
Molecular determinants of Akt-induced keratinocyte transformation

Activation of the Hexosamine Pathway Leads toPhosphorylation of Insulin Receptor Substrate-1 onSer307 and Ser612 and Impairs the Phosphatidylinositol3-Kinase/Akt/Mammalian Target of Rapamycin InsulinBiosynthetic Pathway in RIN Pancreatic �-Cells
FRANCESCO ANDREOZZI, CRISTINA D’ALESSANDRIS, MASSIMO FEDERICI, EMANUELA LARATTA,SILVIA DEL GUERRA, STEFANO DEL PRATO, PIERO MARCHETTI, RENATO LAURO,FRANCESCO PERTICONE, AND GIORGIO SESTI
Department of Experimental and Clinical Medicine (F.A., E.L., F.P., G.S.), University of Catanzaro-Magna Græcia, 88100Catanzaro, Italy; Laboratory of Molecular Medicine (C.D., M.F., R.L.), Department of Internal Medicine, University of Rome-Tor Vergata, 00133 Rome, Italy; and Department of Endocrinology and Metabolism (S.D.G., S.D.P., P.M.), Metabolic Unit,University of Pisa, 56100 Pisa, Italy
Many adverse effects of glucose were attributed to its in-creased routing through the hexosamine pathway (HBP).There is evidence for an autocrine role of the insulin signalingin �-cell function. We tested the hypothesis that activation ofthe HBP induces defects in insulin biosynthesis by affectingthe insulin-mediated protein translation signaling. Exposureof human pancreatic islets and RIN �-cells to glucosamineresulted in reduction in glucose- and insulin-stimulated in-sulin biosynthesis, which in RIN �-cells was associated withimpairment in insulin-stimulated insulin receptor substrate-1(IRS-1) phosphorylation at Tyr608 and Tyr628, which are es-sential for engaging phosphatidylinositol 3-kinase (PI 3-kinase). These changes were accompanied by impaired acti-vation of PI 3-kinase, and activation of Akt/mammalian targetof rapamycin/phosphorylated heat- and acid-stable protein-1/p70S6 kinase pathway. RIN �-cells exposed to high glucoseexhibited increased c-Jun N-terminal kinase (JNK) andERK1/2 activity, which was associated with increased IRS-1
phosphorylation at serine (Ser)307 and Ser612, respectively,that inhibits coupling of IRS-1 to the insulin receptor and isupstream of the inhibition of IRS-1 tyrosine phosphorylation.Azaserine reverted the stimulatory effects of high glucose onJNK and ERK1/2 activity and IRS-1 phosphorylation at Ser307
and Ser612. Glucosamine mimicked the stimulatory effects ofhigh glucose on JNK and ERK1/2 activity and IRS-1 phosphor-ylation at Ser307 and Ser612. Inhibition of JNK and MAPKkinase-1 activity reverted the negative effects of glucosamineon insulin-mediated protein synthesis. These results suggestthat activation of the HBP accounts, in part, for glucose-induced phosphorylation at Ser307 and Ser612 of IRS-1 medi-ated by JNK and ERK1/2, respectively. These changes result inimpaired coupling of IRS-1 and PI 3-kinase, and activation ofthe Akt/mammalian target of rapamycin/phosphorylatedheat- and acid-stable protein-1/p70S6 kinase pathway. (Endo-crinology 145: 2845–2857, 2004)
THE PATHOGENESIS OF type 2 diabetes is characterizedby a combination of peripheral insulin resistance and
a progressive decline in pancreatic �-cell function. Wheninsulin secretion by �-cells fails to compensate for insulinresistance, glucose intolerance and, eventually, overt hyper-glycemia occurs (1). Chronic elevation of glucose concentra-tions deteriorates �-cell function through a process referredto as glucose toxicity (2, 3). Exposure to a hyperglycemicenvironment of human or murine islets and �-cell lines leadsto alterations in insulin gene transcription, insulin biosyn-
thesis, glucose-induced insulin secretion, and �-cell survival(4–9).
Several of the adverse effects of glucose have been attributedto increased routing of carbohydrate through the hexosaminepathway (HBP) (10–12). In the HBP, glutamine/fructose-6-phosphate amidotransferase (GFAT) catalyzes the conversionof fructose-6-phosphate to glucosamine-6-phosphate with glu-tamine acting as an aminodonor. The final product of this path-way is the substrate uridine 5�-diphosphate-N-acetylglu-cosamine (GlcNAc), which is the donor sugar used by theenzyme O-linked N-acetylglucosamine transferase for O-linkedglycosylation of proteins. In this reaction, a single GlcNAc moi-ety is enzymatically attached to the hydroxyl group of either Seror Thr residue (13).
Recently it has been demonstrated that insulin released by�-cells in response to glucose stimulus enhanced its ownbiosynthesis, and exogenous insulin mimicked the stimula-tory effects of glucose on insulin biosynthesis (14–18). Themain signaling pathway used by the insulin receptor forpromoting insulin biosynthesis originates with tyrosine
Abbreviations: ECL, Enhanced chemiluminescence; GFAT, glu-tamine/fructose-6-phosphate amidotransferase; GlcNAc, N-acetyl-glucosamine; HBP, hexosamine pathway; IRS-1, insulin receptorsubstrate-1; JNK, c-Jun N-terminal kinase; MEK, MAPK kinase; mTOR,mammalian target of rapamycin; PHAS-1, phosphorylated heat- andacid-stable protein-1; PI 3-kinase, phosphatidylinositol 3-kinase; RIN, ratinsulinoma; SH2, src homology 2.Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving theendocrine community.
0013-7227/04/$15.00/0 Endocrinology 145(6):2845–2857Printed in U.S.A. Copyright © 2004 by The Endocrine Society
doi: 10.1210/en.2003-0939
2845

phosphorylation of insulin receptor substrate-1 (IRS-1) lead-ing to sequential activation of phosphatidylinositol 3-kinase(PI 3-kinase) and its downstream effector, the Ser/Thr kinaseAkt. Akt plays a central role in promoting protein synthesisby modulating downstream elements involved in the controlof the translation process including the kinase mammaliantarget of rapamycin (mTOR) and its downstream targetsp70S6 kinase and phosphorylated heat- and acid-stableprotein-1 (PHAS-1), also termed eucaryotic initiation factor4E binding protein 1 (18–21). We have recently shown thatexposure to a hyperglycemic environment of human pan-creatic islets or �-cell lines leads to alterations in insulin genetranscription and insulin biosynthesis (9). These abnormal-ities have been associated with decreased insulin receptorexpression and increased relative abundance of the Ex11�
isoform of the insulin receptor in both human pancreaticislets and RIN �-cells (9). These changes were paralleled bydefects in insulin signaling involving IRS-associated PI 3-ki-nase/Akt/PHAS-1 pathway in RIN �-cells. Interestingly, re-expression in RIN �-cells chronically exposed to high glucoseof Ex11�, but not Ex11�, isoform restored insulin mRNAexpression (9). These data suggest that changes in early stepsof the insulin receptor signaling may play a role in deter-mining �-cell dysfunction caused by chronic hyperglycemia.
Serine (Ser) phosphorylation of IRS-1 has been implicated asa negative regulator of insulin signaling and may be a contrib-uting factor in the development of insulin resistance (22–26).Upon phosphorylation on Ser residues, IRS-1 has been shownto have a reduced ability to interact with the insulin receptor,to be phosphorylated on tyrosine residues, and to engage thesrc homology 2 (SH2) domains of the p85 regulatory subunit ofPI 3-kinase (22, 23, 26). Several Ser residues in IRS-1 have beenidentified as negative regulatory sites including Ser307, which isactivated by c-Jun N-terminal kinase (JNK) and Ser612, which isactivated by MAPK (22–25). There is evidence that high glucoseand glucosamine, which is a product of the HBP downstreamto GFAT, induces activation of both JNK and MAPK (27–29).However, it is unknown whether activation of the HBP wouldaffect phosphorylation of IRS-1 at Ser307 and Ser612 residues,thus impairing activation of the insulin biosynthetic pathwayinvolving PI3-kinase/Akt/mTOR. The present study was de-signed to examine the effect of activation of the HBP on insulinbiosynthesis in human pancreatic islets and the �-cell line RIN1046–38, a well-characterized cell model, which exhibits the keyregulatory elements of pancreatic �-cells (9, 30–33). We foundthat exposure of �-cells to glucosamine resulted in hyperphos-phorylation of IRS-1 at Ser307 and Ser612 and inhibition of ty-rosine phosphorylation at positions 608 and 628 (Tyr608 andTyr628), which play a major role in binding the SH2 domains ofp85 subunit of PI 3-kinase (34). These changes in IRS-1 phos-phorylation resulted in reduction in insulin biosynthesis due toimpaired activation of the PI3-kinase/Akt/mTOR proteintranslation initiation pathway.
Materials and MethodsProinsulin biosynthesis in human islet and RIN �-cells
The pancreata were procured through the local organ procurementagency, within the framework of a project aiming to clinical islet allo-transplantation into diabetic patients, approved by the Ethics Commit-tee of the University of Pisa. Human pancreatic islets were obtained by
collagenase digestion and density-gradient purification from pancreataof heart beating multiorgan donors as previously described (8, 9, 31).After isolation, the islets were plated on collagen I-coated dishes, al-lowing the cells to attach to the dishes. Islets were cultured in M199medium containing 10% fetal bovine serum. Two days after plating,when most islets were attached, the islets were incubated for 24 h in thepresence or absence of glucosamine (7.5 mm) in M199 medium contain-ing 0.25% BSA. In experiments with protein kinase inhibitors, these wereadded to cells 30 min before glucosamine addition. Proinsulin biosyn-thesis was determined by preincubating islets for 2 h in modified Krebs-Ringer bicarbonate buffer containing 20 mm HEPES and 0.25% BSA,supplemented with 2.8 mm glucose, minimal essential medium aminoacid solution, minimal essential medium nonessential amino acid so-lution, and l-glutamine. Then the islets were stimulated with glucose (16mm) or insulin (100 nm) for 60 min followed by incubation for 60 minin buffer containing 50 �Ci [35S] methionine (Amersham PharmaciaBiotech, Milan, Italy). Thereafter cells were washed and lysed, andinsulin was immunoprecipitated with antiinsulin antibody. Total la-beled proteins were precipitated in 10% trichloroacetic acid and pelletedby centrifugation, and radioactivity was determined by scintillationcounting. RIN1046–38 rat pancreatic �-cells were kindly provided by Dr.W. L. Chick (Transplantation Biology, BioHybrid Technologies, Inc.,Shrewsbury, MA). Proinsulin biosynthesis was determined in RIN�-cells treated for 15 min with maximally stimulating concentrations ofglucose (3 mm) or insulin (100 nm) according to the procedure abovedescribed. In preliminary experiments, the effect of glucose and glu-cosamine on O-linked GlcNAc modification of proteins in RIN �-cellshad been determined in total cell lysates by immunoblotting with theanti-O-linked GlcNAc monoclonal antibody RL2 (Affinity Bioreagents,Golden, CO). In agreement with our previous results in a different cellmodel (35), exposure of RIN �-cells to different glucose or glucosamineconcentrations for 24 h resulted in a dose-dependent increase in O-Glc-NAcylation levels with maximal effect occurring at 25–33 mm glucose thatwas equivalent to that exerted by 7.5 mm glucosamine (data not shown).
Phospho-specific phosphorylation of IRS-1
RIN 1046–38 �-cells were cultured for 24 h in serum-deprived M-199medium containing 0.1% BSA in the presence or absence of 7.5 mmglucosamine and then incubated in the presence or absence of 100 nminsulin for 5 min. The cells were then washed with ice-cold PBS and lysedfor 1 h at 4 C in lysis buffer [94 mm NaCl, 14 mm Tris (pH 7.6), 0.7 mmMgCl2, 0.7 mm CaCl2, 1.5% Nonidet P-40, 10% glycerol, 2 mm phenyl-methylsulfonyl fluoride, 2 mm Na3VO4, 2 mm EDTA, 10 mm NaPP, 10mm NaF, 8 �g/ml leupeptin]. Insoluble material was removed by cen-trifugation in microfuge for 15 min at 4 C, and the supernatants wereincubated for 16 h at 4 C with anti-IRS-1 (Upstate Biotechnolocy Inc.,Lake Placid, NY). Immune complexes were collected by incubation withprotein A-Sepharose for 2 h at 4 C and resuspended in 2� Laemmlibuffer. Equal amounts of immunoprecipitated proteins were subjectedto SDS-PAGE under reducing conditions. Proteins resolved by SDS-PAGE were electrophoretically transferred to nitrocellulose membrane.The membranes were blotted with antiphosphotyrosine antibody RC20(Transduction Laboratories, Lexington, KY), anti-Ser307 IRS-1, anti-Ser612
IRS-1 (Cell Signaling, Beverly, MA), anti-Tyr608 IRS-1 (Biosource, Cam-arillo, CA), or anti-Tyr628 IRS-1 (Santa Cruz Biotechnology, Santa Cruz,CA). After extensive washings, the blots were incubated with peroxi-dase-conjugated secondary antibodies. To normalize the blots for pro-tein levels, after being immunoblotted with antiphosphospecific anti-bodies, the blots were stripped and reprobed with anti-IRS-1 antibody.Proteins were detected by using enhanced chemiluminescence (ECL)(Amersham Pharmacia Biotech) and band densities were quantified bydensitometry.
IRS-1-associated PI 3-kinase activity
RIN 1046–38 �-cells cultured as described above were incubated inthe presence or absence of 100 nm insulin for 5 min and then lysed. Celllysates were subjected to immunoprecipitation with anti-IRS-1 antibodyand PI 3-kinase activity was assayed in the immunoprecipitates accord-ing to previously described method (9, 31, 35) with phosphatidylinositolused as substrate. The PI 3-phosphate products were visualized byautoradiography, identified by its comigration with a PI 4-phosphate
2846 Endocrinology, June 2004, 145(6):2845–2857 Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis

standard (Calbiochem, La Jolla, CA), and quantified by scanningdensitometry.
Phosphorylation of Akt, tuberin, mTOR, PHAS-1, and p70S6 kinase
After insulin stimulation, RIN 1046–38 �-cells were lysed, and equalamounts of protein from cell lysates were resolved by SDS-PAGE andelectrophoretically transferred to nitrocellulose membranes. The mem-branes were blotted with anti-Ser473 Akt, anti-Thr389 p70S6 kinase, anti-Ser2248 mTOR, or anti-Thr1462 tuberin antibody (Cell Signaling). PHAS-1phosphorylation was detected using a polyclonal anti-PHAS-1 antibody(Santa Cruz Biotechnology) and determining the electrophoretic mo-bility pattern. Blots were visualized using peroxidase-conjugated sec-ondary antibodies followed ECL detection, and band densities werequantified by densitometry.
Phosphorylation of c-Jun, ERK1/2, and Ser307/312
phosphorylation of IRS-1
RIN 1046–38 �-cells were cultured for 24 h in serum-free mediumwith the indicated concentrations of glucose and glucosamine in thepresence or absence of azaserine (Sigma-Aldrich, St. Louis, MO), thecell-permeable peptide JNK inhibitor I (Calbiochem), and PD98059 (Cal-biochem). Cells were then lysed, and equal amounts of proteins weresubjected to SDS-PAGE and immunoblotted with phospho-specific anti-Ser63cJun, anti-Thr202/Tyr204 ERK1/2, anti-Ser307 IRS-1, or Ser612 IRS-1antibody (Cell Signaling). To normalize the blots for protein levels, afterbeing immunoblotted with antiphospho-specific antibody, the blotswere stripped and reprobed with appropriate primary antibody. Pro-teins were detected by using ECL, and band densities were quantifiedby densitometry.
Statistical analysis
All results are given as means � sem. Differences between valueswere evaluated by one-way ANOVA or unpaired Student’s t test asappropriate. Values of P � 0.05 were considered statistically different (*,P � 0.05; **, P � 0.01; ***, P � 0.001).
ResultsImpaired insulin biosynthesis stimulated by glucose orinsulin in human pancreatic islets and RIN �-cells exposedto glucosamine
To examine the possible involvement of the HBP in toxiceffect of glucose, human pancreatic islets and RIN �-cellswere incubated in the presence or absence of glucosamineand then stimulated with glucose or exogenous insulin. Inhuman pancreatic islets, specific insulin biosynthesis, i.e. af-ter normalization to total protein biosynthesis, increased by2-fold (P � 0.01) upon stimulation with glucose and by2.3-fold (P � 0.01) upon stimulation with insulin (Fig. 1).Insulin biosynthesis in human pancreatic islets exposed toglucosamine was reduced by 39% upon glucose stimulation(P � 0.01) and by 24% upon insulin stimulation (P � 0.01) ascompared with untreated control human islets. Only a lim-ited number of human pancreatic islets can be obtained fromdonors; thus, to further investigate the molecular mecha-nisms by which glucosamine would affect glucose- andinsulin-stimulated insulin biosynthesis, we used the pan-
FIG. 1. Effect of glucosamine on glucose- and insulin-stimulated insulin biosynthesis in human pancreatic islets. Human islets were incubatedfor 24 h in serum-free medium in the indicated combinations of glucosamine with or without the kinase inhibitors PD98059 and JNK inhibitorI. The cells were then stimulated with glucose and insulin, as described in Materials and Methods, in the presence or absence of the indicatedinhibitors. Specific insulin biosynthesis was calculated as the ratio of insulin to total protein biosynthesis, and values are given in percentagesrelative to those obtained under basal unstimulated conditions, which were set to 100%. Each bar represents the mean � SEM of threeindependent experiments. Differences between values were evaluated by one-way ANOVA.
Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis Endocrinology, June 2004, 145(6):2845–2857 2847

creatic RIN �-cell line that exhibits the key regulatory eleb-ments of the pancreatic �-cell. In control RIN �-cells, specificinsulin biosynthesis increased by 1.9-fold (P � 0.001) uponstimulation with glucose and by 2.0-fold (P � 0.001) uponstimulation with insulin (Fig. 2). Insulin biosynthesis in RIN�-cells exposed to glucosamine was reduced by 40% uponglucose stimulation (P � 0.001) and by 45% upon insulinstimulation (P � 0.01) as compared with untreated control�-cells. To examine whether glucosamine affects cell sur-vival, apoptosis was evaluated by flow cytometry by mea-suring the number of hypodiploid events after propidiumiodide staining of fixed cells. Exposure of RIN �-cells toglucosamine in the presence of 0.2% BSA for 24 h resulted inapoptosis of less than 10% of total cells. These data wereconfirmed by assessment of caspase-3 activity, which servesas a biochemical marker for the execution phase of apoptosis(data not shown). To analyze the intracellular signaling thatin human pancreatic islets and RIN �-cells contributes toregulate insulin biosynthesis in response to both stimuli, wecombined the stimulation with glucose or insulin with thecotreatment with inhibitors of the insulin receptor tyrosinekinase [hydroxy-2-naphthalenylmethylphosphonic acid trisace-toxymethyl ester (HNMPA)], PI 3-kinase (wortmannin andLY294002), and mTOR (rapamycin). As shown in Figs. 1 and 2,HNMPA, wortmannin, LY294002, and rapamycin inhibited bothglucose- and insulin-stimulated insulin biosynthesis in both hu-man pancreatic islets and RIN �-cells. These data are consistent
with previous studies showing a role for the insulin receptor,PI3-kinase, and mTOR in the signaling pathway involved in in-sulin biosynthesis by pancreatic �-cells in response to secretedinsulin or exogenous insulin (15, 16).
Impaired tyrosine phosphorylation of IRS-1 and IRS-2, PI3-kinase activity, and Akt activation in RIN �-cells exposedto glucosamine or high-glucose concentrations
To test the hypothesis that reduced insulin biosynthesisinduced by glucosamine treatment was accompanied by al-terations in early steps in insulin signaling, tyrosine phos-phorylation of both IRS-1 and IRS-2 was determined in ly-sates from glucosamine- and high glucose-treated RIN�-cells by immunoprecipitation with anti-IRS-1 antibody fol-lowed by immunoblotting with antiphosphotyrosine anti-body. As shown in Fig. 3A, exposure of the cells to highglucose concentrations resulted in a 47% reduction of insulin-stimulated tyrosine phosphorylation of IRS-1 as comparedwith control RIN �-cells. Likewise, insulin-stimulated PI3-kinase activity associated with IRS-1 was reduced by 36%in RIN �-cells exposed to high glucose concentrations ascompared with control RIN �-cells (Fig. 3B). Accordingly,exposure of the cells to glucosamine resulted in a 38% re-duction of insulin-stimulated tyrosine phosphorylation ofIRS-1 as compared with control RIN �-cells. Insulin-stimu-lated PI 3-kinase activity associated with IRS-1 was reduced
FIG. 2. Effect of glucosamine on glucose- and insulin-stimulated insulin biosynthesis in RIN �-cells. RIN �-cells were incubated for 24 h inserum-free medium in the indicated combinations of glucosamine with or without the kinase inhibitors PD98059 and JNK inhibitor I. The cellswere then stimulated with glucose and insulin, as described in Materials and Methods, in the presence or absence of the indicated inhibitors.Specific insulin biosynthesis was calculated as the ratio of insulin to total protein biosynthesis, and values are given in percentages relativeto those obtained under basal unstimulated conditions, which were set to 100%. Each bar represents the mean � SEM of six independentexperiments carried out in triplicate. Differences between values were evaluated by one-way ANOVA.
2848 Endocrinology, June 2004, 145(6):2845–2857 Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis

by 40% in RIN �-cells exposed to glucosamine as comparedwith control RIN �-cells (Fig. 3B). Expression of IRS-1 did notdiffer between RIN �-cells exposed to glucosamine or highglucose concentrations and control RIN �-cells as detected byimmunoblotting (Fig. 3A).
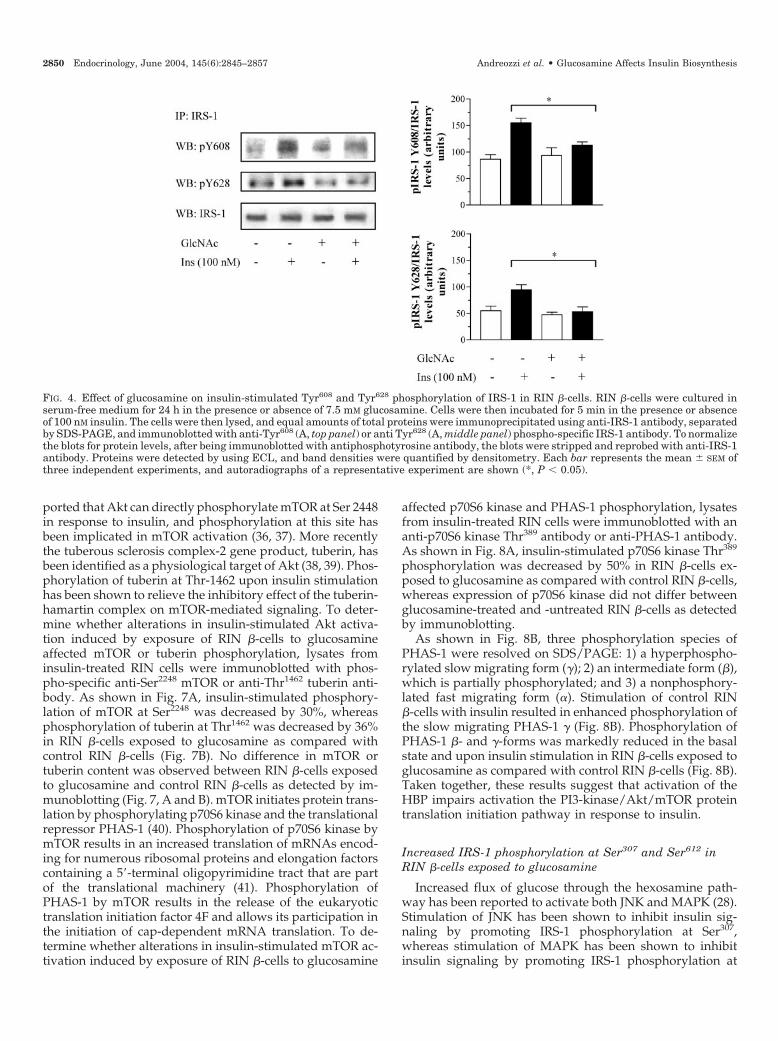
There is evidence that tyrosine residues in the YXXM mo-tifs at positions 608 and 628 (Tyr608 and Tyr628) play a majorrole in engaging the tandem SH2 domains of p85 subunit ofPI 3-kinase (34). To determine whether alterations in insulin-stimulated PI 3-kinase activation induced by exposure of RIN�-cells to glucosamine affected Tyr608 and Tyr628 phosphor-ylation of IRS-1, lysates from insulin-treated RIN �-cells wereimmunoprecipitated with anti-IRS-1 antibody followed byimmunoblotting with phospho-specific anti-Tyr608 and anti-Tyr628 IRS-1 antibody. As shown in Fig. 4, insulin-stimulatedTyr608 and Tyr628 phosphorylation of IRS-1 was decreased by30 and 46%, respectively, in RIN �-cells exposed to glu-cosamine as compared with control RIN �-cells. Exposure ofcells to glucosamine also resulted in a 39% reduction ofinsulin-stimulated tyrosine phosphorylation of IRS-2 as com-pared with control RIN �-cells (Fig. 5A). Insulin-stimulatedPI 3-kinase activity immunoprecipitated with anti-phospho-tyrosine antibody was reduced by 38% in RIN �-cells ex-posed to glucosamine as compared with control RIN �-cells
(Fig. 5B). The Ser/Thr kinase Akt is a critical downstreameffector of PI 3-kinase that mediates numerous biologicaleffects induced by insulin including protein synthesis. Todetermine whether alterations in insulin-stimulated PI 3-kinase activation induced by exposure of RIN �-cells to glu-cosamine affected Akt phosphorylation, lysates from insulin-treated RIN cells were immunoblotted with phospho-specific anti-Ser473 antibody. Insulin-induced Ser473 Aktphosphorylation was reduced by 37% (Fig. 6). No differencein Akt content was observed between RIN �-cells exposed toglucosamine and control RIN �-cells as detected by immu-noblotting (Fig. 6). Taken together, these results suggest thatactivation of the HBP impairs the insulin-stimulated tyro-sine phosphorylation cascade leading to activation of PI3-kinase/Akt signaling pathway.
Impaired activation of the mTOR/p70S6 kinase/PHAS-1protein translation pathway in RIN �-cells exposedto glucosamine
Akt promotes protein synthesis by modulating down-stream proteins involved in the control of the translationprocess including the kinase mTOR and its downstreamtargets p70S6 kinase and PHAS-1 (19–21). It has been re-
FIG. 3. Effect of glucosamine on insulin-stimulated tyrosine phosphorylation of IRS-1 and IRS-1-associated PI 3-kinase activity RIN �-cells.RIN �-cells were cultured in serum-free medium for 24 h in the presence or absence of 7.5 mM glucosamine. Cells were then incubated for 5min in the presence or absence of 100 nM insulin. The cells were then lysed, and equal amounts of total proteins were immunoprecipitated withanti-IRS-1 antibody, subjected to SDS-PAGE, and immunoblotted with anti-pY (phosphotyrosine) antibody (A, top panel). Kinase assays wereperformed on anti-IRS-1 immunoprecipitates (B) using phosphatidylinositol as an in vitro substrate. Each bar represents the mean � SEM ofat least three independent experiments. To normalize the blots for protein levels, after being immunoblotted with antiphosphotyrosine antibody,the blots were stripped and reprobed with anti-IRS-1 antibody (A, lower panel). Proteins were detected by using ECL, and band densities werequantified by densitometry. Each bar represents the mean � SEM of at least three independent experiments, and autoradiographs of arepresentative experiment are shown. *, P � 0.05; **, P � 0.01.
Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis Endocrinology, June 2004, 145(6):2845–2857 2849
ported that Akt can directly phosphorylate mTOR at Ser 2448in response to insulin, and phosphorylation at this site hasbeen implicated in mTOR activation (36, 37). More recentlythe tuberous sclerosis complex-2 gene product, tuberin, hasbeen identified as a physiological target of Akt (38, 39). Phos-phorylation of tuberin at Thr-1462 upon insulin stimulationhas been shown to relieve the inhibitory effect of the tuberin-hamartin complex on mTOR-mediated signaling. To deter-mine whether alterations in insulin-stimulated Akt activa-tion induced by exposure of RIN �-cells to glucosamineaffected mTOR or tuberin phosphorylation, lysates frominsulin-treated RIN cells were immunoblotted with phos-pho-specific anti-Ser2248 mTOR or anti-Thr1462 tuberin anti-body. As shown in Fig. 7A, insulin-stimulated phosphory-lation of mTOR at Ser2248 was decreased by 30%, whereasphosphorylation of tuberin at Thr1462 was decreased by 36%in RIN �-cells exposed to glucosamine as compared withcontrol RIN �-cells (Fig. 7B). No difference in mTOR ortuberin content was observed between RIN �-cells exposedto glucosamine and control RIN �-cells as detected by im-munoblotting (Fig. 7, A and B). mTOR initiates protein trans-lation by phosphorylating p70S6 kinase and the translationalrepressor PHAS-1 (40). Phosphorylation of p70S6 kinase bymTOR results in an increased translation of mRNAs encod-ing for numerous ribosomal proteins and elongation factorscontaining a 5�-terminal oligopyrimidine tract that are partof the translational machinery (41). Phosphorylation ofPHAS-1 by mTOR results in the release of the eukaryotictranslation initiation factor 4F and allows its participation inthe initiation of cap-dependent mRNA translation. To de-termine whether alterations in insulin-stimulated mTOR ac-tivation induced by exposure of RIN �-cells to glucosamine
affected p70S6 kinase and PHAS-1 phosphorylation, lysatesfrom insulin-treated RIN cells were immunoblotted with ananti-p70S6 kinase Thr389 antibody or anti-PHAS-1 antibody.As shown in Fig. 8A, insulin-stimulated p70S6 kinase Thr389
phosphorylation was decreased by 50% in RIN �-cells ex-posed to glucosamine as compared with control RIN �-cells,whereas expression of p70S6 kinase did not differ betweenglucosamine-treated and -untreated RIN �-cells as detectedby immunoblotting.
As shown in Fig. 8B, three phosphorylation species ofPHAS-1 were resolved on SDS/PAGE: 1) a hyperphospho-rylated slow migrating form (�); 2) an intermediate form (�),which is partially phosphorylated; and 3) a nonphosphory-lated fast migrating form (�). Stimulation of control RIN�-cells with insulin resulted in enhanced phosphorylation ofthe slow migrating PHAS-1 � (Fig. 8B). Phosphorylation ofPHAS-1 �- and �-forms was markedly reduced in the basalstate and upon insulin stimulation in RIN �-cells exposed toglucosamine as compared with control RIN �-cells (Fig. 8B).Taken together, these results suggest that activation of theHBP impairs activation the PI3-kinase/Akt/mTOR proteintranslation initiation pathway in response to insulin.
Increased IRS-1 phosphorylation at Ser307 and Ser612 inRIN �-cells exposed to glucosamine
Increased flux of glucose through the hexosamine path-way has been reported to activate both JNK and MAPK (28).Stimulation of JNK has been shown to inhibit insulin sig-naling by promoting IRS-1 phosphorylation at Ser307,whereas stimulation of MAPK has been shown to inhibitinsulin signaling by promoting IRS-1 phosphorylation at
FIG. 4. Effect of glucosamine on insulin-stimulated Tyr608 and Tyr628 phosphorylation of IRS-1 in RIN �-cells. RIN �-cells were cultured inserum-free medium for 24 h in the presence or absence of 7.5 mM glucosamine. Cells were then incubated for 5 min in the presence or absenceof 100 nM insulin. The cells were then lysed, and equal amounts of total proteins were immunoprecipitated using anti-IRS-1 antibody, separatedby SDS-PAGE, and immunoblotted with anti-Tyr608 (A, top panel) or anti Tyr628 (A, middle panel) phospho-specific IRS-1 antibody. To normalizethe blots for protein levels, after being immunoblotted with antiphosphotyrosine antibody, the blots were stripped and reprobed with anti-IRS-1antibody. Proteins were detected by using ECL, and band densities were quantified by densitometry. Each bar represents the mean � SEM ofthree independent experiments, and autoradiographs of a representative experiment are shown (*, P � 0.05).
2850 Endocrinology, June 2004, 145(6):2845–2857 Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis
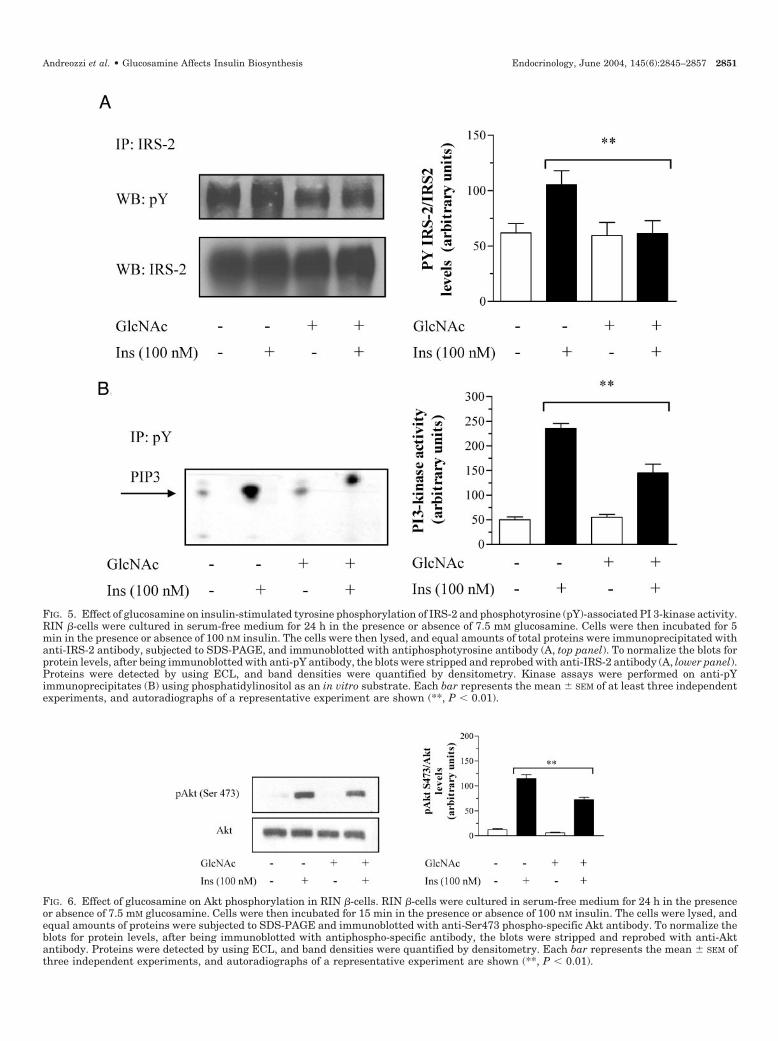
FIG. 6. Effect of glucosamine on Akt phosphorylation in RIN �-cells. RIN �-cells were cultured in serum-free medium for 24 h in the presenceor absence of 7.5 mM glucosamine. Cells were then incubated for 15 min in the presence or absence of 100 nM insulin. The cells were lysed, andequal amounts of proteins were subjected to SDS-PAGE and immunoblotted with anti-Ser473 phospho-specific Akt antibody. To normalize theblots for protein levels, after being immunoblotted with antiphospho-specific antibody, the blots were stripped and reprobed with anti-Aktantibody. Proteins were detected by using ECL, and band densities were quantified by densitometry. Each bar represents the mean � SEM ofthree independent experiments, and autoradiographs of a representative experiment are shown (**, P � 0.01).
FIG. 5. Effect of glucosamine on insulin-stimulated tyrosine phosphorylation of IRS-2 and phosphotyrosine (pY)-associated PI 3-kinase activity.RIN �-cells were cultured in serum-free medium for 24 h in the presence or absence of 7.5 mM glucosamine. Cells were then incubated for 5min in the presence or absence of 100 nM insulin. The cells were then lysed, and equal amounts of total proteins were immunoprecipitated withanti-IRS-2 antibody, subjected to SDS-PAGE, and immunoblotted with antiphosphotyrosine antibody (A, top panel). To normalize the blots forprotein levels, after being immunoblotted with anti-pY antibody, the blots were stripped and reprobed with anti-IRS-2 antibody (A, lower panel).Proteins were detected by using ECL, and band densities were quantified by densitometry. Kinase assays were performed on anti-pYimmunoprecipitates (B) using phosphatidylinositol as an in vitro substrate. Each bar represents the mean � SEM of at least three independentexperiments, and autoradiographs of a representative experiment are shown (**, P � 0.01).
Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis Endocrinology, June 2004, 145(6):2845–2857 2851

Ser612. We therefore investigated whether activation of theHBP induces IRS-1 phosphorylation at Ser307 and Ser612 byactivating JNK and MAPK, respectively. As shown in Fig. 9,A–C, exposure of RIN �-cells to high glucose resulted in anincreased phosphorylation of both JNK and the transcriptionfactor c-Jun, a physiological substrate of JNK as well as theERK-1 and -2 members of MAPK family. In addition, expo-sure of RIN �-cells to high glucose increased phosphoryla-tion of both IRS-1 Ser307 and IRS-1 Ser612 (Fig. 10, A and B),causing a decrease in the electrophoretic mobility shift ofIRS-1 (Fig. 10C). Inhibition of hexosamine biosynthesis by the
GFAT inhibitor azaserine reversed the stimulatory effects ofglucose on JNK, c-Jun, and ERK1/2 activation (Fig. 9, A–C)as well as IRS-1 phosphorylation at Ser307 and Ser612 (Fig. 10,A and B). Accordingly, exposure of RIN �-cells to glu-cosamine resulted in an increased phosphorylation of JNK,c-Jun, and ERK1/2 as compared with control RIN �-cells(Fig. 9, A–C). Exposure of RIN �-cells to glucosamine in-creased phosphorylation of both IRS-1 Ser307 and IRS-1Ser612, which was reversed by treatment with either a cell-permeable JNK inhibitor or PD98059, a reversible MAPKkinase (MEK)1 inhibitor, the enzyme that directly activates
FIG. 7. Effect of glucosamine on phosphorylation of mTOR and tuberin in RIN �-cells. RIN �-cells were cultured in serum-free medium for 24 hin the presence or absence of 7.5 mM glucosamine. Cells were then incubated 15 min in the presence or absence of 100 nM insulin. The cellswere lysed, and equal amounts of proteins were subjected to SDS-PAGE and immunoblotted with phosphor-specific anti-Ser2248 mTOR (A) oranti-Thr1462 tuberin (B) antibody. To normalize the blots for protein levels, after being immunoblotted with antiphosphospecific antibodies, theblots were stripped and reprobed with anti-mTOR or antituberin antibody. Proteins were detected by using ECL, and band densities werequantified by densitometry. Each bar represents the mean � SEM of three independent experiments, and autoradiographs of a representativeexperiment are shown (*, P � 0.05; **, P � 0.01).
FIG. 8. Effect of glucosamine on phosphorylation of p70S6 kinase and PHAS-1 in RIN �-cells. RIN �-cells were cultured in serum-free mediumfor 24 h in the presence or absence of 7.5 mM glucosamine. Cells were then incubated for 30 min in the presence or absence of 100 nM insulin.The cells were lysed, and equal amounts of proteins were subjected to SDS-PAGE and immunoblotted with phospho-specific anti-Thr389 p70S6kinase (A) or anti-PHAS-1 (B) antibody. To normalize the blots for protein levels, after being immunoblotted with antiphospho-specific antibody,the blots were stripped and reprobed with anti-p70S6 kinase antibody (A). Proteins were detected by using ECL, and band densities werequantified by densitometry. Each bar represents the mean � SEM of three independent experiments, and autoradiographs of a representativeexperiment are shown (*, P � 0.05; **P � 0.01).
2852 Endocrinology, June 2004, 145(6):2845–2857 Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis

ERK1/2, respectively (Fig. 10, A and B). Taken together,these results suggest that increased flux of glucose throughthe HBP induces IRS-1 phosphorylation at Ser307 and Ser612
by activation of JNK and ERK1/2, respectively, which can beeffectively reversed by their respective inhibitors.
To further demonstrate that activation of JNK and ERK1/2is involved in inhibitory effect of glucosamine on the insulinbiosynthetic pathway, we decided to determine whether JNKand MEK1 inhibitors can reverse impaired insulin biosyn-thesis induced by glucosamine in human pancreatic islets
and RIN �-cells. Treatment of human pancreatic islets withJNK inhibitor I partially reversed the inhibitory effect ofglucosamine, causing an increase of insulin biosynthesis by1.45-fold (P � 0.05) upon glucose stimulation and 1.25-foldupon insulin stimulation (P � 0.05) (Fig. 1). PD98059 re-versed the inhibitory effect of glucosamine causing an in-crease of insulin biosynthesis by 1.3-fold (P � 0.05) uponglucose stimulation and 1.2-fold (P � 0.01) upon insulinstimulation (Fig. 1). Cotreatment of human pancreatic isletswith both JNK inhibitor and PD98059 completely reversed
FIG. 9. Effect of high glucose or glucosamine on activation of JNK, c-Jun, and ERK1/2 in RIN �-cells. RIN �-cells were incubated for 24 h inserum-free medium in the indicated combinations of glucose with or without azaserine and glucosamine with or without cell-permeable peptideJNK inhibitor or PD98059. Cells were then lysed, and equal amounts of total proteins were subjected to SDS-PAGE and immunoblotted withphospho-specific anti-JNK, anti-Ser63cJun (used as an indirect measure of JNK activity) (B), or anti-Thr202/Tyr204 ERK1/2 (C). To normalizethe blots for protein levels, after being immunoblotted with antiphospho-specific antibody, the blots were stripped and reprobed with appropriateprimary antibody. Proteins were detected by using ECL, and band densities were quantified by densitometry. Each bar represents the mean �SEM of three independent experiments, and autoradiographs of a representative experiment are shown (*, P � 0.05; **, P � 0.01; ***, P � 0.001).
Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis Endocrinology, June 2004, 145(6):2845–2857 2853

the inhibitory effect of glucosamine on insulin biosynthe-sis stimulated by glucose or insulin (Fig. 1). Likewise,treatment of RIN �-cells with JNK inhibitor partially re-versed the inhibitory effect of glucosamine, causing anincrease of insulin biosynthesis by 1.35-fold (P � 0.05)upon glucose stimulation and 1.58-fold (P � 0.01) uponinsulin stimulation (Fig. 2). PD98059 reversed the inhibi-tory effect of glucosamine, causing an increase of insulinbiosynthesis by 1.47-fold (P � 0.05) upon glucose stimu-lation and 1.55-fold (P � 0.01) upon insulin stimulation(Fig. 2). Cotreatment of RIN �-cells with both JNK inhib-itor and PD98059 completely reversed the inhibitory effectof glucosamine on insulin biosynthesis stimulated by glu-cose or insulin (Fig. 2). These data are consistent with theconclusion that impairment in the insulin biosyntheticpathway involving IRS-1/PI 3-kinase/Akt/mTOR causedby activation of the hexosamine pathway is mediated, atleast in part, through IRS-1 phosphorylation at Ser307 andSer612 induced by JNK and ERK1/2, respectively.
Discussion
Glucose is the main physiological nutrient regulator of�-cell function including insulin secretion and biosynthesis.However, chronic hyperglycemia is accompanied by a de-cline in glucose-stimulated insulin secretion and biosynthe-sis, a phenomenon referred to as glucose toxicity (2–9). Anincreased flux of glucose through the HBP has been hypoth-esized to mediate many of the adverse effects of chronichyperglycemia (10–12). Recent investigations (14–18) haveprovided functional and genetic evidence for an autocrinerole of the insulin signaling cascade in �-cell growth andfunction. Because activation of the HBP has been reported toalter insulin signaling in target tissues such as skeletal mus-cle, adipocytes, and endothelium (35, 42, 43), we tested thehypothesis that activation of this metabolic pathway inducesdefects in insulin biosynthesis in both human pancreaticislets and the RIN pancreatic �-cell line by affecting theinsulin-mediated protein translation signaling. Our initial
FIG. 10. Effect of high glucose or glucosamine on phosphorylation of IRS-1 at Ser307 and Ser612 and on IRS-1 mobility shift in RIN �-cells. RIN�-cells were incubated for 24 h in serum-free medium in the indicated combinations of glucose with or without azaserine and glucosamine withor without cell-permeable peptide JNK inhibitor or PD98059. Cells were then lysed, and lysates were immunoprecipitated using anti-IRS-1antibody, separated by SDS-PAGE, and immunoblotted with phospho-specific anti-Ser307 IRS-1 (A), anti-Ser612 IRS-1 (B), and anti-IRS-1 (C)antibodies. To normalize the blots for protein levels after being immunoblotted with antiphospho-specific antibody, the blots were stripped andreprobed with anti-IRS-1 antibody. Proteins were detected by using ECL, and band densities were quantified by densitometry. Each barrepresents the mean � SEM of three independent experiments, and autoradiographs of a representative experiment are shown (*, P � 0.05; **,P � 0.01; ***, P � 0.001).
2854 Endocrinology, June 2004, 145(6):2845–2857 Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis

approach was to determine whether insulin secreted by�-cells contributes to its biosynthesis via an insulin receptor-mediated mechanism and whether this effect could be mim-icked by exogenous insulin. Consistent with previous studies(16, 18), we found that glucose and exogenous insulin equallystimulate insulin biosynthesis in both human pancreatic is-lets and RIN �-cells, and inhibitors of the insulin receptortyrosine kinase, PI 3-kinase, and mTOR abolished bothglucose- and insulin-stimulated insulin biosynthesis. To ex-clude the possibility that the observed stimulatory effects ofglucose and insulin were a result of recovery from a lack ofglucose and amino acids such as leucine, which has beenshown to be essential for glucose and insulin signaling
through mTOR (19), in vitro incubations were routinely per-formed in medium containing both glucose and amino acids,and labeled methionine was used as tracer instead of themost commonly used leucine. Exposure of human pancreaticislets and RIN �-cells to glucosamine resulted in a markedreduction in glucose- and insulin-stimulated insulin biosyn-thesis as compared with control �-cells. In the presence ofglucosamine, we observed in RIN �-cells a significant im-pairment in insulin-stimulated total tyrosine phosphoryla-tion of IRS-1 and IRS-2 as well as a reduction in specificphosphorylation of tyrosine residues of IRS-1 at positions 608and 628, which have an important role for engaging the SH2domains of the p85 regulatory subunit of PI 3-kinase (34).
FIG. 11. Schematic diagram of the hypothesized effects of high glucose and glucosamine on insulin-mediated protein synthesis in pancreatic�-cells. Exposure of pancreatic �-cells to high glucose or glucosamine may induces defects in insulin biosynthesis by affecting the insulin-mediated protein translation signaling through the activation of the HBP. Increased routing of glucose through HBP was associated withimpairment in insulin-stimulated IRS-1 phosphorylation at Tyr608 and Tyr628, which are essential for engaging PI 3-kinase. Glucosaminemimicked the stimulatory effects of high glucose on JNK and ERK1/2 activity and IRS-1 phosphorylation at Ser307 and Ser612. These resultssuggest that activation of the HBP accounts, in part, for glucose-induced phosphorylation at Ser307 and Ser612 of IRS-1 mediated by JNK andERK1/2, respectively. These changes result in impaired coupling of IRS-1 and PI 3-kinase, impaired activation of PI 3-kinase, and impairedactivation of the Akt/mTOR/PHAS-1/p70S6 kinase protein translation initiation pathway.
Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis Endocrinology, June 2004, 145(6):2845–2857 2855

These changes were accompanied by a significant impair-ment activation of IRS-1-associated PI 3-kinase, and the se-quential activation of Akt.
The initial signaling events that are thought to be respon-sible for initiating mRNA translation involve activation ofp70S6 kinase and PHAS-1 through a mTOR-mediated path-way (19–21). Akt promotes protein synthesis by modulatingmTOR activity by a direct and indirect way. Akt phosphor-ylates mTOR at Ser2448, and this event has been correlatedwith mTOR activity, although there is also evidence thatsubstitution of Ser2448 by alanine does not affect the ability ofmTOR to activate p70S6 kinase (44). More recently it has beenproposed an alternative way by which Akt regulates mTOR.Indeed, it has been demonstrated that Akt stimulates tuberinat Thr1462 upon insulin stimulation, thus relieving the inhib-itory function of the tuberin-hamartin complex on the acti-vation of mTOR and its downstream targets p70S6 kinaseand PHAS-1 (38, 39). Interestingly, we found that exposureof RIN �-cells to glucosamine resulted in a significant im-pairment in insulin-stimulated phosphorylation of bothmTOR at Ser2448 and tuberin at Thr1462. These changes wereaccompanied by a significant impairment of the sequentialactivation of p70S6 kinase and PHAS-1. These results areconsistent with the idea that activation of the HBP mightinterferes with insulin protein biosynthesis by affecting atmultiple steps the Akt/mTOR/PHAS-1/p70S6 kinase pro-tein translation initiation pathway.
A growing body of evidence has implicated Ser phosphor-ylation of IRS-1 as a mechanism of insulin resistance (22–26).Several studies have shown that enhanced Ser phosphory-lation of IRS-1 stimulated by a variety of factors interfereswith both the ability of this substrate to be tyrosine phos-phorylated upon insulin stimulation and reduces its abilityto engage the p85 regulatory subunit of PI 3-kinase. Morerecent investigations (22–25) have characterized several spe-cific Ser phosphorylation sites in IRS-1 as responsible forthese inhibitory effects and have identified the activatingkinases including JNK and ERK1/2. Activation of JNK hasbeen shown to result in stimulation of Ser307 of IRS-1,whereas activation of ERK1/2 has been shown to result in anincreased phosphorylation of Ser612. Consistent with previ-ous studies, we found that RIN �-cells exposed to high glu-cose exhibited increased JNK and ERK1/2 activity, whichwas associated with a concomitant increase in IRS-1 phos-phorylation on Ser307 and Ser612, respectively. Interestingly,azaserine, a GFAT inhibitor that blocks glucose flux throughthe HBP, inhibited the stimulatory effects of glucose on JNKand ERK1/2 activity and reverted the enhanced Ser307 andSer612 phosphorylation of IRS-1 stimulated by high glucose.Glucosamine mimicked the stimulatory effects of high glu-cose on JNK and ERK1/2 activity as well as IRS-1 phosphor-ylation on Ser307 and Ser612. These results indicate that acti-vation of the HBP accounts, in part, for glucose-inducedphosphorylation at Ser307 and Ser612 of IRS-1 mediated byJNK and ERK1/2, respectively, and thus for inhibition ofinsulin protein translation signaling involving the PI 3-kinase/Akt/mTOR pathway. We further demonstrated thecause-effect relationship between these two events by usinginhibitors of JNK and MEK1. Indeed, we found that inhibi-tion of JNK and MEK1 activity prevented the negative effects
of glucosamine on insulin protein translation signaling byreverting its adverse effect of insulin biosynthesis in bothhuman pancreatic islets and the RIN pancreatic �-cell line.
In summary, the present study has confirmed previous find-ings demonstrating that insulin promotes its own biosynthesisby an autocrine pathway involving the insulin receptor kinase/IRS-1/PI3-kinase/Akt/mTOR protein translation pathway.We show that activation of the HBP exerts an inhibitory effecton insulin biosynthesis and, for the first time, have correlatedthese changes with activation of JNK and ERK1/2. Our datasuggest that the uncoupling of IRS-1 and PI 3-kinase in glu-cosamine-treated RIN �-cells may be linked to an increasedphosphorylation at Ser307 and Ser612 of IRS-1 mediated byJNK and ERK1/2, respectively. These changes were associ-ated with a concomitant reduction in phosphorylation ofTyr608 and Tyr628 in two YXXM motifs essential for engagingp85 regulatory subunit of PI 3-kinase, resulting in impair-ment in binding of IRS-1 to p85 subunit, activation of IRS-1-associated PI 3-kinase, and sequential activation of theAkt/mTOR/PHAS-1/p70S6 kinase protein translation ini-tiation pathway (Fig. 11). Obviously, we cannot exclude thepossibility that other Ser/Thr kinase may phosphorylateIRS-1 under the conditions used in the present study, leadingto impairment in the activation of downstream protein trans-lation pathway. Notwithstanding, the present results suggestthat glucose-induced activation of JNK and ERK1/2 might bean important negative regulator for insulin biosyntheticpathway, and thus inhibiting these two kinases might be aplausible pharmacological target to treat pancreatic �-cellsdysfunction associated with type 2 diabetes.
Acknowledgments
Received July 24, 2003. Accepted February 26, 2004.Address all correspondence and requests for reprints to: Giorgio
Sesti, M.D., Dipartimento di Medicina Sperimentale e Clinica, Universitadi Catanzaro-Magna Græcia, Via Tommaso Campanella, 115, 88100Catanzaro, Italy. E-mail: [email protected].
F.A. and C.D.A. contributed equally to this manuscript.This work was supported in part by Grant E.1309 from Telethon-Italy
(to G.S.), Grant QLG1-CT-1999-00674 from the European Community“EuroDiabetesGene” (to G.S.), and Grant PRIN-COFIN from Ministerodell’Istruzione, dell’Universita e della Ricerca (to G.S. and R.L.).
References
1. Weyer C, Bogardus C, Mott DM, Pratley RE 1999 The natural history of insulinsecretory dysfunction and insulin resistance in the pathogenesis of type 2diabetes mellitus. J Clin Invest 104:787–794
2. Rossetti L, Giaccari A, DeFronzo RA 1990 Glucose toxicity. Diabetes Care13:610–630
3. Eizirik DL, Korbutt GS, Hellerstrom C 1992 Prolonged exposure of humanpancreatic islets to high glucose concentrations in vitro impairs the �-cellfunction. J Clin Invest 90:1263–1268
4. Sharma A, Olson LK, Robertson R, Stein R 1995 The reduction of insulin genetranscription in HIT-T15b cells chronically exposed to high glucose concen-tration is associated with the loss of RIPE3b1 and SFT-1 transcription factorexpression. Mol Endocrinol 9:1127–1134
5. Ling Z, Pipeleers DG 1996 Prolonged exposure of human � cells to elevatedglucose levels results in sustained cellular activation leading to a loss of glucoseregulation. J Clin Invest 98:2805–2812
6. Moran A, Zhang HJ, Olson LK, Harmon JS, Poitout V, Robertson RP 1997Differentiation of glucose toxicity from beta cell exhaustion during the evo-lution of defective insulin gene expression in the pancreatic islet cell line,HIT-T15. J Clin Invest 99:534–539
7. Marshak S, Leibowitz G, Bertuzzi F, Socci C, Kaiser N, Gross DJ, Cerasi E,Melloul D 1999 Impaired �-cell functions induced by chronic exposure ofcultured human pancreatic islets to high glucose. Diabetes 48:1230–1236
2856 Endocrinology, June 2004, 145(6):2845–2857 Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis

8. Federici M, Hribal M, Perego L, Ranalli M, Caradonna Z, Perego C, UselliniL, Nano R, Bonini P, Bertuzzi F, Marlier LNJL, Davalli AM, Carandente O,Pontiroli AE, Melino G, Marchetti P, Lauro R, Sesti G, Folli F 2001 Highglucose causes apoptosis in cultured human pancreatic islets of Langerhans:a potential role for regulation of specific Bcl family genes toward an apoptoticcell death program. Diabetes 50:1290–1301
9. Hribal ML, Perego L, Lovari S, Andreozzi F, Menghini R, Perego C, Finzi G,Usellini L, Placidi C, Capella C, Guzzi V, Lauro D, Bertuzzi F, Davalli A,Pozza G, Pontiroli A, Federici M, Lauro R, Brunetti A, Folli F, Sesti G 2003Chronic hyperglycemia impairs insulin secretion by affecting insulin receptorexpression, splicing, and signaling in RIN �-cell line and human islets ofLangerhans. FASEB J 17:1340–1342
10. Marshall S, Bacote V, Traxinger RR 1991 Discovery of a metabolic pathwaymediating glucose-induced desensitization of the glucose transport system.J Biol Chem 266:4706–4712
11. Giaccari A, Morviducci L, Zorretta D, Sbraccia P, Legnetti F, Caiola S,Buongiorno A, Bonadonna RC, Tamburano G 1995 In vivo effects of glu-cosamine on insulin secretion and insulin sensitivity in the rat: possible rel-evance to the maladaptive responses to chronic hyperglycaemia. Diabetologia38:518–524
12. Kaneto H, Xu G, Song K-H, Suzuma K, Bonner-Weir S, Sharma A, Weir GC2001 Activation of the hexosamine pathway leads to deterioration of pancreatic�-cell function through the induction of oxidative stress. J Biol Chem 276:31099–31104
13. Wells L, Vosseller K, Hart GW 2001 Glycosylation of nucleocytoplasmicproteins: signal transduction and O-GlcNAc. Science 291:2376–2378
14. Xu GG, Rothenberg PL 1998 Insulin receptor signaling in the �-cell influencesinsulin gene expression and insulin content: evidence for autocrine �-cellregulation. Diabetes 47:1243–1252
15. Leibiger IB, Leibiger B, Moede T, Berggren PO 1998 Exocytosis of insulinpromotes insulin gene transcription via the insulin receptor/PI-3 kinase/p70s6 kinase and CaM kinase pathways. Mol Cell 1:933–938
16. Xu G, Marshall CA, Lin TA, Kwon G, Munivenkatappa RB, Hill JR, Law-rence Jr JC, McDaniel ML 1998 Insulin mediates glucose-stimulated phos-phorylation of PHAS-1 by pancreatic � cells. An insulin-receptor mechanismfor autoregulation of protein synthesis by translation. J Biol Chem 273:4485–4491
17. Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR1999 Tissue-specific knockout of the insulin receptor in pancreatic � cellscreates an insulin secretory defect similar to that in type 2 diabetes. Cell96:329–339
18. Leibiger B, Wahlander K, Berggren PO, Leibiger IB 2000 Glucose-stimulatedinsulin biosynthesis depends on insulin-stimulated insulin gene transcription.J Biol Chem 275:30153–30156
19. McDaniel ML, Marshall CA, Pappan KL, Kwon G 2002 Metabolic and au-tocrine regulation of the mammalian target of rapamycin by pancreatic �-cells.Diabetes 51:2877–2885
20. Brown EJ, Beal PA, Keith, CT, Chen J, Shin TB, Schreiber SL 1995 Controlof p70 S6 kinase by kinase activity of FRAP in vivo. Nature 377:441–446
21. Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N 1998 4E-BP1,a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev 12:502–513
22. De Fea K, Roth RA 1997 Modulation of insulin receptor substrate-1 tyrosinephosphorylation and function by mitogen-activated protein kinase. J BiolChem 272:31400–31406
23. De Fea K, Roth RA 1997 Protein kinase C modulation of insulin receptorsubstrate-1 tyrosine phosphorylation requires serine 612. Biochemistry 36:12939–12947
24. Aguirre V, Uchida T, Yenush L, Davis R, White MF 2000 The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulinreceptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 275:9047–9054
25. Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF 2002Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactionswith the insulin receptor and inhibits insulin action. J Biol Chem 277:1531–1537
26. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM
1996 IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity inTNF-�- and obesity-induced insulin resistance. Science 271:665–668
27. Frodin M, Sekine N, Roche E, Filloux C, Prentki M, Wollheim CB, VanObberghen E 1995 Glucose, other secretagogues, and nerve growth factorstimulate mitogen-activated protein kinase in the insulin-secreting �-cell line,INS-1. J Biol Chem 270:7882–7889
28. Liu W, Schoenkerman A, Lowe Jr WL 2000 Activation of members of themitogen-activated protein kinase family by glucose in endothelial cells. Am JPhysiol Endocrinol Metab 279:E782–E790
29. Ho FM, Liu SH, Liau CS, Huang PJ, Lin-Shiau SY 2000 High glucose-inducedapoptosis in human endothelial cells is mediated by sequential activations ofc-Jun NH2-terminal kinase and caspase-3. Circulation 101:2618–2624
30. Clark SA, Burnham BL, Chick WL 1990 Modulation of glucose-induced in-sulin secretion from a rat clonal �-cell line. Endocrinology 127:2779–2788
31. Federici M, Hribal M, Ranalli M, Marselli L, Porzio O, Lauro D, Borboni P,Lauro R, Marchetti P, Melino G, Sesti G 2001 The common Arg972 poly-morphism in insulin receptor substrate-1 causes apoptosis of human pancre-atic islets. FASEB J 15:22–24
32. Porzio O, Federici M, Hribal ML, Lauro D, Accili D, Lauro R, Borboni P, SestiG 1999 The Gly972-�Arg amino acid polymorphism in IRS-1 impairs insulinsecretion in pancreatic � cells. J Clin Invest 104:357–364
33. Borboni P, Porzio O, Pierucci D, Cicconi S, Magnaterra R, Federici M, SestiG, Lauro D, D’Agata V, Cavallaro S, Marlier LN 1999 Molecular and func-tional characterization of pituitary adenylate cyclase-activating polypeptide(PACAP-38)/vasoactive intestinal polypeptide receptors in pancreatic �-cellsand effects of PACAP-38 on components of the insulin secretory system.Endocrinology 140:5530–5537
34. Esposito DL, Li Y, Cama A, Quon MJ 2001 Tyr(612) and Tyr(632) in humaninsulin receptor substrate-1 are important for full activation of insulin-stim-ulated phosphatidylinositol 3-kinase activity and translocation of GLUT4 inadipose cells. Endocrinology 142:2833–2840
35. Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D,Sbraccia P, Spagnoli LG, Sesti G, Lauro R 2002 Insulin-dependent activationof endothelial nitric oxide synthase is impaired by O-linked glycosylationmodification of signaling proteins in human coronary endothelial cells. Cir-culation 106:466–472
36. Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR 1999 Mammaliantarget of rapamycin is a direct target for protein kinase B: identification of aconvergence point for opposing effects of insulin and amino-acid deficiencyon protein translation. Biochem J 344:427–431
37. Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence Jr JC 1998 Evidence ofinsulin-stimulated phosphorylation and activation of the mammalian target ofrapamycin mediated by a protein kinase B signaling pathway. Proc Natl AcadSci USA 95:7772–7777
38. Inoki K, Li Y, Zhu T, Wu J, Guan KL 2002 TSC2 is phosphorylated andinhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648–657
39. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC 2002 Identificationof the tuberous sclerosis complex-2 tumor suppressor gene product tuberin asa target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10:151–162
40. Gingras A, Raught B, Sonenberg N 2001 Regulation of translation initiationby FRAP/mTOR. Genes Dev 15:807–826
41. Fumagalli S, Thomas GI 2000 S6 phosphorylation and signal transduction. In:Sonenberg N, Hershey J, Mathews M, eds. Translational control. Cold springHarbor, NY: Cold Spring Harbor Laboratory Press; 695–717
42. Patti ME, Virkamaki A, Landaker EJ, Kahn CR, Yki-Jarvinen H 1999 Acti-vation of the hexosamine pathway by glucosamine in vivo induces insulinresistance of early postreceptor insulin signaling events in skeletal muscle.Diabetes 48:1562–1571
43. Vosseller K, Wells L, Lane MD, Hart GW 2002 Elevated nucleocytoplasmicglycosylation by O-GlcNAc results in insulin resistance associated with defectsin Akt activation in 3T3–L1 adipocytes. Proc Natl Acad Sci USA 99:5313–5318
44. Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM,Abraham RT 2000 A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 60:3504–3513
Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving theendocrine community.
Andreozzi et al. • Glucosamine Affects Insulin Biosynthesis Endocrinology, June 2004, 145(6):2845–2857 2857
Copyright © 2022 FDOKUMEN




















