







Bahasa
Halaman
Hukum


Vol 4 Issue 3, 2010
_________________________________________________________
www.currentbiotica.com
285
ISSN 0973-4031
Modification of alkaline lysis method for rapid plasmid DNA isolation
S.V. Ramu, K.C. Babitha, K.S. Shailesh, Karaba N Nataraja and M. Udayakumar*
Department of Crop Physiology, University of Agricultural Sciences,
GKVK, Bangalore – 560 065, INDIA
*Corresponding author : [email protected]
ABSTRACT
Isolation of quality plasmid DNA from bacterial cells is critical in molecular biology
experiments. Although efficient protocol for plasmid isolation are developed and standardized,
rapid and cost effective protocols are desirable while handling a large number of samples. In
present study we are reporting a simple procedure for extracting naked plasmid DNA which can
be used for a large number of samples with sufficient purity. The method involves denaturation
of high molecular weight chromosomal DNA using strong alkali, during neutralization process,
chromosomal DNA renatures, and proteins denature to form an insoluble clot in the presence of
choatropic agent guanidine hydrochloride, leaving plasmid DNA in the supernatant. The
modified protocol which is less expensive compared to the commercial plasmid DNA isolation
kit available, yields quality plasmid DNA. We tested the yield and quality of different kinds of
plasmid extracted from bacteria. The results suggest that the protocol can be effectively used for
molecular cloning, sequencing, and transfection.
KEYWORDS: Alkaline lysis, Guanidine hydrochloride, Plasmid extraction
INTRODUCTION
Bacterial plasmid DNA is widely
used as cloning vehicles in recombinant
DNA research. The isolation of plasmid
DNA is a critical step for many procedures
such as cloning, DNA sequencing,
transfection, and gene therapy. Most of the
applications require high purity plasmid
DNA. Certain methods like isolation by
anion-exchange columns, even though
widely used, are relatively expensive. The
method that use silica oxide as the DNA-
binding matrix has the limitation, as
bacterial lipopolysaccharide or endotoxin is
co-purified which can interfere with
downstream applications. The
lipopolysaccharide content in plasmid
preparation can heavily influence the
efficiency of transfection (Clevell, and
Helinski, 1969, Colman et.al 1978, Barnes,
1977).
Certain applications such as
recombinant bacterial colony screening to
permit analysis of many clones in a short
period, relatively less purified DNA can be
used (Telford et al.1977, Eckhardt et al.
1978 and Mickel et al 1977). Quick plasmid
DNA isolation methods involve boiling and
alkaline lysis (Holmes and Quigley 1981;
Sambrook and Russell 2001) and a few
other reliable protocols have multiple steps
and are time consuming (Mak et al. 1991;
Tarczynski et al. 1994; Wang et al. 1995;

Vol 4 Issue 3, 2010
_________________________________________________________
www.currentbiotica.com
286
ISSN 0973-4031
Aranishi 2002). A rapid protocol using
sucrose (8%) and triton X-100 (5%)
described by Liu and Mishra (1995) has
disadvantages. The high concentrations of
Triton X-100 may interfere with DNA
migration in agarose gel electrophoresis or
restriction digestion even after a 10-fold
dilution as suggested by the authors, and the
small volume of DNA used for restriction
digestion may be inadequate in many
circumstances. One-step procedure for
screening recombinant plasmids by size was
reported (Beuken et al.1998). Recently, a
rapid plasmid extraction method by the
direct boiling of E. coli cells, using 0.1%
Triton X-100 avoiding isopropanol/ethanol
precipitation and drying steps used in
standard plasmid preparation has been
reported (Ouyang et al., 2008). However,
the ratio of optical density (OD 260/280nm)
of extracted DNA was in the range of 2.7 to
3.4, suggesting the presence of impurities.
In this study we are reporting another
modified method for plasmid DNA
extraction from E. coli using Guanidine HCl
which is simple enough to isolate plasmid
vectors of different sizes and can yield
plasmid DNA in a form equally as pure as
the commercially available kits. The
procedure described is more versatile, cost
effective and rapid.
MATERIALS AND METHODS
The research experiments were
conducted at the Department of Crop
Physiology, University of Agricultural
Sciences, Bangalore during 2009-2010.
Cell strains and media:
Escherichia coli (DH5α) cultures
harboring different plasmids were grown in
Lauria (L) broth containing yeast extract
5gL-1
, NaCl 10gL-1
and tryptone 10gL-1
(Sambrook et.al, 1989) with appropriate
antibiotics (Kanamycin 50µg/mL;
Ampicilin 100µg/mL, and Chloramphenicol
25µg/mL). Different vectors used are
presented in the table 1 and figure 1a.
Procedure for extraction of plasmid DNA:
About 1.5 mL of bacterial cultures
grown in 3ml of L-broth containing
appropriate antibiotics was transferred to
microfuge tubes and spun at 12,000xg for 60
seconds to pellet down the cells. The
remaining 1.5ml culture was transferred to
the same tube after discarding the
supernatant and centrifuged to collect cells.
The supernatant was carefully removed with
a fine-tip pipette and the pellet was
thoroughly resuspended in 200ul of Solution
I (Sterile distilled water and RNase A
solution – (2.5mg/ml), stored at 4°C) by
vortexing. To the reaction mixture, 200µL
of Solution II (Alkaline SDS solution – 1%
NaOH and 2 % sodium dodecyl sulfate
(SDS), stored at room temperature) was
added and the tube was gently inverted to
get a clear suspension. To the suspension,
350 µL of Solution III (3M Guanidine HCl
(Sigma Aldrich Chemical Company, USA)
prepared in water and stored at room
temperature.) was added and the contents of
the tube were gently mixed by inverting for
a few seconds. The tube then was
centrifuged for 5-10 min at 12,000xg and the
clear supernatant was transferred to a fresh
centrifuge tube. To the reaction mix, equal
volumes of absolute alcohol was added and
incubated at room temperature for 10 min.
Generally any DNA would get precipitated
if the alcohol concentration is above 66%
and hence alcohol twice the volume of the
supernatant is generally added. On the other
hand isopropanol at a final concentration of

Vol 4 Issue 3, 2010
_________________________________________________________
www.currentbiotica.com
287
ISSN 0973-4031
30-50% could be conveniently used). . The
DNA pellet from the reaction mixture was
collected by centrifugation (10 min at
12000xg) and the supernatant was removed
by pipetting. The pellet was washed with
chilled ethanol (70%, v/v) by centrifuging at
12,000xg for 5 min. The DNA pellet was
dried at 650C for 3 to 5 min or air dried until
the ethanol evaporated. The purified DNA
was dissolved in 30-50µl of Tris or TE
buffer (10mM, pH 8 according to the pellet
size and used for analysis. The isolated
DNA was quantified using
spectrophotometer (UV-VIS, Simadzu,
Japan) and also by analyzed by agarose gel
electrophoresis (Sambrook et al., 1989). The
efficiency of the protocol was compared
with plasmid isolation from kit (Kit product
code PLN70-1KT Sigma, USA)
To check the quality of DNA
isolated, restriction analysis was done at
370C for 1 hr using specific restriction
enzymes (SacI, XhoI and SmaI, MBI,
Fermentas,USA). The identity of the
plasmids was confirmed by amplifying the
gene of interest by PCR using specific
primers. All the PCRs were carried out
under the following conditions; 940C for 4
min, 940Cfor 1min, 56-57
0C for 1 min and
720C for 1 min, followed by 25 cycles and
final extension of 720Cfor 8min.
RESULTS AND DISCUSSION
Exposure of bacterial suspensions to
strong anionic detergent at high pH
denatures chromosomal DNA and proteins.
There is a narrow range of pH (about 12.0-
12.5) within which denaturation of linear
DNA but not covalently coiled circular
DNA (CCC-DNA) occurs and that this
property can be used for purifying CCC-
DNA. In present protocol the cells were
suspended in sterile distilled water without
considering the pH of the solution. Plasmid-
containing cells were lysed completely with
strong detergent, SDS and alkali NaOH
(1%).
In molecular cloning, chaotropic
agents like guanidine HCL are used to
destroy the 3-dimensional structures of
proteins which convert most proteins to a
randomly coiled state (Tanford 1968.,
Gordon 1972). This strong denaturant can
solubilize insoluble or denatured proteins
such as inclusion bodies (Mukhopadhyay
1997, Rudloph, and Lilie1996). This can be
used as the first step in refolding proteins
(Levine et al.1995) or enzymes into their
active form (Mukhopadhyay 1997).
Guanidine HCl, an inhibitor of RNase, is
used in the isolation of RNA to dissociate
the nucleoprotein into its nucleic acid and
protein moieties (Cox 1968). Highly
concentrated (6 - 8 M) Guanidine HCl
solutions are used to denature native
globular proteins. It apparently disrupts
hydrogen bonds which hold the protein in its
unique structure. However, there also is
evidence suggesting that guanidine HCl may
disrupt hydrophobic interactions by
promoting the solubility of hydrophobic
residues in aqueous solutions (Gordon et al.
1972).
In the protocol reported here, we
used 3M Guanidium HCl to dissociate the
nucleoprotein and other protein moieties.
Alkaline extraction method is designed to
prevent the generation of the "irreversibly
denatured" form, at the same time; the
extracts must be alkaline enough for
denaturation of chromosomal DNA to occur.
The medium concentration of guanidine HCl
caused precipitation of protein-SDS
complexes (Kay et.al 1952 and Marko et.al
1951) and of high molecular weight RNA
(Crestfield et.al 1955). In this way, most of
Vol 4 Issue 3, 2010
_________________________________________________________
www.currentbiotica.com
288
ISSN 0973-4031
the three major contaminating
macromolecules are co-precipitated and
removed by a single centrifugation in a
bench-top centrifuge at room temperature.
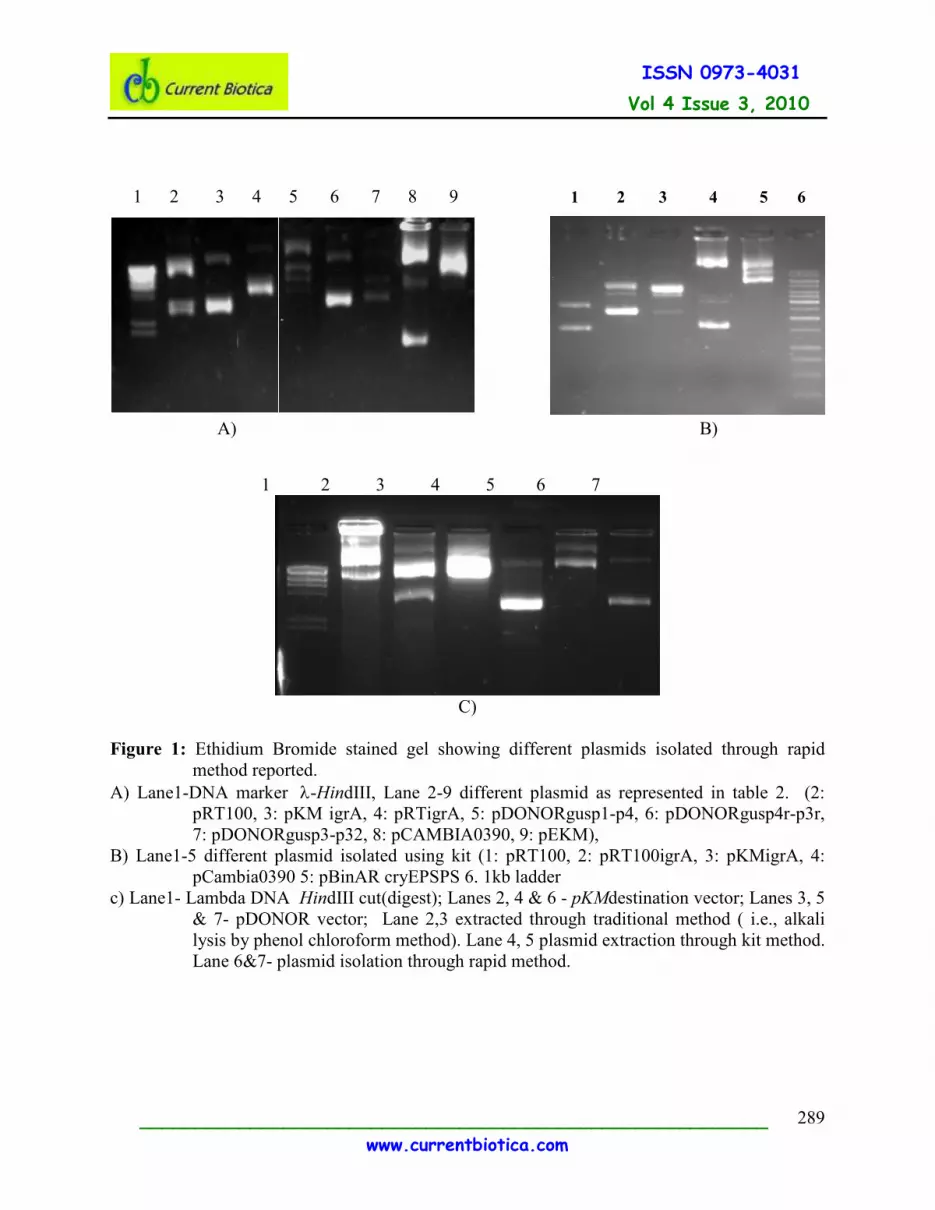
As shown in the figure 1a ,1b and 1c pure
plasmid DNA was recovered from the
supernatant by ethanol precipitation.
The data revealed that the present
protocol can yield good quantity of plasmid
DNA (Table 1), and the optical density (OD)
ratios at 260/280mn wavelength indicated
good quality DNA as compared with that of
Kit method. The OD ratio at 260/280shows
that the presences of impurities are
significantly less compared to the method
described by Ouyang et al. (2008). The
obtained plasmid DNA is suitable for
digestion by restriction enzymes (Figure 3).
The target genes in the respective plasmid
DNA was confirmed through PCR
amplification using gene specific primers,
which also suggest the suitability of plasmid
isolated for further applications (Figure 2).
The protocol described here is a cost
effective. Each isolated plasmid sample
approximately cost about 35-40
INR/sample, where as many commercial kits
are cost about 150 INR /sample (Sigma
Plasmid kit) and 125 INR/ sample (Qiagen
Plasmid kit), which offer great economy for
routine recombinant plasmid isolation in
terms of daily use. Each isolated plasmid
sample approximately cost about two-three
times lesser than many commercial kits. The
protocol developed in the present study is
simple, reliable, rapid and cost effective for
plasmid isolation from bacterial cells.
ACKNOWLEDGEMENT: Authors are
thankful to Dr. Purshotham and Dr. Kiran
Ghnati Seeds company, Mansanto,
Bangalore respectively for reviewing the
manuscript and their critical comments. .
Table 1: Quantification of naked plasmid DNA. All plasmid DNAs were extracted
through the method reported in this study and quantified using spectrophotometer
Plasmids isolated using
modified method
Plasmids isolated using
DNA extraction kit Sl.No Sample ID
Plasmid
~size(kb)with
insert ng. µL-1
260/280 ng. µL-1
260/280
1 pRT1002x35s
3.7 272.38 1.47 150.21 1.87
2 pKM igrA
5 300.33 2.19 200.12 1.92
3 pRTigrA
5.8 143.3 1.87 100.23 1.84
4 pDonor Gus P1-P4 6.0 162.7 2.07 200.22 1.96
5 pDonor Gus P4r-
P3r 6.1 171.12 1.94 153.32 1.89
6 pDonor Gus P3-P2 6.0 522.94 2.23 195.65 1.79
7 pCAMBIA0390
4.2 1483.4 2.06 154.89 1.93
8 pEKM
15 150.3 2.08 156.23 1.87
Vol 4 Issue 3, 2010
_________________________________________________________
www.currentbiotica.com
289
ISSN 0973-4031
1 2 3 4 5 6 7 8 9
A) B)
1 2 3 4 5 6 7
C)
Figure 1: Ethidium Bromide stained gel showing different plasmids isolated through rapid
method reported.
A) Lane1-DNA marker λ-HindIII, Lane 2-9 different plasmid as represented in table 2. (2:
pRT100, 3: pKM igrA, 4: pRTigrA, 5: pDONORgusp1-p4, 6: pDONORgusp4r-p3r,
7: pDONORgusp3-p32, 8: pCAMBIA0390, 9: pEKM),
B) Lane1-5 different plasmid isolated using kit (1: pRT100, 2: pRT100igrA, 3: pKMigrA, 4:
pCambia0390 5: pBinAR cryEPSPS 6. 1kb ladder
c) Lane1- Lambda DNA HindIII cut(digest); Lanes 2, 4 & 6 - pKMdestination vector; Lanes 3, 5
& 7- pDONOR vector; Lane 2,3 extracted through traditional method ( i.e., alkali
lysis by phenol chloroform method). Lane 4, 5 plasmid extraction through kit method.
Lane 6&7- plasmid isolation through rapid method.
1 2 3 4 5 6

Vol 4 Issue 3, 2010
_________________________________________________________
www.currentbiotica.com
290
ISSN 0973-4031
1 2 3 1kb 4 5 6 B1 B2 B3
Figure 2: PCR amplification of various DNA inserts within plasmid vector using gene specific
primers in the binary vector carrying three different genes.
Lanes. 1, 2, & 3 from pKMNWBGW isolated through the method proposed in this paper.
Lanes 4, 5 & 6 from the pKMNWBGW isolated through kit method.
Lanes B1, B2 & B3 are the negative controls for the specific primers.
Lane 1&4 amplified using Forward: ggggatccagcagtcgaggggagcgagc and Reverse:
gggatatcgactccatgtacgctacgccg primers. Lane 2&5 are amplified using Forward:
5’ctcgaggcaccaatctagacctc3’ and Reverse: 5’ccatggttttttctctacctctctc3’ primers. Lane
3&6 were amplified using Forward: ccatgggacttttctagttgca and Reverse:
5’ggggtaccttatttgattttctaaag3’ primers.
Figure 3: Restriction digestion of plasmid DNAs extracted from (by) rapid method.
Lane –1 plasmid DNA uncut of pKMNWBGW, 2, 3 and 4: digested plasmid of pKMNWBGW
with restriction enzyme SacI, lane 5 – 1 kb ladder, lane 6 uncut plasmid DNA of pDONOR221,
lane 7- digested plasmid DNA using XhoI –SmaI.
2.2kb
1.8kb
1 2 3 4 5 6 7
1.1kb

Vol 4 Issue 3, 2010
_________________________________________________________
www.currentbiotica.com
291
ISSN 0973-4031
REFERENCES
Aranishi, F. ( 2002) Flexprep: scale-flexible
rapid plasmid preparation for analysis
of recombinant clones. Mol.
Biotechnol., 21(1): 39–41.
Barnes, W.M. (1977), Plasmid detection and
sizing in single colony lysates,
Science., 195(4276) : 393-394.
Beuken, e., Vink, c. and Bruggeman, C.A.
(1998) One-step procedure for
screening recombinant plasmids by
size. Biotechniques., 24(5): 748– 750.
Clevell, D.B. and Helinski, D.R. (1969),
Supercoiled Circular DNA-Protein
Complex In Escherichia Coli:
Purification And Induced Conversion
To An Open Circular DNA Form,
Proc. Nat. Acad. Sci. USA., 62: 1159-
1166.
Colman, A., Byers, M.J., Primrose, S.B. and
Lyons, A. (1978) Rapid purification of
plasmid DNAs by hydroxyapatite
chromatographyEur. J. Biochem., 91:
303-310.
Cox.R.A, (1968): The use of guanidium
chloride in the isolation of nucleic
acids. Methods Enzymology., 12: 120-
129.
Crestfield, A.M., Smith, K.C. and Allen,
F.W. Y(1955), The Preparation And
Characterization of Ribonucleic Acids
From Yeast, J. Biol. Chem., 21: 6185-
193.
Eckhardt, T. (1978), A rapid method for the
identification of plasmid
deoxyribonucleic acid in bacteria.
Plasmid, 1: 584-588.
Gordon J.A (1972): Denaturation of
globular proteins. Interaction of
guanidium salts with three proteins.
Biochemistry, 11: 1862-1870.
Holmes, D.S. and Quigley, M. (1981) A
rapid boiling method for the
preparation of bacterial plasmids.
Anal. Biochem., 114(1): 193–197.
Kay, E.R.M., Simmons, N.S. and Dounce,
A.L. (1952), An improved preparation
of sodium deoxyribonucleate J. Am.
Chem. Soc., 74:1724-1726.
Lan Ouyang, Liang Qin, Zhiwu Xu, Jianguo
He and Qiuyun Liu. (2008) A rapid
plasmid preparation method by the
direct boiling of Escherichia coli cells.
J. Rapid Methods & Automation
Microb., 16: 22–29.
Levine, A.D., et al. (1995), High Level
Expression and Refolding of Mouse
Interleukin 4 Synthesized in
Escherichia coli. J. Biol. Chem., 270:
7445-7452
Liu, Z. and Mishra, N.C. ( 1995) Single-tube
method for plasmid miniprep from
large numbers of clones for direct
screening by size or restriction
digestion. Biotechniques., 18(2):214–
217.
Mak, Y.M., Sornarajah, R. and Ho, K.K.
(1991) A rapid procedure for detecting
recombinant plasmids using butanol
extraction. Biotechniques., 11(16):
723.

Vol 4 Issue 3, 2010
_________________________________________________________
www.currentbiotica.com
292
ISSN 0973-4031
Marko, A.M. and Butler, G.C. (1951) The
Isolation Of Sodium
Desoxyribonucleate With Sodium
Dodecyl Sulfate J. Biol. Chem., 190:
165-176.
Mickel, S., Arena, V. Jr. and Bauer, W.
(1977) Physical properties and gel
electrophoresis behavior of R12-
derived plasmid DNAs, Nucleic Acids
Research., 4: 1465-1482.
Mukhopadhyay, A. (1997) Inclusion bodies
and purification of proteins in
biologically active forms Adv.
Biochem. Eng. Biotechnol., 56:61-109.
Rudloph, R. and Lilie, H. (1996), In vitro
folding of inclusion body proteins
FASEB J., 10: 49-56
Sambrook, J. and Russell, D.W. (2001)
Molecular Cloning: A Laboratory
Manual, Cold Spring Harbor
Laboratory Press, Cold Spring Harbor,
NY.3rd Ed, pp. 1.32, 1.35, 1.38.
Tanford.C. (1968): Protein denaturation.
Adv. Protein chemistry., 23: 121-282.
Tarczynski, M.C., Meyer, W.J., Min, J.J.,
Wood, K.A. and Hellwig, R.J. (1994)
Two minute miniprep method for
plasmid DNA isolation.
Biotechniques., 16(3): 514–519.
Telford, J., Boseley, P, Schaffner, W. and
Birnstiel, M. (1977), Novel screening
procedure for recombinant plasmids.
Science., 195: 391-393.
Wang, K., Gan, L. and Hood, L. (1995) A
microtiter plate-based highthroughput
DNA purification method. Anal.
Biochem., 226(1): 85–90.
[MS received 18 August 2010; Accepted 12 November 2010]
Disclaimer: Statements, information, scientific names, spellings, inferences, products, style, etc. mentioned in Current
Biotica are attributed to the authors and do in no way imply endorsement/concurrence by Current Biotica. Queries related to
articles should be directed to authors and not to editorial board.
Top Related
Copyright © 2022 FDOKUMEN

