
An Overview of Phenserine Tartrate, A Novel Acetylcholinesterase Inhibitor for the Treatment of...
10
Current Alzheimer Research, 2005, 2, 281-290 281 1567-2050/05 $50.00+.00 ©2005 Bentham Science Publishers Ltd. An Overview of Phenserine Tartrate, A Novel Acetylcholinesterase Inhibi- tor for the Treatment of Alzheimer’s Disease Nigel H. Greig 1, *, Kumar Sambamurti 2 , Qian-sheng Yu 1 , Arnold Brossi 3 , Gosse B. Bruinsma 4 and Debomoy K. Lahiri 5 1 Drug Design & Development Section, Laboratory of Neurosciences, National Institute on Aging, National Institutes of Health, Baltimore, MD, USA; 2 Department of Physiology & Neuroscience, Medical University of South Carolina, Charleston, SC, USA; 3 Laboratory of Natural Products, School of Pharmacy, University of N. Carolina, Chapel Hill, NC, USA ; 4 Axonyx Inc., New York, NY; 5 Department of Psychiatry, Indiana University School of Medicine, Indianapo- lis, IN, USA Abstract: Existing cholinesterase (ChE) inhibitor therapies for Alzheimer’s disease (AD), while effective in improving cognitive, behavioral and functional impairments, do not alter disease progression. Novel drug design studies have fo- cused on the classical ChE inhibitor, (-)-physostigmine, producing alterations in chemical composition and three- dimensional structure, which may offer an improved therapeutic index. The phenylcarbamate derivative, (-)-phenserine, is a selective, non-competitive inhibitor of acetylcholinesterase (AChE). In vivo , (-)-phenserine produces rapid, potent, and long-lasting AChE inhibition. As a possible result of its preferential brain selectivity, (-)-phenserine is significantly less toxic than (-)-physostigmine. In studies using the Stone maze paradigm, (-)-phenserine has been shown to improve cogni- tive performance in both young learning-impaired and elderly rats. In addition to reducing inactivation of acetylcholine in the brain, (-)-phenserine appears to have a second mode of action. Reduced secretion of beta-amyloid (Aβ) has been ob- served in cell lines exposed to (-)-phenserine, occurring through translational regulation of beta-amyloid precursor protein (β-APP) mRNA via a non-cholinergic mechanism. These in vitro findings appear to translate in vivo into animal models and humans. In a small study of patients with AD, (-)-phenserine treatment tended to reduce β-APP and Aβ levels in plasma samples. Clinical studies also reveal that (-)-phenserine (5–10 mg b.i.d.) had a favorable safety and pharmacologi- cal profile, produced significant improvements in cognitive function and was well tolerated in patients with AD treated for 12 weeks. Further randomized, double-blind, placebo-controlled Phase III studies assessing the efficacy, safety/tolerability and potential disease-modifying effects of (-)-phenserine in patients with AD are currently ongoing. Keywords: Alzheimer’s disease, phenserine, cholinesterase inhibitor, beta-amyloid, cognitive function, disease modification. INTRODUCTION Alzheimer’s disease (AD) is the most common form of dementia [1,2], accounting for approximately 70% of de- mentia cases in most industrialized countries [3]. Worldwide, the disease is estimated to affect more than 15 million indi- viduals [4] In the United States alone, there were more than 4.5 million afflicted in 2000 – a number anticipated to in- crease to over 13 million by 2050 [5]. Patients with AD ex- perience increasing deterioration as the disease progresses, resulting in memory impairments, behavioral abnormalities, and increasing difficulty in maintaining independence and performing activities of daily living [6]. Eventually, all pa- tients become dependent on the care of others [7]. The progressive neurodegeneration of AD is character- ized pathologically by extracellular deposits of insoluble beta-amyloid (Aβ) protein in senile plaques, perivascular deposits of A β, and intracellular formation of neurofibrillary tangles composed of tau protein. Furthermore, loss of *Address correspondence to this author at the Chief, Drug Design & Devel- opment Section, Laboratory of Neurosciences, Intramural Research Pro- gram, National Institute on Aging, National Institutes of Health, Baltimore, MD 21224, USA; Tel: 410-558-8278; Fax: 410-558-8323; E-mail: [email protected] cholinergic neurons in areas of the brain associated with higher mental functioning, particularly the neocortex and hippocampus, is a key characteristic [8,9]. This cholinergic degeneration is one of the earliest observable changes that occur during the course of AD, with the degree of choliner- gic deficit correlating directly to the severity of memory im- pairment [10]. The ‘cholinergic hypothesis’ of AD was the basis for the development of treatment approaches designed to facilitate the activity of the surviving cholinergic system [11] The cholinesterase (ChE) inhibitors, widely used in AD, prevent the inactivation of the neurotransmitter, acetylcholine (ACh) after its release from the pre-synaptic neuron [12] thereby increasing the ability of ACh to stimulate nicotinic and mus- carinic ACh receptors [12]. The post-synaptic element of the cholinergic system appears to be maintained well into the disease process [11]. Currently available ChE inhibitors (donepezil, rivastigmine and galantamine) have been shown to be effective in providing symptomatic benefit related to the improvement of cognitive, behavioral and functional impairments associated with AD [13]. In addition, some studies have also suggested that ChE inhibitors may slow cognitive decline or functional deterioration temporarily [13]. However, there is as yet no conclusive evidence that
Transcript of An Overview of Phenserine Tartrate, A Novel Acetylcholinesterase Inhibitor for the Treatment of...

Current Alzheimer Research, 2005, 2, 281-290 281
1567-2050/05 $50.00+.00 ©2005 Bentham Science Publishers Ltd.
An Overview of Phenserine Tartrate, A Novel Acetylcholinesterase Inhibi-tor for the Treatment of Alzheimer’s Disease
Nigel H. Greig1,*, Kumar Sambamurti2, Qian-sheng Yu1, Arnold Brossi3, Gosse B. Bruinsma4 andDebomoy K. Lahiri5
1Drug Design & Development Section, Laboratory of Neurosciences, National Institute on Aging, National Institutes ofHealth, Baltimore, MD, USA; 2Department of Physiology & Neuroscience, Medical University of South Carolina,Charleston, SC, USA; 3Laboratory of Natural Products, School of Pharmacy, University of N. Carolina, Chapel Hill,NC, USA ; 4Axonyx Inc., New York, NY; 5Department of Psychiatry, Indiana University School of Medicine, Indianapo-lis, IN, USA
Abstract: Existing cholinesterase (ChE) inhibitor therapies for Alzheimer’s disease (AD), while effective in improvingcognitive, behavioral and functional impairments, do not alter disease progression. Novel drug design studies have fo-cused on the classical ChE inhibitor, (-)-physostigmine, producing alterations in chemical composition and three-dimensional structure, which may offer an improved therapeutic index. The phenylcarbamate derivative, (-)-phenserine, isa selective, non-competitive inhibitor of acetylcholinesterase (AChE). In vivo, (-)-phenserine produces rapid, potent, andlong-lasting AChE inhibition. As a possible result of its preferential brain selectivity, (-)-phenserine is significantly lesstoxic than (-)-physostigmine. In studies using the Stone maze paradigm, (-)-phenserine has been shown to improve cogni-tive performance in both young learning-impaired and elderly rats. In addition to reducing inactivation of acetylcholine inthe brain, (-)-phenserine appears to have a second mode of action. Reduced secretion of beta-amyloid (Aβ) has been ob-served in cell lines exposed to (-)-phenserine, occurring through translational regulation of beta-amyloid precursor protein(β-APP) mRNA via a non-cholinergic mechanism. These in vitro findings appear to translate in vivo into animal modelsand humans. In a small study of patients with AD, (-)-phenserine treatment tended to reduce β-APP and Aβ levels inplasma samples. Clinical studies also reveal that (-)-phenserine (5–10 mg b.i.d.) had a favorable safety and pharmacologi-cal profile, produced significant improvements in cognitive function and was well tolerated in patients with AD treated for12 weeks. Further randomized, double-blind, placebo-controlled Phase III studies assessing the efficacy, safety/tolerabilityand potential disease-modifying effects of (-)-phenserine in patients with AD are currently ongoing.
Keywords: Alzheimer’s disease, phenserine, cholinesterase inhibitor, beta-amyloid, cognitive function, disease modification.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form ofdementia [1,2], accounting for approximately 70% of de-mentia cases in most industrialized countries [3]. Worldwide,the disease is estimated to affect more than 15 million indi-viduals [4] In the United States alone, there were more than4.5 million afflicted in 2000 – a number anticipated to in-crease to over 13 million by 2050 [5]. Patients with AD ex-perience increasing deterioration as the disease progresses,resulting in memory impairments, behavioral abnormalities,and increasing difficulty in maintaining independence andperforming activities of daily living [6]. Eventually, all pa-tients become dependent on the care of others [7].
The progressive neurodegeneration of AD is character-ized pathologically by extracellular deposits of insolublebeta-amyloid (Aβ) protein in senile plaques, perivasculardeposits of Aβ, and intracellular formation of neurofibrillarytangles composed of tau protein. Furthermore, loss of
*Address correspondence to this author at the Chief, Drug Design & Devel-opment Section, Laboratory of Neurosciences, Intramural Research Pro-gram, National Institute on Aging, National Institutes of Health, Baltimore,MD 21224, USA; Tel: 410-558-8278; Fax: 410-558-8323; E-mail:[email protected]
cholinergic neurons in areas of the brain associated withhigher mental functioning, particularly the neocortex andhippocampus, is a key characteristic [8,9]. This cholinergicdegeneration is one of the earliest observable changes thatoccur during the course of AD, with the degree of choliner-gic deficit correlating directly to the severity of memory im-pairment [10].
The ‘cholinergic hypothesis’ of AD was the basis for thedevelopment of treatment approaches designed to facilitatethe activity of the surviving cholinergic system [11] Thecholinesterase (ChE) inhibitors, widely used in AD, preventthe inactivation of the neurotransmitter, acetylcholine (ACh)after its release from the pre-synaptic neuron [12] therebyincreasing the ability of ACh to stimulate nicotinic and mus-carinic ACh receptors [12]. The post-synaptic element of thecholinergic system appears to be maintained well into thedisease process [11]. Currently available ChE inhibitors(donepezil, rivastigmine and galantamine) have been shownto be effective in providing symptomatic benefit related tothe improvement of cognitive, behavioral and functionalimpairments associated with AD [13]. In addition, somestudies have also suggested that ChE inhibitors may slowcognitive decline or functional deterioration temporarily[13]. However, there is as yet no conclusive evidence that

282 Current Alzheimer Research, 2005, Vol. 2, No. 3 Greig et al.
any of the available agents alter the underlying neuro-pathological process underpinning AD [14]. Hence, thesearch for a cure and preventative therapy for AD continues.
In this regard, Aβ has been a primary research target, andstrategies to lower its rate of production or increase its me-tabolism and clearance from the brain have represented amajor focus of current AD research [15]. Likewise, the spe-cific molecular form that represents the toxic species of Aβand mechanism(s) via which it induces cellular dysfunctionand apoptosis remain focuses of research and debate [16],and likely will yield targets of therapeutic potential. It isclear that neuropathological events that underpin the devel-opment of AD which were once considered separate, arecomplexly linked. Biochemical cascades that induce theawry production of Aβ may alter tau posphorylation ormechanisms regulating ACh synthesis. In a reciprocal man-ner, alterations in cholinergic function feed back on beta-amyloid precursor protein (β-APP) processing to yield amy-loidogenic and non-amyloidogenic products [9]. Such inter-plays are numerous, simple as well as complex, and providemechanisms through which selective drugs with a primaryaction on one target, can have secondary and tertiary actionson others that may be valuable or adverse [17]. The plethoraof targets, together with regulatory interactions betweenthem, additionally, opens the unusual scenario of designingagents, whether purposefully or unintentionally, along theconcept of multi-potent compounds that combine within asingle molecule the necessary characteristics to hit two verydifferent targets associated with the same disease process. Anumber of experimental agents have combined anticho-linesterase activity with an additional action [18–20].
Numerous backbones have acted as pharmacophores inthe design and development of anticholinesterases. Indeed,the chemical structures of those currently approved for thetreatment of AD are diverse [21]. Our drug design studieshave focused on the classical carbamate ChE inhibitor,(-)-physostigmine, and have incorporated features to improveits therapeutic index by providing preferential brain distribu-tion to limit systemic cholinergic actions, extending its shortduration of action, improving oral bioavailability and in-creasing acetylcholinesterase (AChE) selectivity to reduceactivity against butyrylcholinesterase (BuChE). The pharma-cological role of BuChE has been less well characterizedthan AChE. Once considered to be physiologically irrele-vant, as a lack of BuChE activity consequent to mutations inhumans is not associated with any abnormality [22], the con-servation of its expression throughout invertebrate, verte-brate and mammalian species [23], together with the exis-tence of an AChE knockout mouse that survives with normallevels of BuChE [24] are clear indicators of its physiologicalrelevance. It has been proposed, although not proven, that foroptimal clinical utility of anticholinesterases, AChE selec-tivity would minimize co-inhibition of both enzymes, whichhas been associated with potential liability (an increase inperipheral side effects without increase in central therapeuticeffects) [25]. (-)-Phenserine tartrate, a phenylcarbamateanalogue of (-)-physostigmine, was a product of these studiesand is a long-acting and centrally active inhibitor of AChE.In addition to being potent inhibitor of AChE, initial studiesboth in vitro [26] and in vivo [27,28] have demonstrated that(-)-phenserine inhibits the formation of β-APP, the source of
neurotoxic Aβ peptide. This property appears to be inde-pendent of its cholinergic action and provides (-)-phenserinethe potential to act beyond the symptomatic treatment asso-ciated with existing anticholinesterases and play a role indisease modification The present article summarizes thepharmacological, preclinical and clinical profiles of(-)-phenserine, a potential new treatment for patients withAD.
CHEMISTRY
(-)-Phenserine is a crystalline compound that was pre-pared and chosen from extensive medicinal chemistry studiesfocused on optimizing the structure/activity relation of hexa-hydropyrrolo[2,3b]indole carbamates and their ring B and Chetero analogues (reviewed in Greig et al. 1995a; Brossi etal. 1996 and described in detail by Yu et al. 1997, 1998,1999, 2001) [29–34]. Interestingly, its first reported synthe-sis was by Polonovski [35] for no pharmacological reason,but to elucidate the structure of (-)-physostigmine that wasunknown at the time. Thereafter, it was forgotten. Synthe-sized amongst numerous novel carbamates [29, 30 ibid], itwas selected for preclinical evaluation. An unsubstitutedphenylcarbamate of (-)-physostigmine, (-)-phenserine is usedbiologically as a L(+)-tartrate salt to aid its aqueous solubil-ity, with the chemical composition: C20H23N3O2 • C4H6O6.The chemical structure of (-)-phenserine tartrate is shown in(Fig. 1). Of particular note, it has a chiral center in the 3aposition and exists as the chirally pure (3aS)-, (-)-, enanti-omer. The unnatural, (3aR)-, (+)-, enantiomer is devoid ofanticholinesterase action.
Fig. (1). Chemical structure of (-)-phenserine L(+)-tartrate. Thetricyclic backbone is numbered A, B and C from left to right.
Interaction with AChE
AChE and BuChE have been fully sequenced and sharesome 65% amino acid sequence homology although they areencoded by unrelated genes on different chromosomes; spe-cifically on 7 (7q22) and 3 (3q26) in humans, respectively[23]. During recent years, detailed X-ray diffraction studiesof AChE and specific inhibitors of the enzyme, together withextensive biochemical research and site-directed mutagenesisstudies, has elucidated the three-dimensional interaction ofdifferent types of ChE inhibitor, as well as substrates thatmimic ACh, within AChE and BuChE [36]. The active sitesfor ACh and ChE inhibitor interactions for both AChE andBuChE are buried within a primarily hydrophobic 20 Å deepgorge [37], into which ACh diffuses and is cleaved. Electro-static charges associated with seven negatively chargedamino acid residues close to the gorge opening of AChE trapor steer charged ligands towards and into its entrance[38,39]. Once ACh enters the aromatic active-site gorge ofeither ChE, it binds at two locations: i) to a catalytic region
O
HN O
N8
N1
O OH
OHHO
OOH

An Overview of Phenserine Tartrate Current Alzheimer Research, 2005, Vol. 2, No. 3 283
near the base of the gorge, termed the acyl pocket – attract-ing the acetyl moiety of ACh , and ii) to a choline bindingsite midway up, attracting the choline function. The quater-nary choline of ACh interacts with Trp84 in Torpedo AChE(Trp86 in human AChE) and, to a lesser degree, with Phe330
within the choline binding site [40]. This interaction orien-tates ACh to support the approach and nucleophilic attack onits carbonyl group by key amino acids within the acylpocket. This is initiated via electron transfer between themembers of a catalytic triad; occurring through Ser200 inTorpedo AChE (Ser203 in human enzyme), it involves theimidazole ring of His and the carboxylic group of Glu. Aquaternary transition state between the Ser and ACh results,which rapidly collapses into an acetylated-enzyme interme-diate and released choline. Speedy hydrolysis of the ace-tylester reactivates the enzyme to allow it to efficientlycleave further ACh: some 104 ACh molecules per second[37, 41]. The same sequence of events occurs with BuChE-induced metabolism of ACh.
(-)- Phenserine interacts with the same binding domainsdescribed for ACh with a high selectivity towards AChE.This selectivity is achieved by capitalizing on key differ-ences between the three-dimensional structures of the gorgesof the two ChEs, resulting from their highly similar but di-vergent amino acid sequences. The described struc-ture/activity studies honed the three-dimensional configura-tion of hexahydropyrrolo[2,3b]indole carbamates to provide(-)-phenserine and analogues that preferentially fit withinAChE, and cymserine analogues that fit BuChE [21, 29, 33,34]. Amongst many differentiating amino acids, substratebinding within AChE is constrained by the presence of twophenylalanines (Phe288 and Phe290 in Torpedo, equivalent toPhe295 and Phe297 in human enzyme), whose large aromaticresidues protrude into the gorge. The switching of these totwo smaller aliphatic amino acids, Val and Leu, providesBuChE slightly different properties and a greater spacewithin its acyl pocket to permit the binding of larger sub-strates, exemplified by butyrylcholine, and a more promiscu-ous enzymatic activity. The use of site-directed mutagenesisat these sites broadens the specificity of AChE to resembleBuChE [42].
For (-)-phenserine, under physiological conditions, thebasic N1(CH3)
group in the C ring (Fig. 1) (PKa = ~8.46)gains a H+ to form a quaternary ammonium group. In a man-ner similar to ACh, positively charged (-)-phenserine isdrawn into the enzyme gorge by electrostatic field forces,wherein it interacts with the exact same binding domains asACh to form an inhibitor–enzyme complex [34]. The C ringorientates towards the choline binding domain, allowing thephenylcarbamate moiety to approach the acyl one to supportthe nucleophilic attack of the catalytic triad, via Ser, onthe carbonyl function of (-)-phenserine (Fig. 2). A(-)-phenserine-AChE intermediate likely exists in a tetrahe-dral configuration that then rapidly collapses to a carbamy-lated drug-enzyme complex with the tricyclic residue of(-)-phenserine (the A,B,C ring moiety) acting as a leavinggroup. This complex is considerably more stable than theacetyl-enzyme complex associated with ACh and, conse-quentially, the AChE is reversibly inactivated for a long du-ration. The AChE-selectivity and long-duration of inhibitionassociated with (-)-phenserine likely derives from the inter-
action of its phenylcarbamate with the protruding phenyla-lanines at the gorge base. Both hydrophobic and π electronbonding interactions, resulting from π-π stacking of thephenyl group of the carbamate between the flanking phenylmoieties of the phenylalanines stabilize this complex (Fig. 2)[34].
Fig. (2). Molecular model of the binding interactions between(-)-phenserine and the acyl and choline domains with AChE.(-)-Phenserine, 14.6 Å length, docks within a 20 Å deep gorgewithin AChE with the N1(CH3) orientating toward Trp86, the phen-ylcarbamate lying between Phe297 and Phe295 to allow the nucleo-phic attack of Ser203 on the carbonyl function of (-)-phenserine [34].Electron transfer between the catalytic triad of amino acids is shownin green.
PRECLINICAL PROFILE
Enzyme Kinetics, Pharmacokinetics and Pharmacody-namics
Classical enzyme kinetic studies have demonstrated that(-)-phenserine is a selective pure non-competitive inhibitorof AChE [43–45]. At high concentrations (-)-phenserine in-teracts with an additional binding domain within AChE, theperipheral anionic site, which is not present in BuChE and isclose to the gorge mouth. This is the preferential binding siteof donepezil and a domain that has involvement in AChEAβinteractions that augment aggregation [46,47]. Enzyme ki-netics of (-)-phenserine were found to be similar in humansand rats, and for AChE derived from erythrocyte and brainpreparations. In erythrocytes, IC50 (concentration required toinhibit 50% enzyme activity) values were 22 nM for AChEand 1,560 nM for BuChE, while in brain corresponding val-ues were 36 and 2,500 nM, respectively [21]. Thus,(-)-phenserine is 70-fold selective for AChE over BuChE. Incontrast, (-)-physostigmine lacks AChE selectively, witherythrocyte-derived IC50 values of 28 nM for AChE and16 nM for BuChE [21].
In-vivo studies have demonstrated rapid, potent and long-lasting AChE inhibition following administration of(-)-phenserine, which is in accord with its rapid carbamyla-
284 Current Alzheimer Research, 2005, Vol. 2, No. 3 Greig et al.
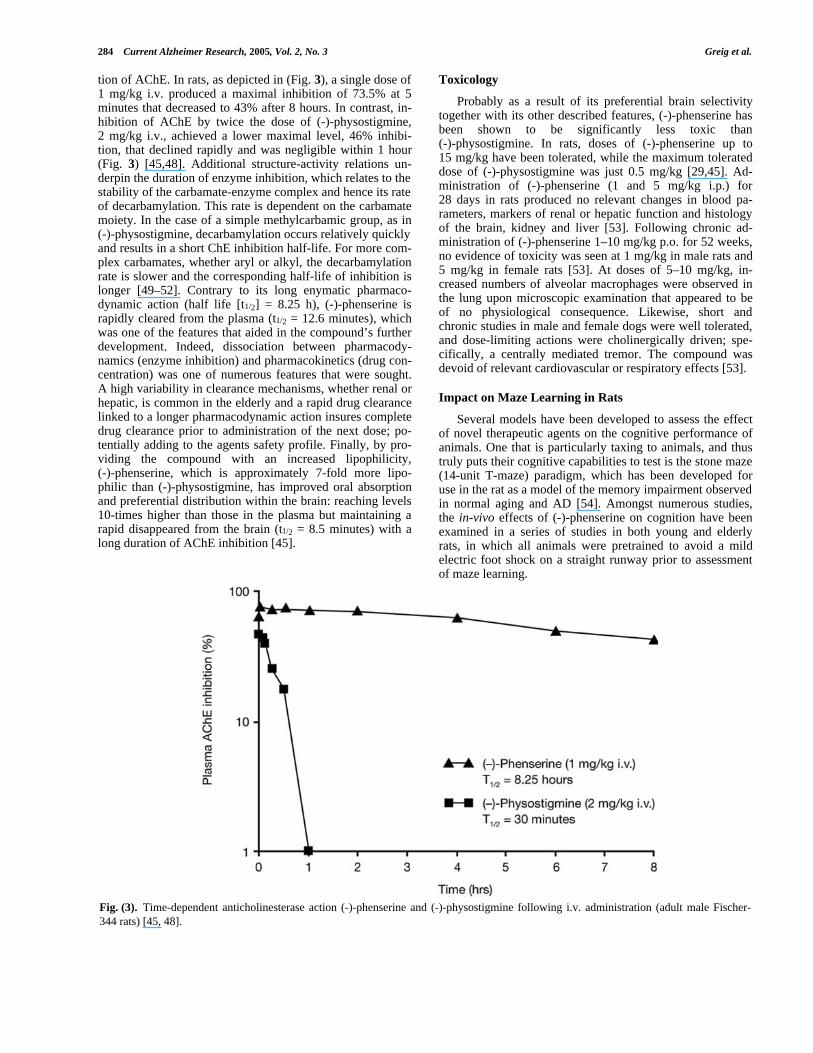
tion of AChE. In rats, as depicted in (Fig. 3), a single dose of1 mg/kg i.v. produced a maximal inhibition of 73.5% at 5minutes that decreased to 43% after 8 hours. In contrast, in-hibition of AChE by twice the dose of (-)-physostigmine,2 mg/kg i.v., achieved a lower maximal level, 46% inhibi-tion, that declined rapidly and was negligible within 1 hour(Fig. 3) [45,48]. Additional structure-activity relations un-derpin the duration of enzyme inhibition, which relates to thestability of the carbamate-enzyme complex and hence its rateof decarbamylation. This rate is dependent on the carbamatemoiety. In the case of a simple methylcarbamic group, as in(-)-physostigmine, decarbamylation occurs relatively quicklyand results in a short ChE inhibition half-life. For more com-plex carbamates, whether aryl or alkyl, the decarbamylationrate is slower and the corresponding half-life of inhibition islonger [49–52]. Contrary to its long enymatic pharmaco-dynamic action (half life [t1/2] = 8.25 h), (-)-phenserine israpidly cleared from the plasma (t1/2 = 12.6 minutes), whichwas one of the features that aided in the compound’s furtherdevelopment. Indeed, dissociation between pharmacody-namics (enzyme inhibition) and pharmacokinetics (drug con-centration) was one of numerous features that were sought.A high variability in clearance mechanisms, whether renal orhepatic, is common in the elderly and a rapid drug clearancelinked to a longer pharmacodynamic action insures completedrug clearance prior to administration of the next dose; po-tentially adding to the agents safety profile. Finally, by pro-viding the compound with an increased lipophilicity,(-)-phenserine, which is approximately 7-fold more lipo-philic than (-)-physostigmine, has improved oral absorptionand preferential distribution within the brain: reaching levels10-times higher than those in the plasma but maintaining arapid disappeared from the brain (t1/2 = 8.5 minutes) with along duration of AChE inhibition [45].
Toxicology
Probably as a result of its preferential brain selectivitytogether with its other described features, (-)-phenserine hasbeen shown to be significantly less toxic than(-)-physostigmine. In rats, doses of (-)-phenserine up to15 mg/kg have been tolerated, while the maximum tolerateddose of (-)-physostigmine was just 0.5 mg/kg [29,45]. Ad-ministration of (-)-phenserine (1 and 5 mg/kg i.p.) for28 days in rats produced no relevant changes in blood pa-rameters, markers of renal or hepatic function and histologyof the brain, kidney and liver [53]. Following chronic ad-ministration of (-)-phenserine 1–10 mg/kg p.o. for 52 weeks,no evidence of toxicity was seen at 1 mg/kg in male rats and5 mg/kg in female rats [53]. At doses of 5–10 mg/kg, in-creased numbers of alveolar macrophages were observed inthe lung upon microscopic examination that appeared to beof no physiological consequence. Likewise, short andchronic studies in male and female dogs were well tolerated,and dose-limiting actions were cholinergically driven; spe-cifically, a centrally mediated tremor. The compound wasdevoid of relevant cardiovascular or respiratory effects [53].
Impact on Maze Learning in Rats
Several models have been developed to assess the effectof novel therapeutic agents on the cognitive performance ofanimals. One that is particularly taxing to animals, and thustruly puts their cognitive capabilities to test is the stone maze(14-unit T-maze) paradigm, which has been developed foruse in the rat as a model of the memory impairment observedin normal aging and AD [54]. Amongst numerous studies,the in-vivo effects of (-)-phenserine on cognition have beenexamined in a series of studies in both young and elderlyrats, in which all animals were pretrained to avoid a mildelectric foot shock on a straight runway prior to assessmentof maze learning.
Fig. (3). Time-dependent anticholinesterase action (-)-phenserine and (-)-physostigmine following i.v. administration (adult male Fischer-344 rats) [45, 48].

An Overview of Phenserine Tartrate Current Alzheimer Research, 2005, Vol. 2, No. 3 285
Scopolamine is a classical muscarinic receptor antagonistthat is known to impair memory acquisition and cognitiveperformance in humans as well as rodents [54]. The effect of(-)-phenserine on scopolamine-induced learning impairmentwas assessed in young male Fischer-344 rats [48]. Prior totraining on a 14-unit T-maze, animals were injected i.p.either twice with saline to act as controls, or with scopola-mine to induce learning impairment followed by(-)-phenserine 1.5–10 mg/kg. Compared with controls, alldoses of (-)-phenserine (except 7.5 mg/kg) significantly(p<0.05) ameliorated error performance, runtime and shockfrequency and duration in learning-impaired rats. Although ahigher incidence of centrally mediated actions, such aschewing, grooming and tremor was seen with the two high-est (-)-phenserine doses used (7.5 and 10.0 mg/kg), classicalperipherally-mediated cholinergic effects such as salivationand diarrhea were not observed.
A marked age-related decline in learning performancehas been demonstrated in various rodent species, including inFischer-344 rats [54]. Consequently, the actions of a shortcourse of (-)-phenserine were assessed in aged (21−22month-old) male Fischer-344 rats [55]. Treatment with(-)-phenserine (1 to 3 mg/kg) prior to training on a 14-unit T-maze significantly (p<0.05) reduced errors, and in somecases additionally lowered runtime and shock frequency andduration, with greatest effects seen at 1 and 2 mg/kg dosescompared with untreated aged controls. No side effects werenoted with (-)-phenserine 1–2 mg/kg across the 5 days oftreatment; although a fine tremor was detected in some ratsadministered 3 mg/kg. In further studies, a dose-responsecurve which fell as low as 0.25 mg/kg proved effective inachieving improved cognitive performance [56].
The glutamatergic system has been implicated in memoryacquisition, and inhibition of the N-methyl-D-aspartate
(NMDA) subtype of glutamate receptor has been shown toreduce learning performance in young rats [54]. The effect of(-)-phenserine was examined in a study of young rats treatedwith a high-affinity competitive antagonist of the NMDAreceptor, 3(+) 2-carboxypiperzin-4-yl propyl phosphonicacid (CPP) to induce learning deficits [56]. Prior to trainingon a 14-unit T-maze, young male Fischer-344 rats were in-jected i.p. with saline, CPP alone, or CPP plus (-)-phenserine0.25–0.75 mg/kg. Compared with untreated controls, thenumber of errors was significantly (p>0.05) increased inanimals treated with CPP alone. In the learning impairedrats, (-)-phenserine significantly (p>0.05) reduced the num-ber of errors made relative to rats receiving CPP alone (Fig.4). Similarly, runtime and shock duration were significantly(p>0.05) reduced in the control and (-)-phenserine groupscompared with CPP alone. These results show that(-)-phenserine is effective in overcoming non-cholinergic-induced memory impairments in rats, and may be of signifi-cance in AD since deficits in the glutamatergic system havealso been observed in the disease.
Modulation of β-APP and Aβ Peptide
The neurotoxic peptide Aβ is a major component of theextraneuronal plaques that pathologically characterize AD.Aβ is derived from β-APP, a 110−120 kDa glycoprotein thatconsists of a large exocytoplasmic domain, a transmembranedomain and a relatively short cytoplasmic tail. Reduction inthe levels of Aβ in the brain is a major focus in the designand development of effective therapies for AD [9,15]. Theeffect of (-)-phenserine on β-APP and Aβ has been examinedin vitro and in vivo.
In human neuroblastoma cell lines (SK-N-SH and SH-SY-5Y), (-)-phenserine dose-dependently reduced the secre-tion of soluble β-APP and Aβ, without causing cellular tox-
Fig. (4). Effect of phenserine on number of errors (per block of three trials) in learning impaired rats [56].

286 Current Alzheimer Research, 2005, Vol. 2, No. 3 Greig et al.
icity [26,57]. Alteration of β-APP protein expression by(-)-phenserine occurred without a change in β-APP mRNAlevels, indicating a post-transcriptional mechanism throughregulation of β-APP mRNA translation. Studies involvinghuman (U-138) and rat (PC12) cell lines indicate that(-)-phenserine modulates β-APP translation via regulatoryelements in the promoter and/or 5’-untranslated region(5’-UTR) of the mRNA, sites which are also modulated bycytokines and growth factors [58, 59]. Modulatory actionsappear to be selective to the β-APP gene, since the 5’-UTRof β-APP mRNA is highly conserved in mammalian species[60], differs dramatically from amyloid precursor-like pro-teins [60], and contains a unique CAGA box in amyloidplaque producing species [60]. Reduced production and se-cretion of β-APP and Aβ in human neuroblastoma cells (SK-N-SH) treated with (-)-phenserine occurred in a concentra-tion- and time-dependent manner as can be seen in (Fig. 5).Alteration of β-APP and Aβ levels occurred via a non-cholinergic mechanism. Actions were achieved despite inhi-bition of the classical pathways associated with cholinergicstimulation, and a similar effect could be achieved with the(+) enantiomer of phenserine, which lacks anticholinesteraseactivity [26].
Studies have investigated whether the effects of (-)- and(+)-phenserine on β-APP and Aβ levels observed in cellculture also translate into animals [26,28,57,61]. Adult maleSprague-Dawley rats, all with forebrain lesions of the nu-cleus basalis of Meynert that resulted in an immediate andlong-lasting increase in the synthesis and secretion of β-APPin the cortex, hippocampus and cerebrospinal fluid (CSF),were administered (-)-phenserine 2.5 mg/kg s.c. twice daily.As indicated in (Fig. 6), levels of secreted β-APP in the CSFin lesioned (-)-phenserine-treated animals were not signifi-
cantly different from those in sham-operated, vehicle-treatedanimals, and were significantly (p<0.0004) lower than inlesioned untreated animals [26,28,57]. Additionally,(-)-phenserine treatment led to a significant (p<0.05) reduc-tion in secreted β-APP in the CSF in sham-operated animalscompared with untreated controls. Interestingly, the classicalChE inhibitor, diisoprophyl fluorophosphate (DFP), amongstmany others, lacked this action [28]. Quantitative analysis ofβ-APP in the parietal cortex of the same rats confirmed thesefindings. In a similar manner, in both normal as well as intransgenic mice that over express human β-APP and Aβ,treatment with (-)-phenserine 2.5 mg/kg i.p. daily for as littleas 3 weeks significantly reduced β-APP levels in brain andCSF, in addition to mouse and human Aβ levels, respectively[61]. Thus far, no toxicological issues have been identifiedwith the reductions in β-APP and Aβ levels in rodent anddog studies that have extended beyond a year.
Plasma samples obtained in clinical trials of(-)-phenserine have been retrospectively examined to deter-mine the effects on β-APP and Aβ levels in a small blindedgroup of patients with AD [61]. In patients treated with(-)-phenserine for 3 or 6 weeks, levels of β-APP in theplasma have indicated a trend towards overall reduction(p=0.06) compared with placebo-treated controls. Also, inpatients treated with (-)-phenserine for 3 or 12 weeks, overallplasma levels of Aβ showed a trend towards reduction com-pared with placebo treatment, and the rate of increase in Aβobserved from 3 to 12 weeks appeared slower among pa-tients treated with (-)-phenserine versus controls [60]. Thedescribed trends, observed in a small number of availablesamples, provided a sufficiently positive signal to instigate aseparate 6 month duration, randomized, placebo controlled,double-blind trial to assess the actions of (-)-phenserine on
Fig. (5). Time-and concentration-dependent actions of -(-) phenserine on intracellular levels and levels of secreted β-APP and secreted Aβ inSK-N-SH human neuroblastoma cells, assessed by Western blot. -(+) phenserine had a similar action on β-APP and Aβ levels to those shownhere [26, 57].

An Overview of Phenserine Tartrate Current Alzheimer Research, 2005, Vol. 2, No. 3 287
CSF and plasma levels of Aβ and β-APP. While the trial isstill ongoing, an interim analysis on CSF levels of Aβ (1- 42)from the first 37 patients showed that treatment with (-)-phenserine (15mg BID) resulted in a 58 pg/ml differencefrom placebo following 6 months of treatment. There wassufficient information in this interim analysis to concludethat continued enrollment to the 15mg dose group was justi-fied. The clinical studies final results may not only furtherelucidate the action of the compound on disease progressionbut may, additionally, provide associations with cognitivemeasures as well as brain structural imaging studies that arebeing assessed within the same subjects.
CLINICAL PROFILE
Safety, Pharmacokinetics and Pharmacodynamics
In humans, (-)-phenserine produces long-acting, reversi-ble inhibition of AChE [21]. In a single-dose oral bioavail-ability study in healthy young subjects, (-)-phenserine 10 mgwas shown to be rapidly absorbed and eliminated, and therewas a direct association between plasma drug concentrationand erythrocyte AChE inhibition activity [53]. The safetyand pharmacokinetics of single- and multiple-dose(-)-phenserine have been examined in Phase I, blinded, pla-cebo-controlled studies involving healthy elderly volunteers(aged 55–80 years).
In one study, in 32 elderly subjects administered a singledose of (-)-phenserine 5, 10 or 20 p.o. without a titrationschedule, doses of 5 and 10 mg were well tolerated and themaximum-tolerated dose was found to be 10 mg (with dose-limiting side effects of nausea and vomiting occurring at20 mg and above) [53]. No clinically significant changes invital signs or laboratory. However, parameters, were de-tected at any dose studied. Oral bioavailability of
(-)-phenserine was high and predictable. Plasma levels of(-)-phenserine 5–20 mg increased non-linearly with dose andwere maximal in 1.25–1.5 hours: Cmax was 0.46, 1.33 and6.73 ng/mL at 5, 10 and 20 mg doses, respectively. A 4-foldincrease in dose from 5 to 20 mg led to an approximate 25-fold increase in area under curve in 24 hours (AUC0–24).Peak erythrocyte AChE inhibition was seen 2 hours afteradministration of (-)-phenserine 5–20 mg, with approxi-mately 12%, 23% and 46% inhibition at the 5, 10 and 20 mgdose levels, respectively. This effect was linearly related to(-)-phenserine plasma concentration. The level of inhibitionachieved in brain was not determined. Animal studies indi-cated that the level of inhibition will be higher than deter-mined for erythrocytes [45] and is a focus of an ongoingclinical trial.
In 32 elderly individuals administered (-)-phenserine 5and 10 mg q.d. and b.i.d. for 6 days, no dose-limiting or seri-ous adverse events (AEs) occurred and no clinically signifi-cant changes in laboratory or electrocardiogram (ECG)parameters were identified [53]. (-)-phenserine 5 mg q.d.reached a peak plasma concentration approximately 1.5hours after dosing (Cmax 0.34 on Day 1). Peak plasma con-centration increased with dose and was similar for subjectstreated once or twice daily. Consistent AUC0–24 values dur-ing treatment with (-)-phenserine for 6 days confirmed a lackof drug accumulation with repeated dosing [53], as was pro-posed in drug design studies. Erythrocyte AChE inhibitionwas approximately 15% and 30% in the 5 and 10 mg dosegroups, respectively, and was unaffected by dose regimen.
The interaction potential of (-)-phenserine with othermedications has not yet been fully characterized and repre-sents a major focus of ongoing studies. Metabolism involvesN-demethylation and hydroxylation and occurs predomi-nantly through the CYP 3A4 pathway [53].
Fig. (6). Action of -(-) phenserine on CSF levels of secreted β-APP in naïve animals and those with forebrain lesions [27, 28, 57].
288 Current Alzheimer Research, 2005, Vol. 2, No. 3 Greig et al.
Efficacy and Safety/Tolerability of (-)-Phenserine in Pa-tients with AD
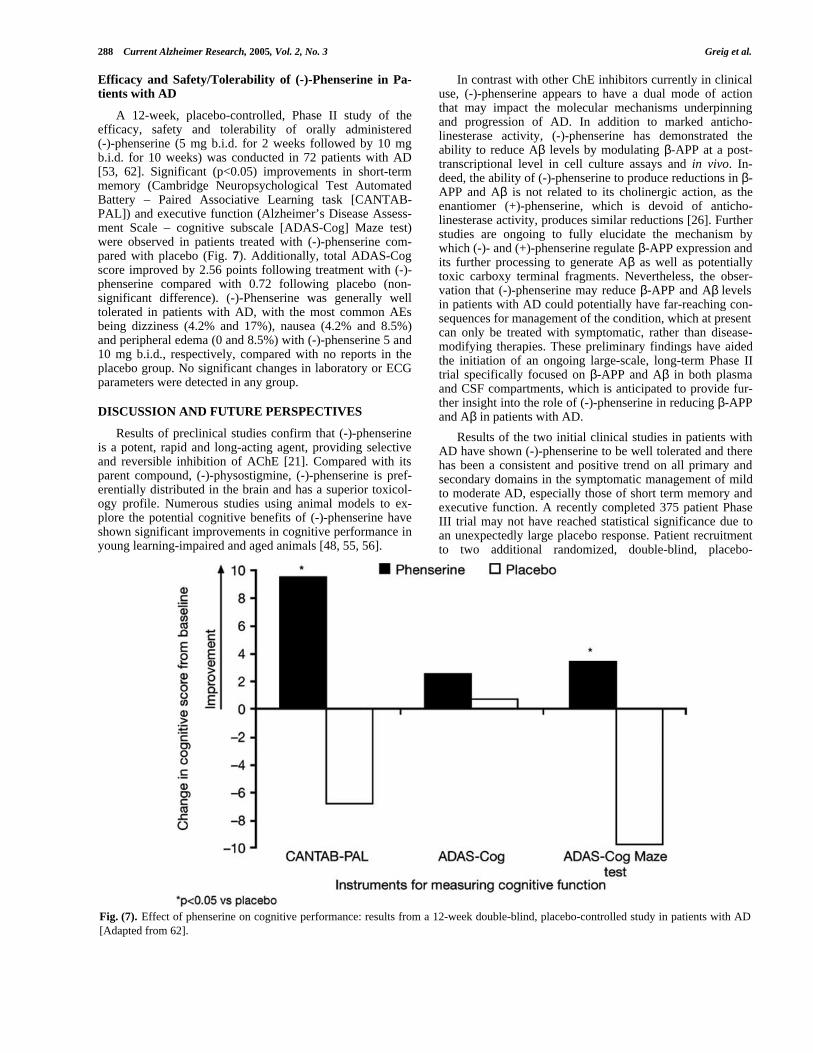
A 12-week, placebo-controlled, Phase II study of theefficacy, safety and tolerability of orally administered(-)-phenserine (5 mg b.i.d. for 2 weeks followed by 10 mgb.i.d. for 10 weeks) was conducted in 72 patients with AD[53, 62]. Significant (p<0.05) improvements in short-termmemory (Cambridge Neuropsychological Test AutomatedBattery – Paired Associative Learning task [CANTAB-PAL]) and executive function (Alzheimer’s Disease Assess-ment Scale – cognitive subscale [ADAS-Cog] Maze test)were observed in patients treated with (-)-phenserine com-pared with placebo (Fig. 7). Additionally, total ADAS-Cogscore improved by 2.56 points following treatment with (-)-phenserine compared with 0.72 following placebo (non-significant difference). (-)-Phenserine was generally welltolerated in patients with AD, with the most common AEsbeing dizziness (4.2% and 17%), nausea (4.2% and 8.5%)and peripheral edema (0 and 8.5%) with (-)-phenserine 5 and10 mg b.i.d., respectively, compared with no reports in theplacebo group. No significant changes in laboratory or ECGparameters were detected in any group.
DISCUSSION AND FUTURE PERSPECTIVES
Results of preclinical studies confirm that (-)-phenserineis a potent, rapid and long-acting agent, providing selectiveand reversible inhibition of AChE [21]. Compared with itsparent compound, (-)-physostigmine, (-)-phenserine is pref-erentially distributed in the brain and has a superior toxicol-ogy profile. Numerous studies using animal models to ex-plore the potential cognitive benefits of (-)-phenserine haveshown significant improvements in cognitive performance inyoung learning-impaired and aged animals [48, 55, 56].
In contrast with other ChE inhibitors currently in clinicaluse, (-)-phenserine appears to have a dual mode of actionthat may impact the molecular mechanisms underpinningand progression of AD. In addition to marked anticho-linesterase activity, (-)-phenserine has demonstrated theability to reduce Aβ levels by modulating β-APP at a post-transcriptional level in cell culture assays and in vivo. In-deed, the ability of (-)-phenserine to produce reductions in β-APP and Aβ is not related to its cholinergic action, as theenantiomer (+)-phenserine, which is devoid of anticho-linesterase activity, produces similar reductions [26]. Furtherstudies are ongoing to fully elucidate the mechanism bywhich (-)- and (+)-phenserine regulate β-APP expression andits further processing to generate Aβ as well as potentiallytoxic carboxy terminal fragments. Nevertheless, the obser-vation that (-)-phenserine may reduce β-APP and Aβ levelsin patients with AD could potentially have far-reaching con-sequences for management of the condition, which at presentcan only be treated with symptomatic, rather than disease-modifying therapies. These preliminary findings have aidedthe initiation of an ongoing large-scale, long-term Phase IItrial specifically focused on β-APP and Aβ in both plasmaand CSF compartments, which is anticipated to provide fur-ther insight into the role of (-)-phenserine in reducing β-APPand Aβ in patients with AD.
Results of the two initial clinical studies in patients withAD have shown (-)-phenserine to be well tolerated and therehas been a consistent and positive trend on all primary andsecondary domains in the symptomatic management of mildto moderate AD, especially those of short term memory andexecutive function. A recently completed 375 patient PhaseIII trial may not have reached statistical significance due toan unexpectedly large placebo response. Patient recruitmentto two additional randomized, double-blind, placebo-
Fig. (7). Effect of phenserine on cognitive performance: results from a 12-week double-blind, placebo-controlled study in patients with AD[Adapted from 62].

An Overview of Phenserine Tartrate Current Alzheimer Research, 2005, Vol. 2, No. 3 289
controlled Phase III trials using the same immediate releaseformulation of (-)-phenserine has been halted to take advan-tage of a reformulation initiative that is being pursued inorder to achieve higher overall treatment exposure during thedosing interval.
ACKNOWLEDGEMENT
The authors are indebted to Emma Potts for her assis-tance in preparing this article. NHG and QSY are supportedby the Intramural Research Program of the National Instituteon Aging, NIH. KS and DKL are supported by grants fromNIH in addition to funding from Axonyx Inc.
ABBREVIATIONS
Aβ = beta-Amyloid
ACh = Acetylcholine
AChE = Acetylcholinesterase
AD = Alzheimer’s disease
ADAS-Cog = Alzheimer’s Disease AssessmentScale – cognitive subscale
AEs = Adverse events
β-APP = beta-Amyloid precursor protein
BuChE = Butyrylcholinesterase
CANTAB-PAL = Cambridge Neuropsychological TestAutomated Battery – Paired Associa-tive Learning
ChE = Cholinesterase
CPP = 3(+) 2-Carboxypiperzin-4-yl propylphosphonic acid
CSF = Cerebrospinal fluid
DFP = Diisopropyl fluorophosphate
ECG = Electrocardiogram
NMDA = N-methyl-D-aspartate
REFERENCES
[1] Cummings JL and Mendez MF. Alzheimer’s disease: cognitive andbehavioural pharmacotherapy. Connecticut Medicine 61: 543–552(1997).
[2] Small GW, Rabins PV, Barry PP, Buckholtz NS, DeKosky ST,Ferris SH, et al. Diagnosis and treatment of Alzheimer disease andrelated disorders: consensus statement of the American Associationfor Geriatric Psychiatry, the Alzheimer’s Association, and theAmerican Geriatrics Society. JAMA 278: 1363–1371 (1997).
[3] Geldmacher DS and Whitehouse Jr PJ. Differential diagnosis ofAlzheimer’s disease. Neurology 48: S2–9 (1997).
[4] Katzman R and Kawas C. In: ‘Alzheimer Disease’. (Ed: Terry RD,Katzman R and Bick KL), Raven Press Ltd, New York (1994).
[5] Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alz-heimer disease in the US population: prevalence estimates usingthe 2000 census. Arch Neurol. 60(8):1119–1122 (2003).
[6] Caro J, Ward A, Ishak K, Migliaccio-Walle K, Getsios D, Papado-poulos G, Torfs K. To what degree does cognitive impairment inAlzheimer’s disease predict dependence of patients on caregivers?BMC Neurol 2(1):6 (2002).
[7] Torti FM Jr, Gwyther LP, Reed SD, Friedman JY, Schulman KA.A multinational review of recent trends and reports in dementiacaregiver burden. Alzheimer Dis Assoc Disord. 18(2): 99–109(2004).
[8] Selkoe DJ. Translating cell biology into therapeutic advances inAlzheimer’s disease. Nature 399 (6738 Suppl): A23–31 (1999).
[9] Sambamurti K, Greig NH and Lahiri DK. Advances in the cellularand molecular biology of the beta-amyloid protein in Alzheimer'sdisease. Neuromolecular Med 1: 1–31 (2002).
[10] Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT andDelon MR. Alzheimer's disease and senile dementia: loss of neu-rons in the basal forebrain. Science 215: 1237–1239 (1982).
[11] Bartus R, Dean R, Beer B, Lippa A. The cholinergic hypothesis ofgeriatric memory dysfunction. Science 217: 408–14 (1982).
[12] Weinstock M. Selectivity of cholinesterase inhibition. Drugs12(4):307–323 (1999).
[13] Cummings JL. Cognitive and behavioral heterogeneity in Alz-heimer’s disease: seeking the neurobiological basis. Neurobiologyand Aging 21: 845–861 (2000).
[14] Bullock R. Future directions in the treatment of Alzheimer's dis-ease. Expert Opin Investig Drugs 13(4): 303–304 (2004).
[15] Lahiri DK, Farlow MR, Sambamurti K, Greig NH, Giacobini E andSchneider LS. A critical analysis of new molecular targets andstrategies for drug developments in Alzheimer's disease. Curr DrugTargets 4: 97–112 (2003a).
[16] Lahiri DK and Greig NH. Lethal weapon: amyloid β-peptide, rolein the oxidative stress and neurodegeneration of Alzheimer’s dis-ease. Neurobiol Aging 25: 581–587 (2004).
[17] Samuels SC, Grossman H. Emerging therapeutics for Alzheimer'sdisease: an avenue of hope. CNS Spectr 8(11): 834–845 (2003).
[18] McKenna MT, Proctor GR, Young LC and Harvey AL. Noveltacrine analogues for potential use against Alzheimer's disease:potent and selective acetylcholinesterase inhibitors and 5-HT up-take inhibitors. J Med Chem 40: 3516–3523 (1997).
[19] Bruhlmann C, Ooms F, Carrupt PA, Testa B, Catto M, Leonetti F,et al. Coumarins derivatives as dual inhibitors of acetylcho-linesterase and monoamine oxidase. J Med Chem 44: 3195–3198(2001).
[20] Piazzi L, Rampa A, Bisi A, Gobbi S, Belluti F, Cavalli A, et al. 3-(4-[[Benzyl(methyl)amino] methyl]phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and ace-tylcholinesterase-induced beta-amyloid aggregation: a dual func-tion lead for Alzheimer's disease therapy. J Med Chem 46:2279–2282 (2003).
[21] Greig NH, Sambamurti K, Yu QS, Perry TA, Holloway HW,Haberman F, et al. In: ‘Butyrylcholinesterase its function and in-hibitors’ (Ed: Giacobini E), Martin Dunitz, London, p. 69–90(2003).
[22] Primo-Parmo SL, Bartels CF, Wiersema B, van der Spek AF, InnisJW and La Du BN. Characterization of 12 silent alleles of the hu-man butyrylcholinesterase (BCHE) gene. Am J Hum Genet 58:52–64 (1996).
[23] Soreq H and Zakut H. In: ‘Human Cholinesterases and Anticho-linesterases’. Academic Press, NY (1993).
[24] Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG andLockridge O. Acetylcholinesterase knockouts establish centralcholinergic pathways and can use butyrylcholinesterase to hydro-lyze acetylcholine. Neuroscience 110: 627–639 (2002).
[25] Liston DR, Nielsen JA, Villalobos A, Chapin D, Jones SB, Hub-bard ST, et al. Pharmacology of selective acetylcholinesterase in-hibitors: implications for use in Alzheimer’s disease. Eur J Phar-macol 486: 9–19 (2004).
[26] Shaw KT, Utsuki T, Rogers J, Yu QS, Sambamurti K, Brossi A, etal. Phenserine regulates translation of beta-amyloid precursor pro-tein mRNA by a putative interleukin-1 responsive element, a targetfor drug development. Proc Natl Acad Sci USA 98: 7605–7610(2001).
[27] Greig NG, Ingram D, Wallace WC, Utsuki T, Yu Q, HollowayHW, et al. In: ‘Alzheimer’s disease: molecular biology to therapy’.(Ed: Becker B, Giacobini E and Robert P), Birkhauser, Boston, p.231–237 (1996).
[28] Haroutunian V, Greig N, Pei XF, Utsuki T, Gluck R, Acevedo LD,et al. Pharmacological modulation of Alzheimer’s beta-amyloidprecursor protein in the CSF of rats with forebrain cholinergic sys-tem lesions. Mol Brain Res 46: 161–168 (1997).
[29] Greig NH, Pei X-F, Soncrant TT, Ingram DK, Brossi A. Phenserineand ring C hetero-analogues: drug candidates for the treatment ofAlzheimer’s disease. Medicinal Res Rev 15: 3–31 (1995a).

290 Current Alzheimer Research, 2005, Vol. 2, No. 3 Greig et al.
[30] Brossi A, Pei X-F and Greig NH. Phenserine, a novel anticho-linesterase related to physostigmine: total synthesis, and biologicalproperties. Austr J Chem 49: 171–190 (1996).
[31] Yu QS, Pei XF, Holloway HW, Greig NH and Brossi A. Totalsyntheses and anticholinesterase activities of (3aS)-N(8)-norphysostigmine, (3aS)-N(8)-norphenserine, their antipodal iso-mers, and other N(8)-substituted analogues. J Med Chem 40:2895–2901 (1997).
[32] Yu QS, Greig NH, Holloway HW and Brossi A. Syntheses andanticholinesterase activities of (3aS)-N1,N8-bisnorphenserine,(3aS)-N1,N8-bisnorphysostigmine, their antipodal isomers, andother potential metabolites of phenserine. J Med Chem 41:2371–2379 (1998).
[33] Yu QS, Holloway HW, Utsuki T, Brossi A and Greig NH. Synthe-sis of novel phenserine-based-selective inhibitors of butyrylcho-linesterase for Alzheimer’s disease. J Med Chem 42: 1855–1861(1999).
[34] Yu QS, Holloway HW, Flippen-Anderson JL, Hoffman B, BrossiA and Greig NH. Methyl analogues of the experimental Alzheimerdrug phenserine: synthesis and structure/activity relationships foracetyl- and butyrylcholinesterase inhibitory action. J Med Chem44: 4062–4071 (2001).
[35] Polonovski M. Bl Soc Chim Fr 1916;19:47; summarized in Beil-stein E 1954. II;23:333.
[36] Taylor P and Radic Z. The cholinesterases: from genes to proteins.Annu Rev Pharmacol Toxicol 34: 281–320 (1994).
[37] Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L,et al. Atomic structure of acetylcholinesterase from Torpedo cali-fornica: a prototypic acetylcholine-binding protein. Science 253:872–879 (1991).
[38] Ripoll DR, Faerman CH, Axelsen PH, Silman I and Sussman JL.An electrostatic mechanism for substrate guidance down the aro-matic gorge of acetylcholinesterase. Proc Natl Acad Sci USA 90:5128–5132 (1993).
[39] Tan RC, Truong TN, McCammon JA and Sussman JL. Acetylcho-linesterase: Electrostatic Steering Increases the Rate of LigandBinding. Biochemistry 32: 401–403 (1993).
[40] Soreq H and Seidman S. Acetylcholinesterase - New roles for anold actor. Nat Rev Neurosci 2: 294–302 (2001).
[41] Quinn DM. Acetylcholinesterase: enzyme structure, reaction dy-namics and virtual transition states. Chem Rev 87: 955–979 (1987).
[42] Harel M, Sussman JL, Krejci E, Bon S, Chanal P, Massoulie J, etal., Conversion of acetylcholinesterase to butyrylcholinesterase:modelling and mutagenesis. Proc Natl Acad Sci USA 89:10827–10831 (1992).
[43] Al-Jafari AA, Kamal MA, Greig NH, Alhomida AS and Perry ER.Kinetics of human erythrocyte acetylcholinesterase inhibition by anovel derivative of physostigmine: phenserine. Biochem BiophysRes Comm 248: 180–185 (1998).
[44] Al-Jafari AA, Kamal MA, Alhomida AS and Greig NH. Kinetics ofrat brain acetylcholinesterase inhibition by two experimental Alz-heimer’s disease drugs, phenserine and tolserine. J Biochem MolBio Biophysiol 4: 323–335 (2000).
[45] Greig NH, De Micheli E, Holloway HW, Yu QS, Utsuki T, PerryTA, et al. The experimental Alzheimer drug phenserine: preclinicalpharmacokinetics and pharmacodynamics. Acta Neurol Scand 102(Suppl. 176): 74–84 (2000).
[46] Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vicente M,Linker C, et al. Acetylcholinesterase accelerates assembly of amy-loid-β-peptides into Alzheimer’s fibrils: Possible role of the pe-ripheral site of the enzyme. Neuron 16: 881–891 (1996).
[47] Inestrosa NC and Alarcon R. Molecular interactions of acetylcho-linesterase with senile plaques. J Physiol Paris 92: 341–344 (1998).
[48] Iijima S, Greig NH, Garofalo P, Spangler EL, Heller B, Brossi A,et al. Phenserine: a physostigmine derivative that is a long-acting
inhibitor of cholinesterase and demonstrates a wide dose range forattenuating a scolpolamine-induced learning impairment of rats in a14-unit T-maze. Psychopharmacology (Berl) 112: 415–420 (1993).
[49] Brufani M, Castellano C, Marta M, Oliverio A, Pagella PG, PavoneF, et al. A long-lasting cholinesterase inhibitor affecting neural andbehavioural processes. Pharmacol Biochem Behav 26: 625–629(1987).
[50] Yu QS, Liu C, Brzostowska M, Chrisey L, Brossi A, Grieg NH, etal. Physovenines: efficient synthesis of (-)- and (+)-physovenineand synthesis of carbamate analogues of (-)-physovenine. Anticho-linesterase activity and analgesic properties of optically activephysovenines. Helv Chim Acta 74: 761–764 (1991).
[51] Yu QS, Greig NH, Holloway HW, Flippen-Anderson F and BrossiA. (-)-(3aS)-Eseroline carbamate (II), a potent cholinesterase in-hibitor and close analogue of physostigmine: reanalysis. MedChem Res 10: 186–199 (2000).
[52] Pomponi M, Giardina B, Gatta F and Marta M. Physostigmine andtetrahydroaminoacridine analogs as alternative drugs for the treat-ment of Alzheimer’s disease. Med Chem Res 2: 306–327 (1992).
[53] Data on File, Axonyx Corporation (2004).[54] Ingram DK, Spangler EL, Iijima S, Ikari H, Kuo H, Greig NH, et
al. Rodent models of memory dysfunction in Alzheimer’s diseaseand normal aging: moving beyond the cholinergic hypothesis. LifeSci 55: 2037–2049 (1994).
[55] Ikari H, Spangler EL, Greig NH, Pei XF, Brossi A, Speer D, et al.Maze learning in aged rats is enhanced by phenserine, a novel anti-cholinesterase. Neuroreport 6: 481–484 (1995).
[56] Patel N, Spangler EL, Greig NH, Yu Q-S, Ingram DK and MeyerRC. Phenserine, a novel acetylcholinesterase inhibitor, attenuatesimpaired learning of rats in a 14-unit T-maze induced by blockadeof the N-methyl-D-aspartate receptor. Neuro Report 9: 171–176(1998).
[57] Greig NH, Utsuki T, Yu QS, Holloway HW, Perry T, Giordano T,et al. (-) Phenserine, an experimental Alzheimer’s disease acetyl-cholinesterase inhibitor that effects cognition and beta-amyloidprecursor protein translation. Poster 32I. 8th International Mont-real-Springfield Symposium on Advances in Alzheimer Therapy,April 14-17, 2004, pp. 64.
[58] Lahiri DK, Ge Y-W, Farlow MR, Chen D, Sambamurti K, GreigNH, et al. Modifying amyloid precursor protein expression toregulate amyloid _-peptide: actions of phenserine, transforminggrowth factor and cytokines in cell culture. Poster 48I. 8th Interna-tional Montreal-Springfield Symposium on Advances in AlzheimerTherapy, April 14-17, 2004, pp. 102
[59] Lahiri DK, Ge Y-W, Rogers JT and Greig NH. Role of cytokines inthe regulation of APP gene expression in Alzheimer’s disease:identification of a 5’-UTR-binding nuclear factor. J Alz Dis 5:81–90 (2003b).
[60] Maloney B, Ge Y, Greig NH and Lahiri DK. Presence of a “CAGAbox” in the APP gene unique to amyloid-forming species and ab-sent in all APLP-1/2 genes: implications in Alzheimer’s disease.FASEB J 18: 1288–1290 (2004).
[61] Sambamurti K, Taylor S, Chung P, Lamb B, Chen D, Lahiri DK, etal. Effect of (-) phenserine on the levels of amyloid precursor pro-tein (APP) and amyloid β-peptide in cell culture and transgenicmice expressing human APP. Poster 70I. 8th International Mont-real-Springfield Symposium on Advances in Alzheimer Therapy,April 14-17, 2004, pp. 102
[62] Harrison JE, Kirby L, Baumer B, Eisner LS, Safirstein B, Birrell J,et al. A double blind, randomized, placebo-controlled study of (-)-phenserine in Alzheimer’s disease. Poster. Poster 36I. 8th Interna-tional Montreal-Springfield Symposium on Advances in AlzheimerTherapy, April 14-17, 2004, pp. 68




















