
GARCH-Based Identification and Estimation of Triangular Systems

Altering the Ordering and Disordering of a Triangular
Nanographene at Room Temperature H. S. Wong*1,2, X. Feng3, Z. Y. Yang1, K. Müllen3, N. Chandrasekhar2, C. Durkan1* 1: Nanoscience Centre, University of Cambridge, 11 JJ Thomson Avenue, Cambridge,
CB3 0FF, UK 2: Institute of Materials Research and Engineering, 3 Research Link, Singapore 117602 3: Max-Planck-Institut für Polymerforschung, Postfach 3148, 55021 Mainz, Germany
*: corresponding authors, email: [email protected], [email protected] Abstract
Molecular self-organisation has the potential to serve as an efficient and versatile tool for the spontaneous creation of low-dimensional nanostructures on surfaces. We demonstrate how the subtle balance between intermolecular interactions and molecule-surface interactions can be altered by modifying the environment or through manipulation by means of the tip in a scanning tunnelling microscope (STM) at room temperature. We show how this leads to the distinctive ordering and disordering of a triangular nanographene molecule, the Trizigzag-hexa-peri-hexabenzocoronenes-phenyl-6 (TrizigzagHBC-Ph6), on two different surfaces: graphite and Au(111). The assembly of submonolayer films on graphite reveals a six-fold packing symmetry under UHV conditions, whereas at the graphite-phenyloctane interface, they reorganise into a four-fold packing symmetry, mediated by the solvent molecules. On Au(111) under UHV conditions in the multilayer films we investigated, although disorder prevails with the molecules being randomly distributed, their packing behaviour can be altered by the scanning motion of the tip. The asymmetric diode-like current-voltage characteristics of the molecules are retained when deposited on both substrates. This study highlights the importance of the surrounding medium and any external stimulus in influencing the molecular organisation process, and offers a unique approach for controlling the assembly of molecules at a desired location on a substrate. Introduction

With the initial realization that molecules can behave as current-carrying
entities, investigations of molecular electronic junctions, in which molecules and
molecular assemblies operate as semiconductors, constitute a major part of the active
field of molecular electronics (1-3). The eventual goal of integrating molecules into
future electronics devices motivates the study of molecular self-organisation, which is
promising in serving as a natural means of constructing and engineering complex
architectures on substrates, based on the interplay between various non-covalent
interactions working simultaneously to build functional molecular nanostructures in a
bottom-up approach.
In particular, the adsorption behaviour of the family of aromatic molecules with
π-electron characteristics is being extensively pursued (4-6), given their relatively high
charge-carrier mobility (7-9) and importance as building blocks for various electronic
and optoelectronic devices (6,10-14). These “nanographenes” are also perceived as
potential precursors for growing graphene as well as for fabricating graphene-based
materials (15,16). The Trizigzag-hexa-peri-hexabenzocoronenes-phenyl-6
(TrizigzagHBC-Ph6 as shown in Fig. 1) under investigation in this article is one of the
triangular nanographenes (17), which are large polyaromatic hydrocarbons, comprising
sp2 carbon, bringing about a highly delocalized π-electron system. Its three-fold
symmetry with a helical packing structure provides them an optimal local arrangement
for charge transport (8). Given their relatively large dimension and deviations from
planarity, ordering on surfaces depends on the substrate and the inherent molecular
structure in a complex manner, since the molecules interact simultaneously with a large
number of surface atoms. As such, the resulting packing may be less restricted by the
periodicity of the substrate lattice, especially when external influences come into play,
which is the topic of this article.
Despite the possibility of attaining a large variety of ordered and disordered
two-dimensional monolayers on different surfaces through molecular design (15,18), it
remains a challenge to alter the original as-deposited pattern of any given molecule into
other forms of arrangement, due to its interaction with the substrate. In this article, we
study how an assembly, when deposited on substrates (Au(111) or graphite), can be
affected by the surrounding medium (ultrahigh vacuum or co-deposited with phenyl
octane) and an external force (scanning tip). Understanding the mechanisms controlling
the self-organisation process, by disrupting the equilibrium interactions, is pivotal in

constructing a wider range of surface molecular architectures catered for specific
applications.
Experimental
0.01 mg of TrizigzagHBC-Ph6 in powder form was dissolved in 10 ml of 1-
phenyloctane (b.p. 262˚C) and underwent sonication for 2 minutes. The resulting
solution was yellowish. The synthesis of TrizgzagHBC-Ph6 is described elsewhere (17).
The two substrates used here, HOPG (highly oriented pyrolytic graphite) and
Au(111) were prepared as follows: a HOPG substrate was freshly cleaved with adhesive
tape in air immediately before the molecular solution was deposited. The Au(111) on
mica substrate was cleaned in UHV (Ultra High Vacuum) by several cycles of Ar+ ion
sputtering at an ion energy of 800 eV and subsequent annealing at 450˚C to ensure the
surface was in good condition, i.e. a condition whereby the herringbone surface
reconstruction was observed under UHVSTM. A drop of solution was subsequently
deposited onto each substrate under ambient conditions at which point the samples were
placed in the load-lock of the vacuum system and pumped down overnight to a pressure
of 10-8 mbar and then heated to 80˚C to aid removal of any residual solvent. All UHV
measurements were performed at room temperature and at a pressure of the order of 10-
10 mbar with an Omicron UHV STM/AFM system. The STM tips were prepared by
electrochemically etching a 0.25 mm diameter tungsten wire with 3M NaOH. Liquid
STM imaging on HOPG was performed under ambient conditions with a home-built
STM, using mechanically-formed Pt/Ir tips. In this case, the solution was dropped onto
the substrate in the vicinity of the STM tip, forming a meniscus on the tip, which was
therefore immersed in the solution. The droplet size was observed to only marginally
shrink during the several hours of experimentation.
Each plot of I-V (Current-Voltage) data, taken in UHV, is an average of at least
ten voltage sweeps, with 500 data points each. While performing the spectroscopic
measurements, the tunnelling gap was held constant by freezing the feedback loop
while a voltage ramp was applied and the tunnelling current was recorded as a function
of the applied bias voltage. All STM images and I-V curves were analysed with WSxM
(19) without filtering.

For the optimization of the TrizigzagHBC-Ph6 molecular structure as shown in
Fig. 1, we used the molecular simulation package TINKER 5.1 (20) incorporating the
MM3 force field model (21), which is suitable for π-systems, using periodic boundary
conditions and a combination of simulated annealing and geometry optimization
techniques.
Results and Discussion
Molecules on Graphite in UHV
Figs. 2a and b show the resulting assembly of the molecules on HOPG, imaged
in UHV, having a six-fold packing symmetry, and despite the lack of intermolecular
resolution, the molecules do have a triangular appearance. From the 3-D representation
in Fig. 2b, it can be seen that the molecules appear to be sitting on the surface. The
blurring of the areas outside the assembly is indicative of a 2-d gas phase. This is strong
evidence that the substrate-molecule interaction is rather weak, despite the fact that the
spacing and arrangement of carbon atoms within the molecule is somewhat similar to
that of the graphite atomic lattice. One would therefore have expected a natural
tendency for the molecules to order in a manner that is commensurate with the
orientation of the underlying graphite lattice as illustrated in the proposed model in Fig.
2c. The Van der Waals interaction between the molecules is that of H-H while that
between the molecules and substrate is a C-C attractive interaction, both of which are
non-covalent. The weak ordering of the molecules is then in this case dictated by inter-
molecular interactions, and the symmetry of the assembly is dominated by the
molecular symmetry rather than by that of the HOPG substrate, even though they are
nominally similar.
Molecules on Graphite in Solvent

At the phenyloctane-graphite interface, the Trizigzag-Ph6 molecules arrange
themselves into a four-fold packing symmetry (Fig. 3a). This demonstrates the breaking
of commensurability between the molecular ordering and both the orientation of the
surface atomic lattice and the internal molecular symmetry, caused by the solvent
molecules. The possible molecular arrangement has been proposed in Fig. 3b for the
four-fold packing symmetry. The co-deposition of molecules with phenyloctane has
resulted in a stabilised structure. Repeated imaging over the same area showed no
alteration with time.
This disparity in molecular packing under different environments allows us to
infer that in both cases, the molecule-substrate interaction is too weak to direct the
ordering, but that the addition of a polar solvent has a marked effect and can alter the
packing symmetry between six-fold and four-fold. In the latter case, the four-fold
symmetry is not commensurate with the substrate and deviates from the molecular six-
fold symmetry.
The I-V characteristics (Fig. 2d) were measured through the conjugated cores
of the TrizigzagHBC in UHV, showing an asymmetry, i.e. a lower current for positive
bias (the voltage is applied to the sample), whereas the I-V profile on bare graphite is
symmetric. The asymmetric I-V characteristic of nanographene belonging to the
polycyclic aromatic hydrocarbon (PAH) family has been relatively well-studied,
explained by the resonant amplification of the tunnelling current through the HOMO
levels at negative sample bias. This is the result of a non-symmetric equilibrium Fermi
level of the molecule in the HOMO-LUMO gap (22). At the same time, the HOMO
levels are generally more delocalized and consequently exhibit greater electron
transmission across the molecule in tunnelling experiments. Conduction is therefore
dominated by the HOMO levels. This behaviour is analogous to a switch where at
positive sample bias, the trizigzagHBC molecules were driven to the OFF conductance
state, and at negative sample bias, they were switched to the ON conductance state
(23,24).
Molecules on Au(111) in UHV

On Au(111), the lateral interaction between the molecules is perturbed by the
relatively stronger molecule-surface interactions than in HOPG, due to the increased
electron density in Au(111), such that higher coverage without any ordering is observed
(Fig.4a), with the same solution deposition procedure. The films studied here are
therefore multilayers, and it is not possible to determine which is the layer in contact
with gold. Given the quantity of material deposited, we expect the film to be no more
than three monolayers thick on average. The molecules appear mobile during imaging
at room temperature, and upon increasing the tip-sample separation, individual
molecules can be observed randomly distributed on the surface, appearing as white dots
(Fig.4b). There is no apparent difference in the ordering for the different layers
observed, suggesting that the disorder is present even for the first layer. The observed
mobility of the TrizigzagHBC molecules also suggests that the molecule-substrate
interaction is non-covalent. The molecules interact with the gold surface via the
formation of a non-covalent π- interaction between their aromatic π-electrons and the
electron deficient metal surface.
Besides thermal activation, the scanning tip of the STM can induce mobility of
molecules on the surface leading to the lowering of surface energy through
rearrangement of molecules towards a smoother configuration. This is shown through
the evolution of the film structure during repeated imaging. This is somewhat akin to
the Ostwald ripening process, which describes the growth of larger domains at the
expense of smaller domains, although strictly that process relates to well-ordered
crystals, whereas our domains are disordered. The thermodynamic driving force is the
reduction of the circumference-to-area ratio and thereby the lowering of the interfacial
or line energy. The series of 11 images presented in Fig. 5 demonstrates how the process
of surface-energy minimisation of TrizigzagHBC on Au(111) is induced by the
scanning tip, and how the molecular layer appears to be more continuous.
The “hole” or cavity feature indicated by the large circle (Fig. 5) is seen to
evolve gradually from an irregular shaped cavity to a smoother circular form, and
eventually to merge with the nearest hole to form a larger hole, with a consequent
reduction in their joint circumference. An initial smaller cavity indicated by the smaller
circle shrinks with progressive scans and eventually disappears. Larger cavities are seen
to enlarge at the expense of smaller cavities. There is hence a critical cavity
area/dimension such that any cavity smaller than the critical area will be consumed and

cavities larger than the critical area will expand and coalesce with neighbouring ones.
The diffusion of cavities towards the right region is due to a slight thermal drift of the
tip leftwards in the course of repeated scans. The energy barrier is due to nearby
molecules which need to move into the cavity in order to let a particular molecule pass
through. The overall process results in a change in the cavity domain diameters from
an average of 5 to 8 nm to that of 12 to 20 nm. In simple terms, a molecule in contact
with a neighbour is in a lower energy configuration than those not in contact. Therefore
it is the desire of every molecule to achieve as many neighbours as they can possibly
have. Since the molecules along the circumference of a cavity have fewer neighbours
than molecules within the continuous film, they are therefore in a higher energy state.
In order to minimize the overall surface energy within the system, the number of
boundary molecules (on the cavity circumference) must be minimised, and this is
achieved by forming a smaller number of larger cavities.
To ascertain the point that the ripening process was accelerated by the scanning
tip, a similar region was imaged before and after the process (Fig. 6a & b). The features
around the scanned region remain relatively unaffected (as depicted by a circle);
validating that with minimal perturbation by the tip, the molecules remain more or less
at the same position after a longer lapse period in the course of 11 scans. The I-V
characteristic curves (Fig. 6c) were recorded in the cavity and on the molecular cluster
as indicated. Similar to Fig. 2c, an asymmetry in I-V profile is recorded since the
molecule is the same and the molecule-substrate interaction is relatively weak.
However, this asymmetry, which is the characteristic electronic profile of the
TrizigzagHBC, is recorded in both regions. Using this information, we can therefore
infer that even in the cavity region, there is a monolayer of molecules on top of the gold,
and that in the surrounding regions, there is a bilayer. After normalising (by multiplying
the lower-current curve by a factor of 2), both curves show perfect overlap. The higher
current for a given voltage in the cavity region is naturally because conduction through
one layer shows a lower resistance than through two layers.
Next, we examined a region in the vicinity of a step edge subjected to the same
conditions. From the series of images (Fig. 7), it can be seen that with repeated scans,
some cavities diffuse out to the edge, where they open up leading to a concave trench
along the edge, which gradually flattens and smoothens out. This is analogous to the
interaction of a bubble at the surface of a liquid. As before, holes within the scanned

region were either annihilated or merged to form larger ones and irregularly-shaped
holes slowly evolved into a more circular and smooth form. It should be noted that the
appearance of this edge is not consistent with that of a step edge on Au(111), so we
conclude that what we are observing is a step in the molecular film, which is several
layers thick in this region of the surface. A look at the line profile across the step and
cavities in Fig. 7m shows that the height across the step is the same as the depth of the
cavity, which is approximately 0.3 nm. The molecular height appears to be large
because the six phenyl rings which are the side groups of the molecule are not flat due
to the steric hindrance and repulsion from the hydrogen atoms. This can be clearly
observed from the simulated molecular structure in Fig. 1 where the six phenyl rings
are twisted and not in-plane with the TrizigzagHBC core.
Another point to note is the possibility of molecular diffusion over a terrace. In
general, molecules will have a lower energy barrier for diffusion along an edge or
crossing of a corner, and crossing an edge is often an energetically more costly process
than terrace diffusion due to the existence of the Schwoebel-Ehrlich barrier (25,26) at
the step edge. Therefore we do not expect that molecules residing on the upper terrace
would cross the step edge and get to the lower terrace during diffusion. Having said
that, the molecules are still expected to have a strong preference to adsorb at both the
upper and the lower step edge, which has frequently been observed for surface
adsorbates.
As observed in Fig. 7, after a sequence of 12 images, the cavities in the vicinity
of the step were either consumed or diffused away, implying that the molecules were
attracted to the step edge. The region around the step edge “thickens” by the molecules
after only a few scans and remains stable. This is due to the enhanced interaction
between the molecules and step edges by the Smoluchowski effect (27) where the
electron distribution at the step was smoothed by a charge transfer from the top to the
bottom of the step edge. This has been reported in STM experiments where benzene
molecules were shown to have adsorbed in two rows, one at the top and one at the
bottom of a Cu(111) step at 77 K thereby setting up adsorption sites for subsequent
rows of molecules (28,29). Again, in order to minimise the interfacial energy, any
irregularities at the edge are replaced by a smoother form (the Euler–Lagrange equation
validates that smooth shapes minimise surface area) (30). Ostwald ripening, which is a

manifestation of the minimisation of surface energy, has so far been reported by some
as changes in the size of ordered molecular clusters itself (31,32) rather than on the
dynamics of cavities in a disordered monolayer.
Conclusion
In summary, we demonstrate that at the graphite-phenyl octane interface,
TrizigzagHBC-Ph6 molecules are packed in a four-fold symmetric arrangement,
dominated by the balance of intermolecular interaction and solvent-mediation,
independent of the graphite lattice symmetry. The delicate balance of interaction forces
could however be altered by the removal of solvent which consequently enhanced the
molecule-molecule interaction. This lead to the transformation of ordering from the
original four-fold to that of a six-fold hexagonal symmetry which was coincidentally
commensurate with the hexagonal close packed structure of the graphite lattice, but was
in fact determined by the molecular symmetry, as evidenced by the appearance of a 2-
d gas phase elsewhere on the surface. When the same molecule is deposited on Au(111)
and scanned in UHV, a disordered multilayer structure is observed with a random
distribution of the molecules over the substrate. Amidst such disordering, a
phenomenon governed by the minimisation of surface energy through a process where
the molecules rearrange themselves causing large holes to grow at the expense of
smaller holes, could be induced by the scanning tip of the STM.
Ultimately, for the successful implementation of these self-organised patterns
in practice, methods to convert these desired molecular arrangements into more rigid
and robust structures need to be developed while retaining their precise spatial
organization. All these are essential requirements for any nanofabrication methodology
that aims to promote further miniaturization of future electronic components especially
those based mostly on carbon.
Acknowledgment HSW would like to acknowledge the scholarship from Astar (Singapore). References

1) Jorge M. Seminario, “Molecular Electronics – Approaching Reality,” Nature Materials 4, 111 (2005) 2) C. Joachim, J. K. Gimzewski, A. Aviram, “Electronics using hybrid-molecular and mono-molecular devices,” Nature 408, 541 (2000) 3) J. V. Barth, G. Costantini, K. Kern, “Engineering atomic and molecular nanostructures at surfaces,” Nature 437, 671 (2005) 4) J. Wu, A. Fechtenkötter, J. Gauss, M. D. Watson, M. Kastler, C. Fechtenkötter, M. Wagner, K. Müllen, “Controlled Self-Assembly of Hexa-peri hexabenzocoronenes in Solution,” J. Am. Chem. Soc. 126, 11311 (2004) 5) S. E. Stein and R. L. Brown, “π-Electron Properties of Large Condensed Polyaromatic Hydrocarbons,” J. Am. Chem. Soc. 109, 3721 (1987) 6) K. Müllen and J. P. Rabe, “Nanographenes as Active Components of Single Molecule Electronics and How a Scanning Tunneling Microscope Puts Them To Work,” Acc. Chem. Res. 41, 4, 511 (2008) 7) W. Pisula, M. Kastler, D. Wasserfallen, M. Mondeshki, J. Piris, I. Schnell and K. Mullen, “Relation between supramolecular order and charge carrier mobility of branched alkyl hexa-peri-hexabenzocoronenes,” Chem. Mater. 18, 3634 (2006)
8) X. Feng, V. Marcon, W. Pisula, M. Ryan Hansen, J. Kirkpatrick, F. Grozema, D. Andrienko, K. Kremer and Klaus Müllen, “Towards high charge-carrier mobilities by rational design of the shape and periphery of discotics,” Nature Materials 8, 421 (2009)
9) M. Duati, C. Grave, N. Tcbeborateva, J. Wu, K. Müllen, A. Shaporenko, M. Zharnikov, J. K. Kriebel, G. M. Whitesides, and M. A. Rampi, “Electron Transport Across Hexa-peri-hexabenzocoronene Units in a Metal–Self-Assembled Monolayer–Metal Junction,” Adv. Mater. 18, 329 (2006) 10) A. Kraft, A. C. Grimsdale, A. B. Holmes, “Electroluminescent conjugated polymers - seeing polymers in a new light,” Angew. Chem. Int. Ed. 37, 402 (1998) 11) L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend, J. D. MacKenzie, “Self-Organized Discotic Liquid Crystals for High-Efficiency Organic Photovoltaics,” Science 293, 1119 (2001) 12) M. G. Debije, J. Piris, M. P. de Haas, J. M. Warman, Z. Tomović, C. D. Simpson, M. D. Watson, K. Müllen, “The Optical and Charge Transport Properties of Discotic Materials with Large Aromatic Hydrocarbon Cores,” J. Am. Chem. Soc. 126, 4641 (2004) 13) I. Diez-Perez, Z. Li, J. Hihath, J. Li, C. Zhang, X. Yang, L. Zang, Y. Dai, X. Feng, K. Muellen, N. Tao, “Gate-controlled electron transport in coronenes as a bottom-up approach towards graphene transistors,” Nature Comms. 1, 31 (2010)

14) J. Wu, W. Pisula, and K. Mullen, “Graphenes as Potential Material for Electronics,” Chem. Rev. 107, 718 (2007) 15) M. D. Watson, A. Fechtenkötter, and Klaus Müllen, “Big Is Beautiful-“Aromaticity” Revisited from the Viewpoint of Macromolecular and Supramolecular Benzene Chemistry,” Chem. Rev. 101, 1267 (2001) 16) S. Müller and K. Müllen, “Expanding benzene to giant graphenes: towards molecular devices,” Phil. Trans. R. Soc. A 365, 1453 (2007) 17) X. Feng, J. Wu, M. Ai, W. Pisula, L. Zhi, J. P. Rabe, K. Müllen, “Triangle-Shaped Polycyclic Aromatic Hydrocarbons,” Angew. Chem. Int. Ed. 46, 3033 (2007) 18) L. Gross, F. Moresco, P. Ruffieux, A. Gourdon, C. Joachim, K.-H. Rieder, “Tailoring molecular self-organization by chemical synthesis: Hexaphenylbenzene, hexa-peri-hexabenzocoronene, and derivatives on Cu (111),” Phys. Rev. B 71, 165428 (2005) 19) I. Horcas, R. Fernandez, J.M. Gomez-Rodriguez, J. Colchero, J. Gomez-Herrero and A. M. Baro, Rev. Sci. Instrum. 78, 013705 (2007) 20) J. W. Ponder and F. M. Richard, “An efficient newton-like method for molecular mechanics energy minimization of large molecules,” J. Comput. Chem. 8, 1016 (1987) 21) N. L. Allinger, Y. H. Yuh and J.-H. Lii, “Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 1,” J. Am. Chem. Soc. 111, 8551 (1989) 22) S. Datta, W. Tian, S. Hong, R. Reifenberger, J. I. Henderson, and C. P. Kubiak, “Current-Voltage Characteristics of Self-Assembled Monolayers by Scanning Tunneling Microscopy,” Phys. Rev. Lett. 79, 2530 (1997) 23) A. Stabel, P. Herwig, K. Müllen, J. P. Rabe, “Diodelike Current-Voltage Curves for a Single Molecule-Tunneling Spectroscopy with Submolecular Resolution of an Alkylated, peri- Condensed Hexabenzocoronene,” Angew. Chem. Int. Ed. Engl. 34, 1609 (1994) 24) F. Jäckel, M. D. Watson, K. Müllen, J. P. Rabe, “Prototypical Single-Molecule Chemical-Field-Effect Transistor with Nanometer-Sized Gates,” Phys. Rev. Lett. 92, 188303 (2004) 25) S. C. Wang and G. Ehrlich, “Atom incorporation at surface clusters: An atomic view,” Phys. Rev. Lett. 67, 2509 (1991) 26) S. C. Wang and G. Ehrlich, “Adatom motion to lattice steps: A direct view,” Phys. Rev. Lett. 70, 41 (1993) 27) R. Smoluchowski, “Anisotrophy of the electronic work function of metals,” Phys. Rev., 60, 661 (1941)

28) M. M. Kamna, S. J. Stranick, P. S. Weiss, “Imaging substrate-mediated interactions,” Science 274, 118 (1996) 29) E. C. H. Sykes, P. Han, S. A. Kandel, G. S. McCarty, P. S. Weiss, “Substrate mediated interactions and intermolecular forces between molecules adsorbed on surfaces,” Acc. Chem. Res. 36, 945 (2003) 30) R. Courant and D. Hilbert, “Methods of Mathematical Physics: Vol I,” Interscience Publishers, New York (1953) 31) Y-R. Ma, P. Moriarty, P. H. Beton, “Disorder-Order Ripening of C60 Islands,” Phys. Rev. Lett. 78, 13, 2588 (1997) 32) E. Barrena, C. Ocal, M. Salmeron, “Evolution of the structure and mechanical stability of self-assembled alkanethiol islands on Au(111) due to diffusion and ripening,” J. Chem. Phys. 111, 21 (1999)

Figure 1: Optimized molecular structure of the TrizigzagHBC-Ph6
(a)

-1.5 -1.0 -0.5 0.0 0.5 1.0 1.5-3.0-2.5-2.0-1.5-1.0-0.50.00.51.01.5
Curr
ent (
nA)
Voltage (V)
HOPG TrizigzagHBC
(d)
(b)
(c)

Figure 2: (a) After removal of solvent, the TrizigzagHBC on HOPG assumes an ordered six-fold hexagonal packing in UHV (inset: 2D FFT showing a hexagonal symmetry. The slight distortion is due to thermal drift) (0.7V, 0.12 nA). (b) 3-D representation of Fig 2(a), showing molecular rows. (c) The proposed packing model for molecular arrangement on the graphite substrate. The molecular packing orientation is similar to the orientation of the graphite atomic lattice. (d) Current-voltage characteristic curves of TrizigzagHBC on graphite. Both molecule and bare HOPG were probed. An asymmetric curve is observed when measured from the molecule. (setpoint parameters: 0.7V, 0.1nA)
(a)

Figure 3: (a) TrizigzagHBC in phenyl octane on HOPG as-deposited showing a square packing at the phenyl octane-graphite interface (inset: 2DFFT showing a four fold, square, symmetry) (0.8V, 0.15 nA). (b) The proposed packing model for molecular arrangement on an area indicated by the green box in (a). This square molecular packing does not coincide with the hexagonal atomic lattice of the graphite surface (0.8V, 0.15nA).
(a)

Figure 4: (a) TrizigzagHBC on Au(111) as-deposited showing a larger coverage than on graphite but without any ordering (0.8V, 0.3 nA). (b) An area indicated by a blue box in (a) showing randomly arranged individual molecules, appearing as white spots (0.8V, 0.1 nA).

Figure 5: A series of eleven 100x100 nm2 images (A) to (K) at the same region illustrating the Oswald ripening phenomenon occurring on the terrace region upon lowering the tip-sample distance. The feature in the large blue circle serve as a marker showing how it evolved from an irregular shape to a circular form, slowly diffusing to the right and eventually annihilate with another cavity. The smaller green circle depicts three initial small cavities which reduce in size and eventually being consumed completely. Note that the holes diffuse to the right and an increased coverage is observed on the left (0.6V, 0.3 nA). (L) the enlarged region after 11 scans (0.8V 0.2nA). The drifting of holes to the right region is due to the drifting of the tip to the left during continuous scanning.
A B C D
E F G H
I J K L
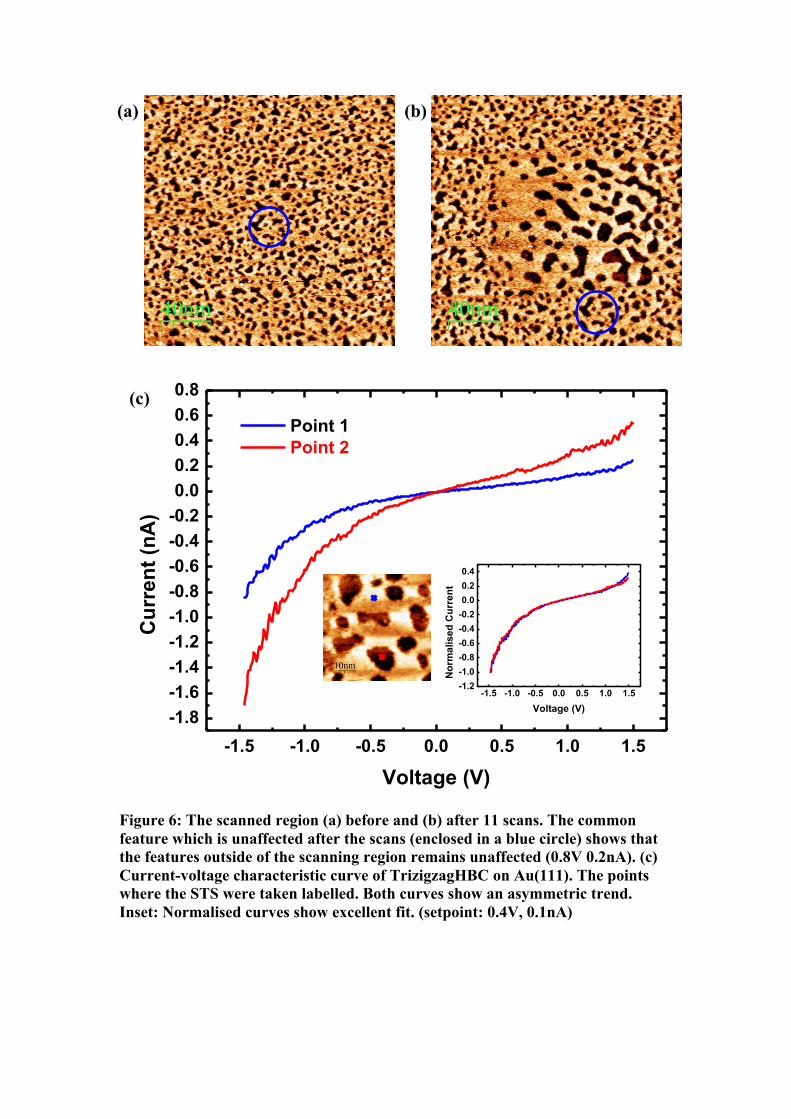
Figure 6: The scanned region (a) before and (b) after 11 scans. The common feature which is unaffected after the scans (enclosed in a blue circle) shows that the features outside of the scanning region remains unaffected (0.8V 0.2nA). (c) Current-voltage characteristic curve of TrizigzagHBC on Au(111). The points where the STS were taken labelled. Both curves show an asymmetric trend. Inset: Normalised curves show excellent fit. (setpoint: 0.4V, 0.1nA)
(b) (a)
-1.5 -1.0 -0.5 0.0 0.5 1.0 1.5-1.8-1.6-1.4-1.2-1.0-0.8-0.6-0.4-0.20.00.20.40.60.8
Cur
rent
(nA
)
Voltage (V)
Point 1 Point 2
-1.5 -1.0 -0.5 0.0 0.5 1.0 1.5-1.2-1.0-0.8-0.6-0.4-0.20.00.20.4
Nor
mal
ised
Cur
rent
Voltage (V)
(c)

A B C D
E F G H
0 10 20 30 40 50 60
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
Heig
ht (n
m)
Lateral Distance (nm)
(M)
I J K L

Figure 7: A series of twelve 100x100 nm2 images (A) to (L) at the same region (with some drifts) demonstrating the Oswald ripening phenomenon occurring directly in the vicinity of a step. Besides the annihilation of smaller cavities into bigger ones, the cavities are seen migrating towards the step edges as depicted by the circle. (0.6V, 0.3 nA). (M) Line profile across the molecular step as indicated by a line on (L). The height across the step and the depth of the cavity is approximately 0.3 nm.
Copyright © 2022 FDOKUMEN




















