
Annotating discourse markers in spontaneous speech corpora ...
Upload
khangminh22Category
view
1download
0
MS_Piano A Software Tool for Annotating Peaks in CID TandemMass Spectra of Peptides and N‑GlycopeptidesXiaoyu Yang Pedatsur Neta Yuri A Mirokhin Dmitrii V Tchekhovskoi Concepcion A RemorozaMeghan C Burke Yuxue Liang Sanford P Markey and Stephen E Stein
Cite This httpsdoiorg101021acsjproteome1c00324 Read Online
ACCESS Metrics amp More Article Recommendations sı Supporting Information
ABSTRACT Annotating product ion peaks in tandem mass spectra is essential forevaluating spectral quality and validating peptide identification This task is more complexfor glycopeptides and is crucial for the confident determination of glycosylation sites inglycoproteins MS_Piano (Mass Spectrum Peptide Annotation) software was developed forreliable annotation of peaks in collision induced dissociation (CID) tandem mass spectra ofpeptides or N-glycopeptides for given peptide sequences charge states and optionalmodifications The program annotates each peak in high or low resolution spectra withpossible product ion(s) and the mass difference between the measured and theoretical mzvalues Spectral quality is measured by two major parameters the ratio between the sum ofunannotated vs all peak intensities in the top 20 peaks and the intensity of the highestunannotated peak The product ions of peptides glycans and glycopeptides in spectra are labeled in different class-type colors tofacilitate interpretation MS_Piano assists validating peptide and N-glycopeptide identification from database and library searchesand provides quality control and optimizes search reliability in custom developed peptide mass spectral libraries The software isfreely available in exe and dll formats for the Windows operating system
KEYWORDS peak annotation peptide fragmentation glycopeptide fragmentation proteomics glycoproteomics mass spectrometrysoftware peptide identification glycopeptide identification
INTRODUCTION
Tandem high and low resolution mass spectrometry havebecome routine for identifying peptides their modificationsand precursor proteins in proteomic studies1minus4 This techniqueis also used to identify glycosylation sites in glycoproteins incomplex biological samples56 Two principal means ofidentifying peptides and glycopeptides in bottom-up proteomicstudies are by searching protein sequence databases7minus1034minus36
and mass spectral libraries11minus15 Combining these methods canenhance results16 However sometimes high scoring spectrahave unassigned ions due to contamination or incorrectidentification This is especially true for data collected fromhighly complex protein digests containing thousands ofspectra each of which may contain peaks from cofragmenta-tion of ions present in mz fragmentation windows In suchcases annotating peaks in mass spectra with reliablecharacterized product ions can provide additional confidenceof correct and context-consistent identification This isparticularly useful for library searching where spectral qualitycan be ensured by identifying the origin of all peaks in eachlibrary spectrum prior to its use as a reliable reference Priorannotation enables users to quickly understand the significanceof any differences in query and library spectraSoftware tools for automatically annotating product ion
peaks in tandem mass spectra have been limited and fewsoftware tools have been reported One of these is an online
tool MS-Product17 Such programs usually provide usefulpredicted fragmentation ions but do not annotate exper-imental spectra verify identification correctness or evaluatespectrum quality They require users to manually match thetheoretical mz values with the experimental ones or writeadditional programs for implementing this match Otherstudies have noted commonly encountered product ions1819
but these are not available in currently used software Severalsoftware programs are available (eg free software PDV30
TOPPView31 and the commercial software ProteomeDiscoverer32) as spectral visualization tools but these do notprovide glycopeptide product ion annotationHere we present our newly developed software tool
MS_Piano (Mass Spectrum Peptide Annotation) that rapidlyannotates peaks in tandem mass spectra of both peptides andN-glycopeptides with product ions based on peptide sequencecharge state and modifications using a variety of rulesdeveloped to minimize errors This software also provides ameasure of spectral quality to evaluate the validity of
Received April 19 2021
Technical Notepubsacsorgjpr
Not subject to US Copyright PublishedXXXX by American Chemical Society
Ahttpsdoiorg101021acsjproteome1c00324
J Proteome Res XXXX XXX XXXminusXXX
Dow
nloa
ded
via
NA
TL
IN
ST O
F ST
AN
DA
RD
S amp
TE
CH
NO
LO
GY
on
July
29
202
1 at
17
583
6 (U
TC
)Se
e ht
tps
pub
sac
sor
gsh
arin
ggui
delin
es f
or o
ptio
ns o
n ho
w to
legi
timat
ely
shar
e pu
blis
hed
artic
les
identifications It is easy to use for proteomic andglycoproteomic data analysis and freely available as a Windowsapplication in both exe and dll formats
SOFTWARE FEATURESMS_Piano annotates peaks in a tandem high or low resolutionmass spectrum of a peptide or N-glycopeptide based on apresumed sequence charge state and optional modificationsPeptide Annotation
Peaks in a tandem mass spectrum are annotated with precursor(p) product ion y b a immonium ions18 internal fragmentions neutral losses or gains from the precursor and productions as well as their isotopic ions A peak is not labeled as aninternal fragment if it is assigned with any of the above ions forsimplicity and to avoid internal fragment complexityMS_Piano follows common peptide fragmentation andproduct ion nomenclature2829 and excludes c x and z ionsfor annotating collision induced dissociation (CID) spectraProduct ion annotations and examples are shown in Figure 1The software provides more than 100 built-in modificationsand used more than 800 modifications from unimod20 Userscan add their own modifications in a separate fileldquomod_addedtxtrdquo Detailed information on name formulaand amino acid residues of all the built-in modificationsimmonium and fragment ions neutral losses and gains iTRAQ(isobaric tags for relative and absolute quantitation) and TMT(Tandem Mass Tag) fragments are provided in the SupportingInformation (Table S1minusS5)For each peptidersquos sequence charge state and optional
modifications MS_Piano uses the example format in Figure 2An example of an annotated high-resolution mass spectrum
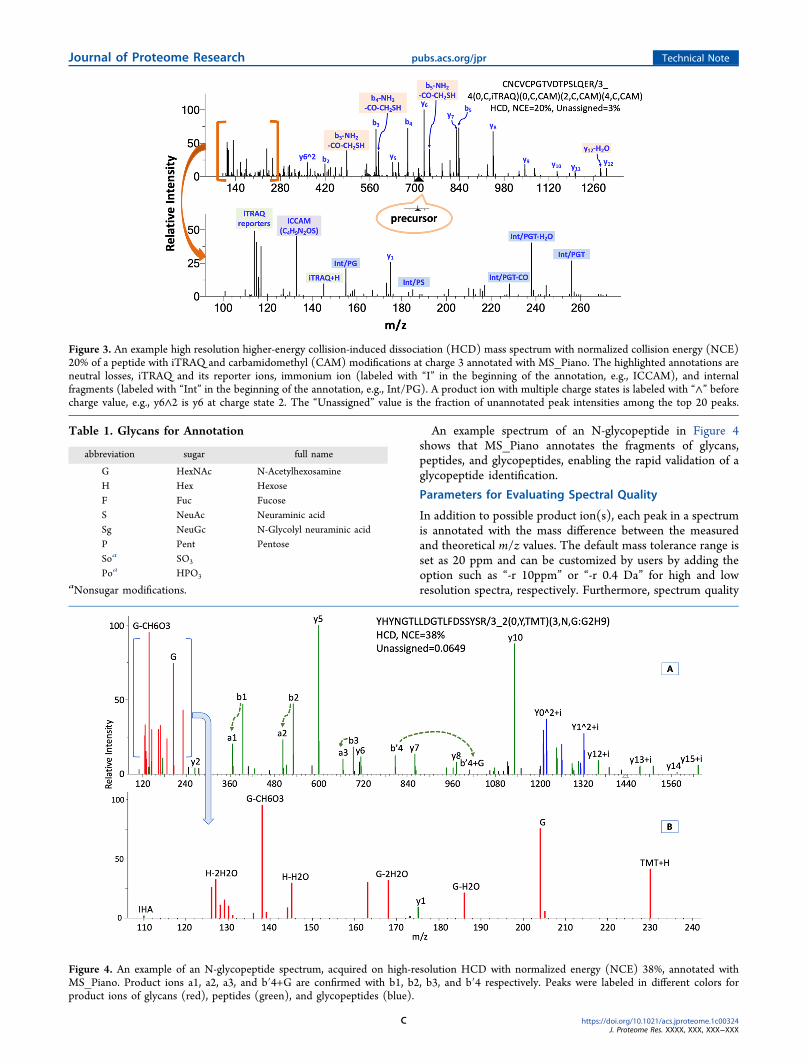
in Figure 3 shows that MS_Piano annotates not only y bimmonium and internal fragment ions but also iTRAQ and its
reporters the neutral losses of b y and internal fragmentsTwo examples of low-resolution ion trap mass spectraannotated with MS_Piano are shown in Figure S1 in theSupporting InformationGlycopeptide Annotation
We extended MS_Piano to annotate peaks of CID tandemmass spectra of N-glycopeptides We use 6 commonmonosaccharides found in mammals as basic units for glycancomposition (Table 1) These short symbols enable theircompact representation on computer screens We also use ldquoGrdquoto distinguish glycosylation from other peptide modificationsFor example in Figure 4 YHYNGTLLDGTLFDSSYSR3_2(0YTMT)(3NGG2H9) G2H9 is the glycan on aminoacid Asparagine (N) at location 3 counting from N-terminusstarting from 0In addition to the above-described product ions and
fragments for peptide annotation the following 3 production types were added for N-glycopeptide annotation
1 Peptides Product ions yprime bprime and aprime ions are y b and aions that have lost N-glycosylation respectively Theseions plus various sugars are also used for annotation Forexample in Figure 4 bprime4 is b4 ion without glycan G2H9and bprime4+G is bprime4 with a G modification
2 Glycans Glycans and those with various losses eg GG-H2O G-2H2O in Figure 4 and GHS in Figure 6
3 Glycopeptides Peptide sequence with various glycansFor brevity the capital letter Y is followed by numbers(Y0 to Y10) to annotate common N-glycopeptidefragments from high-mannose glycans (Table 2) egisotopic Y0 at charge 2 (annotated as ldquoY0and2+irdquo) inFigure 4
The presence of mz = 1970445 indicates the loss ofC2H9NO3 loss from NeuAc21 the loss of C2H6O3 CH6O3 andC2H4O2 serve to annotate HexNac22 After careful manualexamination the following annotations were added to thesoftware C7H5O2 C8H7O3 C11H5NO C9H7O4 C9H10NO4and C9H11O6As a critical step in MS_Piano when a peak in a spectrum
could be annotated with multiple product ions they areprioritized in the following order precursor (p) Yglycopeptides glycan oxonium ions y or b yprime or bprime immoniumions or fragments from modifications a or aprime ions Prioritizingpeak annotations facilitates optimal library searching results
Figure 1 Varieties of product ions and fragments used in MS_Piano (c x and z ions are not used for annotating CID mass spectra)
Figure 2 An example format for a peptide sequence charge state andoptional modifications
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
B
An example spectrum of an N-glycopeptide in Figure 4shows that MS_Piano annotates the fragments of glycanspeptides and glycopeptides enabling the rapid validation of aglycopeptide identification
Parameters for Evaluating Spectral Quality
In addition to possible product ion(s) each peak in a spectrumis annotated with the mass difference between the measuredand theoretical mz values The default mass tolerance range isset as 20 ppm and can be customized by users by adding theoption such as ldquo-r 10ppmrdquo or ldquo-r 04 Dardquo for high and lowresolution spectra respectively Furthermore spectrum quality
Figure 3 An example high resolution higher-energy collision-induced dissociation (HCD) mass spectrum with normalized collision energy (NCE)20 of a peptide with iTRAQ and carbamidomethyl (CAM) modifications at charge 3 annotated with MS_Piano The highlighted annotations areneutral losses iTRAQ and its reporter ions immonium ion (labeled with ldquoIrdquo in the beginning of the annotation eg ICCAM) and internalfragments (labeled with ldquoIntrdquo in the beginning of the annotation eg IntPG) A product ion with multiple charge states is labeled with ldquoandrdquo beforecharge value eg y6and2 is y6 at charge state 2 The ldquoUnassignedrdquo value is the fraction of unannotated peak intensities among the top 20 peaks
Table 1 Glycans for Annotation
abbreviation sugar full name
G HexNAc N-AcetylhexosamineH Hex HexoseF Fuc FucoseS NeuAc Neuraminic acidSg NeuGc N-Glycolyl neuraminic acidP Pent PentoseSoa SO3
Poa HPO3aNonsugar modifications
Figure 4 An example of an N-glycopeptide spectrum acquired on high-resolution HCD with normalized energy (NCE) 38 annotated withMS_Piano Product ions a1 a2 a3 and bprime4+G are confirmed with b1 b2 b3 and bprime4 respectively Peaks were labeled in different colors forproduct ions of glycans (red) peptides (green) and glycopeptides (blue)
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
C
is measured with the fraction of unannotated peak intensitiesamong the top 20 peaks (Unassigned in Figures 3 and 4) or allthe peaks (Unassigned_all) the intensity of the highestunassigned peak (max_unassigned_ab of base peak) andnumber of unassigned peaks in top 20 peaks (top_20_-num_unassigned_peaks) or number of unassigned peaks(num_unassigned_peaks) etc By using these parametersusers can easily assess spectrum quality to verify peptideidentification
SOFTWARE DESCRIPTIONMS_Piano was developed in Microsoft Visual C++ 2015 and isreleased in 2 formats exe and dll (peptide annotation only)The exe software can be directly used without installation onthe Windows operating system The command is simple egMS_Piano Cinmsp CoutmspData Input and Output
The NIST text file format (msp) is used for input and outputof mz vs intensity lists The following screenshots (Figure 5)illustrate an example spectrum of a peptide in an msp fileannotated with MS_Piano software In the input file thefollowing information is required for annotation peptide
sequence modifications (optional) charge state precursor mz value (Parent optional) number of peaks (Num peaks) anda peak list with mz and intensity values The peptideinformation is listed in the Name line In the output fileMS_Piano annotates peaks with product ions and massdifference between experimental and theoretical mz valueThe output file also provides the parameters described aboveto indicate spectral quality
Visualization
Tandem mass spectra with peaks annotated with reliableproduct ions in the output file can be viewed (Figures 4 6) inthe MS Search program12 to facilitate validation and generatefigures for publications and presentations The annotated peakswith fragments of peptides glycans and glycopeptides in amass spectrum can be displayed in different colors by adjustingcolor and fonts in the MS Search version 25 or later
SOFTWARE PERFORMANCE TESTING ANDAPPLICATION
MS_Pianoexe program was tested for performance on adesktop computer Windows 10 Enterprise with Intel(R)Core(TM) i7minus6700 CPU 340 GHz with 64 GB memoryAs an example of performance MS_Piano took lt15 h toannotate 1 million high resolution mass spectra of peptidesfrom protein digest samples by processing 4 files in paralleleach containing 250 000 identified spectra Peptide length was10minus30 charge state from 2 to 5 with 0minus4 modifications and30minus500 peaks in 95 of these testing spectra These spectrawere first converted to msp files (mass vs intensity peak lists intext format) from the MS-GF+9 searching results Forglycopeptide spectra the annotation time increases with thenumber of sugars in the glycans MS_Piano was tested andrefined by annotating the msp files converted from MS-GF+9
searching output files of all the data generated in Study 3 of theClinical Proteomic Tumor Analysis Consortium (NCINIH)
Table 2 N-Glycopeptide Fragments for Annotation
abbreviation fragments
Y0 no glycansY1 Y0+GY2 Y0+G2Y3 Y0+G2HY4 Y0+G2H2Y5 Y0+G2H3Y6 Y0+G2H4Y7 Y0+G2H5Y8 Y0+G2H6Y9 Y0+G2H7
Figure 5 An example spectrum of a peptide in an msp file annotated with MS_Piano
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
D
ldquoUnassigned rdquo and other parameters described abovetogether with MS-GF+ searching score were used to evaluatespectral quality and identification This software has annotated90 784 MS2 spectra of all dipeptides all tryptic tripeptides and1828 bioactive commercial peptides (purity gt90) in theNIST Tandem Mass Spectral Library2324 It has been used inannotating mass spectra of digests from iTRAQ and TMTlabeled proteins25 and human hair26 It is also used routinely toannotate high resolution mass spectra and evaluate spectralquality for optimal search for refining the NIST PeptideTandem Mass Spectral Library13 with more than 43 millionspectra MS_Piano has been used to annotate mass spectra ofprotein digests of single glycoproteins (Figure 6) including theSpike Protein in SARS-CoV-2
DISCUSSION AND CONCLUSIONSMS_Piano enables the annotation of tandem mass spectra ofboth peptides and N-glycopeptides that contain virtually allknown modifications A key feature of MS_Piano is itscapability to annotate one million peptide spectra in lt2 h withreliable product ions including some common but usuallyneglected fragments The annotated spectra can be viewedwith diverse ions displayed in different colors in NIST MSSearch12 browser for enhanced visual examination Annotatedspectra can be used to validate spectra and verify peptideidentified by sequence database searching directly orembedded in other software packages to implement variousproteomics data analysis software27 The output msp files ofspectra annotated with MS_Piano can be used on Skyline33
platform directly MS_Piano provides a metric for spectrumquality and a reportable filter for constructing peptide massspectral libraries The software is helpful for understandingpeptide fragmentation pathways Its different formats provideflexibility for biologists and chemists to use exe directly or foradvanced programmers to embed a functional dll into otherprograms
Due to the format complexity of raw data acquired on massspectrometers from different manufactures and the peak listslacking in searching results from various peptide andglycopeptide search engines the simple text format (msp) isused for input but users need to convert their files to thisformat to use this program However files can be combinedfrom searching results from libraries search engines and denovo sequencing for annotation We developed an msp fileconverter (free software convert2 msp also available at thefollowing MS_Piano download website) which quicklyconverts the results from free protein and glycoproteinsequence searching engine MSFragger34 and pGlyco35
respectively to msp files and automatically connects toMS_Piano for spectral annotation to facilitate building-your-own libraries More capabilities such as taking mzXML andpepXML together with raw data as input files annotating massspectra of negative mode ETD and O-glycopeptides will beadded to the software Software instructions and examples canbe freely downloaded at httpschemdatanistgovdokuwikidokuphpid=peptidewms_piano
ASSOCIATED CONTENT
sı Supporting Information
The Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjproteome1c00324
Product ions used in MS_Piano Table S1 Peptidemodifications Table S2 Immonium and fragment ionsTable S3 Neutral losses Table S4 iTRAQ fragmentsTable S5 TMT fragments Figure S1 Two examples oflow-resolution ion trap mass spectra of a peptideBradykinin with sequence RPPGFSPFR at charge 2and 3 respectively annotated with MS_Piano (XLSX)
Figure 6 An example of energy dependence fragmentation of an N-glycopeptide (in Alpha-1-acid glycoprotein in plasma) in charge 3 annotatedwith MS_Piano Peaks in 4 high-resolution HCD spectra at different collision energies were labeled in different colors for product ions of glycans(red) peptides (green) and glycopeptides (blue)
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
E
AUTHOR INFORMATIONCorresponding Author
Xiaoyu Yang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0003-3371-9567 Email xiaoyuyangnistgov
Authors
Pedatsur Neta minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Yuri A Mirokhin minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Dmitrii V Tchekhovskoi minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Concepcion A Remoroza minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0003-1540-1635
Meghan C Burke minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0001-7231-0655
Yuxue Liang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0002-6430-915X
Sanford P Markey minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Stephen E Stein minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Complete contact information is available athttpspubsacsorg101021acsjproteome1c00324
Notes
Certain commercial software or instruments are identified inthis paper in order to specify the experimental procedureadequately Such identification is not intended to implyrecommendation or endorsement by the National Institute ofStandards and Technology nor is it intended to imply that thesoftware or instruments identified are necessarily the bestavailable for the purposeThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors thank Drs Lewis Geer Guanghui Wang OlegToropov Zheng Zhang Sergey Sheetlin and William Wallacewho have provided suggestions useful discussions feedbackand technical support that have helped the improvement of thesoftware
REFERENCES(1) Patterson S D Aebersold R H Proteomics the first decadeand beyond Nat Genet 2003 33 (Suppl) 311minus323(2) Han X Aslanian A Yates J R 3rd Mass spectrometry forproteomics Curr Opin Chem Biol 2008 12 (5) 483minus490(3) Yu Q Paulo J A Naverrete-Perea J McAlister G CCanterbury J D Bailey D J Robitaille A M Huguet R
Zabrouskov V Gygi S P Schweppe D K Benchmarking theOrbitrap Tribrid Eclipse for Next Generation Multiplexed Proteo-mics Anal Chem 2020 92 (9) 6478minus6485(4) Adhikari S Nice E C Deutsch E W Lane L Omenn G SPennington S R Paik Y K Overall C M Corrales F J CristeaI M Van Eyk J E Uhleacuten M Lindskog C Chan D W BairochA Waddington J C Justice J L LaBaer J Rodriguez H He FKostrzewa M Ping P Gundry R L Stewart P Srivastava SSrivastava S Nogueira F C S Domont G B Vandenbrouck YLam M P Y Wennersten S Vizcaino J A Wilkins M SchwenkJ M Lundberg E Bandeira N Marko-Varga G Weintraub S TPineau C Kusebauch U Moritz R L Ahn S B Palmblad MSnyder M P Aebersold R Baker M S A high-stringency blueprintof the human proteome Nat Commun 2020 11 (1) 5301(5) Sun S Shah P Eshghi S T Yang W Trikannad N YangS Chen L Aiyetan P Houmlti N Zhang Z Chan D W Zhang HComprehensive analysis of protein glycosylation by solid-phaseextraction of N-linked glycans and glycosite-containing peptidesNat Biotechnol 2016 34 (1) 84minus88(6) Watanabe Y Allen J D Wrapp D McLellan J S CrispinM Site-specific glycan analysis of the SARS-CoV-2 spike Science2020 369 (6501) 330minus333(7) Perkins D N Pappin D J Creasy D M Cottrell J SProbability-based protein identification by searching sequencedatabases using mass spectrometry data Electrophoresis 1999 20(18) 3551minus3567(8) Geer L Y Markey S P Kowalak J A Wagner L Xu MMaynard D M Yang X Shi W Bryant S H Open massspectrometry search algorithm J Proteome Res 2004 3 (5) 958minus964(9) Byonic httpswwwproteinmetricscom (accessed in Decem-ber 2020)(10) Kim S Pevzner P A MS-GF+ makes progress towards auniversal database search tool for proteomics Nat Commun 2014 55277(11) Toghi Eshghi S Shah P Yang W Li X Zhang HGPQuest A Spectral Library Matching Algorithm for Site-SpecificAssignment of Tandem Mass Spectra to Intact N-glycopeptides AnalChem 2015 87 (10) 5181minus8(12) NIST MS Search browser httpschemdatanistgovdokuwikidokuphpid=peptidewnistmssearch (accessed in Decem-ber 2020)(13) NIST Libraries of Peptide Tandem Mass Spectra httpschemdatanistgovdokuwikidokuphpid=peptidewstart (accessedin December 2020)(14) Lam H Deutsch E W Eddes J S Eng J K King NStein S E Aebersold R Development and validation of a spectrallibrary searching method for peptide identification from MSMSProteomics 2007 7 (5) 655minus667(15) Lam H Deutsch E W Eddes J S Eng J K Stein S EAebersold R Building consensus spectral libraries for peptideidentification in proteomics Nat Methods 2008 5 (10) 873minus875(16) Fernaacutendez-Costa C Martiacutenez-Bartolomeacute S McClatchy DYates J R 3rd Improving Proteomics Data Reproducibility with aDual-Search Strategy Anal Chem 2020 92 (2) 1697minus1701(17) MS-Product httpsprospector2ucsfeduprospectorcgi-binmsformcgiform=msproduct (accessed in December 2020)(18) Liang Y Neta P Yang X Stein S E Collision-InducedDissociation of Deprotonated Peptides Relative Abundance of Side-Chain Neutral Losses Residue-Specific Product Ions and Compar-ison with Protonated Peptides J Am Soc Mass Spectrom 2018 29(3) 463minus469(19) Kilpatrick L E Neta P Yang X Simoacuten-Manso Y LiangY Stein S E Formation of y + 10 and y + 11 ions in the collision-induced dissociation of peptide ions J Am Soc Mass Spectrom 201223 (4) 655minus663(20) Unimod httpswwwunimodorgdownloadshtml (accessedin December 2020)(21) Kleikamp H B C Lin Y M McMillan D G G GeelhoedJ S Naus-Wiezer S N H van Baarlen P Saha C Louwen R
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
F
Sorokin D Y van Loosdrecht M C M et al Tackling the chemicaldiversity of microbial nonulosonic acidsA universal large-scalesurvey approach Chem Sci 2020 11 3074minus3080(22) Yu J Schorlemer M Gomez Toledo A Pett C SihlbomC Larson G Westerlind U Nilsson J Distinctive MSMSFragmentation Pathways of Glycopeptide-Generated Oxonium IonsProvide Evidence of the Glycan Structure Chem - Eur J 2016 22(3) 1114minus1124(23) Yang X Neta P Stein S E Quality control for buildinglibraries from electrospray ionization tandem mass spectra AnalChem 2014 86 (13) 6393minus6400(24) Yang X Neta P Stein S E Extending a Tandem MassSpectral Library to Include MS2 Spectra of Fragment Ions ProducedIn-Source and MSn Spectra J Am Soc Mass Spectrom 2017 28 (11)2280minus2287(25) Zhang Z Yang X Mirokhin Y A Tchekhovskoi D V JiW Markey S P Roth J Neta P Hizal D B Bowen M A SteinS E Interconversion of Peptide Mass Spectral Libraries Derivatizedwith iTRAQ or TMT Labels J Proteome Res 2016 15 (9) 3180minus3187(26) Zhang Z Burke M C Wallace W E Liang Y Sheetlin SL Mirokhin Y A Tchekhovskoi D V Stein S E SensitiveMethod for the Confident Identification of Genetically VariantPeptides in Human Hair Keratin J Forensic Sci 2020 65 (2) 406minus420(27) Bittremieux W spectrum_utils A Python Package for MassSpectrometry Data Processing and Visualization Anal Chem 202092 (1) 659minus661(28) Roepstorff P Fohlman J Proposal for a commonnomenclature for sequence ions in mass spectra of peptides BiomedMass Spectrom 1984 11 (11) 601(29) Johnson R S Martin S A Biemann K Stults J T WatsonJ T Novel fragmentation process of peptides by collision-induceddecomposition in a tandem mass spectrometer differentiation ofleucine and isoleucine Anal Chem 1987 59 (21) 2621minus2655(30) Li K Vaudel M Zhang B Ren Y Wen B PDV anintegrative proteomics data viewer Bioinformatics 2019 35 (7)1249minus1251(31) Sturm M Kohlbacher O TOPPView an open-source viewerfor mass spectrometry data J Proteome Res 2009 8 (7) 3760minus3763(32) Proteome Discoverer httpswwwthermofishercomusenhomeindustrialmass-spectrometryliquid-chromatography-mass-spectrometry-lc-mslc-ms-softwaremulti-omics-data-analysisproteome-discoverer-softwarehtml (accessed in June 2020)(33) MacLean B Tomazela D M Shulman N Chambers MFinney G L Frewen B Kern R Tabb D L Liebler D CMacCoss M J Skyline an open source document editor for creatingand analyzing targeted proteomics experiments Bioinformatics 201026 (7) 966minus968(34) Kong A T Leprevost F V Avtonomov D MMellacheruvu D Nesvizhskii A I MSFragger ultrafast andcomprehensive peptide identification in mass spectrometry-basedproteomics Nat Methods 2017 14 (5) 513minus520(35) Liu M Q Zeng W F Fang P Cao W Q Liu C Yan GQ Zhang Y Peng C Wu J Q Zhang X J Tu H J Chi HSun R X Cao Y Dong M Q Jiang B Y Huang J M Shen HL Wong C C L He S M Yang P Y pGlyco 20 enablesprecision N-glycoproteomics with comprehensive quality control andone-step mass spectrometry for intact glycopeptide identification NatCommun 2017 8 (1) 438(36) Polasky D A Yu F Teo G C Nesvizhskii A I Fast andcomprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco Nat Methods 2020 17 (11) 1125minus1132
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
G

identifications It is easy to use for proteomic andglycoproteomic data analysis and freely available as a Windowsapplication in both exe and dll formats
SOFTWARE FEATURESMS_Piano annotates peaks in a tandem high or low resolutionmass spectrum of a peptide or N-glycopeptide based on apresumed sequence charge state and optional modificationsPeptide Annotation
Peaks in a tandem mass spectrum are annotated with precursor(p) product ion y b a immonium ions18 internal fragmentions neutral losses or gains from the precursor and productions as well as their isotopic ions A peak is not labeled as aninternal fragment if it is assigned with any of the above ions forsimplicity and to avoid internal fragment complexityMS_Piano follows common peptide fragmentation andproduct ion nomenclature2829 and excludes c x and z ionsfor annotating collision induced dissociation (CID) spectraProduct ion annotations and examples are shown in Figure 1The software provides more than 100 built-in modificationsand used more than 800 modifications from unimod20 Userscan add their own modifications in a separate fileldquomod_addedtxtrdquo Detailed information on name formulaand amino acid residues of all the built-in modificationsimmonium and fragment ions neutral losses and gains iTRAQ(isobaric tags for relative and absolute quantitation) and TMT(Tandem Mass Tag) fragments are provided in the SupportingInformation (Table S1minusS5)For each peptidersquos sequence charge state and optional
modifications MS_Piano uses the example format in Figure 2An example of an annotated high-resolution mass spectrum
in Figure 3 shows that MS_Piano annotates not only y bimmonium and internal fragment ions but also iTRAQ and its
reporters the neutral losses of b y and internal fragmentsTwo examples of low-resolution ion trap mass spectraannotated with MS_Piano are shown in Figure S1 in theSupporting InformationGlycopeptide Annotation
We extended MS_Piano to annotate peaks of CID tandemmass spectra of N-glycopeptides We use 6 commonmonosaccharides found in mammals as basic units for glycancomposition (Table 1) These short symbols enable theircompact representation on computer screens We also use ldquoGrdquoto distinguish glycosylation from other peptide modificationsFor example in Figure 4 YHYNGTLLDGTLFDSSYSR3_2(0YTMT)(3NGG2H9) G2H9 is the glycan on aminoacid Asparagine (N) at location 3 counting from N-terminusstarting from 0In addition to the above-described product ions and
fragments for peptide annotation the following 3 production types were added for N-glycopeptide annotation
1 Peptides Product ions yprime bprime and aprime ions are y b and aions that have lost N-glycosylation respectively Theseions plus various sugars are also used for annotation Forexample in Figure 4 bprime4 is b4 ion without glycan G2H9and bprime4+G is bprime4 with a G modification
2 Glycans Glycans and those with various losses eg GG-H2O G-2H2O in Figure 4 and GHS in Figure 6
3 Glycopeptides Peptide sequence with various glycansFor brevity the capital letter Y is followed by numbers(Y0 to Y10) to annotate common N-glycopeptidefragments from high-mannose glycans (Table 2) egisotopic Y0 at charge 2 (annotated as ldquoY0and2+irdquo) inFigure 4
The presence of mz = 1970445 indicates the loss ofC2H9NO3 loss from NeuAc21 the loss of C2H6O3 CH6O3 andC2H4O2 serve to annotate HexNac22 After careful manualexamination the following annotations were added to thesoftware C7H5O2 C8H7O3 C11H5NO C9H7O4 C9H10NO4and C9H11O6As a critical step in MS_Piano when a peak in a spectrum
could be annotated with multiple product ions they areprioritized in the following order precursor (p) Yglycopeptides glycan oxonium ions y or b yprime or bprime immoniumions or fragments from modifications a or aprime ions Prioritizingpeak annotations facilitates optimal library searching results
Figure 1 Varieties of product ions and fragments used in MS_Piano (c x and z ions are not used for annotating CID mass spectra)
Figure 2 An example format for a peptide sequence charge state andoptional modifications
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
B
An example spectrum of an N-glycopeptide in Figure 4shows that MS_Piano annotates the fragments of glycanspeptides and glycopeptides enabling the rapid validation of aglycopeptide identification
Parameters for Evaluating Spectral Quality
In addition to possible product ion(s) each peak in a spectrumis annotated with the mass difference between the measuredand theoretical mz values The default mass tolerance range isset as 20 ppm and can be customized by users by adding theoption such as ldquo-r 10ppmrdquo or ldquo-r 04 Dardquo for high and lowresolution spectra respectively Furthermore spectrum quality
Figure 3 An example high resolution higher-energy collision-induced dissociation (HCD) mass spectrum with normalized collision energy (NCE)20 of a peptide with iTRAQ and carbamidomethyl (CAM) modifications at charge 3 annotated with MS_Piano The highlighted annotations areneutral losses iTRAQ and its reporter ions immonium ion (labeled with ldquoIrdquo in the beginning of the annotation eg ICCAM) and internalfragments (labeled with ldquoIntrdquo in the beginning of the annotation eg IntPG) A product ion with multiple charge states is labeled with ldquoandrdquo beforecharge value eg y6and2 is y6 at charge state 2 The ldquoUnassignedrdquo value is the fraction of unannotated peak intensities among the top 20 peaks
Table 1 Glycans for Annotation
abbreviation sugar full name
G HexNAc N-AcetylhexosamineH Hex HexoseF Fuc FucoseS NeuAc Neuraminic acidSg NeuGc N-Glycolyl neuraminic acidP Pent PentoseSoa SO3
Poa HPO3aNonsugar modifications
Figure 4 An example of an N-glycopeptide spectrum acquired on high-resolution HCD with normalized energy (NCE) 38 annotated withMS_Piano Product ions a1 a2 a3 and bprime4+G are confirmed with b1 b2 b3 and bprime4 respectively Peaks were labeled in different colors forproduct ions of glycans (red) peptides (green) and glycopeptides (blue)
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
C
is measured with the fraction of unannotated peak intensitiesamong the top 20 peaks (Unassigned in Figures 3 and 4) or allthe peaks (Unassigned_all) the intensity of the highestunassigned peak (max_unassigned_ab of base peak) andnumber of unassigned peaks in top 20 peaks (top_20_-num_unassigned_peaks) or number of unassigned peaks(num_unassigned_peaks) etc By using these parametersusers can easily assess spectrum quality to verify peptideidentification
SOFTWARE DESCRIPTIONMS_Piano was developed in Microsoft Visual C++ 2015 and isreleased in 2 formats exe and dll (peptide annotation only)The exe software can be directly used without installation onthe Windows operating system The command is simple egMS_Piano Cinmsp CoutmspData Input and Output
The NIST text file format (msp) is used for input and outputof mz vs intensity lists The following screenshots (Figure 5)illustrate an example spectrum of a peptide in an msp fileannotated with MS_Piano software In the input file thefollowing information is required for annotation peptide
sequence modifications (optional) charge state precursor mz value (Parent optional) number of peaks (Num peaks) anda peak list with mz and intensity values The peptideinformation is listed in the Name line In the output fileMS_Piano annotates peaks with product ions and massdifference between experimental and theoretical mz valueThe output file also provides the parameters described aboveto indicate spectral quality
Visualization
Tandem mass spectra with peaks annotated with reliableproduct ions in the output file can be viewed (Figures 4 6) inthe MS Search program12 to facilitate validation and generatefigures for publications and presentations The annotated peakswith fragments of peptides glycans and glycopeptides in amass spectrum can be displayed in different colors by adjustingcolor and fonts in the MS Search version 25 or later
SOFTWARE PERFORMANCE TESTING ANDAPPLICATION
MS_Pianoexe program was tested for performance on adesktop computer Windows 10 Enterprise with Intel(R)Core(TM) i7minus6700 CPU 340 GHz with 64 GB memoryAs an example of performance MS_Piano took lt15 h toannotate 1 million high resolution mass spectra of peptidesfrom protein digest samples by processing 4 files in paralleleach containing 250 000 identified spectra Peptide length was10minus30 charge state from 2 to 5 with 0minus4 modifications and30minus500 peaks in 95 of these testing spectra These spectrawere first converted to msp files (mass vs intensity peak lists intext format) from the MS-GF+9 searching results Forglycopeptide spectra the annotation time increases with thenumber of sugars in the glycans MS_Piano was tested andrefined by annotating the msp files converted from MS-GF+9
searching output files of all the data generated in Study 3 of theClinical Proteomic Tumor Analysis Consortium (NCINIH)
Table 2 N-Glycopeptide Fragments for Annotation
abbreviation fragments
Y0 no glycansY1 Y0+GY2 Y0+G2Y3 Y0+G2HY4 Y0+G2H2Y5 Y0+G2H3Y6 Y0+G2H4Y7 Y0+G2H5Y8 Y0+G2H6Y9 Y0+G2H7
Figure 5 An example spectrum of a peptide in an msp file annotated with MS_Piano
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
D
ldquoUnassigned rdquo and other parameters described abovetogether with MS-GF+ searching score were used to evaluatespectral quality and identification This software has annotated90 784 MS2 spectra of all dipeptides all tryptic tripeptides and1828 bioactive commercial peptides (purity gt90) in theNIST Tandem Mass Spectral Library2324 It has been used inannotating mass spectra of digests from iTRAQ and TMTlabeled proteins25 and human hair26 It is also used routinely toannotate high resolution mass spectra and evaluate spectralquality for optimal search for refining the NIST PeptideTandem Mass Spectral Library13 with more than 43 millionspectra MS_Piano has been used to annotate mass spectra ofprotein digests of single glycoproteins (Figure 6) including theSpike Protein in SARS-CoV-2
DISCUSSION AND CONCLUSIONSMS_Piano enables the annotation of tandem mass spectra ofboth peptides and N-glycopeptides that contain virtually allknown modifications A key feature of MS_Piano is itscapability to annotate one million peptide spectra in lt2 h withreliable product ions including some common but usuallyneglected fragments The annotated spectra can be viewedwith diverse ions displayed in different colors in NIST MSSearch12 browser for enhanced visual examination Annotatedspectra can be used to validate spectra and verify peptideidentified by sequence database searching directly orembedded in other software packages to implement variousproteomics data analysis software27 The output msp files ofspectra annotated with MS_Piano can be used on Skyline33
platform directly MS_Piano provides a metric for spectrumquality and a reportable filter for constructing peptide massspectral libraries The software is helpful for understandingpeptide fragmentation pathways Its different formats provideflexibility for biologists and chemists to use exe directly or foradvanced programmers to embed a functional dll into otherprograms
Due to the format complexity of raw data acquired on massspectrometers from different manufactures and the peak listslacking in searching results from various peptide andglycopeptide search engines the simple text format (msp) isused for input but users need to convert their files to thisformat to use this program However files can be combinedfrom searching results from libraries search engines and denovo sequencing for annotation We developed an msp fileconverter (free software convert2 msp also available at thefollowing MS_Piano download website) which quicklyconverts the results from free protein and glycoproteinsequence searching engine MSFragger34 and pGlyco35
respectively to msp files and automatically connects toMS_Piano for spectral annotation to facilitate building-your-own libraries More capabilities such as taking mzXML andpepXML together with raw data as input files annotating massspectra of negative mode ETD and O-glycopeptides will beadded to the software Software instructions and examples canbe freely downloaded at httpschemdatanistgovdokuwikidokuphpid=peptidewms_piano
ASSOCIATED CONTENT
sı Supporting Information
The Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjproteome1c00324
Product ions used in MS_Piano Table S1 Peptidemodifications Table S2 Immonium and fragment ionsTable S3 Neutral losses Table S4 iTRAQ fragmentsTable S5 TMT fragments Figure S1 Two examples oflow-resolution ion trap mass spectra of a peptideBradykinin with sequence RPPGFSPFR at charge 2and 3 respectively annotated with MS_Piano (XLSX)
Figure 6 An example of energy dependence fragmentation of an N-glycopeptide (in Alpha-1-acid glycoprotein in plasma) in charge 3 annotatedwith MS_Piano Peaks in 4 high-resolution HCD spectra at different collision energies were labeled in different colors for product ions of glycans(red) peptides (green) and glycopeptides (blue)
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
E
AUTHOR INFORMATIONCorresponding Author
Xiaoyu Yang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0003-3371-9567 Email xiaoyuyangnistgov
Authors
Pedatsur Neta minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Yuri A Mirokhin minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Dmitrii V Tchekhovskoi minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Concepcion A Remoroza minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0003-1540-1635
Meghan C Burke minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0001-7231-0655
Yuxue Liang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0002-6430-915X
Sanford P Markey minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Stephen E Stein minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Complete contact information is available athttpspubsacsorg101021acsjproteome1c00324
Notes
Certain commercial software or instruments are identified inthis paper in order to specify the experimental procedureadequately Such identification is not intended to implyrecommendation or endorsement by the National Institute ofStandards and Technology nor is it intended to imply that thesoftware or instruments identified are necessarily the bestavailable for the purposeThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors thank Drs Lewis Geer Guanghui Wang OlegToropov Zheng Zhang Sergey Sheetlin and William Wallacewho have provided suggestions useful discussions feedbackand technical support that have helped the improvement of thesoftware
REFERENCES(1) Patterson S D Aebersold R H Proteomics the first decadeand beyond Nat Genet 2003 33 (Suppl) 311minus323(2) Han X Aslanian A Yates J R 3rd Mass spectrometry forproteomics Curr Opin Chem Biol 2008 12 (5) 483minus490(3) Yu Q Paulo J A Naverrete-Perea J McAlister G CCanterbury J D Bailey D J Robitaille A M Huguet R
Zabrouskov V Gygi S P Schweppe D K Benchmarking theOrbitrap Tribrid Eclipse for Next Generation Multiplexed Proteo-mics Anal Chem 2020 92 (9) 6478minus6485(4) Adhikari S Nice E C Deutsch E W Lane L Omenn G SPennington S R Paik Y K Overall C M Corrales F J CristeaI M Van Eyk J E Uhleacuten M Lindskog C Chan D W BairochA Waddington J C Justice J L LaBaer J Rodriguez H He FKostrzewa M Ping P Gundry R L Stewart P Srivastava SSrivastava S Nogueira F C S Domont G B Vandenbrouck YLam M P Y Wennersten S Vizcaino J A Wilkins M SchwenkJ M Lundberg E Bandeira N Marko-Varga G Weintraub S TPineau C Kusebauch U Moritz R L Ahn S B Palmblad MSnyder M P Aebersold R Baker M S A high-stringency blueprintof the human proteome Nat Commun 2020 11 (1) 5301(5) Sun S Shah P Eshghi S T Yang W Trikannad N YangS Chen L Aiyetan P Houmlti N Zhang Z Chan D W Zhang HComprehensive analysis of protein glycosylation by solid-phaseextraction of N-linked glycans and glycosite-containing peptidesNat Biotechnol 2016 34 (1) 84minus88(6) Watanabe Y Allen J D Wrapp D McLellan J S CrispinM Site-specific glycan analysis of the SARS-CoV-2 spike Science2020 369 (6501) 330minus333(7) Perkins D N Pappin D J Creasy D M Cottrell J SProbability-based protein identification by searching sequencedatabases using mass spectrometry data Electrophoresis 1999 20(18) 3551minus3567(8) Geer L Y Markey S P Kowalak J A Wagner L Xu MMaynard D M Yang X Shi W Bryant S H Open massspectrometry search algorithm J Proteome Res 2004 3 (5) 958minus964(9) Byonic httpswwwproteinmetricscom (accessed in Decem-ber 2020)(10) Kim S Pevzner P A MS-GF+ makes progress towards auniversal database search tool for proteomics Nat Commun 2014 55277(11) Toghi Eshghi S Shah P Yang W Li X Zhang HGPQuest A Spectral Library Matching Algorithm for Site-SpecificAssignment of Tandem Mass Spectra to Intact N-glycopeptides AnalChem 2015 87 (10) 5181minus8(12) NIST MS Search browser httpschemdatanistgovdokuwikidokuphpid=peptidewnistmssearch (accessed in Decem-ber 2020)(13) NIST Libraries of Peptide Tandem Mass Spectra httpschemdatanistgovdokuwikidokuphpid=peptidewstart (accessedin December 2020)(14) Lam H Deutsch E W Eddes J S Eng J K King NStein S E Aebersold R Development and validation of a spectrallibrary searching method for peptide identification from MSMSProteomics 2007 7 (5) 655minus667(15) Lam H Deutsch E W Eddes J S Eng J K Stein S EAebersold R Building consensus spectral libraries for peptideidentification in proteomics Nat Methods 2008 5 (10) 873minus875(16) Fernaacutendez-Costa C Martiacutenez-Bartolomeacute S McClatchy DYates J R 3rd Improving Proteomics Data Reproducibility with aDual-Search Strategy Anal Chem 2020 92 (2) 1697minus1701(17) MS-Product httpsprospector2ucsfeduprospectorcgi-binmsformcgiform=msproduct (accessed in December 2020)(18) Liang Y Neta P Yang X Stein S E Collision-InducedDissociation of Deprotonated Peptides Relative Abundance of Side-Chain Neutral Losses Residue-Specific Product Ions and Compar-ison with Protonated Peptides J Am Soc Mass Spectrom 2018 29(3) 463minus469(19) Kilpatrick L E Neta P Yang X Simoacuten-Manso Y LiangY Stein S E Formation of y + 10 and y + 11 ions in the collision-induced dissociation of peptide ions J Am Soc Mass Spectrom 201223 (4) 655minus663(20) Unimod httpswwwunimodorgdownloadshtml (accessedin December 2020)(21) Kleikamp H B C Lin Y M McMillan D G G GeelhoedJ S Naus-Wiezer S N H van Baarlen P Saha C Louwen R
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
F
Sorokin D Y van Loosdrecht M C M et al Tackling the chemicaldiversity of microbial nonulosonic acidsA universal large-scalesurvey approach Chem Sci 2020 11 3074minus3080(22) Yu J Schorlemer M Gomez Toledo A Pett C SihlbomC Larson G Westerlind U Nilsson J Distinctive MSMSFragmentation Pathways of Glycopeptide-Generated Oxonium IonsProvide Evidence of the Glycan Structure Chem - Eur J 2016 22(3) 1114minus1124(23) Yang X Neta P Stein S E Quality control for buildinglibraries from electrospray ionization tandem mass spectra AnalChem 2014 86 (13) 6393minus6400(24) Yang X Neta P Stein S E Extending a Tandem MassSpectral Library to Include MS2 Spectra of Fragment Ions ProducedIn-Source and MSn Spectra J Am Soc Mass Spectrom 2017 28 (11)2280minus2287(25) Zhang Z Yang X Mirokhin Y A Tchekhovskoi D V JiW Markey S P Roth J Neta P Hizal D B Bowen M A SteinS E Interconversion of Peptide Mass Spectral Libraries Derivatizedwith iTRAQ or TMT Labels J Proteome Res 2016 15 (9) 3180minus3187(26) Zhang Z Burke M C Wallace W E Liang Y Sheetlin SL Mirokhin Y A Tchekhovskoi D V Stein S E SensitiveMethod for the Confident Identification of Genetically VariantPeptides in Human Hair Keratin J Forensic Sci 2020 65 (2) 406minus420(27) Bittremieux W spectrum_utils A Python Package for MassSpectrometry Data Processing and Visualization Anal Chem 202092 (1) 659minus661(28) Roepstorff P Fohlman J Proposal for a commonnomenclature for sequence ions in mass spectra of peptides BiomedMass Spectrom 1984 11 (11) 601(29) Johnson R S Martin S A Biemann K Stults J T WatsonJ T Novel fragmentation process of peptides by collision-induceddecomposition in a tandem mass spectrometer differentiation ofleucine and isoleucine Anal Chem 1987 59 (21) 2621minus2655(30) Li K Vaudel M Zhang B Ren Y Wen B PDV anintegrative proteomics data viewer Bioinformatics 2019 35 (7)1249minus1251(31) Sturm M Kohlbacher O TOPPView an open-source viewerfor mass spectrometry data J Proteome Res 2009 8 (7) 3760minus3763(32) Proteome Discoverer httpswwwthermofishercomusenhomeindustrialmass-spectrometryliquid-chromatography-mass-spectrometry-lc-mslc-ms-softwaremulti-omics-data-analysisproteome-discoverer-softwarehtml (accessed in June 2020)(33) MacLean B Tomazela D M Shulman N Chambers MFinney G L Frewen B Kern R Tabb D L Liebler D CMacCoss M J Skyline an open source document editor for creatingand analyzing targeted proteomics experiments Bioinformatics 201026 (7) 966minus968(34) Kong A T Leprevost F V Avtonomov D MMellacheruvu D Nesvizhskii A I MSFragger ultrafast andcomprehensive peptide identification in mass spectrometry-basedproteomics Nat Methods 2017 14 (5) 513minus520(35) Liu M Q Zeng W F Fang P Cao W Q Liu C Yan GQ Zhang Y Peng C Wu J Q Zhang X J Tu H J Chi HSun R X Cao Y Dong M Q Jiang B Y Huang J M Shen HL Wong C C L He S M Yang P Y pGlyco 20 enablesprecision N-glycoproteomics with comprehensive quality control andone-step mass spectrometry for intact glycopeptide identification NatCommun 2017 8 (1) 438(36) Polasky D A Yu F Teo G C Nesvizhskii A I Fast andcomprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco Nat Methods 2020 17 (11) 1125minus1132
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
G
An example spectrum of an N-glycopeptide in Figure 4shows that MS_Piano annotates the fragments of glycanspeptides and glycopeptides enabling the rapid validation of aglycopeptide identification
Parameters for Evaluating Spectral Quality
In addition to possible product ion(s) each peak in a spectrumis annotated with the mass difference between the measuredand theoretical mz values The default mass tolerance range isset as 20 ppm and can be customized by users by adding theoption such as ldquo-r 10ppmrdquo or ldquo-r 04 Dardquo for high and lowresolution spectra respectively Furthermore spectrum quality
Figure 3 An example high resolution higher-energy collision-induced dissociation (HCD) mass spectrum with normalized collision energy (NCE)20 of a peptide with iTRAQ and carbamidomethyl (CAM) modifications at charge 3 annotated with MS_Piano The highlighted annotations areneutral losses iTRAQ and its reporter ions immonium ion (labeled with ldquoIrdquo in the beginning of the annotation eg ICCAM) and internalfragments (labeled with ldquoIntrdquo in the beginning of the annotation eg IntPG) A product ion with multiple charge states is labeled with ldquoandrdquo beforecharge value eg y6and2 is y6 at charge state 2 The ldquoUnassignedrdquo value is the fraction of unannotated peak intensities among the top 20 peaks
Table 1 Glycans for Annotation
abbreviation sugar full name
G HexNAc N-AcetylhexosamineH Hex HexoseF Fuc FucoseS NeuAc Neuraminic acidSg NeuGc N-Glycolyl neuraminic acidP Pent PentoseSoa SO3
Poa HPO3aNonsugar modifications
Figure 4 An example of an N-glycopeptide spectrum acquired on high-resolution HCD with normalized energy (NCE) 38 annotated withMS_Piano Product ions a1 a2 a3 and bprime4+G are confirmed with b1 b2 b3 and bprime4 respectively Peaks were labeled in different colors forproduct ions of glycans (red) peptides (green) and glycopeptides (blue)
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
C
is measured with the fraction of unannotated peak intensitiesamong the top 20 peaks (Unassigned in Figures 3 and 4) or allthe peaks (Unassigned_all) the intensity of the highestunassigned peak (max_unassigned_ab of base peak) andnumber of unassigned peaks in top 20 peaks (top_20_-num_unassigned_peaks) or number of unassigned peaks(num_unassigned_peaks) etc By using these parametersusers can easily assess spectrum quality to verify peptideidentification
SOFTWARE DESCRIPTIONMS_Piano was developed in Microsoft Visual C++ 2015 and isreleased in 2 formats exe and dll (peptide annotation only)The exe software can be directly used without installation onthe Windows operating system The command is simple egMS_Piano Cinmsp CoutmspData Input and Output
The NIST text file format (msp) is used for input and outputof mz vs intensity lists The following screenshots (Figure 5)illustrate an example spectrum of a peptide in an msp fileannotated with MS_Piano software In the input file thefollowing information is required for annotation peptide
sequence modifications (optional) charge state precursor mz value (Parent optional) number of peaks (Num peaks) anda peak list with mz and intensity values The peptideinformation is listed in the Name line In the output fileMS_Piano annotates peaks with product ions and massdifference between experimental and theoretical mz valueThe output file also provides the parameters described aboveto indicate spectral quality
Visualization
Tandem mass spectra with peaks annotated with reliableproduct ions in the output file can be viewed (Figures 4 6) inthe MS Search program12 to facilitate validation and generatefigures for publications and presentations The annotated peakswith fragments of peptides glycans and glycopeptides in amass spectrum can be displayed in different colors by adjustingcolor and fonts in the MS Search version 25 or later
SOFTWARE PERFORMANCE TESTING ANDAPPLICATION
MS_Pianoexe program was tested for performance on adesktop computer Windows 10 Enterprise with Intel(R)Core(TM) i7minus6700 CPU 340 GHz with 64 GB memoryAs an example of performance MS_Piano took lt15 h toannotate 1 million high resolution mass spectra of peptidesfrom protein digest samples by processing 4 files in paralleleach containing 250 000 identified spectra Peptide length was10minus30 charge state from 2 to 5 with 0minus4 modifications and30minus500 peaks in 95 of these testing spectra These spectrawere first converted to msp files (mass vs intensity peak lists intext format) from the MS-GF+9 searching results Forglycopeptide spectra the annotation time increases with thenumber of sugars in the glycans MS_Piano was tested andrefined by annotating the msp files converted from MS-GF+9
searching output files of all the data generated in Study 3 of theClinical Proteomic Tumor Analysis Consortium (NCINIH)
Table 2 N-Glycopeptide Fragments for Annotation
abbreviation fragments
Y0 no glycansY1 Y0+GY2 Y0+G2Y3 Y0+G2HY4 Y0+G2H2Y5 Y0+G2H3Y6 Y0+G2H4Y7 Y0+G2H5Y8 Y0+G2H6Y9 Y0+G2H7
Figure 5 An example spectrum of a peptide in an msp file annotated with MS_Piano
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
D
ldquoUnassigned rdquo and other parameters described abovetogether with MS-GF+ searching score were used to evaluatespectral quality and identification This software has annotated90 784 MS2 spectra of all dipeptides all tryptic tripeptides and1828 bioactive commercial peptides (purity gt90) in theNIST Tandem Mass Spectral Library2324 It has been used inannotating mass spectra of digests from iTRAQ and TMTlabeled proteins25 and human hair26 It is also used routinely toannotate high resolution mass spectra and evaluate spectralquality for optimal search for refining the NIST PeptideTandem Mass Spectral Library13 with more than 43 millionspectra MS_Piano has been used to annotate mass spectra ofprotein digests of single glycoproteins (Figure 6) including theSpike Protein in SARS-CoV-2
DISCUSSION AND CONCLUSIONSMS_Piano enables the annotation of tandem mass spectra ofboth peptides and N-glycopeptides that contain virtually allknown modifications A key feature of MS_Piano is itscapability to annotate one million peptide spectra in lt2 h withreliable product ions including some common but usuallyneglected fragments The annotated spectra can be viewedwith diverse ions displayed in different colors in NIST MSSearch12 browser for enhanced visual examination Annotatedspectra can be used to validate spectra and verify peptideidentified by sequence database searching directly orembedded in other software packages to implement variousproteomics data analysis software27 The output msp files ofspectra annotated with MS_Piano can be used on Skyline33
platform directly MS_Piano provides a metric for spectrumquality and a reportable filter for constructing peptide massspectral libraries The software is helpful for understandingpeptide fragmentation pathways Its different formats provideflexibility for biologists and chemists to use exe directly or foradvanced programmers to embed a functional dll into otherprograms
Due to the format complexity of raw data acquired on massspectrometers from different manufactures and the peak listslacking in searching results from various peptide andglycopeptide search engines the simple text format (msp) isused for input but users need to convert their files to thisformat to use this program However files can be combinedfrom searching results from libraries search engines and denovo sequencing for annotation We developed an msp fileconverter (free software convert2 msp also available at thefollowing MS_Piano download website) which quicklyconverts the results from free protein and glycoproteinsequence searching engine MSFragger34 and pGlyco35
respectively to msp files and automatically connects toMS_Piano for spectral annotation to facilitate building-your-own libraries More capabilities such as taking mzXML andpepXML together with raw data as input files annotating massspectra of negative mode ETD and O-glycopeptides will beadded to the software Software instructions and examples canbe freely downloaded at httpschemdatanistgovdokuwikidokuphpid=peptidewms_piano
ASSOCIATED CONTENT
sı Supporting Information
The Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjproteome1c00324
Product ions used in MS_Piano Table S1 Peptidemodifications Table S2 Immonium and fragment ionsTable S3 Neutral losses Table S4 iTRAQ fragmentsTable S5 TMT fragments Figure S1 Two examples oflow-resolution ion trap mass spectra of a peptideBradykinin with sequence RPPGFSPFR at charge 2and 3 respectively annotated with MS_Piano (XLSX)
Figure 6 An example of energy dependence fragmentation of an N-glycopeptide (in Alpha-1-acid glycoprotein in plasma) in charge 3 annotatedwith MS_Piano Peaks in 4 high-resolution HCD spectra at different collision energies were labeled in different colors for product ions of glycans(red) peptides (green) and glycopeptides (blue)
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
E
AUTHOR INFORMATIONCorresponding Author
Xiaoyu Yang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0003-3371-9567 Email xiaoyuyangnistgov
Authors
Pedatsur Neta minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Yuri A Mirokhin minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Dmitrii V Tchekhovskoi minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Concepcion A Remoroza minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0003-1540-1635
Meghan C Burke minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0001-7231-0655
Yuxue Liang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0002-6430-915X
Sanford P Markey minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Stephen E Stein minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Complete contact information is available athttpspubsacsorg101021acsjproteome1c00324
Notes
Certain commercial software or instruments are identified inthis paper in order to specify the experimental procedureadequately Such identification is not intended to implyrecommendation or endorsement by the National Institute ofStandards and Technology nor is it intended to imply that thesoftware or instruments identified are necessarily the bestavailable for the purposeThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors thank Drs Lewis Geer Guanghui Wang OlegToropov Zheng Zhang Sergey Sheetlin and William Wallacewho have provided suggestions useful discussions feedbackand technical support that have helped the improvement of thesoftware
REFERENCES(1) Patterson S D Aebersold R H Proteomics the first decadeand beyond Nat Genet 2003 33 (Suppl) 311minus323(2) Han X Aslanian A Yates J R 3rd Mass spectrometry forproteomics Curr Opin Chem Biol 2008 12 (5) 483minus490(3) Yu Q Paulo J A Naverrete-Perea J McAlister G CCanterbury J D Bailey D J Robitaille A M Huguet R
Zabrouskov V Gygi S P Schweppe D K Benchmarking theOrbitrap Tribrid Eclipse for Next Generation Multiplexed Proteo-mics Anal Chem 2020 92 (9) 6478minus6485(4) Adhikari S Nice E C Deutsch E W Lane L Omenn G SPennington S R Paik Y K Overall C M Corrales F J CristeaI M Van Eyk J E Uhleacuten M Lindskog C Chan D W BairochA Waddington J C Justice J L LaBaer J Rodriguez H He FKostrzewa M Ping P Gundry R L Stewart P Srivastava SSrivastava S Nogueira F C S Domont G B Vandenbrouck YLam M P Y Wennersten S Vizcaino J A Wilkins M SchwenkJ M Lundberg E Bandeira N Marko-Varga G Weintraub S TPineau C Kusebauch U Moritz R L Ahn S B Palmblad MSnyder M P Aebersold R Baker M S A high-stringency blueprintof the human proteome Nat Commun 2020 11 (1) 5301(5) Sun S Shah P Eshghi S T Yang W Trikannad N YangS Chen L Aiyetan P Houmlti N Zhang Z Chan D W Zhang HComprehensive analysis of protein glycosylation by solid-phaseextraction of N-linked glycans and glycosite-containing peptidesNat Biotechnol 2016 34 (1) 84minus88(6) Watanabe Y Allen J D Wrapp D McLellan J S CrispinM Site-specific glycan analysis of the SARS-CoV-2 spike Science2020 369 (6501) 330minus333(7) Perkins D N Pappin D J Creasy D M Cottrell J SProbability-based protein identification by searching sequencedatabases using mass spectrometry data Electrophoresis 1999 20(18) 3551minus3567(8) Geer L Y Markey S P Kowalak J A Wagner L Xu MMaynard D M Yang X Shi W Bryant S H Open massspectrometry search algorithm J Proteome Res 2004 3 (5) 958minus964(9) Byonic httpswwwproteinmetricscom (accessed in Decem-ber 2020)(10) Kim S Pevzner P A MS-GF+ makes progress towards auniversal database search tool for proteomics Nat Commun 2014 55277(11) Toghi Eshghi S Shah P Yang W Li X Zhang HGPQuest A Spectral Library Matching Algorithm for Site-SpecificAssignment of Tandem Mass Spectra to Intact N-glycopeptides AnalChem 2015 87 (10) 5181minus8(12) NIST MS Search browser httpschemdatanistgovdokuwikidokuphpid=peptidewnistmssearch (accessed in Decem-ber 2020)(13) NIST Libraries of Peptide Tandem Mass Spectra httpschemdatanistgovdokuwikidokuphpid=peptidewstart (accessedin December 2020)(14) Lam H Deutsch E W Eddes J S Eng J K King NStein S E Aebersold R Development and validation of a spectrallibrary searching method for peptide identification from MSMSProteomics 2007 7 (5) 655minus667(15) Lam H Deutsch E W Eddes J S Eng J K Stein S EAebersold R Building consensus spectral libraries for peptideidentification in proteomics Nat Methods 2008 5 (10) 873minus875(16) Fernaacutendez-Costa C Martiacutenez-Bartolomeacute S McClatchy DYates J R 3rd Improving Proteomics Data Reproducibility with aDual-Search Strategy Anal Chem 2020 92 (2) 1697minus1701(17) MS-Product httpsprospector2ucsfeduprospectorcgi-binmsformcgiform=msproduct (accessed in December 2020)(18) Liang Y Neta P Yang X Stein S E Collision-InducedDissociation of Deprotonated Peptides Relative Abundance of Side-Chain Neutral Losses Residue-Specific Product Ions and Compar-ison with Protonated Peptides J Am Soc Mass Spectrom 2018 29(3) 463minus469(19) Kilpatrick L E Neta P Yang X Simoacuten-Manso Y LiangY Stein S E Formation of y + 10 and y + 11 ions in the collision-induced dissociation of peptide ions J Am Soc Mass Spectrom 201223 (4) 655minus663(20) Unimod httpswwwunimodorgdownloadshtml (accessedin December 2020)(21) Kleikamp H B C Lin Y M McMillan D G G GeelhoedJ S Naus-Wiezer S N H van Baarlen P Saha C Louwen R
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
F
Sorokin D Y van Loosdrecht M C M et al Tackling the chemicaldiversity of microbial nonulosonic acidsA universal large-scalesurvey approach Chem Sci 2020 11 3074minus3080(22) Yu J Schorlemer M Gomez Toledo A Pett C SihlbomC Larson G Westerlind U Nilsson J Distinctive MSMSFragmentation Pathways of Glycopeptide-Generated Oxonium IonsProvide Evidence of the Glycan Structure Chem - Eur J 2016 22(3) 1114minus1124(23) Yang X Neta P Stein S E Quality control for buildinglibraries from electrospray ionization tandem mass spectra AnalChem 2014 86 (13) 6393minus6400(24) Yang X Neta P Stein S E Extending a Tandem MassSpectral Library to Include MS2 Spectra of Fragment Ions ProducedIn-Source and MSn Spectra J Am Soc Mass Spectrom 2017 28 (11)2280minus2287(25) Zhang Z Yang X Mirokhin Y A Tchekhovskoi D V JiW Markey S P Roth J Neta P Hizal D B Bowen M A SteinS E Interconversion of Peptide Mass Spectral Libraries Derivatizedwith iTRAQ or TMT Labels J Proteome Res 2016 15 (9) 3180minus3187(26) Zhang Z Burke M C Wallace W E Liang Y Sheetlin SL Mirokhin Y A Tchekhovskoi D V Stein S E SensitiveMethod for the Confident Identification of Genetically VariantPeptides in Human Hair Keratin J Forensic Sci 2020 65 (2) 406minus420(27) Bittremieux W spectrum_utils A Python Package for MassSpectrometry Data Processing and Visualization Anal Chem 202092 (1) 659minus661(28) Roepstorff P Fohlman J Proposal for a commonnomenclature for sequence ions in mass spectra of peptides BiomedMass Spectrom 1984 11 (11) 601(29) Johnson R S Martin S A Biemann K Stults J T WatsonJ T Novel fragmentation process of peptides by collision-induceddecomposition in a tandem mass spectrometer differentiation ofleucine and isoleucine Anal Chem 1987 59 (21) 2621minus2655(30) Li K Vaudel M Zhang B Ren Y Wen B PDV anintegrative proteomics data viewer Bioinformatics 2019 35 (7)1249minus1251(31) Sturm M Kohlbacher O TOPPView an open-source viewerfor mass spectrometry data J Proteome Res 2009 8 (7) 3760minus3763(32) Proteome Discoverer httpswwwthermofishercomusenhomeindustrialmass-spectrometryliquid-chromatography-mass-spectrometry-lc-mslc-ms-softwaremulti-omics-data-analysisproteome-discoverer-softwarehtml (accessed in June 2020)(33) MacLean B Tomazela D M Shulman N Chambers MFinney G L Frewen B Kern R Tabb D L Liebler D CMacCoss M J Skyline an open source document editor for creatingand analyzing targeted proteomics experiments Bioinformatics 201026 (7) 966minus968(34) Kong A T Leprevost F V Avtonomov D MMellacheruvu D Nesvizhskii A I MSFragger ultrafast andcomprehensive peptide identification in mass spectrometry-basedproteomics Nat Methods 2017 14 (5) 513minus520(35) Liu M Q Zeng W F Fang P Cao W Q Liu C Yan GQ Zhang Y Peng C Wu J Q Zhang X J Tu H J Chi HSun R X Cao Y Dong M Q Jiang B Y Huang J M Shen HL Wong C C L He S M Yang P Y pGlyco 20 enablesprecision N-glycoproteomics with comprehensive quality control andone-step mass spectrometry for intact glycopeptide identification NatCommun 2017 8 (1) 438(36) Polasky D A Yu F Teo G C Nesvizhskii A I Fast andcomprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco Nat Methods 2020 17 (11) 1125minus1132
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
G

is measured with the fraction of unannotated peak intensitiesamong the top 20 peaks (Unassigned in Figures 3 and 4) or allthe peaks (Unassigned_all) the intensity of the highestunassigned peak (max_unassigned_ab of base peak) andnumber of unassigned peaks in top 20 peaks (top_20_-num_unassigned_peaks) or number of unassigned peaks(num_unassigned_peaks) etc By using these parametersusers can easily assess spectrum quality to verify peptideidentification
SOFTWARE DESCRIPTIONMS_Piano was developed in Microsoft Visual C++ 2015 and isreleased in 2 formats exe and dll (peptide annotation only)The exe software can be directly used without installation onthe Windows operating system The command is simple egMS_Piano Cinmsp CoutmspData Input and Output
The NIST text file format (msp) is used for input and outputof mz vs intensity lists The following screenshots (Figure 5)illustrate an example spectrum of a peptide in an msp fileannotated with MS_Piano software In the input file thefollowing information is required for annotation peptide
sequence modifications (optional) charge state precursor mz value (Parent optional) number of peaks (Num peaks) anda peak list with mz and intensity values The peptideinformation is listed in the Name line In the output fileMS_Piano annotates peaks with product ions and massdifference between experimental and theoretical mz valueThe output file also provides the parameters described aboveto indicate spectral quality
Visualization
Tandem mass spectra with peaks annotated with reliableproduct ions in the output file can be viewed (Figures 4 6) inthe MS Search program12 to facilitate validation and generatefigures for publications and presentations The annotated peakswith fragments of peptides glycans and glycopeptides in amass spectrum can be displayed in different colors by adjustingcolor and fonts in the MS Search version 25 or later
SOFTWARE PERFORMANCE TESTING ANDAPPLICATION
MS_Pianoexe program was tested for performance on adesktop computer Windows 10 Enterprise with Intel(R)Core(TM) i7minus6700 CPU 340 GHz with 64 GB memoryAs an example of performance MS_Piano took lt15 h toannotate 1 million high resolution mass spectra of peptidesfrom protein digest samples by processing 4 files in paralleleach containing 250 000 identified spectra Peptide length was10minus30 charge state from 2 to 5 with 0minus4 modifications and30minus500 peaks in 95 of these testing spectra These spectrawere first converted to msp files (mass vs intensity peak lists intext format) from the MS-GF+9 searching results Forglycopeptide spectra the annotation time increases with thenumber of sugars in the glycans MS_Piano was tested andrefined by annotating the msp files converted from MS-GF+9
searching output files of all the data generated in Study 3 of theClinical Proteomic Tumor Analysis Consortium (NCINIH)
Table 2 N-Glycopeptide Fragments for Annotation
abbreviation fragments
Y0 no glycansY1 Y0+GY2 Y0+G2Y3 Y0+G2HY4 Y0+G2H2Y5 Y0+G2H3Y6 Y0+G2H4Y7 Y0+G2H5Y8 Y0+G2H6Y9 Y0+G2H7
Figure 5 An example spectrum of a peptide in an msp file annotated with MS_Piano
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
D
ldquoUnassigned rdquo and other parameters described abovetogether with MS-GF+ searching score were used to evaluatespectral quality and identification This software has annotated90 784 MS2 spectra of all dipeptides all tryptic tripeptides and1828 bioactive commercial peptides (purity gt90) in theNIST Tandem Mass Spectral Library2324 It has been used inannotating mass spectra of digests from iTRAQ and TMTlabeled proteins25 and human hair26 It is also used routinely toannotate high resolution mass spectra and evaluate spectralquality for optimal search for refining the NIST PeptideTandem Mass Spectral Library13 with more than 43 millionspectra MS_Piano has been used to annotate mass spectra ofprotein digests of single glycoproteins (Figure 6) including theSpike Protein in SARS-CoV-2
DISCUSSION AND CONCLUSIONSMS_Piano enables the annotation of tandem mass spectra ofboth peptides and N-glycopeptides that contain virtually allknown modifications A key feature of MS_Piano is itscapability to annotate one million peptide spectra in lt2 h withreliable product ions including some common but usuallyneglected fragments The annotated spectra can be viewedwith diverse ions displayed in different colors in NIST MSSearch12 browser for enhanced visual examination Annotatedspectra can be used to validate spectra and verify peptideidentified by sequence database searching directly orembedded in other software packages to implement variousproteomics data analysis software27 The output msp files ofspectra annotated with MS_Piano can be used on Skyline33
platform directly MS_Piano provides a metric for spectrumquality and a reportable filter for constructing peptide massspectral libraries The software is helpful for understandingpeptide fragmentation pathways Its different formats provideflexibility for biologists and chemists to use exe directly or foradvanced programmers to embed a functional dll into otherprograms
Due to the format complexity of raw data acquired on massspectrometers from different manufactures and the peak listslacking in searching results from various peptide andglycopeptide search engines the simple text format (msp) isused for input but users need to convert their files to thisformat to use this program However files can be combinedfrom searching results from libraries search engines and denovo sequencing for annotation We developed an msp fileconverter (free software convert2 msp also available at thefollowing MS_Piano download website) which quicklyconverts the results from free protein and glycoproteinsequence searching engine MSFragger34 and pGlyco35
respectively to msp files and automatically connects toMS_Piano for spectral annotation to facilitate building-your-own libraries More capabilities such as taking mzXML andpepXML together with raw data as input files annotating massspectra of negative mode ETD and O-glycopeptides will beadded to the software Software instructions and examples canbe freely downloaded at httpschemdatanistgovdokuwikidokuphpid=peptidewms_piano
ASSOCIATED CONTENT
sı Supporting Information
The Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjproteome1c00324
Product ions used in MS_Piano Table S1 Peptidemodifications Table S2 Immonium and fragment ionsTable S3 Neutral losses Table S4 iTRAQ fragmentsTable S5 TMT fragments Figure S1 Two examples oflow-resolution ion trap mass spectra of a peptideBradykinin with sequence RPPGFSPFR at charge 2and 3 respectively annotated with MS_Piano (XLSX)
Figure 6 An example of energy dependence fragmentation of an N-glycopeptide (in Alpha-1-acid glycoprotein in plasma) in charge 3 annotatedwith MS_Piano Peaks in 4 high-resolution HCD spectra at different collision energies were labeled in different colors for product ions of glycans(red) peptides (green) and glycopeptides (blue)
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
E
AUTHOR INFORMATIONCorresponding Author
Xiaoyu Yang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0003-3371-9567 Email xiaoyuyangnistgov
Authors
Pedatsur Neta minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Yuri A Mirokhin minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Dmitrii V Tchekhovskoi minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Concepcion A Remoroza minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0003-1540-1635
Meghan C Burke minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0001-7231-0655
Yuxue Liang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0002-6430-915X
Sanford P Markey minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Stephen E Stein minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Complete contact information is available athttpspubsacsorg101021acsjproteome1c00324
Notes
Certain commercial software or instruments are identified inthis paper in order to specify the experimental procedureadequately Such identification is not intended to implyrecommendation or endorsement by the National Institute ofStandards and Technology nor is it intended to imply that thesoftware or instruments identified are necessarily the bestavailable for the purposeThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors thank Drs Lewis Geer Guanghui Wang OlegToropov Zheng Zhang Sergey Sheetlin and William Wallacewho have provided suggestions useful discussions feedbackand technical support that have helped the improvement of thesoftware
REFERENCES(1) Patterson S D Aebersold R H Proteomics the first decadeand beyond Nat Genet 2003 33 (Suppl) 311minus323(2) Han X Aslanian A Yates J R 3rd Mass spectrometry forproteomics Curr Opin Chem Biol 2008 12 (5) 483minus490(3) Yu Q Paulo J A Naverrete-Perea J McAlister G CCanterbury J D Bailey D J Robitaille A M Huguet R
Zabrouskov V Gygi S P Schweppe D K Benchmarking theOrbitrap Tribrid Eclipse for Next Generation Multiplexed Proteo-mics Anal Chem 2020 92 (9) 6478minus6485(4) Adhikari S Nice E C Deutsch E W Lane L Omenn G SPennington S R Paik Y K Overall C M Corrales F J CristeaI M Van Eyk J E Uhleacuten M Lindskog C Chan D W BairochA Waddington J C Justice J L LaBaer J Rodriguez H He FKostrzewa M Ping P Gundry R L Stewart P Srivastava SSrivastava S Nogueira F C S Domont G B Vandenbrouck YLam M P Y Wennersten S Vizcaino J A Wilkins M SchwenkJ M Lundberg E Bandeira N Marko-Varga G Weintraub S TPineau C Kusebauch U Moritz R L Ahn S B Palmblad MSnyder M P Aebersold R Baker M S A high-stringency blueprintof the human proteome Nat Commun 2020 11 (1) 5301(5) Sun S Shah P Eshghi S T Yang W Trikannad N YangS Chen L Aiyetan P Houmlti N Zhang Z Chan D W Zhang HComprehensive analysis of protein glycosylation by solid-phaseextraction of N-linked glycans and glycosite-containing peptidesNat Biotechnol 2016 34 (1) 84minus88(6) Watanabe Y Allen J D Wrapp D McLellan J S CrispinM Site-specific glycan analysis of the SARS-CoV-2 spike Science2020 369 (6501) 330minus333(7) Perkins D N Pappin D J Creasy D M Cottrell J SProbability-based protein identification by searching sequencedatabases using mass spectrometry data Electrophoresis 1999 20(18) 3551minus3567(8) Geer L Y Markey S P Kowalak J A Wagner L Xu MMaynard D M Yang X Shi W Bryant S H Open massspectrometry search algorithm J Proteome Res 2004 3 (5) 958minus964(9) Byonic httpswwwproteinmetricscom (accessed in Decem-ber 2020)(10) Kim S Pevzner P A MS-GF+ makes progress towards auniversal database search tool for proteomics Nat Commun 2014 55277(11) Toghi Eshghi S Shah P Yang W Li X Zhang HGPQuest A Spectral Library Matching Algorithm for Site-SpecificAssignment of Tandem Mass Spectra to Intact N-glycopeptides AnalChem 2015 87 (10) 5181minus8(12) NIST MS Search browser httpschemdatanistgovdokuwikidokuphpid=peptidewnistmssearch (accessed in Decem-ber 2020)(13) NIST Libraries of Peptide Tandem Mass Spectra httpschemdatanistgovdokuwikidokuphpid=peptidewstart (accessedin December 2020)(14) Lam H Deutsch E W Eddes J S Eng J K King NStein S E Aebersold R Development and validation of a spectrallibrary searching method for peptide identification from MSMSProteomics 2007 7 (5) 655minus667(15) Lam H Deutsch E W Eddes J S Eng J K Stein S EAebersold R Building consensus spectral libraries for peptideidentification in proteomics Nat Methods 2008 5 (10) 873minus875(16) Fernaacutendez-Costa C Martiacutenez-Bartolomeacute S McClatchy DYates J R 3rd Improving Proteomics Data Reproducibility with aDual-Search Strategy Anal Chem 2020 92 (2) 1697minus1701(17) MS-Product httpsprospector2ucsfeduprospectorcgi-binmsformcgiform=msproduct (accessed in December 2020)(18) Liang Y Neta P Yang X Stein S E Collision-InducedDissociation of Deprotonated Peptides Relative Abundance of Side-Chain Neutral Losses Residue-Specific Product Ions and Compar-ison with Protonated Peptides J Am Soc Mass Spectrom 2018 29(3) 463minus469(19) Kilpatrick L E Neta P Yang X Simoacuten-Manso Y LiangY Stein S E Formation of y + 10 and y + 11 ions in the collision-induced dissociation of peptide ions J Am Soc Mass Spectrom 201223 (4) 655minus663(20) Unimod httpswwwunimodorgdownloadshtml (accessedin December 2020)(21) Kleikamp H B C Lin Y M McMillan D G G GeelhoedJ S Naus-Wiezer S N H van Baarlen P Saha C Louwen R
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
F
Sorokin D Y van Loosdrecht M C M et al Tackling the chemicaldiversity of microbial nonulosonic acidsA universal large-scalesurvey approach Chem Sci 2020 11 3074minus3080(22) Yu J Schorlemer M Gomez Toledo A Pett C SihlbomC Larson G Westerlind U Nilsson J Distinctive MSMSFragmentation Pathways of Glycopeptide-Generated Oxonium IonsProvide Evidence of the Glycan Structure Chem - Eur J 2016 22(3) 1114minus1124(23) Yang X Neta P Stein S E Quality control for buildinglibraries from electrospray ionization tandem mass spectra AnalChem 2014 86 (13) 6393minus6400(24) Yang X Neta P Stein S E Extending a Tandem MassSpectral Library to Include MS2 Spectra of Fragment Ions ProducedIn-Source and MSn Spectra J Am Soc Mass Spectrom 2017 28 (11)2280minus2287(25) Zhang Z Yang X Mirokhin Y A Tchekhovskoi D V JiW Markey S P Roth J Neta P Hizal D B Bowen M A SteinS E Interconversion of Peptide Mass Spectral Libraries Derivatizedwith iTRAQ or TMT Labels J Proteome Res 2016 15 (9) 3180minus3187(26) Zhang Z Burke M C Wallace W E Liang Y Sheetlin SL Mirokhin Y A Tchekhovskoi D V Stein S E SensitiveMethod for the Confident Identification of Genetically VariantPeptides in Human Hair Keratin J Forensic Sci 2020 65 (2) 406minus420(27) Bittremieux W spectrum_utils A Python Package for MassSpectrometry Data Processing and Visualization Anal Chem 202092 (1) 659minus661(28) Roepstorff P Fohlman J Proposal for a commonnomenclature for sequence ions in mass spectra of peptides BiomedMass Spectrom 1984 11 (11) 601(29) Johnson R S Martin S A Biemann K Stults J T WatsonJ T Novel fragmentation process of peptides by collision-induceddecomposition in a tandem mass spectrometer differentiation ofleucine and isoleucine Anal Chem 1987 59 (21) 2621minus2655(30) Li K Vaudel M Zhang B Ren Y Wen B PDV anintegrative proteomics data viewer Bioinformatics 2019 35 (7)1249minus1251(31) Sturm M Kohlbacher O TOPPView an open-source viewerfor mass spectrometry data J Proteome Res 2009 8 (7) 3760minus3763(32) Proteome Discoverer httpswwwthermofishercomusenhomeindustrialmass-spectrometryliquid-chromatography-mass-spectrometry-lc-mslc-ms-softwaremulti-omics-data-analysisproteome-discoverer-softwarehtml (accessed in June 2020)(33) MacLean B Tomazela D M Shulman N Chambers MFinney G L Frewen B Kern R Tabb D L Liebler D CMacCoss M J Skyline an open source document editor for creatingand analyzing targeted proteomics experiments Bioinformatics 201026 (7) 966minus968(34) Kong A T Leprevost F V Avtonomov D MMellacheruvu D Nesvizhskii A I MSFragger ultrafast andcomprehensive peptide identification in mass spectrometry-basedproteomics Nat Methods 2017 14 (5) 513minus520(35) Liu M Q Zeng W F Fang P Cao W Q Liu C Yan GQ Zhang Y Peng C Wu J Q Zhang X J Tu H J Chi HSun R X Cao Y Dong M Q Jiang B Y Huang J M Shen HL Wong C C L He S M Yang P Y pGlyco 20 enablesprecision N-glycoproteomics with comprehensive quality control andone-step mass spectrometry for intact glycopeptide identification NatCommun 2017 8 (1) 438(36) Polasky D A Yu F Teo G C Nesvizhskii A I Fast andcomprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco Nat Methods 2020 17 (11) 1125minus1132
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
G

ldquoUnassigned rdquo and other parameters described abovetogether with MS-GF+ searching score were used to evaluatespectral quality and identification This software has annotated90 784 MS2 spectra of all dipeptides all tryptic tripeptides and1828 bioactive commercial peptides (purity gt90) in theNIST Tandem Mass Spectral Library2324 It has been used inannotating mass spectra of digests from iTRAQ and TMTlabeled proteins25 and human hair26 It is also used routinely toannotate high resolution mass spectra and evaluate spectralquality for optimal search for refining the NIST PeptideTandem Mass Spectral Library13 with more than 43 millionspectra MS_Piano has been used to annotate mass spectra ofprotein digests of single glycoproteins (Figure 6) including theSpike Protein in SARS-CoV-2
DISCUSSION AND CONCLUSIONSMS_Piano enables the annotation of tandem mass spectra ofboth peptides and N-glycopeptides that contain virtually allknown modifications A key feature of MS_Piano is itscapability to annotate one million peptide spectra in lt2 h withreliable product ions including some common but usuallyneglected fragments The annotated spectra can be viewedwith diverse ions displayed in different colors in NIST MSSearch12 browser for enhanced visual examination Annotatedspectra can be used to validate spectra and verify peptideidentified by sequence database searching directly orembedded in other software packages to implement variousproteomics data analysis software27 The output msp files ofspectra annotated with MS_Piano can be used on Skyline33
platform directly MS_Piano provides a metric for spectrumquality and a reportable filter for constructing peptide massspectral libraries The software is helpful for understandingpeptide fragmentation pathways Its different formats provideflexibility for biologists and chemists to use exe directly or foradvanced programmers to embed a functional dll into otherprograms
Due to the format complexity of raw data acquired on massspectrometers from different manufactures and the peak listslacking in searching results from various peptide andglycopeptide search engines the simple text format (msp) isused for input but users need to convert their files to thisformat to use this program However files can be combinedfrom searching results from libraries search engines and denovo sequencing for annotation We developed an msp fileconverter (free software convert2 msp also available at thefollowing MS_Piano download website) which quicklyconverts the results from free protein and glycoproteinsequence searching engine MSFragger34 and pGlyco35
respectively to msp files and automatically connects toMS_Piano for spectral annotation to facilitate building-your-own libraries More capabilities such as taking mzXML andpepXML together with raw data as input files annotating massspectra of negative mode ETD and O-glycopeptides will beadded to the software Software instructions and examples canbe freely downloaded at httpschemdatanistgovdokuwikidokuphpid=peptidewms_piano
ASSOCIATED CONTENT
sı Supporting Information
The Supporting Information is available free of charge athttpspubsacsorgdoi101021acsjproteome1c00324
Product ions used in MS_Piano Table S1 Peptidemodifications Table S2 Immonium and fragment ionsTable S3 Neutral losses Table S4 iTRAQ fragmentsTable S5 TMT fragments Figure S1 Two examples oflow-resolution ion trap mass spectra of a peptideBradykinin with sequence RPPGFSPFR at charge 2and 3 respectively annotated with MS_Piano (XLSX)
Figure 6 An example of energy dependence fragmentation of an N-glycopeptide (in Alpha-1-acid glycoprotein in plasma) in charge 3 annotatedwith MS_Piano Peaks in 4 high-resolution HCD spectra at different collision energies were labeled in different colors for product ions of glycans(red) peptides (green) and glycopeptides (blue)
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
E
AUTHOR INFORMATIONCorresponding Author
Xiaoyu Yang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0003-3371-9567 Email xiaoyuyangnistgov
Authors
Pedatsur Neta minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Yuri A Mirokhin minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Dmitrii V Tchekhovskoi minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Concepcion A Remoroza minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0003-1540-1635
Meghan C Burke minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0001-7231-0655
Yuxue Liang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0002-6430-915X
Sanford P Markey minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Stephen E Stein minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Complete contact information is available athttpspubsacsorg101021acsjproteome1c00324
Notes
Certain commercial software or instruments are identified inthis paper in order to specify the experimental procedureadequately Such identification is not intended to implyrecommendation or endorsement by the National Institute ofStandards and Technology nor is it intended to imply that thesoftware or instruments identified are necessarily the bestavailable for the purposeThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors thank Drs Lewis Geer Guanghui Wang OlegToropov Zheng Zhang Sergey Sheetlin and William Wallacewho have provided suggestions useful discussions feedbackand technical support that have helped the improvement of thesoftware
REFERENCES(1) Patterson S D Aebersold R H Proteomics the first decadeand beyond Nat Genet 2003 33 (Suppl) 311minus323(2) Han X Aslanian A Yates J R 3rd Mass spectrometry forproteomics Curr Opin Chem Biol 2008 12 (5) 483minus490(3) Yu Q Paulo J A Naverrete-Perea J McAlister G CCanterbury J D Bailey D J Robitaille A M Huguet R
Zabrouskov V Gygi S P Schweppe D K Benchmarking theOrbitrap Tribrid Eclipse for Next Generation Multiplexed Proteo-mics Anal Chem 2020 92 (9) 6478minus6485(4) Adhikari S Nice E C Deutsch E W Lane L Omenn G SPennington S R Paik Y K Overall C M Corrales F J CristeaI M Van Eyk J E Uhleacuten M Lindskog C Chan D W BairochA Waddington J C Justice J L LaBaer J Rodriguez H He FKostrzewa M Ping P Gundry R L Stewart P Srivastava SSrivastava S Nogueira F C S Domont G B Vandenbrouck YLam M P Y Wennersten S Vizcaino J A Wilkins M SchwenkJ M Lundberg E Bandeira N Marko-Varga G Weintraub S TPineau C Kusebauch U Moritz R L Ahn S B Palmblad MSnyder M P Aebersold R Baker M S A high-stringency blueprintof the human proteome Nat Commun 2020 11 (1) 5301(5) Sun S Shah P Eshghi S T Yang W Trikannad N YangS Chen L Aiyetan P Houmlti N Zhang Z Chan D W Zhang HComprehensive analysis of protein glycosylation by solid-phaseextraction of N-linked glycans and glycosite-containing peptidesNat Biotechnol 2016 34 (1) 84minus88(6) Watanabe Y Allen J D Wrapp D McLellan J S CrispinM Site-specific glycan analysis of the SARS-CoV-2 spike Science2020 369 (6501) 330minus333(7) Perkins D N Pappin D J Creasy D M Cottrell J SProbability-based protein identification by searching sequencedatabases using mass spectrometry data Electrophoresis 1999 20(18) 3551minus3567(8) Geer L Y Markey S P Kowalak J A Wagner L Xu MMaynard D M Yang X Shi W Bryant S H Open massspectrometry search algorithm J Proteome Res 2004 3 (5) 958minus964(9) Byonic httpswwwproteinmetricscom (accessed in Decem-ber 2020)(10) Kim S Pevzner P A MS-GF+ makes progress towards auniversal database search tool for proteomics Nat Commun 2014 55277(11) Toghi Eshghi S Shah P Yang W Li X Zhang HGPQuest A Spectral Library Matching Algorithm for Site-SpecificAssignment of Tandem Mass Spectra to Intact N-glycopeptides AnalChem 2015 87 (10) 5181minus8(12) NIST MS Search browser httpschemdatanistgovdokuwikidokuphpid=peptidewnistmssearch (accessed in Decem-ber 2020)(13) NIST Libraries of Peptide Tandem Mass Spectra httpschemdatanistgovdokuwikidokuphpid=peptidewstart (accessedin December 2020)(14) Lam H Deutsch E W Eddes J S Eng J K King NStein S E Aebersold R Development and validation of a spectrallibrary searching method for peptide identification from MSMSProteomics 2007 7 (5) 655minus667(15) Lam H Deutsch E W Eddes J S Eng J K Stein S EAebersold R Building consensus spectral libraries for peptideidentification in proteomics Nat Methods 2008 5 (10) 873minus875(16) Fernaacutendez-Costa C Martiacutenez-Bartolomeacute S McClatchy DYates J R 3rd Improving Proteomics Data Reproducibility with aDual-Search Strategy Anal Chem 2020 92 (2) 1697minus1701(17) MS-Product httpsprospector2ucsfeduprospectorcgi-binmsformcgiform=msproduct (accessed in December 2020)(18) Liang Y Neta P Yang X Stein S E Collision-InducedDissociation of Deprotonated Peptides Relative Abundance of Side-Chain Neutral Losses Residue-Specific Product Ions and Compar-ison with Protonated Peptides J Am Soc Mass Spectrom 2018 29(3) 463minus469(19) Kilpatrick L E Neta P Yang X Simoacuten-Manso Y LiangY Stein S E Formation of y + 10 and y + 11 ions in the collision-induced dissociation of peptide ions J Am Soc Mass Spectrom 201223 (4) 655minus663(20) Unimod httpswwwunimodorgdownloadshtml (accessedin December 2020)(21) Kleikamp H B C Lin Y M McMillan D G G GeelhoedJ S Naus-Wiezer S N H van Baarlen P Saha C Louwen R
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
F
Sorokin D Y van Loosdrecht M C M et al Tackling the chemicaldiversity of microbial nonulosonic acidsA universal large-scalesurvey approach Chem Sci 2020 11 3074minus3080(22) Yu J Schorlemer M Gomez Toledo A Pett C SihlbomC Larson G Westerlind U Nilsson J Distinctive MSMSFragmentation Pathways of Glycopeptide-Generated Oxonium IonsProvide Evidence of the Glycan Structure Chem - Eur J 2016 22(3) 1114minus1124(23) Yang X Neta P Stein S E Quality control for buildinglibraries from electrospray ionization tandem mass spectra AnalChem 2014 86 (13) 6393minus6400(24) Yang X Neta P Stein S E Extending a Tandem MassSpectral Library to Include MS2 Spectra of Fragment Ions ProducedIn-Source and MSn Spectra J Am Soc Mass Spectrom 2017 28 (11)2280minus2287(25) Zhang Z Yang X Mirokhin Y A Tchekhovskoi D V JiW Markey S P Roth J Neta P Hizal D B Bowen M A SteinS E Interconversion of Peptide Mass Spectral Libraries Derivatizedwith iTRAQ or TMT Labels J Proteome Res 2016 15 (9) 3180minus3187(26) Zhang Z Burke M C Wallace W E Liang Y Sheetlin SL Mirokhin Y A Tchekhovskoi D V Stein S E SensitiveMethod for the Confident Identification of Genetically VariantPeptides in Human Hair Keratin J Forensic Sci 2020 65 (2) 406minus420(27) Bittremieux W spectrum_utils A Python Package for MassSpectrometry Data Processing and Visualization Anal Chem 202092 (1) 659minus661(28) Roepstorff P Fohlman J Proposal for a commonnomenclature for sequence ions in mass spectra of peptides BiomedMass Spectrom 1984 11 (11) 601(29) Johnson R S Martin S A Biemann K Stults J T WatsonJ T Novel fragmentation process of peptides by collision-induceddecomposition in a tandem mass spectrometer differentiation ofleucine and isoleucine Anal Chem 1987 59 (21) 2621minus2655(30) Li K Vaudel M Zhang B Ren Y Wen B PDV anintegrative proteomics data viewer Bioinformatics 2019 35 (7)1249minus1251(31) Sturm M Kohlbacher O TOPPView an open-source viewerfor mass spectrometry data J Proteome Res 2009 8 (7) 3760minus3763(32) Proteome Discoverer httpswwwthermofishercomusenhomeindustrialmass-spectrometryliquid-chromatography-mass-spectrometry-lc-mslc-ms-softwaremulti-omics-data-analysisproteome-discoverer-softwarehtml (accessed in June 2020)(33) MacLean B Tomazela D M Shulman N Chambers MFinney G L Frewen B Kern R Tabb D L Liebler D CMacCoss M J Skyline an open source document editor for creatingand analyzing targeted proteomics experiments Bioinformatics 201026 (7) 966minus968(34) Kong A T Leprevost F V Avtonomov D MMellacheruvu D Nesvizhskii A I MSFragger ultrafast andcomprehensive peptide identification in mass spectrometry-basedproteomics Nat Methods 2017 14 (5) 513minus520(35) Liu M Q Zeng W F Fang P Cao W Q Liu C Yan GQ Zhang Y Peng C Wu J Q Zhang X J Tu H J Chi HSun R X Cao Y Dong M Q Jiang B Y Huang J M Shen HL Wong C C L He S M Yang P Y pGlyco 20 enablesprecision N-glycoproteomics with comprehensive quality control andone-step mass spectrometry for intact glycopeptide identification NatCommun 2017 8 (1) 438(36) Polasky D A Yu F Teo G C Nesvizhskii A I Fast andcomprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco Nat Methods 2020 17 (11) 1125minus1132
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
G

AUTHOR INFORMATIONCorresponding Author
Xiaoyu Yang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0003-3371-9567 Email xiaoyuyangnistgov
Authors
Pedatsur Neta minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Yuri A Mirokhin minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Dmitrii V Tchekhovskoi minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Concepcion A Remoroza minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0003-1540-1635
Meghan C Burke minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States orcidorg0000-0001-7231-0655
Yuxue Liang minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States orcidorg0000-0002-6430-915X
Sanford P Markey minus Mass Spectrometry Data CenterNational Institute of Standards and TechnologyGaithersburg Maryland 20899 United States
Stephen E Stein minus Mass Spectrometry Data Center NationalInstitute of Standards and Technology GaithersburgMaryland 20899 United States
Complete contact information is available athttpspubsacsorg101021acsjproteome1c00324
Notes
Certain commercial software or instruments are identified inthis paper in order to specify the experimental procedureadequately Such identification is not intended to implyrecommendation or endorsement by the National Institute ofStandards and Technology nor is it intended to imply that thesoftware or instruments identified are necessarily the bestavailable for the purposeThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors thank Drs Lewis Geer Guanghui Wang OlegToropov Zheng Zhang Sergey Sheetlin and William Wallacewho have provided suggestions useful discussions feedbackand technical support that have helped the improvement of thesoftware
REFERENCES(1) Patterson S D Aebersold R H Proteomics the first decadeand beyond Nat Genet 2003 33 (Suppl) 311minus323(2) Han X Aslanian A Yates J R 3rd Mass spectrometry forproteomics Curr Opin Chem Biol 2008 12 (5) 483minus490(3) Yu Q Paulo J A Naverrete-Perea J McAlister G CCanterbury J D Bailey D J Robitaille A M Huguet R
Zabrouskov V Gygi S P Schweppe D K Benchmarking theOrbitrap Tribrid Eclipse for Next Generation Multiplexed Proteo-mics Anal Chem 2020 92 (9) 6478minus6485(4) Adhikari S Nice E C Deutsch E W Lane L Omenn G SPennington S R Paik Y K Overall C M Corrales F J CristeaI M Van Eyk J E Uhleacuten M Lindskog C Chan D W BairochA Waddington J C Justice J L LaBaer J Rodriguez H He FKostrzewa M Ping P Gundry R L Stewart P Srivastava SSrivastava S Nogueira F C S Domont G B Vandenbrouck YLam M P Y Wennersten S Vizcaino J A Wilkins M SchwenkJ M Lundberg E Bandeira N Marko-Varga G Weintraub S TPineau C Kusebauch U Moritz R L Ahn S B Palmblad MSnyder M P Aebersold R Baker M S A high-stringency blueprintof the human proteome Nat Commun 2020 11 (1) 5301(5) Sun S Shah P Eshghi S T Yang W Trikannad N YangS Chen L Aiyetan P Houmlti N Zhang Z Chan D W Zhang HComprehensive analysis of protein glycosylation by solid-phaseextraction of N-linked glycans and glycosite-containing peptidesNat Biotechnol 2016 34 (1) 84minus88(6) Watanabe Y Allen J D Wrapp D McLellan J S CrispinM Site-specific glycan analysis of the SARS-CoV-2 spike Science2020 369 (6501) 330minus333(7) Perkins D N Pappin D J Creasy D M Cottrell J SProbability-based protein identification by searching sequencedatabases using mass spectrometry data Electrophoresis 1999 20(18) 3551minus3567(8) Geer L Y Markey S P Kowalak J A Wagner L Xu MMaynard D M Yang X Shi W Bryant S H Open massspectrometry search algorithm J Proteome Res 2004 3 (5) 958minus964(9) Byonic httpswwwproteinmetricscom (accessed in Decem-ber 2020)(10) Kim S Pevzner P A MS-GF+ makes progress towards auniversal database search tool for proteomics Nat Commun 2014 55277(11) Toghi Eshghi S Shah P Yang W Li X Zhang HGPQuest A Spectral Library Matching Algorithm for Site-SpecificAssignment of Tandem Mass Spectra to Intact N-glycopeptides AnalChem 2015 87 (10) 5181minus8(12) NIST MS Search browser httpschemdatanistgovdokuwikidokuphpid=peptidewnistmssearch (accessed in Decem-ber 2020)(13) NIST Libraries of Peptide Tandem Mass Spectra httpschemdatanistgovdokuwikidokuphpid=peptidewstart (accessedin December 2020)(14) Lam H Deutsch E W Eddes J S Eng J K King NStein S E Aebersold R Development and validation of a spectrallibrary searching method for peptide identification from MSMSProteomics 2007 7 (5) 655minus667(15) Lam H Deutsch E W Eddes J S Eng J K Stein S EAebersold R Building consensus spectral libraries for peptideidentification in proteomics Nat Methods 2008 5 (10) 873minus875(16) Fernaacutendez-Costa C Martiacutenez-Bartolomeacute S McClatchy DYates J R 3rd Improving Proteomics Data Reproducibility with aDual-Search Strategy Anal Chem 2020 92 (2) 1697minus1701(17) MS-Product httpsprospector2ucsfeduprospectorcgi-binmsformcgiform=msproduct (accessed in December 2020)(18) Liang Y Neta P Yang X Stein S E Collision-InducedDissociation of Deprotonated Peptides Relative Abundance of Side-Chain Neutral Losses Residue-Specific Product Ions and Compar-ison with Protonated Peptides J Am Soc Mass Spectrom 2018 29(3) 463minus469(19) Kilpatrick L E Neta P Yang X Simoacuten-Manso Y LiangY Stein S E Formation of y + 10 and y + 11 ions in the collision-induced dissociation of peptide ions J Am Soc Mass Spectrom 201223 (4) 655minus663(20) Unimod httpswwwunimodorgdownloadshtml (accessedin December 2020)(21) Kleikamp H B C Lin Y M McMillan D G G GeelhoedJ S Naus-Wiezer S N H van Baarlen P Saha C Louwen R
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
F
Sorokin D Y van Loosdrecht M C M et al Tackling the chemicaldiversity of microbial nonulosonic acidsA universal large-scalesurvey approach Chem Sci 2020 11 3074minus3080(22) Yu J Schorlemer M Gomez Toledo A Pett C SihlbomC Larson G Westerlind U Nilsson J Distinctive MSMSFragmentation Pathways of Glycopeptide-Generated Oxonium IonsProvide Evidence of the Glycan Structure Chem - Eur J 2016 22(3) 1114minus1124(23) Yang X Neta P Stein S E Quality control for buildinglibraries from electrospray ionization tandem mass spectra AnalChem 2014 86 (13) 6393minus6400(24) Yang X Neta P Stein S E Extending a Tandem MassSpectral Library to Include MS2 Spectra of Fragment Ions ProducedIn-Source and MSn Spectra J Am Soc Mass Spectrom 2017 28 (11)2280minus2287(25) Zhang Z Yang X Mirokhin Y A Tchekhovskoi D V JiW Markey S P Roth J Neta P Hizal D B Bowen M A SteinS E Interconversion of Peptide Mass Spectral Libraries Derivatizedwith iTRAQ or TMT Labels J Proteome Res 2016 15 (9) 3180minus3187(26) Zhang Z Burke M C Wallace W E Liang Y Sheetlin SL Mirokhin Y A Tchekhovskoi D V Stein S E SensitiveMethod for the Confident Identification of Genetically VariantPeptides in Human Hair Keratin J Forensic Sci 2020 65 (2) 406minus420(27) Bittremieux W spectrum_utils A Python Package for MassSpectrometry Data Processing and Visualization Anal Chem 202092 (1) 659minus661(28) Roepstorff P Fohlman J Proposal for a commonnomenclature for sequence ions in mass spectra of peptides BiomedMass Spectrom 1984 11 (11) 601(29) Johnson R S Martin S A Biemann K Stults J T WatsonJ T Novel fragmentation process of peptides by collision-induceddecomposition in a tandem mass spectrometer differentiation ofleucine and isoleucine Anal Chem 1987 59 (21) 2621minus2655(30) Li K Vaudel M Zhang B Ren Y Wen B PDV anintegrative proteomics data viewer Bioinformatics 2019 35 (7)1249minus1251(31) Sturm M Kohlbacher O TOPPView an open-source viewerfor mass spectrometry data J Proteome Res 2009 8 (7) 3760minus3763(32) Proteome Discoverer httpswwwthermofishercomusenhomeindustrialmass-spectrometryliquid-chromatography-mass-spectrometry-lc-mslc-ms-softwaremulti-omics-data-analysisproteome-discoverer-softwarehtml (accessed in June 2020)(33) MacLean B Tomazela D M Shulman N Chambers MFinney G L Frewen B Kern R Tabb D L Liebler D CMacCoss M J Skyline an open source document editor for creatingand analyzing targeted proteomics experiments Bioinformatics 201026 (7) 966minus968(34) Kong A T Leprevost F V Avtonomov D MMellacheruvu D Nesvizhskii A I MSFragger ultrafast andcomprehensive peptide identification in mass spectrometry-basedproteomics Nat Methods 2017 14 (5) 513minus520(35) Liu M Q Zeng W F Fang P Cao W Q Liu C Yan GQ Zhang Y Peng C Wu J Q Zhang X J Tu H J Chi HSun R X Cao Y Dong M Q Jiang B Y Huang J M Shen HL Wong C C L He S M Yang P Y pGlyco 20 enablesprecision N-glycoproteomics with comprehensive quality control andone-step mass spectrometry for intact glycopeptide identification NatCommun 2017 8 (1) 438(36) Polasky D A Yu F Teo G C Nesvizhskii A I Fast andcomprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco Nat Methods 2020 17 (11) 1125minus1132
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
G

Sorokin D Y van Loosdrecht M C M et al Tackling the chemicaldiversity of microbial nonulosonic acidsA universal large-scalesurvey approach Chem Sci 2020 11 3074minus3080(22) Yu J Schorlemer M Gomez Toledo A Pett C SihlbomC Larson G Westerlind U Nilsson J Distinctive MSMSFragmentation Pathways of Glycopeptide-Generated Oxonium IonsProvide Evidence of the Glycan Structure Chem - Eur J 2016 22(3) 1114minus1124(23) Yang X Neta P Stein S E Quality control for buildinglibraries from electrospray ionization tandem mass spectra AnalChem 2014 86 (13) 6393minus6400(24) Yang X Neta P Stein S E Extending a Tandem MassSpectral Library to Include MS2 Spectra of Fragment Ions ProducedIn-Source and MSn Spectra J Am Soc Mass Spectrom 2017 28 (11)2280minus2287(25) Zhang Z Yang X Mirokhin Y A Tchekhovskoi D V JiW Markey S P Roth J Neta P Hizal D B Bowen M A SteinS E Interconversion of Peptide Mass Spectral Libraries Derivatizedwith iTRAQ or TMT Labels J Proteome Res 2016 15 (9) 3180minus3187(26) Zhang Z Burke M C Wallace W E Liang Y Sheetlin SL Mirokhin Y A Tchekhovskoi D V Stein S E SensitiveMethod for the Confident Identification of Genetically VariantPeptides in Human Hair Keratin J Forensic Sci 2020 65 (2) 406minus420(27) Bittremieux W spectrum_utils A Python Package for MassSpectrometry Data Processing and Visualization Anal Chem 202092 (1) 659minus661(28) Roepstorff P Fohlman J Proposal for a commonnomenclature for sequence ions in mass spectra of peptides BiomedMass Spectrom 1984 11 (11) 601(29) Johnson R S Martin S A Biemann K Stults J T WatsonJ T Novel fragmentation process of peptides by collision-induceddecomposition in a tandem mass spectrometer differentiation ofleucine and isoleucine Anal Chem 1987 59 (21) 2621minus2655(30) Li K Vaudel M Zhang B Ren Y Wen B PDV anintegrative proteomics data viewer Bioinformatics 2019 35 (7)1249minus1251(31) Sturm M Kohlbacher O TOPPView an open-source viewerfor mass spectrometry data J Proteome Res 2009 8 (7) 3760minus3763(32) Proteome Discoverer httpswwwthermofishercomusenhomeindustrialmass-spectrometryliquid-chromatography-mass-spectrometry-lc-mslc-ms-softwaremulti-omics-data-analysisproteome-discoverer-softwarehtml (accessed in June 2020)(33) MacLean B Tomazela D M Shulman N Chambers MFinney G L Frewen B Kern R Tabb D L Liebler D CMacCoss M J Skyline an open source document editor for creatingand analyzing targeted proteomics experiments Bioinformatics 201026 (7) 966minus968(34) Kong A T Leprevost F V Avtonomov D MMellacheruvu D Nesvizhskii A I MSFragger ultrafast andcomprehensive peptide identification in mass spectrometry-basedproteomics Nat Methods 2017 14 (5) 513minus520(35) Liu M Q Zeng W F Fang P Cao W Q Liu C Yan GQ Zhang Y Peng C Wu J Q Zhang X J Tu H J Chi HSun R X Cao Y Dong M Q Jiang B Y Huang J M Shen HL Wong C C L He S M Yang P Y pGlyco 20 enablesprecision N-glycoproteomics with comprehensive quality control andone-step mass spectrometry for intact glycopeptide identification NatCommun 2017 8 (1) 438(36) Polasky D A Yu F Teo G C Nesvizhskii A I Fast andcomprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco Nat Methods 2020 17 (11) 1125minus1132
Journal of Proteome Research pubsacsorgjpr Technical Note
httpsdoiorg101021acsjproteome1c00324J Proteome Res XXXX XXX XXXminusXXX
G
Copyright © 2022 FDOKUMEN











![Cashier, 1 copy for the CID/EPS-in-Charge, and ] copy for the ...](https://static.fdokumen.com/doc/165x107/631a86b35d5809cabd0f7028/cashier-1-copy-for-the-cideps-in-charge-and-copy-for-the-.jpg)

![•“Longinos en el Poema de Mio Cid: espejos, identidades e ideología”, Olivar 10 (2007), pp. 265-276. [ISSN 1515-1115]](https://static.fdokumen.com/doc/165x107/6320dacbc5de3ed8a70deb60/longinos-en-el-poema-de-mio-cid-espejos-identidades-e-ideologia-olivar.jpg)






