
High Rates of O’Nyong Nyong and Chikungunya Virus Transmission in Coastal Kenya
Upload
independentCategory
view
0download
0
Elsevier Editorial System(tm) for Infection, Genetics and Evolution Manuscript Draft Manuscript Number: MEEGID-D-14-00393R1 Title: Phylogeny of Dengue and Chikungunya viruses in Al Hodayda governorate, Yemen. Article Type: Research paper Keywords: DENV, CHIKV, phylogeny Corresponding Author: Dr. massimo ciccozzi, Corresponding Author's Institution: National Institute of Health First Author: massimo ciccozzi Order of Authors: massimo ciccozzi; Alessandra Lo Presti; Eleonora Cella; Marta Giovanetti; Alessia Lai; Gamal El-Sawaf; Giovanni Faggioni; Fenicia Vescio; Ranya Al Ameri; Riccardo De Santis; Ghada Helaly; Alice Pomponi; Dalia Metwally; Massimo Fantini; Hussein Qadi; Gianguglielmo Zehender; Florigio Lista; Giovanni Rezza Abstract: Yemen, which is located in the southwestern end of the Arabian Peninsula, is one of countries most affected by recurrent epidemics caused by emerging vector-borne viruses. Dengue virus (DENV) outbreaks have been reported with increasing frequency in several governorates since the year 2000, and the chikungunya virus (CHIKV) has been also responsible of large outbreaks and it is now a major public health problem in Yemen. We report the results of the phylogenetic analysis of DENV-2 and CHIKV isolates (NS1 and E1 genes, respectively) detected in an outbreak occurred in al-Hodayda in 2012. Estimates of the introduction date of CHIKV and DENV-2, and the phylogeographic analysis of DENV-2 are also presented. Phylogenetic analysis showed that the Yemen isolates of DENV belonged to the lineage 2 Cosmopolitan subtype, whereas CHIKV isolates from Yemen belonged to the ECSA genotype. All the CHIKV isolates from Yemen were statistically supported and dated back to the year 2010 (95% HPD: 2009-2011); these sequences showed an alanine in the aminoacid position 226 of the E1 protein. Phylogeographic analysis of DENV-2 virus showed that cluster 1, which included Yemen isolates, dated back to 2003 Burkina Faso strains (95%HPD 1999-2007). The Yemen, cluster dated back to 2011 (95% HPD 2009-2012). Our study sheds light on the global spatiotemporal dynamics of DENV-2 and CHIKV in Yemen. This study reinforces both the need to monitor the spread of CHIKV and DENV, and to apply significant measures for vector control.

Michel Tibayrenc
UM1-CNRS 5290-IRD 224, MIVEGEC/IDVEGEC
(Infectious Diseases and Vectors, Ecology, Genetics, Evolution and Control),
Centre National de la Recherche Scientifique (CNRS), IRD Center,
BP 64501, 34394 Montpellier Cedex 5, France,
Email: [email protected]
Dear Editor
We submitted the revised version of the paper entitled “Phylogeny of Dengue and Chikungunya
viruses in Al Hodayda governorate, Yemen” . We answered point by point all the questions by
the reviewers. All authors have seen and approved this version of the manuscript.
Thank you for considering this paper for publication in your journal.
Sincerely,
Massimo Ciccozzi
Department of Infectious Parasitic and Immunomediated Diseases, Reference Centre on Phylogeny,
Molecular Epidemiology and Microbial Evolution (FEMEM)/ Epidemiology Unit, National
Institute of Health, Rome, Italy
e-mail: [email protected]; Phone number: +390649903187
Cover Letter

Reviewers' comments:
Reviewer #1: The authors have reported a phylogenetic analysis of dengue and chikungunya viruses
recovered in Yemen. This is ,essentially, a descriptive report that does not provide any surprises and
does not appear to provide any insights into the how and why of global movement of arboviruses
and their establishment of transmission cycles at new localities. The analyses appear to have been
undertaken with a rigor beyond that required for a study of this kind e.g. Fig S1 - but for which the
authors might be commended.
Q: The Reviewer could find no rationale for the analysis of dengue NS1 genes rather than E genes,
particularly as the database of E gene sequences is much larger than that of NS1 sequences.
A: We agree in part with the Reviewer about the database. Indeed NS1 was used for DENV and
E gene for CHIKV and both the dataset were built used specific criteria as reported in material
and methods.
Q:The Abstract could be shorter and the question being asked and the significance of the
conclusion(s) made clearer.
A: As suggested by the Review we modified the abstract.
Q: The manuscript was easy to read and to comprehend although there are some grammatical issues
that should be addressed e.g. the appropriate use of "the". However, the manuscript is too long for
the information conveyed.
A:Done.
Q:The authors should consult the Guide to Authors for the correct use of capitals when writing the
name of a virus. Dengue is a disease not a virus e.g. a mosquito transmits dengue viruses, not
dengue.
A:Thanks to the Review. We have corrected as requested.
Q:The figures are clear but there are more figures in the text than are necessary. A phylogenetic tree
for DENV sequences and another for CHIKV sequences would have been adequate.
Some of the data relating to times to recent ancestors have probably been over-interpreted.
A:We disagree with the Review, alla the figures in the manuscript are important to describe the
outbreaks of these two viruses, indeed we put as supplementary figures the only two not full
necessary.
Reviewer #2: The manuscript described the genetic analysis of Dengue and Chikungunya viruses in
Al Hodayda governorate, Yemen. Even the data are not new, information of these viruses in Yemen
is limited. Therefore, it is worthy to publish it in the journal.
Two comments:
Q: 1. English has some grammatical error. The manuscript should be checked by a native English
speaker.
A:Done.
Q: 2. Some parts of data were published in EID journal. Authors did cite this paper in the
manuscript. A complete citation such as title, year... is needed.
A:Done.
*Reply to Reviewers

Highlights
Phylogenetic analysis of DENV-2 and CHIKV isolates detected in Yemen was reported.
Estimates of introduction date of CHIKV and DENV-2 was presented.
Phylogeographic analysis of DENV-2 is presented.
This study reinforces the need to monitor the spread of CHIKV and DENV
*Highlights (for review)

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
1
Phylogeny of Dengue and Chikungunya viruses in Al Hodayda governorate, Yemen.
Massimo Ciccozzi1,2*
, Alessandra Lo Presti1, Eleonora Cella
1, Marta Giovanetti
1, Alessia Lai
3,
Gamal El-Sawaf4, Giovanni Faggioni
5, Fenicia Vescio
1, Ranya Al Ameri
6, Riccardo De Santis
5,
Ghada Helaly4, Alice Pomponi
5, Dalia Metwally
4, Massimo Fantini
7, Hussein Qadi
6, Gianguglielmo
Zehender3, Florigio Lista
5, Giovanni Rezza
1.
1Department of Infectious, Parasitic and Immunomediated Diseases, Istituto Superiore di Sanità,
Rome, Italy
2University Hospital Campus Bio-Medico
3Department of Biomedical and Clinical Science, Infectious Diseases and Immunopathology
Section, „L. Sacco‟ Hospital, University of Milan, Milan, Italy
4Medical Research Institute, Alexandria University, Egypt
5Histology and Molecular Biology Section, Army Medical and Veterinary Research Center, Rome,
Italy
6University of Sana‟a, Republic of Yemen
7Department of Clinical Sciences and Translational Medicine, University of Rome “Tor Vergata”,
Rome, Italy
*Corresponding author: Massimo Ciccozzi
Department of Infectious Parasitic and Immunomediated Diseases, Istituto Superiore di Sanità,
Rome, Italy
e-mail: [email protected] ; Phone number: +390649903187
*ManuscriptClick here to view linked References

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
2
ABSTRACT
Yemen, which is located in the southwestern end of the Arabian Peninsula, is one of countries most
affected by recurrent epidemics caused by emerging vector-borne viruses. Dengue virus (DENV)
outbreaks have been reported with increasing frequency in several governorates since the year 2000,
and the chikungunya virus (CHIKV) has been also responsible of large outbreaks and it is now a
major public health problem in Yemen. We report the results of the phylogenetic analysis of
DENV-2 and CHIKV isolates (NS1 and E1 genes, respectively) detected in an outbreak occurred in
al-Hodayda in 2012. Estimates of the introduction date of CHIKV and DENV-2, and the
phylogeographic analysis of DENV-2 are also presented. Phylogenetic analysis showed that the
Yemen isolates of DENV belonged to the lineage 2 Cosmopolitan subtype, whereas CHIKV
isolates from Yemen belonged to the ECSA genotype. All the CHIKV isolates from Yemen were
statistically supported and dated back to the year 2010 (95% HPD: 2009-2011); these sequences
showed an alanine in the aminoacid position 226 of the E1 protein. Phylogeographic analysis of
DENV-2 virus showed that cluster 1, which included Yemen isolates, dated back to 2003 Burkina
Faso strains (95%HPD 1999-2007). The Yemen, cluster dated back to 2011 (95% HPD 2009-2012).
Our study sheds light on the global spatiotemporal dynamics of DENV-2 and CHIKV in Yemen.
This study reinforces both the need to monitor the spread of CHIKV and DENV, and to apply
significant measures for vector control.
Keywords: DENV, CHIKV, phylogeny
1. Introduction

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
3
Arbovirus related infections are not uncommon in the Arabian peninsula as well as in other
countries in the Middle East and North Africa (Hotez et al., 2012). Dengue (DEN) outbreaks have
been reported in Djibouti and in the Arabian peninsula since 1990 (Ooi and Gubler, 2011), and the
Dengue virus (DENV) is now endemic in western and southern regions of Saudi Arabia (Ahmed,
2010).
Yemen, which is located in the southwestern end of the Arabian Peninsula, is one of countries most
affected by recurrent epidemics caused by emerging vector borne viruses. Outbreaks of DEN fever
have been reported with increasing frequency since the year 2000 in several governorates, and
Chikungunya virus (CHIKV) has been also responsible of large outbreaks (EpiSouth, 2011) and it is
now a major public health problem in Yemen (Zayed et al. 2012).
There is some evidence that several DENV serotypes circulated in Yemen over the last decades.
DENV-3 has been the predominant serotype for a long time. In fact, DENV-3 caused a major
outbreak in the western governorate of al-Hudayda in 2005 (WHO, 2009). The same serotype was
detected in travelers from Yemen (Ravanini et al., 2010) and during an outbreak in the southern
district of Hadramouuth (Ghouth et al., 2012). However, the identification of hemorrhagic
manifestations in patients infected with the DENV-3 genotype in Al Mukalla, the capital city of
Hadramouth governorate, suggested previous circulation of other DENV genotypes (Madani et al.,
2013). Moreover, high antibody titers against all four DENV serotypes, consistent with a secondary
heterotypic infection, in a Yemenite traveler affected by hemorrhagic DEN in 1983, may also
suggest that multiple introductions with further circulation of different DENV serotypes occurred
even in past decades (Jimenez-Lucho et al., 1984).
A study conducted in the governorate of Al-Hodayda in 2011 which involved 47 patients with
Dengue-like illness, detected DENV-1 in two cases and DENV-3 in one case, while no DENV-2
positive samples were identified (Rezza et al., 2014).

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
4
During a recent outbreak of DENV-like illness in the same region in 2012, we found co-circulation
of several DENV and CHIKV genotypes (Rezza et al., 2014). DENV-2 was largely predominant
(41 cases out of 55 tested DENV RNA positive), but DENV-1 was also found in 2 cases. Thus,
whether DENV 2 serotype was newly introduced in Yemen in 2012 remained undefined.
Hereby we report the results of the phylogenetic analysis of DENV-2 and CHIKV isolates detected
in the al-Hodayda outbreak. Estimates of the introduction date of CHIKV and DENV-2, and the
phylogeographic analysis of DENV-2 is also presented.
2. Materials and methods
2.1 Patients, serum samples and genomic RNA
Four-hundred patients were consecutive enrolled by five hospital centers (Renal Center,
Maritime College, Al Rasheed, AI-Thawra, Al Salakhana) located in AI-Hodayda, Yemen
between January 2011 and June 2012. The case definition was fever (>37.5°C) and at least two
of the following symptoms: headache, joint pain, muscle pain, skin rash. Serum samples were
collected within four days from the date of hospital admission, and stored and shipped at -
20°C. Of the study participants, fifty-five were PCR positive for DENV RNA of which twelve
were untyped, forty-one were positive for DENV 2 and two were positive for DENV 1. Eleven
sera were PCR positive for CHIKV RNA. The study has been approved by local ethical
committee.
2.2 Analysis of nucleic acids
Samples tested positive with high viral RNA titer were chosen for sequencing analysis. Nine sera
which tested positive for DENV serotype 2 and eight sera which tested positive for CHIKV
underwent amplification and sequencing reactions for NS1 and E1 genes, respectively. The

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
5
amplification of DENV NS1 gene serotype 2, was performed using the oligonucleotide 5'-
GATAGTGGTTGCGTTGTGAGC-3' (forward) and 5'-AGCTGTGACCAAGGAGTTGAC-3'
(reverse). The CHIKV E1 gene, was amplified using the oligonucleotides 5'-
AGCGAAGCACATGTGGAGAAG-3' (forward) and 5-TTAGTGCCTGCTGAACGACAC-3
(reverse) . All the amplifications were performed by using the Superscript III One step RT-PCR kit
(Invitrogen, CA). The reaction conditions were 50°C x 30 minutes, 95°C x 2 minutes followed by
10 cycles of 95°C x 10 seconds, 54°C x 30 (T - 0.5 °C each cycle) seconds, 68°C x 1 minute, then
25 cycles of 95°C x 10 seconds, 54°C x 30 seconds, 68°C x 1 minute. The PCR products were
visualized on 1% agarose gel and purified by spin column (Macherey Nagel, Germany). Sequencing
reactions were carried out on the Ceq 8000 automated DNA sequencer (CEQ 8000, Beckman
Coulter), according to manufacturer‟s instructions. The obtained sequences for CHIKV and Dengue
viruses have been deposited in GenBank under the following accession numbers: from KJ742803 to
KJ742819.
2.3 Viruses dataset
Two dataset for DENV were built. The first one included 9 Dengue virus NS1 gene isolates plus 49
reference sequences downloaded from GenBank (http://www.ncbi.nlm.nih.gov/), which was used to
perform the lineage and subtype assignment. The second dataset included 9 Dengue 2 virus NS1
gene isolates previously classified as subtype Cosmopolitan plus 72 Dengue 2 virus NS1 gene
Cosmopolitan subtype sequences downloaded from NCBI (http://www.ncbi.nlm.nih.gov/). The
following inclusion criteria are used for sequences: (1) sequences already published in peer-
reviewed journals (2) no uncertainty about the subtype assignment; (3) known sampling date and
location. This dataset was built to estimate of the Dengue 2 virus Cosmopolitan subtype NS1 gene
mean evolutionary rate and to perform the phylogeography.

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
6
Two different datasets for CHIKV were built. The first one included 8 CHIKV partial E1 gene
isolates from Yemen and 111 reference sequences downloaded from GenBank
(http://www.ncbi.nlm.nih.gov/genbank/) and was used to assign the genotype.
The second dataset included 126 CHIKV partial E1 sequences and was used to co-estimate the
evolutionary rate and time-scaled phylogeny. Collection dates of the sequences ranged from 1953 to
2012. Starting from the Bayesian tree of the second dataset, a clade including 70 sequences (8
isolates from Yemen, 6 from Buthan, 18 from India, 1 from France that was a case imported from
India, 3 from Singapore, 7 from Sri Lanka; 1 from Japan that was a Japanese returning from Sri
Lanka, 2 from Italy, 1 from Malaysia, 1 from Bangladesh, 4 from Thailand, 8 from Cambodia, 2
from Kenya, 2 from Comoros, 1 from Seychelles, 1 from Mauritius, 1 from Reunion, 3 from
China), was highlighted and used to study both the phylogenetic relationships between Yemen and
other countries and to date the epidemic. Moreover, the presence of the mutation A226V was
investigated.
The sequences were chosen on the basis of the same criteria described above for DENV.
The sequences of all the datasets were aligned using Clustal X (Ciccozzi et al., 2013) and manually
edited by Bioedit software v. 7.2.5. Modeltest 3.7 was used (Ciccozzi et al., 2013) for all the
datasets to select the simplest evolutionary model that adequately fitted the sequence data.
2.4 Likelihood mapping
The phylogenetic signal of each dataset was investigated by means of the likelihood mapping
analysis of 10,000 random quartets generated using TreePuzzle as already described (Zehender et
al., 2011; Ciccozzi et al, 2013). Groups of four randomly chosen sequences (quartets) were
evaluated. For each quartet the three possible unrooted trees were reconstructed using the maximum
likelihood approach under the selected substitution model. The posterior probabilities of each tree
were then plotted on a triangular surface so that fully resolved trees fall into the corners and the

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
7
unresolved quartets in the centre of the triangle (a star-tree). When using this strategy, if more than
30% of the dots fall into the centre of the triangle, the data are considered unreliable for the
purposes of phylogenetic inference.
2.5 Phylogenetic analysis
Maximum likelihood (ML) phylogenetic trees were estimated, on the first dataset for both Dengue
and CHIKV viruses, using the best fitting substitution model selected with ModelTest (Ciccozzi et
al., 2013), (TrN + I + G for Dengue virus and HKY + I+ G for CHIKV virus) with Phyml 3.0
(http://www.atgc-montpellier.fr/phyml/). The statistical robustness and reliability of the branching
order within the phylogenetic tree was confirmed with the bootstrap analysis (bootstrap value
≥70%).
2.6 Evolutionary rate estimate
The evolutionary rate was estimated by using a Bayesian MCMC approach (Beast v. 1.7.4,
http://beast.bio.ed.ac.uk) (Drummond et al., 2005; Drummond and Rambaut, 2007) implementing
the model selected with ModelTest using both a strict and an uncorrelated log-normal relaxed clock
model. The data set used were the second ones for both Dengue and Chikungunya viruses.
As coalescent priors, three parametric demographic models of population growth (constant size,
exponential, expansion growth) and a Bayesian skyline plot (BSP, a non-parametric piecewise-
constant model) were compared. The best fitting models were selected using a Bayes Factor (BF
with marginal likelihoods). In accordance with Kass and Raftery (Kass and Raftery, 1995), the
strength of the evidence against H0 was evaluated as follows: 2 ln BF < 2, no evidence; 2–6, weak
evidence; 6–10, strong evidence; > 10, very strong evidence. A negative value indicates evidence in
favor of H0. Only values of ≥ 6 were considered significant.

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
8
Chains were conducted for at least 50 x 106
generations, and sampled every 5000 steps and for 150
106
generations, sampling every 15,000th generation for DENV and CHIKV respectively.
Convergence of the MCMC was assessed by calculating the ESS for each parameter. Only
parameter estimates with ESS‟s of > 250 were accepted. Maximum clade credibility trees were
obtained from the trees posterior distributions with the Tree-Annotator software v 1.7.4, included in
the Beast package (Drummond et al., 2005; Drummond and Rambaut, 2007). Statistical support for
specific monophyletic clades was assessed by calculating the posterior probability.
2.7 Phylogeographic analysis
The continuous-time Markov Chain (MCC) process over discrete sampling locations implemented
in BEAST (Drummond and Rambaut, 2007) was used for the geographical analysis, implementing
the Bayesian Stochastic Search Variable Selection (BSSVS) model which allows the diffusion rates
to be zero with a positive prior probability. Comparison of the posterior and prior probabilities of
the individual rates being zero provided a formal BF for testing the significance of the linkage
between locations. This analysis was performed only for the Dengue 2 virus NS1 region (second
dataset).
The sampling locations were, Yemen, Australia, Brunei Darussalam, Burkina Faso, China, East
Timor, Guam, Indonesia, India, Pakistan, Singapore, Sri Lanka, and Vietnam.
The final trees were manipulated in FigTree v. 1.4 for display. The most probable location of each
node was highlighted by labeling the branches with different colors.
3. Results
3.1 Likelihood mapping analysis

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
9
The phylogenetic noise of each data set was investigated by means of likelihood mapping. For the
Dengue virus polyprotein the percentage of dots falling in the central area of the triangles was 1.2%
and 2.0% for the first and second dataset, respectively (Fig. S1); since none of the dataset showed
more that 30% of noise, all of them contained sufficient phylogenetic signal. For the chikungunya
virus in each data set the percentage of dots falling in the central area of the triangles was 14.8 %
for the first dataset and 18.3 % for the second dataset (Fig. S2). As none of the datasets showed
more than 30% of noise, all of them showed a sufficient phylogenetic signal.
3.2 Phylogenetic analysis
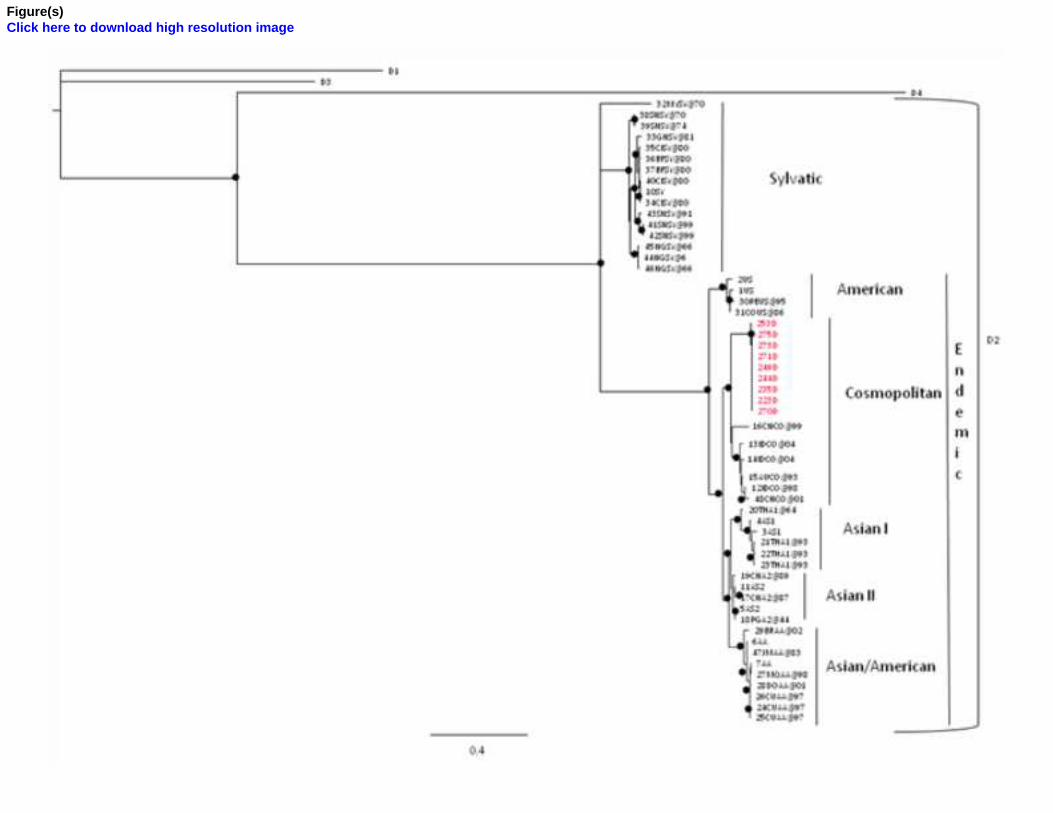
The ML tree for DENV, showed distinct clusters of the different lineages and subtypes. The Yemen
isolates belonged to the lineage 2 Cosmopolitan subtype (Fig. 1). All the isolates clustered in the
same statistically supported clade, indicating a unique epidemic. Maximum likelihood (ML)
phylogenetic analysis for Chikungunya virus (Fig. 2) showed three distinct genotypes: West Africa,
Asia and East-central– South – African (ECSA) genotype. All the eight sequences from Yemen
belonged to the ECSA genotype and clustered inside the clade which include also the viruses
isolated during the Indian Ocean outbreak.
3.3 Evolutionary rate estimate
Bayes Factor analysis for Dengue virus showed that the relaxed clock fitted the data significantly
better than the strict clock (2lnBF between the strict and relaxed clock was 15 in favor of the
second). Under the relaxed clock, the BF analysis showed that the BSP was better than the other
models (2lnBF >24). The estimated mean value of DENV-2 Cosmopolitan subtype NS1 gene
evolutionary rate was 8.23 x 10-4
sub/site/year (95%HPD 6.62 x10-4
-1.01 x10-3
).

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
10
Bayes Factor analysis for CHIKV, showed that the relaxed clock fitted the data significantly better
than the strict clock (2lnBF between the strict and relaxed clock was 90 in favor of the second).
Under the relaxed clock, the BF analysis showed that the BSP was better than the other models
(2lnBF > 100). The estimated mean value of the CHIKV E1 evolutionary rate, was 1.037 x 10-3
substitution/site/year (95% highest posterior density interval HPD: 5.15 x 10-4
- 1.716 x 10-3
).
Fig. 3 highlighted the Bayesian maximum clade credibility tree of the 70 CHIKV E1 sequences
which included the eight isolates from Yemen. Clade I was statistically supported and included five
sequences from India, the eight isolates from Yemen, a sequence from France that was a case
imported from India (from Rajasthan - Acc. Number FR846304) and six sequences from Buthan.
All the sequences from Yemen were included in a statistically supported cluster and dated back to
the year 2010 (95% HPD: 2009-2011).
3.4 Phylogeographic analysis of DENV-2 virus
Fig. 4 shows the time and location-annotated Bayesian maximum clade credibility tree of the
DENV-2 Cosmopolitan subtype NS1 gene sequences. The most probable locations of the internal
nodes are indicated by different colors, and the time to most recent common ancestor (tMRCA)
estimates under a relaxed molecular clock model are highlighted. The phylogeographic analysis
showed that the root of the tree dated back 1964 (HPD 95% 1939-1984) and highlighted three
distinct clades (A, B and C). Clade A originated in Indonesia and included sequences from
Australia, Brunei Darussalam, China, East Timor, Guam, Indonesia, Singapore, Vietnam. Clade B
originated in India and included sequences from China, India, Pakistan and Sri Lanka. Clade C
originated in Indonesia and included sequences from Burkina Faso, Indonesia and Yemen. All
these Clade has been statistically supported by a state probability ≥ 0.35.
Phylogeographic reconstruction indicated the most probable location of cluster 1 within Clade C,
which included the Yemen isolates and Burkina Faso sequences, probably was Burkina Faso and

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
11
dated back to 2003 (95%HPD 1999-2007). Moreover the Yemen cluster alone dated back to 2011
(95% HPD 2009-2012).
3.5 CHIKV A226V mutation analysis
The presence of the mutation A226V in the 70 CHIKV E1 sequences (which also included the eight
isolates from Yemen) reported in Figure 3 was investigated. The valine in the aminoacid position
226 of the E1 sequences was identified in the cluster which included the sequences from Reunion
and Mauritius and in the clade which included the sequences from India (isolated in 2007), Italy
(collected in 2007), Sri Lanka (collected in 2008), Malaysia (collected in 2008), Bangladesh
(collected in 2008), Singapore (collected in 2008), Thailand (collected in 2008- 2009), China
(collected in 2010), Cambodia (collected in 2011). All the other sequences did not showed the
mutation A226V. Thence, the CHIKV strains which circulated in Yemen showed an alanine in the
aminoacid position 226 of the E1 protein.
4. Discussion
We recently described a simultaneous outbreak of DENV and CHIKV in Al-Hodayda, a port city
located in Western Yemen, in 2012. Although there was some evidence that DENV-1 serotype, but
not DENV-2, was already circulating in 2011, we found that DENV-2 largely predominated in 2012
(Rezza et al., 2014). This finding suggested that the DENV-2 serotype could have been newly
introduced in the area of Al-Hodayda, causing to an explosive outbreak. Actually, our evolutionary
analysis confirmed that DENV-2 was introduced late in 2011, which is consistent with molecular
epidemiology data. With regard to CHIKV, we estimated that its introduction dates back to 2010,
which is rather consistent with the reports of a massive epidemic that occurred in the area of Al-
Hodayda in 2011 (EpiSouth, 2011). In particular, it should be mentioned that, in the same year,

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
12
CHIKV was detected by PCR in pools of Aedes aegypti mosquitoes collected in the governorate of
Al-Hodayda (Zayed et al., 2012).
These findings suggest that vector-borne viruses, such as DENV and CHIKV, are continuously
reintroduced in Yemen. Specifically, the site of our study, Al Hodayda, is a port city located along
the Red Sea coast, in the so-called Tihamah (“hot lands” or “hot earth”). Red sea ports, with
movement of refugees and intense human activities, are the entry gates for infected humans and
vectors, representing also a favorable habitat for mosquitoes. Furthermore, water scarcity and lack
of infrastructure in peri-urban areas require storage of water for household, and water containers
also favor the breeding habitat of mosquitoes (Rezza et al., 2014). To this regard, a case-control
study conducted in Jeddah in 2004 identified a series of local and individual factors, such as the
presence of stagnant water in indoor drainage holes, indoor larvae near construction sites, and older
age, associated with mosquito-borne infections. On the other hand, face-to-face health education
significantly decreased the risk of infection (Kholedi et al., 2012).
Our phylogeographic analyses, based on DENV-2 Cosmopolitan subtype NS1 gene sequences
sampled from different countries worldwide, suggested that Burkina Faso acted as viral source
populations for the Yemen epidemic strain. This could be explained by the importation of travel-
associated Dengue cases from Africa (Jimenez-Lucho et al., 1984; Ravanini et al., 2010; Nicoletti et
al., 2008). Although the highest similarity was found with Burkina Faso strains, an alternative
hypothesis is that Yemen strains originated from India or Indonesia. The interpretation of this
analysis needs caution, since it relies on a limited number of data included in the web and then
biased by the lack of information on many epidemics occurred in tropical ares of Africa and Asia.
With regard to CHIKV virus, Yemen strains were similar to ECSA strains causing the Indian Ocean
outbreak after 2004. The lack of the A226V mutation is consistent with the predominance of Aedes
aegypti as CHIKV virus vector in Yemen (Zayed et al., 2012).

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
13
In conclusion, the geographic spread of the DENV-2 virus strains and the dates of introduction (the
time-scaled phylogeny) of DENV-2 and CHIKV viruses in Yemen were estimated thorough a
sophisticated Bayesian statistical inference framework. Further understanding of the key source
areas of viral dispersal and the factors underlying the emergence and spread of fast evolving viruses
can help directing epidemiological surveillance efforts. This study reinforces the need to monitor
the spread of CHIKV and DENV viruses, and to reinforce vector control measures. This is a critical
area for future research that may ultimately contribute to improve public health measures against
vectorborne infections.
References
Ahmed MM., 2010. Clinical profile of Dengue fever infection in King Abdul Aziz University
Hospital Saudi Arabia. J Infect Dev. 4, 503-510.
Ciccozzi M., Callegaro A., lo Presti A., Cella E., Giovanetti M., Salpini R., Babakir-mina M.,
Farina C., Maggiolo F., Perno C.F., Ciotti M., 2013. When phylogenetic analysis complements the
epidemiological investigation: a case of HIV-2 infection, Italy. New Microbiol. 36(1), 93-6.
Drummond, A.J., Rambaut, A., Shapiro, B., Pybus, O.G., 2005. Bayesian coalescent inference of
past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
14
Drummond, A.J., Rambaut, A., 2007. BEAST: bayesian evolutionary analysis by sampling trees.
BMC Evol. Biol. 7, 214.
EpiSouth., 2011. EpiSouthWeekly Epi Bulletin 155 (http://www.episouth.org).
Ghouth ASB, Amarasingh, A, Letson W., 2012. Dengue outbreak in Hadramouth, Yemen, 2010: an
epidemiological perspective. Am J Trop Med Hyg. 86, 1072-1076.
Hotez PJ, Savioli L, Fenwick A., 2012. Neglected tropical diseases of the Middle East and North
Africa: review of their prevalence, distribution, and opportunities for control. PLOS neglected
Tropical Diseases. 6, e1475.
Jimenez-Lucho VE, Fisher EJ, Saravolatz LD., 1984. Dengue with hemorrhagic manifestations: an
imported case from the Middle East. Am J Trop Med Hyg. 33, 650-653.
Kholedi AA, Balubaid O, Milaat W, Kabbash IA, Ibrahim A., 2012. Factors associated with the
spread of Dengue fever in Jeddah Governorate, Saudi Arabia. East Mediterr Health. 18, 15-23.
Madani TA, El-Tayeb ME, Al-Bar HMS, Azhar EI, Kao M, Alshoeb HO, Bamoosa AR., 2013.
Outbreak of viral hemorrhagic fever caused by Dengue virus type 3 in Al-Mukalla, Yemen. BMC
Infect Dis. 13, 136.
Nicoletti L, Ciccozzi M, Marchi A, Fiorentini C, Martucci P, D'Ancona F, Ciofi degli Atti M,
Pompa MG, Rezza G, Ciufolini MG., 2008. Chikungunya and Dengue Viruses in Travelers. Emerg
Infect Dis. Jan. 14(1), 177–178.
Ooi EE, Gubler DJ., 2011. Dengue and Dengue hemorrhagic fever (Ch. 75). In: Guerrant RL,
Walker DH, eds. Tropical infectious diseases: principles, pathogens and practice. 3rd
edition
Saunders Elsevier.

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
15
Ravanini P, Huhtamo E, Hasu E, Rosa F, Costantino S, Crobu MG, Ilaria V, Nicosia AM, Garavelli
PL, Vapalahti O., 2010. Imported Dengue virus serotype 3, Yemen to Italy, 2010. Em Infect Dis.
17, 929-931.
Rezza G., El-Sawaf G., Faggioni G., Vescio F., Al Ameri R., De Santis R., Helaly G., Pomponi A.,
Metwally D., Fantini M., Qadi H., Ciccozzi M., Lista F., 2014. Co-circulation of Dengue and
Chikungunya Viruses, Al Hudaydah, Yemen, 2012. Emerg Infect Dis. 20(8), 1351-4.
World Health Organization., 2009. Dengue: guidelines for diagnosis, treatment, prevention and
control. New Ed. Geneva, Switzerland.
Zayed A, Awash AA, Esmal MA, Al-Mohamadi HA, Al-Salwai M, Al-Jasari A, Medhat
I, Morales-Betoulle ME, Mnzava A., 2012. Detection of Chikungunya virus in Aedes Aegypti
during the 2011 outbreak in Al Hodayda, Yemen. Acta Topica. 123, 62-66.
Zehender G, Ebranati E, Bernini F, Lo Presti A, Rezza G, Delogu M, Galli M, Ciccozzi M., 2011.
Phylogeography and epidemiological history of West Nile virus genotype 1a in Europe and the
Mediterranean basin. Infect Genet Evol. 11(3), 646-53.
Figure legends
Fig. 1. Maximum likelihood tree including 9 isolates of Dengue NS1 gene plus 49 reference
sequences. The isolate analyzed are in red. The tree was rooted by the midpoint rooting. The scale
bar at the bottom indicating 0.4 nucleotide substitutions per site. The • along the branches
represents significant statistical support for the clusters subtending that branch (p<0.001 in the zero-
branch-length test and bootstrap support ≥ 70%). Different lineages and subtypes are indicated by
bracket.
Fig. 2. Maximum likelihood phylogenetic analysis of 8 CHIKV partial E1 isolates from Yemen
and 111 reference sequences. The scale bar at the bottom indicates 0.02 nucleotide substitutions
per site. The • along the branch represents significant statistical support for the clade subtending
that branch (bootstrap ≥ 70%). The isolates from Yemen are indicated in bold.

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
16
Fig. 3. Bayesian phylogenetic tree of 70 CHIKV partial E1 sequences . One • along the branches
represents significant statistical support for the clade subtending that branch (posterior probabilities
≥ 98%). Years are reported on the scale axis below. Isolates from Yemen are colored in red. The
cluster including the sequences Yemen is zoomed and his date is reported in the node. The Valine
or the Alanine in the aminoacid position 226 of the E1 sequences was indicated.
Fig.4. Bayesian phylogeographic tree of Dengue 2 cosmopolitan NS1 region sequences. The
significant statistical support for the clade or cluster subtending the branch (posterior probability ≥
98%) is indicated with a black circle along that branch. Geographic locations showed with different
colors in the tree are represent in the legend on the left. In the right corner there is the zoom of the
Yemen clade. The time of the most recent common ancestor, and the credibility interval based on
95% highest posterior density interval (95% HPD) is also reported.
Fig. S1. Likelihood mapping of the first (A), second (B) Dengue NS1 dataset. The dots inside the
triangles represents the posterior probabilities of the possible unrooted topologies for each quartet.
Numbers indicate the percentage of dots in the centre of the triangle corresponding to phylogenetic
noise (star-like trees).
Fig. S2. Likelihood mapping of CHIKV partial E1 sequences of the first (a) and second (b)
datasets. The dots inside the triangles represent the posterior probabilities of the possible unrooted
topologies for each quartet. Numbers indicate the percentage of dots in the centre of the triangle
corresponding to phylogenetic noise (star-like trees).
Figure(s)Click here to download high resolution image

Figure(s)Click here to download high resolution image

Figure(s)Click here to download high resolution image

Figure(s)Click here to download high resolution image

Supplementary MaterialClick here to download Supplementary Material: Fig. S1.tif

Supplementary MaterialClick here to download Supplementary Material: Fig. S2.tif
Copyright © 2022 FDOKUMEN




















