
Comparison of Exit Moment Spectra for Extrinsic Metric Balls
Upload
independentCategory
view
4download
0
ORIGINAL PAPER
A triterpenediol from Boswellia serrata induces apoptosis throughboth the intrinsic and extrinsic apoptotic pathways in humanleukemia HL-60 cells
Shashi Bhushan Æ Ajay Kumar Æ Fayaz Malik ÆSamar Singh Andotra Æ Vijay Kumar Sethi ÆIndu Pal Kaur Æ Subhash Chandra Taneja ÆGhulam Nabi Qazi Æ Jaswant Singh
� Springer Science+Business Media, LLC 2007
Abstract A triterpenediol (TPD) comprising of isomeric
mixture of 3a, 24-dihydroxyurs-12-ene and 3a, 24-di-
hydroxyolean-12-ene from Boswellia serrata induces
apoptosis in cancer cells. An attempt was made in this
study to investigate the mechanism of cell death by TPD in
human leukemia HL-60 cells. It inhibited cell proliferation
with IC50 ~ 12 lg/ml and produced apoptosis as measured
by various biological end points e.g. increased sub-G0
DNA fraction, DNA ladder formation, enhanced Annex-
inV-FITC binding of the cells. Further, initial events
involved massive reactive oxygen species (ROS) and nitric
oxide (NO) formation, which were significantly inhibited
by their respective inhibitors. Persistent high levels of NO
and ROS caused Bcl-2 cleavage and translocation of Bax to
mitochondria, which lead to loss of mitochondrial mem-
brane potential (Dwm) and release of cytochrome c, AIF,
Smac/DIABLO to the cytosol. These events were associ-
ated with decreased expression of survivin and ICAD with
attendant activation of caspases leading to PARP cleavage.
Furthermore, TPD up regulated the expression of cell death
receptors DR4 and TNF-R1 level, leading to caspase-8
activation. These studies thus demonstrate that TPD
produces oxidative stress in cancer cells that triggers self-
demise by ROS and NO regulated activation of both the
intrinsic and extrinsic signaling cascades.
Keywords Boswellia serrata � Triterpenediol �Apoptosis � HL-60 cells � RNOS � cytochrome c
Abbreviations
DAF-2-DA Diaminofluoresceine-2-diacetate
DCFH-DA 2,7-Dichlorofluoresceine diacetate
LPS Lipopolysaccharide
Dwm Mitochondrial membrane potential
MTT 3-(4,5-Dimethylthiazole-2-yl)-2,5-
diphenyltetrazolium bromide
PBMC Peripheral blood mononuclear cell
NAC N-acetyl-cysteine
iNOS Inducible nitric oxide synthase
PI Propidium iodide
PTP Mitochondrial permeability transition pore
NO Nitric oxide
Rh-123 Rhodamine-123
sMIT s-Methylisothiourea
AIF Apoptosis inducing factor
PMSF Phenylmethanesulfonyl fluoride
ICAD Inhibitor of caspase activating DNAse
Introduction
More than 50% of anti-cancer drugs are directly or indi-
rectly derived from medicinal plants, which still continue
to provide essential source of novel discovery leads [1].
The gum resin of Boswellia serrata, a kind of deciduous
tree grown in the dry part of China and India, has tradi-
tionally been used for the treatment of inflammatory and
arthritic diseases [2]. The pentacyclic triterpenic acids
S. Bhushan � A. Kumar � F. Malik � S. S. Andotra �V. K. Sethi � S. C. Taneja � G. N. Qazi � J. Singh (&)
Division of Pharmacology, Indian Institute of Integrative
Medicine, Canal Road, Jammu 180001, India
e-mail: [email protected]
I. P. Kaur
University Institute of Pharmaceutical Sciences, Panjab
University, Chandigarh 160014, India
123
Apoptosis
DOI 10.1007/s10495-007-0105-5

called boswellic acid such as b-boswellic acid, 11-keto-b-
boswellic acid, 3-O-acetyl-b-boswellic acid and 3-O-ace-
tyl-11-keto-b-boswellic acid in the gum resin are the major
chemical constituents reported for their anti-inflammatory,
anti-proliferative and anticancer activities [3–4]. Structure
activity relationship indicated that pentacyclic ring skele-
ton of boswellic acid is important for anti-topoisomerase
activity [4]. We further isolated another pentacyclic tri-
terpenediol from Boswellia serrata, which exists in nature
as an isomeric mixture of 3a, 24-dihydroxyurs-12-ene and
3a, 24-dihydroxyolean-12-ene (TPD). One of the isomers
3a, 24-dihydroxyurs-12-ene has been earlier isolated and
reported as natural triterpenediol from the gum resin of
Boswellia serrata [5], the same when chemically synthe-
sized was found to contain an isomeric mixture of 3a, 24-
dihydroxyurs-12-ene and 3a, 24-dihydroxyolean-12-ene
(TPD) as it exists in nature in the gum resin of Boswellia
serrata [6]. Some of our preliminary studies have indicated
that TPD is able to inhibit proliferation of a large number
of human cancer cell lines. It was therefore our interest to
find out in-depth the mechanism of action of cell cyto-
toxicity by TPD, particularly in the induction of pro-
grammed cell death (apoptosis). Because deregulation of
apoptosis is the hallmark of all cancer cells and the agents
that activate programmed cell death in cancer cells could
be valuable anticancer therapeutics [7]. Anti-cancer drugs
act through different pathways converging ultimately into
activation of apoptosis in cancer cells leading to cell
cytotoxicity. Recent studies have amply documented that
two major pathways are involved in the regulation of
apoptosis. The drugs may kill cells either by activation of
extrinsic or intrinsic apoptotic pathways. The extrinsic
apoptotic pathway involves cell surface death receptors,
such as Fas/CD95 and TNFR1, which upon activation up
regulate down stream signaling cascade leading to the
activation of caspase-8 [8]. The intrinsic pathway is
dependent on various cell stress stimuli leading to altered
ratio of Bcl-2 family members affecting cytochrome c and
apoptotic protease activating factor-1 (Apaf-1) release that
leads to caspase-9 activation [9]. The active forms of
caspase-8 and caspase-9 may activate downstream effec-
tors caspases-3, -6, and 7. Thus, enable the cleavage of
several intracellular polypeptides as well as activation of
DNase that leads to DNA fragmentation.
Growing evidence suggests that cancer cells exhibit
increased intrinsic ROS stress, due in part to oncogenic
stimulation, increased metabolic activity, and mitochon-
drial malfunction [10]. Since the mitochondrial respiratory
chain (electron transport complexes) is a major source of
ROS generation in the cells, the vulnerability of the
mitochondrial DNA to ROS-mediated damage appears to
be a mechanism to amplify ROS stress in cancer cells
[11]. The escalated ROS generation in cancer cells serves
as an endogenous source of DNA-damaging agents that
promote genetic instability. Many drugs would act as pro-
oxidant, which initially may involve generation of free
radicals such as reactive oxygen/nitrogen species eventu-
ally leading to the activation of apoptosis [12–13]. Several
anti-cancer drugs doxorubicin, mitomycin c, etoposide and
cisplatin are superoxide generating agents [14]. The anti-
estrogen tamoxifen, increasingly used alongside the other
breast cancer therapies, has also been shown to induce
oxidative stress within carcinoma cells in vitro [15]. We
thus became interested to know the mechanism of cancer
cells killing by TPD so that the lead might be developed
into a novel antineoplastic therapeutic. We here report for
the first time the cell cytotoxicity by TPD in human leu-
kemia HL-60 cells and the molecular mechanisms involved
therein. Our studies provide a deeper insight into the events
leading to TPD induced apoptosis in HL-60 cells. The
observed apoptotic activity of TPD is associated with the
early generation of ROS and NO leading simultaneously to
loss of mitochondrial membrane potential, release of pro-
apoptotic factors and activation of caspases, which alto-
gether account for apoptotic cell death.
Materials and methods
Chemicals and antibodies
RPMI-1640 medium, diaminofluoresceine-2-diacetate
(DCF-DA), 2¢, 7¢-dichlorofluoresceine diacetate (DCF-DA),
rhodamine-123 (Rh-123), s-methylisothiourea (sMIT),
propidium iodide (PI), DNase-free RNase, proteinaseK,
3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bro-
mide (MTT), Hoechst 33258, diaminofluorescine 2-diace-
tate (DAF-2DA), ascorbate, PMSF, cyclosporine A, LPS,
eukaryotic protease inhibitor cocktail, N-acetyl cysteine
(NAC), tiron, penicillin, streptomycin, L-glutamine, pyruvic
acid, camptothecin, histopaque-1077 and-1119 were pur-
chased from Sigma chemical Co. Fetal bovine serum was
obtained from GIBCO Invitrogen Corporation (#16000-044,
lot No. 1237517) USA. AnnexinV-FITC apoptosis detection
kit, Caspase-8, -9 inhibitors and ApoAlert caspases assay
kits were from B.D. Clontech, USA. Mouse anti-human
antibodies to Bax (#SC20067), PARP-1 (#SC8007), Bcl-2
(#SC7382), b-Actin (#SC-47778), TNF-R1 (#SC8436),
Survivin (#SC-17779), p21 (#SC-817), goat anti-human
antibodies to DR4 (#SC-6824), ICAD (#SC-6866), goat
anti-rabbit IgG-HRP (#SC2301) and goat anti-mouse IgG-
HRP (#SC2031) were from Santa Cruz Biotechnology,
USA. Rabbit anti-human antibodies to AIF (#PC536),
Smac/DIABLO (#PC-574) and rabbit anti-goat IgG-HRP
(#401504) were from Calbiochem, USA. Mouse anti-human
Apoptosis
123

antibodies to cytochrome c (#556433, clone 7H8.2C12) and
rabbit anti-mouse antibody to iNOS (#N32030-150) were
from BD Biosciences, Pharmingen, USA. Electrophoresis
reagents, Protein estimation kit and protein markers were
from Bio-Rad Laboratories, USA. Hyper film and ECL Plus
western blotting detection kit were from Amersham Bio-
sciences, UK.
Isolation of bioactive triterpenediol 3a, 24-
dihydroxyurs-12-ene and 3a, 24-dihydroxyolean-
12-ene (TPD)
Boswellia serrata gum resin (1 kg) collected from the
forests of Madhya Pradesh, India was cleansed and made
free of extraneous impurities. The gum was extracted with
3 · 3L of ethyl alcohol (95%) at room temperature. The
combined extract after filtration was concentrated under
reduced pressure. The concentrated extract (530 g) was
diluted with 3% potassium hydroxide solution (3 L) and
the mixture stirred for one hour. The aqueous extract thus
obtained was extracted with (2 · 2 L) hexane and then
with 10% ethyl acetate in hexane (1 · 2 L). The combined
organic solvents were washed with water, dried over
anhydrous sodium sulphate and concentrated under re-
duced pressure to give 130 g of viscous liquid, comprising
the neutral components of the gum resin. A portion of the
concentrated extract (20 g) was subjected to repeated col-
umn chromatography over silica gel. Gradient elution with
hexane/ethyl acetate furnished besides other compounds, a
compound with a single spot on thin layer chromatography
(400 mg) mp 182�C, [a] D + 63 (0.26% yield). It was as an
inseparable mixture of two isomers identified by 13CNMR
and 1 HNMR [5]. The ratio of the isomeric mixture was
found to be (80:20), as determined by reverse phase HPLC
on C-18 column (acetonitrile: water; 95:5) and detection at
210 nm. The isomeric mixture may also be obtained by
semi-synthetic route from boswellic acids as described
below.
Preparation of 3a, 24-dihydroxyurs-12-ene and 3a,
24-dihydroxyolean-12-ene (TPD) from boswellic acid
The natural isomeric mixture of (a + b) boswellic acid
(R = H) was first converted to methyl ester (R = Me,
Scheme 1) by diazomethane generated in situ. Thus a
mixture of (a + b) of boswellic acid (230 mg) was dis-
solved in dry solvent ether and diazomethane solution in
diethyl ether was slowly added till completion of reaction
as monitored by TLC. After the removal of the solvent and
excess of diazomethane, the mixture comprising the methyl
esters was re-dissolved in dry solvent ether at room tem-
perature and 20 mg of lithium aluminum hydride (LiAlH4)
was added to the stirring solution under nitrogen atmo-
sphere and stirred for one hour. The excess of LiAlH4 was
neutralized by adding ethyl acetate and the organic layer
washed with water. Further concentrating the solvent gave
the desired product 3a, 24-dihydroxyurs-12-ene and 3a,
24-dihydroxyolean-12-ene in almost quantitative yield
(98%). It was crystallized from hexane-ethyl acetate to get
white crystals of TPD. The product gave similar physico-
chemical and spectra as of the one obtained directly from
the gum resin described above.
Cell culture, growth conditions and treatment
Human promyelocytic leukemia cell line HL-60, human
acute lymphoblastic leukemia cell line MOLT-4, normal
monkey kidney cell line CV-1, human breast cancer cell
line MCF-7 were procured from National Centre for Cell
Sciences (NCCS), Pune, India. Human lung carcinoma cell
line A549, human colon cancer cell line HCT-15, human
cervix carcinoma cell line HeLa and human colon cancer
cell line SW-620 procured from National Cancer Institute,
Frederick, U.S.A. Human PBMC were prepared from the
blood of normal human by using Sigma’s Histopaque-1077
and 1119 solution [16]. Cells were grown in RPMI-1640/
MEM medium containing 10% FCS, 100 unit pencillin/
100 lg streptomycin per ml medium. Cells were grown in
CO2 incubator (Thermocon Electron Corporation, USA) at
37�C with 98% humidity and 5% CO2 gas environment.
Cells were treated with TPD dissolved in DMSO while the
untreated control cultures received only the vehicle
(DMSO, <0.2%).
Preparation of mice peritoneal macrophages
Mice peritoneal macrophages were isolated and cultured as
described earlier [17]. Mice were sacrificed and peritoneal
Scheme 1
Apoptosis
123

exudates cells isolated. The cells were washed twice with
RPMI and finally adjusted to 3 · 106 cells/ml of complete
RPMI medium. Cells were allowed to adhere for 3 h and
non-adherent cells were washed off with PBS. The adher-
ent cells were then treated with the TPD and LPS in the
presence and absence of sMIT (1 mM) for 24 h and the
lysate analyzed for the iNOS expression.
Cell proliferation assay
Cells from suspension and adherent SW-620/MCF-7 cul-
tures grown in 96-well plates were exposed to indicate
concentrations of TPD for 48 h thereafter, 20 ll of MTT
solution (2.5 mg/ml) was added to each well and incubated
at 37�C for 2 h. The plates were centrifuged and the
supernatant was discarded while the MTT-formazon crys-
tals were dissolved in 100 ll DMSO. The OD measured at
570 nm with reference wavelength of 620 nm [18].
DNA fragmentation
Cells (2 · 106) in culture were treated with different con-
centrations of TPD. Cells were lysed, genomic DNA ex-
tracted and electrophoresed as described earlier [18].
Hoechst 33258 staining of cells for nuclear morphology
HL-60 cells (2 · 106 cells/3 ml) were grown in 6 well
plates and treated with TPD for 6 h. Cells were collected,
washed, fixed and stained with Hoechst-33258 and
observed under inverted fluorescence microscope
(Olympus1X70) [18].
Flow cytometric analysis of apoptosis and necrosis
The extent of apoptosis was determined using FITC-la-
beled AnnexinV antibody by flow cytometry [19]. Cells
(1 · 106) were treated with different concentrations of
TPD for 6 h, washed and stained with AnnexinV-FITC
antibody and PI as per the instructions given by the man-
ufacturer. The cells were scanned for fluorescence intensity
in FL-1 (FITC) and FL-2 (PI) channels. The fraction of cell
population in different quadrants was analyzed using
quadrant statistics. Cells in the lower right quadrant rep-
resented apoptosis and in the upper right quadrant repre-
sented necrosis or post apoptotic necrosis [18].
Flow cytometric analysis of intracellular nitric oxide
using DAF-2-DA
Intracellular nitric oxide was measured by employing a low
molecular weight fluorescent probe diaminofluoresceine 2-
diacetate (DAF-2-DA), which is membrane permeable and
usually serves as a reporter of nitric oxide synthase activity.
Immediately after NO is generated inside the cells, it binds
with the chromophore to yield strong fluorescent signal that
can be measured in the green (FL-1) channel of the flow
cytometer [20]. Cells (1 · 106/2 ml/well of 12-well plate)
were pre-incubated for 30 min with DAF-2-DA (10 lM),
and then incubated together with TPD in the presence and
absence of sMIT (1 mM). Cells were collected, washed in
PBS and analyzed on flow cytometer in FL-1 channel for
evaluation of NO positive cell population [18].
Flow cytometric measurement of intracellular
peroxides (ROS) in HL-60 cells
The level of intracellular peroxides was determined by using
2,7-dichlorofluoresceindiacetate [21, 22]. Cells (1 · 106/
2 ml/12 well plate) were incubated with TPD in the pres-
ence and absence of anti-oxidant for 6 h and finally treated
with DCFH-DA (5 lM) for 30 min. Cells were washed in
PBS and incubated with propidium iodide (5 lg/ml) for
15 min. The green DCF-fluorescence was analyzed in the
FL-1 channel (Excitation k 488 nm; Emission k 535 nm)
and PI fluorescence was analyzed in the FL-2 channel.
Flow cytometric determination of mitochondrial
membrane potential
Changes in mitochondrial transmembrane potential (Dwm)
as a result of mitochondrial perturbation were measured
after staining with Rhodamine-123 [18]. Cells (1 · 106/
2 ml/12 well plates) were treated with TPD in the presence
and absence of different ROS/NO inhibitors for 6 h. Rho-
damine-123 (5 lM) was added 1 h before the termination
of experiment. Cells were collected, washed in PBS and
incubated with propidium iodide (5 lg/ml) for 15 min. The
decrease in fluorescence intensity because of mitochondrial
membrane potential loss was analyzed in FL-1 channel.
Cell cycle analysis by flow cytometry
Cells (1 · 106/ml) were treated with different concentra-
tions of TPD for 24 h, fixed in cold 70% alcohol in PBS,
washed, digested with DNase free RNase (400 lg/ml) at
37�C for 45 min and stained with propidium iodide [18].
Cells were analyzed immediately on a SLR flow-cytometer
(Becton Dickinson, USA). The fluorescence intensity of
sub-G0 cell fraction represents the apoptotic cell popula-
tion [23].
Caspase assays
Cells were incubated with TPD in the presence and absence
of ascorbate (5 mM), sMIT (1 mM) and NAC (5 mM) for
Apoptosis
123

indicated time period. Cells were collected, washed in PBS
and lysed in cell lysis buffer. Activities of caspase-3, -8,
and -9/6 in the cell lysates were determined fluorimetrically
using BD Apoalert caspase assay kits as per instructions of
the manufacturer. Specific inhibitors were used as negative
control to determine whether fluorescence intensity chan-
ges were specific for the activity of caspases. The peptide
based inhibitors used were DEVD-CHO for caspase-3,
IETD-fmk for caspase-8 and LEHD-CHO for caspase-9
[18].
Preparation of total cell lysates for immunoblotting
Cells (3 · 106) were collected, washed with cold PBS and
incubated with cold lysis buffer (50 mM Tris pH8.0,
150 mM NaCl, 5 mM EDTA, 1% v/v Nonidet P-40, 1 mM
PMSF and 1% (v/v) eukaryotic protease inhibitor cocktail)
for 30 min on ice [24]. Cells were centrifuged at 12000 · g
for10 min at 4�C and the supernatant was collected as
whole cell lysates for western blot analysis of various
proteins.
Preparation of cytosolic and mitochondrial lysates
Cytosolic fractions for analysis of cytochrome c, Smac/
DIABLO and Bcl-2 immunoblotting were obtained by
selective plasma membrane permeabilization with digito-
nin [25]. Briefly, 2 · 106 cells were lysed for 1–2 min in
lysis buffer (75 mM NaCl, 8 mM Na2HPO4, 1 mM
NaH2PO4, 1 mM EDTA, and 350 lg/ml digitonin and 1%
(v/v) eukaryotic protease inhibitor cocktail). The lysates
were centrifuged at 12,000 · g for 1 min, and the super-
natant collected as cytosolic fraction and stored at –70�C.
Residual pellet was lysed with whole cell lysis buffer by
incubating on ice for 30 min [24]. After centrifugation at
12,000 · g for 10 min at 4�C, cell lysates were transferred
to fresh tubes and stored at –70�C as mitochondrial frac-
tion.
Preparation of nuclear fraction for AIF immunoblotting
HL-60 cells (1 · 107) pellet were washed in cold PBS and
suspended in 400 ll ice-cold hypotonic buffer (10 mM
HEPES/KOH pH 7.9, 2 mM MgCl2, 0.1 mM EDTA,
10 mM KCl, 1 mM dithiotheritol, 0.5 mM PMSF, 1% (v/
v) eukaryotic protease inhibitor cocktail) for 10 min on ice.
Suspension was vortexes and centrifuged at 15,000 · g for
30 s at 4�C. The supernatant was discarded and the cell
pellet was gently resuspended in 100 ll of ice cold saline
buffer (50 mM HEPES/KOH pH7.9, 50 mM KCL,
300 mM NaCl, 0.1 mM EDTA, 10% glycerol, 1 mM DTT,
0.5 mM PMSF, 1% (v/v) eukaryotic protease inhibitor
cocktail) on ice for 20 min. Cells suspension was vortexes
and centrifuged at 15000 · g for 5 min at 4�C [26]. The
supernatant was stored at –70�C as nuclear lysate.
Western blots analysis
The protein lysates prepared were subjected to discontin-
uous SDS-PAGE analysis. Proteins aliquots (50 lg) were
resolved on SDS-PAGE and then electro transferred to
PVDF membrane overnight at 4�C at 30 V. Non-specific
binding was blocked by incubation with 5% non-fat milk in
Tris-buffered saline containing 0.1% Tween-20 (TBST) for
1 h at room temperature. The blots were probed with
respective primary antibodies for 2 h and washed three
times with TBST [27]. The blots were then incubated with
horseradish peroxidase conjugated mouse or rabbit sec-
ondary antibodies for 1 h, washed again three times with
TBST and signals detected using ECL plus chemilumi-
nescence’s kit on X-ray film.
Protein measurement
Protein was measured employing Bio-Rad protein assay kit
using bovine serum albumin as standard.
Statistical analysis
Data expressed as Mean ± SD, unless otherwise indicated.
Comparisons were made between control and treated
groups unless otherwise indicated using unpaired Student’s
t-test and p-values < 0.01 was considered significant.
Results
Inhibition of cell proliferation by TPD
Using a conventional tetrazolium-based colorimetric cell
proliferation assay, we evaluated the cytotoxicity of TPD in
different cancer cells as described earlier [18]. Treatment
of cells with TPD for 48 h produced concentration
dependent inhibition of cell proliferation of HL-60, Molt-4
and SW-620 with IC50 value of approx. 12 lg/ml while
MCF-7 cells were relatively more sensitive with IC50 of
5 lg/ml (Fig. 1A).
TPD induces DNA fragmentation typical of apoptosis
To investigate whether TPD treatment induced DNA
fragmentation, genomic DNA was isolated from TPD
treated HL-60 cells. Apoptosis typically involves intra-
nucleosomal chromatin cleavage by endonucleases in
multiples of 180 bp leading to DNA fragmentation result-
ing in a typical DNA laddering. Cells treated with TPD for
Apoptosis
123

18 h exhibited typical DNA ladder. At low concentration,
DNA ladder was barely visible and this became increas-
ingly prominent in cells treated with higher concentration
of TPD (Fig. 1B). The DNA ladder was diffused inter-
spersed with smear indicative of some post-apoptotic
necrosis in both HL-60 and SW-620 cells treated with
higher concentration of TPD. DNA isolated from untreated
and treated human PBMC or normal monkey kidney CV-1
cells did not show any DNA ladder (Fig. 1B).
TPD induced altered nuclear morphology is rescued by
NAC
Nuclei of untreated HL-60 cells appeared round in shape,
while treatment with TPD resulted in nuclear condensation
and formation of apoptotic bodies. The morphological
changes were accompanied by increase in number of
scattered apoptotic bodies. Pretreatment of cells with
sMIT, an inhibitor of iNOS, and NAC an ROS scavenger
Fig. 1 (A) Influence of TPD on proliferation of various human
cancer cell lines. The cells grown in 96-well culture plate were treated
with different concentration of TPD for 48 h, thereafter cultures
incubated with MTT for 2 h at 37�C. Other conditions are described
in Material and Method. Data are Mean ± SD (n = 8 wells) and
representative of two similar experiments. (B) TPD induced DNA
fragmentation in human cancer and normal cell lines. Genomic DNA
was extracted from cells treated with different concentrations of TPD
for 18 h. (i) HL-60 cells (ii) normal human PBMC and normal
monkey kidney CV-1 cells (iii) SW-620 cells. Other conditions are
described in Materials and Methods. The data are representative one
of two similar experiments. (C) Influence of TPD on nuclear
morphology of HL-60 cells. The cells were treated with 30 lg/ml
TPD for 6 h and stained with Hoechst 33258 and visualized for
nuclear morphology and apoptotic bodies. (a) Untreated control show
normal nuclear morphology; (b) Cells treated with TPD indicate
condensed nuclei, scattered and typical apoptotic bodies formation
(arrows); The TPD induced morphological changes were returned
back by incubation with (c) 5 mM NAC and (d) 1 mM sMIT
Apoptosis
123

rescued the formation of apoptotic bodies in TPD treated
cells (Fig. 1C).
Flow cytometric estimation of TPD induced apoptosis
and necrosis
Exposure of phosphotidyl serine on the surface of cells is
an early event in the onset of apoptosis, which has strong
binding affinity for AnnexinV in the presence of calcium.
HL-60 and Molt-4 cells were incubated with different
concentration of TPD and cells were stained with Annex-
inV-FITC and PI to assess the apoptotic and necrotic cell
population (Fig. 2). TPD produced dose dependant in-
crease in the apoptotic cell population. The basal apoptotic
population in the untreated culture was 3%, which in-
creased to 23% at 50 lg/ml within a short period of 6 h.
Camptothecin, used as positive control, produced about
14% apoptotic population during the same exposure period.
Apoptosis thus appeared to be the primary mode of cell
death induced by TPD.
TPD induces early generation of intracellular nitric
oxide
The extent of NO generation in cells was analyzed by flow
cytometry after staining with DAF-2DA (FL-1). It was
observed that NO levels increased significantly with
increasing concentrations of TPD. Inducible nitric oxide
Fig. 2 Flow cytometric
analysis of TPD induced
apoptosis in HL-60 and MOLT-
4 cells using AnnexinV-FITC/
PI. Cells were incubated with
indicated concentrations of TPD
for 6 h and stained with
annexinV-FITC/PI to analyze
apoptotic and necrotic cell
populations as described in
Materials and Methods. Data are
representative of one of two
similar experiments
Apoptosis
123

synthase (iNOS) inhibitor, sMIT, suppressed remarkably
the NO levels from 84% to 38% (Fig. 3A). The increase in
NO level was also observed in Molt-4 and MCF-7 cells,
which were again protected by sMIT. Whether the increase
in NO is because of iNOS induction, the western blot
analysis was carried out using rabbit anti-mouse antibody
to iNOS, which is also supposed to detect human form,
could not detect the iNOS expression in HL-60 cells [28].
However, when the same antibody was used against TPD
treated mouse macrophages, it was found that TPD induced
the expression of iNOs several fold (Fig. 3B), thereby
indirectly suggesting that NO is derived from iNOS
expression in HL-60 cells.
TPD mediated early ROS generation and influence of
inhibitors
Cells were incubated with TPD for 6 h and analyzed by
flow cytometry after double staining with DCFH-DA and
PI [29]. There was hardly any DCF fluorescence in
the untreated HL-60 cells. However, cells treated with
30 lg/ml of TPD, observed a significant increase in DCF
Fig. 3 (A) TPD induces early
intracellular nitric oxide
generation as shown in cancer
cell lines HL-60, MOLT-4 and
MCF-7. Cells (1 · 106) were
exposed to TPD and NO probe
DAF-2DA for 6 h. The cells
were analyzed by flow-
cytometery for NO fluorescence
in FL-1 channel. Data are
representative of one of two
similar experiments. (B)
Immunoblot analysis of iNOS in
mice peritoneal macrophages
treated with TPD (10, 30 lg)
and LPS (1, 5 lg) with or
without sMIT (1 mM) for 24 h.
Total cell lysates were prepared
and 50 lg protein samples were
loaded on SDS-PAGE gel for
western blot analysis of iNOS
using anti-mouse antibody as
described in Material and
Methods. Data are
representative of one of two
similar experiments
Apoptosis
123

positive cell population by about 75% (Fig. 4A). Ascor-
bate, NAC, tiron and trolox significantly suppressed the
pro-oxidant effect of TPD (30 lg/ml) from 75% to 38, 28,
51 and 38%, respectively. Oxidative burst is a character-
istic event produced by TPD not only in HL-60 cells but
also in other tumor cell lines such as A-549, HCT-15,
Molt-4 and MCF-7 cells examined (Fig. 4B).
Loss of mitochondrial membrane potential by TPD
HL-60 cells exposed to TPD for 6 h were analyzed for
mitochondrial membrane potential (Dwm) loss employing
Rh-123 uptake by flow cytometery. In the untreated control
cells, almost all cells were functionally active with high
Rh-123 fluorescence (Fig. 5A). TPD at 30 lg caused
mitochondrial damage and hence the decrease of mito-
chondrial membrane potential by about 40%. The loss of
Dwm is largely due to the opening of mitochondrial per-
meability transition pores (PTP), which conduit the leakage
of cytochrome c and pro-apoptotic proteins from mito-
chondria to the cytosol. Cyclosporine A (CsA) is a potent
inhibitor of PTP where it inhibits the opening of PTP [30].
Cells incubated with CsA (2 lM) restored the energized
state by about 45% while other ROS inhibitors sparingly
protected cells.
RNOS generation is an early event elicited by TPD
We asked if RNOS generation is an early event in the
induction of apoptosis. In fact, NO generation appeared to
be an early event with parallel ROS production. Loss of
mitochondrial membrane potential (Dwm) and onset of
apoptosis happened to be late subsequent events (Fig. 5B).
The relationship between these important critical events
was determined in the gated cell population exposed to
TPD for different time periods and measured by flow
cytometery. At 30 min there was a significant increase in
NO level and ROS generation while loss of mitochondrial
Fig. 4 (A) TPD mediated early
generation of peroxides in HL-
60 cells and modulation by
various anti-oxidants. Cells
(1 · 106) were treated with
30 lg/ml TPD in the presence
and absence of various ROS
inhibitors for 6 h, followed by
incubation with DCFH-DA
(5 lM) for 30 min. Cells were
analyzed for DCF-fluorescence
on flow-cytometer in the FL-1
(DCF-Fluorescence) vs. FL-2
(PI-Fluorescence) channels.
Data are representative of one of
two similar experiments. (B)
Sensitivity of various human
cancer cell lines towards TPD
mediated generation of
peroxides. Cells (1 · 106) after
various treatments with TPD in
the presence and absence of
NAC (5 mM) for 6 h were
incubated with DCFH-DA
(5 lM) for 30 min. Cells were
analyzed for DCF-fluorescence
as described above. Data are
Mean ± S.D. from three similar
experiments. P-value: **<0.001
compared to untreated control;
@ < 0.01 compared to TPD,
30 lg treated cells;
@@ < 0.001 compared to TPD,
30 lg treated cells
Apoptosis
123
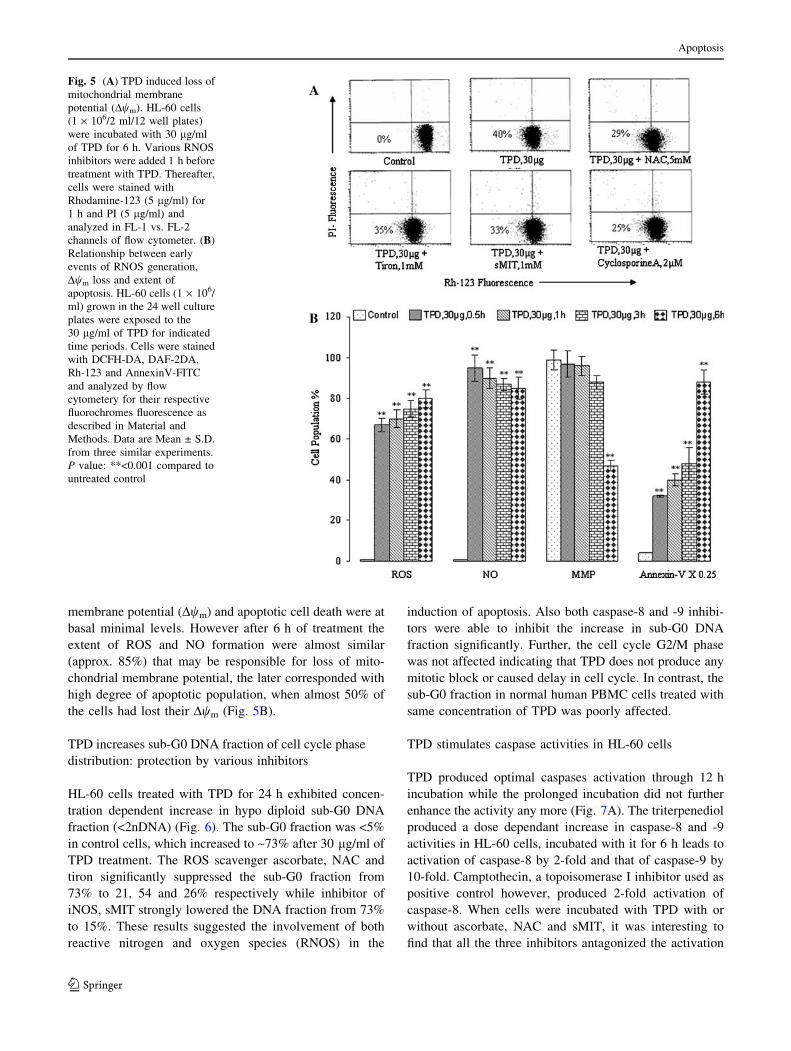
membrane potential (Dwm) and apoptotic cell death were at
basal minimal levels. However after 6 h of treatment the
extent of ROS and NO formation were almost similar
(approx. 85%) that may be responsible for loss of mito-
chondrial membrane potential, the later corresponded with
high degree of apoptotic population, when almost 50% of
the cells had lost their Dwm (Fig. 5B).
TPD increases sub-G0 DNA fraction of cell cycle phase
distribution: protection by various inhibitors
HL-60 cells treated with TPD for 24 h exhibited concen-
tration dependent increase in hypo diploid sub-G0 DNA
fraction (<2nDNA) (Fig. 6). The sub-G0 fraction was <5%
in control cells, which increased to ~73% after 30 lg/ml of
TPD treatment. The ROS scavenger ascorbate, NAC and
tiron significantly suppressed the sub-G0 fraction from
73% to 21, 54 and 26% respectively while inhibitor of
iNOS, sMIT strongly lowered the DNA fraction from 73%
to 15%. These results suggested the involvement of both
reactive nitrogen and oxygen species (RNOS) in the
induction of apoptosis. Also both caspase-8 and -9 inhibi-
tors were able to inhibit the increase in sub-G0 DNA
fraction significantly. Further, the cell cycle G2/M phase
was not affected indicating that TPD does not produce any
mitotic block or caused delay in cell cycle. In contrast, the
sub-G0 fraction in normal human PBMC cells treated with
same concentration of TPD was poorly affected.
TPD stimulates caspase activities in HL-60 cells
TPD produced optimal caspases activation through 12 h
incubation while the prolonged incubation did not further
enhance the activity any more (Fig. 7A). The triterpenediol
produced a dose dependant increase in caspase-8 and -9
activities in HL-60 cells, incubated with it for 6 h leads to
activation of caspase-8 by 2-fold and that of caspase-9 by
10-fold. Camptothecin, a topoisomerase I inhibitor used as
positive control however, produced 2-fold activation of
caspase-8. When cells were incubated with TPD with or
without ascorbate, NAC and sMIT, it was interesting to
find that all the three inhibitors antagonized the activation
Fig. 5 (A) TPD induced loss of
mitochondrial membrane
potential (Dwm). HL-60 cells
(1 · 106/2 ml/12 well plates)
were incubated with 30 lg/ml
of TPD for 6 h. Various RNOS
inhibitors were added 1 h before
treatment with TPD. Thereafter,
cells were stained with
Rhodamine-123 (5 lg/ml) for
1 h and PI (5 lg/ml) and
analyzed in FL-1 vs. FL-2
channels of flow cytometer. (B)
Relationship between early
events of RNOS generation,
Dwm loss and extent of
apoptosis. HL-60 cells (1 · 106/
ml) grown in the 24 well culture
plates were exposed to the
30 lg/ml of TPD for indicated
time periods. Cells were stained
with DCFH-DA, DAF-2DA,
Rh-123 and AnnexinV-FITC
and analyzed by flow
cytometery for their respective
fluorochromes fluorescence as
described in Material and
Methods. Data are Mean ± S.D.
from three similar experiments.
P value: **<0.001 compared to
untreated control
Apoptosis
123

of caspase-8 and -9 equipotently. This again confirms that
pro-oxidant environment induced by TPD is responsible for
sustained stimulation of caspases in leukemia cells. Spe-
cific inhibitors for caspases were utilized in the assay
system to corroborate the specificity of caspase activities
investigated (Fig. 7B).
TPD induced mitochondrial dysfunction is associated
with translocation of pro-apoptotic molecules with
concomitant cleavage of Bcl-2
Members of Bcl-2 family of proteins are known to control
the release of cytochrome c from mitochondria [31].
Therefore we investigated the effect of TPD on Bcl-2
levels in HL-60 cells and it was interesting to find out that a
truncated 23 kDa form of Bcl-2 appeared in cytosolic
fraction (Fig. 8A). The appearance of cleaved Bcl-2 pro-
tein may represent a caspase dependant transformation of
28 kDa (anti-apoptotic) to 23 kDa (pro-apoptotic) Bcl-2
[32, 33]. The altered Bcl-2 level by TPD was significantly
reversed by NAC and sMIT. Further TPD increased the
release of Smac/DIABLO and cytochrome c in to the
cytosol with simultaneous translocation of Bax to mito-
chondria. It is mentioned that Smac/DIABLO is a proa-
poptotic protein, which is released from the mitochondria
during the intrinsic pathway of apoptosis. Translocation of
cytochrome c, Bax, Smac/DIABLO and Bcl-2 cleavage
were both time and concentration dependant and this effect
was significantly attenuated by the NAC and sMIT. TPD
significantly decrease the Bcl-2/Bax ratio (82%) in the
mitochondria from 2.51 to 0.47, which was significantly
reverted back by NAC and sMIT (Fig. 8B).
TPD activates extrinsic signaling cascade by up
regulation of apical death receptors
HL-60 cells treated with TPD exhibited increased expres-
sion of death receptor DR4 and TNF-R1 levels (Fig. 9A, B).
TNF-R1 and DR4 activation seems to be the early events
however, as postulated that NO might be responsible for
TNF-R1 over expression [33], this effect on the contrary
was not inhibited by the sMIT, but in the presence of both
sMIT and NAC, its over expression was inhibited signifi-
cantly (Fig. 9B). Nevertheless, induction of TNF-R1, DR4
and caspase-8 activity indicated that TPD might also be
contributing substantially to the apoptotic death machinery
by activation of extrinsic signaling cascade.
Effect of TPD on other apoptotic related genes
expression
The triterpenediol at 30 lg also increased the release of
apoptosis inducing factor (AIF) into the nuclei of HL-60
cells (Fig. 10A). AIF is a mitochondrial flavoprotein that is
capable of inducing chromatin condensation following an
apoptotic stimulus. Our studies also demonstrate that TPD
induces p21 over expression in HL-60 cells (Fig. 10B). p21
is a cyclin-dependent kinase (CDK) inhibitor, which is best
Fig. 6 DNA Cell cycle
analyses in TPD treated HL-60
cells and influence of RNOS
inhibitors. Cells were exposed
to different concentrations of
TPD for 24 h in the presence
and absence of various
inhibitors. All the inhibitors
were added 1 h before TPD
treatment. Cells were stained
with PI to determine DNA
fluorescence and cell cycle
phase distribution as described
in Materials and Methods.
Fraction of cells for hypo-
diploid (sub-G0, <2n DNA)
population analyzed from FL2-
A vs. cell counts is shown (%).
Data are representative of one of
two similar experiments
Apoptosis
123

known for its ability to regulate cell cycle, besides its ef-
fects on apoptosis and differentiation [34]. The CDK
inhibitor p21 increases intracellular levels of ROS both in
normal fibroblasts and in p53-negative cancer cells [35].
Over expression of p21 was impaired by NAC in the
present studies (Fig. 10B) suggesting that induction of p21
and ROS by TPD are complementary to each other.
Regulation of Survivin, ICAD and PARP levels in HL-
60 cells during TPD treatment
Survivin is a cell cycle regulated inhibitor of apoptosis
protein (IAP), mostly over expressed in most of tumors. It
is highly expressed in HL-60 cells, which TPD decreased
over a period of treatment. TPD mediated release of
SMAC/DIABLO from mitochondria might be expected to
down-regulate the expression of survivin and this was
observed in our studies. However, after exposure of 24 h,
the protein marginally increased, which was rescued effi-
ciently by NAC and sMIT (Fig. 11A). Another important
protein related to DNA fragmentation is caspase-activated
DNase (CAD). CAD complexes with an inhibitor of CAD,
(ICAD) also called DFF-45, in non-apoptotic growing
cells. TPD produced time related decrease in its expression,
which was protected by RNOS inhibitors. ICAD is an
acidic protein, with two recognition sites for caspase 3.
When caspase 3 is activated by apoptotic stimuli, ICAD is
cleaved, resulting in the release of CAD. This released
CAD appears to cause DNA fragmentation in the nucleus
of the cell [36]. Activated caspase-3 also uses poly ADP
ribose polymerase (PARP) as a substrate during DNA re-
pair [37]. TPD at 10 lg and 30 lg induced cleavage of
PARP in HL-60 cells (Fig. 11B). Cleavage of PARP,
116 kDa into 89 kDa, started in less than 3 h and the
uncleaved 116 kDa protein was completely cleaved into
89 kDa through 24 h treatment. Co-treatment with NAC or
sMIT resulted in a significant protection against TPD in-
duced PARP cleavage and ICAD down-regulation
(Fig. 11B).
Discussion
The results of the present study describe the pro-apoptotic
activity of a novel triterpenediol isomeric mixture (TPD),
isolated from the Boswellia serrata on human leukemia
HL-60 cells, for the first time. This is evidenced from the
fact that TPD inhibited cell proliferation and induced
apoptosis measured by various biological end points such
as DNA fragmentation, apoptotic body’s formation and
annexin-V binding in HL-60 cells. All these biological end-
points indicated that the cancer cells death is due to the
induction of apoptosis by TPD while no such effect in
terms of DNA fragmentation was observed in human
PBMC and monkey kidney normal cell line CV-1. Con-
sidering the potential that TPD offers in its development as
anticancer agent, we further sought to understand early
events associated with apoptotic cell death. We observed
Fig. 7 (A) TPD induced differential activation of various caspases in
HL-60 cells. The cells in culture were exposed to 30 lg/ml of TPD
for indicated time periods. The caspases activities were determined
fluorometrically in the cell lysate of HL-60 cells using BD ApoAlert
caspase assay kits as described in Materials and Methods. All assays
were performed according to the instructions provided by the
supplier. Specific inhibitors were used for negative control as
described in Materials and Methods. Data are Mean ± S.D. from
three similar experiments. P values: **<0.001 compared to untreated
control, @@<0.001 compared to TPD treated cells. (B) Influence of
RNOS inhibitors on TDP stimulated caspase activities. HL-60 cells
(2 · 106/2 ml) were exposed to different concentration of TPD for
6 h for assays of caspase-8 and -9 activities. Cultures were pre-
exposed for 1 h to sMIT, NAC and ascorbate prior to TPD treatment.
Camptothecin was used as positive control. Other conditions were
same as described in Fig. 7A
Apoptosis
123

that TPD induced early generation of reactive nitrogen or
oxygen species (RNOS), which may account for various
consequences leading to cell death. Though confronting
Fig. 8 (A) Influence of TPD on
the expression of important
proteins involved in the
initiation of apoptosis. HL-60
cells (2 · 106) were treated with
30 lg/ml of TPD for indicated
time periods in the presence and
absence of NAC and sMIT. b-
actin was used as internal
control to represent the same
amount of proteins applied for
SDS-PAGE. Specific antibodies
were used for detection of
cytochrome c, Smac/DIABLO,
Bax and Bcl-2. (B) Influence of
TPD on the Bcl-2/Bax ratio in
the mitochondrial fraction of
HL-60 cells. Data are
representative of one of two
similar experiments. P values:
**<0.001 compared to untreated
control, @@ < 0.001 compared
to TPD treated cells
Fig. 9 Western blot analysis of TNF-R1 and DR4 were performed in
total cell lysates of TPD treated HL-60 cells. Cells (3 · 106) were
treated with 30 lg/ml of TPD for indicated time periods (A) and for
24 h in the presence and absence of indicated inhibitors (B) Other
conditions were same as described in Fig. 8
Fig. 10 (A) Immunoblot analysis of AIF in nuclear fraction of TPD
treated HL-60 cells. Nuclear fraction was prepared and protein
(50 lg) was resolved on 10% SDS-PAGE gel for western blot
analysis as described in Fig.8. (B) Western blot analysis of p21 was
performed in total cell lysate of HL-60 cells. Cells (2 · 106) were
treated with 30 lg/ml of TPD for indicated time periods in the
presence and absence of NAC and sMIT. Other conditions were same
as described in Fig. 8
Apoptosis
123

opinions have been advanced about the role of NO in cells,
current research however, has shown that NO plays an
important role in regulating electron transport chain [12],
while its overproduction would stimulate formation of a
variety of free oxidant radicals by mitochondria leading to
inhibition of electron transport chain and hence electron
leakage. This would result in mitochondrial oxidative stress
[9, 11], which may lead to apoptosis and cell cytotoxicity.
The production of NO in leukemia cells is presumably
through iNOS induction because several human myeloid
and T lymphoblast leukemia cells are known to generate
NO, under different stimuli [38]. Further, early NO for-
mation is known to stimulate mitochondrial superoxide
generation, which we observed simultaneously in TPD
treated HL-60 cells. Interestingly both the ROS and iNOS
inhibitors had a protective effect on the DNA damage in-
duced by TPD, suggesting that TPD is triggering a pro-
oxidant effect in cancer cells.
The biological consequences of such interactions i.e.
both NO and ROS production can produce deleterious ef-
fects on mitochondrial membrane potential, Dwm [9, 39–
40]. TPD induces loss of mitochondrial membrane poten-
tial more prominently after 6 h treatment that corroborated
with increase in apoptosis. The loss of Dwm by TPD was
inhibited substantially by CsA [41] indicating involvement
of mitochondrial PTP in the release of proteins. Several
studies in the past have suggested that generation of RNOS
and abrogation of mitochondrial membrane potential lead
to the activation of caspases. TPD induces the activation of
caspase-8 and 9 in HL-60 cells, which were inhibited
significantly by ascorbate, NAC and sMIT. It may be
mentioned that TPD increased remarkably caspase -9
activity and its activation is solely regulated by mito-
chondrial release of apoptotic protease activating factor-1
(Apaf-1) and cytochrome c, which is a part of mitochon-
drial dependant apoptosis pathway [7]. TPD also induced
the caspase-8 activation in HL-60 cells because of up-
stream over-expression of cell surface receptors such as
TNF-R1 and DR-4 [42]. TPD induced over-expression of
TNF-R1 and DR4, was significantly inhibited by NAC and
sMIT, suggesting again the involvement of pro-oxidant
effect of TPD in the extrinsic signaling pathway of apop-
tosis in HL-60 cells.
In view of caspase-9 activation by TPD, we further
investigated the role of members of Bcl-2 family of pro-
teins and release of proapoptotic proteins from mitochon-
dria. Bcl-2 family proteins play both pro-apoptotic and
anti-apoptotic role in cancer cells. Normally Bcl-2 level is
much higher in cancer cell. We observed cleavage of the
Bcl-2 in cytosolic fraction of TPD treated HL-60 cells
suggesting that RNOS might have oxidatively modified
Bcl-2 protein rendering it susceptible to proteolytic
cleavage, probably by caspase-3 [43]. This is evident from
the fact that both NAC and sMIT rescued TPD modified
Bcl-2 cleavage in HL-60 cells. The cleavage of Bcl-2
obviously would shift the balance in the direction of
apoptosis. Because Bcl-2 is a substrate for activated cas-
pase-3 and the cleaved product is a 23 kDa Bax like pro-
tein, which in fact is pro-apoptotic in nature. Moreover,
cleavage of Bcl-2 is an early event in apoptosis cascade and
it is an important signal for cytochrome c release upon
specific cellular insults [32, 33]. The drugs enabling Bcl-2
cleavage also are known to accelerate apoptosis in various
cell types [44, 45]. Cleavage of Bcl-2 by TPD changes the
symmetry of mitochondria, which causes the opening of
mitochondrial transpermeable pore and brings about Bax
translocation from cytosol to mitochondria and consequent
release of small molecules like AIF, Smac/DIABLO and
cytochrome c from mitochondria to cytosol. Apoptosis
inducing factor (AIF), a 57 kDa mitochondrial flavoprotein
is released from the mitochondria and translocated to the
nucleus after an apoptotic signal where it is capable of
inducing nuclear chromatin condensation and large scale
DNA fragmentation, associated with increase in ROS for-
mation [46, 47]. Besides AIF translocation, TPD also in-
creased both cytochrome c and Bax levels in cytosolic and
mitochondrial fraction, respectively. Translocation of these
proteins impairs mitochondrial functions and brings the
cells to a ‘‘point of no return’’ to enter apoptosis via acti-
vation of caspases. Along with cytochrome c there is
simultaneous release of Smac/DIABLO in high amounts in
TPD treated cells, which not only promotes the proteolytic
activation of procaspase-3 by neutralizing the activity of
IAP such as survivin [48] but also down regulates the
Fig. 11 (A) Western blot analysis of survivin, ICAD and PARP were
performed in total cell lysates of HL-60 cells for indicated time
periods in the presence and absence of NAC and sMIT. Immuno-
blotting analysis was performed as described in Fig. 8. Data are
representative of one of two similar experiments. (B) Effect of low
concentration of TPD (10 lg/ml) on the PARP cleavage and
protection by RNOS inhibitors. Cells (2 · 106) were treated with of
TPD for indicated time periods in the presence and absence of NAC
and sMIT. Other conditions were same as described in Fig. 8
Apoptosis
123

expression of survivin. Elevated levels of survivin are
reported in cancer cells, where it inhibits the pro-caspases
from becoming functionally active [49]. So, by inhibiting
the survivin, TPD reinforces the caspase activity and finally
enhances apoptosis. It is also reported that p21 increases
intracellular levels of ROS both in normal fibroblasts and
in p53-negative cancer cells [35]. The p21 protein is a
cyclin-dependent kinase (CDK) inhibitor, which is best
known for its ability to regulate the cell cycle [34]. TPD
up-regulation of p21 level in HL-60 cells, suggests that the
triterpenediol may be inhibiting CDKs through induced
level of p21. NAC significantly inhibited the effects of
TPD on p21.
The elevated level of caspase-3 could utilize poly-ADP
ribose polymerase (PARP, 116 kDa), a DNA repair
enzyme as its substrate [37]. As a result a cleaved product
(85 kDa) of PARP was observed in our studies. When
caspase-3 was activated by apoptotic stimuli, ICAD got
cleaved, resulting in the release of CAD. This released
CAD appears to cause DNA fragmentation in nuclei [36].
TPD induced the PARP cleavage and inhibited the over
expression of ICAD in HL-60 cells and this effect again
was substantially protected by the NAC and sMIT. So these
finding suggest that TPD caused early NO and ROS pro-
duction are responsible for the induction of apoptosis
through both intrinsic and extrinsic apoptotic pathway.
Moreover TPD appeared to accelerate apoptosis more
efficiently through intrinsic pathway than the extrinsic one
because factors related to the intrinsic pathway were rela-
tively, strongly affected by sMIT and NAC. Though we
have analyzed the expression of various pro- and anti-
apoptotic proteins altered by TPD, these studies neverthe-
less provide important information about the pro apoptotic
nature of TDP prospecting this candidate for developing
into a potential anti-cancer therapeutic.
Conclusion
In conclusion, TPD, a novel and natural triterpenediol from
Boswellia serrata was found to induce early endogenous
NO and peroxide formation in leukemia HL-60 cells. The
oxidative stress produced by TPD, because of massive
ROS and NO generation, caused activation of the TNF
family proteins (TNF-R1, DR4), leads to caspase-8 acti-
vation and induces apoptosis through extrinsic pathway.
Secondly, TPD caused disruption of mitochondrial mem-
brane potential, rendered Bcl-2 cleavage, Bax transloca-
tion, decrease Bcl-2/Bax ratio, release of AIF, Smac/
DIABLO and cytochrome c from the mitochondria. These
events are accompanied by down regulation of survivin and
activation of caspases -3, 8, -9, which cleave the ICAD and
PARP and finally induce apoptosis through intrinsic path-
way. It was so interesting that TPD did not exert any
cytotoxic effect on the normal cells as confirmed by the
DNA fragmentation and cell cycle analysis in human
PBMC. These results provide the basis for further in-depth
drug targeted studies, while the pro-apoptotic feature of
triterpenediol raises the potential use of TPD as an anti-
tumor agent.
Acknowledgements We are highly thankful to Dr. Sarang Bani and
Sheikh Fayaz (SRF, CSIR) for their help in the use of Flow
Cytometery.
References
1. Lee KH (1999) Anticancer drug design based on plant-derived
natural products. J Biomed Sci 6:236–250
2. Han R (1994) Highlight on the studies of anticancer drugs derived
from plants in China. Stem Cells 12:53–63
3. Safayhi H, Mack T, Sabieraj J, Anazodo MI, Subramanian LR,
Ammon HP (1992) Boswellic acids: novel, specific, non-redox
inhibitors of 5-lipoxygenase. J Pharmacol Exp Ther 26:1143–
1146
4. Hoernlein RF, Orlikowsky TH, Zehrer C et al. (1999) Acetyl-11-
keto-beta-boswellic acid induce apoptosis in HL-60 and CCRF-
CEM cells and inhibit topoisomerase-I. J Pharmacol Exp Ther
288:613–619
5. Mahajan B, Taneja SC, Sethi VK, Dhar KL (1995) Two triterp-
enoids from Boswellia serrata gum resin. Phytochemistry
39:453–455
6. Allan GG (1968) The Stereochemistry of the Boswellic Acids.
Phytochemistry 7:963–973
7. Debatin KM (2000) Activation of apoptosis pathways by anti-
cancer treatment. Toxicol Lett 112/113:41–48
8. Wilson MR (1998) Apoptotic signal transduction: emerging
pathways. Biochem Cell Biol 76:573–582
9. Sun XM, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen
GM (1999) Distinct caspase cascades are initiated in receptor-
mediated and chemical induced apoptosis. J Biol Chem 274:5053–
5060
10. Pelicano H, Carney D, Huang P (2004) ROS stress in cancer cells
and therapeutic implication. Drug Resistance Updates 7:97–110
11. Ghafourifar P, Brimgold U, Klein SD, Richter C (2001) Mito-
chondrial nitric oxide synthase, oxidative stress and apoptosis.
Biol Signals Recept 10:57–65
12. Chun-Qi L, Wogan GN (2005) Nitric oxide as modulator of
apoptosis. Cancer Letters 226:1–15
13. Kellner C, Zunin SJ (2004) Nitric oxide is synthesized in acute
leukemia cell, after exposure to phenolic antioxidants and ini-
tially protects against mitochondrial membrane depolarization.
Cancer Lett 215:43–52
14. Szewczyk A, Wojtczak L (2002) Mitochondria as a pharmaco-
logical target. Pharmacol Rev 54:101–127
15. Ferlini C, Scambia G, Marone M et al. (1999) Tamoxifen induces
oxidative stress and apoptosis in oestrogen receptor negative
human cancer cell lines. Br J Cancer 79:257–263
16. English D, Andersen BR (1974) Single-step separation of red
blood cells, granulocytes and mononuclear leukocyte on discon-
tinuous density gradient of ficoll-hypaque. J Immunol Method
5:249–252
17. Malik F, Singh J, Khajuria A et al (2007) A standardized root
extract of Withania somnifera and its major constituent withan-
olide-A solicit humoral and cell-mediated immune responses by
Apoptosis
123

up regulation of Th1-dominant polarization in BALB/c mice. Life
Sci 80:1525–1538
18. Bhushan S, Singh J, Rao MJ, Saxena AK, Qazi GN (2006) A
novel lignan composition from Cedrus deodara induces apoptosis
and early nitric oxide generation in human leukemia Molt -4 and
HL-60 cells. Nitric oxide 14:72–88
19. Vermes I, Haanen C, Steffen-Nakken H, Reutellingsperger C
(1995) A novel assay for apoptosis, Flow cytometric detection of
phosphatidylserine expression on early apoptotic cells using
fluorescein labeled AnnexinV. J Immunol Meth 184:39–51
20. Kojima H, Sakurai K, Kikuchi K et al (1998) Development of a
fluorescent indicator for the nitric oxide based on the fluorescein
chromophore. Chem Pharm Bull 46:373–375
21. Rothe G, Valet G (1996) Flow cytometric analysis of respiratory
burst activity in phagocytes with hydroethidine and 2¢, 7¢-di-
chlorofluorescein. J Leukocy Biol 47:440–448
22. Royall JA, Ischiropoulos H (1993) Evaluation of 2¢, 7¢-dichlo-
rofluorescein and dihydrorhodamine-123 as fluorescent probes for
intracellular H2O2 in cultured endothelial cells. Arch Biochem
Biophys 302:348–355
23. Yu Z, Li W (2006) Induction of apoptosis by puerarin in colon
cancer HT-29 cells. Cancer Lett 238:53–60
24. Findley HW, Gu L, Yeager AM, Zhou M (1997) Expression and
regulation of Bcl-2, Bcl-xl, and Bax correlate with p53 status and
sensitivity to apoptosis in childhood acute lymphoblastic leuke-
mia. Blood 89:2986–2993
25. Wang Z, Wang S, Dai Y, Grant S (2002) Bryostatin 1 increases
1-D- arabinofuranosylcytosine-induced cytochrome c release and
apoptosis in human leukemia cells ectopically expressing Bcl-xL.
J Pharmacol Exp Ther 301:568–577
26. Shina TY, Kimb SH, Sukb K et al (2005) Anti-allergic effects of
Lycopus lucidus on mast cell-mediated allergy model. Toxicol
Appl Pharmacol 209:255–262
27. Lin HI, Lee YJ, Chen BF et al (2005) Involvement of Bcl-2
family, cytochrome c in caspase 3 in induction of apoptosis by
beauvericin in human non-small cell lung cancer cells. Cancer
Lett 230:248–259
28. Coers W, Timens W, Kempinga C, Klok PA, Moshage H (1998)
Specificity of antibodies to nitric oxide synthase isoforms in
human, guinea pig, rat, and mouse tissues. J Histochem Cyto-
chem 46:1385–1391
29. Borutaite V, Brown GC (2003) Nitric oxide induces apoptosis via
hydrogen peroxide but necrosis via energy and thiol depletion.
Free Radic Biol Med 35:1457–1468
30. Broekemeier KM, Dempsey M, Pfeiffer DR (1989) Cyclosporin
A is a potent inhibitor of the inner membrane permeability
transition in liver mitochondria. J Biol Chem 264:7826–7830
31. Cory. S, Adams JM (2002) Bcl-2 family: regulation of the
cellular life or death switch. NatRevCancer 2:647–656
32. Cheng EH, Kirsh DG, Clem RJ, Ravi RR, Kastan MB, Bedi A
(1997) Conversion of Bcl-2 to a Bax-like death effector by
caspases. Science 278:1966–1968
33. Wan CK, Wang C, Cheung HY, Yang M, Fong WF (2005)
Triptolide induces Bcl-2 cleavage and mitochondria depen-
dant apoptosis in p53-deficient HL-60 cells. Cancer Lett
241:31–41
34. Ghanem L, Steinman R (2005) A proapoptotic function of p21 in
differentiating granulocytes. Leukemia Res 29:1315–1323
35. Macip S, Igarashi M, Fang L et al. (2002) Inhibition of p21-
mediated ROS accumulation can rescue p21-induced senescence.
The EMBO J 21:2180–2188
36. Sakahira H, Enari M, Nagata S (1999) Functional differences of
two forms of the inhibitor of caspase-activated DNase, ICAD-L,
and ICAD-S. J Bio Chem 274:15740–15744
37. Yingchang M, Thomas SD, Xiaohua X, Casson LK, Miller DM,
Bates PJ (2003) Apoptosis in leukemia cell is accompanied by
alterations in the level and localization of nucleolin. J Bio Chem
278:8572–8579
38. Secchiero P, Gonelli A, Celeghini C et al. (2001) Activation of
the nitric oxide synthase pathway represents a key component of
tumor necrosis factor-related apoptosis-inducing ligand mediated
cytotoxicity on hematologic malignancies. Blood 98:2220–2228
39. Richter LC, Vollgraf U (1998) Mode of cell injury and death after
hydrogen peroxide exposure in cultured oligodedendroglia cells.
Exp Cell Res 244:218–229
40. Wink DA, Mitchell JB (1998) Chemical biology of nitric oxide:
insights into regulatory, cytotoxic, and cytoprotective mecha-
nisms of nitric oxide. Free Radic Biol Med 25:434–456
41. Radi R, Cassina A, Hodara R, Quijano C, Castro L (2002) Per-
oxynitrite reactions and formation in mitochondria. Free Radic
Biol Med 33:1451–1464
42. Nagata S, Golstein P (1995) The Fas death factor. Science
267:1449–1456
43. Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A,
Ueno K, Hardwick JM (1997) Conversion of Bcl-2 to a Bax-like
death effector by caspases. Science 278:1966–1968
44. Bello BD, Valentini MA, Zunino F, Comporti M., Maellaro E
(2001) Cleavage of Bcl-2 in oxidant and cisplatin -induced
apoptosis of human melanoma cells. Oncogene 20:4591–4595
45. Fujita N, Tsuruo T (1998) Involvement of Bcl-2 Cleavage in the
Acceleration of VP-16 Induced U937 Cell Apoptosis. Biochem-
ical Biophysical Res Comun 246:484–488
46. Dumont C, Durrbach A, Bidere N et al (2000) Caspase-inde-
pendent commitment phase to apoptosis in activated blood T
lymphocytes: reversibility at low apoptotic insult. Blood
96:1030–1038
47. Apostolova N, Cervera AM, Victor VM et al (2006) Loss of
apoptosis-inducing factor leads to an increase in reactive oxygen
species, and an impairment of respiration that can be reversed by
antioxidants. Cell Death Differ 13:354–357
48. Srinivasula SM, Datta P, Fan XJ, Fernandes-Alnemri T, Huang Z,
Alnemri ES (2000) Molecular determinants of the caspase-pro-
moting activity of Smac/DIABLO and its role in the death
receptor pathway. J Bio Chem 275:36152–36157
49. Sah NK, Khan Z, Khan GJ, Bisen PS (2006) Structural,
functional and therapeutic biology of survivin. Cancer Lett
244:164–171
Apoptosis
123
Copyright © 2022 FDOKUMEN




















