
A computational assay to design an epitope-based peptide vaccine against chikungunya virus
14
Author Proof 1029 10.2217/FVL.12.95 © 2012 Future Medicine Ltd ISSN 1746-0794 Future Virology part of Future Virol. (2012) 7(10), 1029–1042 Chikungunya virus (CHIKV) is an arthropod- borne virus. CHIKV transmission in humans is reported to be carried out by Aedes spp. (e.g. , Aedes albopictus) mosquitoes [1–3] . Clinical symp- toms of chikungunya infection are fever, rash, headache, joint pain or arthralgia, and muscle pain or myalgia [4–6] . The symptoms usually last between 1 and 10 days [4] . Several outbreaks in Africa, India and South-East Asia have been reported previously [7–9] . Recently, from 2004 until now, there have been reports of a reemerged CHIKV infec- tion in several geographic regions [10–14] . The first of its outbreaks was recorded in Kenya in 2004. Similar outbreaks in different islands (e.g., Comoros Islands, southwest Indian Ocean) were also filed in early 2005 [7,15–18] . In 2005–06 it broke out in a massive propor- tion of India. Molecular analysis of the strains isolated on islands in the Indian Ocean and in India revealed that the epidemic was caused by a central/east African genotypic variant of CHIKV [11,12,19–21] . Soon, a large number of travelers from industrialized countries became infected with CHIKV. Hence what started as a mere endemic spark became the equivalent of a global forest fire without much ado [22,23] . CHIKV is an alphavirus and belongs to the Togaviridae family [24] . It possesses a single- strand positive-sense RNA genome. Two of the proteins in CHIKV have been sequenced in abundance to characterize the envelope pro- teins CHIKV- E1 and E2 [24–26] . E1-targeted antibodies are more likely to cross-react with other alphaviruses and thus are nonspecific compared with E2-targeted antibodies [26,27] . Monoclonal antibody-associated competi- tive binding assays identified approximately seven epitopes on the E1 glycoproteins of Sindbis virus (SINV), Simian foamy virus (SFV), Western equine encephalitis virus, and Venezuelan equine encephalitis virus (VEEV). Epitopes for E1 proteins are mostly present on the surface of infected cells instead of the virion surface. In vitro analysis of anti- body reactions to E1 include hemagglutination inhibition (HI), neutralization of virus infec- tivity, and inhibition of fusion. The presence of different functionally distinct domains has been suggested based on this analysis [28–30] . Mapping monoclonal antibody binding sites to specific E1 sequences is always a challenge. One acid-exposed epitope involves E1–157 of SFV [31] . A neutralizing mAb distinguishes A computational assay to design an epitope-based peptide vaccine against chikungunya virus Md. Rezaul Islam 1 , M Sadman Sakib 1 & Aubhishek Zaman* 2 1 Department of Biochemistry & Molecular Biology, University of Dhaka, Dhaka, Bangladesh 2 Department of Genetic Engineering & Biotechnology, University of Dhaka, Dhaka, Bangladesh *Author for correspondence: [email protected] Aim: Chikungunya virus, an arthropod-borne alphavirus, belongs to the Togavirus family. Despite severe epidemic outbreaks on several occasions, not much progress has been made with regard to epitope-based drug design for chikungunya virus. In this study we performed a proteome-wide search to look for a conserved region among the available viral proteins, one which has the capacity to trigger a significant immune response. Materials & methods: The conserved region was analyzed by performing an alignment of sequences collected from sources from varied geographic locations and time periods. Subsequently, the immune parameters for the peptide sequences were determined using several in silico tools and immune databases. Results: Both T-cell immunity and B-cell immunity were checked for the peptides to ensure that they had the capacity to induce both humoral and cell-based immunity. Our study reveals a stretch of conserved region in glycoprotein E2; yet this peptide sequence could interact with as many as seven HLAs and showed population coverage as high as 73.46%. The epitope was further tested for binding against the HLA structure using in silico docking techniques to validate the binding cleft epitope interaction in detail. Conclusion: Although the study requires further in vivo screening, keeping in mind the consistency and reproducibility of the immune system at selecting and reacting to peptide epitopes, this study allows us to claim a novel peptide antigen target in E2 protein with good confidence. Keywords n chikungunya virus n CHIKV n E2 envelope protein n epitope-based drug design Preliminary Communication
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of A computational assay to design an epitope-based peptide vaccine against chikungunya virus

Author Pro
of
102910.2217/FVL.12.95 © 2012 Future Medicine Ltd ISSN 1746-0794
Futu
re V
irolo
gy
part of
Future Virol. (2012) 7(10), 1029–1042
Chikungunya virus (CHIKV) is an arthropod-borne virus. CHIKV transmission in humans is reported to be carried out by Aedes spp. (e.g., Aedes albopictus) mosquitoes [1–3]. Clinical symp-toms of chikungunya infection are fever, rash, headache, joint pain or arthralgia, and muscle pain or myalgia [4–6]. The symptoms usually last between 1 and 10 days [4]. Several outbreaks in Africa, India and South-East Asia have been reported previously [7–9].
Recently, from 2004 until now, there have been reports of a reemerged CHIKV infec-tion in several geographic regions [10–14]. The first of its outbreaks was recorded in Kenya in 2004. Similar outbreaks in different islands (e.g., Comoros Islands, southwest Indian Ocean) were also filed in early 2005 [7,15–18]. In 2005–06 it broke out in a massive propor-tion of India. Molecular analysis of the strains isolated on islands in the Indian Ocean and in India revealed that the epidemic was caused by a central/east African genotypic variant of CHIKV [11,12,19–21]. Soon, a large number of travelers from industrialized countries became infected with CHIKV. Hence what started as a mere endemic spark became the equivalent of a global forest fire without much ado [22,23].
CHIKV is an alphavirus and belongs to the Togaviridae family [24]. It possesses a single-strand positive-sense RNA genome. Two of the proteins in CHIKV have been sequenced in abundance to characterize the envelope pro-teins CHIKV- E1 and E2 [24–26]. E1-targeted antibodies are more likely to cross-react with other alphaviruses and thus are nonspecific compared with E2-targeted antibodies [26,27]. Monoclonal antibody-associated competi-tive binding assays identified approximately seven epitopes on the E1 glycoproteins of Sindbis virus (SINV), Simian foamy virus (SFV), Western equine encephalitis virus, and Venezuelan equine encephalitis virus (VEEV). Epitopes for E1 proteins are mostly present on the surface of infected cells instead of the virion surface. In vitro analysis of anti-body reactions to E1 include hemagglutination inhibition (HI), neutralization of virus infec-tivity, and inhibition of fusion. The presence of different functionally distinct domains has been suggested based on this analysis [28–30]. Mapping monoclonal antibody binding sites to specific E1 sequences is always a challenge. One acid-exposed epitope involves E1–157 of SFV [31]. A neutralizing mAb distinguishes
A computational assay to design an epitope-based peptide vaccine against chikungunya virus
Md. Rezaul Islam1, M Sadman Sakib1 & Aubhishek Zaman*2
1Department of Biochemistry & Molecular Biology, University of Dhaka, Dhaka, Bangladesh2Department of Genetic Engineering & Biotechnology, University of Dhaka, Dhaka, Bangladesh*Author for correspondence: [email protected]
Aim: Chikungunya virus, an arthropod-borne alphavirus, belongs to the Togavirus family. Despite severe epidemic outbreaks on several occasions, not much progress has been made with regard to epitope-based drug design for chikungunya virus. In this study we performed a proteome-wide search to look for a conserved region among the available viral proteins, one which has the capacity to trigger a significant immune response. Materials & methods: The conserved region was analyzed by performing an alignment of sequences collected from sources from varied geographic locations and time periods. Subsequently, the immune parameters for the peptide sequences were determined using several in silico tools and immune databases. Results: Both T-cell immunity and B-cell immunity were checked for the peptides to ensure that they had the capacity to induce both humoral and cell-based immunity. Our study reveals a stretch of conserved region in glycoprotein E2; yet this peptide sequence could interact with as many as seven HLAs and showed population coverage as high as 73.46%. The epitope was further tested for binding against the HLA structure using in silico docking techniques to validate the binding cleft epitope interaction in detail. Conclusion: Although the study requires further in vivo screening, keeping in mind the consistency and reproducibility of the immune system at selecting and reacting to peptide epitopes, this study allows us to claim a novel peptide antigen target in E2 protein with good confidence.
Keywords
n chikungunya virus n CHIKV n E2 envelope protein n epitope-based drug design
Prelim
ina
ry Co
mm
un
ica
tion

Author Pro
of
Future Virol. (2012) 7(10)1030 future science group
strains of SINV differing only at residues 72 and 313 of E1, and a mAb escape mutant for a different epitope has a change at E1–132, suggesting the involvement of these regions in neutralization.
Antibodies to E2 are usually virus-specific. Monoclonal antibody-associated binding assays have identified four to five epitopes on the E2 glycoproteins of SINV, SFV, Ross River Virus (RRV) and VEEV [32]. The foci of in vitro analysis of E2 targeted antibodies include HI, viral infectivity neutralization and inhibition of viral binding to the cell surface. Many anti-E2 monoclonal antibodies have both neutralizing and HI activity, suggesting that these functions overlap. After exposure to acidic pH the conformational change in E2 is less drastic than that in E1. Different viral mutants, naturally occurring variants, mutational approaches and genetically engi-neered viruses have been used to map vari-ous E2-specific epitopes. From these studies, two major neutralizing sites are reported. Overlapping epitopes in the middle of the E2 linear sequence encompass residues 181–216 in SINV, 180–216 in VEEV and 216–251 in RRV and identify one neutralizing site which is hydrophilic region that forms an exterior loop
exposed on the virion surface. Linear deter-minants probably exist in this region because these monoclonal antibodies frequently react in western blots and recognize Î fusion pro-teins, and antibodies to peptides from this region are protective against challenge.
The second neutralizing epitope on E2 appears to be more complicated and is pri-marily conformational. Variation in the amino acid sequence of an epitope affect monoclonal antibody binding, and monoclonal antibod-ies to this epitope sequence do not react with denatured proteins in Western blots. The region defined by this epitope is responsible for SINV binding to heparan sulfate and is nonfunctional if pE2 is not processed [33,34]. Visualization of the binding of mAb to this epitope on SINV and RRV using cryoelectron-microscopy identifies this region of E2 as one of the two knobs on the glycoprotein spikes.
Although much has been done to study the antibody response against CHIKV, not much progress has been achieved in understanding T-cell epitope-based immune elicitation for the virus [35]. Only recently have there been some reports that T-cell immunity and innate immunity lie at the center of protection from CHIKV [36,37]. In studies carried out in other
HRTT AMI L
HPHE I I L Y YY YYPNYQE EWVMHKKE V V L T V P T EGL E V TWGNNE P YKYWPQL S T N T AHGHPHE I I L Y Y E L P TY Y YG
Figure 1. Sequence LOGO identifies conserved region of the E2 protein from 60 different sources. Amino acid sequences in ‘C terminal’ peptides of E2 envelope proteins from different variants vary in few positions. The changed region has been highlighted with circles. The sequence of the epitope is conserved in all 60 sequences. The error bar represents small sample correction; small sample correction rules out the possibility of overestimating the entropy while aligning.
Preliminary Communication Islam, Sakib & Zaman

Author Pro
of
www.futuremedicine.com 1031future science group
viruses such as dengue and influenza, target-ing T cells has been successful [38–40]. T-cell epitope-based techniques might be important for the future, as CHIKV has been reported to show genome microevolution that helps the virus to evade humoral immunity [41].
Although significant studies have been per-formed in other viruses, there have not been a lot of studies carried out to identify conserved linear patches in CHIKV. In this paper we have tried to perform an in silico assay to find peptide signatures in the envelope proteins of the virus. The results should pave the way to designing a peptide vaccine against the virus. The signature motifs for MHC presentation are well conserved in sequence and polar-ity, and thus sequence analysis often gener-ates predictions that are both precise and efficacious.
Methods & materialRetrieving the protein sequencesThe outer membrane protein (E1 and E2 envelope proteins) sequences of CHIKV were retrieved from the NCBI protein database (http://www.ncbi.nlm.nih.gov/protein/) from different isolates. These sequences were col-lected from different parts of the world and deposited in the database at different times. For E1 protein a total of 204 proteins were taken from east, central and South African (ECSA), Chinese, Indian, East Asian and Equatorial lineages. Similarly, a total of 60 sequences for E2 protein were taken from Indian, East Asian, Chinese, Thai, Srilankan, European, North American and Equatorial east, central and South African, with lineages and time ranges as varying as 1953 to 2011 (Supplementary file 3).
Multiple sequence alignmentRetrieved sequences were used as a platform to generate multiple sequence alignments, which were the basis of finding the conserved regions of the protein sequences. The MEGA5 soft-ware package (www.megasoftware.net) was used to build the alignment. The CLUSTALW algorithm with bootstrap 1000 was chosen and different parameters were set at default to gen-erate the alignment. A web logo was also gener-ated for the conserved peptide sequence using WebLogo [42] based on this alignment [42].
Calculation of nonsynonymous–synonymous substitution ratioThe gene sequences of the retrieved enve-lope proteins from different variants were
downloaded from the NCBI GenBank (www.ncbi.nlm.nih.gov/genbank). Some of the genomic sequences deposited were truncated in length and hence they needed to be adjusted to avoid frame-shift errors. The sequences were adjusted to the same ORF by comparison with a reference sequence (NCBI GENBANK ACC no. HM159384). Subsequently a web tool developed by the computational biology unit of Bergen Center for Computational Science, Norway (service.cbu.uib.no/tool/KaKs) was used for Ka/Ks calculation. Ka/Ks <1 indicates purifying selection whereas Ka/Ks > 1 indi-cates positive selection. Ka/Ks = 1 is termed neutral evolution.
Immunogenicity of conserved peptidesTo evaluate the immunogenicity of the con-served peptide, different bio-computational tools were employed. In order to discover the immunogenicity of the conserved pep-tide, firstly a reverse immunogenic approach was employed for the selection of candidate epitopes.
Figure 2. Conserved region of E2 protein sequence in chikungunya virus E1–E2 glycoprotein complex. Yellow surface represents the conserved ‘C terminal’ peptide region of the E2 envelope protein sequence in the E1-E2 glycoprotein complex. The protein data bank database ID of the E1-E2 glycoprotein complex used here is 2XFC, retrieved from RCSB protein data bank.
Computational assay to design a peptide vaccine against CHIKV Preliminary Communication
Author Pro
of
Future Virol. (2012) 7(10)1032 future science group
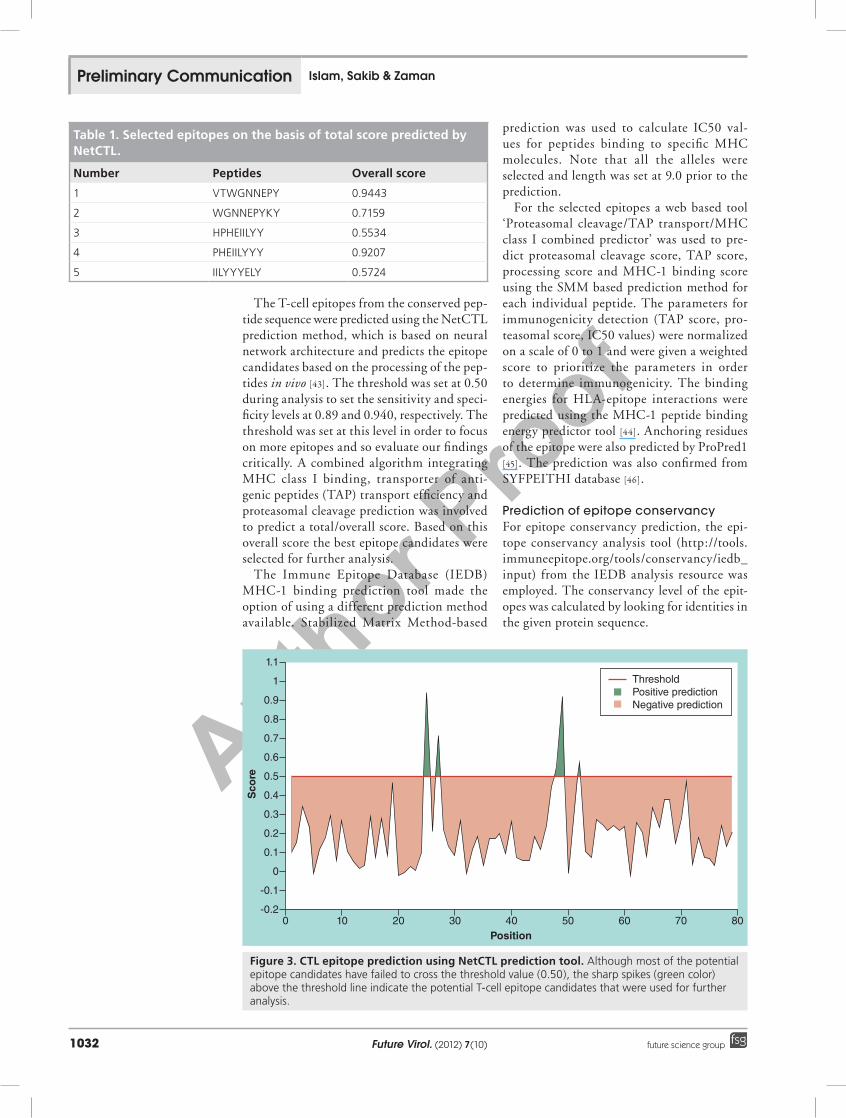
The T-cell epitopes from the conserved pep-tide sequence were predicted using the NetCTL prediction method, which is based on neural network architecture and predicts the epitope candidates based on the processing of the pep-tides in vivo [43]. The threshold was set at 0.50 during analysis to set the sensitivity and speci-ficity levels at 0.89 and 0.940, respectively. The threshold was set at this level in order to focus on more epitopes and so evaluate our findings critically. A combined algorithm integrating MHC class I binding, transporter of anti-genic peptides (TAP) transport efficiency and proteasomal cleavage prediction was involved to predict a total/overall score. Based on this overall score the best epitope candidates were selected for further analysis.
The Immune Epitope Database (IEDB) MHC-1 binding prediction tool made the option of using a different prediction method available. Stabilized Matrix Method-based
prediction was used to calculate IC50 val-ues for peptides binding to specific MHC molecules. Note that all the alleles were selected and length was set at 9.0 prior to the prediction.
For the selected epitopes a web based tool ‘Proteasomal cleavage/TAP transport/MHC class I combined predictor’ was used to pre-dict proteasomal cleavage score, TAP score, processing score and MHC-1 binding score using the SMM based prediction method for each individual peptide. The parameters for immunogenicity detection (TAP score, pro-teasomal score, IC50 values) were normalized on a scale of 0 to 1 and were given a weighted score to prioritize the parameters in order to determine immunogenicity. The binding energies for HLA-epitope interactions were predicted using the MHC-1 peptide binding energy predictor tool [44]. Anchoring residues of the epitope were also predicted by ProPred1 [45]. The prediction was also confirmed from SYFPEITHI database [46].
Prediction of epitope conservancyFor epitope conservancy prediction, the epi-tope conservancy analysis tool (http://tools.immuneepitope.org/tools/conservancy/iedb_input) from the IEDB analysis resource was employed. The conservancy level of the epit-opes was calculated by looking for identities in the given protein sequence.
Table 1. Selected epitopes on the basis of total score predicted by NetCTL.
Number Peptides Overall score
1 VTWGNNEPY 0.9443
2 WGNNEPYKY 0.7159
3 HPHEIILYY 0.5534
4 PHEIILYYY 0.9207
5 IILYYYELY 0.5724
1.1
1
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0
0
-0.1
-0.2
Sco
re
Position10 20 30 40 50 60 70 80
ThresholdPositive predictionNegative prediction
Figure 3. CTL epitope prediction using NetCTL prediction tool. Although most of the potential epitope candidates have failed to cross the threshold value (0.50), the sharp spikes (green color) above the threshold line indicate the potential T-cell epitope candidates that were used for further analysis.
Preliminary Communication Islam, Sakib & Zaman

Author Pro
of
www.futuremedicine.com 1033future science group
Molecular docking study of HLA-epitope interaction Retrieving 3D structure of HLA The 3D structure of HLA B*3501 (PDB ID 3LKN) coordinated with the influenza NP418 epitope from the 1918 strain was downloaded from the Protein Data Bank Database (www.rcsb.org/) and visualized in the PYMOL molecular graphics system. For docking pur-poses the bound influenza NP418 epitope was excluded from the HLA class 1 histocompat-ibility antigen complex (B-35 a-chain and b-2-microglobulin designated as A and B chain respectively). Prior to docking, all the water molecules were removed from the 3D structure of epitope free HLA B*3501.
Design of the 3D epitope structureFor the docking study the HPHEIILYY epitope was chosen because it showed the maximum interactions with different HLAs. The 3D structure of HPHEIILYY was designed using the PEP-FOLD Peptide Structure Prediction server at the RPBS Mobyle Portal [47].
HLA-epitope binding predictionThe AutoDock tool from the MGL software package (version 1.5.6) was employed for dock-ing purpose. Both the protein (HLA B*3501) and ligand (epitope) files were firstly converted to PDBQT format to use them for the dock-ing study. The grid/space box center was set at 3.659, -12.265, and -28.409 A0 in the x, y and z axes respectively, so that the epitope could bind at the binding groove of HLA B*3501. The size was set at 30, 26 and 24 A0 in the x-, y- and z-dimensions, respectively. All the analysis was done at 1.00-A0 spacing. The number of out-puts was set at 10, while the exhaustiveness was kept at the default 8.00. Note all of these above things were done using AutoDock tool. Then in the final stage of docking study, the AutoDock Vina program was used to do the actual docking based on these parameters. All the output files (PDBQT format) were then converted into PDB using OpenBabel (version 2.3.1). The outputs were again visualized in the PYMOL molecular graphics system and the best output was selected on the basis of higher binding energy.
Table 2. Allele selection for different peptide sequences.
Allele Sequence Proteasomescore
TAPscore
MHCscore
Processingscore
Totalscore
MHCIC50
HLA-B*15:17 VTWGNNEPY 1.03 1.36 -1.10 2.39 1.29 12.45
HLA-B*35:01 HPHEIILYY 1.24 1.13 -1.30 2.37 1.07 20.13
HLA-B*15:03 IILYYYELY 1.30 1.29 -1.37 2.59 1.22 23.41
HLA-A*29:02 IILYYYELY 1.30 1.29 -1.45 2.59 1.14 28.20
HLA-B*15:03 VTWGNNEPY 1.03 1.36 -1.46 2.39 0.93 28.74
HLA-B*15:03 WGNNEPYKY 1.38 1.15 -1.70 2.53 0.83 50.20
HLA-A*80:01 HPHEIILYY 1.24 1.13 -1.86 2.37 0.51 72.70
HLA-A*29:02 VTWGNNEPY 1.03 1.36 -1.88 2.39 0.51 76.36
HLA-A*29:02 HPHEIILYY 1.24 1.13 -1.94 2.37 0.43 87.99
HLA-B*35:01 WGNNEPYKY 1.38 1.15 -1.99 2.53 0.54 98.06
HLA-B*53:01 HPHEIILYY 1.24 1.13 -2.02 2.37 0.35 105.53
HLA-B*15:03 HPHEIILYY 1.24 1.13 -2.04 2.37 0.33 110.01
HLA-A*30:02 VTWGNNEPY 1.03 1.36 -2.06 2.39 0.33 114.24
HLA-A*30:02 WGNNEPYKY 1.38 1.15 -2.08 2.53 0.45 120.82
HLA-B*15:03 PHEIILYYY 1.29 1.12 -2.11 2.41 0.31 127.93
HLA-A*80:01 IILYYYELY 1.30 1.29 -2.21 2.59 0.39 161.13
HLA-B*15:02 VTWGNNEPY 1.03 1.36 -2.31 2.39 0.08 205.57
HLA-A*29:02 WGNNEPYKY 1.38 1.15 -2.35 2.53 0.18 223.75
HLA-A*26:02 HPHEIILYY 1.24 1.13 -2.38 2.37 -0.00 237.97
HLA-B*15:02 HPHEIILYY 1.24 1.13 -2.38 2.37 -0.01 241.96
HLA-A*80:01 PHEIILYYY 1.29 1.12 -2.40 2.41 0.02 249.12
TAP: Transporter of antigenic peptides.
Computational assay to design a peptide vaccine against CHIKV Preliminary Communication
Author Pro
of
Future Virol. (2012) 7(10)1034 future science group
ControlTo evaluate our docking study critically and to give it a scientific validation we had to use a control. And for this purpose, the influenza NP418 epitope from the 1918 strain was used as positive control with the ligand for HLA B*3501 protein molecule. The method and the parameters used were the same as those of the previously mentioned study, and a suc-cessful binding of this epitope with the HLA was demonstrated.
A comparative analysis of the best binding energy (Kcal/mol) and the arrangement of the test and the control epitope at the binding groove of MHC allele HLA B*3501 was also performed.
Prediction of population coveragePopulation coverage for individual epitope was predicted by the population coverage tool (http://tools.immuneepitope.org/tools/popu-lation) from the IEDB analysis resource. The allele frequency of the interacting HLA alleles was used to measure the population coverage for the corresponding epitope.
Prediction of B-cell epitope Linear B-cell epitopes were predicted from the given protein sequence using the B-cell epit-ope prediction tool from the IEDB analysis resource. The Kolaskar and Tongaonkar anti-genicity prediction method was employed for
prediction in which, predictions are based on a table that reflects the occurrence of amino acid residues in experimentally known seg-mental epitopes [48]. This method can predict antigenic peptides with approximately 75% accuracy.
Results Evolutionary divergence in chikungunya envelope proteinsE1 and E2 sequences from different strains from different geographic regions were retrieved from the database. These retrieved sequences were used to generate multiple sequence alignments (see Supplementary file 1 & 2). A phylogram generated from these multiple sequence alignments showed significant evo-lutionary divergence among the strains (see Supplementary file 3).
Identification of evolutionarily conserved regions in chikungunya envelope proteinsFrom the output of multiple sequences of E2 proteins, the ‘C terminal’ peptide sequence was found to be more or less conserved in all vari-ants. The C-terminal region spans 114 amino acid residues starting from to 290 to 403 of the reference genome sequence of CHIKV- gb|AFD61558.1 (China 2010) deposited in the NCBI database. There were some vari-able regions in this portion of the sequence. However, the conserved region was chosen as a guide to find an epitope that is situated in the region that is functionally unchanged over the course of evolution. It should be noted that searching for a conserved epitope was not limited to this conserved region alone, as even from an apparently shorter conserved region an epitope, which is of 9–15 amino acid long, could have been designed.
While E2 protein showed significant conser-vancy, E1 had a shorter stretch of 23 conserved amino acids.
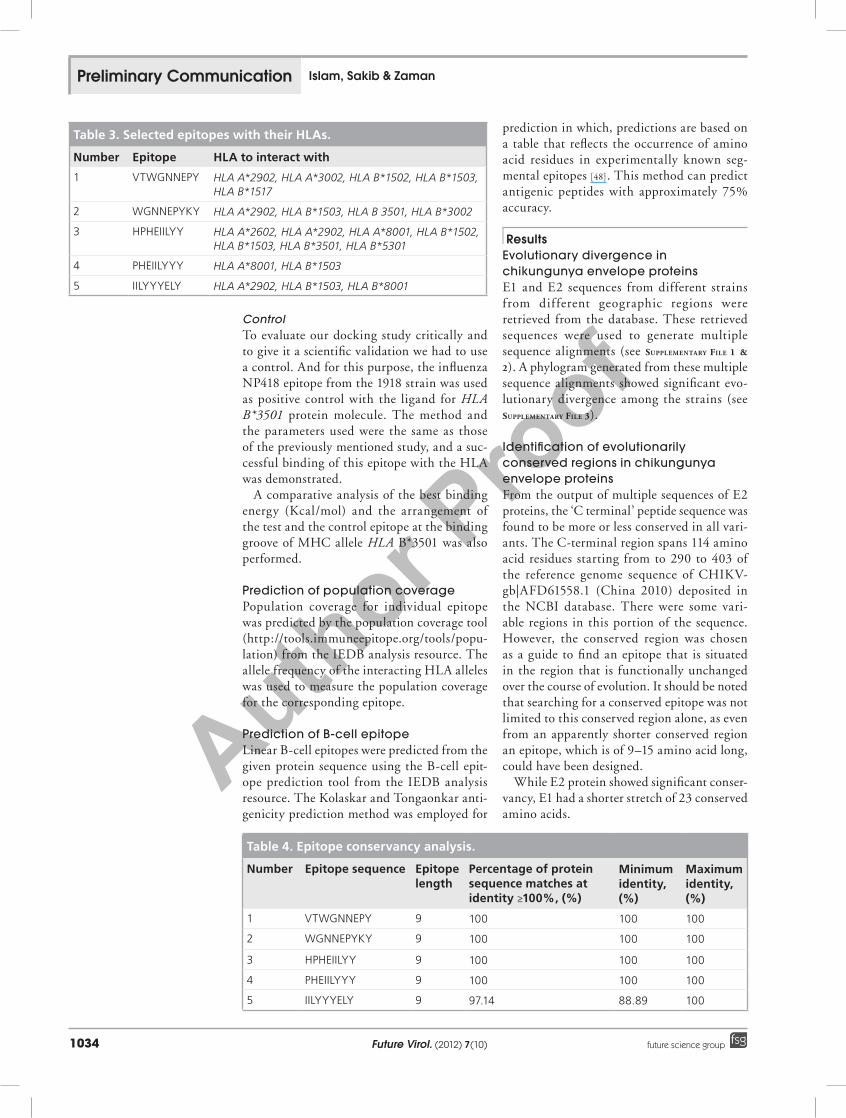
Table 3. Selected epitopes with their HLAs.
Number Epitope HLA to interact with
1 VTWGNNEPY HLA A*2902, HLA A*3002, HLA B*1502, HLA B*1503, HLA B*1517
2 WGNNEPYKY HLA A*2902, HLA B*1503, HLA B 3501, HLA B*3002
3 HPHEIILYY HLA A*2602, HLA A*2902, HLA A*8001, HLA B*1502, HLA B*1503, HLA B*3501, HLA B*5301
4 PHEIILYYY HLA A*8001, HLA B*1503
5 IILYYYELY HLA A*2902, HLA B*1503, HLA B*8001
Table 4. Epitope conservancy analysis.
Number Epitope sequence Epitope length
Percentage of protein sequence matches at identity ≥100%, (%)
Minimum identity, (%)
Maximum identity, (%)
1 VTWGNNEPY 9 100 100 100
2 WGNNEPYKY 9 100 100 100
3 HPHEIILYY 9 100 100 100
4 PHEIILYYY 9 100 100 100
5 IILYYYELY 9 97.14 88.89 100
Preliminary Communication Islam, Sakib & Zaman
Author Pro
of
www.futuremedicine.com 1035future science group
For the E1 protein, a 23 amino acid-long pep-tide ‘EFASAYRAHTASASAKLRVLYQG’ was found to be conserved. Among the predicted overlapping epitopes, only ‘ASAKLRVLY’ sat-isfied the threshold value mentioned before and that too did not interact to as many HLAs as E2 did. That meant that E1 epitope was less promising as a candidate for a universal vac-cine. The population coverage for the inter-acting epitope was also very unimpressive. However, this epitope could be a candidate for a multipeptide vaccine. Therefore, it was
assumed that a peptide sequence generated from E2 had a better chance of being a uni-versal epitope target than E1. Also, literature suggested that immune recognition for E1 epit-opes has been weak (see the Discussion section for further detail). Hence, the E2 conserved sequence was taken and analyzed further. The conserved region of the E2 protein is marked on figure 2 in yellow.
To identify the conservancy of the E2 protein sequences under selective pressure, nonsynonymous to synonymous ratio was
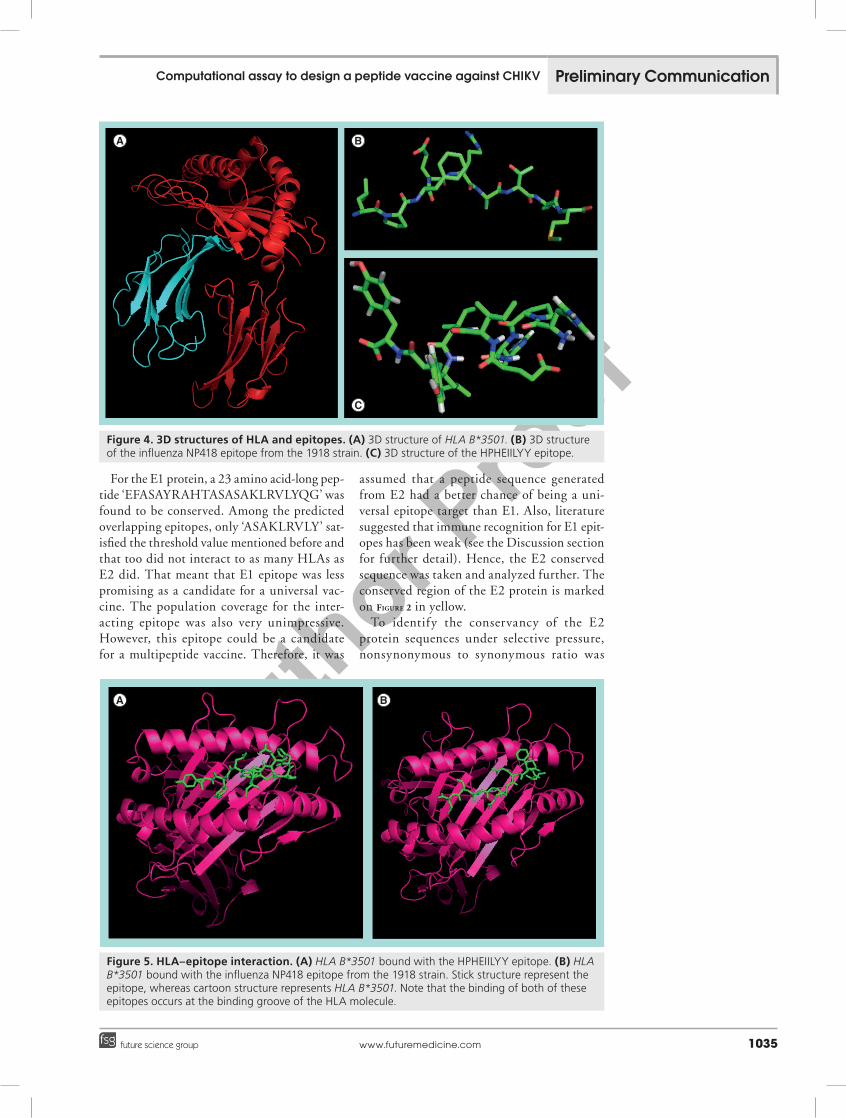
Figure 4. 3D structures of HLA and epitopes. (A) 3D structure of HLA B*3501. (B) 3D structure of the influenza NP418 epitope from the 1918 strain. (C) 3D structure of the HPHEIILYY epitope.
Figure 5. HLA–epitope interaction. (A) HLA B*3501 bound with the HPHEIILYY epitope. (B) HLA B*3501 bound with the influenza NP418 epitope from the 1918 strain. Stick structure represent the epitope, whereas cartoon structure represents HLA B*3501. Note that the binding of both of these epitopes occurs at the binding groove of the HLA molecule.
Computational assay to design a peptide vaccine against CHIKV Preliminary Communication
Author Pro
of
Future Virol. (2012) 7(10)1036 future science group
Cum
ulat
ive
perc
enta
ge o
f PC
Nu
mb
er o
f ep
itop
e h
its/
HL
A c
om
bin
atio
ns
reco
gn
ized
Nu
mb
er o
f ep
itop
e h
its/
HL
A c
om
bin
atio
ns
reco
gn
ized
Nu
mb
er o
f ep
itop
e h
its/
HL
A c
om
bin
atio
ns
reco
gn
ized
Nu
mb
er o
f ep
itop
e h
its/
HL
A c
om
bin
atio
ns
reco
gn
ized
Nu
mb
er o
f ep
itop
e h
its/
HL
A c
om
bin
atio
ns
reco
gn
ized
Nu
mb
er o
f ep
itop
e h
its/
HL
A c
om
bin
atio
ns
reco
gn
ized
Individuals (%)
Individuals (%) Individuals (%) Individuals (%)
Individuals (%) Individuals (%)
90 80 70 60 50 40 30 20 10 0
No
rth
Am
eric
aP
hili
pp
ines
PC
90
PC
90
01
Cumulative PC (%)
90100
80 70 60 50 40 30 20 10 0
Cumulative PC (%)
90100
80 70 60 50 40 30 20 10 0
Cumulative PC (%)
90100
80 70 60 50 40 30 20 10 0
Cumulative PC (%)
90100
80 70 60 50 40 30 20 10 0
Cumulative PC (%)
90100
80 70 60 50 40 30 20 10 0
Cumulative PC (%)
90100
80 70 60 50 40 30 20 10 0
80 70 60 50 40 30 20 10 080 70 60 50 40 30 20 10 070 65 60 55 50 45 40 35 30 25 20 15 10 5 0
65 60 55 50 45 40 35 30 25 20 15 10 5 0 55 50 45 40 35 30 25 20 15 10 5 0
Mal
iK
enya
Sen
egal
US
A
PC
90
PC
90
PC
90P
C90
01
2
01
23
45
67
89
01
23
45
67
89
01
23
45
67
01
23
45
67
810
9
Fig
ure
6. P
op
ula
tio
n c
ove
rag
e b
ased
on
MH
C r
estr
icti
on
dat
a u
sin
g t
he
Imm
un
e Ep
ito
pe
Dat
abas
e an
alys
is r
eso
urc
e. T
he N
orth
Am
eric
an S
eri p
opu
lati
on s
how
s m
axim
um c
umul
ativ
e p
opu
lati
on c
over
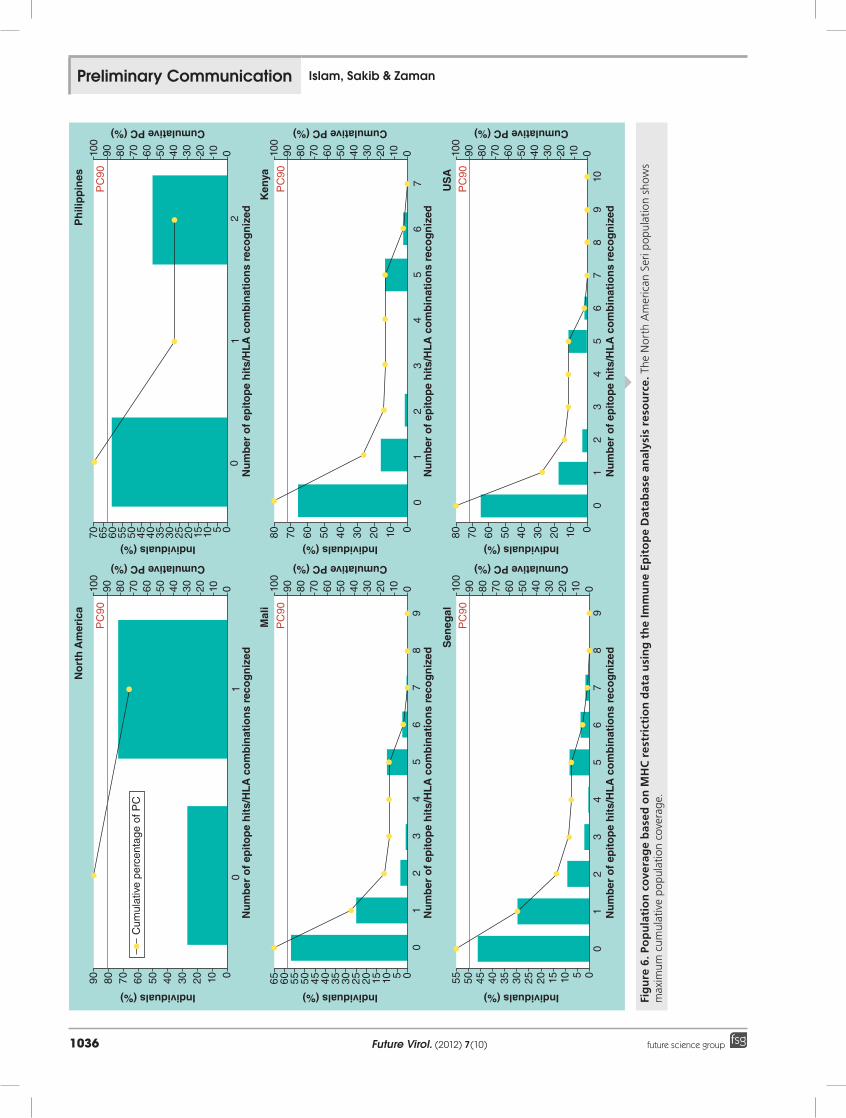
age.
Preliminary Communication Islam, Sakib & Zaman

Author Pro
of
www.futuremedicine.com 1037future science group
Fig
ure
6. P
op
ula
tio
n c
ove
rag
e b
ased
on
MH
C r
estr
icti
on
dat
a u
sin
g Im
mu
ne
Epit
op
e D
atab
ase
anal
ysis
res
ou
rce
(co
nt.
). N
orth
Am
eric
an S
eri p
opu
lati
on s
how
s m
axim
um c
umul
ativ
e p
opu
lati
on c
over
age.
Nu
mb
er o
f ep
itop
e h
its/
HL
A c
om
bin
atio
ns
reco
gn
ized
Nu
mb
er o
f ep
itop
e h
its/
HL
A c
om
bin
atio
ns
reco
gn
ized
Nu
mb
er o
f ep
itop
e h
its/
HL
A c
om
bin
atio
ns
reco
gn
ized
Individuals (%)
Individuals (%) Individuals (%)
Cumulative PC (%)
90100
80 70 60 50 40 30 20 10 0
Cumulative PC (%)
90100
80 70 60 50 40 30 20 10 0
Cumulative PC (%)
90100
80 70 60 50 40 30 20 10 0
80 70 60 50 40 30 20 10 070 65 60 55 50 45 40 35 30 25 20 15 10 5 0
55 50 45 40 35 30 25 20 15 10 5 0
Zim
bab
we
Zam
bia
So
uth
Afr
ica
PC
90
PC
90
PC
90
01
23
45
67
89
01
23
45
67
810
9
01
23
45
67
89
Computational assay to design a peptide vaccine against CHIKV Preliminary Communication
Author Pro
of
Future Virol. (2012) 7(10)1038 future science group
calculated using the Ka/Ks calculation tool developed by the computational biology unit of Bergen Center for Computational Science (see Supplementary file 4). Eight sequences – four with conserved C terminal region and four with full length E2 sequence – were subjected to the Ka/Ks analysis. The results (0.0 for both branches of nodes 1 and 3 – the C-terminal region sequence) show that the selected C-terminal region follows purifying selection and hardly shows any mutation over the evolutionary history.
Immunogenicity of conserved peptidesThe NetCTL prediction tool predicted 79 overlapping potential epitopes from the given peptide sequence, but only five potential T-cell epitope candidates were chosen on the basis of a total score that was a combinatorial result of different parameters mentioned earlier (table 1). A graphical representation also illustrates the best T-cell epitopes predicted by the NetCTL prediction tool (figure 3).
SMM based IEDB MHC-1 binding predic-tion tool retrieved 280 possible MHC-1 allele interactions with the five different T-cell epi-topes those were selected before. The MHC-1 alleles for which the epitopes showed higher affinity (IC50 < 250) were selected for further analysis [49].
The proteasome complex contains enzymes that cleave peptide bonds, converting proteins into peptides. The antigenic peptides from pro-teasome cleavage associate with class I MHC
molecules, and the peptide-MHC complexes are then transported to the cell membrane. TAP transports the peptides to the endoplas-mic reticulum. The predicted proteasomal score, TAP score, processing score and IC50 values are summarized in table 2.
Among the five T-cell epitopes, a 9-mer epi-tope HPHEIILYY was found to interact with HLA A*2602, HLA A*2902, HLA A*8001, HLA B*1502, HLA B*1503, HLA B*3501 and HLA B*5301. No other T-cell epitope inter-acted with as many MHC-1 alleles with as great an affinity as this epitope (table 3).
Epitope conservancy analysis with all of the E2 protein sequences from different strains reveals that, except for ‘IILYYYELY’, all the other four epitopes have a 100% protein sequence match, whereas ‘IILYYYELY’ shows a protein sequence match of 97.14% identity, with minimum and maximum identity of 88.89 and 100%, respectively. The results are summarized in table 4.
Molecular docking study of HLA–epitope interaction AutoDock Vina predicted three possible bind-ing models. On the basis of higher binding energy with HLA B*3501, the best output model for HPHEIILYY epitope was found to have a binding energy of -5.4 kcal/mol.
The 3D structures of HLA and epitope are illustrated in figure 4. All of these figures were generated and captured from the PYMOL molecular graphics system.
Threshold = 1.000
1.6
1.4
1.2
1.0
0.8
0.6
0.4
0.2
00 10 20 30 40 50 60 70 80 90
An
tig
enic
pro
pen
sity
Sequence position
Kolaskar and Tongaonkar antigenicity
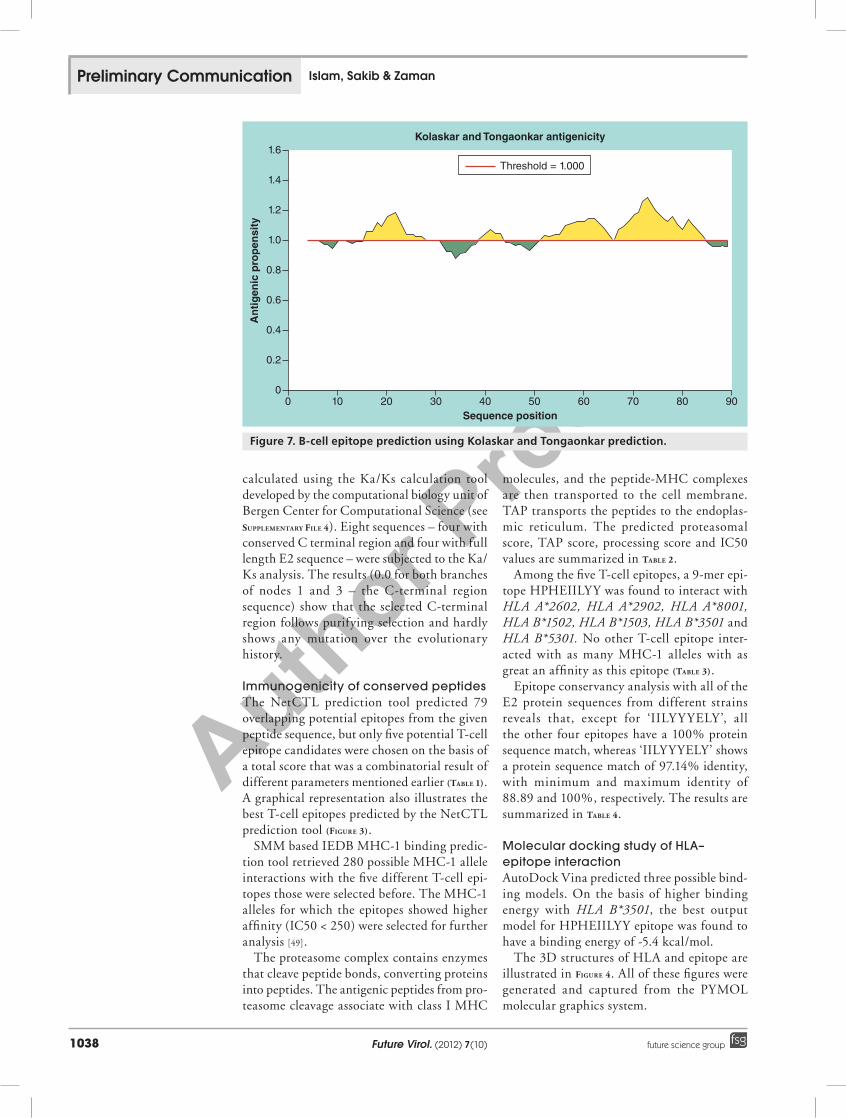
Figure 7. B-cell epitope prediction using Kolaskar and Tongaonkar prediction.
Preliminary Communication Islam, Sakib & Zaman

Author Pro
of
www.futuremedicine.com 1039future science group
For positive control, the best output was found to have a very similar binding affin-ity (-5.2 kcal/mol) to the designed epitope. A comparative look at the binding of these two epitopes is illustrated in figure 5.
Among the nine residues in the epitope, histidine and isoleucine (first and fifth resi-due, respectively) have been predicted to play an anchoring role in the interaction with the MHC molecule by ProPred1. The rest of the residues were found to be located at the bind-ing groove of HLA B*3501. A bulge had been formed to accommodate them in the binding pocket.
Population coverageWe have only represented those populations for which the coverage is greater than 33%. The maximum cumulative population cover-age (73.46%) for five epitopes was found in the North American Seri population. For Senegal and Zambia, the population coverage was over 50% (54.02 and 53.08%, respectively).
For the Philippines, Mali, USA, Kenya, Zimbabwe the cumulative population cover-ages obtained were 42.66, 39.16, 35.01, 33.52 and 40.40%, respectively (figure 6).
Prediction of B-cell epitopefigure 7 shows the antigenic determinant plot; the x-axis shows sequence position and the y-axis shows antigenic propensity. Average antigenic propensity for this protein is 1.045. There are three antigenic determinants in the sequence (table 5). The highest pick is 67–84 amino acid residues, and the sequence is ‘TMTVVVVSVATFILLSMV’. The average for the whole protein is above 1.0; all residues above 1.0 are potentially antigenic.
DiscussionWith the disclosure of huge sequence informa-tion, nowadays epitope-based peptide vaccine design has become a key theme for viral vac-cine preparation. In order to accomplish this intense laboratory experiments and computa-tional prediction have joined hands. Despite its potential as one of the most emerging
cross-host virus with dreaded transmission ability, CHIKV has been left unearthed as far as epitope-based peptide vaccine design is concerned [50,51]. In this study, admittedly in a rather preliminary manner, we have tried to explore this possibility to some extent.
Targeting a T-cell-based epitope was inspired by the fact that the host produces a strong CD8+ T-cell-mediated immune response to infected T cells. CHIKV is no exception to this [36]. There currently exist several vaccine approaches for example live attenuated vac-cine, recombinant CHIKV vaccine, antibod-ies against virus like particles for CHIKV. However, in this study we have taken an alter-native approach; one that has often not been the focus of a great deal of attention; yet on that has not been completely overlooked either, especially in terms of viral diseases [39,40,52].
T-cell epitope-based vaccination is a unique approach. While antibody memory response can be easily dodged by antigenic drift over the course of time, cell-mediated immunity often elicits lasting immunity [38]. A significant sum of resources has already been spent on CHIKV-related vaccine design, yet with very little success to show for it. We believe that, although some of the progresses made in this regard has brought in promises in clinical tri-als, this perspective is very worth trying and attempting.
To be viewed as a good peptide epitope, a sequence has to possess some key properties. First, the epitope has to be fairly well-con-served among the CHIKV protein sequences collected from the database. Second, the epi-tope sequence must have the attributes that ensure T- or B-cell processivity. Third, the processed peptide has to be able to interact with MHC alleles with good affinity. Fourth, the interacting MHC allele has to show good population coverage overall.
The predicted peptide finally fulfilled all the parameters mentioned here. The degree of sequence conservancy of the epitope was 100%, whereas sequences collected from other regions apart for the C-terminal conserved region, even with good T-cell epitope potential,
Table 5. Potential linear peptides predicted to be antigenic determinants.
Number Start position End position Peptide Peptide length
1 16 30 KKEVVLTVPTEGLEV 15
2 52 65 GHPHEIILYYYELY 14
3 67 84 TMTVVVVSVATFILLSMV 18
Computational assay to design a peptide vaccine against CHIKV Preliminary Communication

Author Pro
of
Future Virol. (2012) 7(10)1040 future science group
failed to show 100% conservancy and hence was disregarded as a universal peptide epitope target.
Similar studies were carried out for E1 pro-tein too. For E1 protein, a 23-amino acid pep-tide ‘EFASAYRAHTASASAKLRVLYQG’ was found to be conserved. Among the predicted overlapping epitopes only ‘ASAKLRVLY’ sat-isfied the threshold value set previously. That meant that E1 did not fulfill all the parameters that were necessary and thus was not taken into further consideration. E1 is not a good target for the designing a universal vaccine against CHIKV. Antibodies against E1 are reported to have shown the property of cross-reacting with each other [53,54]. Showing that the immune recognition of the epitope is weaker.
Five potential T-cell epitopes were found in E2 envelope protein, which cross-reacts with seven HLA variants. Also the population cov-erage range from 33.52 to 73.46% in diverse populations. An African population, one of the key regions for the CHIKV epidemic, showed good population coverage for our designed epitope. The most impressive result, how-ever, was for a North American Seri popula-tion. Nonetheless, lack of sequence data from developing countries, which are most known for the spread of CHIKV, prevented us from determining the peptide vaccine efficiency for those populations. In the future it would be interesting to see how efficacious this peptide is within those populations.
A good hit for T-cell epitope potential was observed for an additional sequence away from the C terminal region in the E2 – ‘VTNHKKWQY’. However, when we per-formed an MHC-I binding prediction from the IEDB database, only four alleles (based on IC50 score <250) were found to be interacting with this sequence. On the other hand, the best sequence, ‘HPHEIILYY’, from the conserved region showed seven interactions with different MHC-I alleles. The ‘VTNHKKWQY’ peptide also did not show conservancy in all the available sequences, whereas our small peptide sequence showed 100% conservancy. Hence, the epitope was not taken into consideration further.
Some results were validated using multiple computational tools. For example, the allele frequency necessary for understanding the population coverage of the peptide was cal-culated using the ImmPort database as well as the IEDB database.
We believe that our predicted vaccine, like many other epitope-based vaccines, has the
potential of acting as both a preventive and a therapeutic treatment [55]. Nevertheless, we believe it would be more of a therapeutic vac-cine rather than a preventive one. The principle reason behind this assumption is that the pop-ulation coverage found for the peptide is rather modest. However, the efficacy of the vaccine is often determined in vitro, which is beyond the scope of this study. We would like to reaffirm that the study’s focus remains on finding and validating one or more epitopes computation-ally, and that by doing so we do not claim to have found a definite solution.
Multipeptide vaccination is another dynamic concept. In this study we have found epitopes on both E1 and E2 capable of triggering sig-nificant immune response. Applying several such peptides in combination can be a concept worth researching in the future.
Our results are based on careful sequence analysis and immune data deposited on vari-ous databases. The outcome of the result gives us good confidence to claim that the epitopes found here are good candidates for designing a peptide vaccine able to trigger an efficacious immune response in vivo.
Conclusion & future perspectiveIn vitro and in vivo studies are required to deter-mining the actual effectiveness of the peptide for mounting an immune response. Binding chip assay for HLA and peptide would also be useful to determine the binding affinity of the peptide as a whole. Analyzing the potential of a multipeptide vaccine would also be interesting.
Financial & competing interests disclosureThe authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manu-script. This includes employment, consultancies, honoraria, stock ownership or options, expert testi-mony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research The authors state that they have obtained appropriate insti tutional review board approval or have followed the princi ples outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investi gations involving human sub-jects, informed consent has been obtained from the participants involved.
Preliminary Communication Islam, Sakib & Zaman

Author Pro
of
www.futuremedicine.com 1041future science group
Executive summary
Chikungunya virus as an emerging epidemic n Chikungunya virus (CHIKV) infection has broken out in severe epidemics in multiple occasions. n Treatment options for CHIKV have still not been explored to a good extent; no peptide vaccines have been designed to date. n The study employs an in silico approach to resolve this shortcoming and identify peptide stretches potent enough to recognize different
virus isolates, yet that induces good immune response have been identified.
Peptide-based epitopes can work as vaccine against CHIKV n Immune system recognizes foreign peptides on the basis of their sequences and presents them for to adaptive immune response –
T and B cells. n Although innate immune system also plays a pivotal role in clearing out viral load within the body, it is the adaptive immune system
which plays major role for virus regression. Hence, educating host immune cells with proper peptides sequence is good way of tackling virus infection in the future. n The sequence of the designed peptide has to be in a delicate balance so that it is conserved enough in viruses of the same clade but
varying enough from self cells. Moreover it also would have to have good immune-stimulatory property.
Computational assay predicts probable peptide vaccines n In silico analysis predicted peptide sequence that gave out good immune epitope candidature for T and B cell; five for T and 3for B cells. n Population coverage was also as high as 73.46%, which is quite a high value for an in silico prediction. n Further validation of the work in vivo should be able to improve the coverage even more.
References1. Islam MN, Hossain MA, Khaleque MA et
al. Chikungunya virus infection, a threat to public health. Mymensingh Med. J. 21(2), 372–376 (2012).
2. Zayed A, Awash AA, Esmail MA et al. Detection of chikungunya virus in Aedes aegypti during 2011 outbreak in Al Hodayda, Yemen. Acta Trop. 123(1), 62–66 (2012).
3. Rohani A, Potiwat R, Zamree I, Lee HL, Refractoriness of Aedes aegypti (Linnaeus) to dual infection with dengue and chikungunya virus. Southeast Asian J. Trop. Med. Public Health 40(3), 443–448 (2009).
4. Tang BL. The cell biology of chikungunya virus infection. Cell. Microbiol. 4(9), 1354–1363 (2012).
5. Kucharz EJ, Cebula-Byrska I. Chikungunya fever. Eur. J. Intern. Med. 23(4), 325–329 (2012).
6. Joubert PE, Werneke SW, de la Calle C et al. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 209(5), 1029–1047 (2012).
7. Pellot AS, Alessandri JL, Robin S et al. [Severe forms of chikungunya virus infection in a pediatric intensive care unit on Reunion Island]. Med. Trop. (Mars) 72(Spec No), 88–93 (2012).
8. Ng LF, Ojcius DM. Chikungunya fever – re-emergence of an old disease. Microbes Infect. 11(14–15), 1163–1164 (2009).
9. Her Z, Kam YW, Lin RT, Ng LF. Chikungunya: a bending reality. Microbes Infect. 11(14–15), 1165–1176 (2009).
10. Aoustin T. [Chikungunya and urban sprawl
on Reunion Island]. Med. Trop. (Mars) 72(Spec No), 51–59 (2012).
11. Baville M, Dehecq JS, Reilhes O, Margueron T, Polycarpe D, Filleul L. [New vector control measures implemented between 2005 and 2011 on Reunion Island: lessons learned from chikungunya epidemic]. Med. Trop. (Mars) 72(Spec No), 43–46 (2012).
12. Larrieu S, Balleydier E, Renault P, Baville M, Filleul L. [Epidemiological surveillance du chikungunya on Reunion Island from 2005 to 2011]. Med. Trop. (Mars) 72(Spec No), 38–42 (2012).
13. Long KM, Heise MT. Chikungunya virus transmission – more than meets the eye. J. Infect. Dis. 206(6), 806–807 (2012).
14. Stojcic I. [Chikungunya epidemic in 2005–2006: questions from occupational health professionals]. Med. Trop. (Mars) 72(Spec No), 103–104 (2012).
15. Kurkela S, Sane J, Deren E et al. Chikungunya virus as a causative agent of fever of unknown origin in Finnish travellers to tropics. J. Clin. Virol. 54(3), 289–290 (2012).
16. Gerardin P, Fianu A, Malvy D et al. [Perceived morbidity and community burden of chikungunya in La Reunion]. Med. Trop. (Mars) 72(Spec No), 76–82 (2012).
17. Flahault A, Aumont G, Boisson V et al. An interdisciplinary approach to controlling chikungunya outbreaks on French islands in the south-west Indian ocean. Med. Trop. (Mars) 72(Spec No), 66–71 (2012).
18. Moro ML, Grilli E, Corvetta A et al. Long-term chikungunya infection clinical
manifestations after an outbreak in Italy: a prognostic cohort study. J. Infect. 65(2), 165–172 (2012).
19. Idelson B. [Chikungunya crisis on Reunion Island. Media coverage and conflicting public information]. Med. Trop. (Mars) 72(Spec No), 25–28 (2012).
20. Boisson V, Cresta MP, Thibault L et al. [Chikungunya outbreak on Reunion Island in 2005/2006: role of hospital physicians in raising alert]. Med. Trop. (Mars) 72(Spec No), 19–22 (2012).
21. Jacques C, Bernard-Alex G, Fabrice S. [Lessons learned from the health crisis caused by the chikungunya epidemic on Reunion Island in 2005–2006]. Med. Trop. (Mars) 72(Spec No), 4–5 (2012).
22. Nagpal BN, Saxena R, Srivastava A et al. Retrospective study of chikungunya outbreak in urban areas of India. Indian J. Med. Res. 135, 351–358 (2012).
23. Vilain P, Larrieu S, Renault P, Baville M, Filleul L. How to explain the re-emergence of chikungunya infection in Reunion Island in 2010? Acta Trop. 123(2), 85–90 (2012).
24. Shrinet J, Jain S, Sharma A et al. Genetic characterization of chikungunya virus from New Delhi reveal emergence of a new molecular signature in Indian isolates. Virol. J. 9(1), 100 (2012).
25. Akahata W, Nabel GJ. A specific domain of the chikungunya virus E2 protein regulates particle formation in human cells: implications for alphavirus vaccine design. J. Virol. 86(16), 8879–8883 (2012).
26. Kuo SC, Chen YJ, Wang YM et al. Cell-based analysis of chikungunya virus E1
Computational assay to design a peptide vaccine against CHIKV Preliminary Communication

Author Pro
of
Future Virol. (2012) 7(10)1042 future science group
protein in membrane fusion. J. Biomed. Sci. 19, 44 (2012).
27. Singh RK, Tiwari S, Mishra VK, Tiwari R, Dhole TN. Molecular epidemiology of chikungunya virus: mutation in E1 gene region. J. Virol. Methods. 185(2), 213–220 (2012).
28. Boere WA, Harmsen T, Vinjé J, Benaissa-Trouw BJ, Kraaijeveld CA, Snippe H. Identification of distinct antigenic determinants on Semliki Forest virus by using monoclonal antibodies with different antiviral activities. J. Virol. 52(2), 575–582 (1984).
29. Frazier CL, Shope RE. Detection of antibodies to alphaviruses by enzyme-linked immunosorbent assay. J. Clin. Microbiol. 10(4), 583–585 (1979).
30. Hunt AR, Roehrig JT. Biochemical and biological characteristics of epitopes on the E1 glycoprotein of western equine encephalitis virus. Virology 142(2), 334–346 (1985).
31. Ahn A, Klimjack MR, Chatterjee PK, Kielian M. An epitope of the Semliki Forest virus fusion protein exposed during virus-membrane fusion. J. Virol. 73(12), 10029–10039 (1999).
32. Shukla J, Khan M, Tiwari M et al. Development and evaluation of antigen capture ELISA for early clinical diagnosis of chikungunya. Diagn. Microbiol. Infect. Dis. 65(2), 142–149 (2009).
33. Byrnes AP, Griffin DE. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 72(9), 7349–7356 (1998).
34. Fraser JR, Cunningham AL. Incubation time of epidemic polyarthritis. Med. J. Aust. 1(11), 550–551 (1980).
35. Lee CY, Kam YW, Fric J et al. Chikungunya virus neutralization antigens and direct cell-to-cell transmission are revealed by human antibody-escape mutants. PLoS Pathog. 7(12), e1002390 (2011).
36. Wauquier N, Becquart P, Nkoghe D, Padilla C, Ndjoyi-Mbiguino A, Leroy EM. The acute
phase of chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J. Infect. Dis. 204(1), 115–123 (2011).
37. Hoarau JJ, Jaffar Bandjee MC, Krejbich Trotot P et al. Persistent chronic inflammation and infection by chikungunya arthritogenic alphavirus in spite of a robust host immune response. J. Immunol. 184(10), 5914–5927 (2010).
38. Klavinskis LS, Whitton JL, Oldstone MB. Molecularly engineered vaccine which expresses an immunodominant T-cell epitope induces cytotoxic T lymphocytes that confer protection from lethal virus infection. J. Virol. 63(10), 4311–4316 (1989).
39. Tan PT, Khan AM, August JT. Highly conserved influenza A sequences as T cell epitopes-based vaccine targets to address the viral variability. Hum. Vaccin. 7(4), 402–409 (2011).
40. Olsen LR, Zhang GL, Keskin DB, Reinherz EL, Brusic V. Conservation analysis of dengue virus T-cell epitope-based vaccine candidates using peptide block entropy. Front. Immunol. 2, 69 (2011).
41. Schuffenecker I, Iteman I, Michault A et al. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 3(7), e263 (2006).
42. Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 14(6), 1188–1190 (2004).
43. Larsen MV, Lundegaard C, Lamberth K et al. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur. J. Immunol. 35(8), 2295–2303 (2005).
44. Jojic N, Reyes-Gomez M, Heckerman D, Kadie C, Schueler-Furman O. Learning MHC I–peptide binding. Bioinformatics 22(14), e227–e235 (2006).
45. Singh H, Raghava G. ProPred1: prediction of promiscuous MHC Class-I binding sites. Bioinformatics 19(8), 1009–1014 (2003).
46. Rammensee HG, Bachmann J, Emmerich NP, Bachor OA, Stevanović S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50(3), 213–219 (1999).
47. Maupetit J, Derreumaux P, Tuffery P. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic acids Res. 37(Suppl. 2), W498–W503 (2009).
48. Kolaskar A, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 276(1), 172–174 (1990).
49. De Groot AS, Moise L, McMurry JA et al. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes”. Blood 112(8), 3303–3311 (2008).
50. Senanayake MP, Senanayake SM, Vidanage KK, Gunasena S, Lamabadusuriya SP. Vertical transmission in chikungunya infection. Ceylon Med. J. 54(2), 47–50 (2009).
51. Staples JE, Breiman RF, Powers AM. Chikungunya fever: an epidemiological review of a re-emerging infectious disease. Clin. Infect. Dis. 49(6), 942–948 (2009).
52. Sbai H, Mehta A, DeGroot AS. Use of T cell epitopes for vaccine development. Curr. Drug Targets Infect. Disord. 1(3), 303–313 (2001).
53. Medical Microbiology (Fourth edition). Baron S (Ed.). University of Texas, Medical Branch in Galveston, TX, USA (1996).
54. Griffin DE. Roles and reactivities of antibodies to alphaviruses. Semin. Virol. 6(4), 249–255 (1995).
55. Torresi J, Johnson D, Wedemeyer H. Progress in the development of preventive and therapeutic vaccines for hepatitis C virus. J. Hepatol. 54(6), 1273–1285 (2011).
Preliminary Communication Islam, Sakib & Zaman




















