
Actin Microfilaments Facilitate the Retrograde Transport from the Golgi Complex to the Endoplasmic...
10
Copyright C Munksgaard 2001 Traffic 2001; 2: 717–726 Munksgaard International Publishers ISSN 1398-9219 Actin Microfilaments Facilitate the Retrograde Transport from the Golgi Complex to the Endoplasmic Reticulum in Mammalian Cells Ferran Valderrama 1 , Juan M. Dura ´n 1, *, Teresa Babia ` 1, *, Holger Barth 2 , Jaime Renau-Piqueras 3 and Gustavo Egea 1, ** 1 Departament de Biologia Cel.lular i Anatomia Patolo ` gica, Facultat de Medicina, Institut d’Investigacions Biome ` diques August Pi i Sunyer (IDIBAPS), Universitat de Barcelona, E-08036 Barcelona, Spain 2 Institut für Experimentelle und Klinische Pharmakologie und Toxikologie, Albert-Ludwigs-Universität Freiburg, 79104 Freiburg, Germany 3 Centro de Investigacio ´n, Hospital La Fe, E-46009 Valencia, Spain **Corresponding author: Gustavo Egea, [email protected] The morphology and subcellular positioning of the Golgi complex depend on both microtubule and actin cytoskeletons. In contrast to microtubules, the role of actin cytoskeleton in the secretory pathway in mam- malian cells has not been clearly established. Using cytochalasin D, we have previously shown that micro- filaments are not involved in the endoplasmic retic- ulum–Golgi membrane dynamics. However, it has been reported that, unlike botulinum C2 toxin and la- trunculins, cytochalasin D does not produce net de- polymerization of actin filaments. Therefore, we have reassessed the functional role of actin microfilaments in the early steps of the biosynthetic pathway using C2 toxin and latrunculin B. The anterograde endoplasmic reticulum-to-Golgi transport monitored with the ves- icular stomatitis virus-G protein remained unaltered in cells treated with cytochalasin D, latrunculin B or C2 toxin. Conversely, the brefeldin A-induced Golgi mem- brane fusion into the endoplasmic reticulum, the Golgi- to-endoplasmic reticulum transport of a Shiga toxin mutant form, and the subcellular distribution of the KDEL receptor were all impaired when actin micro- filaments were depolymerized by latrunculin B or C2 toxin. These findings, together with the fact that COPI- coated and uncoated vesicles contain b/g-actin iso- forms, indicate that actin microfilaments are involved in the endoplasmic reticulum/Golgi interface, facilitat- ing the retrograde Golgi-to-endoplasmic reticulum membrane transport, which could be mediated by the orchestrated movement of transport intermediates along microtubule and microfilament tracks. Key words: actin, cytoskeleton, Golgi complex, retro- grade transport, secretory pathway, Shiga *These authors contributed equally to this study. 717 Received 7 June 2001, revised and accepted for publi- cation 13 July 2001 Actin cytoskeleton consists of actin and associated proteins and requires constant reorganization and regulation for its functions in such diverse cellular events as cell-shape changes, cell motility, chemotaxis, development, signal trans- duction, RNA localization, cell polarity and endocytosis (1). The high dynamics and the structural and molecular com- plexity of the actin cytoskeleton make it difficult to study its involvement in these processes. One of the most widely used approaches is to disturb its structure and organization. Sev- eral naturally derived substances interfere with actin cytoskel- eton, including cytochalasins and phalloidins derived from fungi; latrunculin, swinholide and jasplakinolide from sponges (2); and some clostridial ADP-ribosyltransferases (2,3). Al- though they all alter the organization of actin cytoskeleton, their utilization often gives rise to disparate observations. One of the clearest examples are cytochalasins. The disappear- ance of filamentous actin (F-actin) in cells treated with these agents has been attributed to the capping activity on actin filaments, which prevents their assembly (4,5). However, the treatment with cytochalasins does not lead to net depolymer- ization of actin microfilaments, since in cytochalasin D (cyD)- treated cells, the binding of heavy meromyosin chain remains unaltered, and the expected complementary change in the levels of G- and F-actin in cell extracts and in the ratio be- tween Triton-soluble and insoluble actin do not occur (6 and references therein; 7). This is not the case for Clostridium botulinum C2 toxin and latrunculins. The former is a binary toxin that ADP-ribosylates G-actin; the latter is a Red Sea sponge product that binds G-actin noncovalently. Both agents prevent actin assembly (8,9) and disrupt actin fila- ments (7,9,10). Although the involvement of actin microfilaments in mem- brane trafficking is more established in the endocytic path- way (11 and references therein), actin and several actin-as- sociated/binding proteins have also been implicated in the morphology and in the membrane transport to and from the Golgi complex (12–14,15 and references therein, 16–21). However, in both intracellular membrane routes there are dis- parate observations depending on the cell type, the technical approach and the anti-actin agents used (22–25). We recently reported that b- and g-actin isoforms are local- ized to COPI-coated and noncoated transport intermediates (18). This result suggests the involvement of actin in mem-
Transcript of Actin Microfilaments Facilitate the Retrograde Transport from the Golgi Complex to the Endoplasmic...

Copyright C Munksgaard 2001Traffic 2001; 2: 717–726
Munksgaard International Publishers ISSN 1398-9219
Actin Microfilaments Facilitate the RetrogradeTransport from the Golgi Complex to the EndoplasmicReticulum in Mammalian Cells
Ferran Valderrama1, Juan M. Duran1,*,Teresa Babia1,*, Holger Barth2,Jaime Renau-Piqueras3 and Gustavo Egea1,**
1Departament de Biologia Cel.lular i Anatomia Patologica,
Facultat de Medicina, Institut d’Investigacions
Biomediques August Pi i Sunyer (IDIBAPS), Universitat de
Barcelona, E-08036 Barcelona, Spain2Institut für Experimentelle und Klinische Pharmakologie
und Toxikologie, Albert-Ludwigs-Universität Freiburg, 79104
Freiburg, Germany3Centro de Investigacion, Hospital La Fe, E-46009
Valencia, Spain
**Corresponding author: Gustavo Egea,
The morphology and subcellular positioning of theGolgi complex depend on both microtubule and actincytoskeletons. In contrast to microtubules, the role ofactin cytoskeleton in the secretory pathway in mam-malian cells has not been clearly established. Usingcytochalasin D, we have previously shown that micro-filaments are not involved in the endoplasmic retic-ulum–Golgi membrane dynamics. However, it hasbeen reported that, unlike botulinum C2 toxin and la-trunculins, cytochalasin D does not produce net de-polymerization of actin filaments. Therefore, we havereassessed the functional role of actin microfilamentsin the early steps of the biosynthetic pathway using C2toxin and latrunculin B. The anterograde endoplasmicreticulum-to-Golgi transport monitored with the ves-icular stomatitis virus-G protein remained unaltered incells treated with cytochalasin D, latrunculin B or C2toxin. Conversely, the brefeldin A-induced Golgi mem-brane fusion into the endoplasmic reticulum, the Golgi-to-endoplasmic reticulum transport of a Shiga toxinmutant form, and the subcellular distribution of theKDEL receptor were all impaired when actin micro-filaments were depolymerized by latrunculin B or C2toxin. These findings, together with the fact that COPI-coated and uncoated vesicles contain b/g-actin iso-forms, indicate that actin microfilaments are involvedin the endoplasmic reticulum/Golgi interface, facilitat-ing the retrograde Golgi-to-endoplasmic reticulummembrane transport, which could be mediated by theorchestrated movement of transport intermediatesalong microtubule and microfilament tracks.
Key words: actin, cytoskeleton, Golgi complex, retro-grade transport, secretory pathway, Shiga
*These authors contributed equally to this study.
717
Received 7 June 2001, revised and accepted for publi-cation 13 July 2001
Actin cytoskeleton consists of actin and associated proteinsand requires constant reorganization and regulation for itsfunctions in such diverse cellular events as cell-shapechanges, cell motility, chemotaxis, development, signal trans-duction, RNA localization, cell polarity and endocytosis (1).The high dynamics and the structural and molecular com-plexity of the actin cytoskeleton make it difficult to study itsinvolvement in these processes. One of the most widely usedapproaches is to disturb its structure and organization. Sev-eral naturally derived substances interfere with actin cytoskel-eton, including cytochalasins and phalloidins derived fromfungi; latrunculin, swinholide and jasplakinolide from sponges(2); and some clostridial ADP-ribosyltransferases (2,3). Al-though they all alter the organization of actin cytoskeleton,their utilization often gives rise to disparate observations. Oneof the clearest examples are cytochalasins. The disappear-ance of filamentous actin (F-actin) in cells treated with theseagents has been attributed to the capping activity on actinfilaments, which prevents their assembly (4,5). However, thetreatment with cytochalasins does not lead to net depolymer-ization of actin microfilaments, since in cytochalasin D (cyD)-treated cells, the binding of heavy meromyosin chain remainsunaltered, and the expected complementary change in thelevels of G- and F-actin in cell extracts and in the ratio be-tween Triton-soluble and insoluble actin do not occur (6 andreferences therein; 7). This is not the case for Clostridium
botulinum C2 toxin and latrunculins. The former is a binarytoxin that ADP-ribosylates G-actin; the latter is a Red Seasponge product that binds G-actin noncovalently. Bothagents prevent actin assembly (8,9) and disrupt actin fila-ments (7,9,10).
Although the involvement of actin microfilaments in mem-brane trafficking is more established in the endocytic path-way (11 and references therein), actin and several actin-as-sociated/binding proteins have also been implicated in themorphology and in the membrane transport to and from theGolgi complex (12–14,15 and references therein, 16–21).However, in both intracellular membrane routes there are dis-parate observations depending on the cell type, the technicalapproach and the anti-actin agents used (22–25).
We recently reported that b- and g-actin isoforms are local-ized to COPI-coated and noncoated transport intermediates(18). This result suggests the involvement of actin in mem-

Valderrama et al.
brane trafficking in the early steps of the secretory pathway.Using cytochalasin D, we did not observe alterations in theendoplasmic reticulum (ER)–Golgi membrane dynamics, ex-cept for the morphology and subcellular positioning of theGolgi complex (17). We therefore re-examined the role of ac-tin microfilaments in the ER/Golgi membrane dynamicsusing botulinum C2 toxin and latrunculin B as actin-disruptingagents.
Results and Discussion
The Golgi complex is compacted in cells treated with
cytochalasin D (cyD), latrunculin B or C2 toxin, but
only the last two produce net depolymerization of
actin filaments
CyD, C2 toxin and latrunculin B (LT-B) decreased the numberof stress fibers (Figure 1D,F,H, respectively) when comparedto control NRK cells (Figure 1B). Concomitantly, the Golgicomplex showed similar compact morphology (Figure1C,E,G, respectively). When the filamentous actin (F-actin)content was measured in cell extracts from cyD-, C2 toxin-or LT-B-treated cells, only the latter two showed a significantdecrease (Figure 1I). This result is in accordance with pre-vious data showing that cyD does not induce net depolymer-ization of actin filaments (6 and references therein) orchanges in the distribution of actin in Triton X-100-soluble orinsoluble cell fractions (7). Taken together, these results showthat C2 toxin and LT-B produce much higher net depolymer-ization of actin filaments than cyD. We next re-examined thefunctional role of actin microfilaments in the ER-Golgi mem-brane dynamics using C2 toxin and LT-B.
The reassembly of the Golgi complex upon BFA with-
drawal and the ER-to-Golgi transport of the vesicular
stomatitis virus (VSV)- G are unaltered in latrunculin
B- and C2 toxin-treated cells
We reported that microfilaments are not required for theanterograde ER-to-Golgi membrane transport using cyD (17).We thus repeated the same experiments using LT-B and C2toxin. NRK cells were first treated with brefeldin A (BFA, afungal metabolite that induces relocation of Golgi enzymesto the ER), subsequently incubated with LT-B or C2 toxin,and BFA was then withdrawn from the culture medium. Therebuilding of the Golgi complex was visualized by fluor-escence microscopy. In control, C2 toxin- and LT-B-treatedcells, no differences were observed in either the kinetics ofthe reassembly of the Golgi complex (Figure 2) or the ER-to-Golgi transport of the VSV-G glycoprotein (Figure 3). Takentogether, these findings demonstrate that actin microfila-ments are not required for the rebuilding of the Golgi complexor for the anterograde ER-to-Golgi transport.
The Golgi complex disassembly induced by BFA is
delayed in latrunculin B- and C2 toxin-treated cells
We next assessed whether the disassembly of the Golgicomplex induced by BFA was altered when actin cytoskel-eton was disrupted by LT-B or C2 toxin (Figure 4). NRK cells
718 Traffic 2001; 2: 717–726
Figure1: Golgi complex is compacted in NRK cells treatedwith cytochalasin D (cyD), botulinum C2 toxin (C2 toxin) orlatrunculin-B (LT-B) but only C2 toxin and latrunculin B pro-duce net depolymerization of filamentous actin. Double con-focal immunofluorescence microscopy experiments in untreated (Aand B), and cyD- (C and D), C2 toxin- (E and F) or LT-B-treated cells(G and H) stained with anti-Man II antibodies (A, C, E and G) andTRITC-phalloidin (B, D, F and H). The F-actin content was quantifiedusing the fluorescent phalloidin-binding assay (I). The results arethe mean∫SEM of three independent experiments. Bar, 10 mm.

Actin in the ER/Golgi Interface
Figure2: The Golgi complex reassembly upon BFA withdrawal remains unaltered in NRK cells when actin microfilaments aredisrupted with latrunculin-B (LT-B) or botulinum C2 toxin (C2 toxin). Cells were treated first with BFA and then with LT-B (B, E, H)or C2 toxin (C, F, I). Notice the characteristic BFA-induced ER-like staining pattern of the Golgi-resident protein Man II (A, B, C). Next, BFAwas withdrawal from the culture medium and the kinetics of the Golgi reassembly was followed at fluorescence confocal microscopy incontrol (D, G), LT-B- (E, H) and in C2 toxin-treated cells (F, I). As expected, in LT-B and C2 toxin-treated cells, the reassembled Golgi complexshowed a compacted morphology. Bar, 10 mm.
were first incubated with cyD, LT-B or C2 toxin and thentreated with BFA for different times. In control (Figure 4A,E,I)and cyD-treated cells (Figure 4B,F,J) the kinetics of the ap-pearance of the characteristic ER-like staining pattern, con-sistent with the translocation to the ER of the Man II-stainedGolgi complex, was the same. However, in LT-B- (Figure4C,G,K) and C2 toxin-treated cells (Figure 4D,H,L), the BFA-induced disassembly of the Golgi complex was significantlydelayed. In particular, the time for which the morphology ofthe Golgi complex remained unaltered in 50% of the cells inLT-B or C2 toxin-treated cells was practically double that incyD-treated or control cells (20min vs 10min, respectively;Figure 4J). Hence, the results indicate that microfilaments areinvolved in the Golgi-to-ER membrane flux.
The subcellular distribution of the Shiga toxin frag-
ment B and the KDEL receptor is altered in cells with
depolymerized actin
We used Shiga toxin as a marker of the retrograde Golgi-to-ER pathway. Shiga toxin binds to glycolipid GB3, which is
719Traffic 2001; 2: 717–726
expressed in HeLa but not in NRK cells (26), and transportedto the ER via the early/recycling endosomes and the Golgicomplex (27). HeLa cells are also sensitive to botulinum C2toxin (28) and LT-B (7). Cells were incubated with cy3-labeledfragment B of the Shiga toxin bearing the ER-retention KDELmotif (ST-B-KDEL) (29); and its transport to the ER wasmonitored by fluorescence microscopy. Briefly, cells were in-cubated with the toxin at 4 æC, and after 45min, heated to19.5 æC to accumulate the internalized toxin in the early/recyc-ling endosomes (Figure 5A,C). Heating the cells to 37 æC syn-chronized the transport of Shiga toxin to the ER. After 2h at37 æC, in control cells, ST-B-KDEL colocalized with the Golgicomplex stained with anti-giantin antibodies (Figure 5E,F).After 4h at 37 æC, the ST-B-KDEL was seen in the ER, but stillcolocalized with giantin (Figure 5I,J), and after 6h, the ST-B-KDEL was mostly located in the ER (Figure 5M,N). In con-trast, after 4h and 6h of transport, in LT-B treated cells (notshown) and C2 toxin-treated cells (Figure 5K,L,O,P), the ST-B-KDEL labeling was mostly observed in the Golgi complex.We validated the morphological observations by quantifying
Valderrama et al.
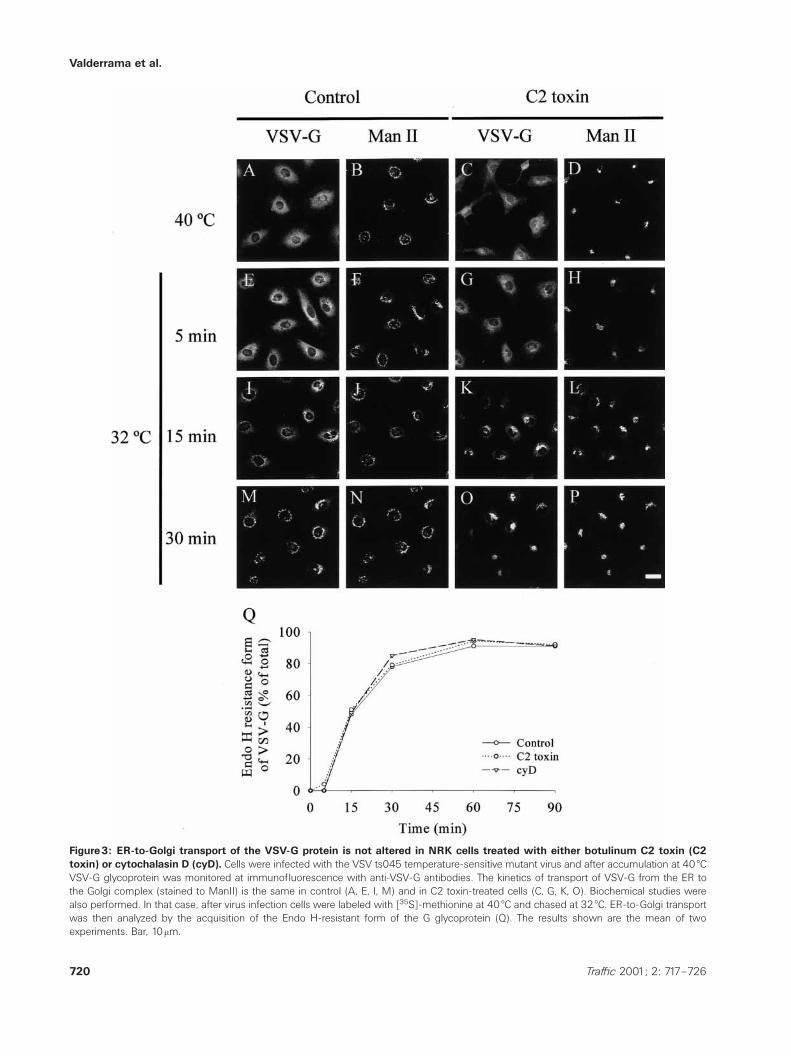
Figure3: ER-to-Golgi transport of the VSV-G protein is not altered in NRK cells treated with either botulinum C2 toxin (C2toxin) or cytochalasin D (cyD). Cells were infected with the VSV ts045 temperature-sensitive mutant virus and after accumulation at 40 æCVSV-G glycoprotein was monitored at immunofluorescence with anti-VSV-G antibodies. The kinetics of transport of VSV-G from the ER tothe Golgi complex (stained to ManII) is the same in control (A, E, I, M) and in C2 toxin-treated cells (C, G, K, O). Biochemical studies werealso performed. In that case, after virus infection cells were labeled with [35S]-methionine at 40 æC and chased at 32 æC. ER-to-Golgi transportwas then analyzed by the acquisition of the Endo H-resistant form of the G glycoprotein (Q). The results shown are the mean of twoexperiments. Bar, 10mm.
720 Traffic 2001; 2: 717–726

Actin in the ER/Golgi Interface
Figure4: Golgi complex disassembly induced by BFA is delayed in NRK cells treated with botulinum C2 toxin (C2 toxin) orlatrunculin-B (LT-B) but not with cytochalasin D (cyD). Confocal immunofluorescence microscopy experiments of the kinetics of thedisassembly of the Man II-stained Golgi complex in control (A, E, I), in cyD- (B, F, J), in LT-B (C, G, K) and in C2 toxin-treated cells (D, H, L).A quantitative analysis of these results is shown in M. The results are the mean∫SEM of three independent experiments; at least 200 cells,randomly chosen, were counted per experimental condition. Bar, 10 mm.
721Traffic 2001; 2: 717–726

Valderrama et al.
Figure5: Golgi-to-ER transport of Shiga toxin B subunit is impaired in botulinum C2 toxin (C2 toxin)-treated HeLa cells. Doubleconfocal immunofluorescence microscopy experiments of the retrograde transport kinetics of the cy3-labeled Shiga toxin fragment B contain-ing the ER-retention KDEL signal (ST-B-Glyc-KDEL/ST-B-KDEL) in untreated (A, B, E, F, I, J, M, N) and C2 toxin-treated cells (C, D, G, H, K,L, O, P). Plasma membrane internalization of the ST-B-Glyc-KDEL was carried out at 19.5 æC to accumulate it in early/recycling endosomesin the absence (A, B) or presence of C2 toxin (C, D). Thereafter, cells were heated to 37 æC (E-P) to induce the synchronic retrograde transportof the ST-B-Glyc-KDEL to the ER through the Golgi complex, which was visualized with anti-giantin antibodies (B, D, F, H, J, L, N, P). (Q)Quantification of cells in which ST-B-Glyc-KDEL was still seen in the Golgi complex. The results are the mean∫SEM of three independentexperiments; at least 200 cells, randomly chosen, were counted per experimental condition. Bar, 10 mm.
722 Traffic 2001; 2: 717–726
Actin in the ER/Golgi Interface
the percentage of cells in which ST-B-KDEL still showed aGolgi-like staining pattern (Figure 5Q). This gives informationon the emptying of the fluorescent ST-B-KDEL from the Golgias a result of its transport to the ER, where it is retained.Unlike cyD-treated cells, C2 toxin- and LT-B-treated cellsshowed a significant retention of ST-B-KDEL in the Golgicomplex (Figure 5Q). It is worth remarking that such retentionwas more efficient when actin microfilaments were disruptedby C2 toxin than by LT-B, possibly attributable to differencesin their induction of net actin depolymerization (Figure 1I).Morphological experiments using native Shiga toxin (ST-B)gave similar results (data not shown). To provide more evi-dence for the role of actin in Golgi-to-ER transport, we nextmeasured N-glycosylation in mutant Shiga toxin B-subunitcontaining an N-glycosylation site and a nonfunctional ver-sion of the KDEL motif (ST-B-Glyc-KDELGL) (29). It is import-ant to take into account that at steady state ST-B-Glyc-KDELGL and native ST-B are located in the Golgi complex,and that the percentage and kinetics of glycosylation of ST-B-Glyc-KDEL and ST-B-Glyc-KDELGL are practically thesame (29). When ST-B-Glyc-KDELGL reaches the ER, it iscore glycosylated. Hence, the measurement of N-glycosyl-ation of ST-B-Glyc-KDELGL is an indicator of the arrival ofShiga toxin at the ER. In C2 toxin-treated cells, glycosylationof ST-B-Glyc-KDELGL underwent a partial inhibition (Figure6). Hence, the morphological and biochemical results dem-onstrate that the arrival of Shiga toxin B subunit at the ER isimpaired when actin microfilaments are disrupted. Conse-quently, Shiga toxin B-subunit would remain for longer eitherin the Golgi complex or in transit between the Golgi complexand the ER. The former suggests a role of actin in the gener-ation of transport intermediates, the latter on the mechan-ism(s) of transport.
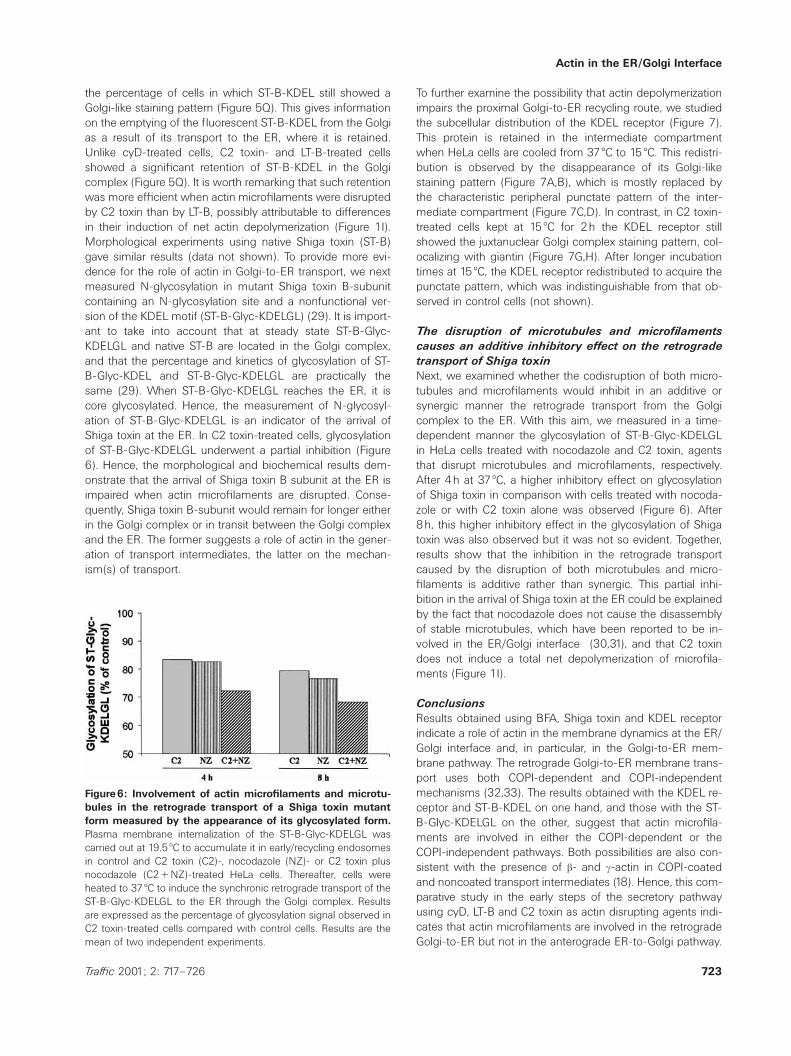
Figure6: Involvement of actin microfilaments and microtu-bules in the retrograde transport of a Shiga toxin mutantform measured by the appearance of its glycosylated form.Plasma membrane internalization of the ST-B-Glyc-KDELGL wascarried out at 19.5 æC to accumulate it in early/recycling endosomesin control and C2 toxin (C2)-, nocodazole (NZ)- or C2 toxin plusnocodazole (C2πNZ)-treated HeLa cells. Thereafter, cells wereheated to 37 æC to induce the synchronic retrograde transport of theST-B-Glyc-KDELGL to the ER through the Golgi complex. Resultsare expressed as the percentage of glycosylation signal observed inC2 toxin-treated cells compared with control cells. Results are themean of two independent experiments.
723Traffic 2001; 2: 717–726
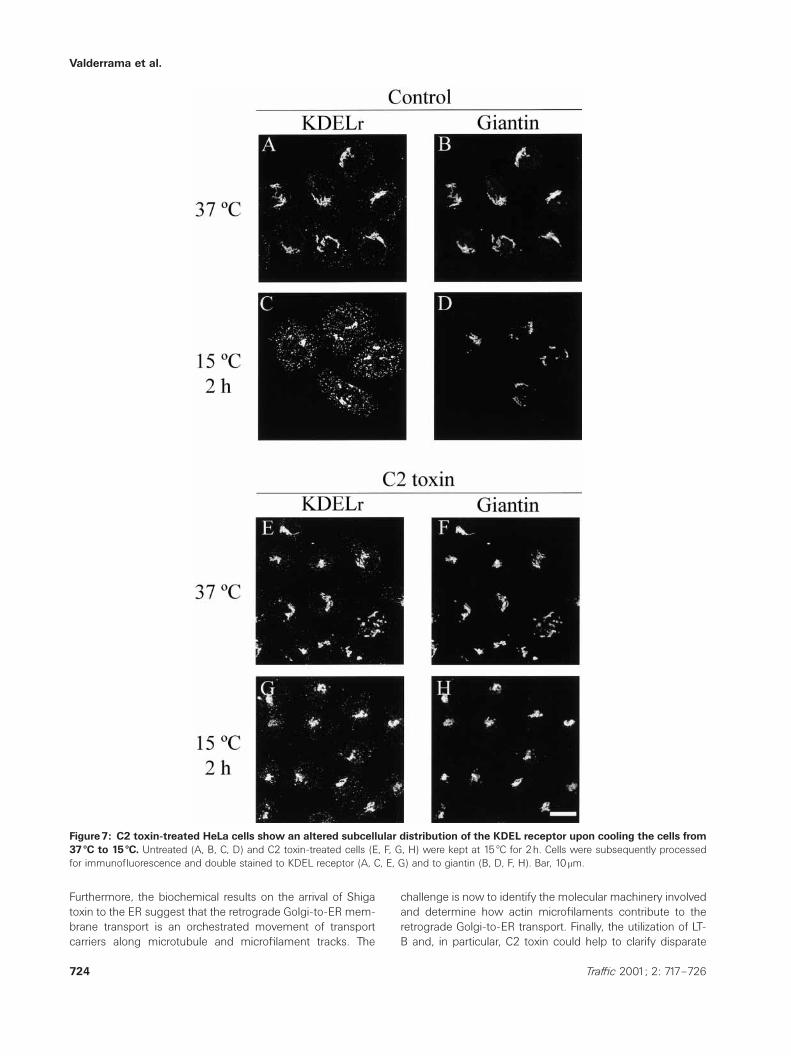
To further examine the possibility that actin depolymerizationimpairs the proximal Golgi-to-ER recycling route, we studiedthe subcellular distribution of the KDEL receptor (Figure 7).This protein is retained in the intermediate compartmentwhen HeLa cells are cooled from 37 æC to 15 æC. This redistri-bution is observed by the disappearance of its Golgi-likestaining pattern (Figure 7A,B), which is mostly replaced bythe characteristic peripheral punctate pattern of the inter-mediate compartment (Figure 7C,D). In contrast, in C2 toxin-treated cells kept at 15 æC for 2h the KDEL receptor stillshowed the juxtanuclear Golgi complex staining pattern, col-ocalizing with giantin (Figure 7G,H). After longer incubationtimes at 15 æC, the KDEL receptor redistributed to acquire thepunctate pattern, which was indistinguishable from that ob-served in control cells (not shown).
The disruption of microtubules and microfilaments
causes an additive inhibitory effect on the retrograde
transport of Shiga toxin
Next, we examined whether the codisruption of both micro-tubules and microfilaments would inhibit in an additive orsynergic manner the retrograde transport from the Golgicomplex to the ER. With this aim, we measured in a time-dependent manner the glycosylation of ST-B-Glyc-KDELGLin HeLa cells treated with nocodazole and C2 toxin, agentsthat disrupt microtubules and microfilaments, respectively.After 4h at 37 æC, a higher inhibitory effect on glycosylationof Shiga toxin in comparison with cells treated with nocoda-zole or with C2 toxin alone was observed (Figure 6). After8h, this higher inhibitory effect in the glycosylation of Shigatoxin was also observed but it was not so evident. Together,results show that the inhibition in the retrograde transportcaused by the disruption of both microtubules and micro-filaments is additive rather than synergic. This partial inhi-bition in the arrival of Shiga toxin at the ER could be explainedby the fact that nocodazole does not cause the disassemblyof stable microtubules, which have been reported to be in-volved in the ER/Golgi interface (30,31), and that C2 toxindoes not induce a total net depolymerization of microfila-ments (Figure 1I).
Conclusions
Results obtained using BFA, Shiga toxin and KDEL receptorindicate a role of actin in the membrane dynamics at the ER/Golgi interface and, in particular, in the Golgi-to-ER mem-brane pathway. The retrograde Golgi-to-ER membrane trans-port uses both COPI-dependent and COPI-independentmechanisms (32,33). The results obtained with the KDEL re-ceptor and ST-B-KDEL on one hand, and those with the ST-B-Glyc-KDELGL on the other, suggest that actin microfila-ments are involved in either the COPI-dependent or theCOPI-independent pathways. Both possibilities are also con-sistent with the presence of b- and g-actin in COPI-coatedand noncoated transport intermediates (18). Hence, this com-parative study in the early steps of the secretory pathwayusing cyD, LT-B and C2 toxin as actin disrupting agents indi-cates that actin microfilaments are involved in the retrogradeGolgi-to-ER but not in the anterograde ER-to-Golgi pathway.
Valderrama et al.
Figure7: C2 toxin-treated HeLa cells show an altered subcellular distribution of the KDEL receptor upon cooling the cells from37 æC to 15 æC. Untreated (A, B, C, D) and C2 toxin-treated cells (E, F, G, H) were kept at 15 æC for 2h. Cells were subsequently processedfor immunofluorescence and double stained to KDEL receptor (A, C, E, G) and to giantin (B, D, F, H). Bar, 10 mm.
Furthermore, the biochemical results on the arrival of Shigatoxin to the ER suggest that the retrograde Golgi-to-ER mem-brane transport is an orchestrated movement of transportcarriers along microtubule and microfilament tracks. The
724 Traffic 2001; 2: 717–726
challenge is now to identify the molecular machinery involvedand determine how actin microfilaments contribute to theretrograde Golgi-to-ER transport. Finally, the utilization of LT-B and, in particular, C2 toxin could help to clarify disparate

Actin in the ER/Golgi Interface
results about the role of actin in the endocytic and bio-synthetic/secretory processes.
Materials and Methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM) and Fetal Calf Serum (FCS)were from Gibco/Brl Life Technologies (Paisley, UK); Endoglycosidase H,secondary TRITC or FITC F(abƒ)2 fragments and 1-deoxymannojirimicin(DMM) were from Boheringer Mannheim (Mannheim, Germany); rabbitanti-mouse IgG was from DAKO (Dako, AS, Denmark); Protein A/G agarosewas from Sta. Cruz Biotechnology Inc. (Sta. Cruz, CA, USA); Pro-Mix L-[35S]-labeling mix from Amersham (Buckinghamshire, UK). All otherchemicals if not otherwise stated were from Sigma Co. (St. Louis, MO,USA).
Cell culture
NRK and HeLa cells were cultured in DMEM supplemented with 10% FCSand 1mM L-glutamine at 37 æC in a humidified atmosphere of 5% CO2.
Treatments with botulinum C2 and Shiga toxins, and latrunculin B
Botulinum C2 toxin (100ng/ml of each C2I and C2II subunits, final concen-tration) was added to cells cultured in DMEM and supplemented with0.5% FCS for at least 4h at 37 æC. Cy3-Shiga toxin B-fragment tagged withKDEL motif (4mg/ml, final concentration) was added to cells (previouslyincubated for 30min in binding medium, FCS-free DMEM) for 45min at4 æC. After the withdrawal of unbound toxin by washing for 5min in ice-cold PBS, cells were incubated with DMEM at 37 æC. Latrunculin-B(500nM, final concentration) was added to cells cultured in DMEM for15min before the experimental procedure.
Immunofluorescence
Indirect immunofluorescence was carried out as previously described (17)and the following antibody dilutions were used: anti-giantin, 1 :500; anti-KDEL receptor, 1 :1000; anti-Man II, 1 :4000, and fluorescent secondaryantibodies, 1 :35. Microscopy and imaging were performed with anOlympus BX60 epifluorescence microscope with a chilled charge-coupleddevice (CCD) camera (Olympus DP50) or with a Leica TCS-NT confocalmicroscope. For the quantitative analysis of the morphological experi-ments, all images were obtained from randomly chosen fields and cap-tured at nonsaturating integration levels with the CCD camera. At least200 cells, randomly chosen, were counted per experimental condition. Theimages were processed on a PC using Adobe Photoshop 5.0.
Virus infection and VSV-G transport assays
The infection with the temperature-sensitive mutant ts045 VSV and theassay of the acquisition of the Endo H-resistance form of the G glyco-protein were performed as described (17). Indirect immunofluorescencetransport of VSV-G from ER-to-Golgi complex was performed as describedelsewhere (34).
Fluorescent phalloidin-binding assay
The F-actin content in untreated and C2 toxin- and cyD-treated cells wasmeasured as previously described (18,24).
Retrograde transport assay of Shiga toxin B-fragment constructs
Mutant Shiga toxin B-subunits (ST-B-Glyc-KDEGL) were iodinated as pre-viously reported (29). HeLa cells were incubated with the iodinated B-subunits mutant (200nM) in DMEM without FCS at 4 æC. Cells were rinsedand incubated at 19.5 æC in DMEM without FCS for 1h, and then with C2toxin (CI and CII subunits, 2mg/ml each) for a further 2h (C2 toxin entersthe cell and is functionally active at 19.5 æC; F. Valderrama and G. Egea,
725Traffic 2001; 2: 717–726
unpublished observations). Thereafter, cells were rinsed and incubatedwith DMEM containing 10% FCS and the mannosidase I inhibitor DMM(1mM, final concentration) at 37 æC. At the indicated time points, cells werelysed in SDS sample buffer. Samples were subsequently run on 10–20%polyacrylamide-SDS gradient gels, analyzed by autoradiography, andquantified with a PhosphorImager using the ImageQuant software (Mol-ecular Dynamics). For the transport experiments with C2 toxin and nocoda-zole, HeLa cells were incubated with iodinated ST-B-Glyc-KDELGL as de-scribed above, but at the 19 æC blockade; besides C2 toxin, nocodazole(30mM, final concentration) was also added.
Acknowledgments
We thank Vivek Malhotra, Maria Antonietta De Matteis, Bruno Goud, andLudger Johannes for helpful discussions, Maite Munoz for technical sup-port, Robin Rycroft for linguistic assistance, and Drs H.-P. Hauri for mono-clonal antibodies to giantin, H.-D. Söling for polyclonal antibodies to KDELreceptor, K. Moremen for polyclonal antibodies to Man II, and LudgerJohannes for the Shiga toxin B-subunit mutants. This work was supportedby grants CICYT SAF 2000–0042, CIRIT ACI99-1, and Gaspar de PortolaAGP 99–06. F.V. and J.M.D. are financed by predoctoral fellowships fromthe University of Barcelona and IDIBAPS, respectively.
References
1. Sheterline P, Clayton J, Sparrow J. Actin. Oxford, UK: Oxford Univer-sity Press; 1999.
2. Spector I, Braet F, Shochet NR, Bubb MR. New anti-actin drugs in thestudy of the organization and function of the actin cytoskeleton.Microsc Res Tech 1999;47:18–37.
3. Aktories K, Wille M, Just I. Clostridial actin-ADP-ribosylating toxins.Curr Top Microbiol Immunol 1992;175:97–113.
4. Casella JF, Flanagan MD, Lin S. Cytochalasin D inhibits actin poly-merization and induces depolymerization of actin filaments formedduring platelet shape change. Nature 1981;293:302–305.
5. Sampath P, Pollard TD. Effects of cytochalasin, phalloidin, and pH onthe elongation of actin filaments. Biochemistry 1991;30:1973–1980.
6. Morris A, Tannenbaum J. Cytochalasin D does not produce net de-polymerization of actin filaments in HEp-2 cells. Nature 1990;287:637–639.
7. Bershadsky AD, Gluck U, Denisenko ON, Sklyarova TV, Spector I, Ben-Ze’ev A. The state of actin assembly regulates actin and vinculin ex-pression by a feedback loop. J Cell Sci 1995;108:1183–1193.
8. Spector I, Shochet NR, Blasberger D, Kashman Y. Latrunculins, novelmarine macrolides that disrupt microfilament organization and affectcell growth: I. Comparison with cytochalasin D. Cell Motil Cytoskeleton1989;13:127–144.
9. Aktories K, Barmann M, Ohishi I, Tsuyama S, Jakobs KH, HabermannE. Botulinum C2 toxin ADP-ribosylates actin. Nature 1986;322:390–392.
10. Coue M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymeriz-ation by latrunculin A. FEBS Lett, 1987;213:316–318.
11. Qualmann B, Kessels MM, Kelly RB. Molecular links between endo-cytosis and the actin cytoskeleton. J Cell Biol 2000;150:F111–F116.
12. Baumann O. The Golgi apparatus in honeybee photoreceptor cells:structural organization and spatial relationship to microtubules and ac-tin filaments. Cell Tissue Res 1998;291:351–361.
13. Buss F, Kendrick-Jones J, Lionne C, Knight AE, Cote GP, Paul LJ. Thelocalization of myosin VI at the Golgi complex and leading edge offibroblasts and its phosphorylation and recruitment into membraneruffles of A431 cells after growth factor stimulation. J Cell Biol1998;143:1535–1545.

Valderrama et al.
14. Godi A, Santone I, Pertile P, Devarajan P, Stabach PR, Morrow JS,Di Tullio G, Polishchuk R, Petrucci TC, Luini A, De Matteis MA. ADPribosylation factor regulates spectrin binding to the Golgi complex.Proc Natl Acad Sci USA 1998;95:8607–8612.
15. Holleran EA, Holzbaur EL. Speculating about spectrin: new insightsinto the Golgi-associated cytoskeleton. Trends Cell Biol 1998;8:26–29.
16. Stankewich MC, Tse WT, Peters LL, Ch’ng Y, John KM, Stabach PR,Devarajan P, Morrow JS, Lux SE. A widely expressed betaIII spectrinassociated with Golgi and cytoplasmic vesicles. Proc Natl Acad SciUSA 1998;95:14158–14163.
17. Valderrama F, Babia T, Ayala I, Kok JW, Renau-Piqueras J, Egea G.Actin microfilaments are essential for the cytological positioning andmorphology of the Golgi complex. Eur J Cell Biol 1998;76:9–17.
18. Valderrama F, Luna A, Babia T, Martınez-Menaguez JA, Ballesta J,Barth H, Chaponnier C, Renau-Piqueras J, Egea G. The Golgi-associ-ated COPI-coated buds and vesicles contain b/g-actin. Proc Natl AcadSci USA 2000;97:1560–1565.
19. di Campli A, Valderrama F, Babia T, De Matteis MA, Luini A, EgeaG. Morphological changes in the Golgi complex correlate with actincytoskeleton rearrangements. Cell Motil Cytoskeleton 1999;43:334–348.
20. Heimann K, Percival JM, Weinberger R, Gunning P, Stow JL. Specificisoforms of actin-binding proteins on distinct populations of Golgi-derived vesicles. J Biol Chem 1999;274:10743–10750.
21. Fucini RV, Navarrete A, Vadakkan C, Lacomis L, Erdjument-BromageH, Tempst P, Stamnes M. Activated ADP-ribosylation factor assemblesdistinct pools of actin on Golgi membranes. J Biol Chem 2000;275:18824–18829.
22. Lamaze C, Fujimoto LM, Yin HL, Schmid SL. The actin cytoskeleton isrequired for receptor-mediated endocytosis in mammalian cells. J BiolChem 1997;272:20332–20335.
23. Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD,Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic andcharacterization of Golgi to plasma membrane transport intermediatesin living cells. J Cell Biol 1998;143:1485–1503.
726 Traffic 2001; 2: 717–726
24. Babia T, Ayala I, Valderrama F, Mato E, Bosch M, Santaren JF, Renau-Piqueras J, Kok JW, Thomson TM, Egea G. N-Ras induces alterationsin Golgi complex architecture and in constitutive protein transport. JCell Sci 1999;112:477–489.
25. Fujimoto LM, Roth R, Heuser JE, Schmid SL. Actin assembly plays avariable, but not obligatory role in receptor-mediated endocytosis inmammalian cells. Traffic 2000;1:161–171.
26. Lingwood CA. Role of verotoxin receptors in pathogenesis. TrendsMicrobiol 1996;4:147–153.
27. Sandvig K, Garred O, Prydz K, Kozlov JV, Hansen SH, van Deurs B.Retrograde transport of endocytosed Shiga toxin to the endoplasmicreticulum. Nature 1992;358:510–512.
28. Barth H, Klingler M, Aktories K, Kinzel V. Clostridium botulinum C2toxin delays entry into mitosis and activation of p34cdc2 kinase andcdc25-C phosphatase in HeLa cells. Infect Immun 1999;67:5083–5090.
29. Johannes L, Tenza D, Antony C, Goud B. Retrograde transport ofKDEL-bearing B-fragment of Shiga toxin. J Biol Chem 1997;272:19554–19561.
30. Mizuno M, Singer SJ. A possible role for stable microtubules in intra-cellular transport from the endoplasmic reticulum to the Golgi com-plex. J Cell Sci 1994;107:1321–1331.
31. Poüs C, Chabin K, Drechou A, Barbot L, Phung-Koskas T, SettegranaC, Bouguet-Kondracki ML, Maurice M, Cassio D, Guyot M, Durand G.Functional specialization of stable and dynamic microtubules in pro-tein traffic in WIF-B cells. J Cell Biol 1998;142:153–165.
32. Girod A, Storrie B, Simpson JC, Johannes L, Goud B, Roberts LM,Lord JM, Nilsson T, Pepperkok R. Evidence for a COP-I-independenttransport route from the Golgi complex to the endoplasmic reticulum.Nat Cell Biol 1999;1:423–430.
33. Storrie B, Pepperkok R, Nilsson T. Breaking the COPI monopoly onGolgi recycling. Trends Cell Biol 2000;10:385–391.
34. Bonatti S, Migliaccio G, Simons K. Palmitylation of viral membraneglycoproteins takes place after exit from the endoplasmic reticulum. JBiol Chem 1989;264:12590–12595.




















