







Bahasa
Halaman
Hukum


Transcriptional re-programming of primarymacrophages reveals distinct apoptotic andanti-tumoral functions of IRF-3 and IRF-7
Delphine Goubau1,2, Raphaelle Romieu-Mourez1,2, Mayra Solis1,2,
Eduardo Hernandez1,2, Thibault Mesplede1,2, Rongtuan Lin1,3,
Douglas Leaman4 and John Hiscott1,2,3
1 Terry Fox Molecular Oncology Group, Lady Davis Institute for Medical Research, McGill
University, Montreal, Que., Canada2 Department of Microbiology and Immunology, McGill University, Montreal, Que., Canada3 Department of Medicine, McGill University, Montreal, Que., Canada4 Department of Biological Sciences, University of Toledo, Toledo, OH, USA
The immunoregulatory transcriptional modulators – IFN-regulatory factor (IRF)-3 and
IRF-7 – possess similar structural features but distinct gene-regulatory potentials. For
example, adenovirus-mediated transduction of the constitutively active form of IRF-3
triggered cell death in primary human MU, whereas expression of active IRF-7 induced a
strong anti-tumoral activity in vitro. To further characterize target genes involved in these
distinct cellular responses, transcriptional profiles of active IRF-3- or IRF-7-transduced
primary human MU were compared and used to direct further mechanistic studies. The
pro-apoptotic BH3-only protein Noxa was identified as a primary IRF-3 target gene and an
essential regulator of IRF-3, dsRNA and vesicular stomatitis virus-induced cell death. The
critical role of IRF-7 and type I IFN production in increasing the immunostimulatory
capacity of MU was also evaluated; IRF-7 increased the expression of a broad range of IFN-
stimulated genes including immunomodulatory cytokines and genes involved in antigen
processing and presentation. Furthermore, active IRF-7 augmented the cross-presentation
capacity and tumoricidal activity of MU and led to an anti-tumor response against the B16
melanoma model in vivo. Altogether, these data further highlight the respective functions
of IRF-3 and IRF-7 to program apoptotic, immune and anti-tumor responses.
Key words: Anti-tumor immunotherapy . Apoptosis . Cross-presentation .
IFN-regulatory factor . Microarray
Supporting Information available online
Introduction
Type I interferons (IFN-a/b) induce the expression of hundreds
of IFN-stimulated genes (ISG), which mediate a broad range of
anti-viral, growth-regulatory and immunomodulatory effects. In
virus-infected cells, these cytokines inhibit many RNA and DNA
viruses at various stages of their replication cycles [1, 2]. During
tumor-cell development, type I IFN exert direct cytotoxic or anti-
proliferative functions and negatively regulate angiogenesis
[3, 4]. Furthermore, direct anti-viral and anti-tumor propertiesCorrespondence: Dr. John Hiscotte-mail: [email protected]
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
Eur. J. Immunol. 2009. 39: 527–540 DOI 10.1002/eji.200838832 Innate Immunity 527

of type I IFN are complemented by their role in immunosurveil-
lance [3]. Type I IFN are produced early during the innate
immune response and have important immunomodulatory effects
on T-lymphocyte and NK-cell activity [5–8], as well as on DC
differentiation and maturation [9–12].
Production of IFN-a/b following infection is dependent on the
activation of the IFN-regulatory factors (IRF)-3 and IRF-7 [13].
These IRF are phosphorylated by the kinases TANK-binding
kinase1 (TBK-1) and IkB kinase-e (IKK-e) following viral infection
and PRR signaling [14, 15] leading to their dimerization, nuclear
translocation and binding to the promoters of type I IFN genes
[16–19]. Although IRF-3 and IRF-7 share highly related structural
and functional characteristics [13, 20, 21], major biological and
functional distinctions can be identified between the two. While
IRF-3 is ubiquitously expressed [22], the basal expression of IRF-7
is low in most cells (with the notable exception of plasmacytoid DC
[23]) and is strongly increased by type I IFN-mediated signaling
[24]. IRF-3 and IRF-7 present both redundant and distinct tran-
scriptional profiles. Qualitative and quantitative differences exist in
type I IFN induction capacities of IRF-3 and IRF-7 upon activation:
IRF-7 can induce the efficient activation of IFNB genes and
multiple IFNA subtypes, whereas IRF-3 is a potent activator of IFNB
genes but not of IFNA genes with the exception of human IFNA1
and mouse Ifna4 genes [21, 24–26]. This observation may account
for the increased vulnerability to viral infections of Irf7�/� mice
compared with Irf3�/� mice [27]. The full range of IRF-3 or IRF-7
target genes has not been compared directly, although indepen-
dent studies in different cell types have suggested that the acti-
vation of IRF-3 or IRF-7 results in a broad perturbation in the
transcriptional profile of cellular genes involved in the anti-viral,
apoptosis and immune responses [28–30]. Generation of consti-
tutively active forms of IRF-3 and IRF-7, that translocate to the
nucleus, bind to the promoters of type I IFN genes and induce IFN-
a/b expression in the absence of viral infections [16–18], have
permitted further insight into the respective cellular functions of
these IRF. The constitutively active IRF-3 5D contains phospho-
mimetic aspartic acid substitutions at aa residues 396, 398, 402,
404 and 405, whereas IRF-7 D247–467 lacks the aa 247–467
region, which encompasses the inhibitory and nuclear export
domains [16–18]. Expression of IRF-3 5D triggered cell death in
numerous cell types, including primary human MF, monocyte-
derived DC, human lung epithelial carcinoma A549 cells, Jurkat
T cells and human embryonic kidney 293 cells [31, 32], whereas
expression of IRF-7 D247–467 in primary human MF induced the
expression of high levels of type I IFN and TRAIL as well as
strong cytostatic activity on different human cancer cells lines
in vitro [32].
In order to investigate the distinct mechanisms underlying
IRF-3 and IRF-7 function, as well as the possible use of these
transcription factors in viral and anti-tumor therapeutic strategies,
whole genome transcriptional profiles of primary human MFtransduced with adenoviral vectors expressing IRF-3 5D (Ad-F3) or
IRF-7 D247–467 (Ad-F7), were used to identify subsets of genes
triggered preferentially by active IRF-3 or IRF-7 expression. The
pro-apoptotic BH3-only protein Noxa was identified as a primary
IRF-3 target gene and an essential regulator of IRF-3, dsRNA and
vesicular stomatitis virus (VSV)-induced cell death. In contrast,
transduction of Ad-F7 was not pro-apoptotic, but rather induced
high levels of type I IFN and immunomodulatory cytokines, as well
as the expression of genes involved in antigen processing and
presentation. Murine MF transduced with Ad-F7 displayed an
increased capacity to cross-present antigens and activate
CD81 T lymphocytes in vitro. The potential immunotherapeutic
use of Ad-F7-transduced MF was further highlighted by their
enhanced tumor inhibitory effects in vivo using the B16 melanoma
model.
Results
Gene expression profiling in Ad-F3 or Ad-F7-transduced MU
To identify genes that may regulate distinct or overlapping
functions of IRF-3 and IRF-7, primary human MF preparations
were transduced with recombinant adenovirus-expressing GFP
(Ad-GFP), Ad-F3 or Ad-F7 vectors and evaluated by microarray
analysis. IRF-3 and IRF-7 transgene expression was readily
detected in transduced MF by immunoblot [32] and by tracking
the nuclear localization of GFP-tagged IRF-3 and IRF-7 proteins
following Ad-vector transduction (Fig. 1A). Seven of the 12
donors with the highest transduction efficiency (46–70% GFP
expression), as measured by flow cytometry (Fig. 1B) were
selected for genome-wide transcriptome analysis using Illumina
bead array. Hierarchical clustering of genes with an adjusted
p-value of o0.01 (1843 genes) showed that non-transduced (NT)
and control adenoviral vector (Ad-GFP) gene expression profiles
were nearly indistinguishable, whereas Ad-F3 and Ad-F7
profiles were distinct from these controls, as well as from each
other (Fig. 1C). Transduction of Ad-F7 in primary MF modulated
the expression of 292 genes with p-value o0.01 and fold-
change Z2.0 and r�2.0 (versus Ad-GFP values) compared
with 72 genes with Ad-F3. A subset of 49 genes was
commonly up-regulated by both active IRF-3 and active IRF-7
(Fig. 1D).
Ad-F3 and Ad-F7 versus Ad-GFP significant genes were
then manually refined and classified according to known
functions and pathways using manual searches in NCBI and the
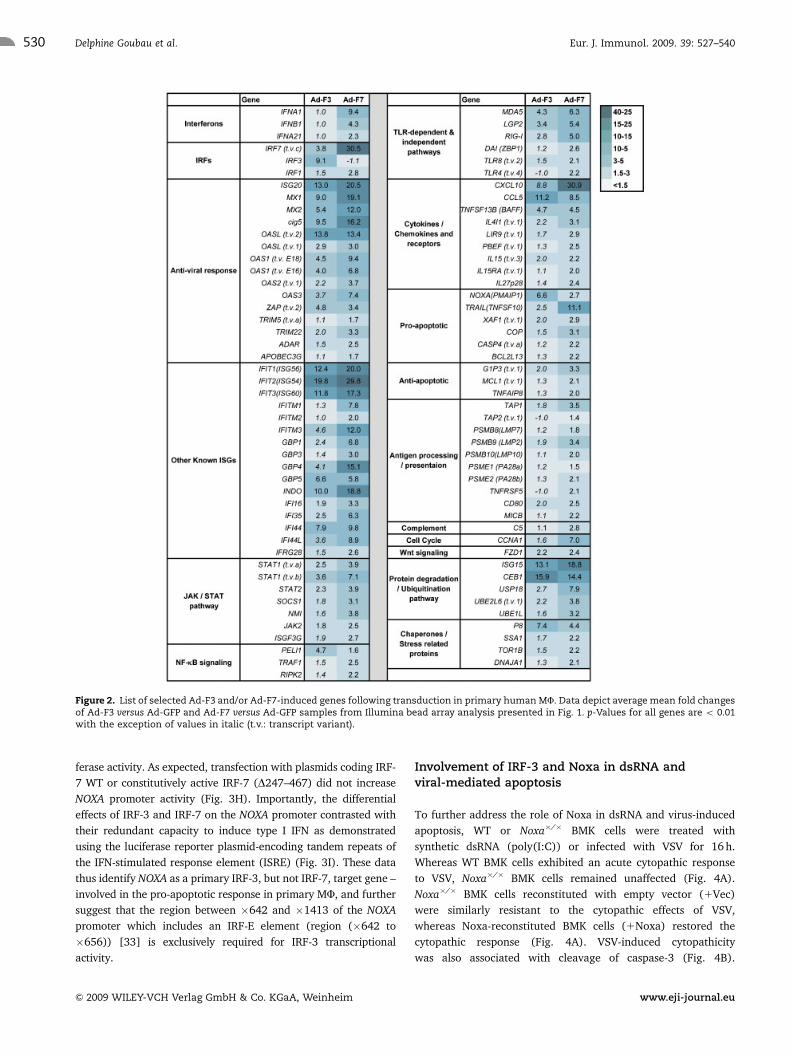
ontology tools DAVID and BIORAG. Figure 2 provides a partial
list of significantly up-regulated genes including genes involved
in innate immunity, apoptosis, antigen processing and presenta-
tion, as well as cytokines and chemokines. Microarray data
were validated by performing real-time PCR on 12 genes (TAP1,
MCL1, RIGI, MDA5, G1P3, TLR8, NOXA, OAS1, RANTES, PELI1,
PSME2 and C5) for 4 donors included in the array (Supporting
Information Fig. 1A). Pearson correlations that averaged
0.88 for all 12 genes (minimum 0.68 and maximum 0.99)
revealed strong linear relationships between real-time PCR
validation and microarray data (Supporting Information
Fig. 1B).
Eur. J. Immunol. 2009. 39: 527–540Delphine Goubau et al.528
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Involvement of Noxa in IRF-3-mediated apoptosis
The gene encoding the pro-apoptotic BH3-only protein Noxa
(Pmaip1) was more strongly induced in Ad-F3 as compared with
Ad-F7-transduced MF (6.6-fold change for Ad-F3 versus 2.7-fold
change for Ad-F7) (Fig. 2), a difference that was confirmed by
real-time PCR (Fig. 3A and Supporting Information Fig. 1A). The
increased expression of Noxa in Ad-F3-transduced MF was
independent of type I IFN, as demonstrated using a neutralizing
antibody for the type I IFN receptor (IFNAR) (Fig. 3A). Because
Noxa is known to induce mitochondrial membrane depolariza-
tion, we tested whether the intrinsic mitochondrial apoptotic
pathway was involved in Ad-F3-induced apoptosis. A 4-fold
increase in mitochondrial depolarization (as assessed by the loss
of MitoProbeTM DilC1(5) staining by flow cytometry) was
observed in MF transduced with Ad-F3 for 24 h, compared with
NT, Ad-GFP and Ad-F7 samples (Fig. 3B). Caspase-3 enzymatic
activity also increased 3.5-fold with Ad-F3 compared with NT
MF, whereas Ad-F7 transduction induced a 1.8-fold increase, as
measured by DEVD-AFC cleavage by fluorometric assay (Fig. 3C).
Disruption of mitochondrial membrane potential and increased
enzymatic activity of caspase-3 observed in Ad-F3-transduced MFcorrelated with increased cleavage of caspase-9 and caspase-3, as
well as the up-regulation of Noxa protein expression by
immunoblot (Fig. 3D). Ad-F7 and IFN-a-2b treatments did not
significantly increase caspase cleavage or Noxa protein levels.
Increased expression of Noxa at the RNA and protein levels, as
well as caspase-3 cleavage and enzymatic activity, was also
observed in Ad-F3-transduced human A549 epithelial carcinoma
cells relative to Ad-F7 and control samples (data not shown).
Importantly, use of pan-caspase inhibitor (Z-VAD-FMK) on these
cells following Ad-F3 transduction reduced caspase-3 enzymatic
activity to levels seen in controls without altering Noxa
expression levels (data not shown).
The direct contribution of Noxa in IRF-3-mediated apoptosis
was tested by assessing the effects of active IRF-3 in Noxa
knock-out and knock-down cells. In Noxa�/�, immortalized
epithelial baby mouse kidney (BMK) cells transduced with Ad-F3,
caspase-3 cleavage and mitochondrial depolarization were
reduced to levels seen in control Ad-GFP-transduced cells (Fig. 3E
and F). Using siRNA targeting, Noxa levels were reduced 60%
following transduction of Ad-F3 in primary human MF and a
corresponding 50% reduction in caspase-3 cleavage was also
observed (Fig. 3G).
Regulation of the NOXA promoter by IRF-3 was next evaluated,
using different promoter deletion constructs linked to a luciferase
reporter gene. The full-length (�1413 to 11) fragment contains
the previously identified IRF-E, CRE and p53 binding sites of the
NOXA promoter [33, 34]. Co-transfection of the plasmid-expres-
sing IRF-3 5D with this 1.4 kb fragment of the NOXA promoter in
human embryonic kidney 293 cells led to a 9-fold increase in
luciferase activity compared with control GFP plasmid or IRF-3 WT
transfected cells (Fig. 3H). Deletion to �677 reduced activation to
3.5-fold following active IRF-3 co-expression, whereas constructs
(�642 to 11) and (�360 to 11) displayed no increase in luci-
Figure 1. Distinct gene expression profiles of Ad-F3 and Ad-F7-transduced MF. Primary human MF from healthy donors weretransduced or not with Ad-GFP, Ad-F3 or Ad-F7. (A) Nuclear transloca-tion of GFP-tagged IRF proteins was monitored 24 h post-transductionusing the nuclear marker To-Pros-3. Confocal fluorescent images shownuclear staining (left), Ad-vector GFP expression (middle) and merge(right). Images are representative of cells for two donors. (B) Cells wereanalyzed by flow cytometry 24 h post-transduction to determine thepercentage of GFP positive cells. MF with the highest transductionefficiency (donors 1–7) were selected for microarray analysis. (C) RNAfrom MF (donors 1–7) was collected 24 h post-transduction, purified,amplified and labeled before Illuminas bead array and data analysiswas carried out as described in Materials and methods. Hierarchical geneaverage linkage clustering was performed using genes with po0.01.Each row represents the relative rank based levels of expression of asingle gene (low expression in light blue and high expression in darkblue) and each column represents a sample (28 samples total). Thesamples include seven donors and four conditions (NT, Ad-GFP, Ad-F3and Ad-F7). (D) Venn diagram represents the number of genes fromarray data with po0.01 and fold-change Z2.0 (up) or r2.0 (down) in Ad-F7 versus Ad-GFP and Ad-F3 versus Ad-GFP-transduced MF.
Eur. J. Immunol. 2009. 39: 527–540 Innate Immunity 529
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
ferase activity. As expected, transfection with plasmids coding IRF-
7 WT or constitutively active IRF-7 (D247–467) did not increase
NOXA promoter activity (Fig. 3H). Importantly, the differential
effects of IRF-3 and IRF-7 on the NOXA promoter contrasted with
their redundant capacity to induce type I IFN as demonstrated
using the luciferase reporter plasmid-encoding tandem repeats of
the IFN-stimulated response element (ISRE) (Fig. 3I). These data
thus identify NOXA as a primary IRF-3, but not IRF-7, target gene –
involved in the pro-apoptotic response in primary MF, and further
suggest that the region between �642 and �1413 of the NOXA
promoter which includes an IRF-E element (region (�642 to
�656)) [33] is exclusively required for IRF-3 transcriptional
activity.
Involvement of IRF-3 and Noxa in dsRNA andviral-mediated apoptosis
To further address the role of Noxa in dsRNA and virus-induced
apoptosis, WT or Noxa�/� BMK cells were treated with
synthetic dsRNA (poly(I:C)) or infected with VSV for 16 h.
Whereas WT BMK cells exhibited an acute cytopathic response
to VSV, Noxa�/� BMK cells remained unaffected (Fig. 4A).
Noxa�/� BMK cells reconstituted with empty vector (1Vec)
were similarly resistant to the cytopathic effects of VSV,
whereas Noxa-reconstituted BMK cells (1Noxa) restored the
cytopathic response (Fig. 4A). VSV-induced cytopathicity
was also associated with cleavage of caspase-3 (Fig. 4B).
Figure 2. List of selected Ad-F3 and/or Ad-F7-induced genes following transduction in primary human MF. Data depict average mean fold changesof Ad-F3 versus Ad-GFP and Ad-F7 versus Ad-GFP samples from Illumina bead array analysis presented in Fig. 1. p-Values for all genes are o 0.01with the exception of values in italic (t.v.: transcript variant).
Eur. J. Immunol. 2009. 39: 527–540Delphine Goubau et al.530
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Figure 3. Involvement of Noxa in IRF-3-mediated apoptosis. (A–D) Primary human MF were transduced or not with Ad-GFP, Ad-F3, Ad-F7 ortreated with IFN-a-2b for (D). (A) Transduced MF were plated with an isotype control (IgG) or a neutralizing antibody for the human IFNAR (anti-IFNAR2). Expression of human NOXA was analyzed 24 h later by real-time PCR. RQ was calculated by normalizing to BETA-ACTIN and Ad-GFP MF.Data are representative of four donors. (B) Cells were harvested at 24 h, incubated with the MitoProbeTM DilC1(5) dye, which accumulates inmitochondria with active membranes, and analyzed by flow cytometry. Data represent mitochondrial depolarization as the percentage of cell thathave lost MitoProbeTM DilC1(5) staining. Data are representative of two donors. (C) Whole-cell extracts (WCE) were prepared at 24 h and incubatedwith the fluorogenic substrate for caspase-3 (Ac-DEVD-AFC). Fold change (7SD; n 5 3) in caspase-3 enzymatic activity is represented by thefluorescence ratio between NT and transduced cells. Data are representative of two donors. (D) WCE were prepared at 24, 48 and 72 h, andsubjected to immunoblot analysis for caspase-3 and caspase-9 cleavage, Noxa expression and tubulin as loading control. Data are representativeof three donors. (E and F) WT and Noxa�/� (noted: �/�) BMK cells were transduced with Ad-GFP, Ad-F3 or Ad-F7. Cells were harvested at 24 h andanalyzed for (E) caspase-3 cleavage by immunoblot and tubulin as loading control and (F) mitochondrial depolarization as described in (B). Data arerepresentative of three independent experiments. (G) Primary human MF were electroporated with control siRNA (lane 1) or Noxa siRNA (lane 2)for 48 h before being transduced with Ad-GFP or Ad-F3. Cells were harvested 24 h post-transduction and subjected to immunoblot analysis as in (D)for caspase-3 cleavage, Noxa and tubulin as loading control. Noxa and cleaved-caspase-3 expression levels were quantified and normalized totubulin levels using the Scion Image 4.0 software. (H and I) 293 cells were transfected with plasmids harboring the luciferase reporter gene underthe control of (H) full length and deletion constructs of the NOXA promoter or of (I) the luciferase reporter plasmid-encoding tandem repeats of theIFN-stimulated response element (ISRE) and indicated IRF-3 or IRF-7-expressing plasmids. Relative luciferase activity (7SD; n 5 3) was analyzed24 h later after normalization with co-transfected Renilla luciferase encoding plasmid. Data are representative of three independent experiments.Casp: caspase.
Eur. J. Immunol. 2009. 39: 527–540 Innate Immunity 531
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Although poly(I:C) treatment did not induce a change in
BMK-cell morphology at this timepoint (Fig. 4A), caspase-3
cleavage was evident in WT cells and Noxa�/�cells reconstituted
with Noxa (1Noxa) (Fig. 4B). We next evaluated if the
ablation of Noxa in BMK cells had an effect on the capacity
of VSV to infect these cells. As demonstrated by RT-PCR
examining the expression of VSV-G mRNA, viral gene expression
occurred regardless of Noxa expression in these cells (Fig. 4C).
Futhermore, VSV-infected Noxa�/� cells continued to shed
virus for many days following infection (D.L., unpublished
observations).
To investigate the role of IRF-3 on cell death, human MF were
treated with the IRF-3 activator, poly(I:C). Increased expression of
ISG56 and Noxa, as well as caspase-3 cleavage, was observed by
immunoblot as early as 2 h post-treatment (data not shown), thus
suggesting a role for IRF-3 in the apoptotic response triggered in
the course of anti-viral defense. To further address this, WT and
Irf3�/� mouse embryonic fibroblasts (MEF) were transfected or
not with poly(I:C) for 20 h. While dsRNA treatment induced
massive cell death in WT MEF, the morphology of treated and non-
treated Irf3�/� MEF was nearly indistinguishable (Fig. 5A).
Observed differences in cytopathology correlated with the 75%
reduction in Noxa mRNA levels following 12 h of poly(I:C) treat-
ment in Irf3�/� MEF compared with WT MEF (Fig. 5B).
ChIP studies were performed to establish a direct association
of IRF-3 with the NOXA promoter in human HT1080 fibro-
sarcoma cells transfected with poly(I:C). A 4-fold increase in IRF-
3 binding to the NOXA promoter was observed within 2 h after
poly(I:C) treatment as compared with the 0 h control
(Fig. 5C). The IFNB promoter used as a positive control also
exhibited an increased IRF-3 association (Fig. 5C). Altogether,
these data support a direct involvement for IRF-3 in Noxa-
mediated apoptosis following viral infection.
Antigen cross-presentation in Ad-F7-transduced MU
From the microarray analysis, Ad-F7 but not Ad-F3 transduction
into primary human MF up-regulated the expression of genes
involved in antigen processing and presentation in the immuno-
logical synapse (Fig. 2). Transduction of active IRF-7 up-regulated
the expression of all three subunits of the immunoproteasome that
replace the catalytic 20S proteasomal-core: LMP2, LMP10 and
LMP7 as well as PA28A and -B subunits of the proteasome activator
(Fig. 2 and Fig. 6A). Furthermore, active IRF-7 induced the
expression of the TAP1 and TAP2 subunits of TAP responsible for
transporting peptides into the lumen of the ER (Fig. 2 and Fig.
6A). Co-stimulatory molecules of the MHC–peptide complex and
MF activation markers CD40, CD80 and CD86 were also up-
regulated in Ad-F7-transduced MF (Fig. 2 and Fig. 6C). The use of
a neutralizing antibody for the IFNAR2 in a second microarray
data set and in real-time PCR analysis following Ad-F7 transduc-
tion demonstrated that type I IFN mediated the increased
expression of genes involved in antigen processing and presenta-
tion (Fig. 6A, Supporting Information Figs. 2 and 3).
Given the increased expression of genes involved in antigen
processing and presentation following Ad-F7 transduction, we
sought to determine if Ad-F7 MF possessed an increased capacity
to cross-present exogenous antigens in the context of MHC class I
and to activate CD81 T cells in vitro. Murine peritoneal MF were
transduced with Ad-vectors, exposed to 0.125–2.0 mg/mL of
soluble chicken egg OVA and co-cultured with syngeneic naıve
OVA-specific MHC class I-restricted CD81 T lymphocytes isolated
from the spleen of transgenic mice that bear an MHC-class-I
Figure 4. Involvement of Noxa in dsRNA and VSV-induced apoptosis.WT and Noxa�/� BMK cells, or Noxa�/� BMK cells stably transfectedwith empty vector (1 Vec) or complemented with a Noxa expressionplasmid (1 Noxa) were treated with synthetic dsRNA (poly(I:C)) orinfected with VSV (MOI 0.1). Cell death was monitored 16 h post-infection by (A) morphological changes or (B) caspase-3 cleavage byimmunoblot analysis of WCE using actin as internal control. (C) Theexpression of VSV-G mRNA was assayed by RT-PCR 24 h after VSVinfection in these cells. Gapdh was used as an internal control. Data arerepresentative of three independent experiments.
Eur. J. Immunol. 2009. 39: 527–540Delphine Goubau et al.532
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

restricted OVA-specific T cell recptor(OT-1) at an effector:target
(E:T) ratio of 1:2. Supernatants were assayed for IL-2 production
by activated OT-I T lymphocytes by ELISA at 48 h. The processing
and presentation of OVA by Ad-F7 MF increased the production of
IL-2 by OT-I cells in a dose-specific response; differences with Ad-
GFP-transduced MF were strongest when 0.5 and 1.0 mg/mL
of OVA were used (p-values 0.0049 and 0.049, respectively)
(Fig. 6B). Similar results were obtained at an E:T ratio of 1:6 (data
not shown). The increased cross-presentation capacity of Ad-F7-
transduced MF was mediated by type I IFN, as blocking the IFN
response using a neutralizing antibody specific for the murine
IFNAR1 strongly inhibited IL-2 production by OT-1 lymphocytes
(p-value 0.0006) (Fig. 6B). Proteasome-mediated proteolysis was
required for the increased antigen processing induced by active
IRF-7 as pre-treatment of cells with a proteasome inhibitor,
lactacystin, before OVA exposure completely blocked the cross-
presentation capacity of Ad-F7 MF (data not shown).
Next, we wanted to determine if the selective effect of Ad-F7
on cross-presentation may be skewed by higher antigen capture.
To evaluate antigen uptake, the capture of red-labeled (using
PKH-26 fluorescent dye) apoptotic CTLL2 cells (CTL line) by the
GFP-expressing MF was monitored by flow cytometry (Support-
ing Information Fig. 4). No significant differences in antigen
capture were observed in MF transduced with Ad-GFP or Ad-F7
(65 and 58% of double-positive (GFP and PKH-26) MF, respec-
tively). Taken together, these data demonstrated that Ad-F7
transduction and subsequent type I IFN production increased the
expression of components of the MHC class I peptide loading
machinery and the cross-presentation capacity of MF.
Increased activation of surrounding MU byAd-F7-transduced MU
As successful immunotherapeutic approaches rely on enhancing
immunogenicity to promote a protective host response to
malignancies, we next evaluated whether Ad-F7 transduction
increased the expression of immunomodulatory genes. Array
results demonstrated that Ad-F7 induced the expression of various
cytokines and chemokines including CXCL10 (IP-10) and CCL5
(RANTES) – monocytic and T-cell chemoattractants, IL15 and
IL15RA – critical regulators of NK and CD81 T-cell proliferation,
activation and survival [35] and TNFSF13B (also known as B cell-
activating factor (BAFF) or BLyS) and pre-B-cell-enhancing factor
(PBEF) – B-cell survival and growth factors [36, 37] (Fig. 2). The
expression of the IL-6/IL-12 familly member, IL27p28, was also
induced by Ad-F7 transduction (Fig. 2). With the exception of
TNFSF13B, expression of these genes was highly dependent on
IFN-a/b triggering of the IFNAR (Supporting Information Fig. 3).
To determine whether Ad-F7-transduced MF could induce the
maturation of surrounding MF through secretion of soluble
factors, primary human MF were transduced with Ad-GFP or
Ad-F7 or pre-treated with recombinant IFN-a-2b for 24 h,
extensively washed and placed in an upper chamber overlaying
non-treated MF for an additional 24 h; up-regulation of
maturation markers CD40, CD80 and CD86 was assessed by real-
time PCR. MF transduced with Ad-F7 (in the upper chamber)
displayed increased expression of CD40, CD80 and CD86, at
levels higher than observed with 1000 IU/mL of IFN-a-2b.
Furthermore, MF in the lower chamber acquired a more mature
phenotype when sharing supernatants with Ad-F7-transduced
MF compared with non-treated or IFN-a-2b-treated MF(Fig. 6C). Importantly, MF in the lower chamber did not express
GFP as asssessed by flow cytometry, demonstrating that increased
expression of co-stimulatory molecules was not a result of
adenovirus transduction in these cells (data not shown). As a
whole, these data demonstrated that Ad-F7-transduced MF were
Figure 5. Role of IRF-3 in dsRNA-mediated apoptosis and binding ofIRF-3 to the Noxa promoter. (A and B) WT and Irf3�/� MEF weretransfected with 10 mg of poly(I:C) using lipofectamine 2000 (Lipo).(A) Cell death was monitored by morphological changes at 20 h and(B) expression levels of murine Noxa and Ifnb mRNA was determined byreal-time PCR 12 h post-transfection. RQ was calculated by normalizingto Gapdh and Lipo samples. (C) IRF-3 binding to the NOXA and IFNBpromoter. HT1080 human fibrosarcoma cells were left untransfected ortransfected with dsRNA using Lipofectin for 2 or 4 h. ChIP studies wereperformed with normal rabbit serum (IgG) or IRF-3 polyclonal Ab. Dataare representative of three independent experiments.
Eur. J. Immunol. 2009. 39: 527–540 Innate Immunity 533
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

capable of inducing the maturation of surrounding MF via the
secretion of immunostimulatory factors.
Increased anti-tumor properties of Ad-F7-transducedmurine MU in vivo
As Ad-F7 was proven efficient in increasing the in vitro cytostatic
activity of primary human MF against breast cancer cell lines
SK-BR-3 and MCF-7 (breast cancer cells) [32], the anti-tumor
activity of Ad-F7-transduced MF was assessed using the
aggressive and poorly immunogenic B16.F0 melanoma model.
First, the effect of IRF-7 on the in vitro inhibitory effect of MF was
tested in co-cultures of Ad-GFP or Ad-F7-transduced syngeneic
peritoneal murine MF with B16.F0 cells at different E:T ratios.
Ad-F7-transduced MF strongly inhibited the proliferation of B16
cells (>70% at E:T ratios of 1:1 and 3:1) after 4 days of co-
culture, whereas GFP-expressing MF displayed a very limited
Figure 6. Increased antigen cross-presentation by active IRF-7-expressing MF. (A) Ad-GFP, Ad-F3, Ad-F7-transduced or IFN-a-2b-treated primaryhuman MF were plated with an isotype control (IgG) or a neutralizing antibody for the human IFNAR (anti-IFNAR2). Expression of human LMP2,LMP7, LMP10, PA28B and TAP1 was analyzed 24 h later by real-time PCR. RQ was calculated by normalizing to GAPDH and Ad-GFP MF. (B) NT, Ad-GFP, Ad-F7-transduced or recombinant murine IFN-a-treated peritoneal MF were plated and cells in the right panel were treated with controlantibody (Ab) or a neutralizing antibody for the murine IFNAR (anti-IFNAR1). Twenty-four hours post-transduction, MF were presented with0.125–2.0 mg/mL (1.0 mg/mL for the right panel) of OVA for 8 h and washed thoroughly. OT-1-derived CD81 T cells were incubated at a 1:2 E:T ratiowith Ad-IRF-transduced MF. Graph represents the average murine IL-2 in pg/mL (7SD) in supernatants collected at 48 h as assayed by ELISA(�po0.05; ��po0.005 with n 5 3). Data are representative of three independent experiments. (C) Primary human MF were transduced with Ad-GFP,Ad-F7 or treated recombinant human IFN-a-2b. 24 h post-treatment, cells were harvested and extensively washed to remove IFN-a-2b from themedia. MF were then placed on 0.02 mm Anopores membranes over non-treated (NT) MF. Twenty four-hours post-co-culture, MF from top andbottom chambers were harvested and the expression of CD40, CD80 and CD86 mRNA was analyzed by real-time PCR as in (A). Data arerepresentative of three independent experiments.
Eur. J. Immunol. 2009. 39: 527–540Delphine Goubau et al.534
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
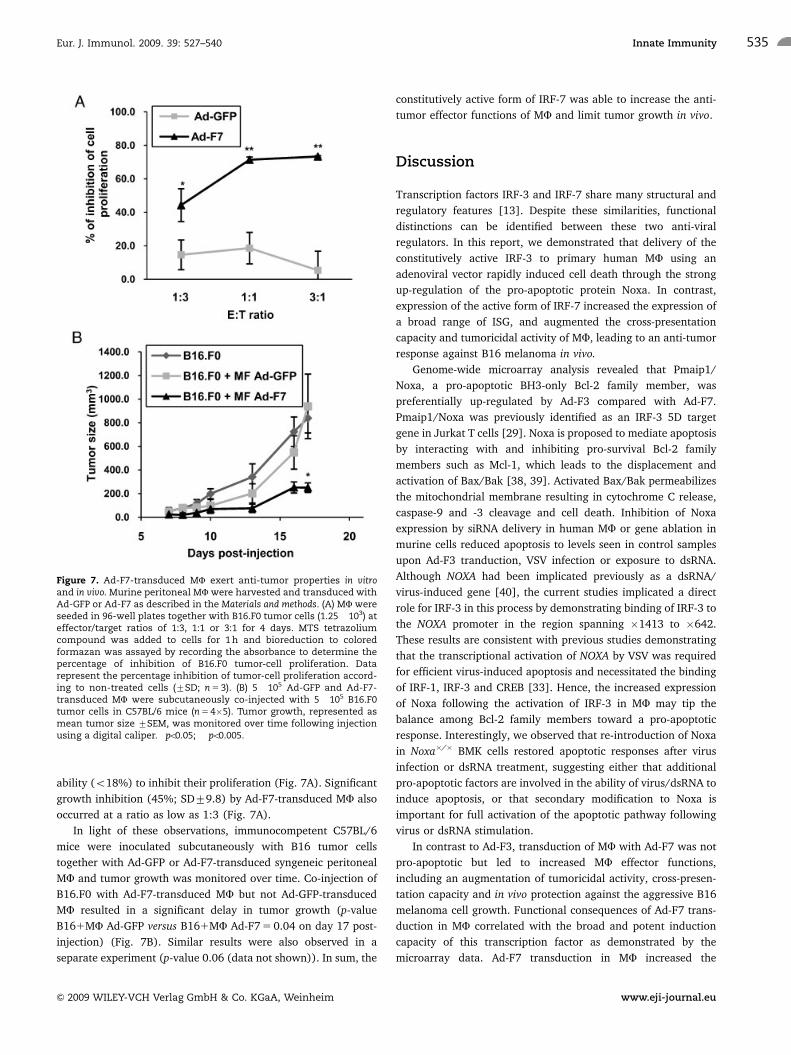
ability (o18%) to inhibit their proliferation (Fig. 7A). Significant
growth inhibition (45%; SD79.8) by Ad-F7-transduced MF also
occurred at a ratio as low as 1:3 (Fig. 7A).
In light of these observations, immunocompetent C57BL/6
mice were inoculated subcutaneously with B16 tumor cells
together with Ad-GFP or Ad-F7-transduced syngeneic peritoneal
MF and tumor growth was monitored over time. Co-injection of
B16.F0 with Ad-F7-transduced MF but not Ad-GFP-transduced
MF resulted in a significant delay in tumor growth (p-value
B161MF Ad-GFP versus B161MF Ad-F7 5 0.04 on day 17 post-
injection) (Fig. 7B). Similar results were also observed in a
separate experiment (p-value 0.06 (data not shown)). In sum, the
constitutively active form of IRF-7 was able to increase the anti-
tumor effector functions of MF and limit tumor growth in vivo.
Discussion
Transcription factors IRF-3 and IRF-7 share many structural and
regulatory features [13]. Despite these similarities, functional
distinctions can be identified between these two anti-viral
regulators. In this report, we demonstrated that delivery of the
constitutively active IRF-3 to primary human MF using an
adenoviral vector rapidly induced cell death through the strong
up-regulation of the pro-apoptotic protein Noxa. In contrast,
expression of the active form of IRF-7 increased the expression of
a broad range of ISG, and augmented the cross-presentation
capacity and tumoricidal activity of MF, leading to an anti-tumor
response against B16 melanoma in vivo.
Genome-wide microarray analysis revealed that Pmaip1/
Noxa, a pro-apoptotic BH3-only Bcl-2 family member, was
preferentially up-regulated by Ad-F3 compared with Ad-F7.
Pmaip1/Noxa was previously identified as an IRF-3 5D target
gene in Jurkat T cells [29]. Noxa is proposed to mediate apoptosis
by interacting with and inhibiting pro-survival Bcl-2 family
members such as Mcl-1, which leads to the displacement and
activation of Bax/Bak [38, 39]. Activated Bax/Bak permeabilizes
the mitochondrial membrane resulting in cytochrome C release,
caspase-9 and -3 cleavage and cell death. Inhibition of Noxa
expression by siRNA delivery in human MF or gene ablation in
murine cells reduced apoptosis to levels seen in control samples
upon Ad-F3 tranduction, VSV infection or exposure to dsRNA.
Although NOXA had been implicated previously as a dsRNA/
virus-induced gene [40], the current studies implicated a direct
role for IRF-3 in this process by demonstrating binding of IRF-3 to
the NOXA promoter in the region spanning �1413 to �642.
These results are consistent with previous studies demonstrating
that the transcriptional activation of NOXA by VSV was required
for efficient virus-induced apoptosis and necessitated the binding
of IRF-1, IRF-3 and CREB [33]. Hence, the increased expression
of Noxa following the activation of IRF-3 in MF may tip the
balance among Bcl-2 family members toward a pro-apoptotic
response. Interestingly, we observed that re-introduction of Noxa
in Noxa�/� BMK cells restored apoptotic responses after virus
infection or dsRNA treatment, suggesting either that additional
pro-apoptotic factors are involved in the ability of virus/dsRNA to
induce apoptosis, or that secondary modification to Noxa is
important for full activation of the apoptotic pathway following
virus or dsRNA stimulation.
In contrast to Ad-F3, transduction of MF with Ad-F7 was not
pro-apoptotic but led to increased MF effector functions,
including an augmentation of tumoricidal activity, cross-presen-
tation capacity and in vivo protection against the aggressive B16
melanoma cell growth. Functional consequences of Ad-F7 trans-
duction in MF correlated with the broad and potent induction
capacity of this transcription factor as demonstrated by the
microarray data. Ad-F7 transduction in MF increased the
Figure 7. Ad-F7-transduced MF exert anti-tumor properties in vitroand in vivo. Murine peritoneal MF were harvested and transduced withAd-GFP or Ad-F7 as described in the Materials and methods. (A) MF wereseeded in 96-well plates together with B16.F0 tumor cells (1.25� 103) ateffector/target ratios of 1:3, 1:1 or 3:1 for 4 days. MTS tetrazoliumcompound was added to cells for 1 h and bioreduction to coloredformazan was assayed by recording the absorbance to determine thepercentage of inhibition of B16.F0 tumor-cell proliferation. Datarepresent the percentage inhibition of tumor-cell proliferation accord-ing to non-treated cells (7SD; n 5 3). (B) 5�105 Ad-GFP and Ad-F7-transduced MF were subcutaneously co-injected with 5�105 B16.F0tumor cells in C57BL/6 mice (n 5 4�5). Tumor growth, represented asmean tumor size 7SEM, was monitored over time following injectionusing a digital caliper. �po0.05; ��po0.005.
Eur. J. Immunol. 2009. 39: 527–540 Innate Immunity 535
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

expression of tumoricidal molecules (TRAIL and IFN-a/b) [32]
and important immunomodulatory cytokines and chemokines
(IP-10, RANTES, IL-15, IL-27p28, BAFF and PBEF). In addition,
transduction with Ad-F7 in MF induced the expression of genes
involved at different steps of MHC class I-mediated antigen
processing, including genes regulating proteasome protein
degradation (LMP2, LMP10 and LMP7), proteasome activation
(PA28-a/b), and peptide translocation to the ER (TAP-1 and -2).
Increased expression of LMP2, LMP7 and LMP10 is particularly
important because these subunits displace the constitutive
proteasomal subunits b1, b2 and b5 and are incorportated
into an alternative form of the proteasome known as the immu-
noproteasome, which alters the catalytic site and possesses an
increased ability to produce peptides with a proper motif
for efficient binding to MHC class I molecule [41–45].
Increased expression of genes involved in antigen processing was
complemented by the up-regulation of T lymphocyte
co-stimulatory molecules CD40, CD80 and CD86, suggesting
that Ad-F7-transduced MF may acquire higher cross-priming
abilities.
Qualitative and quantitative differences in the type I IFN loop
induced following IRF-3 or IRF-7 activation may play a role in their
distinct biological functions. However, our data suggest that the
main mechanism by which IRF-3 or IRF-7 regulate cell viability
may not be, as previously reported, via the IFN-b-mediated up-
regulation of p53 [46] and/or p53-induced Noxa expression [34].
Ad-F7-transduced MF produced higher levels of IFN-b compared
with Ad-F3 MF. In addition, p53 mRNA up-regulation was not
observed in microarray and real-time PCR analyses (data not
shown). These data support work demonstrating that apoptosis
induced by dsRNA or Newcastle Disease Virus was independent of
type I IFN and that the activity of p53 was not required for IRF-3
5D-mediated apoptosis [47]. Furthermore, the expression of Noxa
does not appear to be regulated by the type I IFN loop, because
neutralizing the IFNAR did not alter Noxa expression levels
following Ad-F3 transduction and increased expression of Noxa
following recombinant IFN-a treatment was not observed. This
result is consistent with earlier studies using IFN-a-unresponsive
mutant human cell lines or Jurkat T cells treated with neutralizing
antibodies for IFN-a/b that still exhibited Noxa up-regulation in
response to dsRNA or IRF-3 5D expression, respectively [29, 40].
In contrast, many of the acquired immune and anti-tumor func-
tions of Ad-F7 MF are likely dependent on the type I IFN loop, as
demonstrated by the neutralization of IFNAR, consistent with
previous studies demonstrating that type I IFN favors the differ-
entiation of monocytes to DC [9–12], the generation of the
immunoproteasome following virus infection [48], the cross-
priming of CD81 T cells [49, 50], as well as increased in tumor-
icidal effector functions of DC [51–53].
The consequences of IRF-3- and Noxa-induced cell death,
appear independent of the type I IFN loop, and the relationship to
the control of viral replication and the development of an
immune response remain to be elucidated. One hypothesis is that
Noxa and the mitochondrial apoptotic pathway may be required
for the full activation of IRF-3, as this activation was reduced in
Bax�/�MEF following virus infection [54]. A second hypothesis is
that induction of Noxa by IRF-3 triggers apoptosis in infected
cells in order to clear these rapidly. It was recently reported that
IRF-3-deficient cells do not enter apoptosis when infected with
Sendai virus and continuously produce virus [55]. We have
observed a similar response to VSV in Noxa�/� cells, which fail to
undergo rapid apoptosis following infection and continue to shed
virus for many days thereafter (D.L., unpublished observations).
Together, these findings suggest that IRF-3 and Noxa may control
cell viability in order to avoid in vivo viral persistence.
Our data highlight the importance of the type I IFN loop
induced by IRF-7 in the acquisition of tumoricidal and cross-
priming functions by MF, suggesting their potential in the
development of anti-tumor immunotherapies. Compared with
systemic delivery of IFN-a or pre-treatment of MF with IFN-a, the
intra-tumoral injection of MF transduced with Ad-F7 may result
in an improved immunity and tumor growth inhibition via the
release of type I IFN in the tumor environment, the full induction
of MF effector functions, activation of surrounding APC and
effector immune cells capable of directly mediating tumor cell
killing, including cytotoxic T lymphocytes.
Materials and methods
MU preparation
PBMC were isolated by Ficoll gradient from fresh apheresis with
informed and institutionally approved consent forms from
healthy donors. Monocytes were isolated from PBMC by magnetic
cell sorting using anti-CD14-conjugated microbeads (Stem Cell
Technologies, Vancouver, Canada) and Automacs (Miltenyi
Biotec Auburn, CA, USA) and cultured for 7 days in Iscove media
(Wisent Technologies, Rocklin, CA, USA) supplemented with 2%
human serum type A/B (Wisent Technologies), 700 U/mL of GM-
CSF (a generous gift from Cangene Corporation, Mississauga,
Canada) and antibiotics in gas permeable thermoplastic non-
adherent culture bags (EVAMs, IDM, Paris, France). On day 7,
MF were harvested and re-suspended in complete McCoy’s 5A
medium (supplemented with 10% FBS and antibiotics) (Wisent
Technologies). MF differentiation and purity were analyzed by
flow cytometry as in [32]. Peritoneal MF were exuded from
C57BL/6 (retired breeders) mice (Charles River Laboratories,
Wilmington, MA, USA) by sterile lavage, plated in complete
DMEM (Wisent Technologies), allowed to attach for 2 h and
extensively washed to remove non-adherent cells.
Adenoviral vectors and transduction
Construction, production and purification of the three adenoviral
vectors Ad-GFP, Ad-GFPq/IRF-3 5D (Ad-F3) and Ad-GFPq/IRF-7
D247–467 (Ad-F7), as well as transduction of primary MF and
cell lines used in this study were performed as previously
described in [32].
Eur. J. Immunol. 2009. 39: 527–540Delphine Goubau et al.536
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Mice and cell lines
Retired breeders and 4–6-wk-old female C57BL/6 mice (Charles
River Laboratories) and OT-I mice: C57BL/6-Tg (TcraTcrb)
1100Mjb/J (Jackson Laboratory, Bar Habor, ME, USA) were
handled according to institutionally approved Animal Use
Protocol. Tumor size was measured using an electronic caliper
and calculated using the formula (length�width2/2). The
C57BL/6-derived B16.F0 melanoma cell lines, 293 human
embryonic kidney cells, HT1080 fibrosarcoma cells and WT and
Irf3�/� MEF [26] were cultured in complete DMEM (Wisent
Technologies). WT and Noxa knock-out BMK cells were a gift
from E. White (Rutgers University, Piscataway, NJ, USA), were
established as described previously [56] and cultured in complete
DMEM. Early passage Noxa knock-out BMK cells were stably
transfected with empty pBabepuro plasmid (Noxa�/�1 Vec cells),
or Noxa pBabepuro (Noxa�/�1 Noxa) as described previously
[40]. Results from representative clones are presented.
Reagents
Nuclear staining was done using To-PROs-3 iodide (Molecular
Probes, Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s instructions, and confocal analysis was
done as described in [57]. Apoptotic MF were analyzed by
MitoProbeTM DilC1(5) staining to detect active mitochondrial
membranes (Molecular Probes, Invitrogen). Fourteen percent
acrylamide gels were run when blotting for Noxa (114C307,
Calbiochem, Merck KgaA, Darmstadt, Germany) caspase-3,
cleaved-caspase-3 Asp175, caspase-9 (]9662, ]9661 and ]9502,
respectively, Cell Signaling Technology, Danvers, MA, USA) and
a-tubulin (B-7) (sc-5286, Santa Cruz, Santa Cruz, CA, USA).
Caspase activity was determined as previously described [31]
using Ac-DEVD-AFC (Biomol International, L.P. Plymouth
Meeting, PA, USA). MF were treated with 1000 IU/mL
of recombinant human IFN-a-2b (IntronsA, Schering, Pointe-
Claire, Canada) or 2000 IU/mL of recombinant murine IFN-a(Millipore, Temecula, CA, USA). Human IFNAR was neutralized
using mouse monoclonal antibody against human IFN-a/breceptor chain 2 (anti-IFNAR2) (PBL Biomedical Laboratories,
Piscataway, NJ, USA) or treated with control isotype (IgG2a)
(eBioscience, San Diego, CA, USA) at 20 mg/mL. For cross-
presentation study, purified mouse monoclonal antibodies
specific for the murine IFNAR1 (MARI-5A3) and control antibody
against human IFNGR-1 (GIR-208) (generous gifts from R.D.
Schreiber’s laboratory (St. Louis, MO, USA) [58]) were used at
10 mg/mL. Poly(I:C) (Sigma-Aldrich, St-Louis, MO, USA) and
lipofectamine 2000 (Invitrogen) were also used in this study.
BMK cells were infected with 0.1 MOI of VSV-HR strain (Indiana
serotype). Anopores 0.02 mm membranes (NuncA/S, Roskilde,
Denmark) were used for MF maturation study. Proliferation
assay was performed as described in [32] using CellTiter 96s
Aqueous One Solution Cell Proliferation Assay (MTS assay:
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-
sulfophenyl)-2H-tetrazolium) (Promega, Madison, WI, USA) 4
days after B16 and murine MF co-culture.
RNA preparation and bead array gene expressionanalysis
DNase-treated total RNA was prepared as described in [32].
Sample amplification for microarray analysis was performed using
200 ng of total RNA using Illuminas Total Prep RNA Amplification
Kit (Ambion, Austin, TX, USA). Samples were labeled by
incorporating biotin-16-UTP (Enzo Life Sciences, Farmingdale,
NY, USA) at a 1:1 ratio with unlabeled UTP. Eight hundred and
fifty nanograms of labeled, amplified material was hybridized to
the Illuminas HumanRef-8 BeadChip according to the manufac-
turer’s instructions (Illumina, San Diego, CA, USA). The signal was
then developed with streptavidin-Cy3 (Amersham, GE Healthcare
Bio-sciences, Little Chalfont, UK) and the BeadChip was scanned
with the Illuminas BeadArray reader. After scanning, microarray
data quality was checked by evaluating chip-dependent (biotin,
hybridization, stringency) and sample-dependent (gene intensity,
labeling, background) controls and by comparing sample distribu-
tions and statistics.
Pre-processing, analysis and clustering of microarraydata
Pre-processing was done in R (http://www.r-project.org/) using
the Linear Models for Microarray Analysis (LIMMA) package.
Microarray data were quantile normalized and values smaller
than the background cutoff were replaced with this value. The
24 354-row matrix was reduced by filtering non-expressed genes,
i.e. genes with background value for all samples. A data matrix of
8519 genes (rows) by 28 samples, 4 conditions�7 donors
(columns) was constructed. LIMMA was used to identify
significant differentially expressed genes [59]. Four contrasts of
interest, i.e. NT versus Ad-GFP, Ad-F3 versus Ad-GFP, Ad-F7 versus
Ad-GFP and Ad-F7 versus Ad-F3 were defined. The data were
log2-transformed before fitting a linear model for each gene. To
control the proportion of false-positive calls in all positive calls at
the desired significance level, the p-value was adjusted by
applying a false discovery rate of 0.01. Hierarchical gene average
linkage clustering was performed using Genesis (developed by
Alexander Sturn, [60]). Microarray data have been deposited in
the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.-
gov/geo/) under accession no. GSE12120.
RT-PCR and real-time PCR
Total RNA was reverse transcribed as described in [32]. Real-time
PCR primers were designed using the primer3 website
(primer3_www.cgi v. 0.2) and listed in Supporting Information
Eur. J. Immunol. 2009. 39: 527–540 Innate Immunity 537
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Table 1. Human NOXA, GAPDH and murine Gapdh primers
used are described in [54, 61, 62], respectively. cDNA was
amplified using SyBR Green I PCR master mix (Applied
Biosystems, Foster City, CA, USA) and the data were collected
using the AB 7500 Real-Time PCR System (Applied Biosystems)
and analyzed by Comparative CT Method using the SDS v1.3.1
Relative Quantification (RQ) Software. For semiquantitative
RT-PCR, amplification products were resolved on an agarose
gel and digital image of the ethidium bromide stained bands
inverted for presentation.
ChIP
Cellular proteins were cross-linked to chromatin with 1%
formaldehyde for 30 min at 371C. After sonication, protein–DNA
complexes were immunoprecipitated from nuclear extracts by
using human IRF-3 antiserum (M. David, University of California,
San Francisco, CA, USA) or normal rabbit serum, followed by
capture on protein A sepharose beads (GE Healthcare). After IP
and elution from the resin, DNA–protein cross-linking was
reversed with 250 mM NaCl at 651C for 16 h and the DNA was
purified using the Qiaquick PCR Purification kit (Qiagen). PCR
amplification of NOXA and IFNB gene sequences was performed
using primers detailed in Supporting Information Table 1.
Cross-presentation assay
Twenty-four hours post-transduction and antibody treatments,
MF were presented with OVA (Sigma, Oakville, Canada) for 8 h
and washed thoroughly. CD81 T cells were isolated from the
spleen of OT-I mice and purified using the EasySeps mouse CD8
positive selection kit (Stem Cell Technologies). OT-1 mice
contain a transgenic T-cell receptor designed to recognize OVA
residues 257–264 in the context of H-2 Kb MHC class I molecules.
Supernatants were collected after MF and T-cell co-culture in
96-well plate and assayed by ELISA for IL-2 production
(eBioscience).
Luciferase assays, plasmids and siRNA
For reporter gene assay, 293 cells (1�105 in 24-well plate) were
transfected using lipofectamine 2000 (Invitrogen) with 500 ng of
IRF-3 WT, IRF-3 5D, IRF-7 WT and IRF-7 D247–467 pEGFP-C1
constructs as previously described in [16–18]. Two hundred
nanogram of pRLTK reporter (Renilla luciferase for internal
control) and 200 ng of pGL-3 plasmids harboring the Firefly
luciferase reporter gene expressed under the control of the NOXA
promoter. The full-length (1413 bp) fragment of the NOXA
promoter was amplified by PCR from human Hela cell genomic
DNA, and the PCR product cloned directly into pGEM-T easy
vector (Promega). After sequence confirmation, promoter
sequences were liberated from pGEM-T easy with EcoRI, blunted
with Klenow fragment and ligated into a blunted SacI site in
pGL3-Basic by blunt-end ligation. Proper promoter orientation
was confirmed by restriction analysis and sequencing. The Noxa
promoter/luciferase deletion constructs were generated by PCR
using the �1413 pGL3-Basic-Noxa-promoter as template and the
forward primers (Supporting Information Table 1). A common
reverse primer (Supporting Information Table 1) within the
luciferase gene was used to generate the truncated PCR products,
which were then digested with BglII and NcoI and directionally
ligated into pGL3-basic. Primary human MF were electroporated
as described previously in [62] using Noxa-specific RNAi
sequences described in [33], plated for 48 h and transduced for
24 h with Ad-vectors.
Acknowledgements: Grant support: Canadian Institutes of
Health Research (CIHR), the National Cancer Institute of
Canada, with the support of the Canadian Foundation for AIDS
Research. CIHR Senior Investigator Award (J. Hiscott) and Senior
Chercheur Boursier Award (R. Lin). NIH grant R01 AI068133
(D. Leaman). The authors thank the following people for their
help on specific assays: Alessandra Nardin with primay human
macrophage preparations, Moıra Franc-ois with cross-
presentation assays, Simon Leveille, Moutih Rafei and Tiejun
Zhao for the B16 murine model, Genevieve Boucher and Peter
Wilkinson for microarray data analysis, Tiannan Chen provided
help with Western blots and Guy Klaiman for caspase enzymatic
activity assays.
Conflict of interest: The authors declare no financial or
commercial conflict of interest.
References
1 Stetson, D. B. and Medzhitov, R., Type I interferons in host defense.
Immunity 2006. 25: 373–381.
2 Sadler, A. J. and Williams, B. R., Interferon-inducible antiviral effectors.
Nat. Rev. Immunol. 2008. 8: 559–568.
3 Dunn, G. P., Koebel, C. M. and Schreiber, R. D., Interferons, immunity and
cancer immunoediting. Nat. Rev. Immunol. 2006. 6: 836–848.
4 Ferrantini, M., Capone, I. and Belardelli, F., Interferon-alpha and cancer:
mechanisms of action and new perspectives of clinical use. Biochimie
2007. 89: 884–893.
5 Martinez, J., Huang, X. and Yang, Y., Direct action of type I IFN on NK cells
is required for their activation in response to vaccinia viral infection
in vivo. J. Immunol. 2008. 180: 1592–1597.
6 Sun, S., Zhang, X., Tough, D. F. and Sprent, J., Type I interferon-mediated
stimulation of T cells by CpG DNA. J. Exp. Med. 1998. 188: 2335–2342.
7 Marrack, P., Kappler, J. and Mitchell, T., Type I interferons keep activated
T cells alive. J. Exp. Med. 1999. 189: 521–530.
Eur. J. Immunol. 2009. 39: 527–540Delphine Goubau et al.538
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

8 Kolumam, G. A., Thomas, S., Thompson, L. J., Sprent, J. and Murali-
Krishna, K., Type I interferons act directly on CD8lT cells to allow clonal
expansion and memory formation in response to viral infection. J. Exp.
Med. 2005. 202: 637–650.
9 Santini, S. M., Lapenta, C., Logozzi, M., Parlato, S., Spada, M.,
Di Pucchio, T. and Belardelli, F., Type I interferon as a powerful adjuvant
for monocyte-derived dendritic cell development and activity in vitro and
in Hu-PBL-SCID mice. J. Exp. Med. 2000. 191: 1777–1788.
10 Luft, T., Pang, K. C., Thomas, E., Hertzog, P., Hart, D. N., Trapani, J. and
Cebon, J., Type I IFNs enhance the terminal differentiation of dendritic
cells. J. Immunol. 1998. 161: 1947–1953.
11 Ito, T., Amakawa, R., Inaba, M., Ikehara, S., Inaba, K. and Fukuhara, S.,
Differential regulation of human blood dendritic cell subsets by IFNs.
J. Immunol. 2001. 166: 2961–2969.
12 Blanco, P., Palucka, A. K., Gill, M., Pascual, V. and Banchereau, J.,
Induction of dendritic cell differentiation by IFN-alpha in systemic lupus
erythematosus. Science 2001. 294: 1540–1543.
13 Hiscott, J., Triggering the innate antiviral response through IRF-3
activation. J. Biol. Chem. 2007. 282: 15325–15329.
14 Sharma, S., tenOever, B. R., Grandvaux, N., Zhou, G. P., Lin, R. and
Hiscott, J., Triggering the interferon antiviral response through an IKK-
related pathway. Science 2003. 300: 1148–1151.
15 Fitzgerald, K. A., McWhirter, S. M., Faia, K. L., Rowe, D. C., Latz, E.,
Golenbock, D. T., Coyle, A. J. et al., IKKepsilon and TBK1 are essential
components of the IRF3 signaling pathway. Nat. Immunol. 2003. 4: 491–496.
16 Lin, R., Mamane, Y. and Hiscott, J., Structural and functional analysis of
interferon regulatory factor 3: localization of the transactivation and
autoinhibitory domains. Mol. Cell. Biol. 1999. 19: 2465–2474.
17 Lin, R., Heylbroeck, C., Pitha, P. M. and Hiscott, J., Virus-dependent
phosphorylation of the IRF-3 transcription factor regulates nuclear
translocation, transactivation potential, and proteasome-mediated
degradation. Mol. Cell. Biol. 1998. 18: 2986–2996.
18 Lin, R., Mamane, Y. and Hiscott, J., Multiple regulatory domains control
IRF-7 activity in response to virus infection. J. Biol. Chem. 2000. 275:
34320–34327.
19 Yoneyama, M., Suhara, W., Fukuhara, Y., Fukada, M., Nishida, E. and
Fujita, T., Direct triggering of the type I interferon system by virus
infection: activation of a transcription factor complex containing IRF-3
and CBP/p300. EMBO J. 1998. 17: 1087–1095.
20 Servant, M. J., ten Oever, B. and Lin, R., Review: overlapping and distinct
mechanisms regulating IRF-3 and IRF-7 function. J. Interferon Cytokine Res.
2002. 22: 49–58.
21 Lin, R., Genin, P., Mamane, Y. and Hiscott, J., Selective DNA binding and
association with the CREB binding protein coactivator contribute to
differential activation of alpha/beta interferon genes by interferon
regulatory factors 3 and 7. Mol. Cell. Biol. 2000. 20: 6342–6353.
22 Au, W.-C., Moore, P. A., Lowther, W., Juang, Y.-T. and Pitha, P. M.,
Identification of a member of the interferon regulatory factor family that
binds to the interferon-stimulated response element and activates
expression of interferon-induced genes. Proc. Natl. Acad. Sci. USA 1995.
92: 11657–11661.
23 Izaguirre, A., Barnes, B. J., Amrute, S., Yeow, W. S., Megjugorac, N., Dai, J.,
Feng, D. et al., Comparative analysis of IRf and IFN-alpha expression in
human plasmocytoid and monocyte-dereived dentritic cells. J. Leukoc.
Biol. 2003. 74: 1125–1138.
24 Sato, M., Tanaka, N., Hata, N., Oda, E. and Taniguchi, T., Involvement of
the IRF family transcription factor IRF-3 in virus-induced activation of the
IFN-beta gene. FEBS Lett. 1998. 425: 112–116.
25 Marie, I., Durbin, J. E. and Levy, D. E., Differential viral induction of
distinct interferon-alpha genes by positive feedback through interferon
regulatory factor-7. EMBO J. 1998. 17: 6660–6669.
26 Sato, M., Suemori, H., Hata, N., Asagiri, M., Ogasawara, K., Nakao, K.,
Nakaya, T. et al., Distinct and essential roles of transcription factors IRF-3
and IRF-7 in response to viruses for IFN-alpha/beta gene induction.
Immunity 2000. 13: 539–548.
27 Honda, K., Yanai, H., Negishi, H., Asagiri, M., Sato, M., Mizutani, T.,
Shimada, N. et al., IRF-7 is the master regulator of type-1 interferon-
dependent immune responses. Nature 2005. 434: 772–777.
28 Barnes, B. J., Richards, J., Mancl, M., Hanash, S., Beretta, L. and Pitha,
P. M., Global and distinct targets of IRF-5 and IRF-7 during innate
response to viral infection. J. Biol. Chem. 2004. 279: 45194–45207.
29 Grandvaux, N., Servant, M. J., tenOever, B., Sen, G. C., Balachandran, S.,
Barber, G. N., Lin, R. and Hiscott, J., Transcriptional profiling of interferon
regulatory factor 3 target genes: direct involvement in the regulation of
interferon-stimulated genes. J. Virol. 2002. 76: 5532–5539.
30 Andersen, J., VanScoy, S, Cheng, T. F., Gomez, D. and Reich, N. C., IRF-3-
dependent and augmented target genes during viral infection. Genes
Immun. 2008. 9: 168–175.
31 Heylbroeck, C., Balachandran, S., Servant, M. J., DeLuca, C., Barber, G. N.,
Lin, R. and Hiscott, J., The IRF-3 transcription factor mediates Sendai
virus-induced apoptosis. J. Virol. 2000. 74: 3781–3792.
32 Romieu-Mourez, R., Solis, M., Nardin, A., Goubau, D., Baron-Bodo, V.,
Lin, R., Massie, B. et al., Distinct roles for IFN regulatory factor (IRF)-3 and
IRF-7 in the activation of antitumor properties of human macrophages.
Cancer Res. 2006. 66: 10576–10585.
33 Lallemand, C., Blanchard, B., Palmieri, M., Lebon, P., May, E.
and Tovey, M. G., Single-stranded RNA viruses inactivate the transcrip-
tional activity of p53 but induce NOXA-dependent apoptosis via post-
translational modifications of IRF-1, IRF-3 and CREB. Oncogene 2007. 26:
328–338.
34 Oda, E., Ohki, R., Murasawa, H., Nemoto, J., Shibue, T., Yamashita, T.,
Tokino, T. et al., Noxa, a BH3-only member of the Bcl-2 family and
candidate mediator of p53-induced apoptosis. Science 2000. 288:
1053–1058.
35 Budagian, V., Bulanova, E., Paus, R. and Bulfone-Paus, S., IL-15/IL-15
receptor biology: a guided tour through an expanding universe. Cytokine
Growth Factor Rev. 2006. 17: 259–280.
36 Bossen, C. and Schneider, P., BAFF, APRIL and their receptors: structure,
function and signaling. Semin. Immunol. 2006. 18: 263–275.
37 Luk, T., Malam, Z. and Marshall, J. C., Pre-B cell colony-enhancing factor
(PBEF)/visfatin: a novel mediator of innate immunity. J. Leukoc. Biol. 2008.
83: 804–816.
38 Willis, S. N. and Adams, J. M., Life in the balance: how BH3-only proteins
induce apoptosis. Curr. Opin. Cell Biol. 2005. 17: 617–625.
39 Chen, L., Willis, S. N., Wei, A., Smith, B. J., Fletcher, J. I., Hinds, M. G.,
Colman, P. M. et al., Differential targeting of prosurvival Bcl-2 proteins by
their BH3-only ligands allows complementary apoptotic function. Mol.
Cell 2005. 17: 393–403.
40 Sun, Y. and Leaman, D. W., Involvement of Noxa in cellular apoptotic
responses to interferon, double-stranded RNA, and virus infection. J. Biol.
Chem. 2005. 280: 15561–15568.
41 Ortiz-Navarrete, V., Seelig, A., Gernold, M., Frentzel, S., Kloetzel, P. M. and
Hammerling, G. J., Subunit of the ‘20S’ proteasome (multicatalytic
proteinase) encoded by the major histocompatibility complex. Nature
1991. 353: 662–664.
Eur. J. Immunol. 2009. 39: 527–540 Innate Immunity 539
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

42 Martinez, C. K. and Monaco, J. J., Homology of proteasome subunits to a
major histocompatibility complex-linked LMP gene. Nature 1991. 353:
664–667.
43 Kelly, A., Powis, S. H., Glynne, R., Radley, E., Beck, S. and Trowsdale, J.,
Second proteasome-related gene in the human MHC class II region.
Nature 1991. 353: 667–668.
44 Kesmir, C., van Noort, V., de Boer, R. J. and Hogeweg, P., Bioinformatic
analysis of functional differences between the immunoproteasome and
the constitutive proteasome. Immunogenetics 2003. 55: 437–449.
45 Strehl, B., Textoris-Taube, K., Jakel, S., Voigt, A., Henklein, P., Steinhoff,
U., Kloetzel, P. M. and Kuckelkorn, U., Antitopes define preferential
proteasomal cleavage site usage. J. Biol. Chem. 2008. 283: 17891–17897.
46 Takaoka, A., Hayakawa, S., Yanai, H., Stoiber, D., Negishi, H., Kikuchi, H.,
Sasaki, S. et al., Integration of interferon-alpha/beta signalling to p53
responses in tumour suppression and antiviral defence. Nature 2003. 424:
516–523.
47 Weaver, B. K., Ando, O., Kumar, K. P. and Reich, N. C., Apoptosis
is promoted by the dsRNA-activated factor (DRAF1) during viral
infection independent of the action of interferon or p53. FASEB J. 2001.
15: 501–515.
48 Shin, E. C., Seifert, U., Kato, T., Rice, C. M., Feinstone, S. M., Kloetzel, P. M.
and Rehermann, B., Virus-induced type I IFN stimulates generation of
immunoproteasomes at the site of infection. J. Clin. Invest. 2006. 116:
3006–3014.
49 Le Bon, A. and Tough, D. F., Type I interferon as a stimulus for cross-
priming. Cytokine Growth Factor Rev. 2008. 19: 33–40.
50 Le Bon, A. and Tough, D. F., Links between innate and adaptive immunity
via type I interferon. Curr. Opin. Immunol. 2002. 14: 432–436.
51 Banchereau, J., Ueno, H., Dhodapkar, M., Connolly, J., Finholt, J. P.,
Klechevsky, E., Blanck, J. P. et al., Immune and clinical outcomes in
patients with stage IV melanoma vaccinated with peptide-pulsed
dendritic cells derived from CD341 progenitors and activated with type
I interferon. J. Immunother. 2005. 28: 505–516.
52 Papewalis, C., Jacobs, B., Wuttke, M., Ullrich, E., Baehring, T., Fenk, R.,
Willenberg, H. S. et al., IFN-alpha skews monocytes into CD561-
expressing dendritic cells with potent functional activities in vitro and
in vivo. J. Immunol. 2008. 180: 1462–1470.
53 Fanger, N. A, Maliszewski, C. R., Schooley, K. and Griffith, T. S., Human
dendritic cells mediate cellular apoptosis via tumor necrosis factor-
related apoptosis-inducing ligand (TRAIL). J. Exp. Med. 1999. 190:
1155–1164.
54 Sharif-Askari, E., Nakhaei, P., Oliere, S., Tumilasci, V., Hernandez, E.,
Wilkinson, P., Lin, R. et al., Bax-dependent mitochondrial membrane
permeabilization enhances IRF3-mediated innate immune response
during VSV infection. Virology 2007. 365: 20–33.
55 Peters, K., Chattopadhyay, S. and Sen G. C., IRF-3 activation by Sendai
virus infection is required for cellular apoptosis and avoidance of
persistence. J. Virol. 2008. 82: 3500–3508.
56 Degenhardt, K., Sundararajan, R., Lindsten, T., Thompson, C. and
White, E., Bax and Bak independently promote cytochrome C release
from mitochondria. J. Biol. Chem. 2002. 277: 14127–14134.
57 Lin, R., Lacoste, J., Nakhaei, P., Sun, Q., Yang, L., Paz, S., Wilkinson, P.
et al., Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular
complex from the mitochondrial outer membrane by hepatitis C virus
NS3-4A proteolytic cleavage. J. Virol. 2006. 80: 6072–6083.
58 Sheehan, K. C., Lai, K. S., Dunn, G. P., Bruce, A. T., Diamond, M. S., Heutel,
J. D., Dungo-Arthur, C. et al., Blocking monoclonal antibodies specific for
mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by
in vivo hydrodynamic transfection. J. Interferon Cytokine Res. 2006. 26: 804–819.
59 Smyth, G. K., Linear models and empirical Bayes methods for assessing
differential expression in microarray experiments. Stat. Appl. Genet. Mol.
Biol. 2004. 3: 1–26.
60 Sturn, A., Quackenbush, J. and Trajanoski, Z., Genesis: cluster analysis of
microarray data. Bioinformatics 2002. 18: 207–208.
61 Porta, C., Hadj-Slimane, R., Nejmeddine, M., Pampin, M., Tovey, M. G.,
Espert, L., Alvarez, S. and Chelbi-Alix, M. K., Interferons alpha and
gamma induce p53-dependent and p53-independent apoptosis, respec-
tively. Oncogene 2005. 24: 605–615.
62 Solis, M., Romieu-Mourez, R., Goubau, D., Grandvaux, N., Mesplede, T.,
Julkunen, I., Nardin, A. et al., Involvement of TBK1 and IKKepsilon in
lipopolysaccharide-induced activation of the interferon response in
primary human macrophages. Eur. J. Immunol. 2007. 37: 528–539.
Abbreviations: BMK cells: baby mouse kidney cells � DEVD-AFC:
N-acetyl-Asp-Glu-Val-Asp-AFC (7-amino-4-trifluoromethyl coumarin) �IFNAR: type I IFN receptor � ISG: IFN-stimulated gene � MEF: mouse
embryonic fibroblasts � NT: non-transduced � RQ: relative
quantification � VSV: vesicular stomatitis virus � WCE: whole-cell
extracts
Full correspondence: Dr. John Hiscott, Lady Davis Institute, Jewish
General Hospital, McGill University 3999, Chemin de la Cote Ste-
Catherine Montreal, Quebec H3T 1E2, Canada
Fax: 11-514-340-7576
e-mail: [email protected]
Additional correspondence: Dr. Douglas Leaman, Department of
Biological Sciences, University of Toledo 2801, W. Bancroft Toledo, OH
43606, USA
e-mail: [email protected]
Supporting Information for this article is available at
www.wiley-vch.de/contents/jc_2040/2009/38832_s.pdf
Received: 15/8/2008
Revised: 13/11/2008
Accepted: 28/11/2008
Eur. J. Immunol. 2009. 39: 527–540Delphine Goubau et al.540
& 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
Top Related
Copyright © 2022 FDOKUMEN

