
4-hydroxy-3-methoxy-acetophenone-mediated long-lasting memory recovery, hippocampal neuroprotection,...
10
4-Hydroxy-3-Methoxy-Acetophenone- Mediated Long-Lasting Memory Recovery, Hippocampal Neuroprotection, and Reduction of Glial Cell Activation After Transient Global Cerebral Ischemia in Rats C assia Val erio Romanini, Emilene Dias Fiuza Ferreira, L ıgia Mendes Soares, Amanda Nunes Santiago, Humberto Milani, and R ubia Maria Weffort de Oliveira* Department of Pharmacology and Therapeutics, State University of Maring a, Maring a, Paran a, Brazil 4-Hydroxy-3-methoxy-acetophenone (apocynin) is a nat- urally occurring methoxy-substitute catechol that is iso- lated from the roots of Apocynin cannabinum (Canadian hemp) and Picrorhiza kurroa (Scrophulariaceae). It has been previously shown to have antioxidant and neuropro- tective properties in several models of neurodegenerative disease, including cerebral ischemia. The present study investigates the effects of apocynin on transient global cerebral ischemia (TGCI)-induced retrograde memory deficits in rats. The protective effects of apocynin on neu- rodegeneration and the glial response to TGCI are also evaluated. Rats received a single intraperitoneal injection of apocynin (5 mg/kg) 30 min before TGCI and were tested 7, 14, and 21 days later in the eight-arm aversive radial maze (AvRM). After behavioral testing, the hippo- campi were removed for histological evaluation. The present results confirm that TGCI causes memory impair- ment in the AvRM and that apocynin prevents these memory deficits and attenuates hippocampal neuronal death in a sustained way. Apocynin also decreases OX- 42 and glial fibrillary acidic protein immunoreactivity induced by TGCI. These findings support the potential role of apocynin in preventing neurodegeneration and cognitive impairments following TGCI in rats. The long- term protective effects of apocynin may involve inhibition of the glial response. V C 2015 Wiley Periodicals, Inc. Key words: transient global cerebral ischemia; apocynin; memory recovery; neuroprotection; rats Transient global cerebral ischemia (TGCI) is an immediate and serious outcome observed most frequently after reversible cardiac arrest, dysrhythmias, or severe hypotensive shock (Garcia, 1992). Patients who survive long after cardiac arrest may present a broad range of neu- rologic and cognitive deficits, loss of memory (amnesia) being one of the most salient, enduring, and disabling sequelae, with consequent vocational impairments and a decrease in quality of life (Moulaert et al., 2009, 2010; Peskine et al., 2010). Neurodegeneration in the hippo- campus has commonly been associated with memory dysfunction after TGCI (Zola-Morgan et al., 1986; Bach- evalier and Meunier, 1996), although damage to other brain areas, including the retrosplenial cortex and the dorsomedial thalamus, may also be critically involved (Horstmann et al., 2010). Currently, there is no effective drug treatment for preventing neuronal injury and related functional deficits caused by cerebral ischemia. 4-Hydroxy-3-methoxy-acetophenone (apocynin) is a naturally occurring methoxy-substitute catechol isolated from the roots of Apocynin cannabinum (Canadian hemp) and Picrorhiza kurroa (Scrophulariaceae; a plant used in Ayurvedic medicine for its antioxidant and anti- inflammatory properties; Sun et al., 2008). Experimental reports indicate that apocynin has therapeutic potential against several neurodegenerative processes, including Alzheimer’s disease (Lull et al., 2011), Parkinson’s disease (Philippens et al., 2013), and brain trauma (Ferreira et al., 2013). With regard to cerebral ischemic processes, many studies have evaluated the effects of apocynin on the his- tological and biochemical changes in animal models of focal cerebral ischemia (for review see Carbone et al., 2014), but only a few have assessed neurological/behav- ioral end points within a postischemic period of no longer than 4 days. For example, apocynin administration before ischemia was shown to improve neurological scores and Contract grant sponsor: Conselho Nacional de Desenvolvimento Cient ıfico e Tecnol ogico; Contract grant sponsor: Universidade Estadual de Maring a; Contract grant sponsor: Fundac¸ ~ ao Arauc aria *Correspondence to: R ubia Maria Weffort de Oliveira, Department of Pharmacology and Therapeutics, State University of Maring a, Maring a, Paran a, Brazil. E-mail: [email protected] Received 30 April 2014; Revised 31 December 2014; Accepted 22 January 2015 Published online 00 Month 2015 in Wiley Online Library (wileyonlinelibrary.com). DOI: 10.1002/jnr.23575 V C 2015 Wiley Periodicals, Inc. Journal of Neuroscience Research 00:00–00 (2015)
-
Upload
independent -
Category
Documents
-
view
6 -
download
0
Transcript of 4-hydroxy-3-methoxy-acetophenone-mediated long-lasting memory recovery, hippocampal neuroprotection,...

4-Hydroxy-3-Methoxy-Acetophenone-Mediated Long-Lasting Memory Recovery,Hippocampal Neuroprotection, andReduction of Glial Cell Activation AfterTransient Global Cerebral Ischemia in Rats
C�assia Val�erio Romanini, Emilene Dias Fiuza Ferreira, L�ıgia Mendes Soares,Amanda Nunes Santiago, Humberto Milani, andR�ubia Maria Weffort de Oliveira*
Department of Pharmacology and Therapeutics, State University of Maring�a, Maring�a, Paran�a, Brazil
4-Hydroxy-3-methoxy-acetophenone (apocynin) is a nat-urally occurring methoxy-substitute catechol that is iso-lated from the roots of Apocynin cannabinum (Canadianhemp) and Picrorhiza kurroa (Scrophulariaceae). It hasbeen previously shown to have antioxidant and neuropro-tective properties in several models of neurodegenerativedisease, including cerebral ischemia. The present studyinvestigates the effects of apocynin on transient globalcerebral ischemia (TGCI)-induced retrograde memorydeficits in rats. The protective effects of apocynin on neu-rodegeneration and the glial response to TGCI are alsoevaluated. Rats received a single intraperitoneal injectionof apocynin (5 mg/kg) 30 min before TGCI and weretested 7, 14, and 21 days later in the eight-arm aversiveradial maze (AvRM). After behavioral testing, the hippo-campi were removed for histological evaluation. Thepresent results confirm that TGCI causes memory impair-ment in the AvRM and that apocynin prevents thesememory deficits and attenuates hippocampal neuronaldeath in a sustained way. Apocynin also decreases OX-42 and glial fibrillary acidic protein immunoreactivityinduced by TGCI. These findings support the potentialrole of apocynin in preventing neurodegeneration andcognitive impairments following TGCI in rats. The long-term protective effects of apocynin may involve inhibitionof the glial response. VC 2015 Wiley Periodicals, Inc.
Key words: transient global cerebral ischemia; apocynin;memory recovery; neuroprotection; rats
Transient global cerebral ischemia (TGCI) is animmediate and serious outcome observed most frequentlyafter reversible cardiac arrest, dysrhythmias, or severehypotensive shock (Garcia, 1992). Patients who survivelong after cardiac arrest may present a broad range of neu-rologic and cognitive deficits, loss of memory (amnesia)being one of the most salient, enduring, and disablingsequelae, with consequent vocational impairments and adecrease in quality of life (Moulaert et al., 2009, 2010;
Peskine et al., 2010). Neurodegeneration in the hippo-campus has commonly been associated with memorydysfunction after TGCI (Zola-Morgan et al., 1986; Bach-evalier and Meunier, 1996), although damage to otherbrain areas, including the retrosplenial cortex and thedorsomedial thalamus, may also be critically involved(Horstmann et al., 2010). Currently, there is no effectivedrug treatment for preventing neuronal injury and relatedfunctional deficits caused by cerebral ischemia.
4-Hydroxy-3-methoxy-acetophenone (apocynin) isa naturally occurring methoxy-substitute catechol isolatedfrom the roots of Apocynin cannabinum (Canadian hemp)and Picrorhiza kurroa (Scrophulariaceae; a plant used inAyurvedic medicine for its antioxidant and anti-inflammatory properties; Sun et al., 2008). Experimentalreports indicate that apocynin has therapeutic potentialagainst several neurodegenerative processes, includingAlzheimer’s disease (Lull et al., 2011), Parkinson’s disease(Philippens et al., 2013), and brain trauma (Ferreira et al.,2013). With regard to cerebral ischemic processes, manystudies have evaluated the effects of apocynin on the his-tological and biochemical changes in animal models offocal cerebral ischemia (for review see Carbone et al.,2014), but only a few have assessed neurological/behav-ioral end points within a postischemic period of no longerthan 4 days. For example, apocynin administration beforeischemia was shown to improve neurological scores and
Contract grant sponsor: Conselho Nacional de Desenvolvimento
Cient�ıfico e Tecnol�ogico; Contract grant sponsor: Universidade Estadual
de Maring�a; Contract grant sponsor: Fundac~ao Arauc�aria
*Correspondence to: R�ubia Maria Weffort de Oliveira, Department of
Pharmacology and Therapeutics, State University of Maring�a, Maring�a,Paran�a, Brazil. E-mail: [email protected]
Received 30 April 2014; Revised 31 December 2014; Accepted 22
January 2015
Published online 00 Month 2015 in Wiley Online Library
(wileyonlinelibrary.com). DOI: 10.1002/jnr.23575
VC 2015 Wiley Periodicals, Inc.
Journal of Neuroscience Research 00:00–00 (2015)

reduce infarct volume measured 24 hr after middle cere-bral artery occlusion (MCAO) in rats (Zhao et al., 2010;Genovese et al., 2011) and mice (Tang et al., 2008). Evenless research has been carried out with animal models ofTGCI. Wang et al. (2006) used a postischemic period of4 days and demonstrated the antioxidant and neuropro-tective effects of apocynin administered either 30 minbefore or 5 min after TGCI in gerbils. To date, only onestudy has shown that apocynin, besides increasing neuronsurvival rates and improving motor coordination, amelio-rates spatial learning and memory in mice tested in theMorris water maze from 3 to 7 days after occlusion of thecommon carotid arteries (Shen et al., 2011). These datasuggest that apocynin might have therapeutic potential fortreating the outcome of cerebral ischemia. Indeed, theputative efficacy of drugs in providing functional recoveryafter brain ischemia should be confirmed in the preclinicalsetting only after testing in different behavioral paradigmsand/or animal models of cerebral ischemia, together withthe assessment of long-term functional end points (Cor-bertt and Nurse, 1998; Freret et al., 2011). To the best ofour knowledge, this aspect of preclinical investigation islacking for apocynin. Moreover, neither the rat four-vessel occlusion (4-VO) model of TGCI nor the radialmaze model of learning and memory has been used toevaluate the efficacy of apocynin as a potential neuropro-tective agent.
Therefore, the present study evaluates whetherapocynin prevents neuronal damage and provides long-lasting memory recovery in rats subjected to the 4-VOmodel of TGCI and tests a novel version of the eight-armradial maze task. Because microglial cells and astrocytesplay a fundamental role in TGCI-induced neuronal loss(Takuma et al., 2004; Duan et al., 2011; Yasuda et al.,2011), we also examined the effects of apocynin on glialresponse by determining OX-42 and glial fibrillary acidicprotein (GFAP) hippocampal immunoreactivities.
MATERIALS AND METHODS
Animals
Sixty-six adult, male Wistar rats (inbred strain; 270–300 g, 3 months old) were acquired from the vivarium of theState University of Maring�a. Prior to any experimental manipu-lation, the rats were acclimated for 1–2 weeks in the local vivar-ium and maintained under a controlled temperature(22�C 6 1�C) on a 12-hr light/dark cycle (lights on at 7:00AM). Animals received food and water ad libitum throughoutthe study period. All experimental procedures adhered to theethical principles of the Brazilian College of Animal Experi-mentation and were approved by the Ethics Committee onAnimal Experimentation of the State University of Maring�a(CEAE No. 069/2013). All efforts were made to minimize ani-mal suffering.
Treatment
Apocynin (5 mg/kg; Sigma, St. Louis, MO) or vehicle(0.9% saline) was administered i.p. 30 min before the occlusionof both common carotid arteries. The dose used was from pre-
vious work that showed the neuroprotective effects of apocyninin gerbils (Wang et al., 2006) and rats (Ferreira et al., 2013).
Surgery
TGCI was performed as described elsewhere (Pulsinelliand Brierley, 1979), with modifications. The animals wereanesthetized with a halothane/oxygen mixture (Tanohalo;Crist�alia, Itapira, S~ao Paulo, Brazil) delivered through a univer-sal vaporizer that was regulated by minimal burble flow (0.5 lit-ers/min) to maintain anesthesia, which was evaluated bypinching the animal’s tail. The head of the animal was fixed ina stereotaxic frame for bilateral electrocoagulation of the verte-bral arteries at the level of the first cervical vertebra. An incisionwas then made on the ventral neck to expose the commoncarotid arteries, which were loosely tied with silk thread. Fourhours later, the silk thread was carefully tightened for 15 min inconscious, spontaneously ventilating animals, thus completingthe surgical procedure. Throughout occlusion and during thefirst hour of reperfusion, rats were maintained in a warmingbox (inner temperature 30�C 6 1�C) to avoid ischemia-induced brain hypothermia. During surgery, rectal temperaturewas maintained at 37�C 6 0.5�C with a heating pad and moni-tored with a digital thermometer (APPA MT-520; Minipa, S~aoPaulo, Brazil) coupled to a rectal probe (Minipa Electronics,Houston, TX). Loss of the righting reflex within 2 min ofcarotid artery occlusion, unresponsiveness to gentle touch,mydriasis, and tonic extension of the paws were consideredindicative of effective ischemia. Animals that did not lose therighting reflex or recovered it before the 15-min time pointwere excluded from the study. Sham-operated animals weresubjected to the same surgical intervention with the exceptionthat both the vertebral and the carotid arteries were notoccluded.
Behavioral Testing
Aversive radial maze. Inspired by the circular plat-form task (Barnes, 1988), the aversive radial maze (AvRM) is anonfood-rewarded, eight-arm radial maze that works on thebasis of the rat’s natural behavior of avoiding open, wide, andilluminated areas and its preference for darkened and enclosedplaces, i.e., a goal box located just beneath the opening at thedistal end of a given arm of the maze. AvRM task originaldevelopment details and conceptualization have been describedelsewhere (Paganelli et al., 2004) and subsequently have beenimproved for the task’s allocentric spatial characteristics (Bene-toli et al., 2007). The AvRM consists of a central, polygonalplatform (71 cm across, 16 sides) elevated 90 cm above the floorwith eight arms (55 3 15 cm) that radiate outward from alter-nate sides. At the end of each arm is a 9-cm diameter openingthat provides access to a goal box. Among the eight arms, how-ever, only one contains the true goal box (i.e., a darkened,closed-ended box, 23 3 11 3 9.5 cm), which can be movedfrom one arm to the other between trials. In the remainingseven arms, the boxes are open ended; that is, they have wallslike the true goal box but lack the bottom (i.e., a false goal box).
Training and retention trials. Rats were eval-uated in the AvRM task to assess their ability to remember thespatial location of the goal box (refuge) that was learned during
2 Romanini et al.
Journal of Neuroscience Research

the training phase, prior to TGCI or sham operation. Trainingwas given for 15 days (one session per day, three trials per ses-sion), the details of which have been reported elsewhere (Fer-nandes et al., 2008; Correia et al., 2013). After the animals hadlearned the task up to the asymptotic level of performance, theywere subjected to TGCI or sham surgery and allowed torecover from surgical stress for 7 days. Rats were subjected toretention memory trials (RMTs) 7, 14, and 21 days afterTGCI. Retrograde memory was measured by three parameters:1) the latency to find the goal box, 2) the number of referencememory errors, and 3) the number of working memory errors.A “reference memory error” was counted when the rat visitedan arm that contained a false goal box for the first time. A“working memory error” was counted when the animalreturned to an arm that had been previously visited during thattrial. An arm was considered visited when the rat had enteredhalfway down its length. The animal was considered to haveleft an arm when it had placed all four paws on the centralplatform.
Histology
Animals’ brains were removed and assayed for the histo-logical assessment of neurodegeneration by Nissl staining at 24hr, 72 hr, and 21 days after the surgery. A post hoc histologicalgroup was generated at 7 days for the immunohistochemicalassays. Coronal brain sections were obtained at a stereotaxiclevel 23.60 to 24.30 mm posterior to bregma (Franklin andPaxinos, 1997). The histological analysis was performed blindto the treatment groups.
Nissl Staining
The rats were deeply anesthetized with 50 mg/kgsodium thiopental (Thiopentax; Crist�alia) and transcardiallyperfused with 0.9% saline, followed by Bouin’s fixative. Afterdecapitation, the head was immersed in crushed ice (1–2�C)for 2 hr to avoid the appearance of dark neurons, which couldconfound the actual extent of neurodegeneration. The brainwas carefully removed and postfixed in Bouin’s solution for3 days. With a rotating microtome (RM2445; Leica, Goettin-gen, Germany), 7-mm paraffin-embedded coronal sectionswere cut and distributed into four sets of slides that containedfour coronal sections each, 70 mm apart. After standard dehy-dration and diaphanization procedures, slides that containedadjacent sections were immersed in distilled water and sub-merged in 0.2% cresyl violet solution (Nissl staining) for5 min. The slides were then rinsed in distilled water, dehy-drated in a graded series of ethanol (70%, 80%, 90%, and100%), cleared in xylene, and coverslipped with xylene byusing Permount (Fisher Scientific, S~ao Paulo, Brazil).
Three adjacent coronal sections were selected for bilateralcounts of normal-looking pyramidal neurons in the hippocam-pus. The number of cells that presented a well-delimited, spher-ical form with distinct nucleus and nucleolus was obtained witha BX-41 microscope (3400; Olympus, Tokyo, Japan). Intact-appearing neurons were counted in the CA1, CA2, CA3, andCA4 pyramidal layers under an identical microscopic lightintensity. Neurons that had shrunken cell bodies or surroundingempty spaces were considered destined to have died and
excluded from the analysis. For each individual, the valuesobtained in the three coronal sections were averaged and usedto represent the data (mean 6 SEM) for each group.
Immunohistochemistry
Immunohistochemistry for OX-42 and GFAP was con-ducted 7 days after TGCI to assess the expression of micro-glial cells and astrocytes, respectively. Animals were deeplyanesthetized and transcardially perfused with 0.1 Mphosphate-buffered saline (PBS), followed by 4% paraformal-dehyde in 0.2 M phosphate buffer. The brains were removed,postfixed in the same fixative for 2 hr, and cryoprotected byimmersion in 30% sucrose for 48 hr. Frozen tissue was seriallysectioned on a cryostat (Criocut 1800; Reichert-Jung, Heidel-berg, Germany) into 30-mm coronal sections that werecollected in sextuplicate in Eppendorf tubes containing anti-freeze solution.
Free-floating sections were quenched in 1% H2O2 for 30min and then blocked with 2% bovine serum albumin in 0.1 MPBS for 60 min. The sections were incubated overnight atroom temperature with rabbit polyclonal antibodies (1:500;Santa Cruz Biotechnology, Santa Cruz, CA) in PBS that con-tained 0.3% Triton X-100. Sections were then incubated withbiotinylated secondary antibodies (1:500; Santa Cruz Biotech-nology) for 2 hr and further incubated in ABC solution (Vec-tastain Elite ABC kit; Vector Laboratories, Burlingame, CA)for 2 hr. The peroxidase reaction was performed with 3,30-dia-minobenzidine (DAB; Sigma) and 0.05% H2O2. For the detec-tion of OX-42, NiCl2 was added to the DAB solution toincrease the staining contrast. The sections were mounted ongelatin-coated slides, air dried, cleared with xylene, and cover-slipped with Permount.
Because clusters of activated microglia and astrocytesaccumulated predominantly at the location of ischemic injury,color images of the CA1 hippocampal region from both brainhemispheres were captured with a 340 objective and a camera(QColor; Olympus) coupled to the microscope. The region ofinterest was selected, including a previously defined area locatedbetween the CA1 pyramidal cell layer and lacunosum molecularlay of the hippocampus (Franklin and Paxinos, 1997) in bothbrain hemispheres. The images were then converted to grayscale, and the background was subtracted. OX-42 and GFAPimmunoreactivity was evaluated by measuring the integratedoptical density (IOD) in ImageJ (NIH, Bethesda, MD) in threeadjacent coronal sections 150 mm apart. A mean value of themeasurements was obtained.
Statistical Analysis
The behavioral and histological data were examined forassumptions of normality and homocedasticity (between-groupand within-group homogeneity of variance). The behavioraldata did not satisfactorily fit either to a normal distribution or tohomocedasticity. The data were then further analyzed to searchfor a covariance model to which they could better fit. Follow-ing several information criteria, Akaike’s information criterion,Akaike’s information criterion corrected, Bayesian informationcriterion, and the likelihood ratio test, the model of nonstruc-tured covariance was used, and a two-way repeated-measures
Apocynin and Transient Global Cerebral Ischemia 3
Journal of Neuroscience Research

ANOVA was performed to quantify memory performanceacross the various days of testing or RMTs, with group as thebetween-subjects factor and trial (testing day) as the within-subjects factor. One-way ANOVA was used for between-group comparisons of memory performance, represented astotal latency and total number of errors. If a main effect of groupwas found, then Tukey-Kramer’s multiple-range test was usedto distinguish between them. Paired Student’s t-test was appliedfor within-group comparisons of memory performance meas-ured prior to and after ischemia. The histological data werenormalized to the mean values of the sham group. The datawere analyzed by using the generalized linear model with aPoisson distribution to model the count data (i.e., number ofcells and Nissl staining) and a g distribution for continuousmeasurements (i.e., IOD, OX-42, and GFAP). P< 0.05 wasconsidered statistically significant.
RESULTS
Mortality Rate
One hundred nine rats were initially included in theexperiment. Overall, 32 rats (29.4%) were lost during theexperiments because of 1) anesthesia (8/109, 7.30%), 2)accidental rupture of the carotid artery after tighteningthe occluder thread (5/101, 4.95%), 3) convulsion or pul-monary hemorrhage during or soon after occlusion (6/96,6.25%), and 4) death within the first hours to 7 days afterischemia (13/90, 14.40%).
Learning and Retrograde Memory in the AvRM
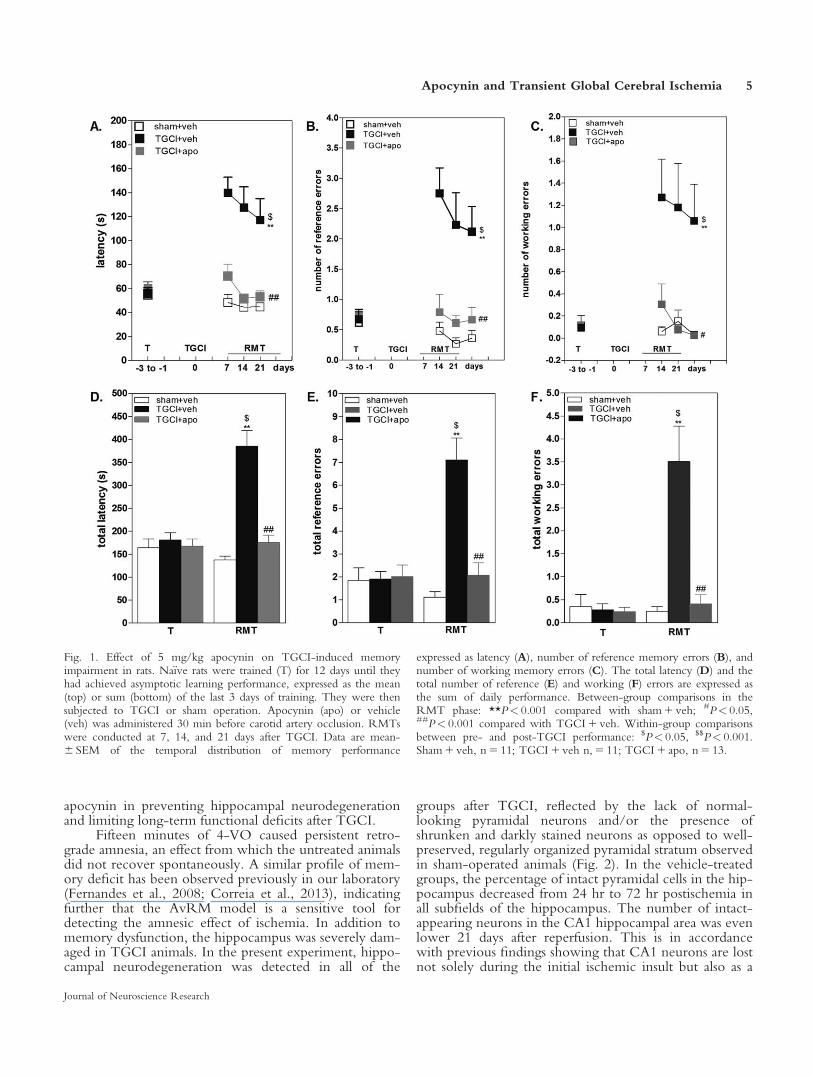
Figure 1 shows the effect of apocynin on retro-grade amnesia caused by TGCI in rats. Retrogradememory reflects the ability of rats to remember the goalbox location that was learned during preoperative train-ing, and the loss of that ability is referred to as retrogradeamnesia. By analyzing the temporal distribution of per-formance across days 7, 14, and 21, repeated-measuresANOVA detected a highly significant main effect ofgroup for the parameters latency, number of referenceerrors, and number of working errors (F2,72 5 19.48–45.34, P< 0.0001). A main effect of trial was notdetected for any of the three parameters (F2,72 5 0.81–2.80, P> 0.05). Compared with the sham 1 vehiclegroup, the TGCI 1 vehicle group took significantlymore time to find the goal box (i.e., latency) and com-mitted more reference and working memory errors(P< 0.0001). When memory performance was expressedas the total latency and the total number of errors, theeffect of TGCI was clearly evident (F2,34 5 16.66–36.10,P< 0.0001, TGCI 1 vehicle vs. sham 1 vehicle). Theanalysis of within-group effects indicated that the totallatency and the total number of errors increased consis-tently from the pre- to postsurgery phases in theTGCI 1 vehicle group (paired t-test, t 5 –4.11 to25.69, P< 0.001) but remained unchanged in thesham 1 vehicle group (t 5 0.53–1.42, P> 0.05). Thesedata indicate that ischemic rats “forgot” the previouslylearned task (i.e., retrograde amnesia). This memorydeficit caused by TGCI was significantly reduced in the
TGCI group that was treated with apocynin, reflectedby all three parameters (Fig. 1A–C, P< 0.0001–0.0004;Fig. 1 D–F, P< 0.0001–0.0002; TGCI 1 apocynin vs.TGCI 1 vehicle). Latency, number of working errors,and number of reference errors were reduced by apocy-nin to the levels of the sham 1 vehicle group (Fig. 1;P> 0.05; TGCI 1 apocynin vs. sham 1 vehicle).
Nissl Staining
TGCI resulted in severe neurodegeneration in thehippocampus, indicated by the presence of shrunken anddarkly stained neurons (Fig. 2). Significant cell loss wasdetected in CA1, CA2, CA3, and CA4 hippocampal areasat 24 hr (v2 5 441.81 to 77.74, P< 0.0001), 72 hr(v2 5 3,863.34 to 406.95, P< 0.0001), and 21 days(v2 5 5,615.53 to 88.92, P< 0.0001) in TGCI 1 vehicleanimals compared with the sham 1 vehicle group (Fig. 2).The number of intact-appearing neurons in the CA1 hip-pocampal area was significantly lower at 72 hr and at 21days compared with the number at 24 hr after reperfusion(v2 5 584.64, P< 0.0001). The number of intact-appearing neurons in CA2, CA3, and CA4 also decreasedfrom 24 to 72 hr (v2 5 392.70 to 81.22, P< 0.0001).However, a significant increase in the number of viablecells was observed in CA2, CA3, and CA4 from 72 hruntil 21 days (P< 0.0001).
At 24 hr, apocynin treatment resulted in significantneuroprotection, as seen from an increase in the numberof intact-appearing hippocampal cells in the CA1(P 5 0.0001) and CA4 (P 5 0.006) hippocampal areas ofTGCI animals compared with the TGCI 1 vehicle group.At 72 hr and at 21 days of reperfusion, apocynin treat-ment prevented hippocampal cell loss in all CA1, CA2,CA3, and CA4 hippocampal subfields of the TGCI groupcompared with controls (P 5 0.0001–0.009).
Immunohistochemistry
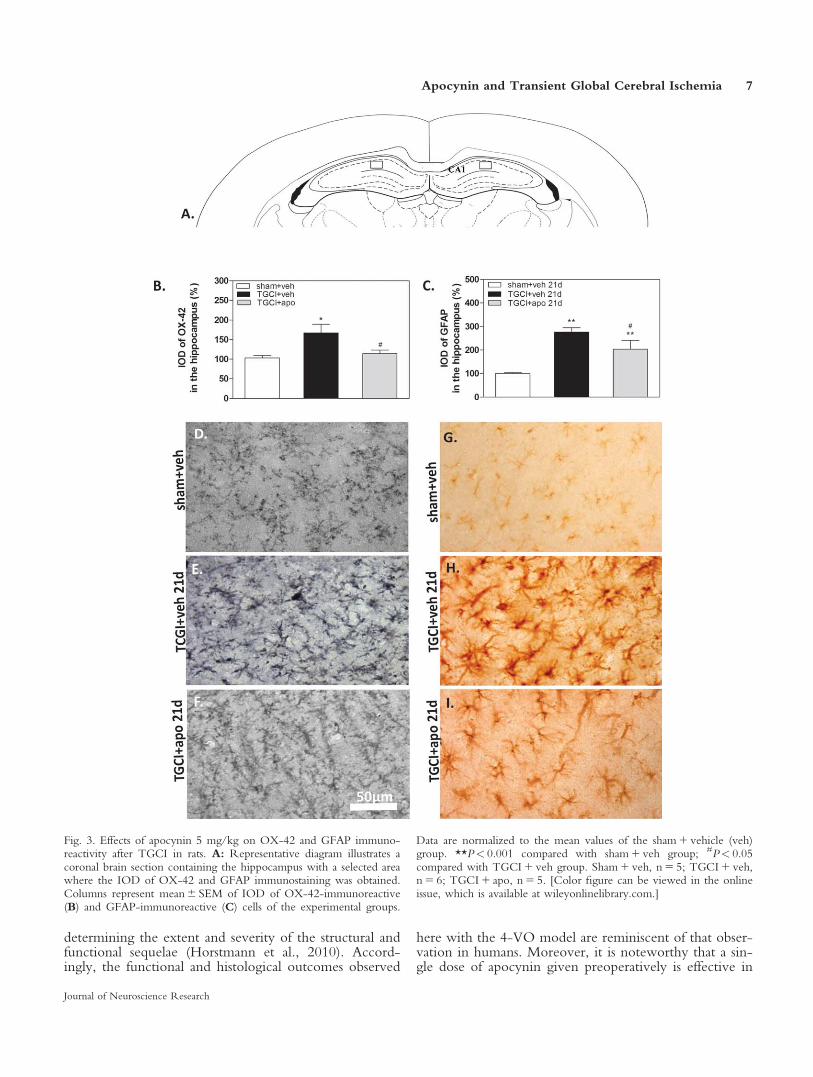
Figure 3 shows OX-42 and GFAP immunoreactivityexpressed as a percentage of IOD. Significant increases inthe immunoreactivity of both OX-42 (v2 5 9.32,P 5 0.009) and GFAP (v2 5 20.00, P< 0.0001) weredetected in the TGCI 1 vehicle group compared with thesham 1 vehicle group. A significant increase in GFAP-positive cells was also detected in the TGCI 1 apocyningroup compared with controls (P< 0.001). Apocynin sig-nificantly decreased OX-42 (P 5 0.009) and GFAP(P 5 0.02) immunoreactivity in TGCI animals comparedwith TGCI animals that received vehicle.
DISCUSSION
The novel finding of the present study is that a singledose of apocynin (5 mg/kg) prevents TGCI-induced ret-rograde amnesia, an effect that was maintained up to 21days after the insult. Moreover, apocynin reduced bothhippocampal cell death and glial cell activation, the latereffect reflected by decreases in OX-42 and GFAP immu-noreactivity in ischemic animals. These findings add togrowing evidence indicating an important role for
4 Romanini et al.
Journal of Neuroscience Research
apocynin in preventing hippocampal neurodegenerationand limiting long-term functional deficits after TGCI.
Fifteen minutes of 4-VO caused persistent retro-grade amnesia, an effect from which the untreated animalsdid not recover spontaneously. A similar profile of mem-ory deficit has been observed previously in our laboratory(Fernandes et al., 2008; Correia et al., 2013), indicatingfurther that the AvRM model is a sensitive tool fordetecting the amnesic effect of ischemia. In addition tomemory dysfunction, the hippocampus was severely dam-aged in TGCI animals. In the present experiment, hippo-campal neurodegeneration was detected in all of the
groups after TGCI, reflected by the lack of normal-looking pyramidal neurons and/or the presence ofshrunken and darkly stained neurons as opposed to well-preserved, regularly organized pyramidal stratum observedin sham-operated animals (Fig. 2). In the vehicle-treatedgroups, the percentage of intact pyramidal cells in the hip-pocampus decreased from 24 hr to 72 hr postischemia inall subfields of the hippocampus. The number of intact-appearing neurons in the CA1 hippocampal area was evenlower 21 days after reperfusion. This is in accordancewith previous findings showing that CA1 neurons are lostnot solely during the initial ischemic insult but also as a
Fig. 1. Effect of 5 mg/kg apocynin on TGCI-induced memoryimpairment in rats. Na€ıve rats were trained (T) for 12 days until theyhad achieved asymptotic learning performance, expressed as the mean(top) or sum (bottom) of the last 3 days of training. They were thensubjected to TGCI or sham operation. Apocynin (apo) or vehicle(veh) was administered 30 min before carotid artery occlusion. RMTswere conducted at 7, 14, and 21 days after TGCI. Data are mean-6 SEM of the temporal distribution of memory performance
expressed as latency (A), number of reference memory errors (B), andnumber of working memory errors (C). The total latency (D) and thetotal number of reference (E) and working (F) errors are expressed asthe sum of daily performance. Between-group comparisons in theRMT phase: **P< 0.001 compared with sham 1 veh; #P< 0.05,##P< 0.001 compared with TGCI 1 veh. Within-group comparisonsbetween pre- and post-TGCI performance: $P< 0.05, $$P< 0.001.Sham 1 veh, n 5 11; TGCI 1 veh n, 5 11; TGCI 1 apo, n 5 13.
Apocynin and Transient Global Cerebral Ischemia 5
Journal of Neuroscience Research
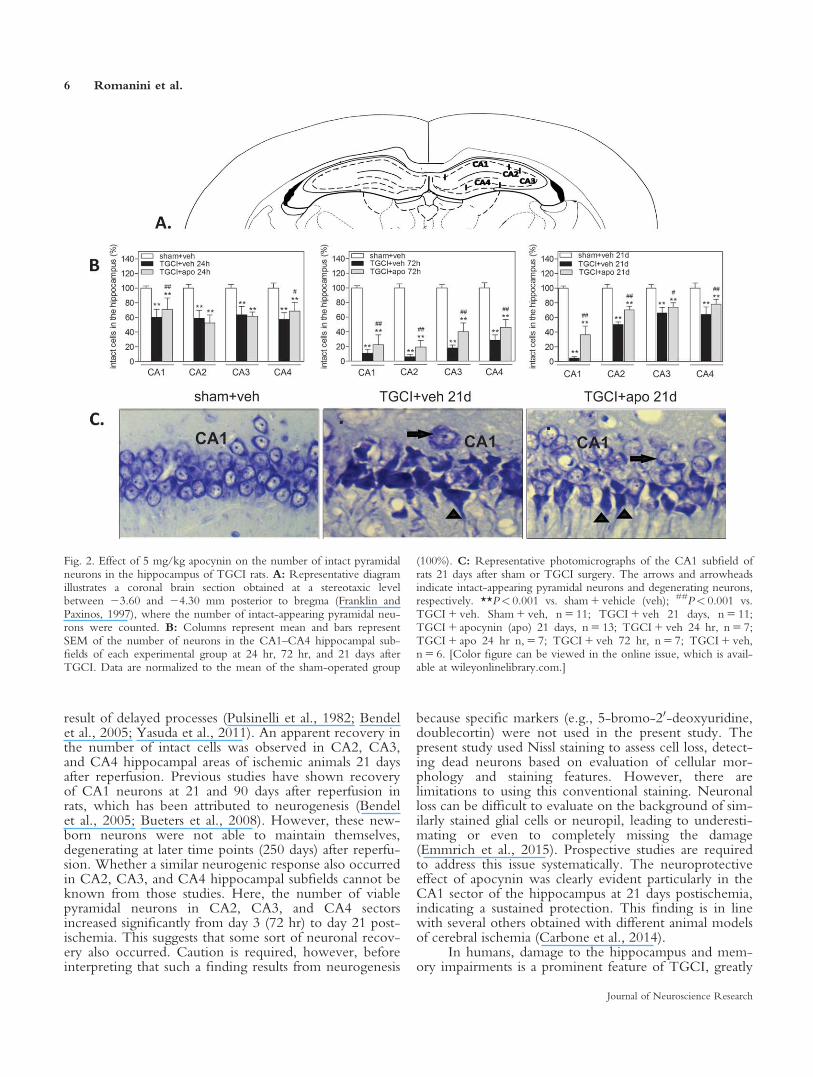
result of delayed processes (Pulsinelli et al., 1982; Bendelet al., 2005; Yasuda et al., 2011). An apparent recovery inthe number of intact cells was observed in CA2, CA3,and CA4 hippocampal areas of ischemic animals 21 daysafter reperfusion. Previous studies have shown recoveryof CA1 neurons at 21 and 90 days after reperfusion inrats, which has been attributed to neurogenesis (Bendelet al., 2005; Bueters et al., 2008). However, these new-born neurons were not able to maintain themselves,degenerating at later time points (250 days) after reperfu-sion. Whether a similar neurogenic response also occurredin CA2, CA3, and CA4 hippocampal subfields cannot beknown from those studies. Here, the number of viablepyramidal neurons in CA2, CA3, and CA4 sectorsincreased significantly from day 3 (72 hr) to day 21 post-ischemia. This suggests that some sort of neuronal recov-ery also occurred. Caution is required, however, beforeinterpreting that such a finding results from neurogenesis
because specific markers (e.g., 5-bromo-20-deoxyuridine,doublecortin) were not used in the present study. Thepresent study used Nissl staining to assess cell loss, detect-ing dead neurons based on evaluation of cellular mor-phology and staining features. However, there arelimitations to using this conventional staining. Neuronalloss can be difficult to evaluate on the background of sim-ilarly stained glial cells or neuropil, leading to underesti-mating or even to completely missing the damage(Emmrich et al., 2015). Prospective studies are requiredto address this issue systematically. The neuroprotectiveeffect of apocynin was clearly evident particularly in theCA1 sector of the hippocampus at 21 days postischemia,indicating a sustained protection. This finding is in linewith several others obtained with different animal modelsof cerebral ischemia (Carbone et al., 2014).
In humans, damage to the hippocampus and mem-ory impairments is a prominent feature of TGCI, greatly
Fig. 2. Effect of 5 mg/kg apocynin on the number of intact pyramidalneurons in the hippocampus of TGCI rats. A: Representative diagramillustrates a coronal brain section obtained at a stereotaxic levelbetween 23.60 and 24.30 mm posterior to bregma (Franklin andPaxinos, 1997), where the number of intact-appearing pyramidal neu-rons were counted. B: Columns represent mean and bars representSEM of the number of neurons in the CA1–CA4 hippocampal sub-fields of each experimental group at 24 hr, 72 hr, and 21 days afterTGCI. Data are normalized to the mean of the sham-operated group
(100%). C: Representative photomicrographs of the CA1 subfield ofrats 21 days after sham or TGCI surgery. The arrows and arrowheadsindicate intact-appearing pyramidal neurons and degenerating neurons,respectively. **P< 0.001 vs. sham 1 vehicle (veh); ##P< 0.001 vs.TGCI 1 veh. Sham 1 veh, n 5 11; TGCI 1 veh 21 days, n 5 11;TGCI 1 apocynin (apo) 21 days, n 5 13; TGCI 1 veh 24 hr, n 5 7;TGCI 1 apo 24 hr n, 5 7; TGCI 1 veh 72 hr, n 5 7; TGCI 1 veh,n 5 6. [Color figure can be viewed in the online issue, which is avail-able at wileyonlinelibrary.com.]
6 Romanini et al.
Journal of Neuroscience Research
determining the extent and severity of the structural andfunctional sequelae (Horstmann et al., 2010). Accord-ingly, the functional and histological outcomes observed
here with the 4-VO model are reminiscent of that obser-vation in humans. Moreover, it is noteworthy that a sin-gle dose of apocynin given preoperatively is effective in
Fig. 3. Effects of apocynin 5 mg/kg on OX-42 and GFAP immuno-reactivity after TGCI in rats. A: Representative diagram illustrates acoronal brain section containing the hippocampus with a selected areawhere the IOD of OX-42 and GFAP immunostaining was obtained.Columns represent mean 6 SEM of IOD of OX-42-immunoreactive(B) and GFAP-immunoreactive (C) cells of the experimental groups.
Data are normalized to the mean values of the sham 1 vehicle (veh)group. **P< 0.001 compared with sham 1 veh group; #P< 0.05compared with TGCI 1 veh group. Sham 1 veh, n 5 5; TGCI 1 veh,n 5 6; TGCI 1 apo, n 5 5. [Color figure can be viewed in the onlineissue, which is available at wileyonlinelibrary.com.]
Apocynin and Transient Global Cerebral Ischemia 7
Journal of Neuroscience Research

preventing retrograde amnesia, restoring the performanceof ischemic rats to the level of the sham-operated groupfor at least 21 days postischemia. Indeed, apocynin hasbeen shown to alleviate functional deficits after braindamage in other experimental conditions. Apocyninreduced brain edema and learning deficit in rats with dif-fuse traumatic brain injury (TBI; Song et al., 2013). Inmice subjected to TBI, apocynin decreased both sensori-motor deficits and apoptosis (Lu et al., 2014) as well asobject recognition memory deficit (Ferreira et al., 2013).Furthermore, apocynin attenuated learning impairmentand oxidative stress in a rat model of intermittent hypoxia(Hui-Guo et al., 2010). Shen et al. (2011) observed thatapocynin reduced learning/memory impairment and con-ferred hippocampal and cortical neuroprotection in micesubjected to TGCI.
The extent to which hippocampal neuroprotectiondetermines the beneficial effect of apocynin on learningand memory recovery is also unclear. It should be notedthat more than 60% of cell loss persisted in the hippocam-pal CA1 sector of ischemic rats treated with apocynin.Such findings suggest dissociation between histologicalneuroprotection and learning/memory recovery follow-ing TGCI. Notably, dysfunction of complex behaviorsand recovery of function may reflect alterations at the sub-cellular, synaptic, or electrophysiologic levels or evenwidespread morphological changes that would not berestricted to a given brain structure, such as the hippocam-pus (de la Tremblaye and Plamondon, 2011).
Another question that arose in the present study ishow a single dose of apocynin given before TGCI abol-ished the retrograde amnesia measured from 7 to 21 dayslater. It is unlikely that apocynin acted by directly affect-ing memory processes. A more reasonable explanation isthat apocynin acted to stop some cellular/biochemicalpathological change triggered by ischemia, an action thatmay have occurred within the first minutes or hours afterischemia/reperfusion. Accordingly, a single dose of apoc-ynin delivered 10 min prior to ischemia in mice providedlearning improvement and neuronal rescue assessed 7 dayslater (Shen et al., 2011), or, when given 30 min beforeischemia, it reduced both infarct size and neurologicalscores assessed 24 hr after MCAO in mice (Tang et al.,2008) or rats (Zhao et al., 2010; Genovese et al., 2011).That the beneficial effects of apocynin on structural andfunctional end points are reflected in the earliest phase ofischemic brain injury is further indicated by the observa-tion that apocynin blood concentration in Sprague Daw-ley rats decreased rapidly and almost completely within 40min of reaching the maximal plasma level at approxi-mately 2 or 5 min following intravenous or intragastricadministration, respectively (Wang et al., 2013).
With regard to such a pharmacokinetic profile ofapocynin, given that the generation of reactive oxygenspecies (ROS) is one of the earliest intracellular eventsafter ischemia/reperfusion (Dirnagl, 2012), and given thatboth lipid peroxidation and protein carbonylation takeplace as early as 2 hr after reperfusion (Lehotsk�y et al.,2004), we can speculate that the long-lasting antiamnesic
effect of apocynin might be related to its well-characterized antioxidant properties. Apocynin is a potentinhibitor of NADPH oxidase (NOX), an enzyme thatcatalyzes the reduction of molecular oxygen to generateROS (Stolk et al., 1994; Carbone et al., 2014). FiveNOX enzyme isoforms (NOX1–NOX5) have beenidentified. NOX is also localized in microglial cells andmay contribute to neurodegeneration that results fromneuroinflammation (Yoshioka et al., 2011; Nayerniaet al., 2014). Localization studies have shown that theNOX2 and NOX4 isoforms are expressed in the hippo-campus and cerebral cortex (Serrano et al., 2003; Nayer-nia et al., 2014). NOX2 appears to be important inoxidative stress-induced neuronal death following brainischemia, reflected by a dramatic reduction of infarct sizein NOX2 knockout mice (Chen et al., 2009) and NOX2inhibitor-treated animals (Zhang et al., 2009). Moreover,NOX2 activation in the rat hippocampal CA1 region hasbeen shown to occur as early as 30 min after reperfusion,reaching a peak 3 hr later; its inhibition resulted indecreased oxidative stress in the hippocampus and amelio-rated cognitive impairment in ischemic mice tested in theMorris water maze (Raz et al., 2010). A recent studyshowed that ROS produced by NOX1 activation alsoaccumulated in hippocampal neurons and damagednuclear DNA after chronic cerebral hypoperfusion in rats(Choi et al., 2012). In addition, both NOX1 inhibitionby adenoassociated virus-mediated NOX1 knockdownand apocynin reduced ROS generation, oxidative DNAdamage, hippocampal degeneration, and cognitiveimpairments. Collectively, these findings indicate thatNOX1 and NOX2 serve as pivotal upstream processes inneurodegeneration and cognitive impairment induced byexperimental cerebral ischemia. Remaining to be investi-gated, however, is how the inhibition of NOX by apocy-nin in the early phase of ischemia/reperfusion convertedinto sustained memory recovery (or its preservation) sev-eral weeks later.
Regarding the effect of apocynin in reducingischemia-induced glial cell activation, the question arises ofwhether an anti-inflammatory action contributed to its finalhistological and behavioral effects. Glial cells (i.e., microglialcells and astrocytes) are capable of secreting a range ofproinflammatory factors, including prostaglandins, chemo-kines, cytokines, proteinases, ROS, and nitric oxide withina few minutes following cerebral ischemia (Yenari et al.,2010; Yasuda et al., 2011; Choi et al., 2012). Therefore,intervening in the glial response in the early stage followingischemia may culminate in a beneficial effect on neuroin-flammation induced by TGCI. Similarly to the presentdata, it has been observed that TGCI-induced microglialactivation and neuronal death were reduced by apocynin inthe gerbil hippocampus (Wang et al., 2006) as well as in themouse striatum (Yoshioka et al., 2011). Tetracyclines given30 min after TGCI increased the number of surviving hip-pocampal neurons and decreased microglia activation ingerbils, measured 6 days after the insult (Yrj€anheikki et al.,1998). Moreover, the protective effects of cannabidiolagainst hippocampal neuronal death were detected 7 days
8 Romanini et al.
Journal of Neuroscience Research

after bilateral common carotid artery occlusion in mice andattributed to the inhibition of reactive astrogliosis (Schiavonet al., 2014). It is unknown whether apocynin-mediatedinhibition of the glial response is a consequence or the causeof the reduced neuronal damage induced by apocynin. Inthis regard, Genovese et al. (2011) have shown that apocy-nin (5 mg/kg, 5 min before reperfusion) significantlydecreases interleukin-1b expression and inhibits the nuclearfactor-jB (NFjB) pathway by preventing inhibitor ofNFjB degradation in the hippocampus of rats subjected toMCAO. These effects were accompanied by a reduction inthe levels of apoptosis measured by TUNEL and proapop-totic molecule B-cell lymphoma-2 (Bcl-2)-associated X(Bax) protein and by an increase in the level of the antia-poptotic molecule Bcl-2. The authors concluded that thebeneficial effect of apocynin on brain ischemia was the shiftof the Bcl-2/Bax balance toward an antiapoptotic state,enhancing the survival of neuronal cells. However, addi-tional experimentation will be required to identify the roleof the glial response and downstream mediators in the neu-roprotective effects of apocynin after TGCI.
Finally, the time elapsed between the onset of ische-mia and the beginning of an effective treatment (i.e., thetherapeutic time window) determined the success of phar-macotherapy against ischemic brain damage. In this regard,it has been argued the putative therapeutic value of apocy-nin is limited (Carbone et al., 2014) because almost all stud-ies investigating the effects of apocynin in animal models ofcerebral ischemia, including the present one, used a preop-erative treatment protocol. To the best of our knowledge,only one study administered apocynin (2.5 mg/kg) at 1 hrafter MCAO in mice, the results of which were negativefor infarct size, neurological deficits, and mortality rate.Moderate benefits were achieved, however, when apocy-nin was given 30 min before MCAO (Jackman et al.,2009). The effectiveness of apocynin restricted to preisch-emic treatment does not invalidate, however, a possibleclinical utility for this substance. Although cerebral hypoxiaor ischemia are most frequently unpredictable events,hypoxic/ischemic brain damage is also likely to occur dur-ing surgical or diagnostic procedures that reduce or inter-rupt cerebral blood flow (Sudo et al., 2001; Neuloh et al.,2007; Sala et al., 2007). Therefore, the therapeutic potentialof apocynin should be considered for acute, prophylacticuse in high-risk populations.
In conclusion, a single dose of apocynin is effectivein preventing the retrograde amnesia caused by TGCI inrats. This behavioral effect was concomitant with reduc-tion in hippocampal neurodegeneration and activation ofglial cell response. It remains to be determined whetherthe efficacy of apocynin restricted to a preischemicadministration protocol indicates a limitation for its clini-cal utility in the setting of cerebral ischemia.
ACKNOWLEDGMENTS
The authors thank Marco Alberto Trombelli for histechnical support. The authors have no conflicts ofinterest.
REFERENCES
Bachevalier J, Meunier M. 1996. Cerebral ischemia: are the memory def-
icits associated with hippocampal cell loss? Hippocampus 6:553–560.
Barnes CA. 1988. Spatial learning and memory processes: the search for their
neurobiological mechanisms in the rat. Trends Neurosci 11:163–169.
Bendel O, Bueters T, von Euler M, Ove Ogren S, Sandin J, von Euler
G. 2005. Reappearance of hippocampal CA1 neurons after ischemia is
associated with recovery of learning and memory. J Cereb Blood Flow
Metab 25:1586–1595.
Benetoli A, Dutra AM, Paganelli RA, Senda DM, Franzin S, Milani H.
2007. Tacrolimus (FK506) reduces hippocampal damage but fails to
prevent learning and memory deficits after transient, global cerebral
ischemia in rats. Pharmacol Biochem Behav 88:28–38.
Bueters T, von Euler M, Bendel O, von Euler G. 2008. Degeneration of
newly formed CA1 neurons following global ischemia in the rat. Exp
Neurol 209:114–124.
Carbone F, Camillo Teixeira P, Braunersreuther V, Mach F, Vuilleumier
N, Montecucco F. 2014. Pathophysiology and treatments of oxidative
injury in ischemic stroke: focus on the phagocytic NADPH oxidase
(NOX) 2. Antioxid Redox Signal (in press).
Chen H, Song YS, Chan PH. 2009. Inhibition of NADPH oxidase is
neuroprotective after ischemia–reperfusion. J Cereb Blood Flow Metab
29:1262–1272.
Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. 2012. Inhibition of
NADPH oxidase promotes alternative and anti-inflammatory microglial
activation during neuroinflammation. J Neurochem 120:292–301.
Corbett D, Nurse S. 1998. The problem of assessing effective neuropro-
tection in experimental cerebral ischemia. Prog Neurobiol 54:531–548.
Correia Bacarin C, Mori MA, Dias Fiuza Ferreira E, Val�erio Romanini
C, Weffort de Oliveira RM, Milani H. 2013. Fish oil provides robust
and sustained memory recovery after cerebral ischemia: influence of
treatment regimen. Physiol Behav 119:61–71.
Dirnagl U. 2012. Pathobiology of injury after stroke: the neurovascular
unit and beyond. Ann N Y Acad Sci 1268:21–25.
Duan YL, Wang SY, Zeng QW, Su DS, Li W, Wang XR, Zhao Z.
2011. Astroglial reaction to delta opioid peptide [D-Ala2,D-Leu5]
enkephalin confers neuroprotection against global ischemia in the adult
rat hippocampus. Neuroscience 192:81–90.
Emmrich JV, Ejaz S, Neher JJ, Williamson DJ, Baron JC. 2015. Regional
distribution of selective neuronal loss and microglial activation across
the MCA territory after transient focal ischemia: quantitative versus
semiquantitative systematic immunohistochemical assessment. J Cereb
Blood Flow Metab 35:20–27.
Fernandes JS, Mori MA, Ekuni R, Oliveira RM, Milani H. 2008. Long-
term treatment with fish oil prevents memory impairments but not hip-
pocampal damage in rats subjected to transient, global cerebral ischemia.
Nutr Res 28:798–808.
Ferreira AP, Rodrigues FS, Della-Pace ID, Mota BC, Oliveira SM,
Velho Gewehr CD, Bobinski F, de Oliveira CV, Brum JS, Oliveira
MS, Furian AF, de Barros CS, Ferreira J, Santos AR, Fighera MR,
Royes LF. 2013. The effect of NADPH-oxidase inhibitor apocynin on
cognitive impairment induced by moderate lateral fluid percussion
injury: role of inflammatory and oxidative brain damage. Neurochem
Int 63:583–593.
Franklin KBJ, Paxinos G. 1997. The mouse brain in stereotaxic coordi-
nates, 2nd ed. San Diego: Academic Press.
Freret T, Schumann-Bard P, Boulouard M, Bouet V. 2011. On the
importance of long-term functional assessment after stroke to improve
translation from bench to bedside. Exp Transl Stroke Med 3:1–5.
Garcia JH. The evolution of brain infarcts: a review. 1992.
J Neuropathol Exp Neurol 51:387–393.
Genovese T, Mazzon E, Paterniti I, Esposito E, Bramanti P, Cuzzocrea
S. 2011. Modulation of NADPH oxidase activation in cerebral ische-
mia/reperfusion injury in rats. Brain Res 1372:92–102.
Apocynin and Transient Global Cerebral Ischemia 9
Journal of Neuroscience Research

Horstmann A, Frisch S, Jentzsch RT, M€uller K, Villringer A, Schroeter
ML. 2010. Resuscitating the heart but losing the brain: brain atrophy
in the aftermath of cardiac arrest. Neurology 74:306–312.
Hui-Guo L, Kui L, Yan-ning Z, Yong-jian X. 2010. Apocynin attenuate
spatial learning deficits and oxidative responses to intermittent hypoxia.
Sleep Med 11:205–212.
Jackman KA, Miller AA, De Silva TM, Crack PJ, Drummond GR,
Sobey CG. 2009. Reduction of cerebral infarct volume by apocynin
requires pretreatment and is absent in Nox2-deficient mice. Br J Phar-
macol 156:680–688.
Lu XY, Wang HD, Xu JG, Ding K, Li T. 2014. NADPH oxidase inhi-
bition improves neurological outcome in experimental traumatic brain
injury. Neurochem Int 69:14–19.
Lull ME, Levesque S, Surace MJ, Block ML. 2011. Chronic apocynin
treatment attenuates beta amyloid plaque size and microglial number in
hAPP(751)(SL) mice. PLoS One 6:e20153.
Moulaert VR, Verbunt JA, van Heugten CM, Wade DT. 2009. Cogni-
tive impairments in survivors of out-of-hospital cardiac arrest: a system-
atic review. Resuscitation. 80:297–305.
Moulaert VR, Wachelder EM, Verbunt JA, Wade DT, van Heugten
CM. 2010. Determinants of quality of life in survivors of cardiac arrest.
J Rehabil Med 42:553–558.
Nayernia Z, Jaquet V, Krause KH. 2014. New insights on NOX
enzymes in the central nervous system. Antioxid Redox Signal 20:
2815–2837.
Neuloh G, Simon M, Schramm J. 2007. Stroke prevention during sur-
gery for deep-seated gliomas. Neurophysiol Clin 37:383–389.
Paganelli RA, Benetolli A, Lima KCM, Cestari-Junior LA, Favero-Filho
LA, Milani H. 2004. A novel version of the 8-arm radial maze: effects
of cerebral ischemia on learning and memory. J Neurosci Methods 132:
9–18.
Peskine A, Picq C, Pradat-Diehl P. 2010. Neurological sequelae after
cerebral anoxia. Brain Inj 24:755–761.
Philippens IH, Wubben JA, Finsen B, ‘t Hart BA. 2013. Oral treatment
with the NADPH oxidase antagonist apocynin mitigates clinical and
pathological features of parkinsonism in the MPTP marmoset model.
J Neuroimmune Pharmacol 8:715–726.
Pulsinelli WA, Brierley JB. 1979. A new model of bilateral hemispheric
ischemia in the unanesthetized rat. Stroke 10:267–272.
Pulsinelli WA, Brierley JB, Plum F. 1982. Temporal profile of neuronal
damage in a model of transient forebrain ischemia. Ann Neurol 11:
491–498.
Raz L, Zhang QG, Zhou CF, Han D, Gulati P, Yang LC, Yang F,
Wang RM, Brann DW. 2010. Role of Rac1 GTPase in NADPH oxi-
dase activation and cognitive impairment following cerebral ischemia in
the rat. PLoS One 7:e12606.
Sala F, Beltramello A, Gerosa M. 2007. Neuroprotective role of neuro-
physiological monitoring during endovascular procedures in the brain
and spinal cord. Neurophysiol Clin 37:415–421.
Schiavon AP, Soares LM, Bonato JM, Milani H, Guimar~aes FS, Weffort
de Oliveira RM. 2014. Protective effects of cannabidiol against hippo-
campal cell death and cognitive impairment induced by bilateral com-
mon carotid artery occlusion in mice. Neurotox Res 26:307–316.
Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E. 2003. NADPH
oxidase immunoreactivity in the mouse brain. Brain Res 988:193–198.
Shen J, Bai XY, Qin Y, Jin WW, Zhou JY, Zhou JP, Yan YG, Wang
Q, Bruce IC, Chen JH, Xia Q. 2011. Interrupted reperfusion reduces
the activation of NADPH oxidase after cerebral I/R injury. Free Radic
Biol Med 50:1780–1786.
Song SX, Gao JL, Wang KJ, Li R, Tian YX, Wei JQ, Cui JZ. 2013.
Attenuation of brain edema and spatial learning deficits by the inhibi-
tion of NADPH oxidase activity using apocynin following diffuse trau-
matic brain injury in rats. Mol Med Rep 7:327–331.
Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. 1994. Characteristics
of the inhibition of NADPH oxidase activation in neutrophils by apoc-
ynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 11:
95–102.
Sudo Y, Takahara Y, Nakajima N. 2001. Pulsatile cardiopulmonary
bypass failed to prevent neuropsychological dysfunction. Ann Thorac
Cardiovasc Surg 7:89–93.
Sun AY, Wang Q, Simonyi A, Sun GY. 2008. Botanical phenolics and
brain health. Neuromol Med 10:259–274.
Takuma K, Baba A, Matsuda T. 2004. Astrocyte apoptosis: implications
for neuroprotection. Prog Neurobiol 72:111–127.
Tang XN, Cairns B, Cairns N, Yenari MA. 2008. Apocynin improves
outcome in experimental stroke with a narrow dose range. Neuro-
science 154:556–562.
Wang K, Li L, Song Y, Ye X, Fu S, Jiang J, Li S. 2013. Improvement of
pharmacokinetics behavior of apocynin by nitrone derivatization: com-
parative pharmacokinetics of nitrone–apocynin and its parent apocynin
in rats. PLoS One 30:8:e70189.
Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY.
2006. Apocynin protects against global cerebral ischemia–reperfusion-
induced oxidative stress and injury in the gerbil hippocampus. Brain
Res 1090:182–189.
Yasuda Y, Shimoda T, Uno K, Tateishi N, Furuya S, Tsuchihashi Y,
Kawai Y, Naruse S, Fujita S. 2011. Temporal and sequential changes of
glial cells and cytokine expression during neuronal degeneration after
transient global ischemia in rats. J Neuroinflammation 8:70.
Yenari MA, Kauppinen TM, Swanson RA. 2010. Microglial activation
in stroke: therapeutic targets. Neurotherapeutics 7:378–391.
Yoshioka H, Niizuma K, Katsu M, Okami N, Sakata H, Kim GS,
Narasimhan P, Chan PH. 2011. NADPH oxidase mediates striatal neu-
ronal injury after transient global cerebral ischemia. J Cereb Blood
Flow Metab 31:868–880.
Yrj€anheikki J, Kein€anen R, Pellikka M, H€okfelt T, Koistinaho J.1998.
Tetracyclines inhibit microglial activation and are neuroprotective in
global brain ischemia. Proc Natl Acad Sci U S A 95:15769–15774.
Zhang P, Hou M, Li Y, Xu X, Barsoum M, Chen Y, Bache RJ. 2009.
NADPH oxidase contributes to coronary endothelial dysfunction in the
failing heart. Am J Physiol Heart Circ Physiol 296:H840–H846.
Zhao H, Mayhan WG, Arrick DM, Xiong W, Sun H. 2010. Alcohol-
induced exacerbation of ischemic brain injury: role of NAD(P)H oxi-
dase. Alcohol Clin Exp Res 34:1948–955.
Zola-Morgan S, Squire LR, Amaral DG. 1986. Human amnesia and the
medial temporal region: enduring memory impairment following a
bilateral lesion limited to field CA1 of the hippocampus. J Neurosci 6:
2950–2967.
10 Romanini et al.
Journal of Neuroscience Research




















