
An efficient selection system for packaging herpes simplex virus amplicons
Upload
independentCategory
view
4download
0
doi:10.1006/jmbi.2000.4022 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 301, 817±826
A Ribozyme Derived from the Catalytic Subunit ofRNase P from Escherichia coli is Highly Effective inInhibiting Replication of Herpes Simplex Virus 1
Phong Trang, Ahmed Kilani, Joseph Kim and Fenyong Liu*
Program in Infectious Diseasesand Immunity, Program inComparative BiochemistrySchool of Public HealthUniversity of California,140 Warren Hall, BerkeleyCA 94720, USA
E-mail address of the [email protected]
Abbreviations used: HSV, herpesexternal guide sequences; DMS, dimCAT, chloramphenicol acetyltransfemultiplicity of infection; TK, thymidplaque-forming unit; HCMV, cytom
0022-2836/00/040817±10 $35.00/0
A sequence-speci®c ribozyme (M1GS RNA) derived from the catalyticRNA subunit of RNase P from Escherichia coli was used to target themRNA encoding human herpes simplex virus 1 (HSV-1) major transcrip-tion activator, ICP4. A reduction of more than 80 % in the expressionlevel of ICP4 and a reduction of about 1000-fold in viral growth wereobserved in cells that stably expressed the ribozyme. In contrast, areduction of less than 10 % in ICP4 expression and viral growth wasobserved in cells that either did not express the ribozyme or produced acatalytically inactive ribozyme mutant. Thus, M1GS ribozyme is highlyeffective in inhibiting HSV-1 growth and can be used as a general gene-targeting agent for anti-HSV applications.
# 2000 Academic Press
Keywords: ribozyme; gene targeting; RNase P; herpes simplex virus;antiviral
*Corresponding authorIntroduction
Herpes simplex virus 1 (HSV-1) is the causativeagent of cold sores and may lead to severe morbid-ity or mortality in neonates and immunocompro-mised individuals (Roizman & Sears, 1996;Whitley, 1996). The emergence of drug resistant-strains of HSV-1 has posed a need for the develop-ment of new drugs and novel treatment strategies.
Conventional antisense molecules (Stein &Cheng, 1993) and ribozymes (Rossi, 1999; Wong-Staal et al., 1998) are promising gene-targetingagents for speci®c inhibition of gene expression.For example, both hammerhead and hairpin ribo-zymes have been shown to cleave viral mRNAsequences and inhibit viral replication in cellsinfected with human viruses, while a ribozymederived from a group I intron has been used torepair mutant mRNAs in cells (Lan et al., 1998; Palet al., 1998; Sarver et al., 1990; Yu et al., 1993; zuPutlitz et al., 1999). Thus, ribozymes can be used asa tool in both basic and clinical research, such as in
ing author:
simplex virus; EGS,ethyl sulphate;
rase; MOI,ine kinase; PFU,egalovirus.
studies of tumorigenesis and antiviral genetherapy.
External guide sequences (EGSs) (Forster &Altman, 1990; Yuan et al., 1992) are antisense oli-goribonucleotides that have been used to diminishgene expression in mammalian cells (Kawa et al.,1998; Liu & Altman, 1995; Ma et al., 2000; Plehn-Dujowich & Altman, 1998; Yuan et al., 1992) withthe aid of either ribonuclease P (RNase P) or its cat-alytic RNA subunit from Escherichia coli (M1 RNA)(Altman & Kirsebom, 1999; Frank & Pace, 1998).The EGS-based technology takes advantage ofRNase P or M1 RNA to cleave a targeted mRNAwhen the EGS hybridizes to the target RNA andforms a structure resembling a portion of the natu-ral tRNA substrate for the enzymes (Forster &Altman, 1990; Yuan et al., 1992) (Figure 1(a)). TheEGSs, when expressed separately from the enzyme,have recently been shown to be effective in inhibit-ing the gene expression of herpes simplex virusand in¯uenza virus and in addition, in abolishingthe replication of in¯uenza virus (Kawa et al., 1998;Plehn-Dujowich & Altman, 1998). To increase thetargeting ef®ciency, the EGS can be covalentlylinked to M1 RNA (e.g. its 30 end) to generate asequence-speci®c ribozyme, M1GS RNA (Franket al., 1994; Liu & Altman, 1995) (Figure 1(a)). Wehave shown that M1GS RNA ef®ciently cleavesmRNA substrates that base-pair with the EGSin vitro and that it effectively inhibits theexpression of herpes thymidine kinase in mamma-
# 2000 Academic Press

Figure 1. (a) Schematic of aptRNA substrate and a model sub-strate (EGS:mRNA) for RNase Pand M1 RNA, and a complex(M1GS:mRNA) in which a M1GSRNA hybridizes to a target RNA(mRNA). A part of the EGS:mRNAand M1GS:mRNA complexesresembles the acceptor stem and T-stem domains of a ptRNA (shownin boldtype). (b) Schematic rep-resentation of the substrates usedin the study. The targetedsequences that bind to the guidesequences of the ribozymes arehighlighted and the site of cleavageis marked with a ®lled arrow.
818 Antiviral RNase P Ribozyme
lian cells (Liu & Altman, 1995). Thus, M1GS RNArepresents a new class of ribozyme that can beused for gene-targeting applications. However, ithas not been demonstrated whether this ribozymecan function as an anti-HSV agent and block HSVreplication. In this study, we investigated whetherRNase P ribozyme can effectively block viral repli-cation by targeting the mRNA encoding the HSV-1major transcription activator ICP4. ICP4, animmediate-early (a) protein, is essential for theexpression of most of the viral early (b) and late (g)genes, and for viral growth (DeLuca et al., 1985;Roizman & Sears, 1996). Our results provide the®rst direct evidence that M1GS ribozyme is highlyeffective in inhibiting HSV-1 growth and demon-strate the utility of M1GS ribozyme as a generalgene-targeting agent for anti-HSV application.
Results
Targeted cleavage of ICP4 mRNA sequence byM1GS ribozyme in vitro
Since most mRNA species inside cells areusually associated with proteins and are present ina highly organized and folded conformation, it iscritical to choose a targeting region that is accessi-ble to binding of ribozymes in order to achieve ef®-cient targeting. In vivo mapping with dimethylsulphate (DMS), which has been extensively usedto determine the accessibility of mRNA and struc-ture of RNAs in cells (Ares & Igel, 1990; Liu &Altman, 1995; Zaug & Cech, 1995), was used tomap the accessibility of ICP4 mRNA expressed inHSV-1 infected cells. In this assay, cells wereinfected with HSV-1 and then incubated with cul-ture media that contained DMS. DMS entered thecells and modi®ed the nucleotides of the mRNAregions that were accessible. Total mRNAs weresubsequently isolated from the infected cells andthose regions of the ICP4 mRNA that were
exposed and modi®ed by DMS were mapped byprimer-extension assays in the presence of reversetranscriptase. A position ®ve nucleotides down-stream from the ICP4 translational initiation codonwas chosen as the cleavage site for M1GS RNA.This site appeared to be one of the regions mostaccessible to DMS modi®cation (data not shown).Moreover, its ¯anking sequence exhibited severalsequence features that need to be present in orderto interact with an M1GS ribozyme to achieve ef®-cient cleavage. These features include the require-ment for a guanosine and a pyrimidine base to bethe nucleotide 30 and 50 adjacent to the site of clea-vage, respectively (Liu & Altman, 1996). The inter-actions of these sequence elements with the M1GSribozyme are critical for recognition and cleavageby the enzyme.
Two M1GS ribozymes, M1-ICP4 and C-ICP4,were constructed by covalently linking the30 termini of the M1 and C102 RNAs witha guide sequence of 13 nucleotides that wascomplementary to the targeted mRNA sequence.C102 RNA, an M1 mutant, contained severalpoint mutations (e.g. A347C348! C347U348,C353C354C355G356! G353G354A355U356) at theP4 helix, a part of the catalytic domain (Kim et al.,1997). This mutant has been shown to be at least104-fold less active than M1 RNA in cleaving aptRNA (Kim et al., 1997). The DNA sequences cod-ing for the M1GS ribozymes were generated byPCR using the DNA sequences for M1 RNA andC102 RNA as the templates and primers that con-tained the sequences complementary to the tar-geted region of ICP4 mRNA. These DNAsequences were under the control of the promoterfor T7 RNA polymerase and M1GS RNAs weresynthesized in vitro from these DNA sequences byT7 RNA polymerase. Two RNA substrates, icp32and icp450, which contained the targeted ICP4mRNA sequence of 32 and 450 nucleotides,respectively, were used (Figure 1(b)). In the

Figure 2. Cleavage of icp32 (a) and icp450 (b) and binding of icp32 by M1GS ribozymes (c). ((a) and (b)) (32P)-labeled ICP4 mRNA substrates (10 nM) were incubated alone (lanes 1 and 4) or in the presence of 10 nM of M1-ICP4(lanes 3 and 6) and C-ICP4 (lanes 2 and 5) for 30 minutes in buffer A (50 mM Tris (pH 7.5), 100 mM NH4Cl, and100 mM MgCl2). Cleavage products were separated on 15 % (a) and 4 % (b) polyacrylamide gels that contained 8 Murea. (c) 0.1 nM of substrate icp32 was either incubated alone (lane 7) or in the presence of 0.1 nM of ribozyme C-ICP4 (lane 8) or M1-ICP4 (lane 9) in buffer B (50 mM Tris (pH 7.5), 100 mM NH4Cl, 100 mM CaCl2, 3 % (w/w) gly-cerol, 0.1 % (w/w) xylene cyanol, 0.1 % (w/w) bromophenol blue) for 15 minutes to allow binding and then loadedon a 5 % polyacrylamide gel.
Antiviral RNase P Ribozyme 819
absence of M1 RNAs (Figure 2, lanes 1 and 4), nocleavage of ICP4 mRNA sequence was observed.Cleavage of these two substrates by M1-ICP4yielded cleavage products of 10 and 22 nucleotides,and 150 and 300 nucleotides, respectively, andmore than 50 % of the substrates were cleaved inthis assay (Figure 1(b), Figure 2). In contrast, clea-vage of the same substrates by C-ICP4 was barelydetected (Figure 2, lanes 2 and 5).
Experiments were carried out to determinewhether the differential cleavage ef®cienciesobserved with M1-ICP4 and C-ICP4 could be dueto their different levels of binding af®nity to theICP4 mRNA sequence. The binding between theM1GS and substrate icp32 was assayed using agel-shift assay. The ribozymes were incubated withthe substrates to allow for binding, and the ribo-zyme-icp32 complexes were separated in poly-acrylamide gels under non-denaturing conditions.Similar amounts of complexes formed betweenthese two M1GS ribozymes and ICP4 mRNAsequence were observed when the same amount ofribozymes was used (Figure 2(c), data not shown).Further detailed assays under different concen-trations of the ribozymes and icp32 indicated thatthe binding af®nity of C-ICP4 to substrate icp32,measured as the dissociation constant (Kd), is simi-lar to that of M1-ICP4 (Figure 2(c)). Meanwhile,very little cleavage product was observed in thepresence of C-ICP4, even under high concen-trations of the ribozyme and a prolonged incu-bation period (Figure 2, lanes 2 and 5; data notshown). These observations suggest that themutations in C-ICP4 do not signi®cantly affect the
binding af®nity of the ribozyme to the mRNAsequence but abolish its catalytic activity. Thus, C-ICP4 RNA may be used as a control for the anti-sense effect in our experiments in cultured cells(see below).
Expression of M1GS ribozymes inmammalian cells
The DNA sequences coding for M1-ICP4 and C-ICP4 were cloned into retroviral vector LXSN andplaced under the control of the small nuclear U6RNA promoter, which has previously been shownto express M1GS RNA and other RNAs steadily(Bertrand et al., 1997; Das et al., 1988; Liu &Altman, 1995; Miller & Rosman, 1989; Yuan et al.,1992). This promoter is transcribed by RNA poly-merase III, and its transcripts are highly expressedand primarily localized in the nucleus. To constructcell lines that express M1GS ribozymes, amphotro-pic packaging cells (PA317) (Miller & Rosman,1989) were transfected with LXSN-M1GS DNAs toproduce retroviruses that contained the genes forM1GS RNA. Subsequently, cCRE cells (Danos &Mulligan, 1988) were infected with these retro-viruses, and cells expressing the ribozymes werecloned. An additional cell line was also constructedthat expressed a ribozyme, M1-CAT, which tar-geted the mRNA for chloramphenicol acetyltrans-ferase (CAT) (Liu & Altman, 1995). No cleavage ofsubstrates icp32 and icp450 by M1-CAT wasobserved in vitro (data not shown). This cell linewas used to determine whether M1GS RNA with

820 Antiviral RNase P Ribozyme
an incorrect guide sequence could target ICP4mRNA in tissue culture.
The level of M1GS RNA in each cell clone wasdetermined by Northern analysis with a DNAprobe that is complementary to M1 RNA. Figure 3shows the results of the Northern analyses of thenuclear RNA fractions isolated from a set of thecloned cell lines that expressed the ribozymes. TheM1GS RNAs were exclusively expressed in thenuclei as they were only detected from the nuclearbut not cytoplasmic RNA fractions (data notshown). This is consistent with previous obser-vations in our laboratory as well as others that thetranscripts expressed by the U6 promoter are pri-marily localized in the nuclei (Bertrand et al., 1997;Good et al., 1997; Kawa et al., 1998; Liu & Altman,1995; Yuan et al., 1992). Only the cell lines thatexpressed similar levels of these ribozymes wereused for further studies in tissue culture. The con-structed cell lines and a control cell line in whichcells were transfected with LXSN vector DNAalone were indistinguishable in terms of cellgrowth and viability for up to two months (data
Figure 3. The expression of M1GS ribozymes in cul-tured cells. Northern analyses were carried out usingnuclear RNA fractions isolated from parental cCRE cells(-, lane 2) and cell lines that expressed M1-CAT (lane 1),C-ICP4 (lane 3), and M1-ICP4 ribozymes (lane 4). Equalamounts of each RNA sample (30 mg) were separatedon 2.5 % agarose gels that contained formaldehyde,transferred to a nitrocellulose membrane, and hybri-dized to a (32P)-radiolabeled probe that contained theDNA sequence coding for M1 RNA. The hybridizedproducts corresponding to the full-length retroviral tran-scripts (�6 kb), transcribed from the LTR promoter, areat the top of the gel and are not shown.
not shown), suggesting that the expression of theribozymes did not exhibit signi®cant cytotoxicity.
Inhibition of viral ICP4 mRNA and proteinexpression by M1GS ribozyme
To determine the ef®cacy of the ribozymes ininhibiting ICP4 expression, cells were infected withHSV-1 at a multiplicity of infection (MOI) of 0.05-0.50. Total RNAs were isolated from cells thatwere treated with 100 mg/ml of cycloheximide.Under this condition, only viral immediate early(a) mRNAs were synthesized (Roizman & Sears,1996). The level of ICP4 mRNA was determined byan RNase protection assay and the level of viralimmediate-early a47 mRNA was used as aninternal control (Figure 4(a)). A reduction of about87(�9) % and 6(�3) % (average of three exper-iments) in the level of ICP4 mRNA expression wasobserved in cells that expressed M1-ICP4 and C-ICP4, respectively (Table 1). No reduction in theexpression level of ICP4 mRNA was observed incells that expressed M1-CAT (Table 1). Theseresults suggest that the signi®cant reduction ofICP4 mRNA expression in cells that expressed M1-ICP4 was due to the targeted cleavage by the ribo-zyme. The low level of inhibition found in cellsthat expressed C-ICP4 was probably due to anantisense effect. This is because C-ICP4 exhibits asimilar degree of binding af®nity to the targetsequence as M1-ICP4 but is catalytically inactive.No products of the cleavage of ICP4 mRNA weredetected in our RNase protection assays, presum-ably because these RNAs, which lacked either acap structure or a polyA sequence, were rapidlydegraded by intracellular RNases.
It is expected that the level of ICP4 proteinshould decrease in M1-ICP4-expressing cellsbecause of the decreased level of ICP4 mRNA. Pro-tein extracts were isolated from cells either mockinfected or infected with HSV-1. Viral proteinswere separated electrophoretically in SDS-poly-acrylamide gels and electrically transferred to twoidentical membranes. One of these membranes wasstained with an anti-ICP4 antibody (anti-ICP4)(Figure 4(c)) and the other was stained with amonoclonal antibody against the HSV-1 immedi-ate-early protein ICP27 (anti-ICP27) (Figure 4(d)).The latter was used to detect the expression ofICP27 that served as an internal control for thequantitation of ICP4 protein expression. Previousstudies have shown that the expression of viralICP27 is not affected by a change in viral ICP4expression during early stage of viral infection(DeLuca et al., 1985; Roizman & Sears, 1996). Byusing a chemilluminescent substrate for the anti-body staining, the expression of ICP4 and ICP27protein was quantitated (Figure 4(c)-(d)). Theresults of three independent experiments are sum-marized in Table 1: a reduction of 80(�10) % and7(�3) % in the level of ICP4 protein was observedin cells that expressed M1-ICP4 and C-ICP4,respectively. No signi®cant reduction in the level

Table 1. Levels of inhibition of the expression of viral genes in cells that expressed M1-ICP4, C-ICP4 and M1-CAT, ascompared to the levels of inhibition in cells that did not express a ribozyme (cCRE)
Ribozyme
Viral gene class cCRE M1-CAT C-ICP4 M1-ICP4
ICP4 mRNA a 0% 0% 6% 87 � 9 %TK mRNA b 0% 0% 6% 87 � 9 %ICP4 protein a 0% 2% 7% 80 � 10 %ICP35 protein g 0% 1% 8% 85 � 6 %gB protein g 0% 0% 5% 85 � 7 %
The values shown are the means from triplicate experiments and the values of standard deviation that were less than 5 % are notshown.
Antiviral RNase P Ribozyme 821
of ICP4 protein expression was observed in cellsthat expressed M1-CAT (Table 1). The low level ofreduction in the expression level of ICP4 proteinobserved in cells that expressed C-ICP4 was pre-sumably attributed to the antisense effect of theguide sequence.
Inhibition of viral gene expression andreplication by the ribozyme-mediated reductionof ICP4 expression
ICP4 protein functions as the HSV-1 major tran-scription activator and is required for theexpression of most of the viral early (b) and late (g)genes (DeLuca et al., 1985; Roizman & Sears, 1996).Inhibition of the ICP4 expression is expected toresult in reduction of the expression of these genes.RNase protection analyses were carried out todetermine the expression level of the mRNA cod-ing for thymidine kinase (TK), one of the viralearly (b) genes (Figure 4(b)). Moreover, Westernanalyses were performed to determine theexpression level of viral capsid protein ICP35, aviral late (g) protein (Figure 4(e)). A signi®cantreduction (about 85 %) in the expression level ofthese genes was observed in cells that expressedM1-ICP4 RNA while no signi®cant reduction wasdetected in cells that expressed C-ICP4 RNA(Figure 4(b) and (e), Table 1). Similar results werealso found in the expression of HSV-1 gB, a viralessential glycoprotein and a late (g) protein(Table 1, data not shown). It has been shown thatICP4 expression is required for the expression ofthese genes (DeLuca et al., 1985; Roizman & Sears,1996). These results suggest the inhibition of over-all viral early and late gene expression in the M1-ICP4-expressing cells.
To determine whether viral growth was inhib-ited in the ribozyme-expressing cells, cells wereinfected by HSV-1 at an MOI of 1-5. Virus stockswere prepared by sonication of the infected cul-tures (cells and culture medium together) thatwere harvested at six-hour intervals through 36hours post-infection. Intracellular virus wasreleased by sonication. The count of the plaque-forming unit (PFU) was determined by measure-ment of the viral titer on African green monkeykidney (Vero) cells. The results of the experimentsare summarized in Figure 5. After 18 hours post-
infection, a reduction of at least 1000-fold in viralyield was observed in cells that expressed M1-ICP4while no signi®cant reduction was found in thosethat expressed C-ICP4 (Figure 5). These results areconsistent with the notion that the inhibition ofICP4 expression in M1-ICP4 expressing-cells resultsin the observed reduction of viral early and lategene expression and viral growth.
Discussion
Ribozymes have been shown to be promisingantiviral agents for inhibition of viral geneexpression and replication (Rossi, 1999; Wong-Staal et al., 1998). Ribozyme technology representsan attractive approach for gene inactivation, sinceit exhibits most of the properties of the convention-al antisense targeting method and in addition, cata-lytic and irreversible cleavage of the target RNA.Several criteria must be satis®ed if successful tar-geting with ribozymes is to be achieved. Amongthese are high ef®ciency of cleavage, sequencespeci®city of the ribozymes, and ef®cient deliveryof the reagents. We have constructed an M1GSRNA that targeted the HSV-1 ICP4 mRNA andshown that the ribozyme cleaved the target mRNAsequence ef®ciently in vitro. Moreover, a reductionof about 80 % in the expression level of ICP4 and a1000-fold reduction in viral growth were observedin cells that expressed the ribozyme. In contrast, areduction of less than 10 % in the levels of ICP4expression and viral growth was observed in cellsthat expressed C-ICP4 or M1-CAT. While C-ICP4is catalytically inactive, this ribozyme containedthe identical guide sequence and exhibited similarbinding af®nity to the mRNA sequence as M1-ICP4. Thus, our results suggest that the overallobserved inhibition of viral gene expression andgrowth with M1-ICP4 RNA was primarily due tothe targeted cleavage by the ribozyme, as opposedto the antisense effect or other non-speci®c effectsof the guide sequence of the ribozyme.
The activity of M1GS ribozymes also appears tobe speci®c. First, the expression of the ribozymesdid not exhibit signi®cant cytotoxicity as cellsexpressing ribozymes were indistinguishable fromthe parental cells in terms of cell growth and viabi-lity for up to two months (data not shown). More-
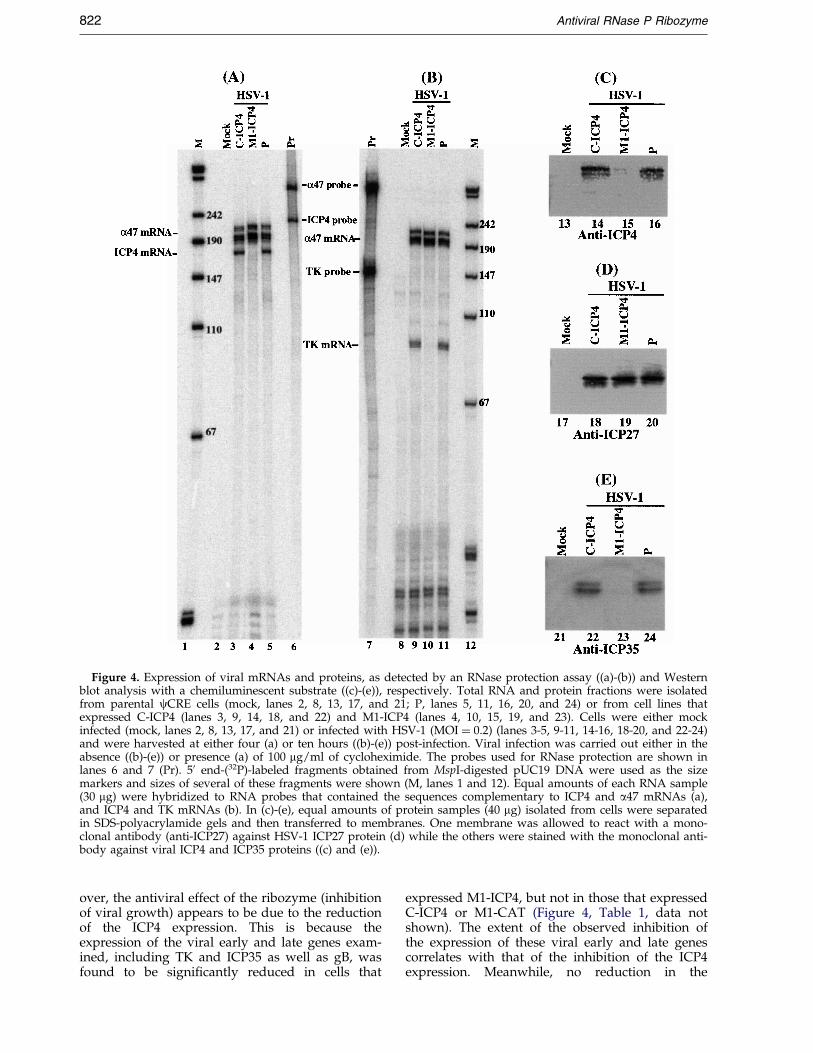
Figure 4. Expression of viral mRNAs and proteins, as detected by an RNase protection assay ((a)-(b)) and Westernblot analysis with a chemiluminescent substrate ((c)-(e)), respectively. Total RNA and protein fractions were isolatedfrom parental cCRE cells (mock, lanes 2, 8, 13, 17, and 21; P, lanes 5, 11, 16, 20, and 24) or from cell lines thatexpressed C-ICP4 (lanes 3, 9, 14, 18, and 22) and M1-ICP4 (lanes 4, 10, 15, 19, and 23). Cells were either mockinfected (mock, lanes 2, 8, 13, 17, and 21) or infected with HSV-1 (MOI � 0.2) (lanes 3-5, 9-11, 14-16, 18-20, and 22-24)and were harvested at either four (a) or ten hours ((b)-(e)) post-infection. Viral infection was carried out either in theabsence ((b)-(e)) or presence (a) of 100 mg/ml of cycloheximide. The probes used for RNase protection are shown inlanes 6 and 7 (Pr). 50 end-(32P)-labeled fragments obtained from MspI-digested pUC19 DNA were used as the sizemarkers and sizes of several of these fragments were shown (M, lanes 1 and 12). Equal amounts of each RNA sample(30 mg) were hybridized to RNA probes that contained the sequences complementary to ICP4 and a47 mRNAs (a),and ICP4 and TK mRNAs (b). In (c)-(e), equal amounts of protein samples (40 mg) isolated from cells were separatedin SDS-polyacrylamide gels and then transferred to membranes. One membrane was allowed to react with a mono-clonal antibody (anti-ICP27) against HSV-1 ICP27 protein (d) while the others were stained with the monoclonal anti-body against viral ICP4 and ICP35 proteins ((c) and (e)).
822 Antiviral RNase P Ribozyme
over, the antiviral effect of the ribozyme (inhibitionof viral growth) appears to be due to the reductionof the ICP4 expression. This is because theexpression of the viral early and late genes exam-ined, including TK and ICP35 as well as gB, wasfound to be signi®cantly reduced in cells that
expressed M1-ICP4, but not in those that expressedC-ICP4 or M1-CAT (Figure 4, Table 1, data notshown). The extent of the observed inhibition ofthe expression of these viral early and late genescorrelates with that of the inhibition of the ICP4expression. Meanwhile, no reduction in the

Figure 5. Growth of HSV in cCRE cells and cell linesthat expressed M1GS RNAs. 5 � 105 cells were infectedwith HSV-1 at a MOI of 2. Virus stocks were preparedfrom the infected cells at six-hour intervals through 36hours post-infection and the PFU count was determinedby measurement of the viral titer on Vero cells. Thesevalues are the means from triplicate experiments. Thestandard deviation is indicated by the error bars.
Antiviral RNase P Ribozyme 823
expression levels of other viral immediate-earlygenes examined (e.g. a47 and ICP27) was found inM1GS-expressing cells when the same amounts ofviral RNA or protein fractions were compared(Figure 4, data not shown). Thus, M1GS ribozymeis highly speci®c in inhibiting the expression of itstarget mRNA.
We showed that the M1GS RNAs introducedinto cultured cells were stably expressed and loca-lized primarily in the nuclei. The expression cas-sette we used to produce these M1GSs is driven bythe promoter for small nuclear U6 RNA. This pro-moter has been extensively used to express func-tional RNAs and ribozymes for gene targetingapplications and the transcript from this promoteris quite stable and primarily localized in the nuclei(Bertrand et al., 1997; Das et al., 1988; Good et al.,1997; Liu & Altman, 1995; Yuan et al., 1992). Thenuclear localization of RNase P ribozymes mayplay a signi®cant role in its ef®cacy in cell culture.This is because the regions of the catalytic M1RNA domain in these M1GS RNAs, which may behomologous to those of the RNA subunit ofhuman RNase P, are believed to interact with cellu-lar proteins, including those associated with RNaseP (Altman & Kirsebom, 1999; Liu & Altman, 1995).Several RNA chaperone proteins have been shownto stimulate the activity of hammerhead and groupI ribozymes by either facilitating folding of theribozyme or dissociation of the product (Bertrand& Rossi, 1995; Herschlag et al., 1995; Tsuchihashiet al., 1993). Further studies on potential inter-actions between M1GS RNAs and cellular proteinswill provide insight into how an M1GS ribozymefunctions in cultured cells.
The M1GS-based technology represents anattractive approach for gene inactivation, since it
generates catalytic and irreversible cleavage of thetarget RNA by using M1 RNA, a highly catalyti-cally active RNA enzyme found in nature (Altman& Kirsebom, 1999; Frank & Pace, 1998). Recent stu-dies in our laboratory have shown that M1GSRNA is also effective in inhibiting gene expressionand replication of human cytomegalovirus(HCMV) (Trang et al., 2000). The level of inhibitionof ICP4 expression observed in this study is similarto that of inhibition of the expression of HCMVIE1/IE2 proteins. However, the level of inhibitionof HSV-1 growth by M1-ICP4 is about ®ve- to ten-fold higher than that of inhibition of HCMV repli-cation (Trang et al., 2000). These results suggestthat the ef®cacy of the ribozyme in inhibiting viralgrowth might be dependent on the nature of thegene target. M1 RNA may further increase itsactivity in cultured cells by interacting with the cel-lular proteins (Altman & Kirsebom, 1999; Liu &Altman, 1995). These properties, as well as thesimple design of the guide sequence, make M1GSan attractive and unique gene-targeting agent,which can be generally used for antiviral as well asother in vivo applications.
HSV-1 is a member of human herpesvirusfamily, which includes seven other differentviruses such as herpes simplex virus 2, VaricellaZoster virus, Epstein-Barr virus, cytomegalovirus,and the recently-discovered Karposi sarcoma-associated herpesvirus (Chang et al., 1994;Roizman & Sears, 1996). All these viruses canengage in lytic replication as well as establishlatent infections. HSV-1 ICP4 encodes the majorviral transcription factor and is among one of the®rst viral proteins expressed during lytic infection(Roizman & Sears, 1996). Its homologous proteinshave been found in every other herpesvirus andare among those of the major targets for drugdevelopment (Anderson et al., 1996; Azad et al.,1993; Cantin et al., 1992; Mulamba et al., 1998). Ourresults indicate that RNase P ribozymes are effec-tive in inhibiting ICP4 expression and viral growthin tissue culture. Future challenges to the use ofM1GS for anti-HSV application is to determinewhether the ribozyme can be delivered in neuronalcells, which are latently infected by the virus(Roizman & Sears, 1996; Whitley, 1996), andwhether the delivered ribozyme can prevent HSVreactivation from latent to lytic infection. More-over, the successful application of the M1GS-basedapproach to other systems will depend critically onthe speci®c sequence and accessibility of the targetsite as well as the intracellular stability of the ribo-zyme. Further studies on these issues will facilitatethe development of M1GS ribozymes for generalgene-targeting applications.
Materials and Methods
Viruses, cells and antibodies
The human 143tkÿ cells, Vero (African green monkeykidney) cells, PA317 cells, and cCRE cells were main-

824 Antiviral RNase P Ribozyme
tained and propagated in Dulbecco's modi®ed Eagle'smedium (DMEM) supplemented with 10 % (w/w) fetalbovine serum. The propagation of HSV-1 (F) in thesecells was carried out as described (Liu & Altman, 1995).The anti-rabbit polyclonal antibody against HSV-1 (F)thymidine kinase was a gift from Dr Bill Summers (YaleUniversity) (Liu & Summers, 1988) and the anti-mousemonoclonal antibody MCA406 against HSV-1 ICP35 pro-tein (Liu & Roizman, 1993) was purchased from Biopro-duct for Sciences Inc. (Indianapolis, IN). The anti-mousemonoclonal antibodies c1101, c1113, and c1123, whichreact with HSV-1 proteins ICP4, ICP27, and gB, respect-ively, were purchased from Goodwin Institute for Can-cer Research (Plantation, FL).
Ribozyme and substrate constructs
Plasmid pFL117 and pC102 contain the DNAsequence coding for M1 RNA and mutant C102 drivenby the T7 RNA polymerase promoter (Kim et al., 1997;Liu & Altman, 1995). Mutant ribozyme C102 containedseveral point mutations (e.g. A347C348! C347U348,C353C354C355G356! G353G354A355U356) at the cata-lytic domain (P4 helix) (Kim et al., 1997). The DNAsequences that encode ribozymes M1-ICP4 and C-ICP4were constructed by PCR using the DNA sequence ofthe ribozymes as the template and oligonucleotidesAF25 (50-GGAATTCTAATACGACTCACTATAG-30) andM1ICP4 (50-CCCGCTCGAGAAAAAATGGTGCATCG-GCGATGGACAGCTATGACCATG-30) as 50 and 30 pri-mers, respectively. The underlined sequence in AF25 rep-resents the promoter sequence for T7 RNA polymerase.The underlined sequence and the bold sequence inM1ICP4 correspond to the 3'ACCA sequence and theguide sequence, respectively. The spacer connecting the30 terminus of M1 RNA and the 50 end of guide sequenceis a 51 nucleotides-long sequence (50-GAAGCTTGA-CCTGCAGGCATGCAAGCTTGGCGTAATCATGGTCA-TAGCTGT-30). The DNA sequence that encodes sub-strate icp32 was constructed by PCR using pGEM3zf(�)as template and oligonucleotide AF25 and sICP4 (50-CGGGATCCGACGCCATCGCCGATGCGGGGCGAT-CCTATAGTGAG-30) as 50 and 30 primers, respectively.The underlined sequence and the bold sequence in sICP4correspond to the sequence complementary to a part ofthe promoter sequence for the T7 RNA polymerase andthe target ICP4 mRNA sequence, respectively. The con-struct pICP4 coding for ICP4 DNA sequence wasa gift from Dr John Blaho (Mount Sinai School ofMedicine).
Cleavage and binding analysis
M1GS RNAs and the ICP4 mRNA substrates (i.e.icp32 and icp450) were synthesized in vitro by T7 RNApolymerase (Promega Inc. Madison, WI) as manufac-ture's recommendations and further puri®ed on 8 %urea/polyacrylamide gels. Subsequently, the M1GSRNAs (10 nM) were mixed with the [32P]-labeled mRNAsubstrate (10 nM). The cleavage reactions were carriedout at 37 �C in a volume of 10 ml for 30 minutes in bufferA (50 mM Tris (pH 7.5), 100 mM NH4Cl, and 100 mMMgCl2) (Kilani & Liu, 1999). Cleavage products wereseparated in denaturing gels and quantitated with aSTORM840 phosphorimager (Molecular Dynamics,Sunnyvale, CA).
The procedures to measure the equilibrium dis-sociation constants (Kd) of the M1GS-icp32 complexes
were modi®ed from Pyle et al. (1990). In brief, variousconcentrations of ribozyme (0.05-50 nM) were pre-incu-bated in buffer B (50 mM Tris (pH 7.5), 100 mM NH4Cl,100 mM CaCl2, 3 % (w/w) glycerol, 0.1 % (w/w) xylenecyanol, 0.1 % (w/w) bromophenol blue) for ten minutesbefore mixing with an equal volume of 0.1-0.5 nM ofRNA substrate preheated under identical conditions. Thesamples were incubated for 15 minutes to allow binding,then loaded on a 5 % polyacrylamide gel, and run at 10W. The electrophoresis running buffer contained100 mM Tris-Hepes (pH 7.5) and 10 mM MgCl2 (Pyleet al., 1990). The amount of the bound complex wasquantitated with a STORM840 Phosphorimager. Thevalue of Kd was then extrapolated from a graph plottingpercentage of product bound versus ribozyme concen-tration. The values obtained were the average of threeexperiments.
Construction of ribozyme-expressing cells
The protocols were modi®ed from Miller and Rosman(Miller & Rosman, 1989). In brief, amphotropic PA317cells were transfected with retroviral vector DNAs(LXSN-M1-ICP4 and LXSN-C-ICP4) with the aid of amammalian transfection kit purchased from GIBCO BRL(Grand Island, NY). At 48 hours post-transfection, cul-ture supernatants that contained retroviruses were col-lected and used to infect cCRE cells. At 48-72 hourspost-infection, cells were incubated in culture mediumthat contained 600 mg/ml neomycin. Cells were sub-sequently selected in the presence of neomycin for twoweeks and neomycin-resistant cells were cloned. Fornorthern analyses of the expression of the ribozymes,both nuclear and cytoplasmic RNA fractions fromM1GS-expression cells were isolated as described (Liu &Roizman, 1991). The RNA fractions were separated in a2.5 % agarose gel that contained formaldehyde, trans-ferred to a nitrocellulose membrane, hybridized with the[32P]-radiolabeled DNA probe that contained the DNAsequence coding for M1 RNA, and ®nally analyzed witha STORM840 phosphorimager. The radiolabeled DNAprobe used to detect M1GS RNAs was synthesized fromplasmid pFL117, by using a random primed labeling kit(Boehringer Manheim, Co., Indianapolis, IN).
Viral infection and preparation of RNA andprotein extracts
A T-25 ¯ask of cells (approximately 106 cells) wereeither mock-infected or infected with HSV-1 in an inocu-lum of 1.5 ml DMEM (GIBCO) supplemented with 1 %fetal calf serum. The multiplicity of infection (MOI) isspeci®ed as that in the Results. After two hours ofexposure of cells to the virus at 37 �C, the inoculum wasreplaced with Dulbecco's modi®ed Eagle's medium sup-plemented with 10 % fetal bovine serum. The infectedcells were incubated for four or ten hours before beingharvested for isolation of viral mRNA or protein. Tomeasure the levels of viral immediate-early (IE) tran-scripts, some of cells were also treated with 100 mg/mlcycloheximide prior to and during infection. Total cellu-lar RNA was isolated from the cells as described (Liu &Roizman, 1991). To prepare protein extracts, cells werewashed twice with phosphate-buffered saline (PBS), andlysed in the disruption buffer (0.05 M Tris (pH 7.0), 8.5 %(w/w) sucrose, 5 % (w/w) b-mercaptoethanol, 2 %(w/w) sodium dodecyl sulphate). The protein samples

Antiviral RNase P Ribozyme 825
were boiled for one minute before electrophoretic separ-ation in SDS-polyacrylamide denaturing gels.
Assays for viral gene expression and growth
At four or ten hours post-infection, cells were har-vested and RNA and protein extracts were prepared asdescribed (Liu & Roizman, 1991). The RNA probes usedto detect the ICP4 mRNA, TK mRNA, and a47 mRNAwere synthesized from pICP4, pTK129 and pTK142,respectively. RNase protection assays were performed asdescribed (Liu & Altman, 1995). The protected RNA pro-ducts were separated in 8 % polyacrylamide denaturinggels and quantitated with a STORM840 phosphorimager.
For Western analyses, the denatured, solubilized poly-peptides from cell lysates were separated on 7.5 % or 9 %(w/w) SDS-polyacrylamide gels cross-linked with N,N`'-methylenebisacylamide. The separated polypeptideswere transferred electrically to nitrocellulose membranesand reacted to the antibodies against HSV-1 ICP4, ICP27,and ICP35. The membranes were subsequently stainedwith a chemiluminescent substrate with the aid of aWestern chemiluminescent substrate kit (Amersham Inc,Arlington Heights, IL) and quantitated with aSTORM840 phosphorimager. Quantitation was per-formed in the linear range of RNA and protein detection.
To determine the level of the inhibition of viralgrowth, 5 � 105 cells were either mock infected orinfected with HSV-1 in an inoculum of 1.5 ml DMEM(GIBCO) supplemented with 1 % fetal calf serum. TheMOI is speci®ed as that in Results. The cells and mediumwere harvested at 6, 12, 18, 24, 30, and 36 hours post-infection and viral stocks were prepared by adding anequal volume of 10 % skim milk, followed by sonication.The titers of the viral stocks were determined by infect-ing 2 � 105 Vero cells in six well plates and counting thenumber of plaques two to three days post-infection, asdescribed (Liu & Roizman, 1991). The values obtainedwere the average from triplicate experiments.
Acknowledgments
The authors are indebted to Dr John Blaho and Dr BillSummers for ICP4 plasmid construct and anti-TK anti-body. Gratitude also goes to Diane Kawa, Edward Nepo-muceno, and Jarone Lee for excellent technicalassistance. P. T., and A. F. K. acknowledge predoctoralfellowship supports from UC-Berkeley. F. L. is a PewScholar in Biomedical Sciences and a recipient of a BasilO'Connor Award (March of Dimes) and a Regent'sJunior Faculty Fellowship (University of California). Theresearch has been supported by grants from State ofCalifornia AIDS research program, March of Dimes, andNIH (AI41927 and GM54875).
References
Altman, S. & Kirsebom, L. A. (1999). Ribonuclease P. InThe RNA World (Gesteland, R. F., Cech, T. R. &Atkins, J. F., eds), 2nd edit., pp. 351-380, ColdSpring Harbor Laboratory Press, Cold SpringHarbor.
Anderson, K. P., Fox, M. C., Brown-Driver, V., Martin,M. J. & Azad, R. F. (1996). Inhibition of humancytomegalovirus immediate-early gene expressionby an antisense oligonucleotide complementary to
immediate-early RNA. Antimicrob. Agents Chemother.40, 2004-2011.
Ares, M. & Igel, A. H. (1990). Lethal and temperature-sensitive mutations and their suppressors identifyan essential structural element in U2 small nuclearRNA. Genes Dev. 4, 2132-45.
Azad, R. F., Driver, V. B., Tanaka, K., Crooke, R. M. &Anderson, K. P. (1993). Antiviral activity of a phos-phorothioate oligonucleotide complementary toRNA of the human cytomegalovirus major immedi-ate-early region. Antimicrob. Agents Chemother. 37,1945-1954.
Bertrand, E., Castanotto, D., Zhou, C., Carbonnelle, C.,Lee, N. S., Good, P., Chatterjee, S., Grange, T.,Pictet, R., Kohn, D., Engelke, D. & Rossi, J. J. (1997).The expression cassette determines the functionalactivity of ribozymes in mammalian cells by con-trolling their intracellular localization. RNA, 3, 75-88.
Bertrand, E. L. & Rossi, J. J. (1995). Facilitation of ham-merhead ribozyme catalysis by the nucleocapsidprotein of HIV-1 and the heterogeneous nuclearribonucleoprotein A1. EMBO J. 13, 2904-2912.
Cantin, E. M., Podsakoff, G., Willey, D. E. & Openshaw,H. (1992). Antiviral effects of herpes simplex virusspeci®c anti-sense nucleic acids. Advan. Exper. Med.Biol. 312, 139-149.
Chang, Y., Cesarman, E., Pessin, M. S., Lee, F.,Culpepper, J., Knowles, D. M. & Moore, P. S.(1994). Identi®cation of herpesvirus-like DNAsequences in AIDS-associated Kaposi's sarcoma.Science, 266, 1865-1869.
Danos, O. & Mulligan, R. C. (1988). Safe and ef®cientgeneration of recombinant retroviruses with ampho-tropic and ecotropic host ranges. Proc. Natl Acad.Sci. USA, 85, 6460-6464.
Das, G., Henning, D., Wright, D. & Reddy, R. (1988).Upstream regulatory elements are necessary andsuf®cient for transcription of a U6 RNA gene byRNA polymerase III. EMBO J. 7, 503-512.
DeLuca, N. A., McCarthy, A. M. & Schaffer, P. A.(1985). Isolation and characterization of deletionmutants of herpes simplex virus type 1 in the geneencoding immediate-early regulatory protein ICP4.J. Virol. 56, 558-570.
Forster, A. C. & Altman, S. (1990). External guidesequences for an RNA enzyme. Science, 249, 783-786.
Frank, D. N. & Pace, N. R. (1998). Ribonuclease P: unityand diversity in a tRNA processing ribozyme.Annu. Rev. Biochem. 67, 153-180.
Frank, D., Harris, M. & Pace, N. R. (1994). Rationaldesign of self-cleaving pre-tRNA-Ribonuclease PRNA conjugates. Biochemistry, 33, 10800-10808.
Good, P. D., Krikos, A. J., Li, S. X., Bertrand, E., Lee,N. S., Giver, L., Ellington, A., Zaia, J. A., Rossi, J. J.& Engelke, D. R. (1997). Expression of small,therapeutic RNAs in human cell nuclei. Gene Ther.4, 45-54.
Herschlag, D., Khosla, M., Tsuchihashi, Z. & Karpel,R. L. (1995). An RNA chaperone activity of non-speci®c RNA binding proteins in hammerhead ribo-zyme catalysis. EMBO J. 13, 2913-2924.
Kawa, D., Wang, J., Yuan, Y. & Liu, F. (1998). Inhibitionof viral gene expression by human ribonuclease P.RNA, 4, 1397-1406.
Kilani, A. F. & Liu, F. (1999). UV crosslink mapping ofthe substrate binding site of RNase P ribozyme to atarget mRNA sequence. RNA, 5, 1235-1247.

826 Antiviral RNase P Ribozyme
Kim, J. J., Kilani, A. F., Zhan, X., Altman, S. & Liu, F.(1997). The protein cofactor allows the sequence ofan RNase P ribozyme to diversify by maintainingthe catalytically active structure of the enzyme.RNA, 3, 613-623.
Lan, N., Howrey, R. P., Lee, S. W., Smith, C. A. &Sullenger, B. A. (1998). Ribozyme-mediated repairof sickle beta-globin mRNAs in erythrocyte precur-sors. Science, 280, 1593-1596.
Liu, F. & Altman, S. (1995). Inhibition of viral geneexpression by the catalytic RNA subunit of RNaseP from Escherichia coli. Genes Dev. 9, 471-480.
Liu, F. & Altman, S. (1996). Requirements for cleavageby a modi®ed RNase P of a small model substrate.Nucl. Acids Res. 24, 2690-2696.
Liu, F. & Roizman, B. (1991). The herpes simplex virus 1gene encoding a protease also contains within itscoding domain the gene encoding the more abun-dant substrate. J. Virol. 65, 5149-5156.
Liu, F. & Roizman, B. (1993). Characterization of theprotease and other products of amino-terminus-proximal cleavage of the herpes simplex virus 1UL26 protein. J. Virol. 67, 1300-1309.
Liu, Q.-Y. & Summers, W. C. (1988). Site-directed muta-genesis of a nucleotide-binding domain in HSV-1thymidine kinase: effects on catalytic activity. Virol-ogy, 163, 638-642.
Ma, M., Benimetskaya, L., Lebedeva, I., Dignam, J.,Takle, G. & Stein, C. A. (2000). Intracellular mRNAcleavage induced through activation of RNase P bynuclease-resistant external guide sequences. NatureBiotechnol. 18, 58-61.
Miller, A. D. & Rosman, G. J. (1989). Improved retro-viral vectors for gene transfer and expression. Bio-Techniques, 7, 980-990.
Mulamba, G. B., Hu, A., Azad, R. F., Anderson, K. P. &Coen, D. M. (1998). Human cytomegalovirusmutant with sequence-dependent resistance to thephosphorothioate oligonucleotide fomivirsen (ISIS2922). Antimicrob. Agents Chemother. 42, 971-973.
Pal, B. K., Scherer, L., Zelby, L., Bertrand, E. & Rossi, J. J.(1998). Monitoring retroviral RNA dimerizationin vivo via hammerhead ribozyme cleavage. J. Virol.72, 8349-8353.
Plehn-Dujowich, D. & Altman, S. (1998). Effective inhi-bition of in¯uenza virus production in culturedcells by external guide sequences and ribonucleaseP. Proc. Natl Acad. Sci. USA, 95, 7327-7332.
Pyle, A. M., McSwiggen, J. A. & Cech, T. R. (1990).Direct measurement of oligonucleotide substrate
binding to wild-type and mutant ribozymes fromTetrahymena. Proc. Natl Acad. Sci. USA, 87, 8187-8191.
Roizman, B. & Sears, A. E. (1996). Herpes simplexviruses and their replication. In Virology. (Fields,B. N. & Knipe, D. M., et al., eds), 3rd edit., pp.2231-2296, Raven Press, New York.
Rossi, J. J. (1999). Ribozymes, genomics and thera-peutics. Chem. Biol. 6, R33-R37.
Sarver, N., Cantin, E. M., Chang, P. S., Zaia, J. A.,Ladne, P. A., Stephens, D. A. & Rossi, J. J. (1990).Ribozymes as potential anti-HIV-1 therapeuticagents. Science, 247, 1222-1225.
Stein, C. A. & Cheng, Y. C. (1993). Antisense oligonu-cleotides as therapeutic agents±is the bullet reallymagical? Science, 261, 1004-1012.
Trang, P., Lee, M., Nepomuceno, E., Kim, J., Zhu, H. &Liu, F. (2000). Effective inhibition of human cytome-galovirus gene expression and replication by a ribo-zyme derived from the catalytic subunit of RNase Pfrom Escherichia coli. Proc. Natl Acad. Sci. USA, 97,5812-5817.
Tsuchihashi, Z., Khosla, M. & Herschlag, D. (1993). Pro-tein enhancement of hammerhead ribozyme cataly-sis. Science, 262, 99-102.
Whitley, R. J. (1996). Herpes simplex viruses. In Virology.(Fields, B. N. & Knipe, D. M., et al., eds), 3rd edit.,pp. 2297-2342, Raven Press, New York.
Wong-Staal, F., Poeschla, E. M. & Looney, D. J. (1998).A controlled, Phase 1 clinical trial to evaluate thesafety and effects in HIV-1 infected humans of auto-logous lymphocytes transduced with a ribozymethat cleaves HIV-1 RNA. Hum. Gene Ther. 9, 2407-2425.
Yu, M., Ojwang, J., Yamada, O., Hampel, A.,Rapapport, J., Looney, D. & Wong-Staal, F. (1993).A hairpin ribozyme inhibits expression of diversestrains of human immunode®ciency virus type 1.Proc. Natl Acad. Sci. USA, 90, 6340-6344.
Yuan, Y., Hwang, E. & Altman, S. (1992). Targeted clea-vage of mRNA by human RNase P. Proc. Natl Acad.Sci. USA, 89, 8006-8010.
Zaug, A. J. & Cech, T. R. (1995). Analysis of the struc-ture of Tetrahymena nuclear RNAs in vivo: telomer-ase RNA, the self-splicing rRNA intron, and U2snRNA. RNA, 1, 363-374.
zu Putlitz, J., Yu, Q., Burke, J. M. & Wands, J. R. (1999).Combinatorial screening and intracellular antiviralactivity of hairpin ribozymes directed against hepa-titis B virus. J. Virol. 73, 5381-5387.
Edited by J. Doudna
(Received 4 May 2000; received in revised form 3 July 2000; accepted 5 July 2000)
Copyright © 2022 FDOKUMEN




















