
Cytotoxic effects of crotamine are mediated through lysosomal membrane permeabilization
Upload
independentCategory
view
5download
0
Activation of Mammalian Target of Rapamycin Controls theLoss of TCR� in Lupus T Cells through HRES-1/Rab4-Regulated Lysosomal Degradation1
David R. Fernandez,*† Tiffany Telarico,*† Eduardo Bonilla,* Qing Li,* Sanjay Banerjee,*Frank A. Middleton,‡ Paul E. Phillips,* Mary K. Crow,§ Stefanie Oess,¶ Werner Muller-Esterl,¶
and Andras Perl2*†
Persistent mitochondrial hyperpolarization (MHP) and enhanced calcium fluxing underlie aberrant T cell activation anddeath pathway selection in systemic lupus erythematosus. Treatment with rapamycin, which effectively controls diseaseactivity, normalizes CD3/CD28-induced calcium fluxing but fails to influence MHP, suggesting that altered calcium fluxingis downstream or independent of mitochondrial dysfunction. In this article, we show that activity of the mammalian targetof rapamycin (mTOR), which is a sensor of the mitochondrial transmembrane potential, is increased in lupus T cells.Activation of mTOR was inducible by NO, a key trigger of MHP, which in turn enhanced the expression of HRES-1/Rab4,a small GTPase that regulates recycling of surface receptors through early endosomes. Expression of HRES-1/Rab4 wasincreased in CD4� lupus T cells, and in accordance with its dominant impact on the endocytic recycling of CD4, it wasinversely correlated with diminished CD4 expression. HRES-1/Rab4 overexpression was also inversely correlated with di-minished TCR� protein levels. Pull-down studies revealed a direct interaction of HRES-1/Rab4 with CD4 and TCR�. Im-portantly, the deficiency of the TCR� chain and of Lck and the compensatory up-regulation of Fc�RI� and Syk, whichmediate enhanced calcium fluxing in lupus T cells, were reversed in patients treated with rapamcyin in vivo. Knockdown ofHRES-1/Rab4 by small interfering RNA and inhibitors of lysosomal function augmented TCR� protein levels in vitro. Theresults suggest that activation of mTOR causes the loss of TCR� in lupus T cells through HRES-1/Rab4-dependent lysosomaldegradation. The Journal of Immunology, 2009, 182: 2063–2073.
S ystemic lupus erythematosus (SLE)3 is an autoimmunedisease of unknown etiology characterized by T and B celldysfunction and production of antinuclear Abs (1). Dys-
regulation of cell death is thought to play a key role in drivingantinuclear Ab production, since the source of immunogenic nu-clear material is necrotic or apoptotic cells in SLE (2). There isenhanced spontaneous apoptosis of circulating T cells in SLE,which has been linked to chronic lymphopenia (3) and compart-
mentalized release of autoantigens (4). Paradoxically, there is de-creased activation-induced T cell death in SLE (5–7), which maycontribute to persistence of autoreactive cells.
The mitochondria play crucial roles in activation and death path-way selection in T lymphocytes (2). Lupus T cells exhibit mito-chondrial dysfunction, which is characterized by the elevation ofthe mitochondrial transmembrane potential (��m) or persistent mi-tochondrial hyperpolarization (MHP) and consequential ATP de-pletion, resulting in decrease of activation-induced apoptosis andpredisposition of T cells for necrosis (6). ATP depletion in lupusT cells was recently confirmed by Krishnan et al. (8). We proposedthat increased release of necrotic materials from T cells could drivedisease pathogenesis by activating macrophages and dendritic cellsand enhancing their capacity to produce NO and IFN-� in SLE (2).Indeed, dendritic cells exposed to necrotic, but not apoptotic, cellsinduce lupus like-disease in MRL mice and accelerate the diseaseof MRL/lpr mice (9).
Enhanced T cell activation-induced calcium fluxing has beenidentified as a central defect in abnormal activation and cytokineproduction by lupus T cells (10). Induction of MHP and mitochon-drial biogenesis by NO augments cytoplasmic calcium levels andregenerates the enhanced rapid calcium signaling profile of lupusT cells (11). Dysregulation of signaling through the TCR has alsobeen shown to be a critical determinant of abnormal calcium flux-ing in SLE (12, 13). The TCR/CD3 �-chain (TCR�) expression isdiminished in SLE T cells, and it is functionally replaced by theFc�R type I �-chain (Fc�RI�), a protein normally found in other celltypes (14). TCR signaling through Fc�RI� and its adaptor proteinSyk is associated with elevated calcium fluxing but only in theabsence of TCR� (12). It has been shown that forced expression of
*Division of Rheumatology, Department of Medicine, †Department of Microbiologyand Immunology, and ‡Genetics Core, Department of Neuroscience and Physiology,State University of New York, Syracuse, NY 13210; §Hospital for Special Surgery,New York, NY 10021; and ¶Department of Biochemistry, University of Frankfurt,Frankfurt, Germany
Received for publication October 27, 2008. Accepted for publication December9, 2008.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This research was supported by National Institutes of Health Grants R01 AI-048079and AI-072648, the Alliance for Lupus Research, and the Central New York Com-munity Foundation.2 Address correspondence and reprint requests to Dr. Andras Perl, Division of Rheu-matology, Department of Medicine, State University of New York, 750 East AdamsStreet, Syracuse, NY 13210. E-mail address: [email protected] Abbreviations used in this paper: SLE, systemic lupus erythematosus; FKBP12, 12-kDaFK506-binding protein; L-NMMA, L-NG-monomethyl arginine citrate; LTR, LysotrackerRed; MHP, mitochondrial hyperpolarization; mTOR, mammalian target of rapamycin;NOSIP, NO synthase inhibitory protein; eNOS, endothelial NO synthase; RA, rheumatoidarthritis; S6K, S6 kinase; siRNA, small interfering RNA; SOD2, superoxide dismutase 2;TAL, transaldolase; TFR, transferrin receptor; VDAC1, voltage-dependent anion channel1; ��m, mitochondrial transmembrane potential.
Copyright © 2009 by The American Association of Immunologists, Inc. 0022-1767/09/$2.00
The Journal of Immunology
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0803600

TCR� is sufficient to reduce calcium influx in SLE T cells tohealthy control levels, illustrating the critical role TCR� plays inthe SLE phenotype (12).
The mammalian target of rapamycin (mTOR) is located in theouter mitochondrial membrane and serves as a sensor of the��m in T cells (15). Although mTOR is highly conserved andcontrols protein translation and other metabolic pathways in allmammalian cells (16), it plays a particularly critical role in Tcell activation. Inhibition of mTOR by rapamycin has beenshown to block T cell function specifically, leading to the ther-apeutic introduction of rapamycin for preventing organ trans-plant rejection (17). Treatment with rapamycin resulted inmarkedly improved disease activity in SLE patients resistant orintolerant to conventional immunosuppressants (18). MHP per-sisted while CD3/CD28-induced calcium fluxing was normal-ized in T cells of rapamycin-treated patients, suggesting thataltered calcium fluxing is downstream or independent of mito-chondrial dysfunction (18). mTOR has recently been localizedto endosomes carrying Rab7 (19) that controls the traffic be-tween early and late endocytic organelles (20).
The classical model of signaling involves membrane-spanningreceptors that, after binding an extracellular ligand at the cell sur-face, activate secondary messengers in the cytosol, enabling thespread of the signal into the nucleus. The signaling machinery canachieve a high order of regulation by exploiting the compartmen-talization and functional specialization of the endocytic pathway(21, 22). Endosomal transport is tightly regulated by the Ras-likesmall Rab GTPases (23). Although the role of receptor recyclingand endosomal trafficking in the immune system is largely un-known, it is likely to be significant. We recently identified HRES-1/Rab4 to have a dominant influence on the expression of CD4,and to a lesser extent on transferrin receptor (TFR), by regulationof their recycling from endosomes in Jurkat and peripheral bloodT cells (24). While the plasma membrane-associated Lck is boundto CD4, an intracellular pool of Lck is associated with TFR-pos-itive recycling endosomes (25, 26). TCR� is also physically asso-ciated with TFR (27). mTOR was shown to modulate the recyclingof TFR (28), which traffics through HRES-1/Rab4-positive endo-somes in Jurkat cells (24). Therefore, we 1) investigated the ge-netic pathways underlying mitochondrial dysfunction in lupus Tcells, 2) determined whether T cell dysfunction was related to anincreased activation state of mTOR, and 3) assessed the role ofRab GTPases in T cell activation.
In the present study, we show that mTOR activity is increasedin lupus T cells. Activation of mTOR was inducible by NO, a keytrigger of MHP and mitochondrial biogenesis (29). In turn, NO-induced stimulation of HRES-1/Rab4 expression was reduced byrapamycin. Thus, NO-dependent MHP lies upstream, whereasenhanced expression of HRES-1/Rab4 lies downstream ofmTOR activation in lupus T cells. Further downstream, CD4,Lck, and TCR� protein levels were depleted, whereas Syk andFc�RI� levels were augmented in lupus T cells, all of whichwere reversed in SLE patients treated with rapamycin in vivo.Pull-down and confocal microscopy studies revealed a directinteraction of HRES-1/Rab4 with TCR�. Depletion of TCR� inlupus T cells was reversed by HRES-1/Rab4 knockdown as wellas by inhibition of lysosomal function in vitro, indicating thatactivation of mTOR causes the loss of TCR� through HRES-1/Rab4-dependent lysosomal degradation.
Materials and MethodsHuman subjects
A total of 44 Caucasian female patients with SLE were investigated. Allpatients satisfied the criteria for a definitive diagnosis (30). Disease
activity was assessed by the SLE Disease Activity Index (SLEDAI)score (31). Six patients were treated with rapamycin 2 mg/day (age:40 � 8.3 years; SLEDAI: 0.8). Among the 38 remaining SLE patientstreated without rapamycin, 28 were receiving prednisone (5–50 mg/day)and immunosuppressive drugs, including hydroxychloroquine (400 mg/day), mycophenolate mofetil (3 g/day), and cyclosporin A (50–100 mg/day). Their mean age was 36.3 � 4.3 years, ranging between 18 and 60(SLEDAI: 1.3 � 0.9). Ten patients (age: 38.5 � 6.4; SLEDAI: 4.8 � 3.8)have been freshly diagnosed and have not been treated with prednisone orcytotoxic drugs. These patients and five additional patients that have re-ceived prednisone or cytotoxic drugs provided cells for microarray analysis(Table S1). As controls, 23 age-matched healthy female subjects and 8female patients with rheumatoid arthritis (RA; age: 51.3 � 6.7 years) (32)were studied. RA patients were treated with methotrexate, cyclosporin A,leflunomide, etanercept, or adalimumab. The study has been approved bythe Institutional Review Board for the Protection of Human Subjects.
Separation and culture of peripheral blood lymphocytes
PBMC were freshly isolated from heparinized venous blood on Ficoll-Hypaque gradient. PBL were separated from monocytes by adherence toautologous serum-coated petri dishes (33). “Untouched” T cells (�95%CD3�) were negatively isolated from PBMC with Dynal Biotech magneticbeads conjugated to IgG Abs for CD14, CD16 HLA class II DR/DP, CD56,and CD235a (catalog no.113-11D; Invitrogen). CD4� T cells (�98%CD4�) were negatively isolated with magnetic beads conjugated to IgGAbs for CD8, CD14, CD16, HLA class II DR/DP, CD56, CDw123, andCD235a (catalog no.113-39D; Invitrogen). The resultant cell populationwas resuspended at 106 cells/ml in RPMI 1640 medium, supplementedwith 10% FCS, 2 mM L-glutamine, 100 IU/ml penicillin, and 100 �g/mlgentamicin in 12-well plates at 37°C in a humidified atmosphere with 5%CO2. In vitro treatments of PBL or negatively isolated T cells was per-formed H2O2 (25 and 50 �M; Sigma-Aldrich), (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate diethylenetriamine(NOC-18, 150 and 300 �M; Calbiochem), rapamycin (50 nM dissolvedin DMSO, catalog no. 9904; Cell Signaling Technology), glutathione(GSH, 10 mM; Sigma-Aldrich), L-NG-monomethyl arginine citrate (L-NMMA,100 �M; Calbiochem), bafilomycin A1, and folimycin (bothfrom Calbiochem).
Flow cytometric analysis of cell viability, ��m, mitochondrialmass, cytoplasmic and mitochondrial calcium levels, and NOproduction
Apoptosis was monitored by staining with annexin V (annexin V-Alexa647; Invitrogen) in parallel with monitoring ��m, mitochondrial mass,ROI, NO, and Ca2� levels as well as expression of surface Ags, as de-scribed in the supplemental materials.4
Microarray analysis of gene expression
RNA was extracted from monocyte-depleted PBL (�80% CD3� T cells)or untouched T cells (�95% CD3� T cells negatively isolated with DynalBiotech kit catalog no. 113-11D). Biotinylated cRNA was produced by invitro transcription and hybridized to Affymetrix HG-U133_Plus_2 chipswith 54,675 probe sets, as described earlier (34, 35). Log2-based normal-ized data on genes present in one of the two study groups (lupus andcontrol PBL or lupus and control T cells) were compared with two-wayANOVA. Values of p were adjusted for multiple comparisons with theHolm formula. Microarray data files have been deposited in the Gene Ex-pression Omnibus MIAME-compliant public database (www.ncbi.nlm.nih.gov/geo under the GEO Series accession no. GSE13887). Lupus-predictorgenes were identified with the k-nearest-neighbor method (Genespringsoftware, version 7.3; Agilent), as described in the supplemental materials.
Western blot analysis
Whole-cell protein lysates were resuspended in SDS-PAGE sample buffer(2 � 105 cells/10 �l), separated on a 12% SDS-polyacrylamide gel, blottedto nitrocellulose, and probed with primary rabbit Abs directed to HRES-1/Rab4 or �-actin, as described previously (24). Primary Abs to superoxidedismutase 2 (SOD2), voltage-dependent anion channel 1 (VDAC1), 12-kDa FK506-binding protein (FKBP12), transaldolase (TAL), endothelialnitric oxide synthase inhibitory protein (NOSIP), p70 S6 kinase (S6K),phospho-S6K (pS6K), 4E-BP1, phospho-4E-BP1, raptor, rictor, mTOR,CD2AP, TCR�, CD4, CD8, Fc�RI�, p56 Lck, Syk, Rab5A, and Rab7, as
4 The online version of this article contains supplemental material.
2064 mTOR CONTROLS TCR� EXPRESSION THROUGH HRES-1/Rab4

well as peroxidase-conjugated secondary Abs and detection by chemilu-minescence using a Kodak Image Station 440CF, are described in the sup-plemental materials.
Detection of the IFN signature by real-time quantitative PCR
RNA was reverse transcribed to cDNA and amplified with primers specificfor the IFN-inducible genes MX1, IFIT1, IFI44, PRKR, and housekeepinggene control hypoxanthine guanine phosphoribosyltransferase 1, as de-scribed earlier (36).
Confocal microscopy
To label acidic lysosomal compartments, negatively isolated T cells wereincubated at 37°C with 5 �M LysoTracker Red (catalog no. L-7528; In-vitrogen) for 30 min in complete RPMI 1640 medium. Cells were washedtwice and resuspended in RPMI 1640 medium and allowed to adhere topoly-L-lysine-coated coverslips for 10 min at room temperature, followedby fixation in a 4% paraformaldehyde-PBS solution. The cells were per-meabilized with a 0.1% saponin/1% FBS/HBSS mixture for 30 min. Pri-mary Abs were directly conjugated with fluorochromes Alexa 488, Alexa555, or Alexa 647 and applied to permeabilized cells for 45 min, washedtwice with permeabilization buffer and twice with HBSS, mounted onslides, and visualized under a Zeiss 510 LSM Meta confocal microscope,as described in the supplemental materials.
Prokaryotic expression of recombinant HRES-1/Rab4 proteinand pull-down assays
Full-length HRES-1/Rab4 protein was expressed as a fusion protein withGST, as described earlier (24). Cell lysates were prepared from Jurkat cellsand PBL. Supernatants were incubated with 5 �g of HRES-1/Rab4-GST-bound agarose beads or 2.6 �g of GST-bound control beads (�100 pmolof each fusion protein) for 2 h at 4°C with and without 1 mM GTP�S(Sigma-Aldrich). The beads were pelleted, washed in lysis buffer, and an-alyzed by SDS-PAGE and Western blot, as described in the supplementalmaterials.
Transfection of small interfering RNA (siRNA)
Up to 107 freshly isolated lymphocytes were electroporated with siRNAspecifc for HRES1/Rab4 nt 377–399 or scrambled siRNA by using theNucleofector protocol for primary human T cells (Amaxa). Expression ofHRES-1/Rab4 and TCR� relative to actin was assayed by Western blot 6,12, 24, 36, 48, and 60 h after transfection, as described in the supplementalmaterials.
Recycling assay
Negatively isolated T cells were stained on ice with allophycocyanin-Cy7-conjugated Ab CD3 (catalog no. 557832; BD Biosciences), Alexa 647-conjugated Ab to CD4 (catalog no. 557707), and PE-conjugated Ab to CD8(catalog no. 555367) for 30 min, then washed three times in ice cold RPMI1640 medium. The cells were resuspended in warm medium and incubatedat 37°C for 3 h, removing samples at 30-min intervals and placing them onice. At the conclusion, the cells were restained with the original Abs on icefor 30 min, then washed three times with ice-cold RPMI 1640 medium andanalyzed by flow cytometry. Recycling is calculated by the increase inmean fluorescence intensity at given time points over the baseline (time 0sample) kept on ice after initial staining throughout the assay. The initialAb incubation step was done with or without 50 �g/ml cycloheximidewithout affecting the results obtained.
Statistical analysis
Statistical analyses were performed using Prism Version 3.0 for Windows(GraphPad). Data were expressed as the mean � SE of individual exper-iments. Changes were considered significant at value of p � 0.05.
ResultsDetection of the mitochondrial and endocytic recycling geneexpression signatures in lupus T cells
The signaling network underlying T cell dysfunction was investi-gated by a series of microarray analysis of gene expression andparallel flow cytometry of mitochondrial function in 15 femalelupus patients relative to 17 female controls. Log2-based normal-ized expression levels of genes present in one of the two studygroups (lupus and control cells) were compared with ANOVA.
Values of p were adjusted for multiple comparisons with the Holmformula by using Genespring software. Similar to previousfindings, mitochondrial mass, ��m, cytoplasmic calcium, andNO production were enhanced in lupus T cells (Fig. S1).Among 10 of 15 lupus patients that have not been exposed toprednisone or cytotoxic drugs (Table S1), expression of 117genes was altered. Eighty-two of these genes had identifiedfunctions. Although many genes participate in multiple signal-ing networks, 22 were involved in metabolic control of ��m, 19in calcium signaling, 12 in cytokine/IFN pathway (37–39), sixin programmed cell death, nine in small GTPase-mediated in-tracellular traffic, eight were transcription factors, and five weresurface receptors/adaptor proteins (Table S2). While lupus Tcells exhibit persistent MHP, exposure of normal human T cellsto CD3/CD28 costimulation or NO elicits transient MHP (29).To distinguish the impact of SLE from that of T cell activation,the influence of CD3/CD28 costimulation and NO on gene ex-pression was assessed in negatively isolated untouched T cellsof four healthy female donors. Expression of 329 genes wasincreased and expression of 173 genes was reduced by CD3/CD28 costimulation (Fig. S2). Expression of 50 genes was in-creased and expression of 204 genes was reduced by exposureto NO donor NOC-18 (Fig. S3). Direction and extent of CD3/CD28 or NO-induced changes in expression of 48 genes cor-related with differences observed in patients with SLE. In con-trast, altered expression of 34 genes could not be attributed toCD3/CD28 or NO, and thus, they may reflect the disease pro-cess in SLE. Expression of 25/34 lupus-specific genes werecorrelated with one or more parameter of mitochondrial dys-function in T cells, whereas 13 of 48 genes coordinately regu-lated by CD3/CD28 stimulation were correlated with mitochon-drial dysfunction (Table S2; �2: 17.27; p � 0.0001). Thisanalysis suggested that lupus-specific changes in gene expression,rather than genes coordinately regulated by CD3/CD28, are moreclosely linked to mitochondrial dysfunction. Among the 34 genessignificantly altered in negatively isolated T cells, 1.8-fold,VDAC1, SOD2, and Rab5A were found to be elevated in nega-tively isolated lupus T cells and predictive of SLE in each lupuspatient using k-nearest-neighbor algorithm (Fig. S3).
FIGURE 1. Western blot detection of SOD2, VDAC1, FKBP12,TAL, and NOSIP in “untouched” T cells of female patients with SLEand age-matched healthy female controls isolated with the negative Tcell isolation kit (Dynal Biotech). The expression of SOD2, VDAC1,FKBP12, TAL, and NOSIP relative to actin was determined in eachdonor by automated densitometry with Kodak Image Station 440CF.Representative blots are shown. The values of p reflect fold changes inprotein levels between 14 SLE patients and 10 controls compared withtwo-tailed t test.
2065The Journal of Immunology
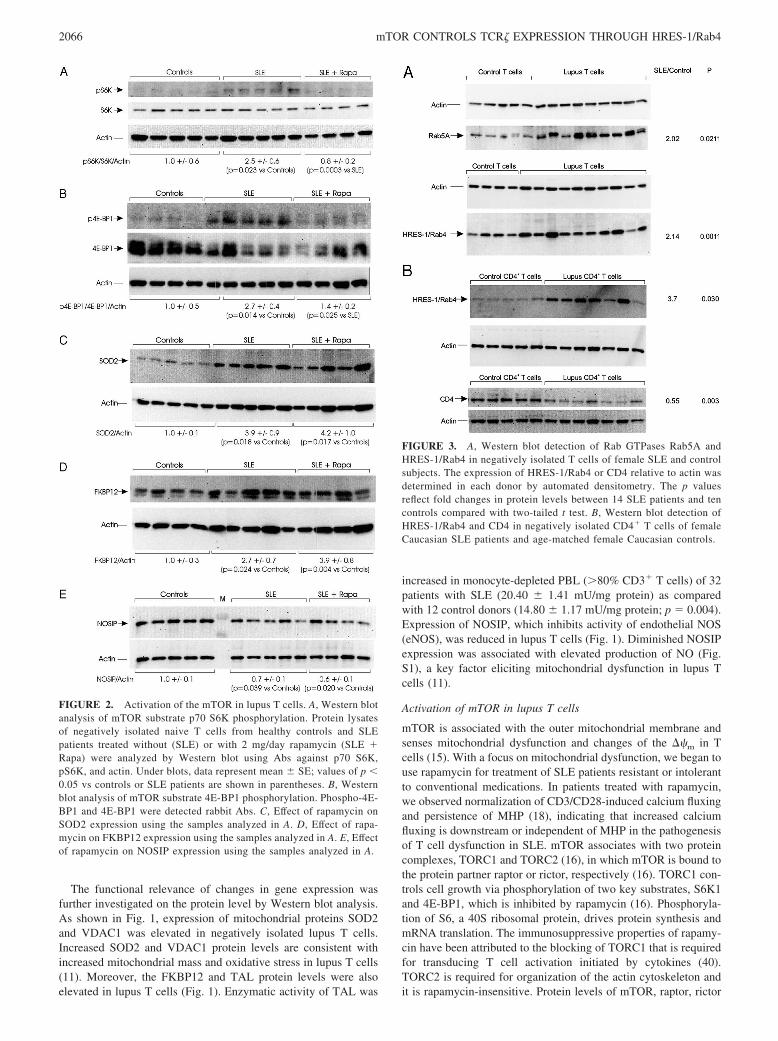
The functional relevance of changes in gene expression wasfurther investigated on the protein level by Western blot analysis.As shown in Fig. 1, expression of mitochondrial proteins SOD2and VDAC1 was elevated in negatively isolated lupus T cells.Increased SOD2 and VDAC1 protein levels are consistent withincreased mitochondrial mass and oxidative stress in lupus T cells(11). Moreover, the FKBP12 and TAL protein levels were alsoelevated in lupus T cells (Fig. 1). Enzymatic activity of TAL was
increased in monocyte-depleted PBL (�80% CD3� T cells) of 32patients with SLE (20.40 � 1.41 mU/mg protein) as comparedwith 12 control donors (14.80 � 1.17 mU/mg protein; p � 0.004).Expression of NOSIP, which inhibits activity of endothelial NOS(eNOS), was reduced in lupus T cells (Fig. 1). Diminished NOSIPexpression was associated with elevated production of NO (Fig.S1), a key factor eliciting mitochondrial dysfunction in lupus Tcells (11).
Activation of mTOR in lupus T cells
mTOR is associated with the outer mitochondrial membrane andsenses mitochondrial dysfunction and changes of the ��m in Tcells (15). With a focus on mitochondrial dysfunction, we began touse rapamycin for treatment of SLE patients resistant or intolerantto conventional medications. In patients treated with rapamycin,we observed normalization of CD3/CD28-induced calcium fluxingand persistence of MHP (18), indicating that increased calciumfluxing is downstream or independent of MHP in the pathogenesisof T cell dysfunction in SLE. mTOR associates with two proteincomplexes, TORC1 and TORC2 (16), in which mTOR is bound tothe protein partner raptor or rictor, respectively (16). TORC1 con-trols cell growth via phosphorylation of two key substrates, S6K1and 4E-BP1, which is inhibited by rapamycin (16). Phosphoryla-tion of S6, a 40S ribosomal protein, drives protein synthesis andmRNA translation. The immunosuppressive properties of rapamy-cin have been attributed to the blocking of TORC1 that is requiredfor transducing T cell activation initiated by cytokines (40).TORC2 is required for organization of the actin cytoskeleton andit is rapamycin-insensitive. Protein levels of mTOR, raptor, rictor
FIGURE 2. Activation of the mTOR in lupus T cells. A, Western blotanalysis of mTOR substrate p70 S6K phosphorylation. Protein lysatesof negatively isolated naive T cells from healthy controls and SLEpatients treated without (SLE) or with 2 mg/day rapamycin (SLE �Rapa) were analyzed by Western blot using Abs against p70 S6K,pS6K, and actin. Under blots, data represent mean � SE; values of p �0.05 vs controls or SLE patients are shown in parentheses. B, Westernblot analysis of mTOR substrate 4E-BP1 phosphorylation. Phospho-4E-BP1 and 4E-BP1 were detected rabbit Abs. C, Effect of rapamycin onSOD2 expression using the samples analyzed in A. D, Effect of rapa-mycin on FKBP12 expression using the samples analyzed in A. E, Effectof rapamycin on NOSIP expression using the samples analyzed in A.
FIGURE 3. A, Western blot detection of Rab GTPases Rab5A andHRES-1/Rab4 in negatively isolated T cells of female SLE and controlsubjects. The expression of HRES-1/Rab4 or CD4 relative to actin wasdetermined in each donor by automated densitometry. The p valuesreflect fold changes in protein levels between 14 SLE patients and tencontrols compared with two-tailed t test. B, Western blot detection ofHRES-1/Rab4 and CD4 in negatively isolated CD4� T cells of femaleCaucasian SLE patients and age-matched female Caucasian controls.
2066 mTOR CONTROLS TCR� EXPRESSION THROUGH HRES-1/Rab4
(data not shown), S6K, and 4E-BP1 were similar in lupus andcontrol T cells (Fig. 2, A and B). However, phosphorylation levelsof mTOR substrates S6K1 and 4E-BP1 were increased 2.5-fold( p � 0.023) and 2.7-fold in lupus T cells ( p � 0.014), respec-tively, which were reversed in rapamycin-treated lupus patients(Fig. 2, A and B). These findings suggest that mTOR kinase ac-tivity is increased in lupus T cells, and it is reversed by rapamycintreatment.
In accordance with the persistence of increased mitochondrialmass and MHP in rapamycin-treated patients (18), elevated ex-pression of SOD2 (Fig. 2C), TAL (data not shown), and FKBP12(Fig. 2D) and diminished expression of NOSIP were unaffected byrapamycin treatment (Fig. 2E).
Activation of endocytic recycling pathway is characterized byoverexpression of Rab5A and HRES-1/Rab4
Microarray analysis of gene expression also showed increasedRab5A levels in negatively isolated lupus T cells (Fig. S3. AndTable S2). The overexpression of Rab5A was confirmed by West-ern blot studies of lupus T cells (Fig. 3A). Rab5A regulates inter-nalization of surface receptors from the plasma membrane to earlyendosomes (22). In contrast, Rab4 is found on endocytic vesiclesthat recycle cargo from the early endosomes (41). The predomi-nant isoform of Rab4 in T cells HRES-1/Rab4 (24) is overex-pressed in lupus T cells (Fig. 3A). With respect to �-actin internalcontrol, HRES-1/Rab4 expression was increased 2.14-fold in T
cells of 26 lupus patients relative to 23 healthy controls ( p �0.0011) Since genetically enforced overexpression of HRES1/Rab4 down-regulated CD4 in normal T cells (24), we investigatedwhether CD4 levels were altered in lupus T cells. CD4 proteinlevels were reduced in negatively isolated untouched lupus T cellsby 46% ( p � 0.045; data not shown). The reduction in CD4 pro-tein levels was not due to diminished numbers of CD4� T cellssince the CD4/CD8 ratio within the CD3� T cell compartment wassimilar between control (3.50; n � 10) and lupus donors (3.45; n �21). Nevertheless, to eliminate the influence of individual varia-tions in CD4/CD8 T cell ration, we also used negatively isolatedCD4 T cells. As shown in Fig. 3B, HRES-1/Rab4 protein levelswere elevated (3.7-fold; p � 0.03), and CD4 protein levels werealso reduced in negatively isolated CD4� T cells (by 45%; p �0.003) from 26 Caucasian female lupus patients relative to 10healthy Caucasian female controls. Protein levels of Rab7, whichregulates transport between early and late endocytic organelles(20), were similar in T cells of control and SLE donors (data notshown).
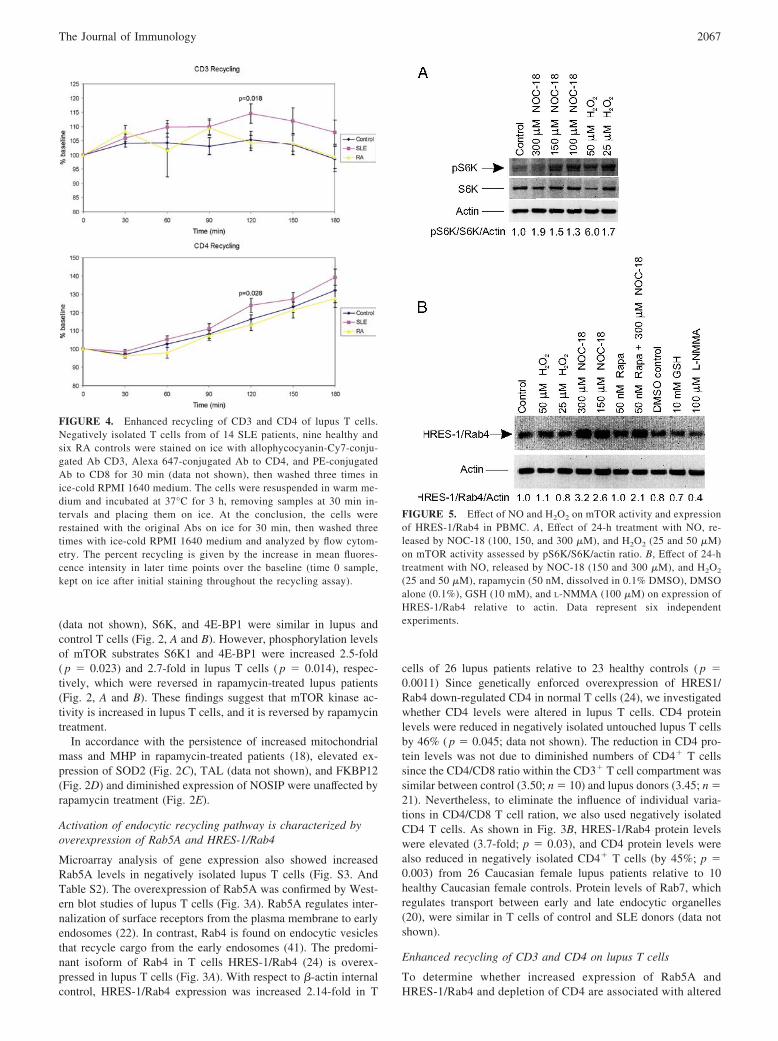
Enhanced recycling of CD3 and CD4 on lupus T cells
To determine whether increased expression of Rab5A andHRES-1/Rab4 and depletion of CD4 are associated with altered
FIGURE 4. Enhanced recycling of CD3 and CD4 of lupus T cells.Negatively isolated T cells from of 14 SLE patients, nine healthy andsix RA controls were stained on ice with allophycocyanin-Cy7-conju-gated Ab CD3, Alexa 647-conjugated Ab to CD4, and PE-conjugatedAb to CD8 for 30 min (data not shown), then washed three times inice-cold RPMI 1640 medium. The cells were resuspended in warm me-dium and incubated at 37°C for 3 h, removing samples at 30 min in-tervals and placing them on ice. At the conclusion, the cells wererestained with the original Abs on ice for 30 min, then washed threetimes with ice-cold RPMI 1640 medium and analyzed by flow cytom-etry. The percent recycling is given by the increase in mean fluores-cence intensity in later time points over the baseline (time 0 sample,kept on ice after initial staining throughout the recycling assay).
FIGURE 5. Effect of NO and H2O2 on mTOR activity and expressionof HRES-1/Rab4 in PBMC. A, Effect of 24-h treatment with NO, re-leased by NOC-18 (100, 150, and 300 �M), and H2O2 (25 and 50 �M)on mTOR activity assessed by pS6K/S6K/actin ratio. B, Effect of 24-htreatment with NO, released by NOC-18 (150 and 300 �M), and H2O2
(25 and 50 �M), rapamycin (50 nM, dissolved in 0.1% DMSO), DMSOalone (0.1%), GSH (10 mM), and L-NMMA (100 �M) on expression ofHRES-1/Rab4 relative to actin. Data represent six independentexperiments.
2067The Journal of Immunology

receptor traffic, constitutive recycling of surface receptors wasinvestigated in negatively isolated T cells from 14 SLE patients,9 healthy and 6 RA controls. The recycling assay was per-formed after staining the cells with Abs against CD3�, CD4,and CD8, then washing the cells and allowing the constitutiveinternalization and recycling of receptors to take place at 37°C.The cells were then restained, and the mean fluorescence inten-sity of the cells, which was assumed to be proportional to thespeed at which internal molecules are brought to the cell sur-face, was determined by flow cytometry. As shown in Fig. 4,recycling of CD3 and CD4 was enhanced on lupus T cells rel-ative to T cells from healthy and RA controls. Surface expres-sion and recycling rates of CD8 were similar in lupus and con-trol T cells (data not shown).
Expression of HRES-1/Rab4 is NO-inducible andrapamycin-sensitive
MHP and increased mitochondrial biogenesis require exposure ofT cells to NO (11). In accordance with previous findings (42),NOC-18 (300 �M), which is capable of slowly releasing NO, andH2O2 (50 �M) stimulated mTOR activity 5.4-fold (n � 7; p �0.005) and 4.5-fold, respectively (n � 7; p � 0.002; representativeWestern blots are shown in Fig. 5). To determine whether in-creased expression of HRES-1/Rab4 is induced by NO or the re-sultant oxidative stress, PBMC were exposed to NOC-18 andH2O2. As shown in Fig. 5, expression of HRES-1/Rab4 was in-duced by 300 �M NOC-18 (n � 6; p � 0.0003) and 50 �M H2O2
(n � 7; p � 0.019). Inhibition of NO synthase by L-NMMA re-duced expression of HRES-1/Rab4 (Fig. 5). In vitro treatment withrapamycin diminished the NO-dependent activation of HRES-1/Rab4 � 55% (n � 7, p � 0.024; Fig. 5).
We previously found that CD3/CD28 elicited NO production inhuman peripheral blood T cells (29). Therefore, CD3/CD28 co-stimulation can be predicted to increase HRES-1/Rab4 protein lev-
els. As NO production is maximal 24 h after T cell stimulation, weobserved a significant (3.3-fold; n � 4, p � 0.03) up-regulation ofHRES-1/Rab4 protein levels relative to �-actin in normal donors48 h after CD3/C28 costimulation. HRES-1/Rab4 protein levelsreturned to baseline 144 h after CD3/C28 costimulation. The spec-ificity of HRES-1/Rab4 stimulation was evidenced by siRNA-me-diated knockdown at 48 and 72 h (Fig. S4). We did not observesignificant knockdown of HRES-1/Rab4 less than 36 h after trans-fection of siRNA and experienced a rebound of HRES-1/Rab4expression 144 h after siRNA treatment, attributable to degrada-tion of siRNA (43).
Rapamycin treatment in vivo reduces the expression of HRES-1/Rab4 and Rab5A and reverses the loss of CD4, Lck, and TCR�in lupus T cells
mTOR was shown to modulate the recycling of GLUT4 (44, 45)and the TFR (28), which traffic through Rab4-positive endo-somes in adipocytes (46) and epithelial cells (41), respectively.However, an interaction of mTOR with Rab4 has not been dem-onstrated previously. In our study, we show that overexpressionof Rab5A (Fig. 6A) and HRES-1/Rab4 was reduced in T cells ofrapamycin-treated lupus patients (Fig. 6B). Since HRES-1/Rab4has a dominant impact on expression of CD4 via controlling itsrecycling and targets CD4 for lysosomal degradation, we alsoexamined CD4 expression in control donors as well as in SLE
FIGURE 6. Expression of Rab5A, HRES-1/Rab4, and CD4 relative toactin in healthy controls and SLE patients treated without (SLE) or with 2mg/day rapamycin (SLE � Rapa). Protein lysates of negatively isolatednaive T cells were analyzed by Western blot. Values of p � 0.05 reflectingfold changes in protein levels are shown.
FIGURE 7. Expression of TCR/CD3�, Lck, Fc�RI�, and Syk in nega-tively isolated naive T cells from healthy controls and SLE patients treatedwithout (SLE) or with 2 mg/day rapamycin (SLE � Rapa). Protein lysateswere analyzed by Western blot. Values of p � 0.05 reflecting fold changesin protein levels are shown.
2068 mTOR CONTROLS TCR� EXPRESSION THROUGH HRES-1/Rab4
patients treated without or with rapamycin. As shown in Fig.6C, CD4 protein levels were reduced in SLE patients, and thesechanges were reversed in T cells from rapamycin-treatedpatients.
The CD4 coreceptor plays essential roles in formation of theimmunological synapse, which is altered in lupus T cells (47, 48).TCR/CD3� levels are depleted in lupus T cells, which leads to acompensatory enhancement of Fc�RI� and Syk expression andplays a critical role in enhanced CD3-induced calcium fluxing(12). Lck levels are also reduced in lupus T cells (47, 49). Lck isconstitutively associated with the CD4 (25, 26, 50), and it isbrought to the immunological synapse via binding to CD4 (51). Asshown previously, HRES-1/Rab4 has a dominant influence on ex-pression of CD4 and, to a lesser extent, on TFR by regulation oftheir recycling from endosomes in Jurkat T cells and primary pe-ripheral blood T cells (24). Expression of CD4 was reduced inlupus T cells and reversed in rapamycin-treated patients (Fig. 6C).Since rapamycin treatment also corrected the enhanced calciumfluxing in lupus T cells (18), we investigated whether this wasmediated through restoration of Lck and CD3�-chain expression.In accordance with previous findings (12, 14), we detected pro-
foundly diminished Lck and TCR� levels in lupus T cells (Fig. 7).Depletion of both Lck and TCR� protein levels was reversed in Tcells of rapamycin-treated lupus patients (Fig. 7). While confirm-ing elevated Fc�RI� and Syk protein levels in lupus T cells, wealso demonstrated their partial reversal in rapamycin-treated pa-tients (Fig. 7).
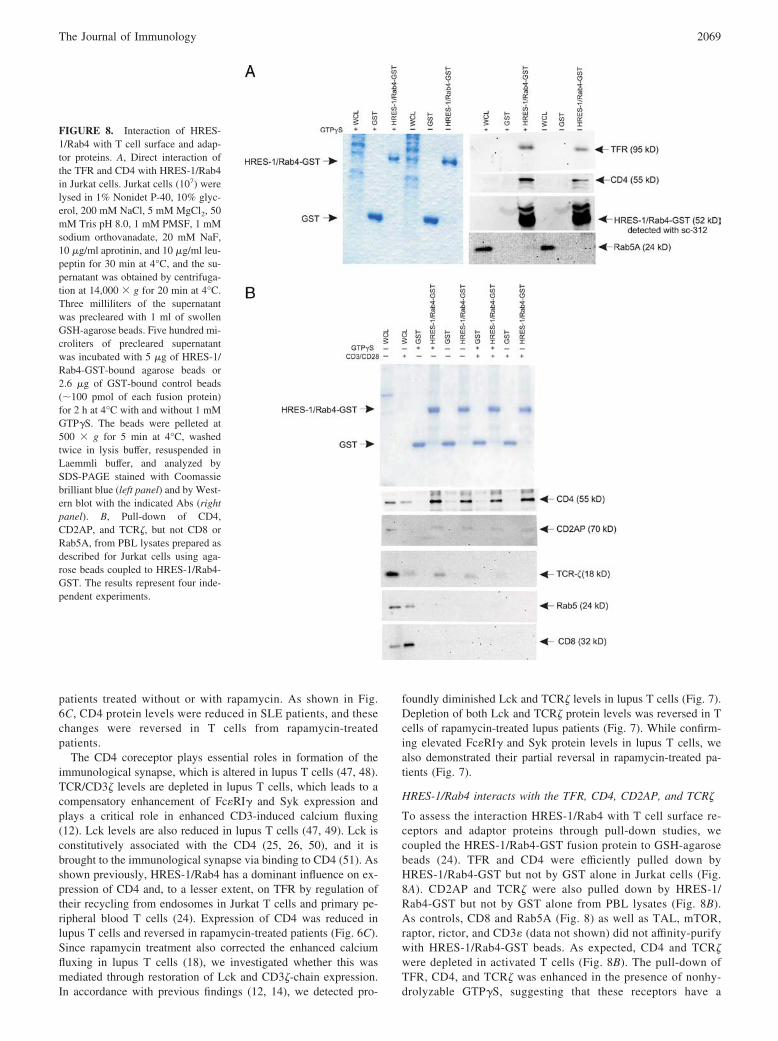
HRES-1/Rab4 interacts with the TFR, CD4, CD2AP, and TCR�
To assess the interaction HRES-1/Rab4 with T cell surface re-ceptors and adaptor proteins through pull-down studies, wecoupled the HRES-1/Rab4-GST fusion protein to GSH-agarosebeads (24). TFR and CD4 were efficiently pulled down byHRES-1/Rab4-GST but not by GST alone in Jurkat cells (Fig.8A). CD2AP and TCR� were also pulled down by HRES-1/Rab4-GST but not by GST alone from PBL lysates (Fig. 8B).As controls, CD8 and Rab5A (Fig. 8) as well as TAL, mTOR,raptor, rictor, and CD3� (data not shown) did not affinity-purifywith HRES-1/Rab4-GST beads. As expected, CD4 and TCR�were depleted in activated T cells (Fig. 8B). The pull-down ofTFR, CD4, and TCR� was enhanced in the presence of nonhy-drolyzable GTP�S, suggesting that these receptors have a
FIGURE 8. Interaction of HRES-1/Rab4 with T cell surface and adap-tor proteins. A, Direct interaction ofthe TFR and CD4 with HRES-1/Rab4in Jurkat cells. Jurkat cells (107) werelysed in 1% Nonidet P-40, 10% glyc-erol, 200 mM NaCl, 5 mM MgCl2, 50mM Tris pH 8.0, 1 mM PMSF, 1 mMsodium orthovanadate, 20 mM NaF,10 �g/ml aprotinin, and 10 �g/ml leu-peptin for 30 min at 4°C, and the su-pernatant was obtained by centrifuga-tion at 14,000 � g for 20 min at 4°C.Three milliliters of the supernatantwas precleared with 1 ml of swollenGSH-agarose beads. Five hundred mi-croliters of precleared supernatantwas incubated with 5 �g of HRES-1/Rab4-GST-bound agarose beads or2.6 �g of GST-bound control beads(�100 pmol of each fusion protein)for 2 h at 4°C with and without 1 mMGTP�S. The beads were pelleted at500 � g for 5 min at 4°C, washedtwice in lysis buffer, resuspended inLaemmli buffer, and analyzed bySDS-PAGE stained with Coomassiebrilliant blue (left panel) and by West-ern blot with the indicated Abs (rightpanel). B, Pull-down of CD4,CD2AP, and TCR�, but not CD8 orRab5A, from PBL lysates prepared asdescribed for Jurkat cells using aga-rose beads coupled to HRES-1/Rab4-GST. The results represent four inde-pendent experiments.
2069The Journal of Immunology

higher affinity for the membrane-associated GTP-bound form(41, 52).
Colocalization of TCR� with HRES-1/Rab4 and lysosomes inlupus T cells
Confocal microscopy of negatively isolated resting T cells fromhealthy controls showed that TCR� and HRES-1/Rab4 werepresent in the cell membrane and intracellular vesicles but not inacidified lysosomes stained with Lysotracker Red (LTR) (Fig. 9).HRES-1/Rab4 was partially associated with lysosomes in normalT cells. The depletion of TCR� in lupus T cells was associated withthe redistribution of the residual of TCR� into HRES-1/Rab4-pos-itive intracellular compartments that appeared to colocalize withlysosomes (Fig. 9). We also observed a partial association ofmTOR with Rab7 and Rab7-HRES-1/Rab4 double-positive endo-somes both in control and lupus T cells (Fig. S5).
Depletion of TCR� in lupus T cells is reversed by HRES-1/Rab4knockdown and inhibition of lysosomal function
Overexpression of HRES-1/Rab4 causes lysosomal degradation ofCD4 (24). Following stimulation by Ag or CD3, the TCR� wasalso shown to be degraded in the lysosome (53, 54). To investigatewhether HRES-1/Rab4 overexpression contributes to depletion ofTCR� in lupus T cells, we reduced HRES-1/Rab4 protein levels bytransfection of siRNA. Knockdown of HRES-1/Rab4 augmentedTCR� protein levels in lupus T cells (Fig. 10A). TCR� levels weresignificantly increased (1.54 � 0.17-fold; p � 0.013) by achieving50% siRNA-mediated knockdown of HRES1/Rab4 36h aftertransfection in five of five SLE patients with baseline HRES1/Rab4 levels elevated 2.1-fold over the mean of10 healthy con-trols. Inhibition of lysosomal function with bafilomycin A1 or fo-limycin also significantly increased TCR� levels in lupus T cells(Fig. 10B).
DiscussionThis study documents increased mTOR activity in lupus T cellsthat contributes to the loss of TCR� through HRES-1/Rab4-regu-lated lysosomal degradation. Upstream of mTOR activation, weidentified NO, which is also a key trigger of MHP and increasedmitochondrial biogenesis in lupus T cells (29). Activation ofmTOR is consistent with its role as a sensor of the ��m (15).Downstream of mTOR activation, we identified increased expres-sion of the small GTPase HRES-1/Rab4, which promotes lysoso-mal degradation of CD4 and TCR� in lupus T cells. Pull-down andconfocal microscopy studies revealed a direct interaction ofHRES-1/Rab4 with TCR�. Depletion of TCR� in lupus T cells wasreversed by HRES-1/Rab4 knockdown as well as by inhibition oflysosomal function in vitro, indicating that activation of mTORcauses the loss of TCR� through HRES-1/Rab4-dependent lyso-somal degradation. Depletion of Lck and TCR� was associatedwith augmented Syk and Fc�RI� levels in lupus T cells (12), all ofwhich were reversed in SLE patients treated with rapamycin invivo. Thus, HRES-1/Rab4-dependent lysosomal degradation ofTCR� is identified as 1) a novel mechanism controlling T cellsignal transduction and 2) a target for pharmacological interven-tion in SLE.
Microarray analysis of gene expression in whole blood orPBMC has been used to identify a variety of genetic signaturesin SLE (55). However, the previous studies had failed to pro-vide subsets of genes linked to mitochondrial dysfunction of Tcells. We chose to perform microarrays on negatively isolateduntouched T cells, providing a distinct data set from those ear-lier studies, without elimination of previously described signa-tures. We verified the existence of the well-described IFN sig-nature of SLE in this subset of cells (37, 38, 56). Whileexposure of PBMC to IFN-� markedly enhanced the expressionof MX1, IFIT1, IFI44, and PRKR, as earlier described (36),IFN-� failed to influence the expression of HRES-1/Rab4 oractivity of mTOR, assessed by S6K phosphorylation (data notshown). Additionally, we identified two novel transcriptionalprograms termed mitochondrial and endocytic recycling signa-tures. The functional relevance of these gene expression signa-tures was validated on the protein level by Western blot anal-ysis and functional studies. Increased expression of themitochondrial proteins VDAC1 and SOD2 is consistent withincreased mitochondrial mass and oxidative stress in lupus Tcells (11). Overexpression of TAL enhanced MHP in Jurkatcells (57) and may also act upstream of MHP of lupus T cells.Reduced expression of NOSIP, which inhibits activity of eNOSand may act upstream of elevated NO production in lupus T
FIGURE 9. Localization of TCR�, HRES-1/Rab4, and lysosomes innegatively isolated resting T cells from healthy controls and lupus patientsusing confocal microscopy. TCR� was visualized with Ab sc-1239 directlyconjugated to Alexa 488 (green). HRES-1/Rab4 was detected with Absc-312 directly conjugated with Alexa 647 (blue). Lysosomes were de-tected with LTR. In control T cells, TCR� and HRES-1/Rab4 were presentin the cell membrane or intracellular vesicles but not in lysosomes stainedwith LTR. In lupus T cells, TCR� was found predominantly in HRES-1/Rab4-positive intracellular membrane clusters that colocalized with lyso-somes. Green color intensities (lower right eight panels) were enhanced tovisualize the localization and compensate for lower expression of TCR� inlupus T cells.
FIGURE 10. Reversal of TCR/CD3� depletion in lupus T cells bydown-regulation of HRES-1/Rab4 expression and inhibitors of lysosomalfunction. A, Effect of siRNA-mediated knock-down of HRES-1/Rab4 onTCR� protein levels in lupus T cells. Cells (107) were electroporated withsiRNA specifc for HRES1/Rab4 nt 377–399 and assayed 36 h later byWestern blot. B, Effect of lysosomal inhibitors bafilomycin A1 and foli-mycin on TCR� levels of lupus T cells. The results represent studies of fivelupus patients.
2070 mTOR CONTROLS TCR� EXPRESSION THROUGH HRES-1/Rab4

cells, was not affected by rapamycin treatment. Thus, TAL andNOSIP control metabolic pathways that regulate ��m and maybe regarded as markers of the mitochondrial signature in lupusT cells.
Overexpression of Rab5A and Rab4, which control internaliza-tion and recycling, respectively, of surface receptors via early en-dosomes (22, 41), were considered markers of an activated recy-cling gene expression signature. The predominant isoform of Rab4in T cells, HRES-1/Rab4, is overexpressed in patients with SLE. Inaccordance with a dominant impact of HRES-1/Rab4 on the en-docytic recycling of CD4 (24), there was an inverse correlationbetween enhanced HRES-1/Rab4 expression and diminished CD4expression in negatively isolated CD4 T cells (R � 0.45, p �0.041). Of note, overexpression of HRES-1/Rab4 was also in-versely correlated with TCR� protein levels (R � 0.66, p �0.0009). These changes in gene expression were associated withenhanced constitutive recycling of CD3� and CD4 in lupus T cellsrelative to healthy and RA disease controls.
We identified the activation of mTOR as a critical checkpointbetween the enhanced mitochondrial and receptor recycling geneexpression signatures. Such gatekeeper function of mTOR is con-sistent with its role in 1) sensing mitochondrial dysfunction andchanges of the ��m in T cells (15) and 2) modulating the traffic ofGLUT4 (44, 45) and TFR (28), which are associated with Rab4-positive endosomes in adipocytes (46) and epithelial cells (41),respectively. Activation of mTOR in lupus T cells was inducibleby NO, a key trigger of MHP and mitochondrial biogenesis (29).In turn, NO-induced stimulation of HRES-1/Rab4 expression wasreduced by rapamycin. Thus, NO-dependent MHP lies upstreamwhile enhanced expression of HRES-1/Rab4 lies downstream ofmTOR activation in lupus T cells.
We found that in vivo rapamycin treatment did not influence NOproduction, which was found to be increased in lupus T cells (Fig.S1). A lack of influence on NO production is consistent with per-sistent MHP and persistently increased mitochondrial biogenesisin T cells of rapamycin-treated lupus patients (18). Rapamycintreatment in vivo reduced the expression of HRES-1/Rab4 andRab5A and reversed the loss of CD4, Lck, and TCR� and theoverexpression of Fc�RI� and Syk in lupus T cells. GST pull-down studies revealed a direct interaction of HRES-1/Rab4 withTFR, CD4, CD2AP, and TCR�. As controls, CD8, Rab5A, TAL,mTOR, raptor, rictor, and CD3� did not affinity-purify withHRES-1/Rab4-GST beads, suggesting that this small GTPase isspecifically associated with recycling endosomes carrying a lim-ited subset of TCR-associated surface proteins, such as CD4 andTFR, the intracellular adaptor CD2AP and the critical signal trans-ducer TCR�. Previous confocal microscopy studies revealed thecolocalization of HRES-1/Rab4 with the TFR and CD4 (24). Inthis study, we demonstrated that TCR� and HRES-1/Rab4 werepresent in the cell membrane or intracellular vesicles but not inlysosomes of negatively isolated resting T cells from healthy con-trols. HRES-1/Rab4 partially colocalized with lysosomes in nor-mal T cells. By contrast, the depleted TCR� in lupus T cells wasredistributed into HRES-1/Rab4-positive intracellular compart-ments that appeared to colocalize with lysosomes. The knockdownof HRES-1/Rab4 and inhibition of lysosomal function increasedTCR� levels in lupus T cells. These observations identified HRES-1/Rab4-dependent lysosomal degradation as a novel mechanismcontributing to the critical loss of TCR� in lupus T cells (10).
Although the lysosomal degradation of the TCR� chain follow-ing stimulation by Ag or CD3 has been proposed by pharmaco-logical inhibitor studies (53, 54), the mechanism leading to suchloss of TCR� has not been previously determined. The present datasuggest that NO, which is produced during T cell activation and
mediates MHP (29), also activates mTOR, enhances HRES-1/Rab4 protein levels and promotes the the lysosomal degradationof TCR�. This study reveals that lysosomal degradation ofTCR� also occurs in untouched lupus T cells due to increasedproduction of NO, activation of mTOR and augmented expres-sion of HRES-1/Rab4.
Development of lupus is determined by an interplay of largenumber of genetic factors (58). Of note, both the eNOS inhibitorNOSIP and HRES-1/Rab4 are located in lupus susceptibility locimapped to 19q13.33 (59–61) and 1q42 (59, 62–65), respectively.We earlier mapped HRES-1 to 1q42 (66) and associated polymor-phisms of the HRES-1 long terminal repeat region with the devel-opment of SLE (67, 68). During this study, reproducible individualvariations were observed in the extent of HRES-1/Rab4 overex-pression among the 26 patients with SLE (mean 2.14-fold rangingfrom 0.9- to 6.2-fold) relative to 23 healthy subjects (mean set at1.0 ranging from 0.45 to 2.16). Variations in gene expression arelikely to be influenced by polymorphic haplotypes of the long ter-minal repeat (69), which harbors an enhancer of HRES-1/Rab4gene transcription (24).
The mechanism by which rapamycin influences HRES-1/Rab4expression is not clear at present. HRES-1/Rab4 mRNA levelswere similar between patients treated with or without rapamycin(data not shown), arguing against an influence on gene transcrip-tion. mTOR works to enhance the translation of proteins generally,but it has been well described that some proteins are up-regulatedmore than others (70), and HRES-1/Rab4 may be one that is af-fected specifically. NO induced both mTOR activity and HRES-1/Rab4 expression, whereas rapamycin inhibited the NO-inducedexpression of HRES-1/Rab4. This suggested that mTOR activity isnecessary for stimulating the expression of HRES-1/Rab4. OurGST pull-down results excluded a direct interaction betweenmTOR, raptor or rictor and HRES-1/Rab4. Colocalization of Rab7and mTOR has recently been described in HEK293 and HeLa cells(19). While Rab4 regulates the recycling of early endosomes, Rab7controls the traffic between early and late endocytic organelles(20). Although protein levels of Rab7 were similar in T cells ofcontrol and SLE donors (data not shown), we observed a partialcolocalization of mTOR with Rab7 and Rab7-HRES-1/Rab4 dou-ble-positive endosomes both in control and lupus T cells (Fig. S5).Thus, mTOR may indirectly interact with HRES-1/Rab4 throughintermediary molecules other than raptor or rictor. In summary, theresults suggest that 1) activation of mTOR causes the loss of TCR�through HRES-1/Rab4-dependent lysosomal degradation and 2)blocking of TCR� degradation may account for the therapeuticefficacy of rapamycin in SLE.
DisclosuresThe authors have no financial conflict of interest.
References1. Nagy, G., A. Koncz, and A. Perl. 2005. T and B cell abnormalities in systemic
lupus erythematosus. Crit. Rev. Immunol. 25: 123–140.2. Perl, A., P. Gergely, Jr., G. Nagy, A. Koncz, and K. Banki. 2004. Mitochondrial
hyperpolarization: a checkpoint of T cell life, death, and autoimmunity. TrendsImmunol. 25: 360–367.
3. Emlen, W., J. A. Niebur, and R. Kadera. 1994. Accelerated in vitro apoptosis oflymphocytes from patients with systemic lupus erythematosus. J. Immunol. 152:3685–3692.
4. Casciola-Rosen, L. A., G. Anhalt, and A. Rosen. 1994. Autoantigens targeted insystemic lupus erythematosus are clustered in two populations of surface struc-tures on apoptotic keratinocytes. J. Exp. Med. 179: 1317–1330.
5. Kovacs, B., D. Vassilopoulos, S. A. Vogelgesang, and G. C. Tsokos. 1996. De-fective CD3-mediated cell death in activated T cells from patients with systemiclupus erythematosus: role of decreased intracellular TNF-�. Clin. Immunol. Im-munopathol. 81: 293–302.
6. Gergely, P. J., C. Grossman, B. Niland, F. Puskas, H. Neupane, F. Allam,K. Banki, P. E. Phillips, and A. Perl. 2002. Mitochondrial hyperpolarization and
2071The Journal of Immunology

ATP depletion in patients with systemic lupus erythematosus. Arthitis Rheum. 46:175–190.
7. Gergely, P. J., B. Niland, N. Gonchoroff, R., Jr., Pullmann, P. E. Phillips, andA. Perl. 2002. Persistent mitochondrial hyperpolarization, increased reactive ox-ygen intermediate production, and cytoplasmic alkalinization characterize alteredIL-10 signaling in patients with systemic lupus erythematosus. J. Immunol. 169:1092–1101.
8. Krishnan, S., J. G. Kiang, C. U. Fisher, M. P. Nambiar, H. T. Nguyen,V. C. Kyttaris, B. Chowdhury, V. Rus, and G. C. Tsokos. 2005. Increasedcaspase-3 expression and activity contribute to reduced CD3� expression in sys-temic lupus erythematosus T cells. J. Immunol. 175: 3417–3423.
9. Ma, L., K. W. Chan, N. J. Trendell-Smith, A. Wu, L. Tian, A. C. Lam,A. K. Chan, C. K. Lo, S. Chik, K. H. Ko, et al. 2005. Systemic autoimmunedisease induced by dendritic cells that have captured necrotic but not apoptoticcells in susceptible mouse strains. Eur. J. Immunol. 35: 3364–3375.
10. Kyttaris, V. C., and G. C. Tsokos. 2004. T lymphocytes in systemic lupus ery-thematosus: an update. Curr. Opin. Rheumatol. 16: 548–552.
11. Nagy, G., M. Barcza, N. Gonchoroff, P. E. Phillips, and A. Perl. 2004. Nitricoxide-dependent mitochondrial biogenesis generates Ca2� signaling profile oflupus T cells. J. Immunol. 173: 3676–3683.
12. Nambiar, M. P., C. U. Fisher, V. G. Warke, S. Krishnan, J. P. Mitchell,N. Delaney, and G. C. Tsokos. 2003. Reconstitution of deficient T cell receptor� chain restores T cell signaling and augments T cell receptor/CD3-induced in-terleukin-2 production in patients with systemic lupus erythematosus. ArthritisRheum. 48: 1948–1955.
13. Kyttaris, V. C., S. Krishnan, and G. C. Tsokos. 2006. Systems biology in sys-temic lupus erythematosus: integrating genes, biology and immune function. Au-toimmunity 39: 705–709.
14. Enyedy, E. J., M. P. Nambiar, S. N. Liossis, G. Dennis, G. M. Kammer, andG. C. Tsokos. 2001. Fc� receptor type I � chain replaces the deficient T cellreceptor � chain in T cells of patients with systemic lupus erythematosus. Ar-thritis Rheum. 44: 1114–1121.
15. Desai, B. N., B. R. Myers, and S. L. Schreiber. 2002. FKBP12-rapamycin-asso-ciated protein associates with mitochondria and senses osmotic stress via mito-chondrial dysfunction. Proc. Natl. Acad. Sci. USA 99: 4319–4324.
16. Sarbassov, D. D., S. M. Ali, and D. M. Sabatini. 2005. Growing roles for themTOR pathway. Curr. Opin. Cell Biol. 17: 596–603.
17. Hartford, C. M., and M. J. Ratain. 2007. Rapamycin: something old, somethingnew, sometimes borrowed and now renewed. Clin. Pharmacol. Ther. 82:381–388.
18. Fernandez, D., E. Bonilla, N. Mirza, and A. Perl. 2006. Rapamycin reducesdisease activity and normalizes T cell activation-induced calcium fluxing in pa-tients with systemic lupus erythematosus. Arthritis Rheum. 54: 2983–2988.
19. Sancak, Y., T. R. Peterson, Y. D. Shaul, R. A. Lindquist, C. C. Thoreen,L. Bar-Peled, and D. M. Sabatini. 2008. The Rag GTPases bind raptor and me-diate amino acid signaling to mTORC1. Science 320: 1496–1501.
20. Grosshans, B. L., D. Ortiz, and P. Novick. 2006. Rabs and their effectors: achiev-ing specificity in membrane traffic. Proc. Natl. Acad. Sci. USA 103:11821–11827.
21. Miaczynska, M., L. Pelkmans, and M. Zerial. 2004. Not just a sink: endosomesin control of signal transduction. Curr. Opin. Cell Biol. 16: 400–406.
22. Rink, J., E. Ghigo, Y. Kalaidzidis, and M. Zerial. 2005. Rab conversion as amechanism of progression from early to late endosomes. Cell 122: 735–749.
23. Touchot, N., P. Chardin, and A. Tavitian. 1987. Four additional members of theras gene superfamily isolated by an oligonucleotide strategy: molecular cloningof YPT-related cDNAs from a rat brain library. Proc. Natl. Acad. Sci. USA 84:8210–8214.
24. Nagy, G., J. Ward, D. D. Mosser, A. Koncz, P. Gergely, C. Stancato, Y. Qian,D. Fernandez, B. Niland, C. E. Grossman, et al. 2006. Regulation of CD4 ex-pression via recycling by HRES-1/RAB4 controls susceptibility to HIV infection.J. Biol. Chem. 281: 34574–34591.
25. Ehrlich, L. I. R., P. J. R. Ebert, M. F. Krummel, A. Weiss, and M. M. Davis.2002. Dynamics of p56lck translocation to the T cell immunological synapsefollowing agonist and antagonist stimulation. Immunity 17: 809–822.
26. Luton, F., V. Legendre, J. P. Gorvel, A. M. Schmitt-Verhulst, and C. Boyer.1997. Tyrosine and serine protein kinase activities associated with ligand- in-duced internalized TCR/CD3 complexes. J. Immunol. 158: 3140–3147.
27. Salmeron, A., A. Borroto, M. Fresno, M. J. Crumpton, S. C. Ley, and B. Alarcon.1995. Transferrin receptor induces tyrosine phosphorylation in T cells and isphysically associated with the TCR �-chain. J. Immunol. 154: 1675–1683.
28. Edinger, A. L., and C. B. Thompson. 2002. Akt maintains cell size and survivalby increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell 13: 2276–2288.
29. Nagy, G., A. Koncz, and A. Perl. 2003. T cell activation-induced mitochondrialhyperpolarization is mediated by Ca2�- and redox-dependent production of nitricoxide. J. Immunol. 171: 5188–5197.
30. Tan, E. M., A. S. Cohen, J. F. Fries, A. T. Masi, D. J. McShane, N. F. Rothfield,J. G. Schaller, N. Talal, and R. J. Winchester. 1982. The 1982 revised criteria forthe classification of systemic lupus erythematosus. Arthritis Rheum. 25:1271–1277.
31. Bombardier, C., D. D. Gladman, M. B. Urowitz, D. Caron, C. H. Chang, and theCommittee on Prognosis Studies in SLE. 1992. Derivation of the SLEDAI: adisease activity index for lupus patients. Arthritis Rheum. 35: 630–640.
32. Arnett, F. C., S. M. Edworthy, D. A. Bloch, D. J. McShane, J. F. Fries,N. S. Cooper, L. A. Healey, S. R. Kaplan, M. H. Liang, and H. S. Luthra. 1988.The American Rheumatism Association 1987 revised criteria for the classifica-tion of rheumatoid arthritis. Arthritis Rheum. 31: 315–324.
33. Perl, A., R. Gonzalez-Cabello, I. Lang, and P. Gergely. 1984. Effector activity ofOKT4� and OKT8� T cell subsets in lectin- dependent cell-mediated cytotox-icity against adherent HEp-2 cells. Cell. Immunol. 84: 185–193.
34. Perl, A., Y. Qian, K. R. Chohan, C. R. Shirley, W. Amidon, S. Banerjee,F. A. Middleton, K. L. Conkrite, M. Barcza, N. Gonchoroff, et al. 2006. Transal-dolase is essential for maintenance of the mitochondrial transmembrane potentialand fertility of spermatozoa. Proc. Natl. Acad. Sci. USA 103: 14813–14818.
35. Qian, Y., S. Banerjee, C. E. Grossman, W. Amidon, G. Nagy, M. Barcza,B. Niland, D. R. Karp, F. A. Middleton, K. Banki, and A. Perl. 2008. Transal-dolase deficiency influences the pentose phosphate pathway, mitochondrial ho-moeostasis and apoptosis signal processing. Biochem. J. 415: 123–134.
36. Hua, J., K. Kirou, C. Lee, and M. K. Crow. 2006. Functional assay of type Iinterferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 54: 1906–1916.
37. Bennett, L., A. K. Palucka, E. Arce, V. Cantrell, J. Borvak, J. Banchereau, andV. Pascual. 2003. Interferon and granulopoiesis signatures in systemic lupus er-ythematosus blood. J. Exp. Med. 197: 711–723.
38. Baechler, E. C., F. M. Batliwalla, G. Karypis, P. M. Gaffney, W. A. Ortmann,K. J. Espe, K. B. Shark, W. J. Grande, K. M. Hughes, V. Kapur, et al. 2003.Interferon-inducible gene expression signature in peripheral blood cells of pa-tients with severe lupus. Proc. Natl. Acad. Sci. USA 100: 2610–2615.
39. Kirou, K. A., C. Lee, S. George, K. Louca, I. G. Papagiannis, M. G. Peterson,N. Ly, R. N. Woodward, K. E. Fry, A. Y. Lau, et al. 2004. Coordinate overex-pression of interferon �-induced genes in systemic lupus erythematosus. ArthritisRheum. 50: 3958–3967.
40. Kirken, R. A., and Y. L. Wang. 2003. Molecular actions of sirolimus: sirolimusand mTor. Transplant. Proc. 35: 227S–230S.
41. van der Sluis, P., M. Hull, P. Webster, P. Male, B. Goud, and I. Mellman. 1992.The small GTP-binding protein rab4 controls an early sorting event on the en-docytic pathway. Cell 70: 729–740.
42. Sarbassov, D. D., and D. M. Sabatini. 2005. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J. Biol. Chem. 280: 39505–39509.
43. Hong, J., Z. Qian, S. Shen, T. Min, C. Tan, J. Xu, Y. Zhao, and W. Huang. 2005.High doses of siRNAs induce eri-1 and adar-1 gene expression and reduce theefficiency of RNA interference in the mouse. Biochem. J. 390: 675–679.
44. Saito, T., C. C. Jones, S. Huang, M. P. Czech, and P. F. Pilch. 2007. The inter-action of Akt with APPL1 is required for insulin-stimulated Glut4 translocation.J. Biol. Chem. 282: 32280–32287.
45. Jiang, X., H. Kenerson, L. Aicher, R. Miyaoka, J. Eary, J. Bissler, andR. S. Yeung. 2008. The tuberous sclerosis complex regulates trafficking of glu-cose transporters and glucose uptake. Am. J. Pathol. 172: 1748–1756.
46. Meusser, B., and T. Sommer. 2004. Vpu-mediated degradation of CD4 reconsti-tuted in yeast reveals mechanistic differences to cellular ER-associated proteindegradation. Mol. Cell 14: 247–258.
47. Krishnan, S., M. P. Nambiar, V. G. Warke, C. U. Fisher, J. Mitchell, N. Delaney,and G. C. Tsokos. 2004. Alterations in lipid raft composition and dynamics con-tribute to abnormal T cell responses in systemic lupus erythematosus. J. Immunol.172: 7821–7831.
48. Jury, E. C., P. S. Kabouridis, F. Flores-Borja, R. A. Mageed, and D. A. Isenberg.2004. Altered lipid raft-associated signaling and ganglioside expression in T lym-phocytes from patients with systemic lupus erythematosus. J. Clin. Invest. 113:1176–1187.
49. Jury, E. C., P. S. Kabouridis, A. Abba, R. A. Mageed, and D. A. Isenberg. 2003.Increased ubiquitination and reduced expression of LCK in T lymphocytes frompatients with systemic lupus erythematosus. Arthritis Rheum. 48: 1343–1354.
50. Thomas, S., R. Kumar, A. Preda-Pais, S. Casares, and T. D. Brumeanu. 2003. Amodel for antigen-specific T-cell anergy: displacement of CD4–p56lck signalo-some from the lipid rafts by a soluble, dimeric peptide-MHC class II chimera.J. Immunol. 170: 5981–5992.
51. Huang, Y., and R. L. Wange. 2004. T cell receptor signaling: beyond complexcomplexes. J. Biol. Chem. 279: 28827–28830.
52. Chamberlain, M. D., T. R. Berry, M. C. Pastor, and D. H. Anderson. 2004. Thep85� subunit of phosphatidylinositol 3-kinase binds to and stimulates theGTPase activity of Rab proteins. J. Biol. Chem. 279: 48607–48614.
53. Valitutti, S., S. Muller, M. Salio, and A. Lanzavecchia. 1997. Degradation of Tcell receptor (TCR)-CD3-� complexes after antigenic stimulation. J. Exp. Med.185: 1859–1864.
54. Naramura, M., I. K. Jang, H. Kole, F. Huang, D. Haines, and H. Gu. 2002. c-Cbland Cbl-b regulate T cell responsiveness by promoting ligand-induced TCRdown-modulation. Nat. Immunol. 3: 1192–1199.
55. Chaussabel, D., C. Quinn, J. Shen, P. Patel, C. Glaser, N. Baldwin, D. Stichweh,D. Blankenship, L. Li, I. Munagala, et al. 2008. A modular analysis frameworkfor blood genomics studies: application to systemic lupus erythematosus. Immu-nity 29: 150–164.
56. Kirou, K. A., C. Lee, S. George, K. Louca, M. G. Peterson, and M. K. Crow.2005. Activation of the interferon � pathway identifies a subgroup of systemiclupus erythematosus patients with distinct serologic features and active disease.Arthritis Rheum. 52: 1491–1503.
57. Banki, K., E. Hutter, N. Gonchoroff, and A. Perl. 1999. Elevation of mitochon-drial transmembrane potential and reactive oxygen intermediate levels are earlyevents and occur independently from activation of caspases in Fas signaling.J. Immunol. 162: 1466–1479.
58. Harley, J., J. Kelly, and K. Kaufman. 2006. Unraveling the genetics of systemiclupus erythematosus. Springer Semin. Immunpathol. 28: 119–130.
59. Moser, K. L., B. R. Neas, J. E. Salmon, H. Yu, C. Gray-McGuire, N. Asundi,G. R. Bruner, J. Fox, J. Kelly, S. Henshall, et al. 1998. Genome scan of human
2072 mTOR CONTROLS TCR� EXPRESSION THROUGH HRES-1/Rab4

systemic lupus erythematosus: evidence for linkage on chromosome 1q in Afri-can-American pedigrees. Proc. Natl. Acad. Sci. USA 95: 14869–14874.
60. Johansson, C. M., R. Zunec, M. A. Garcia, H. R. Scherbarth, G. A. Tate, S. Paira,S. M. Navarro, C. E. Perandones, S. Gamron, A. Alvarellos, et al. 2004. Chro-mosome 17p12–q11 harbors susceptibility loci for systemic lupus erythematosus.Hum. Genet. 115: 230–238.
61. Lindqvist, A. K., K. Steinsson, B. Johanneson, H. Kristjansdottir, A. Arnasson,G. Grondal, I. Jonasson, V. Magnusson, G. Sturfelt, L. Truedsson, et al. 2000. Asusceptibility locus for human systemic lupus erythematosus (hSLE1) on chro-mosome 2q. J. Autoimmun. 14: 169–178.
62. Tsao, B. P., R. M. Cantor, K. C. Kalunian, C.-J. Chen, H. Badsha, R. Singh,D. J. Wallace, R. C. Kitridou, S. Chen, N. Shen, et al. 1997. Evidence for linkageof a candidate chromosome 1 region to human systemic lupus erythematosus.J. Clin. Invest. 99: 725–731.
63. Gaffney, P. M., G. M. Kearns, K. B. Shark, W. A. Ortmann, S. A. Selby,M. L. Malmgren, T. C. Ockenden, R. P. Messner, R. A. King, S. S. Rich, andT. W. Behrens. 1998. A genome-wide search for susceptibility genes in humansystemic lupus erythematosus sib-pair families. Proc. Natl. Acad. Sci. USA 95:14875–14879.
64. Tsao, B. P., R. M. Cantor, J. M. Grossman, N. Shen, N. T. Teophilov,D. J. Wallace, F. C. Arnett, K. Hartung, R. Goldstein, K. C. Kalunian, et al. 1999.
PARP alleles with the linked chromosomal region are associated with systemiclupus erythematosus. J. Clin. Invest. 103: 1135–1140.
65. Shai, R., F. P. Quismorio, Jr., L. Li, O. J. Kwon, J. Morrison, D. J. Wallace,C. M. Neuwelt, C. Brautbar, W. J. Gauderman, and C. O. Jacob. 1999. Genome-wide screen for systemic lupus erythematosus susceptibility genes in multiplexfamilies. Hum. Mol. Genet. 8: 639–644.
66. Perl, A., C. M. Isaacs, R. L. Eddy, M. G. Byers, S. N. Sait, and T. B. Shows.1991. The human T cell leukemia virus-related endogenous sequence (HRES1) islocated on chromosome 1 at q42. Genomics 11: 1172–1173.
67. Magistrelli, C., K. Banki, P. Ferrante, and A. Perl. 1994. Mapping and cloning ofpolymorphic genotypes of the HRES-1 LTR. Arthritis Rheum. 37: S316.
68. Magistrelli, C., E. Samoilova, R. K. Agarwal, K. Banki, P. Ferrante, A. Vladutiu,P. E. Phillips, and A. Perl. 1999. Polymorphic genotypes of the HRES-1 humanendogenous retrovirus locus correlate with systemic lupus erythematosus andautoreactivity. Immunogenetics 49: 829–834.
69. Pullmann, R., Jr., E. Bonilla, P. E. Phillips, F. A. Middleton, and A. Perl. 2008.Haplotypes of the HRES-1 endogenous retrovirus are associated with develop-ment and disease manifestations of systemic lupus erythematosus. ArthritisRheum. 58: 532–540.
70. Ruvinsky, I., and O. Meyuhas. 2006. Ribosomal protein S6 phosphorylation:from protein synthesis to cell size. Trends Biochem. Sci. 31: 342–348.
2073The Journal of Immunology
Copyright © 2022 FDOKUMEN




















