is easierince they are not dependent on antibody production. Currently aumber of approaches have been used to prepare BPA imprintedolymers, which have been applied to the determination [82–85]nd removal [86] of BPA and other phenolic estrogen pollutants innvironmental waters. Novel architectures to develop BPA-MIPs,uch as hybrid molecularly imprinted membranes [86], molecu-arly imprinted polyethersulphone (PES) microspheres [87] or aybrid RAM-MIP [88] have been described in the literature.
Food applications of MIPs have been proposed in a lesser extentreviewed in [89]) although it is an emerging area with promisingevelopments. To the best of our knowledge, only one applica-ion has been developed for the MIP-based extraction of BPA fromood [90]. A combinatorial approach was used to simplify the opti-
isation procedure. Methacrylic acid (MAA) and 4-vinylpyridine4-VP) were assessed as functional monomers, ethylene glycolimethacrylate (EDMA) and trimethylolpropane trimethacrylateTRIM) as cross-linkers, and methanol, acetonitrile and toluenes solvents. The initiator used was 2,2′-azobis methylbutyroni-rile (AIMN) and the template was BPA. Stable isotope labelledompounds like BPA-d16 [88] and structure analogues as p-tert-utylphenol [84], have also been used as templates in otherpproaches to avoid undesirable template leakage from MIP and theonsequent carry over effects. The optimal components selectedere 4-VP (4 mmol) and TRIM (12 mmol), which provided a higheregree of cross-linking, AIMN (0.88 mmol) and toluene (150 mL),hich provided lower non-specific binding. The MIP-based clean-p/extraction/concentration method permitted the determinationf BPA (recovery 78%; RSD 10%, n = 3), free of co-extractives, in theltered aqueous phase of canned foods.
.3. Less common extraction techniques
Miniaturised sorptive extraction techniques such as solid-phaseicroextraction (SPME) and stir bar sorptive extraction (SBSE) have
he capability of improving the isolation and clean-up of contam-nants from food in terms of solvent consumption, automationnd sample handling reduction, however their application to thextraction of BPA from food is still limited to date. Likewise, matrixolid-phase dispersion (MSPD), which has the potential of simpli-ying the extraction of solid samples, has been also scarcely appliedo the extraction of BPA from foods. Below, these applications areommented highlighting their main benefits and drawbacks.
.3.1. Solid-phase microextraction (SPME)The SPME device consists of a fused-silica fibre coated with an
ppropriate stationary phase attached to a modified microsyringe.he extraction can be performed by suspending the fibre in theapour phase above the liquid sample (headspace (HS)-SPME) or by
arried out thermally, by exposing the fibre in the injection port ofgas chromatograph, or chemically, when coupling with LC [91].
HS-SPME followed by GC–MS is ideally suited to the extractionf volatile compounds from foods (reviewed in [92]) since analyte

4 hrom
dtfieLiwdDttttPtBaMp[
m[dimiotdewacew(nssdt(mcidSbstpa[
3
pihcil(
tLoab
adhiGreuPcwa(amoreaLtppcttpc4tuoaafas8r
3
elacobbtc
tp
58 A. Ballesteros-Gómez et al. / J. C
eterminations are practically interference-free, but no applica-ions related to BPA in food have been developed so far. DI-SPMEollowed by GC–MS has been applied to the determination of BPAn aqueous food simulants [93] and water from plastic contain-rs and tableware [94]. DI-SPME has also been proposed prior toC–FL for the fast screening of BPA in canned foods, just analyz-ng the aqueous phase of the can [95]. Different stationary phases
ith the capability to withstand high injector temperatures (poly-imethylsiloxane, PDMS; carboxen/PDMS; PDMS/divinylbenzene,VB; carbowax CW/DVB and polyacrylate, PA) were assessed for
he extraction of BPA by DI-SPME–GC [93,94]. The coating PA gavehe best recoveries on the basis of its similar polarity to BPA. Amonghe coating materials tested for extraction of BPA by DI-SPME–LC,he most polar fibres gave the best results. Recoveries were 6% forDMS-DVB, 7% for PA, 58% for PDMS and 90% for CW, so the lat-er was selected as optimal. The effect of the ionic strength onPA recoveries was significant and the three methods included theddition of salt, namely 7.5% (w/w) [93,95] and 15.4% (w/w) [94].aximum sorption occurred at neutral pH [93,95] and the sorption
rocess was carried out at room temperature [95] or at 50–55 ◦C93,94].
The three methods were sensitive enough for the screening origration assessment of BPA (quantitation limits were 3.8 mg L−1
95] and 0.01–0.02 mg L−1 [93,94]), but they also presented seriousrawbacks. Thus, the automation of SPME with LC–FL through the
nterface supplied by Supelco was not successful, being the off-lineode more efficient that the automated one in terms of sensitiv-
ty and peak resolution [95]. This fact was explained on the basisf the higher concentration factor in the off-line mode (∼67) andhe slower introduction of the analytes into the LC system in theynamic mode, which resulted in broader peaks [95]. In fact, Eis-rt and Pawliszyn [96] later developed the in-tube SPME technique,hich uses an inner-wall coated capillary as the extraction medium
nd it is more suitable for LC automation. In-tube SPE has been suc-essfully applied to the extraction of BPA and other estrogens fromnvironmental waters [97,98], but not in food samples yet. Othereak point of the SPME–LC–FL method was the reproducibility
RSD was 22% for 100 mg L−1 of BPA). With regard to the determi-ation of BPA by SPME–GC–MS, the variability was high betweeneries (mean RSD ∼10–13%) and consequently a calibration for eacheries of analysis was recommended [93,94]. On the other hand,espite only aqueous simulants or drinking water were analyzed,he low recoveries resulting from the effect of matrix components7–65%) [94,95], made advisable the use of the standard addition
ethod for BPA quantitation. The low recoveries were related toompetitiveness between BPA and the matrix components for bind-ng to the limited volume of stationary phase, which led to analyteisplacement [94]. To our knowledge no study related to the use ofPME for extraction of BPA from more complex food matrices haseen reported so far. In fact, the use of DI-SPME in food matriceshould need exhaustive clean-up to ensure the reproducibility andhe robustness of the fibres, which can be affected by adsorption ofroteins or clogged by particles, similarly to that occurring in thenalysis of BPA in other complex matrices such as human plasma99].
.3.2. Stir bar sorptive extraction (SBSE)SBSE uses a stir bar into a sealed glass tube that is coated with
olydimethylsiloxane (PDMS) to extract solutes [100]. In compar-son to SPME, a larger amount of stationary phase (50–250 times
igher) is used and hence better recoveries, sensitivities and sampleapacity are achieved. Like in SPME, the stir bar can be immersednto the liquid sample or can be held in the headspace above theiquid or the solid sample to determine volatiles and semi-volatilesheadspace sorptive extraction, HSSE). Removal of analytes fromwottr
atogr. A 1216 (2009) 449–469
he bar is achieved by GC thermal desorption or elution with aC solvent. The technique was first developed for the extraction ofrganic compounds from aqueous samples, but today a variety ofpplications in the environmental, food and biomedical areas haveeen reported (reviewed in [101]).
Since PDMS is the unique commercially available coating, thepplication of SBSE to polar and semi-polar compounds requireserivatization. Thus, the extraction of BPA from water [102–104]as been carried out by BPA derivatization with acetic anhydride,
mmersion of the bar in the solution and thermal desorption-C–MS analysis. In this way, the LODs for BPA were in the
ange 0.6–2 ng L−1. Despite new SBSE coatings are necessary toxtend the scope of SBSE, only a few ones have been reportedp to now. It is worth noting the ‘dual-phase twisters’ (shortDMS tubes closed at both ends with magnets, with an inneravity that is packed with activated carbon adsorbent), whichere proposed to improve recoveries of polar compounds [105],
nd two selective materials, i.e. a molecularly imprinted polymerMIP) for the extraction of monocrotophos [106] and a restrictedccess media (RAM) for the extraction of caffeine and relatedetabolites [107]. In this respect, a novel SBSE coating based
n polydimethylsiloxane/�-cyclodextrin (PDMS/�-CD) has beenecently developed and applied to the extraction of BPA (and otherstrogenic compounds) from environmental water, drinking waternd leachate from one-off dishware, prior to determination byC–FL [108]. The PDMS/�-CD coating was prepared by a sol–gelechnique, which provided direct chemical binding of the stationaryhase to the glass substrate and resulted in higher stability com-ared to the physical deposition technique frequently employed foroating. The introduction of the cyclodextrin permitted to increasehe recovery of polar compounds due to the high number of func-ional hydroxyl groups in the �-CD molecule and the selectivityrovided by its molecular recognition ability. The extraction wasarried out by immersion of the stir bar in the sample (5 mL) for0 min (non-equilibrium conditions) at room temperature. Desorp-ion of BPA was made with 100 �L of methanol for 2 min underltrasonic agitation (ratio of sample volume over extract volumef 50). The PDMS/�-CD coated stir bar provided better selectivitynd sensitivity for polar compounds than a PDMS coated stir barnd a PDMS–�-CD coated fibre. The lower method performanceor SPME was attributed to the smaller stationary phase volumend the introduction of a third competitive phase in the stirringtep, namely the Teflon coated magnetic bar. The LOD for BPA wasng L−1, with recoveries between 86 and 116% and RSDs in the
ange 0.7–10.7%.
.3.3. Matrix solid-phase dispersion (MSPD)MSPD was first reported in 1989 [109] and it is well suited to the
xtraction of solid, semi-solid and/or highly viscous food and bio-ogical matrices (reviewed in [110,111]). The sample is mixed withsorbent such as C18 bonded silica, sodium sulphate or diatoma-
eous earth, followed by washing with a small volume of solventr packing the dispersant sorbent material into a SPE mini-columnefore elution. MSPD is simple and versatile and offers the possi-ility of performing extraction and clean-up in one step [112,113],his resulting in a drastic shortening of analysis times and solventonsumption.
MSDP has been recently reported for the simultaneous extrac-ion of BPA, nonylphenol and octylphenol from eggs and milk [114]rior to LC/MS/MS determination. The samples (1 g) were blended
ith 1 g of C18 powder just for 5 min, packed and eluted with 10 mLf ethanol. This dispersant agent gave better recoveries for BPAhan graphite carbon black (GCB) because of the strong adsorp-ion of BPA to GCB. The eluent was evaporated to dryness and theesidue was redissolved in dichloromethane/hexane and subject to
A.Ballesteros-G
ómez
etal./J.Chrom
atogr.A1216
(2009)449–469
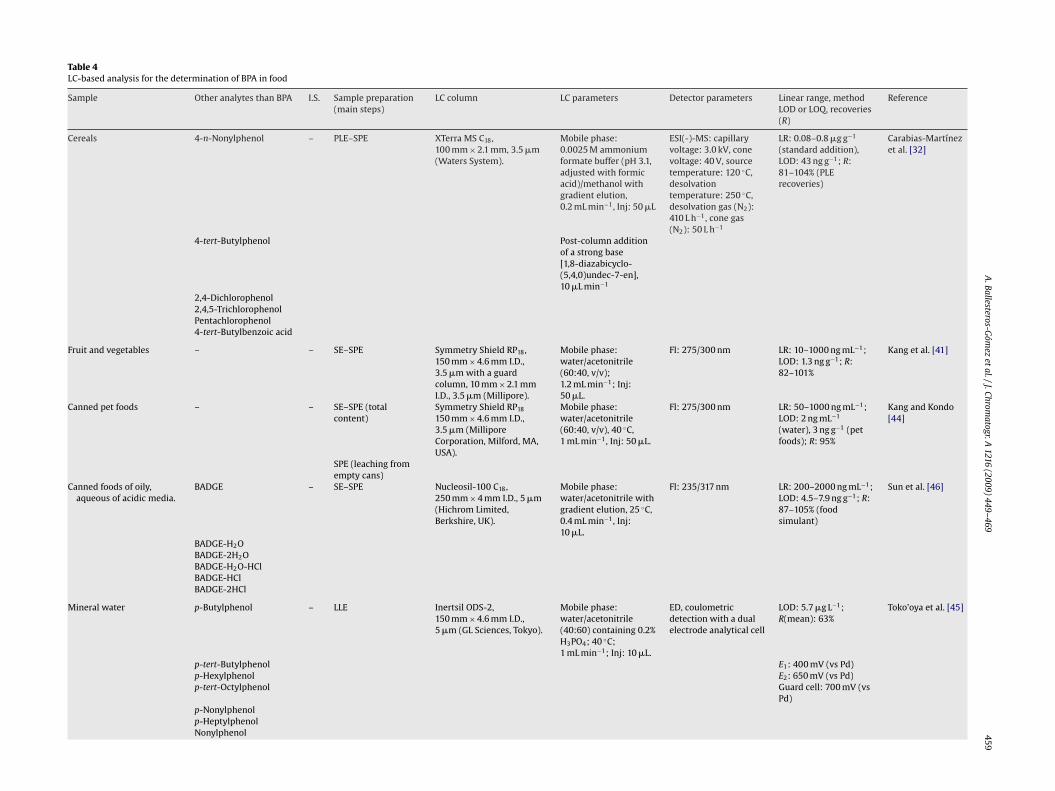
459Table 4LC-based analysis for the determination of BPA in food
Sample Other analytes than BPA I.S. Sample preparation(main steps)
LC column LC parameters Detector parameters Linear range, methodLOD or LOQ, recoveries(R)
Reference
Cereals 4-n-Nonylphenol – PLE–SPE XTerra MS C18,100 mm × 2.1 mm, 3.5 �m(Waters System).
Mobile phase:0.0025 M ammoniumformate buffer (pH 3.1,adjusted with formicacid)/methanol withgradient elution,0.2 mL min−1, Inj: 50 �L
ESI(-)-MS: capillaryvoltage: 3.0 kV, conevoltage: 40 V, sourcetemperature: 120 ◦C,desolvationtemperature: 250 ◦C,desolvation gas (N2):410 L h−1, cone gas(N2): 50 L h−1
LR: 0.08–0.8 �g g−1
(standard addition),LOD: 43 ng g−1; R:81–104% (PLErecoveries)
Carabias-Martínezet al. [32]
4-tert-Butylphenol Post-column additionof a strong base[1,8-diazabicyclo-(5,4,0)undec-7-en],10 �L min−1
2,4-Dichlorophenol2,4,5-TrichlorophenolPentachlorophenol4-tert-Butylbenzoic acid
Fruit and vegetables – – SE–SPE Symmetry Shield RP18,150 mm × 4.6 mm I.D.,3.5 �m with a guardcolumn, 10 mm × 2.1 mmI.D., 3.5 �m (Millipore).
Mobile phase:water/acetonitrile(60:40, v/v);1.2 mL min−1; Inj:50 �L.
Fl: 275/300 nm LR: 10–1000 ng mL−1;LOD: 1.3 ng g−1; R:82–101%
Kang et al. [41]
Canned pet foods – – SE–SPE (totalcontent)
Symmetry Shield RP18
150 mm × 4.6 mm I.D.,3.5 �m (MilliporeCorporation, Milford, MA,USA).
Mobile phase:water/acetonitrile(60:40, v/v), 40 ◦C,1 mL min−1, Inj: 50 �L.
Fl: 275/300 nm LR: 50–1000 ng mL−1;LOD: 2 ng mL−1
(water), 3 ng g−1 (petfoods); R: 95%
Kang and Kondo[44]
SPE (leaching fromempty cans)
Canned foods of oily,aqueous of acidic media.
BADGE – SE–SPE Nucleosil-100 C18,250 mm × 4 mm I.D., 5 �m(Hichrom Limited,Berkshire, UK).
Mobile phase:water/acetonitrile withgradient elution, 25 ◦C,0.4 mL min−1, Inj:10 �L.
Fl: 235/317 nm LR: 200–2000 ng mL−1;LOD: 4.5–7.9 ng g−1; R:87–105% (foodsimulant)
Sun et al. [46]
BADGE-H2OBADGE-2H2OBADGE-H2O-HClBADGE-HClBADGE-2HCl
Mineral water p-Butylphenol – LLE Inertsil ODS-2,150 mm × 4.6 mm I.D.,5 �m (GL Sciences, Tokyo).
Mobile phase:water/acetonitrile(40:60) containing 0.2%H3PO4; 40 ◦C;1 mL min−1; Inj: 10 �L.
ED, coulometricdetection with a dualelectrode analytical cell
LOD: 5.7 �g L−1;R(mean): 63%
Toko’oya et al. [45]
p-tert-Butylphenol E1: 400 mV (vs Pd)p-Hexylphenol E2: 650 mV (vs Pd)p-tert-Octylphenol Guard cell: 700 mV (vs
Pd)p-Nonylphenolp-HeptylphenolNonylphenol
460A
.Ballesteros-Góm
ezet
al./J.Chromatogr.A
1216(2009)
449–469Table 4 (Continued )
Sample Other analytes than BPA I.S. Sample preparation(main steps)
LC column LC parameters Detector parameters Linear range, methodLOD or LOQ, recoveries(R)
Reference
p-Octylphenol
Fish 4-tert-Octylphenol BPA-d16 MAE–SPE Zorbax Eclipse XDB-C18,150 mm × 2.1 mm I.D.
Mobile phase:water/methanol withgradient elution,0.4 mL min−1, Inj: notspecified.
APCI(-)-MS: capillaryvoltage 3 kV, coronacurrent: 40 �A,fragmentor voltage:100 V, drying gas flow5 L min−1, nebulisinggas pressure 60 psi,vapourisingtemperature 280 ◦C
LR:20.7–10044 ng mL−1,LOD: 0.1 ng mL−1
(water), 50 ng g−1
(fish); R: 49–79%(absolute recovery,fish)
Pedersen andLindholst [50]
Meat Octylphenol 4-n-Nonylphenol PLE-SPE Symmetry C18,150 mm × 2.1 mm I.D.,3.5 �m.
Mobile phase: water(0.1%ammonia)/methanolwith gradient elution,0.2 mL min−1, Inj:10 �L.
ESI(-)-MS/MS: capillaryvoltage: 3.5 kV, conevoltage: 70 V, sourcetemperature: 100 ◦C
LR: 1–500 ng mL−1;LOD: 0.3 ng g−1; R:92–97%
Shao et al. [28]
Nonylphenol
Water and soda beverages Octylphenol 4-n-Nonylphenol SPE Symmetry C18,150 mm × 2.1 mm I.D.,3.5 �m.
Mobile phase: water(0.1%ammonia)/methanolwith gradient elution,40 ◦C, 0.2 mL min−1, Inj:10 �L.
ESI(-)-MS/MS Capillaryvoltage: 3.5 kV;desolvationtemperature: 350 ◦C
LR: 1–500 ng mL−1;LOD: 0.01 ng L−1
(drinking water),0.6 ng L−1 (sodabeverage); R: 82–97%
Shao et al. [60]
Nonylphenol
Honey Bisphenol F – SPE Quantitation: CAPCELL PAKC18, 150 mm × 4.6 I.D.,5 �m) (Shiseido Co. Tokyo,Japan).
Mobile phase:water/acetonitrile(70:30, v/v);1.2 mL min−1; Inj:10 �L.
Fl: 275/300 nm LR: 25–500; LOD:2 ng g−1; R:93.9–116.4%
Inoue et al. [62]
Confirmation: CAPCELLPAK MG C18
(250 mm × 2 mm, 5 �m(Shiseido Co. Tokyo, Japan)
Mobile phase: water(0.01 aceticacid)/acetonitrile withgradient elution;0.3 mL min−1; Inj: 5 �L.
ESI(-)-MS: Capillaryvoltage: 3.5 kV;fragmentor voltage:140 V, 220 V;desolvationtemperature: 350 ◦C
–
Milk – BPA-d16 SPE LiChrospher 100 RP-18,250 mm × 4 mm I.D., 5 �m
Mobile phase:water/methanol(30:70, v/v),0.9 mL min−1, Inj: 10 �L
ESI(-)-MS: capillaryvoltage: 3.5 kV, conevoltage: 70 V, sourcetemperature: 500 ◦C
LR: 5–700 ng mL−1;LOD: 0.7 ng mL−1; R:52% (absoluterecovery), 101%(relative recovery)
Maragou et al. [65]
Aqueous portion of cannedfoods
BADGE – SPME XTerra MS C18,100 mm × 4.6 mm, 5 �m(Waters, Milford, MA, USA).
Mobile phase:water/acetonitrile(50:50, v/v);1.2 mL min−1; Inj:20 �L.
Fl: 275/305 nm LR: 10–500 ng mL−1;LOD: 1.1 ng mL−1; R:7–65%
Nerín et al. [95]
BADGE-diolBADGE-2HClBFDGEBFDGE-diol
A.Ballesteros-G
ómez
etal./J.Chrom
atogr.A1216
(2009)449–469
461
Breast milk 4-tert-Octylphenol 13C12-BPA On-line SPE(RAM)
Two ChromolithTMPerformance RP-18,100 mm × 4.6 mm I.D.(Merck KGaA, Germany).
Mobile phase:water/methanol withgradient elution,0.75 mL min−1, Inj:100 �L.
APCI(-)-MS: needlevoltage: −3 V, curtaingas flow: 20 au,collision gas flow: 9 au,nebulizer gas flow:50 au, nebuliser gastemperature: 500 ◦C
LR: 0.1–100 ng mL−1;LOD: 0.28 ng mL−1; R:94% (SPE recoveries)
Ye et al. [73]
ortho-Phenylphenol2,4-Dichlorophenol2,5-Dichlorophenol2,4,5-Trichlorophenol2,4,6-Trichlorophenol2-Hydroxy-4-metoxybenzophenone
Beverages, fruits andvegetables
– – IAC Spherisorb S ODS1,250 mm × 4.6 mm I.D.,5 �m (Knauer, Berlin,Germany).
Mobile phase: 50 mMsodium acetate buffer(adjusted to pH of 4.8with aceticacid)/acetonitrile(60:40, v/v);1 mL min−1; Inj:100 �L.
Fl: 275/305 nm LR: 0.5–100 ng mL−1;LOD: 0.1 ng mL−1,1.1–1.6 ng g−1; R:53–75%
Braunrath andCichna [77]
Wine – – IAC Quantitation: LiChrospher60 RP-Select B,250 mm × 4.0 mm I.D.,5 �m (Merck).
Mobile phase:water:acetonitrile(70:30, v/v);1 mL min−1; Inj:100 �L.
Fl: 275/305 nm LR: 0.5–100 ng mL−1;LOD: 0.1 ng mL−1; R:74–81%
Brenn-Struckhofova andCichna-Markl [79]
Confirmation: HP-HypersilPDS, 250 mm × 4 mm I.D.,5 �m
Water (0.01 M sodiumacetate buffer, pH 4.8adjusted with aceticacid):acetonitrile withgradient elution;0.6 mL min−1; Inj.:100 �L.
ED (coulometricelectrode arraydetection). Potentialsset at 300, 400, 500,550, 600, 650 and750 mV against Pdreference electrodes
–
Eggs and milk Octylphenol 4-n-Nonylphenol MSPD–SPE Symmetry C18,150 mm × 2.1 mm I.D.,3.5 �m
Mobile phase: water(0.1%ammonia)/methanolwith gradient elution,40 ◦C, 0.2 mL min−1, Inj:10 �L
ESI(-)-MS/MS: capillaryvoltage: 3.5 kV, conevoltage: 70 V,Multiplier voltage:650 V, desolvation gasflow (N2): 550 L h−1
source temperature:100 ◦C, desolvationtemperature: 300 ◦C
LR: 1–500 ng mL−1,LOD: 0.1 ng g−1 (eggsand milk); R: 79–83%(eggs), 86–94% (milk)
Shao et al. [114]
Nonylphenol
Abbreviations: PLE, pressurized liquid extraction; SPE, solid-phase extraction; Inj., injection volume, ESI, electrospray ionization, SE, solvent extraction; Fl, fluorescence detection; BADGE, bisphenol A diglycidyl ether; LLE,liquid–liquid extraction; ED, electrochemical detection; MAE, microwave-assisted extraction; APCI, atmospheric pressure chemical ionization; BFDGE, bisphenol F diglycidyl ether; SPME, solid-phase microextraction, RAM,restricted access material.
4 hromatogr. A 1216 (2009) 449–469
SMv
4
sic(ttoLfhdec
4
MacElftg1cpsa
4
wsnstcL
vhm[tf0
tf(emqe
F2t
4
kmoef[U3wam1atgos
shewpwcoBoHatsm
62 A. Ballesteros-Gómez et al. / J. C
PE with aminopropyl to remove the lipidic fraction of the samples.ean recoveries for BPA ranged from 82% to 91% and the mean RSD
alues for eggs and milk were 6% and 4%, respectively.
. Separation and detection
The determination of BPA in foodstuffs requires the use of highlyensitive and selective techniques due to the trace levels at whicht is frequently found (cf. Table 2) and the complexity of food matri-es. Although the SML set by the EU commission is relatively high600 ng g−1), the reported low-dose effects of BPA has given riseo the development of analytical methods with LODs low enougho asses the human exposure at these levels. The determinationf BPA in food is mainly carried out by LC/FL, LC/MS and GC/MS.iquid chromatography offers the advantage of simplicity over GCor which derivatization step is necessary, while the latter providesigher peak resolution. Other techniques like LC–electrochemicaletection (LC–ED) and immunoassays have been used in a lesserxtent. Detailed information about chromatographic and detectiononditions for BPA determination is summarized in Tables 4 and 5.
.1. Liquid chromatography
LC of BPA is usually carried out in reversed-phase C18 columns.obile phases vary according to the detector coupled to LC. Water
nd acetonitrile are the commonest binary solvents when fluores-ence detection is used while water and methanol are preferred forSI-MS and APCI-MS. Elution conditions highly depend on the ana-
ytes to be determined along with BPA and food matrices. Thus, it isrequent to determine BPA with other phenols, endocrine disrup-ors and migrants from food packaging (cf. Table 4) and in this caseradient elution is always performed. Run times range between5 (e.g. [32]) and 40 min (e.g. [60]) depending on the number ofontaminants determined and matrix composition. LC is usuallyerformed at room temperature but temperatures up to 40 ◦C areometimes recommended [e.g. 32 and 60] to reduce analysis timend increase the reproducibility.
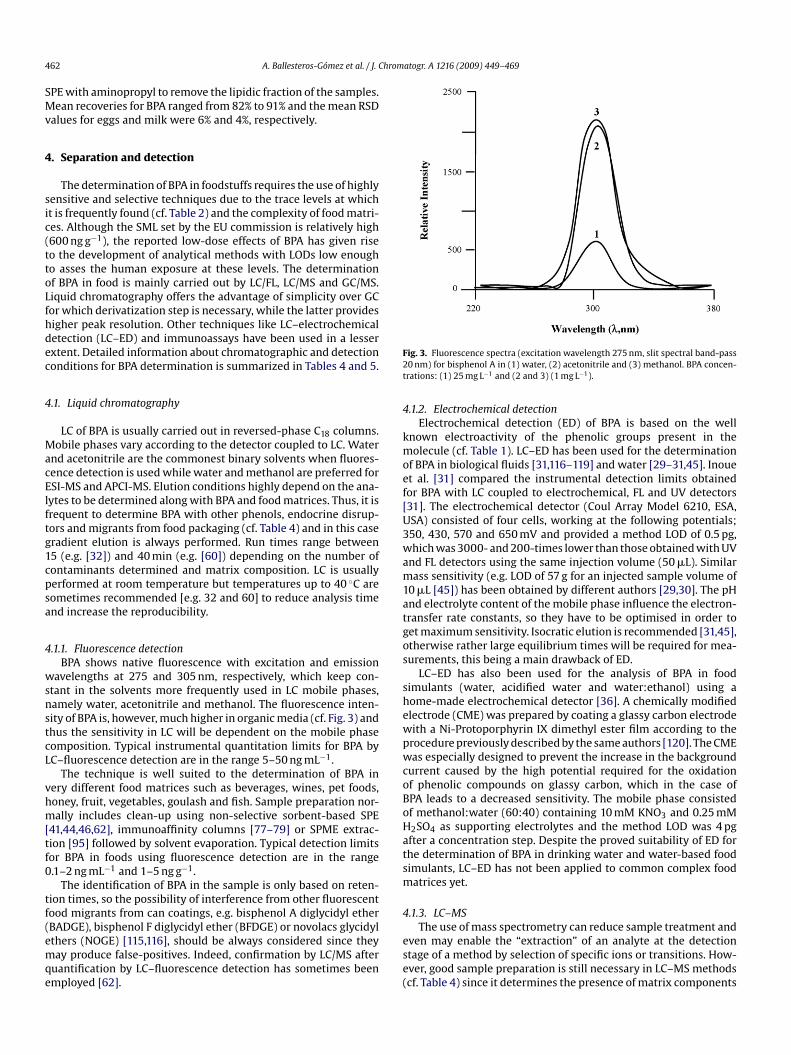
.1.1. Fluorescence detectionBPA shows native fluorescence with excitation and emission
avelengths at 275 and 305 nm, respectively, which keep con-tant in the solvents more frequently used in LC mobile phases,amely water, acetonitrile and methanol. The fluorescence inten-ity of BPA is, however, much higher in organic media (cf. Fig. 3) andhus the sensitivity in LC will be dependent on the mobile phaseomposition. Typical instrumental quantitation limits for BPA byC–fluorescence detection are in the range 5–50 ng mL−1.
The technique is well suited to the determination of BPA inery different food matrices such as beverages, wines, pet foods,oney, fruit, vegetables, goulash and fish. Sample preparation nor-ally includes clean-up using non-selective sorbent-based SPE
41,44,46,62], immunoaffinity columns [77–79] or SPME extrac-ion [95] followed by solvent evaporation. Typical detection limitsor BPA in foods using fluorescence detection are in the range.1–2 ng mL−1 and 1–5 ng g−1.
The identification of BPA in the sample is only based on reten-
ion times, so the possibility of interference from other fluorescentood migrants from can coatings, e.g. bisphenol A diglycidyl etherBADGE), bisphenol F diglycidyl ether (BFDGE) or novolacs glycidylthers (NOGE) [115,116], should be always considered since theyay produce false-positives. Indeed, confirmation by LC/MS afteruantification by LC–fluorescence detection has sometimes beenmployed [62].
4
ese(
ig. 3. Fluorescence spectra (excitation wavelength 275 nm, slit spectral band-pass0 nm) for bisphenol A in (1) water, (2) acetonitrile and (3) methanol. BPA concen-rations: (1) 25 mg L−1 and (2 and 3) (1 mg L−1).
.1.2. Electrochemical detectionElectrochemical detection (ED) of BPA is based on the well
nown electroactivity of the phenolic groups present in theolecule (cf. Table 1). LC–ED has been used for the determination
f BPA in biological fluids [31,116–119] and water [29–31,45]. Inouet al. [31] compared the instrumental detection limits obtainedor BPA with LC coupled to electrochemical, FL and UV detectors31]. The electrochemical detector (Coul Array Model 6210, ESA,SA) consisted of four cells, working at the following potentials;50, 430, 570 and 650 mV and provided a method LOD of 0.5 pg,hich was 3000- and 200-times lower than those obtained with UV
nd FL detectors using the same injection volume (50 �L). Similarass sensitivity (e.g. LOD of 57 g for an injected sample volume of
0 �L [45]) has been obtained by different authors [29,30]. The pHnd electrolyte content of the mobile phase influence the electron-ransfer rate constants, so they have to be optimised in order toet maximum sensitivity. Isocratic elution is recommended [31,45],therwise rather large equilibrium times will be required for mea-urements, this being a main drawback of ED.
LC–ED has also been used for the analysis of BPA in foodimulants (water, acidified water and water:ethanol) using aome-made electrochemical detector [36]. A chemically modifiedlectrode (CME) was prepared by coating a glassy carbon electrodeith a Ni-Protoporphyrin IX dimethyl ester film according to therocedure previously described by the same authors [120]. The CMEas especially designed to prevent the increase in the background
urrent caused by the high potential required for the oxidationf phenolic compounds on glassy carbon, which in the case ofPA leads to a decreased sensitivity. The mobile phase consistedf methanol:water (60:40) containing 10 mM KNO3 and 0.25 mM2SO4 as supporting electrolytes and the method LOD was 4 pgfter a concentration step. Despite the proved suitability of ED forhe determination of BPA in drinking water and water-based foodimulants, LC–ED has not been applied to common complex foodatrices yet.
.1.3. LC–MS
The use of mass spectrometry can reduce sample treatment andven may enable the “extraction” of an analyte at the detectiontage of a method by selection of specific ions or transitions. How-ver, good sample preparation is still necessary in LC–MS methodscf. Table 4) since it determines the presence of matrix components

A.Ballesteros-G
ómez
etal./J.Chrom
atogr.A1216
(2009)449–469
463Table 5GC-based analysis for the determination of BPA in food
Sample Other analytes thanBPA
I.S. Sample preparation(main steps)
Derivatization GC column Temperatureprogram andinjection
MS parameters Linear range, LOD orLOQ, recoveries (R)
Reference
Vegetables fruit, fishsoup and sauces,canned meat,spaghetti and bakedbeans, infant foods,beverages
– BPA-d14 SE or LLE Acetic anhydride DB-5MS,30 m × 0.25 mm
120 ◦C held for2 min, ramped at10 ◦C min−1 to280 ◦C. Inj: splitless
BPA diacetyl: m/z228, m/z 213.
LOQ: 10 ng g−1
(samples <1% fat),20 ng g−1 (samples>1% fat); R: 42–112%.
Thomson etal. [17]
I.D., 0.25 �m (J&W,Folsom, CA, USA).
I.S.: m/z 224
Fish 4-tert-Butylphenol BPA-d16 SE–SPE MTBSTFA, 100 �L tothe dried SPE eluent(75 ◦C, 30 min)
DB-5MS,30 m × 0.25 mm
100 ◦C held for1 min, ramped at3 ◦C min−1 to 200 ◦C,held for 1 min,ramped at20 ◦C min−1 to280 ◦C, held for5 min. Inj: splittingratio 1:10
TBDMS derivateBPA: m/z 441, m/z470
LR: 0.5–50 ng g−1;LOD: 0.41 ng g−1; R:105–120%.
Gyong et al.[38]
4-n-Butylphenol I.D., 0.25 �m (J&W,Folsom, CA, USA)
I.S.: m/z 452, m/z 470
4-Pentylphenol4-n-Hexylphenol4-tert-Octylphenol4-n-HeptylphenolNonylphenol4-Octylphenol
Beverages – BPA-d14 LLE Trifluoroaceticanhydride, 200 �Ladded to the SPEeluent (30 min,room temperature)(60 ◦C, 5 min toremovederivatizationreagent in excess)
HP-5 Trace Analysis,25 m × 0.2 mm I.D.,0.33 �m
100 ◦C held for1 min, ramped at25 ◦C min−1 to150 ◦C, ramped at5 ◦C min−1 to 210 ◦C,ramped at25 ◦C min−1 to280 ◦C, held for2.5 min. Inj: splitless
O-bis(trifluoroacetyl)derivate of BPA: m/z405, m/z 420
LR:0.01–50 �g mL−1;LOD: 1 ng g−1; R:18–97%
Varelis andBalafas [33]
I.S.: m/z 416, m/z 434
Infant formula Daidzein Chrysene-d12 SE–SPE BSTFA:TMCS: DTE(1000:10:2, v/v/w),100 �L added to thedried SPE eluent(80 ◦C, 30 min)
DB-5MS,30 m × 0.25 mm
100 ◦C held for2 min, ramped at10 ◦C min−1 to250 ◦C, ramped at5 ◦C min−1 to 300 ◦C,hold for 5 min. Inj:1 �L, splitless
O-bis(trimethylsilyl)derivate of BPA: m/z372 and m/z 357
LR: 0.01–50 �gmL−1; LOD: 1 ng g−1;R: 18–97%
Kuo andDing [43]
Genistein I.D., 0.25 �m (J&W,Folsom, CA, USA)
I.S.: m/z 284 and m/z269
Seawater and seafood 4-tert-Butylphenol BPA-d14 LLE–SPE (seawater) BSTFA, 100 �L, toevaporated SPEeluate (60 ◦C,30 min)
DB-5MS,30 m × 0.25 mm
Inj: 1 �L O-bis(trimethylsilyl)derivate of BPA: m/z357
R: 74–97% (sweater),92–111% (seafood)
Basheer etal. [51]
4-n-Butylphenol MAE–SPE (seafood) I.D., 0.5 �m (J&W,Folsom, CA, USA)
I.S.: m/z 416 LOD: 6.30 ng L−1
(water), 1.35 ng g−1
(seafood)4-Pentylphenol4-n-Hexylphenol
464
A.Ballesteros-G
ómez
etal./J.Chrom
atogr.A1216
(2009)449–469
Table 5 (Continued )
Sample Other analytes thanBPA
I.S. Sample preparation(main steps)
Derivatization GC column Temperatureprogram andinjection
MS parameters Linear range, LOD orLOQ, recoveries (R)
Reference
4-tert-Octylphenol4-n-Heptylphenol4-Nonylphenol2,4-DichlorophenolPentachlorophenol
Food simulant BADGE – SPME – HP-5 MS,30 m × 0.25 mm,0.25 �m
200 ◦C held for2 min, ramped at10 ◦C min−1 to270 ◦C, hold 15 min.
BPA: m/z 213, m/z228
LR: 0.01–8 �g g−1
(distilled water),0.02–10 �g g−1
(water with 3%acetic acid),0.02–3 �g g−1 (waterwith 10% ethanol)
Salafrancaet al. [93]
LOD: 0.1 ng g−1
(distilled water),1 ng g−1 (water with3% acetic acid),2 ng g−1 (water with10% ethanol)R
Food simulant – – SPME BSTFA:TMCS (99:1,v/v), 7 �L (using thevapour of thederivatizationreagents in theheadspace on thefibre) (65 ◦C, 30 s)
DB-5MS,30 m × 0.25 mm
80 ◦C held for 2 min,ramped at20 ◦C min−1 to280 ◦C, hold 1 min
O-bis(trimethylsilyl)derivate of BPA: m/z372, m/z 357
LR:0.001–100 �g L−1
Chang et al.[94]
I.D., 0.5 �m (J&W,Folsom, CA, USA)
LOD:0.4 ng L−1
R: 13.8%
Abbreviations: SE, solvent extraction; LLE, liquid–liquid extraction; MTBSTFA, N′ ,N′-methyl-(tert-butyldimethylsilyl) trifluoroacetamide; TBDMS, tert-butyldimethylsilyl; BSTFA, N-O-bis(trimethylsilyl) trifluoroacetamide; TMCS,trimethylchlorosilane, DTE, 1,4-dithioerythritol; MAE, microwave-assisted extraction; SPME; solid-phase microextraction; BADGE, bisphenol A diglycidyl ether.

hrom
aqeooCB
ai(AT2ratukyetdi
motbti0tmTttwtot
selhmiw2twa
mftfgUmt2st
aihb
owtBdMast
iua[0rbM[o0
4
tnitseLd
uNmdstictm1a
tiaenmbe
A. Ballesteros-Gómez et al. / J. C
ffecting ionisation efficiency and background noise, and conse-uently detection and quantification limits. Furthermore, cleanxtracts are preferred to extend the column life and spend less timen the instrument maintenance. Anyway, LC–MS based BPA meth-ds offer higher confidence in identification than LC–FL and LC–EC.ompared to GC–MS, the time-consuming derivatization step ofPA is not required.
The LC–MS analysis of BPA is exclusively carried out usingtmospheric pressure ionization interfaces, namely electrosprayonization (ESI) and atmospheric pressure chemical ionizationAPCI), both in negative mode. ESI is more frequently used thanPCI because it generally provides better sensitivity (cf. Table 4).hus, instrumental quantitation limits for BPA of 5 [65] and0.7 ng mL−1 [53] have been reported using ESI(-) and APCI(-),espectively. In both studies, a quadrupolar mass analyzer was usednd the injected sample volume was 10 �L. Using ESI(-) and ariple quadrupole mass analyzer the instrumental LOQ decreasedp to 1 ng mL−1 (injected sample volume: 10 �L) [28,60,73]. To ournowledge, ion-trap instruments have not been used for the anal-sis of BPA in food samples yet, although the analysis of BPA innvironmental samples using ESI(-) ion-trap MS was at least siximes more sensitive than APCI [121], since the fragmentation of theeprotonated molecule (quantitation ion) by loss of CH3
• occurredn APCI even under mild temperature conditions.
The response in ESI-MS for BPA is strongly dependent on theobile phase composition [60,65]. Thus, mobile phases made up
f methanol–water gave higher response for BPA standard solu-ions than those consisting of acetonitrile–water owing to the loweroiling point of the former, which favoured the desolvation ofhe electrospray droplets [65]. The response in acetonitrile–waterncreased by 3-, 4-fold under the addition of modifiers such as.5% [122] and 0.01% of ammonia or 0.01% acetic acid [65]. Con-rary to these studies, the presence of additives in methanol–water
obile phases has been known to decrease the response for BPA.hus, basic additives, which were added to promote deprotona-ion of BPA, resulted in strong ionization suppression [121,123], andhe addition of ammonia (0.01% in water) or acetic acid (0.01% inater) decreased the signal by 1.5- and 3-fold for BPA standard solu-
ions and spiked milk, respectively [65]. However, in some cases thepposite effect has been reported, e.g. the addition of 0.1% ammoniao methanol:water increased the response for BPA [60].
Most MS methods for BPA include the addition of an internaltandard (I.S.) to overcome sample preparation losses and matrixffects (suppression or enhancement of the signal), which lead toow method absolute recoveries. The most used internal standardsave been 4-nonylphenol (when alkylphenols were also deter-ined), deuterated BPA-d16 and isotope labelled 13C12-BPA. The
mportance of using an I.S. was highlighted by Maragou et al. [65],ho overcame the losses caused by signal suppression (around
0%) and those generated during sample preparation (estimatedo be around 28%), by using the internal standard BPA-d16. In thisay, the mean relative recovery of the method was 101% (mean
bsolute recovery of 52%) [65].Independently of the type of analyzer and ionization source, the
ost abundant ion in the BPA mass spectrum, and therefore usedor quantitation purposes, was [M−H]− m/z 227. The mass spec-rum obtained with quadrupole instruments also contained theragment ions m/z 211 and 212, formed by the additional loss of oxy-en, [M−H−O]−, and a methyl radical, [M−H−CH3]•−, respectively.sing mobile phases of acetonitrile–0.01% NH3 in water, an ion with
/z 113 was detected in the BPA mass spectrum, which was relatedo the loss of both acidic protons [M−2H]2−. The [M−H−CH3]•− m/z12 was the most prominent product ion obtained by LC–MS/MS,o it was used for confirmation and/or quantitation of BPA in allhe quoted methods (cf. Table 4). Other fragments of lower relative
r
Ntf
atogr. A 1216 (2009) 449–469 465
bundance were reported in the MS2 ion-trap spectrum, namely theon [M−H−C6H5OH]− m/z 133, resulting from the cleavage of theydroxybenzyl group, and the ion [M−H−C9H10O]− m/z 93, formedy the loss of hydroxyphenyl propyl.
On the other hand, the most abundant ion in the mass spectrumf the commonest internal standard, BPA-d16 (molecular mass 244),as [(M−D2+H2)−H]− m/z 241 instead of [M−D]− m/z 242, owing
o the deuterium/hydrogen exchange in the hydroxyl groups whenPA-d16 was dissolved in a protic medium, which led to its imme-iate transformation in BPA-d14 (molecular weight 242) [124]. TheS/MS spectrum of BPA-d16 showed as base peak the fragment ion
t m/z 223, which was attributed to the loss of the CD3 radical, and aecond fragment at m/z 142 related to the loss of a phenol-d4 groupogether with a deuterium atom derived from a CD3 group [125].
Applications of LC–ESI(-)-MS to the determination of BPAnclude cereals [32] and milk [65], while LC–MS/MS has beensed for the analysis of water and soda beverages [60], meat [28]nd milk and eggs [114], after extraction with PLE [32,28], SPE60] and MSDP [114]. Method detection limits were in the ranges.7–43 ng g−1 and 0.001–0.3 ng g−1 for MS and MS/MS detection,espectively. On the other hand, two methods using APCI(-) haveeen developed for the determination of BPA. The first one usesAE for extraction of BPA from fish followed by LC–MS analysis
50]. The second one determines BPA in breast milk based on ann-line SPE(RAM)–tandem MS system [73]. LODs were in the range.1–50 ng g−1.
.2. GC–MS
GC–MS provides higher resolution and lower detection limitshan LC–MS for the determination of BPA in food, although theeed for a derivatization step makes the GC-based methods labour
ntensive and introduces new sources of errors, mainly due to con-amination. On the other hand, since the presence of lipids canignificantly reduce the analytical performance of GC [126,127],xtensive clean-up is required for fatty foods, such as fish [39]. LikeC/MS methods, the use of an internal standard is common, beingeuterated BPA-d16 and BPA-d14 the most used surrogates.
GC–MS with electron impact (EI) ionization has widely beensed for the confirmation of BPA in food analysis [15,36,39,66,128].o derivatization of BPA is required for this application. The EIass spectrum and the normal fragmentation pathway for BPA are
epicted in Figs. 4a and 5. The base peak in this spectrum corre-ponds to the loss of a methyl group ([C14H13O2]+, m/z 213) fromhe molecular ion ([C15H16O2]•+, m/z 228). This can be rationalisedn terms of resonance stabilization of the resulting tert-benzylicarbocation (cf. fragment 3, Fig. 5). An alternative minor fragmen-ation pathway involves the loss of one of the aryl groups from the
olecular ion to give a tert-benzylic carbocation ([C9H8O]+, m/z35, fragment 4, Fig. 5) and the subsequent loss of methane to givefragment ion at m/z 119 (fragment 5, Fig. 5).
Quantitation of BPA by GC–MS requires the derivatization ofhe analyte in order to improve its separation and detection. Thenclusion of a derivatization step leads to sharper peaks for BPAnd consequently, better separation from other analytes and co-xtracted matrix components, in addition to higher sensitivity (subg g−1 level) [37]. Silylation and acetylation have been by far theost used derivatization procedures, which are usually carried out
y adding 100–200 �L of the corresponding reagent to the driedxtract and allowing the mixture to stand for 30–60 min under
oom temperature or at 65–80 ◦C.Silylation of the active hydrogens of BPA is mainly made using-O-bis(trimethylsilyl) trifluoroacetamide (BSTFA) containing 1% of
rimethylchlorosilane (TMCS) [37,43,51,94]. The addition of TMCSavours the formation of a single derivative, since the reaction
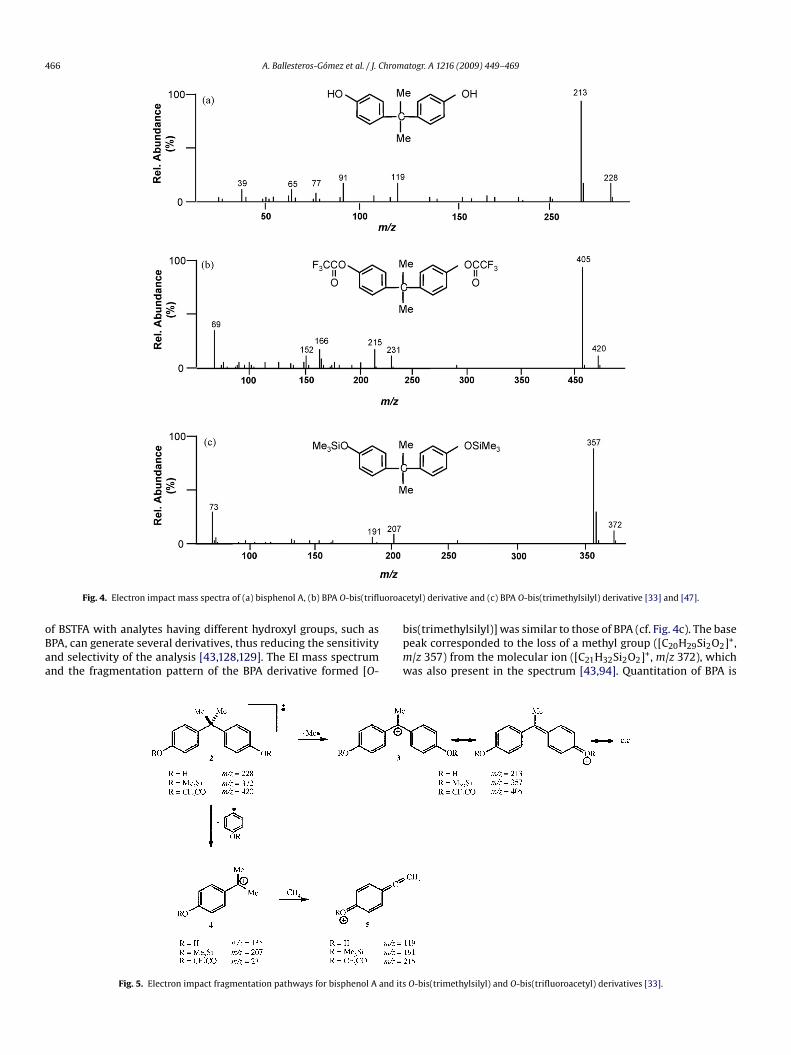
466 A. Ballesteros-Gómez et al. / J. Chromatogr. A 1216 (2009) 449–469
uoroac
oBaa
Fig. 4. Electron impact mass spectra of (a) bisphenol A, (b) BPA O-bis(trifl
f BSTFA with analytes having different hydroxyl groups, such asPA, can generate several derivatives, thus reducing the sensitivitynd selectivity of the analysis [43,128,129]. The EI mass spectrumnd the fragmentation pattern of the BPA derivative formed [O-
bpmw
Fig. 5. Electron impact fragmentation pathways for bisphenol A and its
etyl) derivative and (c) BPA O-bis(trimethylsilyl) derivative [33] and [47].
is(trimethylsilyl)] was similar to those of BPA (cf. Fig. 4c). The baseeak corresponded to the loss of a methyl group ([C20H29Si2O2]+,/z 357) from the molecular ion ([C21H32Si2O2]+, m/z 372), whichas also present in the spectrum [43,94]. Quantitation of BPA is
O-bis(trimethylsilyl) and O-bis(trifluoroacetyl) derivatives [33].

hrom
r3
((rct(tcTtyhdtfdt
[ttptlOconfflto
fgdaitacdoeaas
BBGdhAaw
4
av
[tp[ie
ndmaa5oapotaBacOtgttwap0o
5
tssbtPq
mii(mfaSsoBa
ca
A. Ballesteros-Gómez et al. / J. C
outinely carried out at one (m/z 357) [94] or two (m/z 357 and72) [43] masses of this derivative.
Silylation of BPA has also been carried out using N′,N′-methyl-trimethylsilyl) trifluoroacetamide (MSTFA) and N′,N′-methyl-tert-butyldimethylsilyl) trifluoroacetamide (MTBSTFA) [38]. Botheagents provided BPA derivatives with diagnostic ions for theharacterization of their identities in the mass spectra. The charac-eristic ions for the trimethylsilyl (TMS) and tert-butyldimethylsilylBDMS) BPA derivatives were [M−15]+, at m/z 357 and 441, respec-ively. These ions, which appeared as base peaks in the mass spectra,an be used as quantification ions for BPA in the GC–MS SIM mode.he molecular ions for TMS (m/z 372) and BDMS (m/z 456) deriva-ives were used for BPA confirmation. No difference in reactionields and responses was found between them, but TBDMS gaveigher storage stability (it was stable after 8 days while the TMSerivative decreased to 60% due to hydrolysis at this time). In addi-ion, the retention time for TBDMS derivative was longer than thator TMS (41 and 37 min, respectively, under the experimental con-itions referred in [38]), which resulted in better separation fromhe earlier eluted interferences.
Acetylation of the hydroxyl groups of BPA with acetic anhydride16,17] or trifluoroacetic anhydride [33] is other frequent procedureo obtain BPA derivatives for GC–MS. Fig. 4b includes the mass spec-rum for the O-bis(trifluoroacetyl) derivative and its fragmentationattern. As usual, the base peak in the spectrum corresponded tohe fragment ion [M−15]+ (m/z 405) formed from the ion molecu-ar (m/z 420) by the loss of a methyl group. It was proved that the-bis(trifluoroacetyl) derivative of BPA was more sensitive than theorresponding trimethylsilyl derivative, which was a consequencef the higher molecular mass of the former and the monoisotopicature of fluorine [33]. The higher molecular mass led to more
avourable signal-to-noise ratios and the monoisotopic nature ofuorine prevented the reduction in ion intensity that results fromhe distribution of the current between isotopomeric ions, like itccurs for silicon.
Derivatization of the internal standard (e.g. BPA-d14) has beenound essential to maintain its isotopic purity during chromato-raphic separation on a fused-silica capillary column [33], since itegrades owing to the deuteron–proton (2H–H) exchange in theromatic portion of the molecule. This phenomenon also occursn other monodeuterated phenols [130] and has been attributedo the aromatic electrophilic exchange of deuterium atoms withctive hydrogen atoms located on the internal surface of theolumn [131]. In this respect, although the TMS derivative of BPA-euterated derivatives reduces the loss of isotopic purity, the usef O-bis(trifluoroacetyl) derivative is preferred to prevent 2H–Hxchange in the column, since its stability is higher [33] and inddition, as previously quoted for BPA, its higher molecular massnd the monoisotopic nature of fluorine makes this derivative moreensitive.
GC–MS is the most sensitive technique for the determination ofPA in foods. Stuart et al. [132] compared the instrumental LODs forPA obtained by different techniques, the values being 0.8 ng forC–MS without derivatization, 0.004 ng for GC–MS using methylerivate (0.5 M methanolic solution of phenyltrimethylammoniumydroxide), 1.6 ng for LC–UV (275 nm) and 1.0 ng for LC/ESI(-)-MS.lower instrumental LOD (0.51 pg) has been reported by using an
cetyl derivate [133]. On the other hand, method detection limitsere usually in the ranges 0.4–2 ng g−1 and 0.4–6.3 ng L−1.
.3. Immunochemical methods
Immunochemical methods have widely been utilized in foodnalysis for the detection and quantification of proteins, enzymes,itamins and other naturally occurring substances (reviewed in
pnclo
atogr. A 1216 (2009) 449–469 467
134]). They have also been used for the determination of food con-aminants such as microorganisms, bacterial toxins, mycotoxins,esticides, anabolic hormones and veterinary drugs (reviewed in134,135]). Immunoassays are easy to perform, give good sensitiv-ty and specificity and they require neither qualified personnel norxpensive equipment.
The application of immunochemical techniques to the determi-ation of BPA in foods is rather recent [136–140]. Up to now, theeterminations have focused on the analysis of liquid foods, mainlyilk, water and food simulants, using polyclonal mammalian [136]
nd chicken [137] antibodies in enzyme linked immunosorbentssays (ELISA). The detection limits ranged from 0.05 ng mL−1 to00 ng mL−1, mainly depending on the immunogen and the typef antibody produced. As a small molecule, BPA is not able to initi-te an immune response itself and needs to be conjugated with arotein to form a complete antigen. Consequently, the productionf antibodies for BPA has involved the attachment of the pro-ein bovine serum albumin (BSA) to the derivatized analyte or ton structural analogue (e.g. [4,4-bis(4-hydroxyphenyl)valeric acid,HPVA]) in order to avoid the loss of part of the structural char-cteristics of BPA [138,139]. Kuruto et al. proposed the use of aommercially available ELISA kit (EcoAssay BPA kit supplied bytsuka Pharmaceutical Co., Ltd., Tokyo, Japan) for the determina-
ion of BPA in human colostrum [140]. This kit was able to detectlucoronide-conjugated BPA as well as free BPA. The preparation ofhe sample (1 mL) involved the addition of acetonitrile (3 mL) andhe subsequent centrifugation for protein removal. The supernatantas evaporated to dryness, reconstituted with 1 mL of phosphate
nd subject to SPE (Oasis HLB). The mean recovery of spiked sam-les was 102 ± 19.0% and the detection limit of the method was.3 ng mL−1. The same methodology was applied to determinationf BPA in maternal serum and amniotic fluid [141].
. Conclusions
The determination of BPA in food is a requirement to supporthe enforcement of legislation and assess the risk of human expo-ure to BPA low-doses. At present, LC–fluorescence detection istill frequently used and it gives satisfactory quantitative results,ut LC–MS and GC–MS are becoming more attractive becausehey provide more selective and, therefore, more reliable methods.roper identification of BPA is usually made by GC–MS or LC-tripleuadrupole instruments.
Sample preparation still constitutes the key-step for the deter-ination of BPA in food and it is the origin of the main drawbacks
n the available methodologies, independently of their applicabil-ty to BPA as the only analyte or to a large number of contaminantse.g. BPA along with other phenols, endocrine disruptors and/or
igrants from food packaging). Solvent extraction and SPE are byar the most used extraction techniques for both isolation of BPAnd clean-up of matrix components. Techniques such as SPME andBSE, which offers advantages in terms of solvent consumption,ample size and ease of automation have to overcome some seri-us drawbacks before becoming suitable for the determination ofPA in food. Other alternatives such as MSPD, excellent in specificspects, have not reached widespread utilization yet.
There are two critical aspects in the sample preparation pro-edures developed, which are rather general in food contaminantnalysis, namely the low selectivity of solvents and the exhaustive
urification required to isolate the analyte from matrix compo-ents. As a general rule, repeated solvent extractions are required toompletely isolate BPA from food matrices which involves the use ofarge volumes of organic solvent that subsequently has to be evap-rated for further treatment or analyte concentration. This makes
4 hrom
tIe
pocOfafossmc
wysrarc
A
Sa(
R
68 A. Ballesteros-Gómez et al. / J. C
he respective sample treatments laborious and time-consuming.n this respect, alternative strategies that provide high extractionfficiency for BPA should simplify sample treatment.
Purification of samples becomes essential because matrix com-onents constitute the main source of errors in the determinationf BPA in foods. The effects observed are sample-dependent andonsiderably reduce quantification accuracy in all the techniques.n the other hand, purification of samples is also required for satis-
actory long-term chromatographic system performance during thenalysis of a range of samples. SPE is by far the most used techniqueor sample purification, although proteins and lipids must be previ-usly removed to avoid clogging of the sorbent. The use of selectiveorbents such as RAMs, immunosorbents and MIPs should increaseample throughput and accuracy since they have the potential toake analyte isolation and clean-up in a single-step and to provide
leaner extracts, but their use is still emerging and a rather limited.In our opinion, fluorimetric and mass spectrometric methods
ill continue to play a predominant role in the future for the anal-sis of BPA in foods, although more suitable sample treatmenttrategies should be developed to make these methodologies moreeliable and widely applicable. Research should also focus on cheapnd rapid screening methods that are consistent and robust. In thisespect, ELISA methods, although still an emerging area in BPA-ontaining food analysis, should be a convenient approach.
cknowledgment
The authors gratefully acknowledge financial support frompanish MEC (Project CTQ2005-00643). A. Ballesteros-Gómezcknowledges to the Spanish MEC the doctoral fellowship awardedAP2005-4275).
eferences
[1] E. Burridge, Eur. Chem. News 17 (2003) 14.[2] C.A. Staples, K. Woodburn, N. Caspers, T. Hall, G.M. Klecka, Hum. Ecol. Risk
Asses. 8 (2002) 1083.[3] M. Morgan, Phenol/acetone—facts, figures and future, available online at:
http://www.icis.com/v2/chemicals/9075165/bisphenol-a/uses.html, in: The4th ICIS World Phenol/Acetone Conference, Prague, June 6–7, 2006.
[4] G. Lyons, A. Bisphenol, A Known Endocrine Disruptor, WWF-UK, Surrey, 2000.[5] O.C. Dermer, in: J.J. McKelta, G.E. Weismantel (Eds.), Encyclopedia of Chemical
Processing and Design, Marcel Dekker, New York, 1999, p. 406.[6] Opinion of the Scientific Committee on Food on Bisphenol A, expressed on
17 April 2002, SCF/CS/PM/3936 final, European Commission, Brussels, 2002,available online at http://www.europa.eu/foods/fs/sc/out128 en.pdf.
[7] Opinion of the Scientific Panel on Food Additives, Flavourings, ProcessingAids and Materials in Contact with Food (ACF) on request from the Com-mission related to 2,2-bis(4-hydroxyphenyl)propane (Bisphenol A). Questionnumber EFSA-Q-2005-100, adopted on 29 November 2006. The EFSA Journal428 (2006) 1, available online at http://www.efsa.europa.eu/EFSA/efsa locale-1178620753812 1178620772817.htm.
[8] H. Fromme, T. Küchler, T. Otto, K. Pilz, J. Müller, A. Wenzel, Water Res. 36 (2002)1429.
[9] F.J. Ruiz, S. Rubio, D. Pérez-Bendito, J. Chromatogr. A 1163 (2007) 926.[10] A. Ballesteros-Gómez, F.J. Ruiz, S. Rubio, D. Pérez-Bendito, Anal. Chim. Acta
603 (2007) 51.[11] D.W. Kolpin, E.T. Furlong, M.T. Meyer, E.M. Thurman, S.D. Zaugg, L.B. Barber,
H.T. Buxton, Environ. Sci. Technol. 36 (2002) 1202.[12] R.A. Rudel, J.G. Brody, J.D. Spengler, J. Vallarino, P.W. Geno, G. Sun, A. Yau, J. Air
Waste Manage. Assoc. 51 (2001) 499.[13] R.A. Rudel, D.E. Camann, J.D. Spengler, L.R. Korn, J.G. Brody, Environ. Sci. Tech-
nol. 37 (2003) 4543.[14] N.K. Wilson, J.C. Chuang, M.K. Morgan, R.A. Lordo, L.S. Sheldon, Environ. Res.
103 (2007) 9.[15] J.E. Biles, T.P. McNeal, T.H. Begley, J. Agric. Food Chem. 45 (1997) 4697.[16] A. Goodson, W. Summerfield, I. Cooper, Food Addit. Contam. 19 (2002) 796.[17] B.M. Thomson, P.R. Grounds, Food Addit. Contam. 22 (2005) 65.
[18] L.N. Vandenberg, R. Hauser, M. Marcus, N. Olea, W.V. Welshons, Reprod. Tox-icol. 24 (2007) 139.[19] A.V. Krishnan, P. Stathis, S.F. Permuth, L. Tokes, D. Feldman, Endocrinology 132
(1993) 2279.[20] F.S. Vom Saal, C. Hughes, Environ. Health Perspect. 113 (2005) 926.[21] R.T. Zoeller, R. Bansal, C. Parris, Endocrinology 146 (2005) 607.
atogr. A 1216 (2009) 449–469
[22] Y.B. Wetherill, C.E. Petre, K.R. Monk, A. Puga, K.E. Knudsen, Mol. Cancer Ther.7 (2002) 515.
[23] B.T. Akingbemi, C.M. Sottas, A.I. Koulova, G.R. Klinefelter, M.P. Hardy,Endocrinology 145 (2004) 592.
[24] Integrated Risk Information System (IRIS), Bisphenol A (CASRN 80-05-7), U.S.Environmental Protection Agency, Washington, DC, 1988. Available on line athttp://www.epa.gov/iris/subst/0356.htm.
[25] Commission Directive 2004/19/EC, of 1 March 2004, relating to plastic materi-als and articles intended to come into contact with foodstuffs (Official Journalof the European Communities L71 8).
[26] EC, Regulation (EC) No. 1907/2006 of the European Parliament and of theCouncil of 18 December 2006, concerning the Registration, Evaluation, Autho-risation and Restriction of Chemicals (REACH), establishing a EuropeanAgency, amending Directive 1999/45/EC and Repealing Council Regulation(EEC) No. 793/93 and Commission Regulation (EC) No. 1488/94 as wellas Council Directive 76/769/EEC and Commission Directives 91/155/EEC,93.67/EEC, 93/105/EC and 2000/21/EC, 2006.
[27] J.H. Kang, F. Kondo, Y. Katayama, Toxicology 226 (2006) 79.[28] B. Shao, H. Han, D. Li, Y. Ma, X. Tu, Y. Wu, Food Chem. 105 (2007) 1236.[29] Y. Watabe, T. Kondo, H. Imai, M. Morita, N. Tanaka, K. Hosoya, Anal. Chem. 76
(2004) 105.[30] Y. Watabe, T. Kondo, M. Morita, N. Tanaka, J. Haginaka, K. Hosoya, J. Chro-
matogr. A 1032 (2004) 45.[31] K. Inoue, K. Kato, Y. Yoshimura, T. Makino, H. Nakazawa, J. Chromatogr. B 749
(2000) 17.[32] R. Carabias-Martínez, E. Rodriguez-Gonzalo, P. Revilla-Ruiz, J. Chromatogr. A
1137 (2006) 207.[33] P. Varelis, D. Balafas, J. Chromatogr. A 883 (2000) 163.[34] E.M. Munguía-López, H. Soto-Valdez, J. Agric. Food Chem. 49 (2001) 3666.[35] J. López-Cervantes, P. Paseiro-Losada, Food Addit. Contam. 20 (2003) 596.[36] A. D’Antuono, V. Campo Dall‘Orto, A. Lo Balbo, S. Sobral, I. Rezzano, J. Agric.
Food Chem. 49 (2001) 1098.[37] R.J. Wingender, P. Niketas, C.K. Switala, J. Coat. Technol. 70 (1998) 75.[38] Y. Gyong, J.H. Shin, H.-Y. Kim, J. Khim, M.-K. Lee, J. Hong, Anal. Chim. Acta 603
(2007) 67.[39] E.M. Munguía-López, S. Gerardo-Lugo, E. Peralta, S. Bolumen, H. Soto-Valdez,
Food Addit. Contam. 22 (2005) 892.[40] T. Yoshida, M. Horie, Y. Hoshino, H. Nakazawa, Food Addit. Contam. 18 (2001)
69.[41] J.-H. Kang, F. Kondo, Y. Katayama, Anal. Chim. Acta 555 (2006) 114.[42] J.-H. Kang, F. Kondo, J. Food Prot. 66 (2003) 1439.[43] H.-W. Kuo, W.-H. Ding, J. Chromatogr. A 1027 (2004) 67.[44] J.-H. Kang, F. Kondo, Res. Vet. Sci. 73 (2002) 177.[45] T. Toyo’oka, Y. Oshige, Anal. Sci. 16 (2000) 1071.[46] C. Sun, L.P. Leong, P.J. Barlow, S.H. Chan, B.C. Bloodworth, J. Chromatogr. A 1129
(2006) 145.[47] E.M. Munguía-López, E. Peralta, A. González-León, C. Vargas-Requena, H. Soto-
Valdez, J. Agric. Food Chem. 50 (2002) 7299.[48] J.A. Brotons, M.F. Olea-Serrano, M. Villalobos, V. Pedraza, N. Olea, Environ.
Health Perspect. 103 (1995) 608.[49] C. Perrin, L. Meyer, C. Mujahid, C.J. Blake, Food Chem. 74 (2001) 245.[50] S.N. Pedersen, C. Lindholst, J. Chromatogr. A 864 (1999) 17.[51] C. Basheer, H.K. Lee, K.S. Tan, Mar. Pollut. Bull. 48 (2004) 1145.[52] R. Carabias-Martínez, E. Rodriguez-Gonzalo, P. Revilla-Ruiz, J. Hernández-
Mendez, J. Chromatogr. A 1089 (2005) 1.[53] J.A. Mendiola, M. Herrero, A. Cifuentes, E. Ibánez, J. Chromatogr. A 1152 (2007)
234.[54] S. Tavazzi, E. Benfenati, D. Barcelo, Chromatographia 56 (2002) 463.[55] I. Liska, J. Chromatogr. A 885 (2000) 3.[56] F.J. Dos Ramos, J. Chromatogr. A 880 (2000) 69.[57] V. Ruiz-Gutierrez, M.C. Perez-Camino, J. Chromatogr. A 885 (2000) 321.[58] K. Ridgway, S.P.D. Lalljie, R.M. Smith, J. Chromatogr. A 1153 (2007) 36.[59] V. Camel, Spectrochim, Acta Part B 58 (2003) 1177.[60] B. Shao, H. Han, J. Hu, J. Zhao, G. Wu, Y. Xue, Y. Ma, S. Zhang, Anal. Chim. Acta
530 (2005) 245.[61] R. Kuruto-Niwa, Y. Tateota, Y. Usuki, R. Nozawa, Chemosphere 66 (2007) 1160.[62] K. Inoue, S. Murayama, K. Takeba, Y. Yoshimura, H. Nakazawa, J. Food Compos.
Anal. 16 (2003) 497.[63] A. Covaci, S. Voorspoels, J. Chromatogr. B 827 (2005) 216.[64] C. Lambert, M. Larroque, J. Chromatogr. Sci. 35 (1997) 57.[65] N.C. Maragou, E.N. Lampi, N.S. Thomaidis, M.A. Koupparis, J. Chromatogr. A
1129 (2006) 165.[66] T.P. McNeal, J.E. Biles, T.H. Begley, J.C. Craun, M.L. Hopper, C.A. Sack, ACS Symp.
Ser. 747 (2000) 33.[67] H. Miyakawa, A. Ibe, S. Tabata, Y. Sadamasu, A. Yasui, K. Yasuda, K. Saito, Ann.
Rep. Tokyo Metr. Res. Lab. P.H. 52 (2001) 66.[68] J.H. Kang, F. Kondo, Food Addit. Contam. 19 (2002) 886.[69] M.J. Bogusz, R.D. Maier, K.H. Schiwy-Bochat, U. Kohls, J. Chromatogr. B 683
(1996) 177.
[70] S.H. Ingwersen, Biomed. Chromatogr. 7 (1993) 166.[71] C.P. Desilets, M.A. Rounds, F.E. Regnier, J. Chromatogr. 544 (1991) 25.[72] S. Souverain, S. Rudaz, J.-L. Veuthey, J. Chromatogr. B 801 (2004) 141.[73] X. Ye, Z. Kuklenyik, L.L. Needham, A.M. Calafat, J. Chromatogr. B 831 (2006)110.[74] N. Delaunay, V. Pichon, M.-C. Hennion, J. Chromatogr. A 745 (2000) 15.

hrom
[[[
[[
[
[[
[[
[
[
[
[
[
[
[
[[
[
[[
[
[[[
[
[[[[
[
[
A. Ballesteros-Gómez et al. / J. C
[75] S. Dragacci, F. Grosso, J. Gilbert, J. AOAC Int. 84 (2001) 437.[76] A. Visconti, M. Pascale, G. Centoze, J. Chromatogr. A 864 (1999) 89.[77] R. Braunrath, M. Cichna, J. Chromatogr. A 1062 (2005) 189.[78] R. Braunrath, D. Podlipna, S. Padlesak, M. Cichna-Markl, J. Agric. Food Chem.
53 (2005) 8911.[79] Z. Brenn-Struckhofova, M. Cichna-Markl, Food Addit. Contam. 23 (2006) 1227.[80] M. Zhao, Y. Liu, Y. Li, X. Zhang, W. Chang, J. Chromatogr. B 783 (2003) 401.[81] B. Sellergren (Ed.), Molecularly Imprinted Polymers: Man-Made Mimics of
Antibodies and Their Applications in Analytical Chemistry, Elsevier, Amster-dan, 2001.
[82] J. Ou, L. Hu, L. Hu, X. Li, H. Zou, Talanta 69 (2006) 1001.[83] M. Kawaguchi, Y. Hayatsu, H. Nakata, Y. Ishii, R. Ito, K. Saito, H. Nakazawa, Anal.
Chim. Acta 539 (2005) 83.[84] Y. Watabe, K. Hosoya, N. Tanaka, T. Kubo, M. Morita, J. Chromatogr. A 1073
(2005) 363.[85] K. Takeda, T. Kobayashi, J. Membr. Sci. 275 (2006) 61.[86] Y. Lin, Y. Shi, M. Jiang, Y. Jin, Y. Peng, B. Lu, K. Dai, Environ. Pollut. 153 (2008)
483.[87] K. Yang, Z. Liu, M. Mao, X. Zhang, C. Zhao, N. Nishi, Anal. Chim. Acta 546 (2005)
30.[88] H. Sambe, K. Hoshina, K. Hosoya, J. Haginaka, J. Chromatogr. A 1134 (2006) 16.[89] C. Baggiani, L. Anfossi, C. Giovannoli, Anal. Chim. Acta 591 (2007) 29.[90] A. Martin-Esteban, J.L. Tadeo, Comb. Chem. High Throughput Screening 9
(2006) 747.[91] S. Li, S.G. Weber, Anal. Chem. 69 (1997) 1217.[92] H. Kataoka, H.L. Lord, J. Pawliszyn, J. Chromatogr. A 880 (2000) 35.[93] J. Salafranca, R. Battle, C. Nerín, J. Chromatogr. A 864 (1999) 137.[94] C.-M. Chang, C.-C. Chou, M.-R. Lee, Anal. Chim. Acta 539 (2005) 41.[95] C. Nerín, M.R. Philo, J. Salafranca, L. Castle, J. Chromatogr. A 963 (2002) 375.[96] R. Eisert, J. Pawliszyn, Anal. Chem. 69 (1997) 3140.[97] Y. Wen, B.-S. Zhou, Y. Xu, S.-W. Jin, Y.-Q. Feng, J. Chromatogr. A 1133 (2006) 21.[98] Y. Fan, Y.-Q. Feng, Z.-G. Shi, J.-B. Wang, Anal. Chim. Acta 543 (2005) 1.[99] M. del Olmo, A. Zafra, B. Suárez, A. Gónzalez-Casado, J. Taoufiki, J.L. Vílchez, J.
Chromatogr. B 817 (2005) 167.100] E. Baltussen, P. Sandra, F. David, C. Cramers, J. Microcol. Sep. 11 (1999) 737.101] F. David, P. Sandra, J. Chromatogr. A 1152 (2007) 54.102] M. Kawaguchi, K. Inoue, M. Yoshimura, N. Sakui, N. Okanouchi, R. Ito, Y.
Yoshimura, H. Nakazawa, J. Chromatogr. A 1041 (2004) 19.103] S. Nakamura, S. Daishima, J. Chromatogr. A 1038 (2004) 291.104] M. Kawaguchi, K. Inoue, M. Yoshimura, R. Ito, N. Sakui, N. Okanouchi, H.
Nakazawa, J. Chromatogr. B 805 (2004) 41.105] C. Bicchi, C. Cordero, E. Liberto, P. Rubiolo, B. Sgorbini, F. David, P. Sandra, J.
Chromatogr. A 1094 (2005) 9.106] X. Zhu, J. Cai, J. Yang, Q. Su, Y. Gao, J. Chromatogr. A 1131 (2006) 37.107] J.P. Lambert, W.M. Mullett, E. Kwong, D. Lubda, J. Chromatogr. A 1075 (2005)
43.108] Y. Hu, Y. Zheng, F. Zhu, G. Li, J. Chromatogr. A 1148 (2007) 16.109] S.A. Barker, A.R. Long, C.R. Short, J. Chromatogr. 475 (1989) 353.
[
[
[
atogr. A 1216 (2009) 449–469 469
110] S.A. Barker, J. Biochem. Biophys. Methods 70 (2007) 151.[111] S. Bogialli, A. Di Corcia, J. Biochem. Biophys. Methods 70 (2007) 163.112] S. Bogialli, V. Capitolino, R. Curini, A. Di Corcia, M. Nazzari, M. Sergi, J. Agric.
Food Chem. 52 (2004) 3286.113] M. Fernandez, Y. Picó, J. Manes, J. Chromatogr. A 871 (2000) 43.
[114] B. Shao, H. Han, X. Tun, L. Huang, J. Chromatogr. B 850 (2007) 412.115] M. Biedermann, K. Grob, Food Addit. Contam. 15 (1998) 609.
[116] J. Lintschinger, W. Rauter, Eur. Food Res. Technol. 211 (2000) 211.[117] J. Sajiki, J. Chromatogr. B 755 (2001) 9.[118] J. Sajiki, J. Chromatogr. B 783 (2003) 367.[119] K. Ouchi, S. Watanabe, J. Chromatogr. B 780 (2002) 365.120] V. Campo Dall‘Orto, C. Danilowicz, S. Sobral, A. Lo Balbo, I. Rezzano, Anal. Chim.
Acta 336 (1996) 195.121] H. Gallart-Ayala, E. Moyano, M.T. Galceran, Rapid Commun. Mass Spectrom.
21 (2007) 4039.122] A. Motoyama, A. Suzuki, O. Shirota, R. Namba, Rapid Commun. Mass Spectrom.
13 (1999) 2204.123] T. Benijts, W. Lambert, A. De Leenheer, Anal. Chem. 76 (2004) 704.124] K. Inoue, M. Kawaguchi, Y. Funakoshi, H. Nakazawa, J. Chromatogr. B 798
(2003) 17.125] L. Sabatini, A. Barbieri, F.S. Violante, Rapid. Commun. Mass Spectrom. 19
(2005) 3468.126] G.H. Dodo, M.M. Knigth, J. Chromatogr. A 859 (1999) 235.127] C. Orazio, J. Meadows, S. Kapila, C. Palmer, A.F. Yanders, Chemosphere 18 (1989)
69.128] J.E. Biles, T.P. McNeal, T.H. Begley, H.C. Hollifield, J. Agric. Food Chem. 45 (1997)
3541.129] W.H. Ding, C.C. Chiang, Rapid Commun. Mass Spectrom. 17 (2003) 56.130] M. Mahmoud, J. Chromatogr. A 719 (1996) 474.131] R.W. Alder, R. Baker, J.M. Brown, Mechanisms in Organic Chemistry, Wiley,
New York, 1978, p. 280.132] J.D. Stuart, C.P. Capulong, K.D. Launer, X. Pan, J. Chromatogr. A 1079 (2005)
136.133] X. Jin, G. Jiang, G. Huang, J. Liu, Q. Zhou, Chemosphere 56 (2004) 1113.134] L. Flukal, J. Kás, Trends Anal. Chem. 8 (1989) 112.135] C.A. Spinks, Trends Food Sci. Technol. 11 (2000) 210.136] H. Ohkuma, K. Abe, M. Ito, A. Kokado, A. Kambegawa, M. Maeda, Analyst 127
(2002) 93.137] B. De Meulenaer, K. Baert, H. Lanckriet, V. Van Hoed, A. Huyghebaert, J. Agric.
Food Chem. 50 (2002) 5273.138] M.-P. Zhao, Y.-Z. Li, Z.-Q. Guo, X.-X. Zhang, W.-B. Chang, Talanta 57 (2002)
1205.
139] A. Kim, C.-H. Li, C.-F. Jin, K.W. Lee, S.-H. Lee, K.-J. Shon, N.G. Park, D.-K. Kim,S.-W. Kang, Y.-B. Shim, J.-S. Park, Chemosphere 68 (2007) 1204.140] R. Kuruto-Niwa, Y. Tateoka, Y. Usuki, R. Nozawa, Chemosphere 66 (2007)
1160.141] H. Yamada, I. Furuta, E.H. Kato, S. Takaota, Y. Usuki, G. Kobashi, F. Sata, R. Kishi,
S. Fujimoto, Reprod. Toxicol. 16 (2002) 735.
Copyright © 2022 FDOKUMEN




















