
Aberrant modulation of a delayed rectifier potassium channel by glutamate in Alzheimer's disease
10
Aberrant modulation of a delayed rectifier potassium channel by glutamate in Alzheimer's disease Cornelia Poulopoulou ⁎, Ioannis Markakis, Panagiota Davaki, Eleftheria Tsaltas, Antonis Rombos, Alexandros Hatzimanolis, Dimitrios Vassilopoulos Department of Neurology, Laboratory of Experimental Neurophysiology, Medical School, University of Athens, Eginition Hospital, 72-74 Vas. Sophias Avenue, Athens, Greece abstract article info Article history: Received 18 April 2009 Revised 8 September 2009 Accepted 10 October 2009 Available online 20 October 2009 Keywords: Alzheimer's disease Potassium channels Metabotropic glutamate receptors T lymphocytes In Alzheimer's disease (AD), potassium channel abnormalities have been reported in both neural and peripheral tissues. Herein, using whole-cell patch-clamp, we demonstrate an aberrant glutamate-dependent modulation of K V 1.3 channels in T lymphocytes of AD patients. Although intrinsic K V 1.3 properties in patients were similar to healthy individuals, glutamate (1–1000 μM) failed to yield the hyperpolarizing shift normally observed in K V 1.3 steady-state inactivation (-4.4±2.7 mV in AD vs. -14.3 ± 2.5 mV in controls, 10 μM glutamate), resulting in a 4-fold increase of resting channel activity. Specific agonist and antagonist data indicate that this abnormality is due to dysfunction of cognate group II mGluRs. Given that glutamate is present in plasma and that both mGluRs and K V 1.3 channels regulate T-lymphocyte responsiveness, our finding may account for the presence of immune-associated alterations in AD. Furthermore, if this aberration reflects a corresponding one in neural tissue, it could provide a potential target in AD pathogenesis. © 2009 Elsevier Inc. All rights reserved. Introduction Potassium channels are key determinants of cellular excitability, setting resting membrane potentials and driving repolarization, thus regulating cellular activity in cells as diverse as neurons, glia, and lymphocytes. The properties of these channels are under constant modulation, allowing them to adjust vital cellular signaling processes in response to extracellular cues. The signifi- cance of this mechanism is evidenced by its involvement in neuronal plasticity (Zhang and Linden, 2003): in mammals, in vivo modulation of potassium channel activity regulates cognitive processes such as associative memory and learning (Ghelardini et al., 1998; Giese et al., 2001; Kourich et al., 2001). Such findings prompted investigation for potassium channel abnormalities in Alzheimer's disease (AD), a condition characterized by progressive memory impairment. Indeed, K + channel dysfunction has been demonstrated in fibro- blasts (Etcheberrigaray et al., 1993) and platelets (De Silva et al., 1998) of AD patients. Postmortem studies, with their inherent limi- tations, also show alterations of K + channel expression in AD brains (Ikeda et al., 1991). Furthermore, peptide products of amyloid precursor protein (APP), crucial in AD pathogenesis, reportedly affect K + channel activity (Plant et al., 2005) as well as components of intracellular pathways known to be involved in K + channel modu- lation (Tyszkiewicz and Yan, 2005; Parameshwaran et al., 2008). However, experimental evidence associating AD with specific abnor- malities in K + channel modulation is scanty. We have previously demonstrated that the major CNS neuro- transmitter glutamate (Glu) modulates the activity of the delayed rectifier potassium channel K V 1.3 in human T lymphocytes (Poulopoulou et al., 2005b). Given that (1) K V 1.3 channels are also expressed in the CNS, mainly in microglia, olfactory bulb, and hippocampus (Kues and Wunder, 1992); (2) Glu, which possesses immunoregulatory properties (Boldyrev et al., 2005), modulates neuronal potassium channels; and (3) AD has been associated with both glutamatergic abnormalities (Butterfield and Pocernich, 2003) and immune alterations (Britschgi and Wyss-Coray, 2007), we used T lymphocytes to investigate K V 1.3 channel modulation by Glu in AD patients and control individuals. The intrinsic biophysical properties of K V 1.3 channels in T lymphocytes of healthy individuals have been adequately charac- terized (Cahalan et al., 2001). However, to our knowledge, there is only one study on the properties of these channels in aged subjects (Panyi et al., 1994), while there are no reports on their gating and kinetic features in T lymphocytes of AD patients. Herein, we demonstrate that, while there is no difference in the intrinsic properties of native K V 1.3 channels between AD patients and healthy aged individuals, a single component of Glu-dependent modulation is diminished in all patients examined. Neurobiology of Disease 37 (2010) 339–348 Abbreviations: AD, Alzheimer's disease; Glu, glutamate; mGluRs, metabotropic glutamate receptors; DCG IV, (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine; LY 341495, (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid; PHA, phytohemagglutinin. ⁎ Corresponding author. E-mail address: [email protected] (C. Poulopoulou). Available online on ScienceDirect (www.sciencedirect.com). 0969-9961/$ – see front matter © 2009 Elsevier Inc. All rights reserved. doi:10.1016/j.nbd.2009.10.012 Contents lists available at ScienceDirect Neurobiology of Disease journal homepage: www.elsevier.com/locate/ynbdi
Transcript of Aberrant modulation of a delayed rectifier potassium channel by glutamate in Alzheimer's disease

Aberrant modulation of a delayed rectifier potassium channel by glutamate inAlzheimer's disease
Cornelia Poulopoulou ⁎, Ioannis Markakis, Panagiota Davaki, Eleftheria Tsaltas, Antonis Rombos,Alexandros Hatzimanolis, Dimitrios VassilopoulosDepartment of Neurology, Laboratory of Experimental Neurophysiology, Medical School, University of Athens, Eginition Hospital, 72-74 Vas. Sophias Avenue, Athens, Greece
a b s t r a c ta r t i c l e i n f o
Article history:Received 18 April 2009Revised 8 September 2009Accepted 10 October 2009Available online 20 October 2009
Keywords:Alzheimer's diseasePotassium channelsMetabotropic glutamate receptorsT lymphocytes
In Alzheimer's disease (AD), potassium channel abnormalities have been reported in both neural andperipheral tissues. Herein, using whole-cell patch-clamp, we demonstrate an aberrant glutamate-dependentmodulation of KV1.3 channels in T lymphocytes of AD patients. Although intrinsic KV1.3 properties inpatients were similar to healthy individuals, glutamate (1–1000 μM) failed to yield the hyperpolarizing shiftnormally observed in KV1.3 steady-state inactivation (−4.4±2.7 mV in AD vs. −14.3±2.5 mV in controls,10 μM glutamate), resulting in a 4-fold increase of resting channel activity. Specific agonist and antagonistdata indicate that this abnormality is due to dysfunction of cognate group II mGluRs. Given thatglutamate is present in plasma and that both mGluRs and KV1.3 channels regulate T-lymphocyteresponsiveness, our finding may account for the presence of immune-associated alterations in AD.Furthermore, if this aberration reflects a corresponding one in neural tissue, it could provide a potentialtarget in AD pathogenesis.
© 2009 Elsevier Inc. All rights reserved.
Introduction
Potassium channels are key determinants of cellular excitability,setting resting membrane potentials and driving repolarization,thus regulating cellular activity in cells as diverse as neurons, glia,and lymphocytes. The properties of these channels are underconstant modulation, allowing them to adjust vital cellularsignaling processes in response to extracellular cues. The signifi-cance of this mechanism is evidenced by its involvement inneuronal plasticity (Zhang and Linden, 2003): in mammals,in vivo modulation of potassium channel activity regulates cognitiveprocesses such as associative memory and learning (Ghelardiniet al., 1998; Giese et al., 2001; Kourich et al., 2001). Such findingsprompted investigation for potassium channel abnormalities inAlzheimer's disease (AD), a condition characterized by progressivememory impairment.
Indeed, K+ channel dysfunction has been demonstrated in fibro-blasts (Etcheberrigaray et al., 1993) and platelets (De Silva et al.,1998) of AD patients. Postmortem studies, with their inherent limi-tations, also show alterations of K+ channel expression in AD brains(Ikeda et al., 1991). Furthermore, peptide products of amyloid
precursor protein (APP), crucial in AD pathogenesis, reportedly affectK+ channel activity (Plant et al., 2005) as well as components ofintracellular pathways known to be involved in K+ channel modu-lation (Tyszkiewicz and Yan, 2005; Parameshwaran et al., 2008).However, experimental evidence associating AD with specific abnor-malities in K+ channel modulation is scanty.
We have previously demonstrated that the major CNS neuro-transmitter glutamate (Glu) modulates the activity of the delayedrectifier potassium channel KV1.3 in human T lymphocytes(Poulopoulou et al., 2005b). Given that (1) KV1.3 channels arealso expressed in the CNS, mainly in microglia, olfactory bulb, andhippocampus (Kues and Wunder, 1992); (2) Glu, which possessesimmunoregulatory properties (Boldyrev et al., 2005), modulatesneuronal potassium channels; and (3) AD has been associated withboth glutamatergic abnormalities (Butterfield and Pocernich, 2003)and immune alterations (Britschgi and Wyss-Coray, 2007), we usedT lymphocytes to investigate KV1.3 channel modulation by Glu inAD patients and control individuals.
The intrinsic biophysical properties of KV1.3 channels in Tlymphocytes of healthy individuals have been adequately charac-terized (Cahalan et al., 2001). However, to our knowledge, there isonly one study on the properties of these channels in aged subjects(Panyi et al., 1994), while there are no reports on their gating andkinetic features in T lymphocytes of AD patients. Herein, wedemonstrate that, while there is no difference in the intrinsicproperties of native KV1.3 channels between AD patients and healthyaged individuals, a single component of Glu-dependent modulationis diminished in all patients examined.
Neurobiology of Disease 37 (2010) 339–348
Abbreviations: AD, Alzheimer's disease; Glu, glutamate; mGluRs, metabotropicglutamate receptors; DCG IV, (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine; LY341495, (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoicacid; PHA, phytohemagglutinin.⁎ Corresponding author.
E-mail address: [email protected] (C. Poulopoulou).Available online on ScienceDirect (www.sciencedirect.com).
0969-9961/$ – see front matter © 2009 Elsevier Inc. All rights reserved.doi:10.1016/j.nbd.2009.10.012
Contents lists available at ScienceDirect
Neurobiology of Disease
j ourna l homepage: www.e lsev ie r.com/ locate /ynbd i

Patients and methods
Patients
A total of 40 individuals were recruited in the study: 20patients fulfilled the DSM-IV and NINCDS-ARDA criteria forprobable AD (McKhann et al., 1984) while an equal number ofnon-demented healthy subjects with similar demographic char-acteristics were used as controls (Table 1). Informed consent wasobtained from the recruited subjects and their relatives. Allpatients enrolled in the study had late-onset (N65 years) AD. Theseverity of dementia was assessed as moderate to severe and theaverage duration of disease ranged from 2 to 6 years (mean±SD:4.2±1.9 years). Fourteen out of 20 patients were receivingtreatment with acetyl-cholinesterase inhibitors at the time of thestudy (5 with donepezil, 4 with galantamine, and 5 withrivastigmine), while the remaining 6 had never received similarmedication. Metabolic or other causes of dementia were excludedin the patient group by a complete set of laboratory tests as wellas by computed tomography imaging. Subjects in the control grouphad neither complaints for memory problems nor other evidenceof cognitive dysfunction. Subjects with a history of autoimmunedisorders, diabetes mellitus, cancer, chronic viral disease, or arecent (b3 weeks) history of infection were excluded from thestudy; thus, there was no obvious difference between the twogroups regarding conditions that might affect immune function.Serum biochemical examinations, complete blood counts, andarterial pressure measurements were within normal limits for thecontrol as well as the patient groups.
Cell preparation and proliferation assays
Heparinized blood was obtained by venous puncture andmononuclear cells were immediately separated by Ficoll-Hypaquedensity gradient centrifugation. Adhesion to plastic tissue cultureflasks was used to eliminate the monocyte population (Verheugenand Korn, 1997). For electrophysiological experiments, the purifiedT lymphocytes were kept in RPMI medium and recordings wereperformed on the same day. Before each experiment, they werewashed twice with the extracellular solution used for recordings(see below).
For proliferation assays, T lymphocytes (0.1 ml, 105/well) wereincubated with 4 μg/ml PHA, in Dulbecco's modified Eagle'smedium (DMEM), supplemented with 10% (vol./vol.) fetal bovineserum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/mlstreptomycin at 37 °C and 5% CO2, and assayed for 72 hours usinga colorimetric method (Cell Proliferation ELISA BrdU, Roche AppliedScience, Penzberg, Germany). The standard DMEM containedminimal levels of Glu, as determined by high-performance liquidchromatography. The effect of Glu on stimulated T-lymphocyteproliferation was evaluated, using a Glu-enriched DMEM medium,containing 1 mM of Glu. T-lymphocyte viability was assessed usingTrypan blue exclusion, following cell isolation, as well as in cellcultures and was greater than 90% in all experiments.
Chemicals and salts
Ficoll-Hypaque (Histopaque-1077) and glutamate wereobtained from Sigma Chemical Co. (St Louis, MO, USA). DCG IVand LY 341495 were obtained from Tocris Cookson (Bristol, UK).PHA was purchased from Sigma-Aldrich (St. Louis, MO). RPMI1640 medium was obtained from Gibco/Invitrogen (Paisley, UK).Salts were of analytical grade and obtained from Fluka Biochemica(Switzerland).
Electrophysiological recordings
Membrane currents were measured in the whole-cell configu-ration of the patch-clamp technique (Hamill et al., 1981) using anAxopatch 100B (Axon Instruments, Foster City, CA, USA) amplifier,interfaced to a personal computer with a Digidata 1200 (AxonInstruments) analog-to-digital converter. Pipettes were pulled fromborosilicate glass capillaries (World Precision Instruments, Sarasota,FA) using a two-stage puller (L/M-3P-A, List Medical, Darmstadt,Germany) and had resistances between 3 and 5 MΩ. Sealresistances of 10–20 GΩ were obtained after slight suction at theinterior of the pipette. The extracellular solution contained:(in mM): NaCl 135, KCl 5, CaCl2 1, MgCl2 1, HEPES 10 (pH 7.4,osmolality 280 mOsm/kg). The pipette (recording) solution wascomposed of (in mM): KCl 100, KF 40, CaCl2 1, MgCl2 1, EGTA 10,and HEPES 10 (pH 7.4, osmolality 290 mOsm/kg). Intracellular Ca2+
was buffered to a concentration of ∼100 nM, to prevent activationof calcium-dependent potassium (KCa) channels that are alsopresent on human T lymphocytes (Cahalan et al., 2001). Membranevoltages were corrected for liquid junction potentials. Leak currentswere negligible and no correction was performed. Data were low-pass filtered at 2 kHz. During the measurements, solutions wereapplied by the Super-fast 8 perfusion system (List Medical,Germany) and driven away with the use of a peristaltic pump(List Medical, Germany). All experiments were performed at roomtemperature (20–25 °C).
Voltage protocols
T lymphocytes were allowed to equilibrate for at least20 minutes in the whole-cell configuration before beginning therecordings (Poulopoulou et al., 2005b). The cell membrane wasclamped at a holding potential of −90 mV. Peak currents weremeasured in response to 1000-ms-long voltage steps from −80 to+40 mV in 15-mV increments, given every 60 s. The voltagedependence of steady-state inactivation was estimated by steppingthe membrane from −90 mV to various prepulse potentials(−120 to 0 mV in 15-mV increments) for 100 s and then to a testpulse of +40 mV for 100 ms. Recordings were made both beforeand after (5 min) the addition of Glu (1–1000 μM) to theextracellular medium.
Data analysis
Current measurements were performed using the pClampsoftware (pClamp 6.3, Axon Instruments). Peak conductance wascalculated from maximum current values using the chordequation: gK= Imax/(V−EK), where EK is the Nerst equilibriumpotential for K+ ions. Peak conductance-to-voltage and steady-state inactivation curves were plotted and fitted to Boltzmannfunctions using the Origin technical graphics and data analysisprogram (Microcal, Northampton, MA, USA). The goodness-of-fitdata were evaluated with the Hamilton's R coefficient. Student's t-test for unpaired observations was used for the statisticalevaluation of differences between patient and control data(p=0.05).
Table 1Clinical and demographic characteristics of patients with Alzheimer's disease andhealthy controls.
Group Na Age (years) MMSEb Male/female
AD 20 75.4±4.4 16±3.5 9:11Healthy controls 20 74.8±5.3 28.7±1.3 9:11a Data are expressed as mean±standard deviation; n=number of individuals per
condition.b MMSE: Mini Mental Status Examination.
340 C. Poulopoulou et al. / Neurobiology of Disease 37 (2010) 339–348
Results
Similar KV1.3 gating properties in AD patients and non-dementedindividuals
Using standardized experimental protocols (see methods section),we investigated the basic biophysical features of KV1.3 channels in Tlymphocytes of ADpatients compared to agednon-demented controls.
The recorded currents from T lymphocytes of both groups had areversal potential (Erev) close to the calculated equilibrium potentialfor K+ ions (EK) of our ionic solutions (EK=−85.3 mV) and werecharacterized as KV1.3 based on their total abrogation by 5 nM ofMgTX (Garcia-Calvo et al., 1993) and on the presence of cumulativeinactivation (Marom and Levitan, 1994).
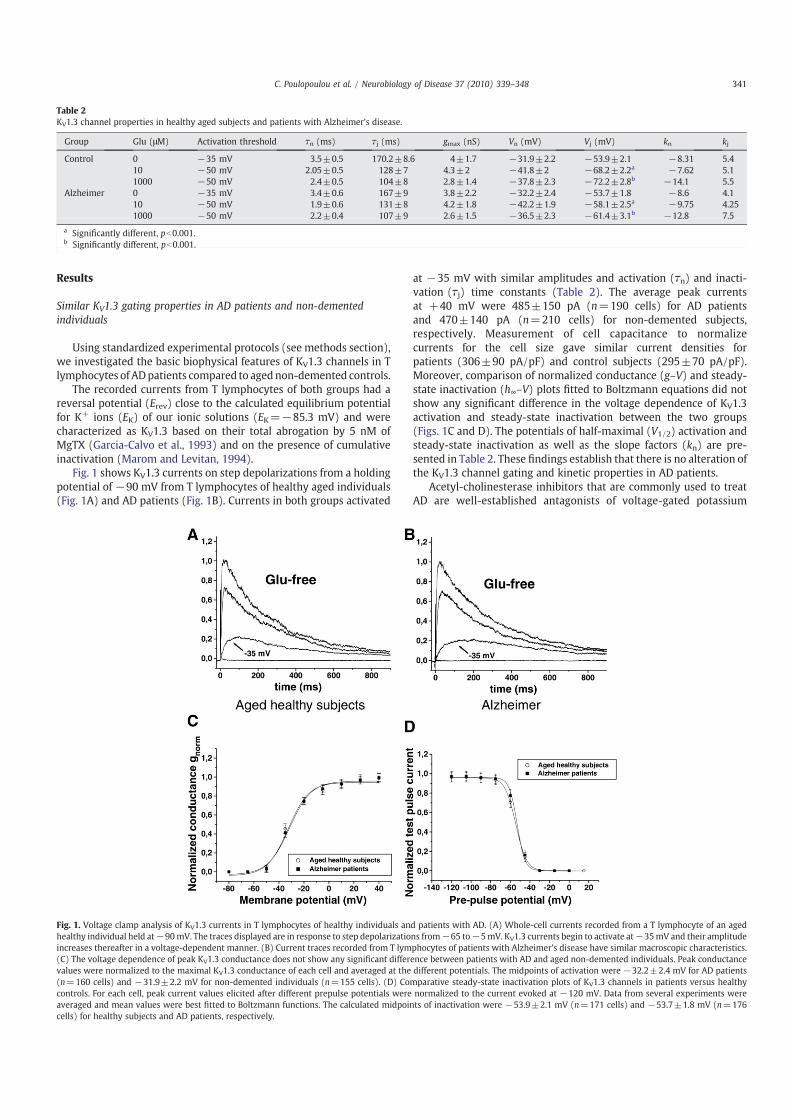
Fig. 1 shows KV1.3 currents on step depolarizations from a holdingpotential of −90 mV from T lymphocytes of healthy aged individuals(Fig. 1A) and AD patients (Fig. 1B). Currents in both groups activated
at −35 mV with similar amplitudes and activation (!n) and inacti-vation (!j) time constants (Table 2). The average peak currentsat +40 mV were 485±150 pA (n=190 cells) for AD patientsand 470±140 pA (n=210 cells) for non-demented subjects,respectively. Measurement of cell capacitance to normalizecurrents for the cell size gave similar current densities forpatients (306±90 pA/pF) and control subjects (295±70 pA/pF).Moreover, comparison of normalized conductance (g–V) and steady-state inactivation (h∞–V) plots fitted to Boltzmann equations did notshow any significant difference in the voltage dependence of KV1.3activation and steady-state inactivation between the two groups(Figs. 1C and D). The potentials of half-maximal (V1/2) activation andsteady-state inactivation as well as the slope factors (kn) are pre-sented in Table 2. These findings establish that there is no alteration ofthe KV1.3 channel gating and kinetic properties in AD patients.
Acetyl-cholinesterase inhibitors that are commonly used to treatAD are well-established antagonists of voltage-gated potassium
Fig. 1. Voltage clamp analysis of KV1.3 currents in T lymphocytes of healthy individuals and patients with AD. (A) Whole-cell currents recorded from a T lymphocyte of an agedhealthy individual held at−90mV. The traces displayed are in response to step depolarizations from−65 to−5mV. KV1.3 currents begin to activate at−35mV and their amplitudeincreases thereafter in a voltage-dependent manner. (B) Current traces recorded from T lymphocytes of patients with Alzheimer's disease have similar macroscopic characteristics.(C) The voltage dependence of peak KV1.3 conductance does not show any significant difference between patients with AD and aged non-demented individuals. Peak conductancevalues were normalized to the maximal KV1.3 conductance of each cell and averaged at the different potentials. The midpoints of activation were −32.2±2.4 mV for AD patients(n=160 cells) and −31.9±2.2 mV for non-demented individuals (n=155 cells). (D) Comparative steady-state inactivation plots of KV1.3 channels in patients versus healthycontrols. For each cell, peak current values elicited after different prepulse potentials were normalized to the current evoked at −120 mV. Data from several experiments wereaveraged and mean values were best fitted to Boltzmann functions. The calculated midpoints of inactivation were −53.9±2.1 mV (n=171 cells) and −53.7±1.8 mV (n=176cells) for healthy subjects and AD patients, respectively.
Table 2KV1.3 channel properties in healthy aged subjects and patients with Alzheimer's disease.
Group Glu (μM) Activation threshold !n (ms) !j (ms) gmax (nS) Vn (mV) Vj (mV) kn kj
Control 0 −35 mV 3.5±0.5 170.2±8.6 4±1.7 −31.9±2.2 −53.9±2.1 −8.31 5.410 −50 mV 2.05±0.5 128±7 4.3±2 −41.8±2 −68.2±2.2a −7.62 5.11000 −50 mV 2.4±0.5 104±8 2.8±1.4 −37.8±2.3 −72.2±2.8b −14.1 5.5
Alzheimer 0 −35 mV 3.4±0.6 167±9 3.8±2.2 −32.2±2.4 −53.7±1.8 −8.6 4.110 −50 mV 1.9±0.6 131±8 4.2±1.8 −42.2±1.9 −58.1±2.5a −9.75 4.251000 −50 mV 2.2±0.4 107±9 2.6±1.5 −36.5±2.3 −61.4±3.1b −12.8 7.5
a Significantly different, pb0.001.b Significantly different, pb0.001.
341C. Poulopoulou et al. / Neurobiology of Disease 37 (2010) 339–348

342 C. Poulopoulou et al. / Neurobiology of Disease 37 (2010) 339–348

channels. Thus, in order to exclude a possible effect of treatment on ourresults, we compared KV1.3 biophysical properties between treatedand untreated AD patients. The potentials of half-maximal activationwere similar in the two patient subgroups (−32.1±2 mV for treatedvs. −32.2±2.1 mV for untreated patients) as were the potentials ofhalf-maximal inactivation (−54±1.7 mV for treated vs. −53.6±1.8 mV for untreated patients). Moreover, there was no significantdifference in the average peak KV1.3 current amplitudes and currentkinetics between the two AD subgroups. This finding argues againstany treatment-related effect on KV1.3 channel properties in AD.
Glu fails to enhance steady-state inactivation of KV1.3 channels inT lymphocytes of AD patients
Having demonstrated that the intrinsic properties of KV1.3channels are similar in AD patients and aged controls, we proceededto examine their modulation by Glu in both groups. Application of Glu(1–1000 μM), to T lymphocytes of control subjects, altered the voltagedependence of gating (activation and steady-state inactivation),activation kinetics, amplitude, and duration of KV1.3 currents aspreviously reported for young healthy individuals (Poulopoulou et al.,2005b). More specifically, in the presence of low Glu concentrations(1–10 μM), KV1.3 currents were activated at a more negative mem-brane potential (−50 mV), with a faster activation rate and increasedamplitude compared to Glu-free solution (compare Fig. 2A1 toFig. 1A). Elevation of Glu above 10 μM and up to 1000 μM (whereits effects saturate) resulted in a dose-dependent decrease in thecurrent amplitude, along with a faster decay rate (Fig. 2A2).Additionally, Glu (1–1000 μM) increased the steady-state inactivationof the current in a dose-dependent manner, as previously reported(Poulopoulou et al., 2005b).
When the effects of Glu on KV1.3 currents of AD patients werestudied, similar alterations in the voltage dependence of activationand current kinetics were observed (Figs. 2A–C). Interestingly,however, differences emerged in the modulation of steady-stateinactivation between the two groups. Specifically, while Glu (1–1000 μM) caused a significant shift in the voltage dependence ofsteady-state inactivation of KV1.3 currents in T lymphocytes of healthyindividuals, this shift was only minimal in T lymphocytes of ADpatients (Fig. 2D). In particular, the overall shift in h∞–V relation for 10and 1000 μM of Glu was only −4.4±2.7 mV and −7.2±2.1 mV,respectively, in AD vs.−14.3±2.5mVand−18.2±2.3mV in controls(pb0.001). Values are summarized in Table 2. The severity of dementiain AD patients tended to be associated to the effect of Glu on KV1.3steady-state inactivation (Fig. 2E).
This abnormality was constantly observed in all cells of all ADpatients and in none of the healthy individuals. It does not seem to beassociated to treatmentwith acetyl-cholinesterase inhibitors, given thatKV1.3 biophysical properties in the presence of Gluwere identical in thesubgroups of treated and untreated AD patients (data not shown).
The altered voltage dependence of steady-state inactivation in ADpatients results in a window current (area under the overlap of g–V
and h–V curves) of 651 nA (n=44 cells) vs. 143 nA in healthyindividuals (n=40 cells) or else in a marked up-regulation of tonicKV1.3 activity in the presence of Glu, as seen in Fig. 3. This is solelyattributed to increased channel availability under these conditions,since the average maximal K+ conductance is similar in the twogroups (∼4 pA/mV) as is the voltage sensitivity (slope) of activationand inactivation. The increase of tonic K+ conductance in AD isbiologically significant, since it is primarily observed in the range ofphysiologically relevant resting potential values and at normalplasma and tissue Glu concentrations (≈10 μM).
The above data are corroborated by the finding that, in thepresence of Glu, differences in KV1.3 current amplitudes between thetwo groups emerged only out of holding potentials around thephysiological range (e.g., −60 mV), where channel inactivation ispresent and not out of our standard holding potential of −90 mV,where channel inactivation is minimal (Fig. 4). This aberration,which was present in all AD patients, would be expected to havemajor implications for the function of KV1.3 channels of AD patients invivo because, at plasma concentrations of Glu, there will be asignificant increase of their potassium conductance around the restingmembrane potential. Given that KV1.3 channels regulate calciuminflux in T lymphocytes following immune stimuli, this increasedpotassium conductance would be reflected on enhanced calcium-dependent processes (e.g., proliferation) of these cells in AD.
Differential effects of Glu on T-lymphocyte proliferation in AD patientsand control individuals
Glu, at concentrations ≥100 μM, has been shown to suppressmitogenic responses of human T lymphocytes through an unidentifiedmechanism (Sommer et al., 1994; Lombardi et al., 2001). Previously,we have proposed that this effect could be, at least in part, attributedto inhibition of KV1.3 currents by Glu, given that the activity of thesechannels regulates T-lymphocyte proliferation. Based on the above,we went on to study the effects of Glu on the mitogenic responses ofcells from AD patients compared to healthy aged individuals.
The mean rate of PHA-induced lymphocyte proliferation in thestandard culture medium (containing minimal Glu concentrations)was slightly elevated in AD patients compared to healthy controls,but the difference was not statistically significant (Fig. 5, inset);however, incubation of cells in modified DMEM containing 1 mM ofGlu revealed a marked difference in the stimulated proliferationbetween the two groups. More specifically, in accordance to previousreports (Lombardi et al., 2001), T lymphocytes of healthy individualsshowed reduced proliferation rates in the Glu-enriched mediumcompared to the standard medium of minimal Glu content.Interestingly, similar treatment of cells from AD patients did notcause any significant change in their proliferation rate (Fig. 5),suggesting an inability of high Glu concentrations to suppresscalcium-dependent T-lymphocyte responses in AD. The above dif-ferences were not due to alterations in cell viability, as demonstratedby Trypan blue exclusion.
Fig. 2. Effects of Glu on KV1.3 currents of healthy individuals and patients with AD. (A) Whole-cell recordings from a T lymphocyte of a healthy individual held at −90 mV andsequentially exposed to 10 (A1) and 1000 μM of Glu (A2). Current traces are in response to depolarizations from −65 to −5 mV (for clarity reasons) and have been normalized totheir peak values in Glu-free solution. Glu (10 μΜ) shifts the activation threshold of KV1.3 currents to more negative potentials (−50 mV vs. −35 mV in Glu-free solution) andaccelerates the rates of current activation and inactivation (A1). At 1000 μM of Glu, current inactivation becomes prominent and amplitudes are markedly reduced (A2). Theactivation rate is similar for both Glu concentrations, indicating that it remains unaffected by dose escalation. (B) Application of 10 (B1) and 1000 μM (B2) of Glu to T lymphocytes ofpatients with Alzheimer's disease has similar effects on KV1.3 current activation and kinetics. (C) Comparative effects of 10 (C1) and 1000 μΜ (C2) of Glu on the conductance-to-voltage relation (g–V) of KV1.3 channels in patients with Alzheimer's disease and healthy aged individuals. Values are the average of several experiments, fitted by a Boltzmannfunction, and have been normalized to themaximum KV1.3 conductance in Glu-free bath. At 10 μΜ of Glu, themidpoints of activation were−41.8±2mV (n=141 cells) for controlscompared to−42.2±1.9 mV (n=178 cells) for patients, while at 1000 μM of Glu, the values for the two groups were−37.8±2.3 mV (n=110 cells) and−36.5±2.3 mV (n=137cells), respectively. (D) Differential effects of Glu on KV1.3 steady-state inactivation, in patients with Alzheimer's disease vs. aged healthy individuals. In healthy subjects, Glu shiftsthe h–V curves to more hyperpolarized potentials, compared to Glu-free solution. This effect is diminished in T lymphocytes of patients with Alzheimer's disease. At 10 μMof Glu, thecalculatedmidpoints of inactivation were−68.2±2.2mV (n=131 cells) for healthy individuals, compared to−58.1±2.5mV (n=132 cells) for patients with AD (pb0.001, D1). At1000 μMof Glu, the values of the same constant were−72.2±2.8mV (n=95 cells) and−61.4±3.1mV (n=120 cells) for control subjects and patients, respectively (pb0.001, D2).(E) Correlation between the severity of dementia and the effect of Glu on KV1.3 steady-state inactivation. Higher MMSE scores in patients with AD tended to be associated with amore hyperpolarized midpoint of KV1.3 inactivation in the presence of 10 μM of Glu (p=0.07, r2=0.34).
343C. Poulopoulou et al. / Neurobiology of Disease 37 (2010) 339–348
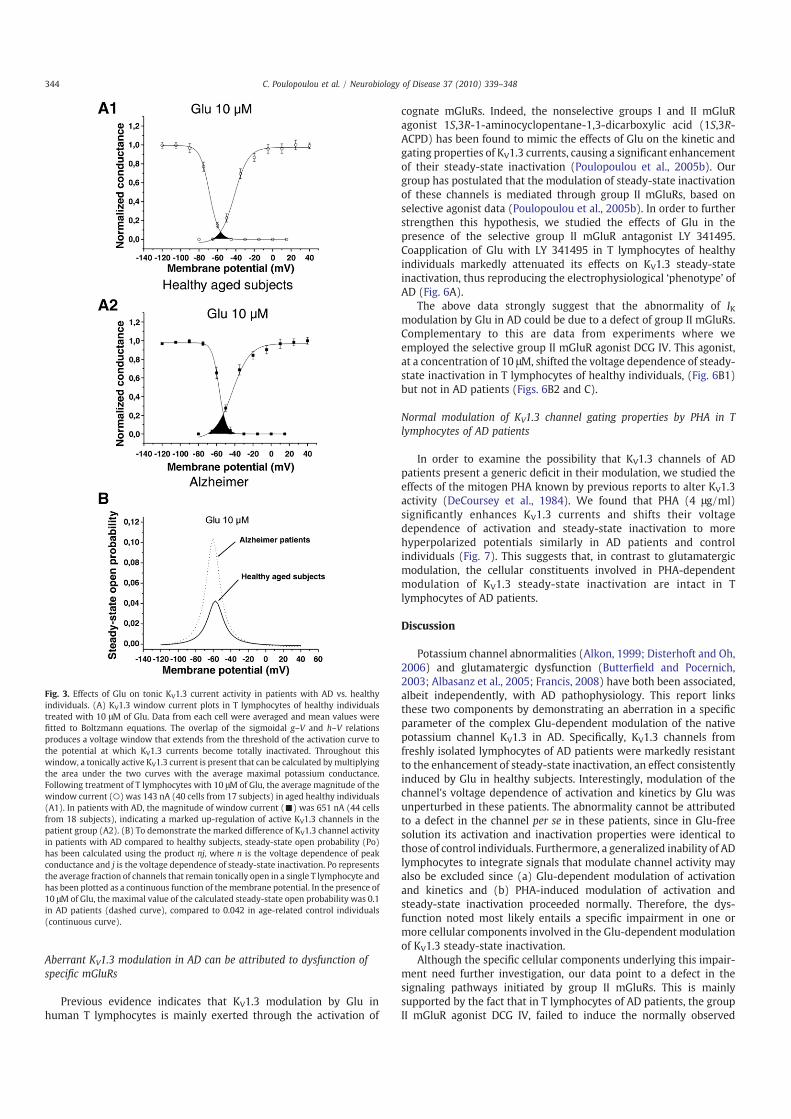
Aberrant KV1.3 modulation in AD can be attributed to dysfunction ofspecific mGluRs
Previous evidence indicates that KV1.3 modulation by Glu inhuman T lymphocytes is mainly exerted through the activation of
cognate mGluRs. Indeed, the nonselective groups I and II mGluRagonist 1S,3R-1-aminocyclopentane-1,3-dicarboxylic acid (1S,3R-ACPD) has been found to mimic the effects of Glu on the kinetic andgating properties of KV1.3 currents, causing a significant enhancementof their steady-state inactivation (Poulopoulou et al., 2005b). Ourgroup has postulated that the modulation of steady-state inactivationof these channels is mediated through group II mGluRs, based onselective agonist data (Poulopoulou et al., 2005b). In order to furtherstrengthen this hypothesis, we studied the effects of Glu in thepresence of the selective group II mGluR antagonist LY 341495.Coapplication of Glu with LY 341495 in T lymphocytes of healthyindividuals markedly attenuated its effects on KV1.3 steady-stateinactivation, thus reproducing the electrophysiological ‘phenotype’ ofAD (Fig. 6A).
The above data strongly suggest that the abnormality of IKmodulation by Glu in AD could be due to a defect of group II mGluRs.Complementary to this are data from experiments where weemployed the selective group II mGluR agonist DCG IV. This agonist,at a concentration of 10 μΜ, shifted the voltage dependence of steady-state inactivation in T lymphocytes of healthy individuals, (Fig. 6B1)but not in AD patients (Figs. 6B2 and C).
Normal modulation of KV1.3 channel gating properties by PHA in Tlymphocytes of AD patients
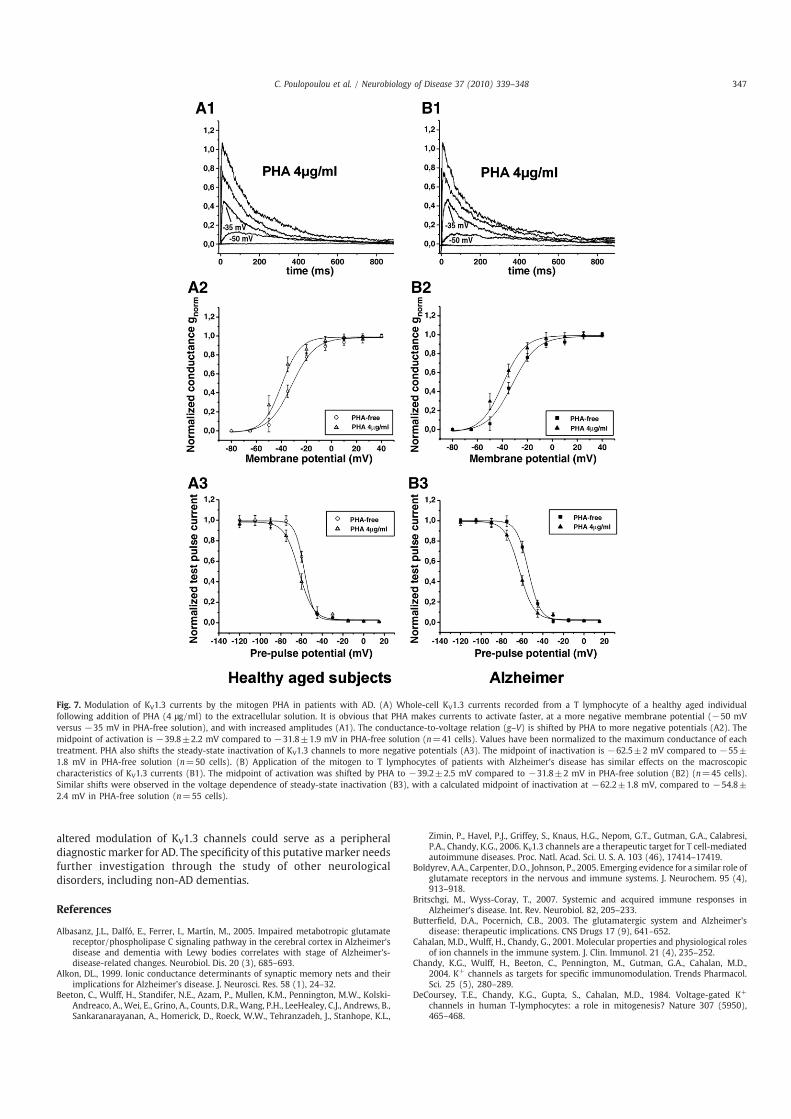
In order to examine the possibility that KV1.3 channels of ADpatients present a generic deficit in their modulation, we studied theeffects of the mitogen PHA known by previous reports to alter KV1.3activity (DeCoursey et al., 1984). We found that PHA (4 μg/ml)significantly enhances KV1.3 currents and shifts their voltagedependence of activation and steady-state inactivation to morehyperpolarized potentials similarly in AD patients and controlindividuals (Fig. 7). This suggests that, in contrast to glutamatergicmodulation, the cellular constituents involved in PHA-dependentmodulation of KV1.3 steady-state inactivation are intact in Tlymphocytes of AD patients.
Discussion
Potassium channel abnormalities (Alkon, 1999; Disterhoft and Oh,2006) and glutamatergic dysfunction (Butterfield and Pocernich,2003; Albasanz et al., 2005; Francis, 2008) have both been associated,albeit independently, with AD pathophysiology. This report linksthese two components by demonstrating an aberration in a specificparameter of the complex Glu-dependent modulation of the nativepotassium channel KV1.3 in AD. Specifically, KV1.3 channels fromfreshly isolated lymphocytes of AD patients were markedly resistantto the enhancement of steady-state inactivation, an effect consistentlyinduced by Glu in healthy subjects. Interestingly, modulation of thechannel's voltage dependence of activation and kinetics by Glu wasunperturbed in these patients. The abnormality cannot be attributedto a defect in the channel per se in these patients, since in Glu-freesolution its activation and inactivation properties were identical tothose of control individuals. Furthermore, a generalized inability of ADlymphocytes to integrate signals that modulate channel activity mayalso be excluded since (a) Glu-dependent modulation of activationand kinetics and (b) PHA-induced modulation of activation andsteady-state inactivation proceeded normally. Therefore, the dys-function noted most likely entails a specific impairment in one ormore cellular components involved in the Glu-dependent modulationof KV1.3 steady-state inactivation.
Although the specific cellular components underlying this impair-ment need further investigation, our data point to a defect in thesignaling pathways initiated by group II mGluRs. This is mainlysupported by the fact that in T lymphocytes of AD patients, the groupII mGluR agonist DCG IV, failed to induce the normally observed
Fig. 3. Effects of Glu on tonic KV1.3 current activity in patients with AD vs. healthyindividuals. (A) KV1.3 window current plots in T lymphocytes of healthy individualstreated with 10 μM of Glu. Data from each cell were averaged and mean values werefitted to Boltzmann equations. The overlap of the sigmoidal g–V and h–V relationsproduces a voltage window that extends from the threshold of the activation curve tothe potential at which KV1.3 currents become totally inactivated. Throughout thiswindow, a tonically active KV1.3 current is present that can be calculated bymultiplyingthe area under the two curves with the average maximal potassium conductance.Following treatment of T lymphocytes with 10 μM of Glu, the average magnitude of thewindow current (○) was 143 nA (40 cells from 17 subjects) in aged healthy individuals(A1). In patients with AD, the magnitude of window current (▪) was 651 nA (44 cellsfrom 18 subjects), indicating a marked up-regulation of active KV1.3 channels in thepatient group (A2). (B) To demonstrate the marked difference of KV1.3 channel activityin patients with AD compared to healthy subjects, steady-state open probability (Po)has been calculated using the product nj, where n is the voltage dependence of peakconductance and j is the voltage dependence of steady-state inactivation. Po representsthe average fraction of channels that remain tonically open in a single T lymphocyte andhas been plotted as a continuous function of the membrane potential. In the presence of10 μM of Glu, the maximal value of the calculated steady-state open probability was 0.1in AD patients (dashed curve), compared to 0.042 in age-related control individuals(continuous curve).
344 C. Poulopoulou et al. / Neurobiology of Disease 37 (2010) 339–348

negative shift in KV1.3 steady-state inactivation; it is furtherstrengthened by the ability of the selective antagonist LY 341495 toreproduce the characteristic AD-related abnormality in T lymphocytesof healthy individuals. Having previously established that theexpression of these receptors in T lymphocytes of AD patients issimilar to that of control individuals (Poulopoulou et al., 2005a), we
are currently examining a possible association of AD with a change intheir functionality or with malfunctioning components in thebiochemical cascade linking mGluR activation to the modulation ofKV1.3 steady-state inactivation. One could speculate on the possiblemolecular mechanisms of mGluR dysfunction in AD, based on dataimplicating amyloid-beta peptides in the impairment of signalingpathways activated by Glu receptors (Tyszkiewicz and Yan, 2005;Parameshwaran et al., 2008; Shankar et al., 2008).
Recent findings indicate that key functions of T lymphocytes aresubject to glutamatergic regulation. Specifically, T cells expressfunctional Glu receptors and transporters, while antigen-presentingcells secrete Glu during their interaction with T lymphocytes in theimmune synapse (Pacheco et al., 2007). This latter finding combinedwith the observation that KV1.3 channels are localized in theimmune synapse during antigen presentation (Beeton et al., 2006)imparts biological significance to the modulation of this channel byGlu in T lymphocytes.
In T lymphocytes, activation of KV1.3 channels sustains thedriving force for calcium entry following stimulation, e.g., antigenpresentation (Chandy et al., 2004). Therefore, the calculated four-foldincrease in tonic KV1.3 channel activity for T lymphocytes of ADpatients in the presence of Glu (due to the decreased steady-stateinactivation) would arm their membrane with an enhanced ability toshunt depolarizing currents. Accordingly, one would predict anincreased driving force for calcium entry in T lymphocytes of ADpatients both in vivo and in vitro (since most lymphocyte culturemedia contain Glu). This prediction is in line with the herein reportedfailure of high Glu concentrations to suppress stimulated T-lymphocyte proliferation in AD patients, as well as with previousstudies reporting enhanced calcium-dependent responses in AD,such as mitogen-induced lymphocyte proliferation (Mattson, 2002;
Fig. 4. Increased KV1.3 current responses of AD patients, in the presence of Glu. (A) Current responses to +40 mV from holding potentials of −90 and −60 mV recorded from a Tlymphocyte of a healthy individual bathed in 10 and 1000 μM of Glu. In the presence of 10 μM of Glu, a decrease in the holding potential from −90 to −60 mV results in a markedreduction of KV1.3 current amplitude due to an increased number of inactivated channels at −60 mV (A1). At 1000 μM of Glu, the number of channels available to activate inresponse to the same depolarizing pulse is minimal, as obvious by the lack of current response when the membrane is stepped to +40mV from a holding potential of−60 mV (A2).(B) Similar recordings performed in a T lymphocyte of a patient with Alzheimer's disease. The diminished ability of Glu to enhance the steady-state inactivation of KV1.3 channels inAD patients is reflected in the increased amplitude of currents elicited from a holding potential of −60 mV. Current responses to +40 mV demonstrate that, in contrast to healthyindividuals, in AD patients, a significant fraction of KV1.3 channels is still available to activate in the presence of 10 and 1000 μM of Glu after being held at −60 mV (B1 and B2).Currents were normalized to the peak current evoked out of −90 mV for each treatment.
Fig. 5. Modulation of T-lymphocyte mitogenic responses by Glu in AD patients. Cellswere activated by PHA (4 μg/ml) in a modified culture medium containing 1 mM of Gluand assayed at 72 hours by a colorimetric BrdU ELISA kit. A marked reduction of PHA-induced proliferationwas observed in T lymphocytes of control individuals (0.58±0.12,n=9 subjects, pb0.01). Conversely, cells from AD patients did not show significantalterations of their proliferation when cultured in the Glu-enriched medium (1.09±0.14, n=8 subjects). Values are mean±SD, normalized to the proliferation that wasobserved in the standard culturemedium (minimal Glu). Inset: Stimulated proliferationrates in the standard culture medium were slightly higher in AD patients but did notdiffer significantly between the two groups. Proliferation index corresponds to the ratioof absorbance for PHA stimulated vs. unstimulated cell cultures.
345C. Poulopoulou et al. / Neurobiology of Disease 37 (2010) 339–348

Schindowski et al., 2007), cytokine levels (Solerte et al., 2000), andexpression of T-cell activation markers (Shalit et al., 1995).
Faulty glutamatergic signaling in the CNS has been postulated as acentral event in AD pathogenesis (Butterfield and Pocernich, 2003;Albasanz et al., 2005). The question arises whether the T-lymphocyteabnormality described here constitutes a peripheral manifestation ofa systemic malfunction in Glu signaling, which in the CNS mayunderlie the cardinal symptoms of AD. In line with this notion ofcommon mechanistic workings of the CNS and the immune systemare the findings that Glu modulates neuronal voltage-gated potas-sium channels (Schrader and Tasker, 1997), while T lymphocytes,microglia, and neurons share common mechanisms of KV1.3 channelmodulation (Fadool and Levitan, 1998). Indeed, the degree of
excitability of olfactory bulb neurons and thus their capacity toencode information (Fadool et al., 2004), as well as the ability ofactivated microglia to kill neurons are both under the regulation ofthe KV1.3 channel (Fordyce et al., 2005).
In summary, we have demonstrated an abnormality in themodulation of the native KV1.3 channel by biologically relevantconcentrations of Glu in AD patients. Although this faulty regulationwas demonstrated in T lymphocytes, it is reasonable to assume that itmay emerge in all instances where this modulatory mechanism isbrought into action for the proper pursuance of physiologicalprocesses. This possibility warrants further investigation in neuraltissue. Furthermore, the consistent presence of this aberration in allAD cells and in none of the controls raises the possibility that the
Fig. 6. Involvement of group II mGluRs in the modulation of KV1.3 steady-state inactivation. (A) No significant effect on steady-state inactivation is observed when Glu (10 μΜ) isapplied in the presence of the selective group II mGluR antagonist LY 341495 (1 μΜ). The midpoint of inactivation was shifted by only −1.2±2.6 mV. Recordings were performedfrom 15 cells of 5 healthy individuals before (○) and after coapplication of Glu (⋆). Prolonged (N10 min) reperfusion of the extracellular solution with Glu (•) shifted the steady-stateinactivation to more negative potentials, as expected. (B) Modulation of KV1.3 steady-state inactivation by the selective group II mGluR agonist DCG IV in AD patients and healthy controls. (B1)Inactivation plots of currents recorded from healthy individuals in the standard bathing solution (○) and following addition of 10 μM of DCG IV (▾). The midpoint of inactivation is shifted to−62.7±3.2 mV compared to −53.8±3 mV in the agonist-free solution (7 individuals, 24 cells). (B2) DCG IV (10 μΜ) fails to enhance KV1.3 steady-state inactivation in AD patients. Themidpoint of inactivation was −55.8±3.7 mV in the presence of DCG IV (C1) compared to −53.8±4.1 mV in the standard bathing solution (8 patients, 25 cells). (C) Differences in the steady-state inactivation between AD patients and healthy individuals become obvious in currents elicited to a +40 mV pulse from a holding potential of −60 mV. In contrast to healthy individuals(C1), current amplitudes in AD patients remain increased (C2). Values were normalized to the peak current evoked out of −90 mV for each treatment.
346 C. Poulopoulou et al. / Neurobiology of Disease 37 (2010) 339–348
altered modulation of KV1.3 channels could serve as a peripheraldiagnostic marker for AD. The specificity of this putativemarker needsfurther investigation through the study of other neurologicaldisorders, including non-AD dementias.
References
Albasanz, J.L., Dalfó, E., Ferrer, I., Martín, M., 2005. Impaired metabotropic glutamatereceptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer'sdisease and dementia with Lewy bodies correlates with stage of Alzheimer's-disease-related changes. Neurobiol. Dis. 20 (3), 685–693.
Alkon, DL., 1999. Ionic conductance determinants of synaptic memory nets and theirimplications for Alzheimer's disease. J. Neurosci. Res. 58 (1), 24–32.
Beeton, C., Wulff, H., Standifer, N.E., Azam, P., Mullen, K.M., Pennington, M.W., Kolski-Andreaco, A., Wei, E., Grino, A., Counts, D.R., Wang, P.H., LeeHealey, C.J., Andrews, B.,Sankaranarayanan, A., Homerick, D., Roeck, W.W., Tehranzadeh, J., Stanhope, K.L.,
Zimin, P., Havel, P.J., Griffey, S., Knaus, H.G., Nepom, G.T., Gutman, G.A., Calabresi,P.A., Chandy, K.G., 2006. Kv1.3 channels are a therapeutic target for T cell-mediatedautoimmune diseases. Proc. Natl. Acad. Sci. U. S. A. 103 (46), 17414–17419.
Boldyrev, A.A., Carpenter, D.O., Johnson, P., 2005. Emerging evidence for a similar role ofglutamate receptors in the nervous and immune systems. J. Neurochem. 95 (4),913–918.
Britschgi, M., Wyss-Coray, T., 2007. Systemic and acquired immune responses inAlzheimer's disease. Int. Rev. Neurobiol. 82, 205–233.
Butterfield, D.A., Pocernich, C.B., 2003. The glutamatergic system and Alzheimer'sdisease: therapeutic implications. CNS Drugs 17 (9), 641–652.
Cahalan, M.D., Wulff, H., Chandy, G., 2001. Molecular properties and physiological rolesof ion channels in the immune system. J. Clin. Immunol. 21 (4), 235–252.
Chandy, K.G., Wulff, H., Beeton, C., Pennington, M., Gutman, G.A., Cahalan, M.D.,2004. K+ channels as targets for specific immunomodulation. Trends Pharmacol.Sci. 25 (5), 280–289.
DeCoursey, T.E., Chandy, K.G., Gupta, S., Cahalan, M.D., 1984. Voltage-gated K+
channels in human T-lymphocytes: a role in mitogenesis? Nature 307 (5950),465–468.
Fig. 7. Modulation of KV1.3 currents by the mitogen PHA in patients with AD. (A) Whole-cell KV1.3 currents recorded from a T lymphocyte of a healthy aged individualfollowing addition of PHA (4 μg/ml) to the extracellular solution. It is obvious that PHA makes currents to activate faster, at a more negative membrane potential (−50 mVversus −35 mV in PHA-free solution), and with increased amplitudes (A1). The conductance-to-voltage relation (g–V) is shifted by PHA to more negative potentials (A2). Themidpoint of activation is −39.8±2.2 mV compared to −31.8±1.9 mV in PHA-free solution (n=41 cells). Values have been normalized to the maximum conductance of eachtreatment. PHA also shifts the steady-state inactivation of KV1.3 channels to more negative potentials (A3). The midpoint of inactivation is −62.5±2 mV compared to −55±1.8 mV in PHA-free solution (n=50 cells). (B) Application of the mitogen to T lymphocytes of patients with Alzheimer's disease has similar effects on the macroscopiccharacteristics of KV1.3 currents (B1). The midpoint of activation was shifted by PHA to −39.2±2.5 mV compared to −31.8±2 mV in PHA-free solution (B2) (n=45 cells).Similar shifts were observed in the voltage dependence of steady-state inactivation (B3), with a calculated midpoint of inactivation at −62.2±1.8 mV, compared to −54.8±2.4 mV in PHA-free solution (n=55 cells).
347C. Poulopoulou et al. / Neurobiology of Disease 37 (2010) 339–348

De Silva, H.A., Aronson, J.K., Grahame-Smith, D.G., Jobst, K.A., Smith, A.D., 1998.Abnormal function of potassium channels in platelets of patients with Alzheimer'sdisease. Lancet 352 (9140), 1590–1593.
Disterhoft, J.F., Oh, M.M., 2006. Pharmacological and molecular enhancement oflearning in aging and Alzheimer's disease. J. Physiol. Paris 99 (2-3), 180–192.
Etcheberrigaray, R., Ito, E., Oka, K., Tofel-Grehl, B., Gibson, G.E., Alkon, D.L., 1993.Potassium channel dysfunction in fibroblasts identifies patients with Alzheimerdisease. Proc. Natl. Acad. Sci. U. S. A. 90 (17), 8209–8213.
Fadool, D.A., Levitan, I.B., 1998. Modulation of olfactory bulb neuron potassium currentby tyrosine phosphorylation. J. Neurosci. 18 (16), 6126–6137.
Fadool, D.A., Tucker, K., Perkins, R., Fasciani, G., Thompson, R.M., Parsons, A.D., Overton,J.M., Koni, P.A., Flavell, R.A., Kaczmarek, L.K., 2004. Kv1.3 channel gene-targeteddeletion produces “super-smeller mice” with altered glomeruli, interactingscaffolding proteins, and biophysics. Neuron 41 (3), 389–404.
Fordyce, C.B., Jagasia, R., Zhu, X., Schlichter, L.C., 2005. Microglia Kv1.3 channelscontribute to their ability to kill neurons. J. Neurosci. 25 (31), 7139–7149.
Francis, P.T., 2008. Glutamatergic approaches to the treatment of cognitive andbehavioural symptoms of Alzheimer's disease. Neurodegener. Dis. 5 (3-4), 241–243.
Garcia-Calvo, M., Leonard, R.J., Novick, J., Stevens, S.P., Schmalhofer, W., Kaczorowski, G.J., Garcia, M.L., 1993. Purification, characterization and biosynthesis of margatoxin,a component of Centruroides margaritatus venom that selectively inhibits voltage-dependent potassium channels. J. Biol. Chem. 268 (25), 18866–18874.
Ghelardini, C., Galeotti, N., Bartolini, A., 1998. Influence of potassium channelmodulators on cognitive processes in mice. Br. J. Pharmacol. 123 (6), 1079–1084.
Giese, K.P., Peters, M., Vernon, J., 2001. Modulation of excitability as a learning andmemory mechanism: a molecular genetic perspective. Physiol. Behav. 73 (5),803–810.
Hamill, O.P., Marty, A., Neher, E., Sakmann, B., Sigworth, F.J., 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-freemembrane patches. Pflügers Arch 391 (2), 85–100.
Ikeda, M., Dewar, D., McCulloch, J., 1991. Selective reduction of [125I]apamin bindingsites in Alzheimer hippocampus: a quantitative autoradiographic study. Brain Res.567 (1), 51–56.
Kourich, S., Mourre, C., Soumireau-Mourat, B., 2001. Kaliotoxin, a KV1.1 and KV1.3channel blocker, improves associative learning in rats. Behav. Brain Res. 120 (1),35–46.
Kues, W.A., Wunder, F., 1992. Heterogeneous expression patterns of mammalianpotassium channel genes in developing and adult rat brain. Eur. J. Neurosci. 4 (12),1296–1308.
Lombardi, G., Dianzani, C., Miglio, G., Canonico, P.L., Fantozzi, R., 2001. Characterizationof ionotropic glutamate receptors in human lymphocytes. Br. J. Pharmacol. 133 (6),936–944.
Marom, S., Levitan, I.B., 1994. State-dependent inactivation of the KV1.3 potassiumchannel. Biophys. J. 67 (2), 579–589.
Mattson, M.P., 2002. Oxidative stress, perturbed calcium homeostasis, and immunedysfunction in Alzheimer's disease. J. Neurovirol. 8 (6), 539–550.
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., Stadlan, E.M., 1984.Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work
Group under the Auspices of Department of Health and Human Services Task Forceon Alzheimer's disease. Neurology 34 (7), 939–944.
Pacheco, R., Gallart, T., Lluis, C., Franco, R., 2007. Role of glutamate on T-cell mediatedimmunity. J. Neuroimmunol. 185 (1-2), 9–19.
Panyi, G., Berecki, G., Gaspar, R., Seres, I., Fulop, T., Damjanovich, S., 1994. Peripheralblood lymphocytes display reduced K+ channel activity in aged humans. Biochem.Biophys. Res. Commun. 199 (2), 519–524.
Parameshwaran, K., Dhanasekaran, M., Suppiramaniam, V., 2008. Amyloid betapeptides and glutamatergic synaptic dysregulation. Exp. Neurol. 210 (1), 7–13.
Plant, L., Webster, N., Boyle, J., Ramsden, M., Freir, D.B., Peers, C., Pearson, H.A., 2005.Amyloid beta peptide as a physiological modulator of neuronal ‘A’-type K+ current.Neurobiol. Aging 27 (11), 1673–1683.
Poulopoulou, C., Davaki, P., Koliaraki, V., Kolovou, D., Markakis, I., Vassilopoulos, D.,2005a. Reduced expression of metabotropic glutamate receptor 2 mRNA in T-cellsof ALS patients. Ann. Neurol. 58 (6), 946–949.
Poulopoulou, C., Markakis, I., Davaki, P., Nikolaou, C., Poulopoulos, A., Raptis, E.,Vassilopoulos, D., 2005b. Modulation of voltage-gated potassium channels inhuman T-lymphocytes by extracellular glutamate. Mol. Pharmacol. 67 (3),856–867.
Schindowski, K., Eckert, A., Peters, J., Gorriz, C., Schramm, U., Weinandi, T., Maurer, K.,Frölich, L., Müller, W.E., 2007. Increased T-cell reactivity and elevated levels of CD8+ memory T-cells in Alzheimer's disease-patients and T-cell hyporeactivity in anAlzheimer's disease-mouse model: implications for immunotherapy. Neuromol.Med. 9 (4), 340–354.
Schrader, L.A., Tasker, J.G., 1997. Modulation of multiple potassium currents bymetabotropic glutamate receptors in neurons of the hypothalamic supraopticnucleus. J. Neurophysiol. 78 (6), 3428–3437.
Shalit, F., Sredni, B., Brodie, C., Kott, E., 1995. T-lymphocyte subpopulations andactivation markers correlate with severity of Alzheimer's disease. Clin. Immunol.Immunopathol. 75 (3), 246–250.
Shankar, G.M., Li, S., Mehta, T.H., Garcia-Munoz, A., Shepardson, N.E., Smith, I., Brett,F.M., Farrell, M.A., Rowan, M.J., Lemere, C.A., Regan, C.M., Walsh, D.M., Sabatini, B.L.,Selkoe, D.J., 2008. Amyloid-beta protein dimers isolated directly from Alzheimer'sbrains impair synaptic plasticity and memory. Nat. Med 14 (8), 837–842.
Solerte, S.B., Ceresini, G., Ferrari, E., Fioravantini, M., 2000. Hemorheological changesand overproduction of cytokines in mild to moderate dementia of the Alzheimer'stype. Neurobiol. Aging 21 (2), 271–281.
Sommer, M.H., Xavier, M.H., Fialho, M.B., Wannmacher, C.M., Wajner, M., 1994. Theinfluence of amino acids onmitogen-activated proliferation of human lymphocytesin vitro. Int. J. Immunopharmacol. 16 (10), 865–872.
Tyszkiewicz, J.P., Yan, Z., 2005. beta-Amyloid peptides impair PKC-dependent functionsof metabotropic glutamate receptors in prefrontal cortical neurons. J. Neurophysiol.93 (6), 3102–3111.
Verheugen, J.A., Korn, H., 1997. A charybdotoxin-insensitive conductance in human T-lymphocytes: T cell membrane potential is set by distinct K+ channels. J. Phys. 503(Pt. 2), 317–331.
Zhang, W., Linden, D.J., 2003. The other side of the engram: experience-driven changesin neuronal intrinsic excitability. Nat. Rev., Neurosci. 4 (11), 885–900.
348 C. Poulopoulou et al. / Neurobiology of Disease 37 (2010) 339–348




















