
Ecdysone agonist inducible transcription in transgenic tobacco plants
Upload
independentCategory
view
1download
0
lable at ScienceDirect
Neuropharmacology 61 (2011) 937e949
Contents lists avai
Neuropharmacology
journal homepage: www.elsevier .com/locate/neuropharm
Agonist-specific voltage sensitivity at the dopamine D2S receptor e Moleculardeterminants and relevance to therapeutic ligands
Kristoffer Sahlholm a,*, Ofra Barchad-Avitzur b, Daniel Marcellino a, Maricel Gómez-Soler c, Kjell Fuxe a,Francisco Ciruela c, Peter Århema
aDepartment of Neuroscience, Retzius väg 8, Karolinska Institutet, 171 77 Stockholm, SwedenbDepartment of Neurobiology, The Hebrew University of Jerusalem, Jerusalem, IsraelcDepartament de Patologia i Terapèutica Experimental, Universitat de Barcelona, Barcelona, Spain
a r t i c l e i n f o
Article history:Received 9 April 2011Received in revised form20 May 2011Accepted 24 June 2011
Keywords:Dopamine receptorDopamine agonistElectrophysiologyVoltage sensitivityG protein-coupled receptorRadioligand
Abbreviations: 3-MT, 3-methoxy-tyramine; AT, amrescent Protein; DMSO, dimethyl sulphoxide; DPAT, NFRET, Fluorescence Resonance Energy transfer; GIRKrectifier potassium channel; GPCR, G protein-coumembrane segment; YFP, Yellow Fluorescent Protein.* Corresponding author. Tel.: þ46 8 524 839 76; fa
E-mail addresses: [email protected] (K. Sahac.il (O. Barchad-Avitzur), [email protected] (hotmail.com (M. Gómez-Soler), [email protected](F. Ciruela), [email protected] (P. Århem).
0028-3908/$ e see front matter � 2011 Elsevier Ltd.doi:10.1016/j.neuropharm.2011.06.022
a b s t r a c t
Voltage sensitivity has been demonstrated for some GPCRs. At the dopamine D2S receptor, this voltagesensitivity is agonist-specific; some agonists, including dopamine, exhibit decreased potency at depo-larized potentials, whereas others are not significantly affected. In the present study, we examined someof the receptoreagonist interactions contributing to these differences, and investigated how dopamineD2S receptor voltage sensitivity affects clinically used dopamine agonists. GIRK channel activation involtage-clamped Xenopus oocytes was used as readout of receptor activation. Structurally distinctagonists and complementary site-directed mutagenesis of the receptor’s binding site were used toinvestigate the role of agonistereceptor interactions. We also confirmed that the depolarization-induceddecrease of dopamine potency in GIRK activation is correlated by decreased binding of radiolabeleddopamine, and by decreased potency in G protein activation. In the mutagenesis experiments,a conserved serine residue as well as the conserved aspartate in the receptor’s binding site were found tobe important for voltage sensitive potency of dopamine. Furthermore, the voltage sensitivity of thereceptor had distinct effects on different therapeutic D2 agonists. Depolarization decreased the potencyof several compounds, whereas for others, efficacy was reduced. For some agonists, both potency andefficacy were diminished, whereas for others still, neither parameter was significantly altered. Thepresent work identifies some of the ligandereceptor interactions which determine agonist-specificeffects of voltage at the dopamine D2S receptor. The observed differences between therapeuticagonists might be clinically relevant, and make them potential tools for investigating the roles ofdopamine D2 receptor voltage sensitivity in native tissue.
� 2011 Elsevier Ltd. All rights reserved.
1. Introduction
The dopamine D2 receptor belongs to the superfamily of Gprotein-coupled receptors (GPCRs) and is the main target for ther-apeutic drugs against several common neurological, neuroendocri-nological, and psychiatric disorders such as Parkinson’s disease,
inotetralin; CFP, Cyan Fluo-,n-dipropyl-2-aminotetralin;, G-protein coupled inwardpled receptor; TM, trans-
x: þ46 8 34 95 44.lholm), [email protected]. Marcellino), maricel_gs@(K. Fuxe), [email protected]
All rights reserved.
hyperprolactinemia and schizophrenia (Kvernmo et al., 2008;Prabhakar and Davis, 2008; Seeman, 2006). Dopamine D2 receptoragonists make up an essential part of the pharmacological inventoryfor treatment, especially for the first two of these disorders. Thedopamine D2 receptor is expressed as two distinct splice variants;D2L and D2S (long and short), differing by a stretch of 29 residuesin the third intracellular loop (Usiello et al., 2000). D2S functionsas an inhibitory auto- and heteroreceptor at dopamine, glutamate,and GABA terminals in the CNS, whereas D2L is consideredto mediate the majority of postsynaptic responses to dopamine(Usiello et al., 2000).
Whereas GPCRs have not traditionally been regarded as sensitiveto membrane potential, increasing evidence to this effect has accu-mulated in recent years (see Mahaut-Smith et al., 2008; Parnas andParnas, 2010). Decreases in agonist binding affinity at Gi/o-coupledM2 muscarinic receptor and glutamate mGluR3 upon membrane

K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949938
depolarization were shown both by electrophysiological assays inXenopus oocytes, using G-protein coupled inward rectifier potassiumchannel (GIRK) opening as readout of receptor activity, as well asby radiolabeled agonist binding experiments in Xenopus oocytes(Ben-Chaim et al., 2003; Ohana et al., 2006). Conversely, for Gq-coupled M1 muscarinic receptor and mGluR1 depolarization causedan increase in agonist binding affinity (Ben-Chaim et al., 2006; Ohanaet al., 2006). In other studies of lysophosphatidic acid receptorsexpressed in oocytes, as well as P2Y1 and other Gq-coupled receptorsin rat megakaryocytes, increases in agonist potency were observedupon depolarization (Martinez-Pinna et al., 2010; Gurung et al., 2008).
We recently demonstrated voltage sensitivity of both splicevariants of the dopamine D2 receptor using the GIRK activationassay (Sahlholm et al., 2008a,b). It was further shown that thissensitivity has distinct consequences for the potencies of differentagonists; whereas potencies of several D2 receptor agonists werereduced upon depolarization from �80 to 0 mV, the EC50s of otheragonists were not significantly affected (Sahlholm et al., 2008c).Such agonist-specific effects of GPCR voltage sensitivity were alsoreported recently for the muscarinic M2 receptor (Navarro-Polancoet al., 2011).
Furthermore, Ben-Chaim et al. (2006) recorded charge move-ment within the M2 receptor, which correlated with the voltage-dependent shift in binding affinity, and Kupchik et al. (2011) aswell as Navarro-Polanco et al. (2011) further demonstrated that thischarge movement is affected by the presence of receptor ligands ina concentration-dependent, ligand-specific manner. These findingssuggest that parts of the receptor itself move upon changes of themembrane potential; presumably, one or several charged, voltage-sensing residues move in response to voltage changes, and thesemovements are relayed to the ligand binding site of the receptor.
The D2 receptor orthosteric ligand binding site is relatively wellcharacterized and much is known about the molecular basis ofligandereceptor interactions (Javitch, 1998; Floresca and Schetz,2004). In light of the previous findings outlined above, we wishedto explore further the relationships between agonistereceptorinteractions and voltage-induced effects on agonist potency. Werestricted our analysis to D2S, since most earlier mutational studieshave involved this splice variant. However, since the D2L and D2Sbinding sites are identical at the level of amino acid sequence, ourfindings are likely applicable also to D2L.
Initially, wewanted to examine whether the mechanism behindthe depolarization-induced potency decrease of dopamine at theD2S receptor indeed involves decreased dopamine binding atdepolarized potentials. We also aimed to confirm that this voltagesensitivity can be observed upstream of effector proteins, at thelevel of G protein activation.
Secondly, we hypothesized that agonist-specific voltage sensi-tivity is dependent on differences in the interactions betweenligand and receptor. Thus, we sought to identify ligandereceptorinteractions responsible for the differential impact of receptorvoltage sensitivity on different agonists at the D2S receptor by site-directed mutagenesis of residues in the ligand binding site, and byfurther variation of agonist structure (see Fig. 1).
Finally, given the prominent role for dopamine receptor agonists intherapy and as pharmacological tools, we wanted to investigatewhether a range of clinically used D2 agonists would show anydifferences in terms of how theyare affected byD2S voltage sensitivity.
2. Material and methods
2.1. Molecular biology
Human GIRK1 (Kir3.1) and GIRK4 (Kir3.4) cDNA (provided by Dr. Terence Hebert,University of Montreal, Canada) were in pCDNA3 (Invitrogen). cDNA encoding thehuman dopamine D2S receptor was in pGEM4Z (a gift from Dr. Marc Caron, Duke
University, North Carolina; used for most oocyte experiments with the wt receptor)or in pXOOM (for oocyte experiments with mutant receptors, radioligand bindingexperiments and for all experiments involving mammalian cells). The dual-purposevector, pXOOM, contains both a CMV promoter for mammalian expression as well as50 and 30 untranslated regions from the Xenopus b-globin gene to increase expres-sion of transcribed cRNA in oocytes (Jespersen et al., 2002), and was a gift fromDr. Søren-Peter Olesen, University of Copenhagen, Denmark.
The pXOOM vector was initially chosen to allow for maximal expression effi-ciency in case of poor expression of the D2S receptor mutants. We found thatinjection of 200 pg cRNA/oocyte provided optimal expression of the wt receptor,showing similar EC50s at 0 and �80 mV for GIRK activation by dopamine andR/S-DPATas when 5 ng/oocyte of cRNA from the pGEM4Z vector was used (Sahlholmet al., 2008c), without compromising oocyte survival. However, we subsequentlyused 200 pg cRNA/oocyte for the receptor mutants as well, as adequate GIRKresponse amplitudes were obtained in all cases. Point mutations of the dopamineD2S receptor were made using the QuickChange Lightning kit (Agilent Technologies),according to the manufacturer’s instructions. All mutations were confirmed by DNAsequencing of the entire insert.
For in vitro transcription, plasmids were linearized with the appropriaterestriction enzymes (GIRK 1/4, NotI; D2S in pGEM4Z, SalI; D2S mutants in pXOOM,XhoI) and transcribed in vitro using the SP6 (pGEM4Z) or T7 (pCDNA3 and pXOOM)mMessage mMachine kits (Ambion). cRNA concentration and purity were deter-mined using a spectrophotometer. Human Gg2 was purchased from the MissouricDNA repository (www.cdna.org), YFP-tagged human Gai1 and human Gb1 were giftsfrom Dr. Jean-Pierre Vilardaga, University of Pittsburgh, Pennsylvania. Cerulean-Gb1
was constructed by PCR insertion of the CFP variant cerulean 50 , in frame with thecoding sequence for Gb1.
2.2. Receptor ligands
Dopamine receptor agonists; apomorphine, bromocriptine, catechol, dopamine,N-propylnorapomorphine (NPA), ropinirole, rotigotine (SigmaeAldrich), piribedil(Tocris), R/S-DPAT from Axon MedChem BV (Groeningen, The Netherlands), lisurideand pergolide were gifts from Schering-Plough. Pramipexole was purchased fromthe Swedish pharmacy as Sifrol� tablets (Boehringer-Ingelheim) and the activesubstance was subsequently purified from the tablets by Axon MedChem BV.
Bromocriptine, NPA, R/S-DPAT, rotigotine, lisuride, pergolide and pramipexolewere dissolved in DMSO, whereas the other agonists were dissolved in distilledwater or directly in recording solution. Agonists were diluted in the recordingsolution to obtain the desired concentrations and superfused at 1.5 ml/min usinga pressure-driven, computer-controlled perfusion system (SmartSquirt 8, AutoMateScientific). The maximum final concentration of DMSO used in any experiment was0.1% v/v.
2.3. Oocyte isolation and injection
Oocytes were surgically removed from female Xenopus laevis toads under tri-caine anaesthesia, in accordance with guidelines from the Swedish National Boardfor Laboratory Animals, as previously described (Sahlholm et al., 2008a). All effortswere made tominimize animal suffering, and to reduce the number of animals used.The oocytes were incubated overnight either at 12 �C (electrophysiology experi-ments) or 18 �C (radioligand binding experiments) in modified Barth’s solution,containing (in mM): 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.015 HEPES, 0.33 Ca(NO3)2, 0.41CaCl2, 0.82 MgSO4, pH 7.6 with NaOH, supplemented with 10 mg/ml pyruvate and10 mg/ml penicillinestreptomycin.
The following day, oocytes were injectedwith cRNA (50 nl/cell) using a Nanojectinjector; (Drummond Scientific) and incubated for 3e8 days in modified Barth’ssolution prior to experiments. For electrophysiology experiments, 1 ng of each GIRKsubunit cRNA and 5 ng (using the pGEM4Z vector) or 200 pg (using the pXOOMvector) of dopamine D2S receptor cRNA were injected per oocyte. For radioligandbinding experiments, the oocytes were injected with 200 pg of GIRK subunit and5 ng of D2S receptor (pXOOM vector) cRNA per oocyte.
2.4. Oocyte electrophysiology
The electrophysiological experiments were performed using a two-electrodevoltage-clamp setup (CA-1 amplifier, Dagan; Digidata 1200 analog/digital converter,Molecular Devices), as described (Sahlholm et al., 2008a). For recording of GIRKcurrents, a high-potassium solution (in mM; 64 NaCl, 25 KCl, 0.8 MgCl2, 0.4 CaCl2, 15HEPES, 1 ascorbic acid; pH 7.4) was used, giving a Kþ reversal potential ofw�40 mV.Ascorbic acid was used in order to prevent the oxidation of dopamine and otheragonists. Single�80 or 0mV pulses were applied from a holding potential of�40mVto study current responses to D2S receptor agonists. Oocytes were clamped at the twopotentials in random order.
Three to six increasing concentrations of the agonist being evaluated wereapplied consecutively at the same intervals at both potentials, ending with a satu-rating concentration. For each oocyte and each holding voltage, the evoked currentresponse for each concentration of agonist tested was normalized to the response to1 mM dopamine obtained in the same oocyte, at the corresponding potential, unless

N
H
N
H
H
S
N
H
HO
HO
N
H
HO
HO
N
H
N
H
O
Br
N
H
O
HN
N
O
O
H
HO
O
N
S
OH
N
H
N
H
N
O
NNNNH
NNNN
N
H
O
NNNN
N
NH2
S
H
N
N
N
N
OO
3
4
5
6
7
NH2
NH2
Aminotetralin Phenethylamine
OH
Catechol
OH
Fig. 1. Structures of the agonists used in the present study. Numbers indicating the naming convention for phenethylamine and aminotetralin (AT) agonists are shown. In thephenethylamine series, the non-hydroxylated (b-phenethylamine), 4-hydroxy- (p-tyramine), and 3-hydroxy-(m-tyramine) molecules were used (data from Sahlholm et al., 2008c;see Table 2), as well as 3, 4-dihydroxyphenethylamine (dopamine), 3-methoxy-4-hydroxyphenethylamine (3-methoxy-tyramine; 3-MT), and N-dipropylated analogs ofm-tyramineand dopamine. Additionally, in the AT series, S-2eAT and S-5eOHeAT and R/S-N-dipropylaminotetralin (DPAT) were used.

K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949940
otherwise stated. The evoked current response was determined by subtracting thebasal (agonist-independent) current. For apomorphine, bromocriptine, lisuride,NPA, pergolide, piribedil and rotigotine, the response to reapplication of agonistfollowing washout was markedly diminished as compared to the response beforeagonist application. For this reason, only one round of consecutive agonist appli-cation (following the reference dopamine application) was made, at a singlepotential, to each oocyte.
2.5. Radioligand binding experiments
Binding of radiolabeled ligand was performed by a procedure developedspecifically for single Xenopus oocytes (Ben-Chaim et al., 2003). We measuredbinding of [3H]dopamine; [Ring-2,5,6-3H] (54 Ci/mmol; PerkinElmer) to oocytesinjected with cRNA encoding the dopamine D2S receptor and GIRK1/2 channelsubunits, at two membrane potentials. Variations in membrane potential wereachieved by changing the [Kþ]out while osmolarity and ionic strength were keptconstant by replacing NaCl with KCl. GIRK channels were coexpressed in order toamplify the difference between the membrane potentials thus achieved. Themembrane potential of the oocytes at the different Kþ solutions was measured witha standard intracellular electrode. In ND96 solution (in mM; 96 NaCl, 2 KCl, 1 CaCl2,1 MgCl2, 5 Hepes-NaOH; pH 7.5), oocytes not injected with GIRK channel cRNAexhibited a resting potential of �31.8 � 3 mV, whereas the resting potential ofoocytes injected with the GIRK channel was �82 � 2.6 mV. In high Kþ solution(similar to ND96 solution but with 2 mM NaCl and 96 mM KCl), the oocytes, eitherinjected or uninjected with GIRK channels, were depolarized to þ3 � 0.75 mV.Single oocytes were incubated for 30 s with the indicated concentrations of radio-ligand, in either ND96 or high Kþ solution. Each oocyte was rapidly dropped intoawashing chamber filled with 4 ml of ice-cold ligand-free ND96 or high Kþ solution.In less than 1 s, the oocyte reached the bottom of the washing chamber and wascollected by a pipette into a vial containing 3 ml of scintillation liquid. Nonspecificbinding was measured similarly but in control oocytes injected only with GIRKsubunits. Specific binding was calculated for each experiment by subtraction of theaverage nonspecific binding at each radioligand concentration and each membranepotential (5e10 oocytes), from the total binding to individual oocytes.
2.6. Cell culture and transfection
Human embryonic kidney (HEK)-293T cells were grown at 37 �C in an atmo-sphere of 5% CO2 in Dulbecco’s modified Eagle’s medium (SigmaeAldrich) supple-mented with 1 mM sodium pyruvate, 2 mM L-glutamine, 100 U/mL streptomycin,100mg/mL penicillin and 5% (v/v) fetal bovine serum. The cells were seeded into six-well plates at approximately 300.000 cells/well and transiently transfected thefollowing day with cDNA (in mg/well) encoding the wt D2S receptor (0.5), Gai1-YFP(2.5), Cerulean-Gb1 (0.5), and wt Gg2 (0.5) using Transfectin (Bio-Rad) according tothemanufacturer’s instructions. 24 h after transfection, the cells were lifted off usingVersene (Gibco), diluted w1/5 and re-seeded onto 18 mm coverslips (VWR) coatedwith poly-D-lysine (SigmaeAldrich), and kept in culture for an additional 2e4 daysprior to experiments.
2.7. Combined FRET and patch-clamp electrophysiology
Coverslips with transfected cells were mounted in a coverslip holder and placedon an inverted Axio Observer microscope (Zeiss) equipped with a 63X oil immersionobjective and a dual-emission photometry system (Till Photonics). The extracellularbuffer was Hank’s Balanced Salt Solution (HBSS) (in mM; 137 NaCl, 0.34 Na2HPO4, 5KCl, 0.44 KH2PO4, 0.5 MgCl2, 0.4 Mg2SO4, 1.26 CaCl2, 10 HEPES, 2 D-glucose and 1ascorbic acid; pH 7.4 with NaOH).
A Polychrome V (Till Photonics) was used as the light source. The sample wasilluminated with excitation light at 436 � 10 nm (beam splitter dichroic long-pass(DCLP) 460 nm). Excitation time was 10 ms at 10 Hz, in order to limit photo-bleaching. Emission light intensities were determined at 535 � 15 and 480 � 20 nm(DCLP 505 nm). No corrections for spillover between channels or direct YFP exci-tation were made. Single cells were selected for recording based on their plasmamembrane expression of the two tagged proteins, as judged by their fluorescence. Incontrol experiments (in the absence of agonist application) photobleaching of YFPand Cerulean followed a time course which was well-described by a mono-exponential function. Recorded fluorescence traces were thus corrected forbleaching by subtracting a monoexponential function extrapolated from the fit over50 s preceding agonist application. In some cases where the bleaching was very slow(typically the Cerulean trace), a monoexponential decay function could not be fittedand the trace was left uncorrected.
Cells selected for recording were voltage-clamped employing the patch-clamptechnique in the whole-cell configuration. An Axopatch 200B amplifier (MolecularDevices) was used for controlling transmembrane voltage. Patchmicropipettes werepulled from borosilicate glass capillaries (GC120F-10, Harvard Apparatus) to havea resistance of 4e10 MUwhen filled with intracellular solution (in mM; 10 NaCl, 120KCl, 5 MgCl2, 1 EGTA, 5 HEPES, 5 ATP, 0.2 GTP (pH 7.2 with NaOH)). Cells werevoltage-clamped at �80 or þ40 mV in random order, and dopamine was appliedusing a pressure-driven, computer-controlled perfusion system (Octaflow, ALA
Science). Three or four increasing concentrations were applied consecutively at thesame intervals at both potentials, ending with a saturating concentration (10 mM).Experiments were carried out at room temperature. For each cell and each holdingvoltage, the evoked current response for dopamine concentration of agonist testedwas normalized to the response to 10 mM dopamine obtained in the same cell.A Digidata 1440 analog/digital converter (Molecular Devices) were used for inter-facing the amplifier, the photometry system, and the perfusion system witha personal computer, which was used to control these systems and to record data.
2.8. Data acquisition and analysis
pCLAMP (molecular devices), Origin (Microcal) and GraphPad Prism (GraphPadSoftware) software were used for data collection and analysis. Variable slopesigmoidal concentrationeresponse curves were fitted to the data using GraphPadPrism (Prism Software). The following equation was used for fitting:
Y ¼ top/(1 þ 10^((log EC50 � X) * n))
where Y is the response as a fraction of 1, top is the maximal response, X is thelogarithm of agonist concentration and n is the Hill slope. Analyses of variance(F-test) were performed on the fitted curves, testing (as indicated) the differencesbetween the estimated log EC50s or the estimated top values at the two trans-membrane voltages. P < 0.05 was chosen as the significance limit.
3. Results
3.1. Voltage sensitivity of dopamine at the D2S receptor is apparentat the level of agonist binding and G protein activation
3.1.1. Voltage sensitivity of [3H]dopamine bindingWe previously demonstrated that depolarization reduces the
potency of dopamine in activating GIRK channels via the dopamineD2S receptor (Sahlholm et al., 2008b,c). To test whether diminishedagonist binding underlies this decrease in potency, binding of [3H]dopamine to oocytes expressing D2S receptors was measured attwo membrane potentials. An example of 40 nM of [3H]dopaminebinding to oocytes expressing D2S receptors and GIRK channelsat �82 mV (ND96) and at 0 mV (high Kþ) is depicted in Fig. 2A. It isseen that the specific binding of [3H]dopamine is voltage-dependent; it is significantly higher in ND96 solution than inhigh Kþ (p ¼ 0.0208, n ¼ 5). Similar results were obtained using80 nM [3H]dopamine. The average results of the various experi-ments were normalized as described in the legend of Fig. 2. It isseen that for both concentrations of [3H]dopamine, the binding isvoltage-dependent; greater at �82 mV that at 0 mV.
3.1.2. Voltage sensitivity of G protein activationAs our previous studies were performed using GIRK channels as
readout, we wanted to confirm that D2S receptor voltage sensitivityhas a similar impact on upstream G protein activation as well.
Therefore we combined electrophysiology with a previouslydescribedmethod for monitoring G protein activation in single cellsbased on Förster Resonance Energy Transfer (FRET), using fluo-rescently tagged G protein subunits (Bünemann et al., 2003). Thesetagged subunits have been shown to retain capability to activatedownstream effectors such as GIRK channels (Bünemann et al.,2003), suggesting that the incorporation of the fluorescent tagsdid not grossly perturb their function.
HEK 293Tcells were transiently transfectedwith cDNA encodingthe wt D2S receptor, Gai1 tagged with Yellow Fluorescent Protein(YFP), Gb1 tagged with Cerulean; a variant of Cyan FluorescentProtein (CFP), and wt Gg2. Single cells were selected for recordingbased on their plasma membrane expression of the two taggedproteins, as judged by their fluorescence. The cells were voltage-clamped using the whole-cell configuration of the patch-clamptechnique, and Cerulean was excited at 436 nm for 10 ms at10 Hz, while Cerulean and YFP fluorescence was simultaneouslyrecorded with photodiodes.

A
B
40 80
0
1000
2000
[Dopamine] nM
Normalizedspecificbinding(dpm)
Total n.s s.b Total n.s s.b0
500
1000
1500ND96 High K
+
*
dpm
*
***
Fig. 2. The binding of [3H]dopamine to oocytes expressing the dopamine D2S receptoris voltage-dependent. (A) An example of 40 nM of [3H]dopamine binding to oocytesexpressing D2SR and GIRK channels at �82 mV (ND96, left) and at 0 mV (high Kþ,right). The results are shown in dpm; total binding (Total, black bars), nonspecificbinding (n.s, gray bars), and specific binding (s.b., white bars). The total binding wassignificantly different from the nonspecific binding (control oocytes injected only withGIRK subunits) for both ND96 (P ¼ 0.0004, n ¼ 5) and at high Kþ (P ¼ 0.0097, n ¼ 6).The difference between the nonspecific binding at ND96 and at high Kþ is notsignificant (P ¼ 0.6275, n ¼ 8). (B) Average specific binding with 40 nM and 80 nM. Tobe able to combine the results of the four experiments, the average data points of eachconcentration in the different experiments were normalized to the results obtainedwith 40 nM at ND96 in one of the experiments. Normalized specific binding of twodifferent concentrations (40 nM and 80 nM) at ND96 (-) and at high Kþ (:). Thespecific binding at ND96 was significantly different than at high Kþ at both concen-trations, as determined by Student’s t-test (40 nM, P ¼ 0.0208; 80 nM, P < 0.0001)(n ¼ 5e19 taken from 4 donors). Data points represent mean � S.E.
K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949 941
Stimulation of the D2S receptor with dopamine increased YFPemission while simultaneously decreasing Cerulean emission(Fig. 3A), indicative of an increase in FRET presumably reflecting Gprotein subunit rearrangement upon activation, as reported fromearlier studies using similar combinations of Gai-coupled receptorsand tagged G protein subunits (Bünemann et al., 2003). The ratio ofYFP to Cerulean emission was plotted as in index of FRET (Fig. 3A).Supporting the notion that the fluorescence responses were indeedinduced by D2S receptor activation, no fluorescence changes wereobserved upon dopamine application to cells coexpressing theadenosine A1 receptor with the fluorescently labeled G proteinsubunits (data not shown).
We have previously shown that the effect of depolarization ondopamine potency at D2S is greater at þ40 than at 0 mV (Sahlholmet al., 2008b); however in oocytes recording at such highly depo-larized potentials is difficult, in part because of slowly activatingendogenous channels (see Sahlholm et al., 2008b). Because of therelatively large scatter in the concentrationeresponse data in the
FRET experiments (see Fig. 3B), we chose þ40 mV as our depolar-ized potential in these experiments to obtain a better resolution ofthe voltage sensitivity. Thus, the cells were voltage-clamped bothat �80 and þ40 mV in random order. Three to four concentrationsof dopamine were consecutively applied at each holding voltage,ending with a response-saturating concentration (10 mM), afterwhich dopamine was washed out.
Concentrationeresponse curves were constructed by plottingthe ratio of the bleach-corrected FRET response to each concen-tration of dopamine in a given cell as a fraction of the response tothe final, response-saturating dopamine concentration applied tothat cell (Fig. 3B). Results were plotted separately for eachmembrane voltage. Depolarization from �80 to þ40 mV induceda significant rightward shift of the concentrationeresponse curveof approximately ten-fold (Fig. 3B). There was no significantdifference between the maximum dopamine-induced FRETchanges between the two voltages, suggesting that dopamineefficacywas not appreciably affected by voltage (Fig. 3C). The extentof the potency shift is similar to what was previously reported forthe D2S receptor using the same agonist and voltage range(Sahlholm et al., 2008b). It thus appears that the results obtainedwhen G protein conformational change in HEK 293T cells is used asreadout, are comparable to those obtained using GIRK channelactivation in Xenopus oocytes. Because of the relatively lowthroughput and response resolution of the FRET assay compared tothe GIRK assay, we chose to conduct our further experiments usingthe latter method.
3.2. Agonistereceptor interactions underlying agonist-specificvoltage effects of D2S receptor voltage sensitivity
The depolarization-induced decrease in dopamine bindingdescribed above is likely to be produced through a conformationalchange in the D2S receptor binding site, perturbing certainligandereceptor interactions. Conversely, agonists whose potenciesare less affected by depolarization might not share these interac-tions. The following set of experiments was designed to testwhether differences between agonists, in terms of voltage-inducedpotency shifts, could be correlated with their differential interac-tions with the D2S receptor.
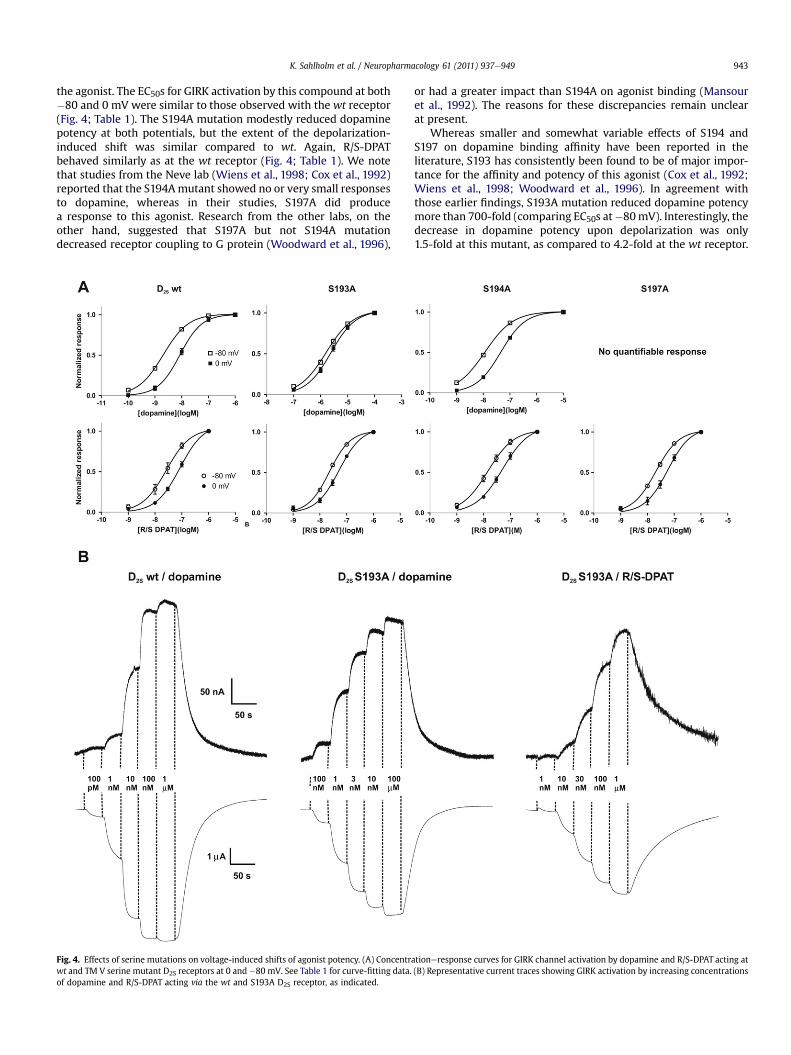
3.2.1. TM V serines e agonist hydroxyl group interactionsWe reported earlier that for some dopamine D2S receptor
agonists, potency was not significantly affected by voltage(Sahlholm et al., 2008c). Specifically, phenethylamines lackinghydroxyl groups on their aromatic rings (b-phenethylamine), orhaving only one hydroxyl group (p- and m-tyramine) did not showany significant potency shifts upon depolarization from �80 mV to0 mV. However, for agonists of the N,n-dipropyl-2-aminotetralin(DPAT) class, voltage sensitive potency was observed regardless ofhydroxylation. We hypothesized that the two hydroxyls of dopa-mine might confer strong voltage sensitivity by locking the agonistinto a sterically constrained network of hydrogen bonds with threeconserved serine residues in transmembrane segment (TM) V ofthe D2S receptor (see Coley et al., 2000), a network of hydrogenbonds which might be prone to perturbation by voltage-inducedreceptor movement. Synergistic effects between agonisthydroxyls and these serine residues in mediating binding affinityhave previously been reported (Coley et al., 2000; Floresca andSchetz, 2004).
To test this hypothesis, we generated three mutants in whicheach individual serine residue had been replaced by alanine; S193A,S194A and S197A. The S197A mutant failed to induce measurableGIRK responses to dopamine, however robust GIRK responses wereobserved when using R/S-DPAT (which lacks hydroxyl groups) as
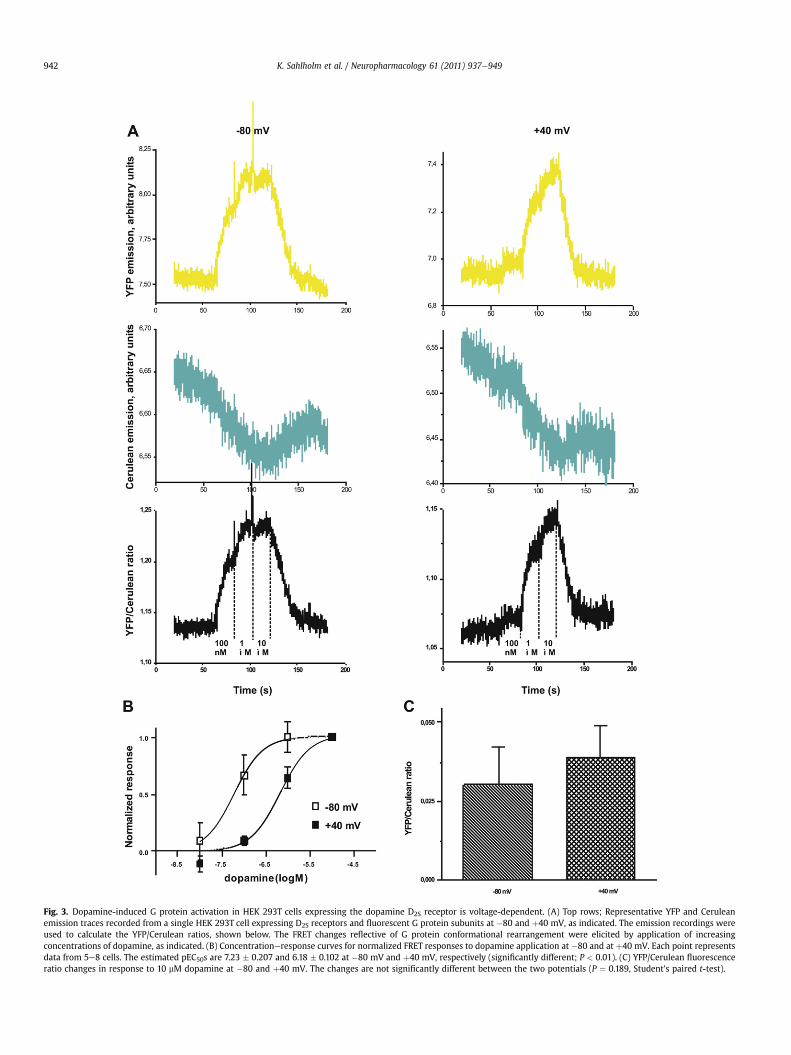
Fig. 3. Dopamine-induced G protein activation in HEK 293T cells expressing the dopamine D2S receptor is voltage-dependent. (A) Top rows; Representative YFP and Ceruleanemission traces recorded from a single HEK 293T cell expressing D2S receptors and fluorescent G protein subunits at �80 and þ40 mV, as indicated. The emission recordings wereused to calculate the YFP/Cerulean ratios, shown below. The FRET changes reflective of G protein conformational rearrangement were elicited by application of increasingconcentrations of dopamine, as indicated. (B) Concentrationeresponse curves for normalized FRET responses to dopamine application at �80 and at þ40 mV. Each point representsdata from 5e8 cells. The estimated pEC50s are 7.23 � 0.207 and 6.18 � 0.102 at �80 mV and þ40 mV, respectively (significantly different; P < 0.01). (C) YFP/Cerulean fluorescenceratio changes in response to 10 mM dopamine at �80 and þ40 mV. The changes are not significantly different between the two potentials (P ¼ 0.189, Student’s paired t-test).
K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949942
K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949 943
the agonist. The EC50s for GIRK activation by this compound at both�80 and 0 mV were similar to those observed with the wt receptor(Fig. 4; Table 1). The S194A mutation modestly reduced dopaminepotency at both potentials, but the extent of the depolarization-induced shift was similar compared to wt. Again, R/S-DPATbehaved similarly as at the wt receptor (Fig. 4; Table 1). We notethat studies from the Neve lab (Wiens et al., 1998; Cox et al., 1992)reported that the S194Amutant showed no or very small responsesto dopamine, whereas in their studies, S197A did producea response to this agonist. Research from the other labs, on theother hand, suggested that S197A but not S194A mutationdecreased receptor coupling to G protein (Woodward et al., 1996),
Fig. 4. Effects of serine mutations on voltage-induced shifts of agonist potency. (A) Concentrwt and TM V serine mutant D2S receptors at 0 and �80 mV. See Table 1 for curve-fitting data.of dopamine and R/S-DPAT acting via the wt and S193A D2S receptor, as indicated.
or had a greater impact than S194A on agonist binding (Mansouret al., 1992). The reasons for these discrepancies remain unclearat present.
Whereas smaller and somewhat variable effects of S194 andS197 on dopamine binding affinity have been reported in theliterature, S193 has consistently been found to be of major impor-tance for the affinity and potency of this agonist (Cox et al., 1992;Wiens et al., 1998; Woodward et al., 1996). In agreement withthose earlier findings, S193A mutation reduced dopamine potencymore than 700-fold (comparing EC50s at�80mV). Interestingly, thedecrease in dopamine potency upon depolarization was only1.5-fold at this mutant, as compared to 4.2-fold at the wt receptor.
ationeresponse curves for GIRK channel activation by dopamine and R/S-DPAT acting at(B) Representative current traces showing GIRK activation by increasing concentrations

Table 1Effects of membrane voltage on the potencies of dopamine, R/S-DPAT, and catechol in activating GIRK via wt and mutant dopamine D2S receptors.
Receptor n Agonist (range of concentrations tested, nM) pEC50 � S.E.M. (EC50, nM) EC50 ratio0 mV/�80 mV
0 mv �80 mV
D2S wt 6 Dopamine (0.1e1000) 8.07 � 0.0389 (8.58) *** 8.69 � 0.0216 (2.06) 4.17D2S S193A 11 Dopamine (100e100 000) 5.61 � 0.0198 (2440) ** 5.79 � 0.0326 (1620) 1.50D2S S194A 4 Dopamine (1e10 000) 7.34 � 0.00499 (4.54) ** 7.94 � 0.0241 (11.5) 3.95D2S S197A e Dopamine e ND ND e
D2S D114A 11 Dopamine (30 000e3 000 000) 3.30 � 0.0640 (502 000) ** 3.07 � 0.0464 (856 000) 0.587D2S wt 4 R/S-DPAT (1e1000) 7.07 � 0.0505 (85.6) *** 7.58 � 0.0782 (26.5) 3.23D2S S193A 13 R/S-DPAT (1e1000) 7.28 � 0.0681 (52.4) ** 7.65 � 0.0203 (22.6) 2.32D2S S194A 4 R/S-DPAT (1e1000) 7.29 � 0.0696 (50.9) ** 7.84 � 0.0154 (14.4) 3.53D2S S197A 4 R/S-DPAT (1e1000) 7.24 � 0.0608 (57.2) ** 7.69 � 0.0154 (20.7) 2.77D2S wt 11 Catechol (30 000e1 000 000) 3.43 � 0.134 (376 000) 3.21 � 0.239 (611 000) 0.615
Asterisks denote statistical significance; **, P < 0.01; ***, P < 0.001 ND; not determined.
K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949944
However, the potencies and voltage-induced potency shiftobserved when using R/S-DPAT as agonist at the S193A mutant wassimilar to wt, suggesting that this serine mutation indeed dimin-ished the effect of voltage on dopamine potency by perturbingspecific agonistereceptor interactions, rather than compromisingthe overall function of the receptor protein (Fig. 4; Table 1).
Exchanging the 3-hydroxyl of dopamine for a methoxy groupgenerates 3-methoxytyramine; a major metabolite of dopamine inthe body which has affinity for dopamine receptors in the mM range(Alachkar et al., 2010). Unlike dopamine, this compound is unableto donate hydrogen bonds at the 3-position. Similar to the tyra-mines, the potency and efficacy of this compound were notsignificantly affected by the depolarization from �80 mV to 0 mV(Table 2).
3.2.2. TM III aspartate e agonist amine moiety interactionTo further investigate the roles of the functional groups of
dopamine in the voltage sensitive interactions of this agonist withthe D2S receptor, we also studied catechol. Catechol has beendescribed as a very weak agonist at the b2 adrenergic receptor(Swaminath et al., 2005) and, lacking the ethylamine side chain ofdopamine (see Fig. 1), is expected to interact with a very restrictedportion of the ligand binding site. We could detect agonism bycatechol at high micromolar concentrations in our GIRK-based D2Sreceptor activation assay (Fig. 5). No current responses wereobserved upon catechol application to oocytes expressing only theD2S receptor or only GIRK 1/4 channels, as for the other agonistsevaluated in this study, and catechol-induced responses in oocytescoexpressing wt D2S receptor and GIRK channels were abolished bycoapplication of 5 mM of the D2 receptor antagonist raclopride (notshown), supporting the notion that catechol indeed elicited GIRKactivation via the D2S receptor. Furthermore, 1 mM catechol failedto evoke detectable GIRK currents in cells coexpressing the S193A
Table 2Effects of voltage on the potencies and efficacies of select phenethylamines and aminote
Agonist (range of concentrations tested, nM) n pEC50 � S.E.M.(EC50, nM)
0 mv �b-phenethylamine a (10e10 000) 4e13 5.51 � 0.139 (3070) 5p-tyramine a (100e1 000 000) 3e7 5.08 � 0.113 (8250) 4m-tyramine a (1e10 000) 3e8 7.00 � 0.122 (101) 73-MT (100e1 000 000) 3e6 5.05 � 0.334 (8890) 5N-dipropyl-dopamine (0.01e100) 4 8.63 � 0.0806 (2.32) *** 9N-dipropyl-m-tyramine (1e10 000) 6 6.98 � 0.0333 (105) *** 7
(S)-AT (200e20 000) 3 5.38 � 0.0634 (4170) 5(S)-5eOHeAT (1e10 000) 5 7.50 � 0.0899 (28.7) * 7
Asterisks denote statistical significance; *, P < 0.05; ***, P < 0.001.a Data from Sahlholm et al., 2008a, with permission from Elsevier.
mutant receptor with GIRK channels (not shown), suggesting thatcatechol acts at the orthosteric binding site of D2S.
However, catechol blocked GIRK channel currents at concentra-tions above 1 mM, at which the GIRK response elicited via D2Sreceptor activation had not yet saturated. For this reason, we wereunable to construct full concentrationeresponse curves for catechol.We thus had to constrain the Hill slope parameter to unity whenanalyzing the data for catechol, in order to obtain a reasonable fit(Fig. 5A). The estimated EC50s for catechol at 0 and �80 mV are notsignificantly different (Table 1), but these estimates are very uncer-tain due to the amount of information missing for the top of thecurves. Notably however, the fractional responses to 100 mM,300 mM, and 1 mM catechol, vs. 1 mM dopamine, were significantlygreater at 0mV than at�80mV (Fig. 5A), suggesting that thepotencyand/or efficacy of catechol might increase with depolarization.
The protonated amine headgroup of dopamine has been shownto interact with an aspartate residue in TM III, D114, which isconserved among amine receptors (Floresca and Schetz, 2004). Wespeculated that the contrasting effect of voltage on catecholpotency, as compared to dopamine, reflected the lack of this ami-neeaspartate interaction. To test this hypothesis, we studied thereceptor mutant D114A. Whereas an early investigation did notdetect specific ligand binding at D114N and D114G mutants(Mansour et al., 1992), a recent study using GIRK activation asreadout was able to detect agonism by dopamine at several D2SD114 mutants, including D114A (Torrice et al., 2009). Consistentwith this later report, dopamine elicited current responses at highmM concentrations in our experiments with the D114A mutant(Fig. 5). Dopamine blocked GIRK currents at concentrations above3 mM, at which the response was near saturation. As, to the best ofthe authors’ knowledge, there is no full D2 receptor agonist whichlacks an amine moiety and which could be used as a reference inthis case, responses were normalized to the current evoked by
tralins in activating GIRK via the wt dopamine D2S receptor.
EC50 ratio0 mV/�80 mV
Relative efficacy � S.E.M.
80 mv 0 mv �80 mV
.59 � 0.0844 (2550) 1.20 0.724 � 0.0874 0.711 � 0.0493
.99 � 0.0936 (10 400) 0.793 0.795 � 0.0430 0.801 � 0.0364
.11 � 0.0772 (78.0) 1.29 0.917 � 0.0553 1.03 � 0.0386
.12 � 0.162 (7590) 1.17 0.584 � 0.0770 0.589 � 0.0462
.41 � 0.0629 (0.388) 6.00 0.894 � 0.0365 0.994 � 0.0258
.75 � 0.0579 (17.9) 5.90 0.708 � 0.0143 *** 0.841 � 0.0198
.39 � 0.150 (4050) 1.03 0.533 � 0.0262 0.497 � 0.0570
.78 � 0.0931 (14.1) 2.03 0.728 � 0.0208 0.777 � 0.0214

Fig. 5. Effects of aspartateeamine interactions on voltage-induced shifts of agonist potency. (A) Concentrationeresponse curves for GIRK channel activation by catechol at the wtreceptor and dopamine at the D114A mutant at 0 and �80 mV. See Table 1 for curve-fitting data. Asterisks in the graph for catechol denote statistical significance, as determined bypaired Student’s t-test; *, P < 0.05; ***. P < 0.001. (B), (C) Thin lines; representative current traces showing GIRK activation at both voltages by increasing concentrations of catecholacting via the wt D2S receptor (B) or dopamine acting via the D114A mutant (C). Thick lines in (B); GIRK current responses to application of 1 mM dopamine, in the same cell.
K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949 945
3 mM dopamine. With this mutant, the estimated EC50s for dopa-mine at 0 and �80 mV are significantly different, showing a left-ward shift with depolarization of about two-fold (Fig. 5A; Table 1),similar to what was observed with catechol at the wt receptor.
3.2.3. Agonist propyl chain interactionsAdditionally, the effects of propyl substitutions at the amine
headgroup were examined. In many dopamine agonists, aminesubstituent propyl chains increase D2 receptor affinity, and arebelieved to interact with a hydrophobic pocket between TM III andTM VII of the receptor (Malmberg et al., 1994). It was previouslyreported that no significant depolarization-induced shift in potencywas observed with m-tyramine in the GIRK activation assay(Sahlholm et al., 2008c). However, we found that addition of twopropyl chains to the amine moiety of this compound results in an
agonist with a pronounced voltage sensitivity, displaying a significantrightward potency shift of near six-fold upon depolarization (seeTable 2). Moreover, the maximum GIRK activation amplitude, ascompared to that elicited by 1 mM dopamine, was significantly lowerat 0 than at �80 mV, suggesting that depolarization diminished theefficacy of this compound. Compared to dopamine, dipropyl-dopamine showed a modest increase in the depolarization-inducedloss of potency, also exhibiting a six-fold potency shift between thetwo voltages (Table 2).
Conversely, removal of the propyl chains of S-DPAT results inS-2-aminotetralin (S-2-AT), which was not significantly affected bydepolarization (Table 2). Because S-2-AT blocked GIRK channels atconcentrations above 20 mM, at which the response had not yetsaturated, Hill slopes were constrained to 1 to enable fitting ofconcentrationeresponse curves. However, S-5eOHeAT did show

Fig. 6. GIRK activation at 0 and �80 mV by a range of therapeutic D2 receptor agonists acting via the wt D2S receptor. (A) Concentrationeresponse curves normalized to the currentproduced by application of 1 mM dopamine. See Table 3 for curve-fitting data. (B), (C) Thin lines; representative current traces showing GIRK activation at both voltages by increasingconcentrations of N-propylnorapomorphine (B), and ropinirole (C). In the case of ropinirole, both traces were recorded in the same cell. Thick lines; GIRK current responses toapplication of 1 mM dopamine, in the same respective cells.
K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949946

Table 3Effects of voltage on potencies and efficacies of therapeutic dopamine agonists in activating GIRK via the wt dopamine D2S receptor.
Agonist (range of concentrations tested, nM) n pEC50 � S.E.M. (EC50, nM) EC50 ratio0 mV/�80 mV
Relative efficacy � S.E.M.
0 mv �80 mv 0 mv �80 mV
Apomorphine (0.01e100) 5 9.33 � 0.132 (0.471) 9.57 � 0.0722 (0.272) 1.73 0.817 � 0.0432 0.841 � 0.0237NPA (0.01e10) 4e5 9.90 � 0.157 (0.127) 9.91 � 0.266 (0.123) 1.03 0.627 � 0.0804 0.627 � 0.0471
Bromocriptine (10e1000) 7 7.14 � 0.0968 (73.2) * 7.43 � 0.0829 (37.4) 1.96 0.479 � 0.0360 * 0.672 � 0.0388Lisuride (1e1000) 5 8.06 � 0.104 (8.71) 8.29 � 0.0786 (5.11) 1.70 0.479 � 0.0299 ** 0.613 � 0.0223Pergolide (0.01e100) 3e6 9.45 � 0.111 (0.354) * 9.78 � 0.0903 (0.165) 2.15 0.685 � 0.0301 ** 0.915 � 0.0338
Pramipexole (0.1e1000) 4 8.34 � 0.156 (4.54) ** 9.02 � 0.178 (0.946) 4.80 0.950 � 0.0604 0.987 � 0.0649Piribedil (1e1000) 3e4 7.73 � 0.0718 (18.6) *** 8.20 � 0.0741 (6.28) 2.96 0.646 � 0.0324 *** 0.913 � 0.0336Ropinirole (0.1e1000) 4 7.87 � 0.181 (13.5) ** 8.53 � 0.117 (2.97) 4.55 0.919 � 0.0747 0.874 � 0.0468Rotigotine (0.1e100) 3e4 9.00 � 0.0849 (1.01) 9.19 � 0.0873 (0.640) 1.58 0.549 � 0.0290 ** 0.688 � 0.0293
Asterisks denote statistical significance; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949 947
a significant rightward shift with depolarization, suggesting thata single hydroxyl group is sufficient for a voltage sensitive ago-nistereceptor interaction in the case of aminotetralins (Table 2).
3.3. Therapeutic dopamine receptor agonists differ in their voltagesensitivities at the D2S receptor
Finally, we investigated how different dopamine D2 receptoragonist therapeutics are affected by D2S receptor voltage sensitivity.Three ergoline compounds; bromocriptine, lisuride and pergolide;two aporphines; apomorphine and n-propylnorapomorphine; andfour structurally unrelated agonists; piribedil, pramipexole, ropi-nirole, and rotigotine, were investigated (see Fig. 1). The relativeefficacies of these agonists versus dopaminewere determined in allexperiments, by measuring the response evoked by a saturatingconcentration (1 mM) of dopamine before obtaining the cumulativeconcentrationeresponse characteristics of the synthetic agonists.Marked differences in the effects of membrane voltage wereobserved between the various compounds (Fig. 6 and Table 3).
For the aporphines, varying themembrane potential from�80mVto 0 mV produced no significant shifts in potency or in efficacy. Thethree ergolines all showed a significant decrease in efficacy withdepolarization, whereas potency decreased about two-fold or less,reaching statistical significance only for bromocriptine and pergolide.Among the other agonist therapeutics tested, ropinirole and prami-pexole resembled dopamine in that depolarization produceda significant rightward shift of about 4.5-fold. The efficacies of theseagonists were not significantly affected by voltage change. Piribedilshowed a three-fold decrease in potency and a significant decreasein efficacy. Despite rotigotine being closely related structurally toS-5eOHeDPAT, whose potency is significantly reduced upon depo-larization (Sahlholm et al., 2008c), it’s potency showed only a non-significant trend to a shift of about 1.5; however its efficacydecreased slightly but significantly.
4. Discussion
In the present study, we first demonstrate that the reducedpotency of dopamine observed in the GIRK channel activation assayat depolarized potentials is correlated with a reduction of D2Sreceptor binding of radiolabeled dopamine. This is in line withprevious findings concerning voltage sensitivity of muscarinic M2and glutamate mGluR3 receptors (Ben-Chaim et al., 2003; Ohanaet al., 2006). We further demonstrate that D2S receptor voltagesensitivity is observed also in mammalian cells, at the level of Gprotein activation.
We move on to show that the voltage sensitivity of dopaminepotency can be perturbed bymutating one of the conserved serines
in TM V, particularly S193, or the conserved aspartate residue in TMIII (D114), which are known to contact the hydroxyl groups andamine moiety of dopamine, respectively (Cox et al., 1992; Coleyet al., 2000; Floresca and Schetz, 2004). The perturbation ofvoltage sensitivity induced at the S193Amutant receptor is agonist-specific, as the voltage sensitivity of an agonist which lackshydroxyl groups is barely affected by this mutation. The presentfindings support the previous suggestion (Sahlholm et al., 2008c)that interactions between the agonist and the TM V serines areimportant for the voltage sensitivity of phenethylamine agonists,but not for agonists of the DPAT class.
We also show that therapeutic dopamine agonist drugs displaysignificant differences in terms of how they are affected by D2Sreceptor voltage sensitivity. Notably, the efficacies of some thera-peutic agonists (the three ergolines, piribedil, and rotigotine), ascompared to a maximally effective concentration of dopamine,were markedly decreased by depolarization. Effects of voltage onGPCR ligand efficacy were previously reported from a study inmegakaryocytes by Gurung et al. (2008), where some antagonistsat the Gq-coupled P2Y1 receptor evoked agonistic responses(intracellular Ca2þ increases) at depolarized potentials, as well as ina recent study by Navarro-Polanco et al. (2011), in which the effi-cacy of the muscarinic M2 receptor agonist pilocarpine wasincreased by depolarization.
Whereas perturbing agonistereceptor interactions by receptormutation or removal of agonist functional groups inevitablydecreases agonist affinity and potency, the results obtained withthe therapeutic agonists further indicate that the mechanismsbehind the agonist-specific effects of receptor voltage sensitivityare not simply related to overall differences in agonist affinity forthe receptor; e.g., such that lower-affinity compounds would beless influenced by receptor voltage sensitivity. In fact, some highpotency dopamine agonists like apomorphine and N-propylnor-apomorphine did not display any significant shifts of potency orefficacy with depolarization, whereas some less potent agonists,e.g., pramipexol and piribedil, showed significant reductions ofpotency (and, in the case of piribedil, efficacy). In the present workwe have not directly investigated the affinity of these agonists, anddirect inference from potency to affinity can sometimes bemisleading. However, the rank order of agonist potencies describedhere correlates well with the relative affinities of these compoundsfor the dopamine D2 receptor, as reported from ligand bindingstudies elsewhere; see e.g., Seeman (2007).
Our findings are in disagreement with the suggestion of Ben-Chaim et al. (2006) and Kupchik et al. (2011); that receptorvoltage sensitivity is caused by a general, depolarization-induceduncoupling of the receptor from its G protein. As pointed outrecently by Navarro-Polanco et al. (2011), such a mechanismwould

K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949948
be expected to affect all agonists similarly. Indeed, we have verifiedearlier (Sahlholm et al., 2008c) that both voltage sensitive andinsensitive behavior can be observed for different agonists evenwhen Gi/o signaling is constrained so as to occur via a single Gprotein subtype.
Similar to Navarro-Polanco et al. (2011), we instead propose thatcertain modes of ligandereceptor interaction, e.g., such as those ofdopamine, pramipexole, and ropinirole in our case, are strongly per-turbed by voltage-induced movements of parts of the receptor’sbinding site, whereas other binding modes, e.g., such as thoseemployed by the aporphines, are not, or less strongly affected.Specifically, for non-propylated phenethylamine agonists, it appearsthat two agonist hydroxyl groups, interacting with the conserved TMV serine residue, S193 (and possibly, with S197), along with theamine-TM III aspartate interaction, are necessary for a strongdepolarization-induced potency decrease. However, the addition ofpropyl chains confers voltage sensitive potency to monohydroxylatedphenethylamine- and unhydroxylated DPAT compounds, suggestinga complex relation between binding mode and voltage sensitivity.Likewise, the aporphines do not show voltage sensitive potencydespite having two hydroxyl groups in positions corresponding tothose of dopamine. Thus, the effect of voltage on agonist potency isnot mediated exclusively via one single liganderesidue interaction,but is rather determined by the whole set of ligandereceptorinteractions.
Although the voltage sensor of GPCRs has yet to be identified,the apparent lack of a single, key binding site residue mediating theeffect of voltage on ligand potency and efficacy is hardly surprising,given the highly allosteric nature of the receptor protein. Indeed,GPCR activation has been proposed to proceed via a “global toggleswitch” mechanism in which allosterically linked conformationalmicroswitches, distributed over the different TMs, synergisticallyinfluence each other’s probability of being switched “on” or “off”(Nygaard et al., 2009).
GPCR voltage sensitivity has been shown to play an importantrole in the control of neurotransmitter release at neuromuscularjunctions in mammals and arthropods (Kupchik et al., 2008, 2011).Such a role is not unlikely for the dopamine D2S receptor, whichfunctions as a presynaptic auto- and heteroreceptor in the CNS(Usiello et al., 2000). As of yet, little is known about other physio-logical functions for GPCR voltage sensitivity. Whereas GPCRregulation of fast transmitter release occurs at a millisecond timescale and was suggested to proceed via direct interaction betweenthe receptor and vesicle-associated proteins, rather than G proteinactivation (Kupchik et al., 2008), a multitude of other physiologicalroles involving G proteins and other downstream signaling mole-cules are also plausible, such as in synaptic plasticity and control ofneuronal firing patterns. For example, voltage sensitivity might berelevant to D2 receptor function in dopaminergic neurons of themidbrain, where GIRK channels mediate autoreceptor-inducedinhibition (Lacey et al., 1987), as well as in pituitary lactotrophs,which are the targets of D2 agonists in the treatment of hyper-prolactinemia (Prabhakar and Davis, 2008). Both of these celltypesexhibit depolarized plateau potentials lasting for several seconds(Mrejeru et al., 2011; Van Goor et al., 2001); sufficient time forchanges in receptor occupancy by agonist to be transmitted to the Gprotein pool.
Notably, a recent study by Moreno-Galindo et al. (2011) revealedthat “voltage-dependent relaxation,” a property of acetylcholine-activated GIRK currents in cardiomyocytes believed to be crucial forthe effects of vagal stimulation on heart function, is a reflection ofmuscarinic M2 receptor voltage sensitivity. Those authors used theagonist-specificity of M2 receptor voltage sensitivity to demonstratea link between the two phenomena. Future investigations of theputative roles for dopamine D2 receptor voltage sensing will thus
likely benefit from our present findings that highly potent and effi-cacious dopamine agonists differ in terms of how they are affected bythe voltage sensitivity of this receptor.
5. Conclusion
In the present work, we showed that the depolarization-induced decrease in dopamine potency observed at the level ofGIRK channel activation correlates with a depolarization-induceddecrease in dopamine binding and G protein activation. We alsoidentified some of the ligandereceptor interactions which deter-mine the effect of receptor voltage sensitivity on agonist potency,and demonstrated that a number of clinically used dopamineagonists differ in terms of the voltage sensitivity of their interac-tions with the dopamine D2S receptor. Our findings bear potentialrelevance to understanding the actions of D2 agonist drugs, and forfuture studies of the physiological roles of dopamine D2 receptorvoltage sensitivity.
Role of the funding source
The funding sources, listed under “Acknowledgments”, had no rolein study design, in the collection, analysis, and interpretation of data,in manuscript writing, or in the decision to submit the manuscript.
Acknowledgments
The present work was supported by grants from the SwedishResearch Council, and the Parkinson Foundation in Sweden, to P.Å.,Åhlén-stiftelsen, the Swedish Society for Medical Research, Gålös-tiftelsen, and Stiftelsen Lars Hiertas Minne, to K.S., and by grantsSAF2008-01462 and Consolider-Ingenio CSD2008-00005, fromMinisterio de Ciencia e Innovación, and ICREA Academia-2010 fromthe Catalan Institution for Research and Advanced Studies, to F.C.The authors wish to thank Dr. Hanna Parnas, The Hebrew Univer-sity of Jerusalem, Israel, for extensive, fruitful discussions anddetailed comments on the manuscript. For cDNA sources, see“Material and methods”, Subsection 2.1.
References
Alachkar, A., Brotchie, J.M., Jones, O.T., 2010. Binding of dopamine and 3-methoxytyramine as l-DOPA metabolites to human alpha(2)-adrenergic anddopaminergic receptors. Neurosci. Res. 67, 245e249.
Ben-Chaim, Y., Tour, O., Dascal, N., Parnas, I., Parnas, H., 2003. The M2 muscarinic G-protein-coupled receptor is voltage-sensitive. J. Biol. Chem. 278, 22482e22491.
Ben-Chaim, Y., Chanda, B., Dascal, N., Bezanilla, F., Parnas, I., Parnas, H., 2006.Movement of ‘gating charge’ is coupled to ligand binding in a G-protein-coupled receptor. Nature 444, 106e109.
Bünemann, M., Frank, M., Lohse, M.J., 2003. Gi protein activation in intact cellsinvolves subunit rearrangement rather than dissociation. Proc. Natl. Acad. Sci. US A 100, 16077e16082.
Coley, C., Woodward, R., Johansson, A.M., Strange, P.G., Naylor, L.H., 2000. Effect ofmultiple serine/alanine mutations in the transmembrane spanning region V ofthe D2 dopamine receptor on ligand binding. J. Neurochem. 74, 358e366.
Cox, B.A., Henningsen, R.A., Spanoyannis, A., Neve, R.L., Neve, K.A., 1992. Contri-butions of conserved serine residues to the interactions of ligands with dopa-mine D2 receptors. J. Neurochem. 59, 627e635.
Floresca, C.Z., Schetz, J.A., 2004. Dopamine receptor microdomains involved inmolecular recognition and the regulation of drug affinity and function. J. Recept.Signal. Transduct. Res. 24, 207e239.
Gurung, I.S., Martinez-Pinna, J., Mahaut-Smith, M.P., 2008. Novel consequences ofvoltage-dependence to G-protein-coupled P2Y1 receptors. Br. J. Pharmacol. 154,882e889.
Javitch, J.A., 1998. Mapping the binding-site crevice of the D2 receptor. Adv. Phar-macol. 42, 412e415.
Jespersen, T., Grunnet, M., Angelo, K., Klaerke, D.A., Olesen, S.P., 2002. Dual-functionvector for protein expression in both mammalian cells and Xenopus laevisoocytes. Biotechniques 32, 536e538.
Kupchik, Y.M., Barchad-Avitzur, O., Wess, J., Ben-Chaim, Y., Parnas, I., Parnas, H.,2011. A novel fast mechanism for GPCR-mediated signal transductiondcontrolof neurotransmitter release. J. Cell Biol. 192, 137e151.

K. Sahlholm et al. / Neuropharmacology 61 (2011) 937e949 949
Kupchik, Y.M., Rashkovan, G., Ohana, L., Keren-Raifman, T., Dascal, N., Parnas, H.,Parnas, I., 2008. Molecular mechanisms that control initiation and terminationof physiological depolarization-evoked transmitter release. Proc. Natl. Acad. Sci.U S A 105, 4435e4440.
Kvernmo, T., Houben, J., Sylte, I., 2008. Receptor-binding and pharmacokineticproperties of dopaminergic agonists. Curr. Top. Med. Chem. 8, 1049e1067.
Lacey, M.G., Mercuri, N.B., North, R.A., 1987. Dopamine acts on D2 receptors toincrease potassium conductance in neurones of the rat substantia nigra zonacompacta. J. Physiol. 392, 397e416.
Mahaut-Smith, M.P., Martinez-Pinna, J., Gurung, I.S., 2008. A role for membranepotential in regulating GPCRs? Trends Pharmacol. Sci. 29, 421e429.
Malmberg, A., Nordvall, G., Johansson, A.M., Mohell, N., Hacksell, U., 1994. Molecularbasis for the binding of 2-aminotetralins to human dopamine D2A and D3receptors. Mol. Pharmacol. 46, 299e312.
Mansour, A., Meng, F., Meador-Woodruff, J.H., Taylor, L.P., Civelli, O., Akil, H., 1992.Site-directed mutagenesis of the human dopamine D2 receptor. Eur. J. Phar-macol. 227, 205e214.
Martinez-Pinna, J., Gurung, I.S., Mahaut-Smith, M.P., Morales, A., 2010. Direct voltagecontrol of endogenous lysophosphatidic acid G-protein-coupled receptors in Xen-opus oocytes. J. Physiol. 588, 1683e1693.
Moreno-Galindo, E.G., Sãnchez-Chapula, J.A., Sachse, F.B., Rodriguez-Paredes, J.A., Tris-tani-Firouzi, M., Navarro-Polanco, R.A., 2011. Relaxation gating of the acetylcholine-activated inward rectifier Kþ current (IKACh) is mediated by intrinsic voltage-sensitivity of the muscarinic receptor. J. Physiol. 589, 1755e1767.
Mrejeru, A., Wei, A., Ramirez, J.M., 2011. Calcium-activated non-selective cationcurrents are involved in generation of tonic and bursting activity in dopamineneurons of the substantia nigra pars compacta. J. Physiol. 589, 2497e2514.
Navarro-Polanco, R.A., Moreno Galindo, E.G., Ferrer-Villada, T., Arias, M., Rigby, J.R.,Sãnchez-Chapula, J.A., Tristani-Firouzi, M., 2011. Conformational changes in theM2 muscarinic receptor induced by membrane voltage and agonist binding.J. Physiol. 589, 1741e1753.
Nygaard, R., Frimurer, T.M., Holst, B., Rosenkilde, M.M., Schwartz, T.W., 2009. Ligandbinding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci.30, 249e259.
Ohana, L., Barchad, O., Parnas, I., Parnas, H., 2006. The metabotropic glutamate G-protein-coupled receptors mGluR3 and mGluR1a are voltage-sensitive. J. Biol.Chem. 281, 24204e24215.
Parnas, I., Parnas, H., 2010. Control of neurotransmitter release: from Ca2þ to voltagedependent G-protein coupled receptors. Pflugers Arch. 460, 975e990.
Prabhakar, V.K., Davis, J.R., 2008. Hyperprolactinaemia. Best Pract. Res. Clin. Obstet.Gynaecol. 22, 341e353.
Sahlholm, K., Nilsson, J., Marcellino, D., Fuxe, K., Arhem, P., 2008a. Voltage-dependence of the human dopamine D2 receptor. Synapse 62, 476e480.
Sahlholm, K., Marcellino, D., Nilsson, J., Fuxe, K., Arhem, P., 2008b. Differentialvoltage-sensitivity of D2-like dopamine receptors. Biochem. Biophys. Res.Commun. 374, 496e501.
Sahlholm, K., Marcellino, D., Nilsson, J., Fuxe, K., Arhem, P., 2008c. Voltage-sensi-tivity at the human dopamine D2S receptor is agonist-specific. Biochem. Bio-phys. Res. Commun. 377, 1216e1221.
Seeman, P., 2006. Targeting the dopamine D2 receptor in schizophrenia. ExpertOpin. Ther. Targets 10, 515e531.
Seeman, P., 2007. Antiparkinson therapeutic potencies correlate with their affinitiesat dopamine D2(High) receptors. Synapse 61, 1013e1018.
Swaminath, G., Deupi, X., Lee, T.W., Zhu, W., Thian, F.S., Kobilka, T.S., Kobilka, B.,2005. Probing the beta2 adrenoceptor binding site with catechol revealsdifferences in binding and activation by agonists and partial agonists. J. Biol.Chem. 280, 22165e22171.
Torrice, M.M., Bower, K.S., Lester, H.A., Dougherty, D.A., 2009. Probing the role of thecation-pi interaction in the binding sites of GPCRs using unnatural amino acids.Proc. Natl. Acad. Sci. U S A 106, 11919e11924.
Usiello, A., Baik, J.H., Rougé-Pont, F., Picetti, R., Dierich, A., LeMeur, M., Piazza, P.V.,Borrelli, E., 2000. Distinct functions of the two isoforms of dopamine D2receptors. Nature 408, 199e203.
Van Goor, F., Zivadinovic, D., Martinez-Fuentes, A.J., Stojilkovic, S.S., 2001. Depen-dence of pituitary hormone secretion on the pattern of spontaneous voltage-gated calcium influx. Cell type-specific action potential secretion coupling.J. Biol. Chem. 276, 33840e33846.
Wiens, B.L., Nelson, C.S., Neve, K.A., 1998. Contribution of serine residues toconstitutive and agonist-induced signaling via the D2S dopamine receptor:evidence for multiple, agonist-specific active conformations. Mol. Pharmacol.54, 435e444.
Woodward, R., Coley, C., Daniell, S., Naylor, L.H., Strange, P.G., 1996. Investigation ofthe role of conserved serine residues in the long form of the rat D2 dopaminereceptor using site-directed mutagenesis. J. Neurochem. 66, 394e402.
Copyright © 2022 FDOKUMEN












![Non-amine dopamine transporter probe [3H]tropoxene distributes to dopamine-rich regions of monkey brain](https://static.fdokumen.com/doc/165x107/63224d2f050768990e0fcb6c/non-amine-dopamine-transporter-probe-3htropoxene-distributes-to-dopamine-rich.jpg)




![In vivo vulnerability to competition by endogenous dopamine: Comparison of the D2 receptor agonist radiotracer (-)-N-[11C]propyl-norapomorphine ([11C]NPA) with the D2 receptor antagonist](https://static.fdokumen.com/doc/165x107/631cb975a1cc32504f0c9f3c/in-vivo-vulnerability-to-competition-by-endogenous-dopamine-comparison-of-the-d2-1675155902.jpg)


