







Bahasa
Halaman
Hukum


doi:10.1182/blood-2008-05-155200Prepublished online December 4, 2008;
Wilkens, Brigitte Schlegelberger, Arnold Ganser and Christopher BaumYang, Jurgen Krauter, Nils von Neuhoff, Michael Heuser, Helmut Diedrich, Gudrun Gohring, Ludwig Zhixiong Li, Gernot Beutel, Mathias Rhein, Johann Meyer, Christian Koenecke, Thomas Neumann, Min High affinity neurotrophin receptors and ligands promote leukemogenesis
(965 articles)Myeloid Neoplasia �Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
digital object identifier (DOIs) and date of initial publication. theindexed by PubMed from initial publication. Citations to Advance online articles must include
final publication). Advance online articles are citable and establish publication priority; they areappeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet
Copyright 2011 by The American Society of Hematology; all rights reserved.20036.the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

1
High affinity neurotrophin receptors and ligands promote
leukemogenesis
Zhixiong Li1, Gernot Beutel2, Mathias Rhein1, Johann Meyer1, Christian Koenecke2, Thomas
Neumann1, Min Yang1, Jürgen Krauter2, Nils von Neuhoff3, Michael Heuser2, Helmut Diedrich2,
Gudrun Göhring3, Ludwig Wilkens3, Brigitte Schlegelberger3, Arnold Ganser2, Christopher Baum1,4
1Department of Experimental Hematology, Hannover Medical School, 30625 Hannover, Germany 2Department of Hematology, Hemostasis, Oncology, and Stem Cell Transplantation, Hannover Medical School, 30625 Hannover, Germany 3Institute of Cell and Molecular Pathology, Hannover Medical School, 30625 Hannover, Germany 4Division of Experimental Hematology, Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio 45229-3039, U.S.A.
ZL, GB, and MR contributed equally to this work.
Corresponding author:
Zhixiong Li, MD or Christopher Baum, MD Department of Experimental Hematology, OE6960 Hannover Medical School Carl-Neuberg-Straße 1 30625 Hannover Germany Phone: +49 511-532-5148 Fax: +49 511-532-5105 E-mail: [email protected] [email protected] Running title: leukemogenesis of TRK signaling
Category: neoplasia Part of the data was presented in an oral session at the 2007 Annual Meeting of the American Society
of Hematology in Atlanta.
Blood First Edition Paper, prepublished online December 4, 2008; DOI 10.1182/blood-2008-05-155200
Copyright © 2008 American Society of Hematology
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

2
Abstract
Neurotrophins (NTs) and their receptors play a key role in neurogenesis and survival. The TRK
(tropomyosin-related kinase) receptor protein tyrosine kinases (TRKA, TRKB, TRKC) are high affinity
NT-receptors that are expressed in a variety of human tissues. Their role in normal and malignant
hematopoiesis is poorly understood. In a prospective study involving 94 adult patients (mean age 54.3
years), we demonstrate for the first time cell surface expression of the three TRKs and constitutive
activation in blasts from patients with de novo or secondary acute leukemia. At least one TRK was
expressed in 55% of the analyzed cases. We establish a clear correlation between the TRK
expression pattern and FAB classification. While only few point mutations were found in TRK
sequences by RT-PCR, we observed co-expression of BDNF (ligand for TRKB) in >50% of TRKB+
cases (16/30). Activation of TRKA or TRKB by NGF and BDNF, respectively, efficiently rescued
murine myeloid cells from irradiation-induced apoptosis. Co-expression of TRKB/BDNF or TRKA/NGF
in murine hematopoietic cells induced leukemia. Moreover, activation of TRKs was important for
survival of both human and murine leukemic cells. Our findings suggest that TRKs play an important
role in leukemogenesis and may serve as a new drug target.
Keyword: acute leukemia, TRKs, BDNF, autocrine loop, protein-tyrosine kinase
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

3
Introduction
Current concepts of leukemogenesis postulate a collaboration of ‘class I’ mutations that result in for
example constitutively activated protein-tyrosine kinases (PTKs) with ‘class II’ mutations of
transcription factors such as AML/Runx or ETS proteins. In this scenario, class I mutations (such as
BCR/ABL and FLT3-ITD) promote proliferation but generally do not inhibit differentiation, while the
reverse is true for class II mutations.1 While the molecular analysis of patient samples has supported
this concept in human acute myeloid leukemia (AML), there still remain a substantial proportion of
patients in whom both types of mutations have not yet been demonstrated.
There is growing evidence for involvement of multiple PTK oncogenes, their immediate downstream
targets (e.g. phosphatidylinositol 3-kinase=PI3K), or of proteins regulating their function in
hematological malignancies.2-5 The fact that cytogenetic remissions can be achieved in the majority of
patients with chronic myeloid leukemia (CML) demonstrates a causal role of the BCR-ABL oncoprotein
in this disease.6 Analysis of activated PTK is also of clinical relevance.7,8 At least one third of AML
patients carry mutated FLT3 alleles and have unfavorable prognosis.8,9 It is thus important to identify
other PTKs that are activated in the remaining patients. Moreover, co-activation of receptor PTKs has
been suggested to be important for tumor development and to affect the tumor cell response to
targeted therapy.10 Oncogenic transformation by PTKs occurs in different ways,11 e.g. by genomic re-
arrangements, such as chromosomal translocations, gain-of function (GOF) mutations, PTK
overexpression or small deletions in receptor PTKs and cytoplasmic PTKs. Autocrine and/or paracrine
loops have been suggested as important mechanisms for aberrant kinase activation in human solid
tumors12 and leukemia,13 and may have therapeutic potential.14 However, few experimental studies
convincingly demonstrate the oncogenic potential of autocrine/paracrine circuits of PTKs in animal
models,12,15 and a prognostic role of autocrine loops in human leukemia has not been demonstrated.
The neurotrophins (NTs), which include nerve growth factor (NGF), brain-derived neurotrophic factor
(BDNF), NT-3, NT-4, and NT-6, play a major role in neuronal survival. NTs are unique in that they
utilize two different classes of receptors: the TRK (tropomyosin-related kinase) receptor protein
tyrosine kinases (TRKA, TRKB, TRKC) and the low affinity NGF receptor (LNGFR=p75NTR),16 a
member of the tumor necrosis factor cytokine receptor family. The biologically active receptors for
NGF, BDNF and NT-3 are TRKA, TRKB, and TRKC, respectively. NT-3 can bind to all of the TRK
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

4
receptors, and NT-4 binds preferentially to TRKB. NT binding to TRK receptors leads to dimerization of
receptors and kinase activation. LNGFR may modulate the activity of this signaling complex but is not
required for its function.16 Of note, human embryonic stem cell survival has recently been shown to be
mediated through activation of TRK receptors by NTs.17 Members of the TRK family have been found
in several non-neural cell types,18,19 and may also play a crucial role in initiation, progression, and
metastasis of many tumors in humans, e.g. neuroblastoma, medullary thyroid carcinoma and breast
cancer.20-23 In addition, some data indicate relevance of TRK receptors as prognostic factors.20 For
example, TRKB is associated with bad prognosis in Wilms´ tumor.24 However, relatively little is known
about the mechanisms of oncogenesis mediated by altered TRK signaling.25,26
Many PTK oncogenes are derived from genes (e.g. Abl, FLT3, c-Kit and PDGFR-ß) normally involved
in the regulation of hematopoiesis or hematopoietic cell function.2 TRK receptors and their respective
ligands are also expressed at various stages of hematopoiesis.27,28 A role for neurotrophins in
hematopoiesis has yet to be confirmed using conditional knockouts. Nevertheless, recent data
suggested important functions of TRK signaling in hematopoiesis. TRKs promote proliferation and
survival of lymphocytes and monocytes/macrophages.29 TRKB expression is greatest in precursor
CD4-CD8- thymocytes and progressively declines throughout the T cell differentiation pathway.30
Importantly, there is increasing evidence for involvement of TRK receptors in leukemogenesis. A
cryptic translocation t(12;15) (p13;q25), which resulted in the chimeric transcript TEL-TrkC, was found
in an AML patient.25,31 Furthermore, a deleted form of TRKA (ΔTrkA), in which 75 amino acids are
lacking in the extracellular domain, was identified in another AML patient.32 In mouse models, we
found that ΔTrkA is a very potent oncogene that transforms cells mainly via PI3K and mTOR.33
Another study revealed the induction of TRKA and a contribution of NGF to survival signaling in
human cord blood cells transduced with retroviral vectors encoding the AML1-ETO oncogene.34 In
addition, we had evidence to suggest that a cytoplasmically deleted form of LNGFR may contribute to
leukemia in mice.35 Furthermore, we observed expression of LNGFR in patients with acute leukemia
(AL), preferentially in common ALL.36 Taken together, these data suggest a previously underestimated
role of NT signaling in leukemogenesis. However, with the exception of one report showing expression
of TRKA mRNA in primary leukemic cells in 44% of AML patients,37 the expression pattern of other
TRKs and NTs and their potential prognostic relevance have not been reported in primary leukemic
cells.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

5
Here, we screened AL patients for expression of TRKA, TRKB and TRKC. A distinct expression
pattern of these receptors in different leukemia subtypes as well as expression of NTs was observed.
Co-expression of TRKB and BDNF was associated with poor outcome and induced leukemia in a
murine model. Moreover, TRK signaling was important for maintenance of leukemic cells in vitro. This
study expands current concepts of leukemogenesis and encourages further evaluation of NT receptor
signaling as a drug target in leukemia therapy.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

6
Methods
TRK and NTs expression in leukemic blasts of AL patients
We studied tumor specimens from AL patients who had been diagnosed in the Hannover Medical
School from 2004 to 2006. Blood and/or bone marrow (BM) samples from AL patients were collected
at diagnosis after informed consent was obtained in accordance with the Declaration of Helsinki.
Mononuclear cells from all samples studied were immediately isolated by centrifugation over Ficoll
gradient and freshly used or stored at –1800C until further use. The following monoclonal antibodies
were used: anti-TRKA (clone H10, Biodesign), anti-TRKB (clone75133, R&D), anti-TRKC (clone
75219, R&D), anti-BDNF (Cat# GF35L, CALBIOCHEM), anti-NT-3 (clone 41512, R&D). A polyclonal
antibody against human NGF (cat# BAF256, R&D) was used for detection of NGF. Antibodies were
validated on cell lines that expressed retrovirally encoded TRK receptors and NTs. The study was
approved by the ethics committee of the Hannover Medical School. The majority of patients were
treated according to previously described protocols.38 For surviving patients, the median follow-up
period after diagnosis was 30 months. In all cases, cytomorphologic classification according to FAB
criteria was made on bone marrow and/or peripheral blood smears.
Retroviral vectors, vector production, retroviral transductions, in vivo tumorigenesis assays,
and tumor phenotyping
A retroviral vector encoding full-length human TRKA was kindly provided by Dr. Gary Reuther
(University of South Florida, Tampa, Florida).32 Plasmid SF91.IRES-EGFP.WPRE, which mediates
efficient transgene expression in hematopoietic cells, has been described.33 The retroviral vector is
referred to as SF91-IE. A PCR fragment containing the cDNA of human TRKB or BDNF was
generated and cloned into the NotI site before the IRES-EGFP cassette of SF91-IE. Resulting vectors
encoding TRKB or BDNF were named SF91-TRKB and SF91-BDNF, respectively. Cell free high-titer
supernatants containing the ecotropic envelope protein were generated as described.33 Retroviral
transductions, in vivo tumorigenesis assays, and tumor phenotyping were performed as previously
described (supplementary information).33
Radiation-induced apoptosis assay and leukemic cell growth assays
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

7
2.5×105 32D cells were starved from IL-3 and serum for 3 hours, placed in 24-well plates, and
exposed to 5 Gy -irradiation. Immediately after irradiation, cells were supplemented with NGF, BDNF
(each 100 ng/mL), IL-3 (2 ng/mL), or no factor. Cell viability was analyzed using the Annexin-V assay.
Cells staining negative for both Annexin-V and propidium iodide were counted as viable cells. To
analyze clonal growth, 2x102 or 2x103 murine cells were plated per dish in M3234 media (StemCell
Technologies, Vancouver, Canada) in the presence of signal transduction inhibitors. The assays were
plated as duplicates or quadruplicates, and colonies were counted on day 6. Mononuclear cells from
patients were cultured in RPMI 1640 supplemented with 10% fetal calf serum (FCS) and exposed to
inhibitors and/or idarubicin.39 Inhibitors K252a, AG879 (Calbiochem, Merck, Bad Soden, Germany)
and anti-human BDNF antibody (Promega, Mannheim, Germany) were used.
Western blot analysis
For signal transduction analysis, leukemic cell extracts were prepared following established
protocols.33 Cell lysates were used as indicated in results. Antibodies were purchased from Santa
Cruz Biotechnology (Santa Cruz, California, U.S.A.).
RNA extraction and reverse transcriptase– polymerase chain reaction (RT-PCR), small
interfering RNA (siRNA)-mediated knockdown, BDNF-ELISA, and statistical analysis
Please refer to supplementary information.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

8
Results
Frequent expression of TRK receptors in leukemic blasts of AL patients
We analyzed expression of TRKs in AL patients. 94 adult patients (42 female, 52 male) with a mean
age of 54.3 years and diagnosis of primary or secondary AML (87%), ALL (12%), or AUL (1%) were
enrolled after informed consent. The patients´ clinical characteristics are summarized in Table 1.
Expression of TRKA, TRKB and TRKC was detected by flow cytometry using monoclonal antibodies. If
>20% of leukemic blasts expressed at least one of the TRK receptors, cases were considered TRK+
(Figure 1A, B, S1).40 Thus, 55% of the analyzed cases expressed at least one TRK receptor, without
statistically meaningful differences in expression rates between AML (43/82) and ALL (8/11). We
observed expression of TRKA on AML blasts, which is in agreement with a previous study that
demonstrated expression of TRKA on the RNA level.37
For the first time, we found expression of TRKB and TRKC in human leukemia. Interestingly, while
TRKB can be expressed alone in blasts, TRKA or TRKC expression always occurred concomitantly
with TRKB. About 18% of leukemia cases co-expressed all TRK receptors. Co-expression of two or
more TRK receptors (i.e. TRKA+TRKB, TRKB+TRKC and TRKA+TRKB+TRKC) was observed in
AML, while ALL blasts exclusively expressed only TRKB. Although TRK mRNA was found in
hematopoietic cells from healthy volunteers,27,37 we did not detect TRK receptors on the surface of
normal mononuclear cells by flow cytometry (data not shown), which is in agreement with previous
studies.41
Correlation of TRK expression and French-American-British (FAB) leukemia classification
Next, we analyzed the relationship between patient age, FAB subtype, white blood cell (WBC) counts,
ECOG status, cytogenetics, FLT3 mutation and TRK expression. In agreement with recent
publications,8 we found internal tandem duplications of the juxtamembrane region of the FLT3 receptor
(FLT3-ITD) in 25% (17/67) of AML patients. There was no significant correlation of patient age, WBC
counts, ECOG status, FLT3-ITD or cytogenetics with TRK expression (Table 1). However, in contrast
to a previous study,37 we established a clear correlation of TRK expression pattern and the FAB
classification (Table 1). In particular, TRKA was expressed in 21 of 34 myelo-monocytic/monocytic
leukemias (AML M4 and M5) (62%) whereas only 5 of 48 non-myelo-monocytic/monocytic leukemias
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

9
(10%) were positive (p<0.001). The same observation was made for TRKB and TRKC (71% vs. 38%,
p<0.005 and 47% vs. 8%, p<0.001, respectively). Co-expression of at least two of the three TRK
receptors was observed in 62% (21/34) of patients with AML M4 and M5, but in only 13% (6/48) of
other subtypes (p<0.001). The latter cases were often secondary AML after a myelodysplastic
syndrome (MDS). Thus, our data show that TRK expression in AML is closely linked to monocytic
differentiation (AML M4 and M5), unless the AL developed on the basis of MDS. Corroboratively, we
found expression of TRKA in >98% of THP1 cells (a commonly used human monoblastic leukemia cell
line) by flow cytometry (data not shown).
Expression of NTs in leukemia and constitutive activation of TRK in leukemic cells
To assess potential mutations and deletions, we sequenced the 2nd Ig-like domain, transmembrane
domain and whole intracellular domain (focusing on the kinase domain) of TRKA, TRKB and TRKC
receptors following RT-PCR of RNA isolated from leukemic cells. Mutations, small deletions and
duplications in these regions have been reported as potential mechanisms for constitutive activation of
TRKs.32,42 RT-PCR and direct sequencing of PCR fragments were successfully performed in 98% and
97% of patients, respectively. Unexpectedly, only four different point mutations (TRKB: T573I, Y707N,
and V684I; TRKC: Y800H) were found in four patients by RT-PCR. We did not observe deletions or
duplications in TRKs. As oncogenic TRKA was originally cloned in a patient with colon carcinoma as a
TPM3/TRK transcript caused by translocation within chromosome 1,43 we also searched for the
TPM3/TRK translocation in our patients, yet without success (n=32).
Therefore, we investigated other major mechanisms by which TRK expression could contribute to
transformation or differentiation. Autocrine and/or paracrine loop has been suggested as an important
mechanism of PTK activation in human cancers.12,13 Elevated expression of TRKB and/or BDNF has
also been reported to occur frequently in multiple myeloma (24 and 12 out of 25 cases studied,
respectively), promoting myeloma cell survival.44 Thus, we next investigated expression of NTs in
blasts from patients with AL. We chose flow cytometry to detect intracellular expression of NGF, BDNF
or NT-3. Co-expression of BDNF (primary ligand for TRKB) was observed in over half of TRKB+ cases
(53.3%, 16/30) (Figure 1C, S1). Moreover, we found expression of NGF or NT-3 in 2 patients
expressing TRKs (Figure S2). Importantly, we observed constitutive phosphorylation of TRKs in all
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

10
analyzed primary leukemia samples (n=13, Figure 1D), suggesting a role of TRK signaling in leukemic
transformation.
Co-expression of TRKB and BDNF is associated with poor survival
The good response rate on days 15 and 21 after induction therapy was not significantly different
according to TRK expression (62% vs. 81% for TRK– and TRK+, respectively, p=0.16). However, we
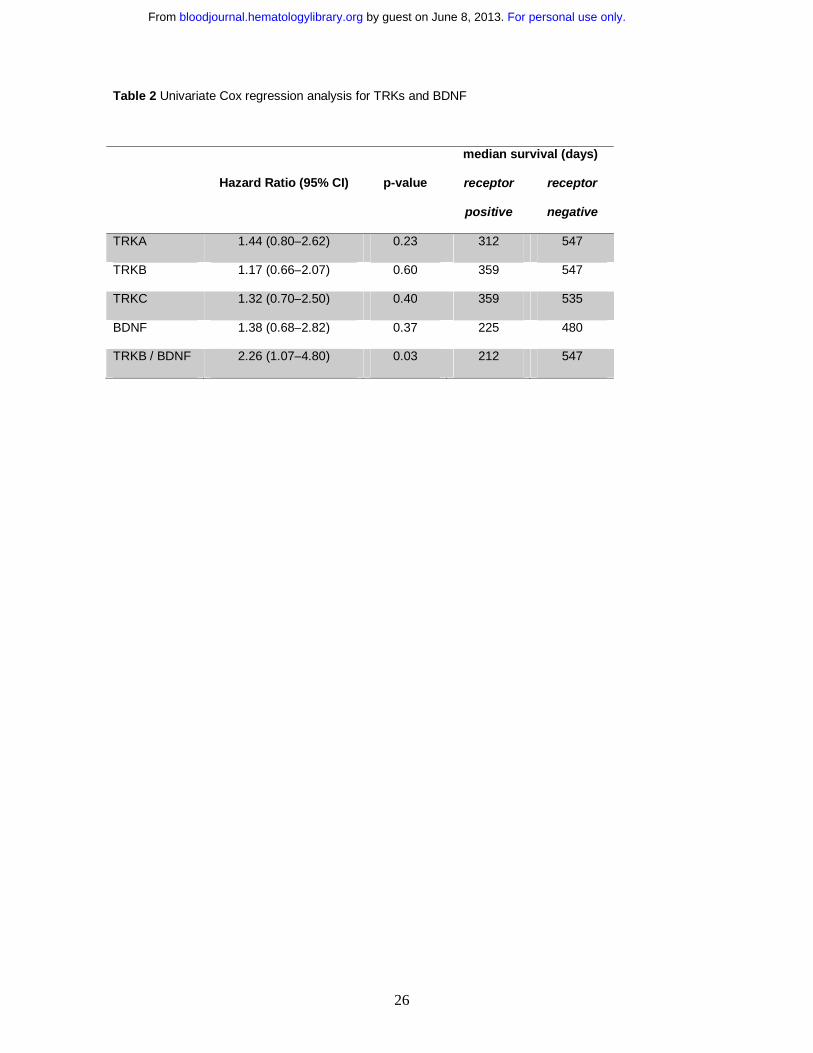
found that expression of TRKs and BDNF was associated with bad prognosis (Table 2). Patients
whose blasts express TRKA had a shorter median survival compared with patients whose blasts do
not (312 vs. 547 days, hazard ratio 1.44, p=0.23). Interestingly, in agreement with other reports,45 it
seems that expression of LNGFR is associated with better prognosis (Figure S3). These differences
may reach statistical significance if investigated in larger cohorts.
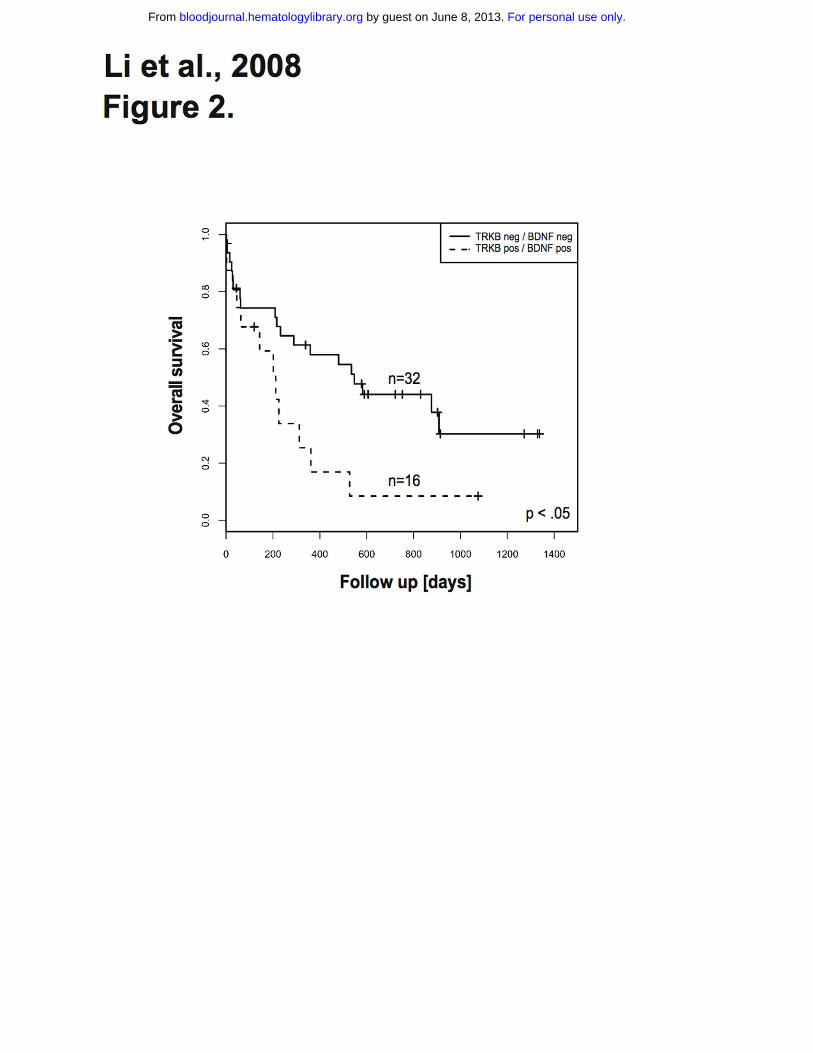
The survival difference was even more pronounced for patients whose blasts co-express TRKB and
BDNF (Table 2). These patients had significantly shorter overall survival when compared with patients
whose blasts express neither TRKB nor BDNF (8% vs. 30% at 3 years, respectively, p<0.05) (Figure
2). However, event free survival displayed no statistical difference. Importantly, there was no
significant difference regarding incidence of FLT3-ITD in both groups of patients. While this finding is
reminiscent of recent data demonstrating association of the TRKB/BDNF autocrine survival pathway
with poor outcome in human solid tumors,24 we are not aware of a previous report demonstrating a
prognostic relevance of an autocrine or paracrine loop in AL.
Activation of TRK signaling protects myeloid cells from apoptosis and supports proliferation
On cell types other than neuronal cells including mast cells, keratinocytes, monocytes and B cells,
TRK signals potentially stimulate cell proliferation through anti-apoptotic effects.18,46 To examine the
role of TRKs in the regulation of apoptosis in myeloid cells, we used 32D cells transduced with
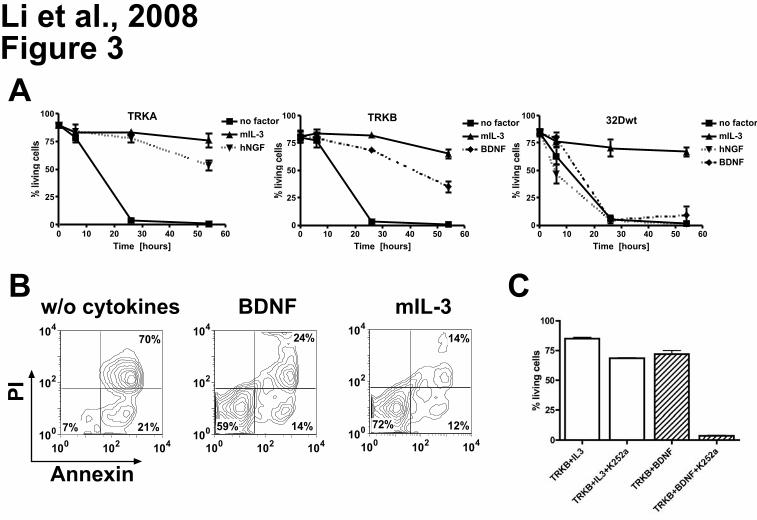
retroviral vectors expressing TRKA or TRKB.33 We tested whether stimulation of 32D/TRKA cells by
NGF could rescue cells from irradiation-induced apoptosis. 32D/TRKA cells were highly sensitive to
irradiation, with less than 10% viable cells 26 hours after irradiation. Upon NGF exposure, up to 88% of
32D/TRKA cells escaped apoptosis (Figure 3A). Similar data were obtained with 32D cells expressing
TRKB after stimulation with BDNF (Figure 3A and 3B). For yet unknown reasons, activation of TRKB
was not as potent as TRKA in protecting the cells from apoptosis at a later time point (54h). Exposure
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

11
to NGF or BDNF rescued less than 8% of non-transduced 32D cells from apoptosis (Figure 3A).
Importantly, treatment of 32D TRKB cells with K252a (a widely used TRK inhibitor17,47,48) significantly
counteracted the anti-apoptotic effect of BDNF activation (Figure 3C), suggesting that kinase activity
of TRKs is absolutely required for their anti-apoptotic function. Moreover, activation of TRKA by NGF
or TRKB by BDNF also supported proliferation of 32D cells in liquid culture (>1 month) and
methycellulose in the absence of IL-3 (data not shown).
Co-expression of TRKA/NGF and TRKB/BDNF efficiently transforms hematopoietic cells and
induces leukemia in mouse models
To address the in vivo leukemogenic potential of autocrine activation of TRKA and TRKB, we first
used a model based on 32D cells as previously described.33 Retroviral vector-mediated expression of
NGF in 32D/TRKA cells caused growth-factor independence in vitro. Even without prior selection for
growth-factor independence in vitro, retroviral vector-mediated co-expression of TRKA and NGF or
TRKB and BDNF in 32D cells elicited a fatal AML in all syngeneic C3H/Hej recipients (n=4/4 for
TRKA/NGF, n=2/2 for TRKB/BDNF). In contrast, only 4 out of 26 animals transplanted with cells
expressing TRKA alone developed leukemia, and none of the animals transplanted with cells
expressing TRKB or NGF or BDNF alone (n=6) developed donor-cell derived leukemia. Interestingly,
in agreement with the less potent anti-apoptotic effect of TRKB observed in 32D cells, transformation
induced by autocrine activation of TRKB required a longer latency in comparison with TRKA/NGF (13
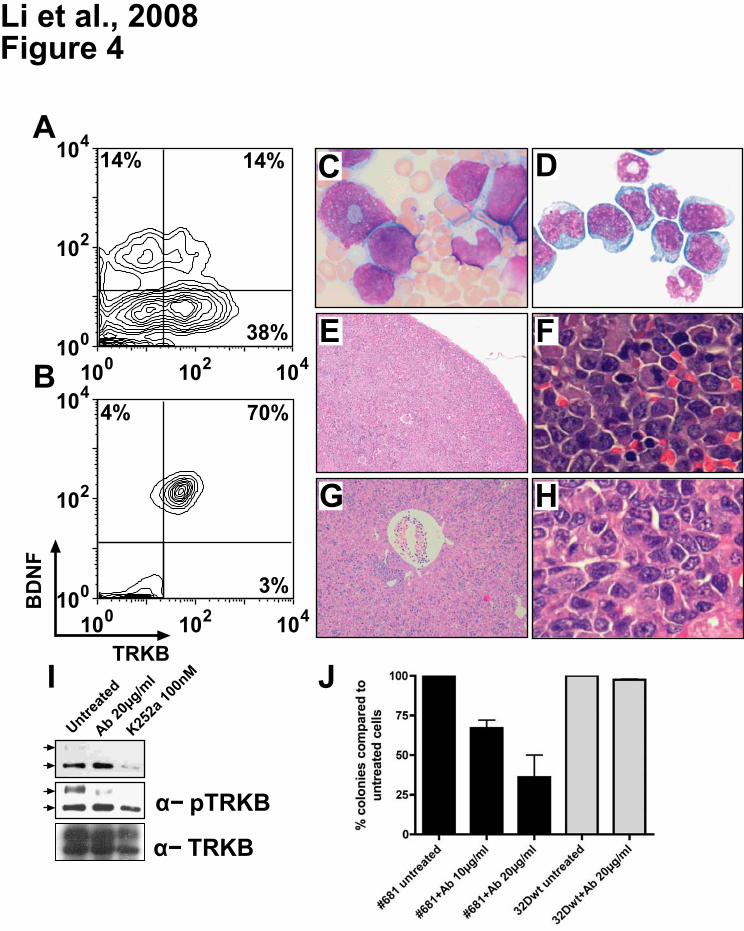
vs. 3.5 weeks). Importantly, while only 14% of cells were positive for TRKB/BDNF on the day of
transplantation (Figure 4A), the majority of leukemic cells recovered from animals with overt disease
co-expressed both vectors (Figure 4B), strongly suggesting selection for leukemic transformation by
autocrine activation. The developing leukemias led to elevated WBC counts (Figure 4C),
splenomegaly, and hepatomegaly. Cytology and histopathology revealed extensive infiltration of blasts
in the BM, spleen, and liver (Figure 4D-H), and in some cases also in the lung, and kidney (data not
shown). As in the patient samples, constitutive activation of TRKB was observed, suggesting a crucial
contribution of TRK signaling to leukemogenesis (Figure 4I).
We next assessed the ability of autocrine activation of TRKB to transform primary murine
hematopoietic cells. Hematopoietic stem/progenitor cells enriched lineage negative (Lin-) BM cells
were transduced with retroviral vectors expressing TRKB (SF91-TRKB), BDNF (SF91-BDNF), or both.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

12
Three of 6 animals co-expressing TRKB and BDNF developed lymphoblastic leukemia within 14
weeks after transplantation (Figure S4). The other 3 remained healthy for another 5 months, but had
no or less than 5% transgene expression. All animals (n=6) transduced with TRKB or BDNF alone
showed normal hematopoiesis. Similarly, we observed development of myeloid leukemia in one out of
3 animals transplanted with Lin- cells co-expressing TRKA and NGF (data not shown). Thus, in
agreement with our clinical observation, activation of TRKs induced both myeloid and lymphoblastic
leukemia in our mouse models. Moreover, our data (patients and mouse) support the view that
LNGFR is not absolutely required for TRK-mediated responses.49
Autocrine loop TRKB/BDNF is a survival factor for murine leukemic cells
Leukemic cells from mouse #681 (Figure 4) grew factor-independently. Importantly, supernatant
collected from #681 cells supported growth of 32D cells expressing TRKB without cytokine
supplementation. BDNF was measured to be >300pg/ml by ELISA (in comparison, concentration of
BDNF in serum of human healthy control was 27pg/ml44) and no membrane-bound BDNF was found
by FACS analysis (data not shown). This strongly suggests existence of a TRKB/BDNF autocrine loop
in #681 cells.
We examined the ability of a neutralizing antibody to BDNF, or the TRK inhibitor K252a, to interfere
with the autocrine activation loop. In the absence of treatment, autophosphorylation of TRKB was
detected in leukemic cells isolated from mouse #681, suggesting that the autocrine loop is
constitutively active. Both anti-BDNF neutralizing antibody and K252a dramatically blocked
phosphorylation of TRKB (Figure 4I). If leukemia development is mediated through activation of TRKs
by NT, then pharmacological inhibition of TRK signaling or blocking the action of NT should reduce
leukemia cell survival. To test this hypothesis, survival of leukemic cells isolated from mouse #681
was measured by colony formation in the presence or absence of BDNF-neutralizing antibody and
K252a.17 Anti-human BDNF antibody showed a dose-dependent effect on leukemic cell survival
(Figure 4J). K252a induced up to 50% growth inhibition of leukemic cells (data not shown). In contrast,
addition of either the anti-BDNF neutralizing antibody (Figure 4J) or K252a did not alter the survival of
control cells. Further, we generated 2 lentiviral vectors expressing siRNAs targeted against different
regions on the TRKB mRNA sequence.50 Compared with control vector transduced cells, siRNA
expression reduced colony formation up to 13 fold, and induced cell death in the absence of IL-3 (data
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

13
not shown). Moreover, leukemic cells from mice co-expressing TRKA/NGF were very sensitive to
treatment of K252a and AG879, a TRKA inhibitor (Figure S5). Collectively, these data reveal that an
autocrine loop involving TRK receptors is a major survival factor for leukemic cells in the murine
model.
TRK signaling is important for survival of human AML cells
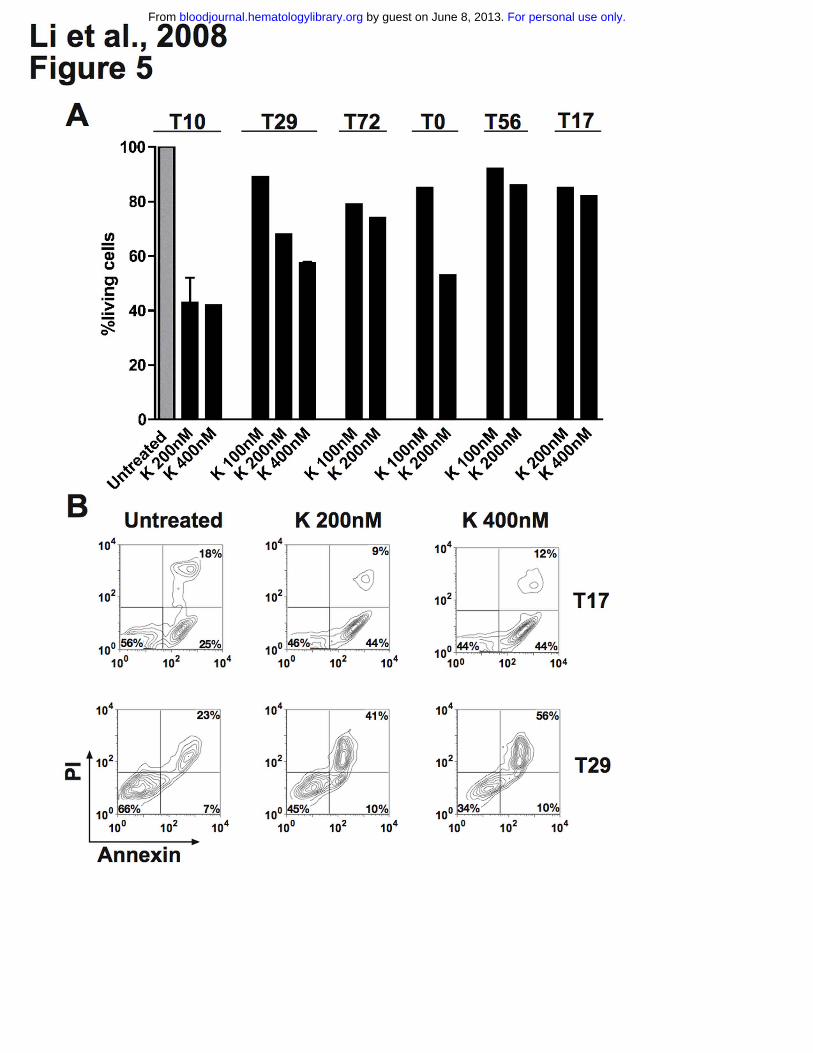
We evaluated apoptosis in cultured leukemic cells obtained from 4 patients with AML. Cells were
cultured in RPMI 1640 (10% FCS) and exposed to the TRK inhibitor K252a (100nM – 400nM) for
approximately 18 hours. We found that K252a induced apoptosis over basal levels in all cases and led
to up to 65% reduction of living cells by FACS analysis (Figure 5A, 5B), while K252a had minimal
effect on leukemic cells from patients T17 and T56 not expressing TRKs (Figure 5A, 5B). Moreover,
we also observed dephosphorylation of TRK proteins after treatment (data not shown), suggesting that
the apoptotic effect of K252a was mainly due to inhibition of TRK signaling. In patient T72 (co-
expressing TRKB/BDNF only), a moderate reduction of living cells was observed upon anti-BDNF
antibody treatment alone or in the combination with K252a (Figure S6). This indicates that survival
activity of a portion of leukemic cells is indeed due to an autocrine loop of TRKB/BDNF. However, no
or less apoptotic effect of anti-BDNF antibody was observed in cells from patients T0 (negative for
BDNF) and T10 (expressing TRKs and BDNF) (Figure S6). In the latter case, it is possible that the
potential effect of the anti-BDNF antibody was compensated by signaling driven from TRKA and/or
TRKC. K252a also enhanced idarubicin-induced apoptosis in leukemic cells from patients T72 and T0
(data not shown). However, cells from patients with ALL (T14 and T20) were resistant or less sensitive
to K252a treatment compared with AML patients (data not shown), suggesting different sensitivities to
TRKs signal transduction inhibition among patients. Generally, the growth inhibition effects of K252a
and anti-BDNF antibody we observed in human leukemic cells (Figure 5, S6) is less strong than in
murine leukemic cells (Figure 4), reflecting the more complex leukemogenesis in humans. However,
our findings collectively suggest that TRK signaling is involved in survival of human blasts, particularly
in AML.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

14
Discussion
NTs and their receptors regulate proliferation, differentiation, and death of normal and neoplastic
neuronal cells and also have been implicated in the formation of various human cancers.16,24 Although
expression of TRKs has been shown in different stages of hematopoiesis, their functional role has
remained largely unclear. By RT-PCR, expression of TRKs and NTs in adult human BM cells was
much lower than in fetal BM cells, suggesting down-regulation in adult hematopoiesis.27 Consistently,
we and others did not observe surface expression of TRKs in normal mononuclear cells.41 In contrast,
expression of TRKA has been shown in different leukemia cell lines.41 Multiple myeloma cells
expressed TRKB, and responded to BDNF by activating MAPK and PI3K/Akt signaling cascades.44 So
far only one group analyzed expression of TRK in human AML, detecting TRKA mRNA in leukemic
cells of less than half of the patients tested.37 However, expression on the protein level and a potential
correlation with leukemia subtypes or prognosis were not assessed. Using stringent criteria, our study
provides direct evidence for frequent and high expression of TRK receptors in human AL. As the TRK
expression pattern did not only depend on the leukemia subtype but was also correlated with
prognosis, we hypothesize that this receptor system may play a pathogenetic role in human leukemia.
The mechanisms underlying aberrant expression of TRKs and BDNF in leukemic cells are unknown.
By real-time RT-PCR, we observed upregulation of TRKA and BDNF but not TRKB and TRKC in
analyzed samples (n=4, data not shown), suggesting that posttranscriptional events may contribute
the expression of TRKB and TRKC.
One characteristic finding in the present study is that TRK expression is associated with
myelomonocytic and monocytic leukemia (Figure 1 and Table 1). It is possible that the expression of
different members of the TRK family simply reflects the cell of origin, without a significant role in tumor
biology. However, although TRKA can be found in B lymphocytes,18 T lymphocytes,29 and
monocytes/macrophages,46,51 we only observed TRKA expression in AML patients by flow cytometry.
Therefore, it is more likely that differential expression of TRK receptors and activation of their
respective signal transduction pathways directly affects the biologic behavior of the cells, which leads
to differentiation, survival, and/or proliferation. For example, tumors with functional TRKB may be
particularly aggressive because TRKB provides a growth advantage and may protect them from
chemotherapy.24 In the present study, expression of TRKs was associated with shorter survival,
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

15
particularly for patients co-expressing TRKB and BDNF. Our data thus support the concept that TRKB
may play an important role in human tumorigenesis.24,52 In neuroblastoma, TRKB expression has been
demonstrated preferentially in aggressive, MYCN-amplified tumors, and is associated with a poor
prognosis. Activation of the BDNF/TRKB signaling pathway in neuroblastoma cells expressing TRKB
increases cell survival,53 and may protect neuroblastoma cells from chemotherapy and thereby
contribute to a more chemoresistant phenotype.54 In a recent functional genomic screen for genes that
suppress anoikis, TRKB was identified as an oncoprotein associated with metastatic capacity.21
Overexpression of TRKB rendered nonmalignant epithelial cells anoikis (apoptosis)-resistant and
highly tumorigenic. Consistent with the model that suppression of anoikis facilitates metastasis, TRKB-
expressing cells formed highly invasive and metastatic tumors in nude mice, with very short
latencies.21 TRKB kinase activity is required and also sufficient for anoikis suppression, tumor
formation, and experimental metastasis.52 Consistently, we found that TRKB kinase activity was
absolutely required for its anti-apoptotic effect (Figure 3D), potentially a major mediator for
transformation induced by TRK signaling. We also demonstrated that targeting TRKs by TRK inhibitor
or siRNA efficiently inhibited growth of leukemic cells in vitro (Figure 4 and 5). These results suggest
that targeting the enzymatic activity of TRKB or its downstream effectors might be beneficial in therapy
of both solid tumors and leukemia.
Autocrine circuits of tyrosine kinases such as FLT313 or KDR14 are potentially involved in human
leukemia. Multiple myeloma cells were found to express TRKB, with potential survival signaling
induced by autocrine BDNF.44 In all of these cases, a prognostic role remains to be determined.
Additionally, there are so far no animal studies which directly confirm their tumorigenic potential,
although blocking autocrine loop function/activity has been shown to be important for treatment of
leukemia in vitro and in animal models.14 Here, we found a high incidence of expression of
TRKB/BDNF and its association with poor prognosis in our patient cohort (Figure 2). Our mouse
models showed direct evidence for the leukemogenic potential of autocrine activation of TRKA/NGF
and TRKB/BDNF (Figure 4). Consistent with recent publications showing <2% of point mutations in
>90 tyrosine kinases (including TRKs) in >300 AML patients,55,56 we found very few point mutations in
TRK receptors, indirectly supporting the hypothesis that autocrine activation represents a major
mechanism for transformation by TRKs, and probably also for many other receptor PTKs. However,
reflecting the finding that NGF and BDNF are also expressed by stromal cells in bone marrow, and the
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

16
recent finding that TRK receptors can also be activated in the absence of NTs,47,57 we do not rule out
the possibility of other mechanisms for activation of TRKs in leukemogenesis, e.g. paracrine or
intracrine mechanism. In fact, we also observed constitutive activation of TRKs in patients expressing
TRKs only (T20, T42 in Figure 1). Interestingly, autocrine activation of TRK has also been reported in
inflammatory cells including monocytes and has been suggested to be an important factor in the
establishment of autoimmune diseases.46 Moreover, recent data demonstrate that NGF is an autocrine
factor essential for the survival of macrophages infected with HIV. These cells take advantage of their
autocrine NGF, survive for a very long period of time and continuously produce virus particles. Thus,
exploring the effects of inhibiting autocrine TRK signaling may open new therapeutic strategies for
both leukemia and latent HIV infection.51
Development of AML is believed to require the co-operation of class I and class II mutations.1 Our
finding that over half of the patients investigated showed expression/mutations of TRKs reduces the
proportion of patients in whom so far no activation of PTKs (class I) has been identified. Therapies
targeting receptor PTKs have provided remarkable responses in both hematologic cancers and solid
tumors, but their clinical efficacy has been limited in many cases, and few, if any, patients are cured.
Recently, Stommel and colleagues have identified obstacles to meaningful response to single-agent
therapies targeting receptor PTKs utilizing a glioblastoma model.10 They found three or more activated
receptor PTKs in each tumor, and up to 10 activated receptor PTKs in some cases. Combinations of
PTK inhibitors, but not single agents, inhibited PI3K signaling and related sequelae. Consistently,
single-agent targeted therapies for AML patients with FLT3-ITD or c-Kit mutations showed only
moderate efficacy in some cases.8 In the present study, we found a significant proportion of AML
patients with both FLT3-ITD and TRK expression. We speculate that assessment of TRK expression
and signaling in these patients, and combination of targeted therapies or multifunctional kinase
inhibitors directed at activated receptor PTKs may improve outcome.58
In summary, we demonstrate that primary AL cells frequently express TRK receptors and BDNF on
the protein level. We found a significant correlation with TRK expression and morphologic subtypes as
established in the FAB classification, and poor outcome in patients with co-expression of TRKB and
BDNF. The leukemogenic activity of this autocrine loop was confirmed in mouse models. Moreover,
activation of TRKs was an important survival factor for leukemic cells from both patients and mice.
Collectively, our findings suggest that TRKs play an important role in leukemogenesis with autocrine
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

17
stimulation as a potential mechanism for oncogenic activity. Thus, TRKs and their downstream
signaling partners might serve as novel therapeutic targets in AL. To improve classification and
treatment of hematological malignancies, we would recommend the prospective assessment of NTs,
their receptors and downstream pathways in larger cohorts and additional malignancies of
hematopoietic origin.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

18
Acknowledgments
This study was supported by the Deutsche Krebshilfe (grant: 10-2090-Li I) and by the Deutsche
Forschungsgemeinschaft (DFG, excellence cluster REBIRTH). CB was also supported by the National
Cancer Institute (R01-CA107492-01A2). MR is a student of the MD/PhD program at Hannover Medical
School (MHH), and received support from the Deutsche José Carreras Leukämie-Stiftung (grant:
DJCLS F05/10). CK was supported by DFG (KO 3582/1-1). We are very grateful to Stefan Bartels and
Ludwig Hoy for help with statistical analysis, Axel Schambach for providing vector backbones, Michael
Morgan for providing reagents and critical reading of this manuscript, Peter Horn and Martin Sauer for
providing cells, Vanessa Prox, Christine Garen, Rene Kirstein, Ellen Neumann, Elke Stürmer, Elvira
Lux, and Cindy Elfers for technical assistance, Rolf Baumann, Hans Grundtke, Jörg Frühauf, Anne
Koop, and Bernd Polivka (all MHH) for irradiation of animals and cells. We also thank Dr. D. Martin-
Zanca for providing cDNA of TPM3/TRK.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

19
Authorship
Z.L., J.M., A.G., and C.B. designed the study. Z.L. and C.B. interpreted the data and wrote the
manuscript. Z.L., M.R., T.N., and M.Y. performed immunophenotyping, mutation analysis, colony
assays, apoptosis assays and animals studies. G.B., C.K., M.H., H.D., and Z.L. collected patients’
data and samples. M.R. and J.M. contributed Western blot analysis. N.N., G.G., L.W., J.K., and B.S.
performed cytogentic and molecular genetic analysis and provided samples. G.B. performed statistical
analysis.
Conflict of interest: The authors declare that no conflict of interest exists.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

20
References
1. Deguchi K, Gilliland DG. Cooperativity between mutations in tyrosine kinases and in hematopoietic transcription factors in AML. Leukemia. 2002;16:740-744.
2. Scheijen B, Griffin JD. Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene. 2002;21:3314-3333.
3. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054-1061.
4. Hu J, Zhou GB, Wang ZY, Chen SJ, Chen Z. Mutant transcription factors and tyrosine kinases as therapeutic targets for leukemias: from acute promyelocytic leukemia to chronic myeloid leukemia and beyond. Adv Cancer Res. 2007;98:191-220.
5. Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172-187.
6. Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408-2417.
7. Boissel N, Leroy H, Brethon B, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia. 2006;20:965-970.
8. Tickenbrock L, Muller-Tidow C, Berdel WE, Serve H. Emerging Flt3 kinase inhibitors in the treatment of leukaemia. Expert Opin Emerg Drugs. 2006;11:153-165.
9. Whitman SP, Ruppert AS, Radmacher MD, et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood. 2008;111:1552-1559.
10. Stommel JM, Kimmelman AC, Ying H, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287-290.
11. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355-365. 12. Jechlinger M, Sommer A, Moriggl R, et al. Autocrine PDGFR signaling promotes
mammary cancer metastasis. J Clin Invest. 2006;116:1561-1570. 13. Zheng R, Levis M, Piloto O, et al. FLT3 ligand causes autocrine signaling in acute
myeloid leukemia cells. Blood. 2004;103:267-274. 14. Dias S, Hattori K, Heissig B, et al. Inhibition of both paracrine and autocrine VEGF/
VEGFR-2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias. Proc Natl Acad Sci U S A. 2001;98:10857-10862.
15. Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913-1925.
16. Lee FS, Kim AH, Khursigara G, Chao MV. The uniqueness of being a neurotrophin receptor. Curr Opin Neurobiol. 2001;11:281-286.
17. Pyle AD, Lock LF, Donovan PJ. Neurotrophins mediate human embryonic stem cell survival. Nat Biotechnol. 2006;24:344-350.
18. Torcia M, Bracci-Laudiero L, Lucibello M, et al. Nerve growth factor is an autocrine survival factor for memory B lymphocytes. Cell. 1996;85:345-356.
19. Pincelli C, Marconi A. Autocrine nerve growth factor in human keratinocytes. J Dermatol Sci. 2000;22:71-79.
20. Descamps S, Pawlowski V, Revillion F, et al. Expression of nerve growth factor receptors and their prognostic value in human breast cancer. Cancer Res. 2001;61:4337-4340.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

21
21. Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430:1034-1039.
22. McGregor LM, McCune BK, Graff JR, et al. Roles of trk family neurotrophin receptors in medullary thyroid carcinoma development and progression. Proc Natl Acad Sci U S A. 1999;96:4540-4545.
23. Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med. 1993;328:847-854.
24. Eggert A, Grotzer MA, Ikegaki N, et al. Expression of the neurotrophin receptor TrkB is associated with unfavorable outcome in Wilms' tumor. J Clin Oncol. 2001;19:689-696.
25. Liu Q, Schwaller J, Kutok J, et al. Signal transduction and transforming properties of the TEL-TRKC fusions associated with t(12;15)(p13;q25) in congenital fibrosarcoma and acute myelogenous leukemia. Embo J. 2000;19:1827-1838.
26. Jin W, Yun C, Kwak MK, Kim TA, Kim SJ. TrkC binds to the type II TGF-beta receptor to suppress TGF-beta signaling. Oncogene. 2007.
27. Labouyrie E, Dubus P, Groppi A, et al. Expression of neurotrophins and their receptors in human bone marrow. Am J Pathol. 1999;154:405-415.
28. Kermani P, Rafii D, Jin DK, et al. Neurotrophins promote revascularization by local recruitment of TrkB+ endothelial cells and systemic mobilization of hematopoietic progenitors. J Clin Invest. 2005;115:653-663.
29. Vega JA, Garcia-Suarez O, Hannestad J, Perez-Perez M, Germana A. Neurotrophins and the immune system. J Anat. 2003;203:1-19.
30. Maroder M, Bellavia D, Meco D, et al. Expression of trKB neurotrophin receptor during T cell development. Role of brain derived neurotrophic factor in immature thymocyte survival. J Immunol. 1996;157:2864-2872.
31. Eguchi M, Eguchi-Ishimae M, Tojo A, et al. Fusion of ETV6 to neurotrophin-3 receptor TRKC in acute myeloid leukemia with t(12;15)(p13;q25). Blood. 1999;93:1355-1363.
32. Reuther GW, Lambert QT, Caligiuri MA, Der CJ. Identification and characterization of an activating TrkA deletion mutation in acute myeloid leukemia. Mol Cell Biol. 2000;20:8655-8666.
33. Meyer J, Rhein M, Schiedlmeier B, et al. Remarkable leukemogenic potency and quality of a constitutively active neurotrophin receptor, deltaTrkA. Leukemia. 2007;21:2171-2180.
34. Mulloy JC, Jankovic V, Wunderlich M, et al. AML1-ETO fusion protein up-regulates TRKA mRNA expression in human CD34+ cells, allowing nerve growth factor-induced expansion. Proc Natl Acad Sci U S A. 2005;102:4016-4021.
35. Li Z, Dullmann J, Schiedlmeier B, et al. Murine leukemia induced by retroviral gene marking. Science. 2002;296:497.
36. Beutel G, Meyer J, Ma L, et al. Expression of the p75 neurotrophin receptor in acute leukaemia. Br J Haematol. 2005;131:67-70.
37. Kaebisch A, Brokt S, Seay U, Lohmeyer J, Jaeger U, Pralle H. Expression of the nerve growth factor receptor c-TRK in human myeloid leukaemia cells. Br J Haematol. 1996;95:102-109.
38. Heuser M, Beutel G, Krauter J, et al. High meningioma 1 (MN1) expression as a predictor for poor outcome in acute myeloid leukemia with normal cytogenetics. Blood. 2006;108:3898-3905.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

22
39. Avellino R, Romano S, Parasole R, et al. Rapamycin stimulates apoptosis of childhood acute lymphoblastic leukemia cells. Blood. 2005;106:1400-1406.
40. Bene MC, Castoldi G, Knapp W, et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia. 1995;9:1783-1786.
41. Chevalier S, Praloran V, Smith C, et al. Expression and functionality of the trkA proto-oncogene product/NGF receptor in undifferentiated hematopoietic cells. Blood. 1994;83:1479-1485.
42. Coulier F, Kumar R, Ernst M, Klein R, Martin-Zanca D, Barbacid M. Human trk oncogenes activated by point mutation, in-frame deletion, and duplication of the tyrosine kinase domain. Mol Cell Biol. 1990;10:4202-4210.
43. Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature. 1986;319:743-748.
44. Pearse RN, Swendeman SL, Li Y, Rafii D, Hempstead BL. A neurotrophin axis in myeloma: TrkB and BDNF promote tumor-cell survival. Blood. 2005;105:4429-4436.
45. Troeger A, Gudowius S, Escherich G, et al. High nerve growth factor receptor (p75NTR) expression is a favourable prognostic factor in paediatric B cell precursor-acute lymphoblastic leukaemia. Br J Haematol. 2007;139:450-457.
46. la Sala A, Corinti S, Federici M, Saragovi HU, Girolomoni G. Ligand activation of nerve growth factor receptor TrkA protects monocytes from apoptosis. J Leukoc Biol. 2000;68:104-110.
47. Lee FS, Chao MV. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci U S A. 2001;98:3555-3560.
48. Loeb DM, Maragos J, Martin-Zanca D, Chao MV, Parada LF, Greene LA. The trk proto-oncogene rescues NGF responsiveness in mutant NGF-nonresponsive PC12 cell lines. Cell. 1991;66:961-966.
49. Glass DJ, Nye SH, Hantzopoulos P, et al. TrkB mediates BDNF/NT-3-dependent survival and proliferation in fibroblasts lacking the low affinity NGF receptor. Cell. 1991;66:405-413.
50. Bartkowska K, Paquin A, Gauthier AS, Kaplan DR, Miller FD. Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development. Development. 2007;134:4369-4380.
51. Garaci E, Caroleo MC, Aloe L, et al. Nerve growth factor is an autocrine factor essential for the survival of macrophages infected with HIV. Proc Natl Acad Sci U S A. 1999;96:14013-14018.
52. Geiger TR, Peeper DS. Critical role for TrkB kinase function in anoikis suppression, tumorigenesis, and metastasis. Cancer Res. 2007;67:6221-6229.
53. Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14:759-767.
54. Ho R, Eggert A, Hishiki T, et al. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer Res. 2002;62:6462-6466.
55. Loriaux MM, Levine RL, Tyner JW, et al. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood. 2008;111:4788-4796.
56. Tomasson MH, Xiang Z, Walgren R, et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood. 2008;111:4797-4808.
57. Rajagopal R, Chen ZY, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci. 2004;24:6650-6658.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

23
58. Zhang W, Konopleva M, Shi YX, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;100:184-198.
59. Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051-1062.
60. Scholl C, Bansal D, Dohner K, et al. The homeobox gene CDX2 is aberrantly expressed in most cases of acute myeloid leukemia and promotes leukemogenesis. J Clin Invest. 2007;117:1037-1048.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
24
Tables
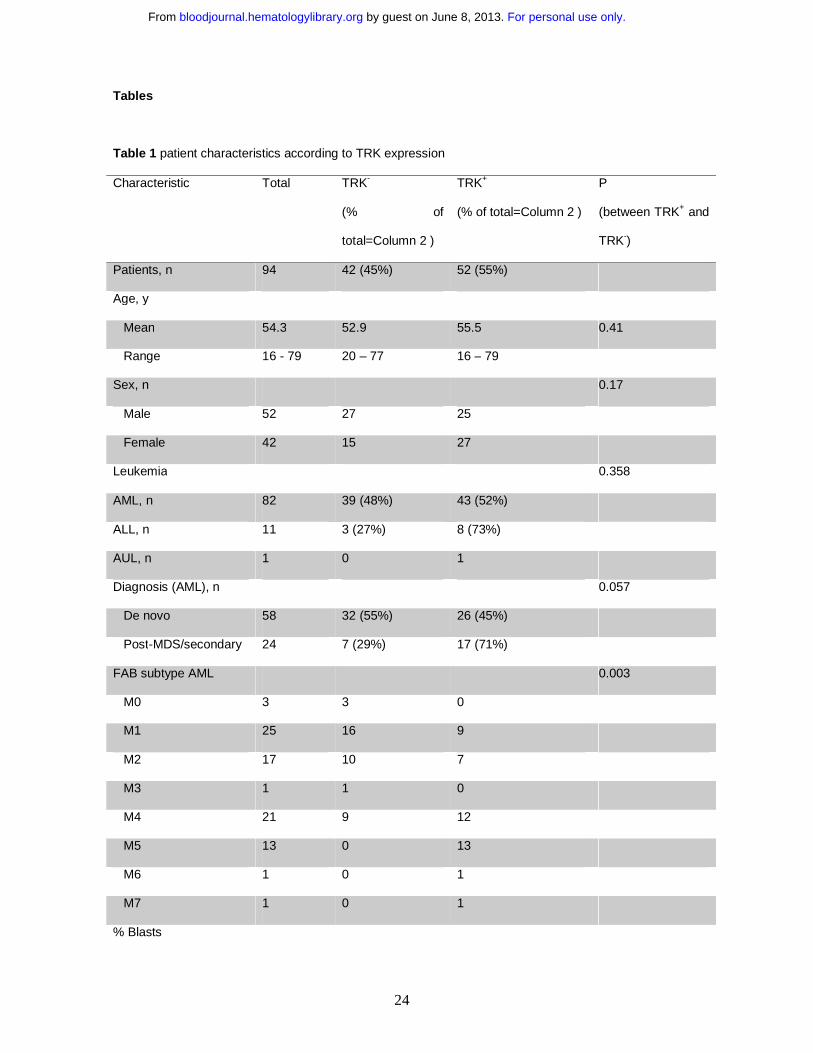
Table 1 patient characteristics according to TRK expression
Characteristic Total TRK-
(% of
total=Column 2 )
TRK+
(% of total=Column 2 )
P
(between TRK+ and
TRK-)
Patients, n 94 42 (45%) 52 (55%)
Age, y
Mean 54.3 52.9 55.5 0.41
Range 16 - 79 20 – 77 16 – 79
Sex, n 0.17
Male 52 27 25
Female 42 15 27
Leukemia 0.358
AML, n 82 39 (48%) 43 (52%)
ALL, n 11 3 (27%) 8 (73%)
AUL, n 1 0 1
Diagnosis (AML), n 0.057
De novo 58 32 (55%) 26 (45%)
Post-MDS/secondary 24 7 (29%) 17 (71%)
FAB subtype AML 0.003
M0 3 3 0
M1 25 16 9
M2 17 10 7
M3 1 1 0
M4 21 9 12
M5 13 0 13
M6 1 0 1
M7 1 0 1
% Blasts
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

25
Bone marrow 73 76 70 0.280
Peripheral blood 44 49 39 0.173
WBC counts, x109/L
Mean 24.4 61.0 0.113
(cutoff: 20.000) 59
Range 0.8 - 159.0 0.6 - 454.0
ECOG status 0.764
0 22 10 12
1 52 25 27
2 13 7 6
3 1 0 1
Cytogenetics 0.481
Normal karyotype 39 17 (44%) 22 (56%)
t(8;21) (q22;q22) 1 1 0
inv(16) (p13q12) 1 1 0
t(15:17)(q22;q11-21) 1 1 0
t(11q23) 2 1 1
Complex karyotype* 21 11 10
Other aberrations 24 8 16
FLT3-ITD-, n (%) 50 26 (52%) 24 (48%) 0.363
FLT3-ITD+, n (%) 17 6 (35%) 11 (65%)
missing 17 (18%)
*Complex karyotype was defined as 3 or more cytogenetic abnormalities in the absence of t(8;21),
inv(16), t(15;17), or t(11q23).60
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
26
Table 2 Univariate Cox regression analysis for TRKs and BDNF
median survival (days)
Hazard Ratio (95% CI) p-value receptor
positive
receptor
negative
TRKA 1.44 (0.80–2.62) 0.23 312 547
TRKB 1.17 (0.66–2.07) 0.60 359 547
TRKC 1.32 (0.70–2.50) 0.40 359 535
BDNF 1.38 (0.68–2.82) 0.37 225 480
TRKB / BDNF 2.26 (1.07–4.80) 0.03 212 547
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

27
Figure legends
Figure 1. Expression of TRK receptors and NTs in patients with AL. (A) Showing the gate for the
blast population. Left panel: Patient T10 (AML M5), middle panel T14 (T-cell ALL), right panel: Patient
T78 (AML M1). (B) Top row showing expression of all three TRK receptors in the blasts of patient T10.
Middle row: patient T14 expressed only TRKB. Bottom row: patient T78 was negative for TRK receptors.
(C) BDNF was also expressed in patients T10 (left panel) and T14 (middle panel), but not in patient T78
(right panel). The isotype controls are shown as insets. (D) Constitutive phosphorylation of p145TRK in
primary leukemic cells. Total cell lysates were blotted and probed with an anti-pTRK (E-6) antibody
detecting phosphorylated forms of all three TRK receptors. The blot was stripped and reprobed with the
anti-TRKB (794) antibody. Please see flow cytometry diagrams in Figures S1 and S3 showing
expression of TRKs and BDNF in the blasts of the patients not shown here.
Figure 2. Kaplan-Meier estimates of overall survival for patients with AML according to TRKs and
BDNF status. Co-expression of TRKB and BDNF was associated with statistically significant poor
outcome. The log-rank test was used to compare differences between survival curves.
Figure 3. Ligand-dependent resistance to radiation-induced apoptosis. (A) 32D cells expressing
TRKA, TRKB, and 32D wild-type cells. Cells were starved for 3 hours and exposed to 5 Gy irradiation.
Cells that were Annexin-V and propidium iodide (PI)-negative were counted as viable cells. Viability was
calculated as the percentage of these cells over the total cell population. (B) BDNF prevented apoptosis
of 32D cells expressing TRKB almost as efficiently as IL-3. Cells were analyzed by flow cytometry 26h
after irradiation. Combined Annexin-V and PI staining was used to distinguish early apoptotic (Annexin-
V+/PI-) and later apoptotic cells (Annexin-V+/PI+). (C) K252a dramatically inhibited anti-apoptotic effects
of BDNF-mediated activation of TRKB, while only slight inhibition was observed if cells were cultured in
the presence of IL-3 alone. Results presented are the mean ± SD of at least 2 independent experiments.
NGF/BDNF concentration was 100 ng/mL, murine IL-3 2ng/ml.
Figure 4. Development of leukemia in mouse #681 transplanted with TRKB and BDNF modified
hematopoietic cells (32D). FACS analysis showing co-expression of TRKB and BDNF before
transplantation (A) and in myeloid blasts recovered from the animal (B). (C) Peripheral blood smear
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

28
showing marked leukocytosis consisting predominantly of immature myeloid cells. (D) Cytospin of BM
showing myeloblasts with an abundant cytoplasm (x 1000). (E, F) Diffuse myeloblastic infiltration in
spleen and complete effacement of its normal structure (x 100, x 1000). (G, H) Extensive infiltration of
leukemic cells in liver, primarily in portal areas, but also diffusely in the sinusoid (x 100, x 1000). (I)
Constitutive activation of TRKB in leukemic cells and blocking of the TRKB/BDNF autocrine loop by anti-
BDNF neutralizing antibody and K252a. Leukemic cells were treated with either 20µg/ml anti-BDNF
antibody or 100nM K252a for 12h. Total cell lysates (300µg) from untreated and treated cells were
immunoprecipitated with the anti-TRK (C-14), separated on SDS-PAGE, blotted and probed with an anti-
pTRK (E-6) antibody (top and middle panels with short and long exposition, respectively). The blot was
stripped and reprobed with the anti-TRKB (H181) antibody. Arrows point to the phosphorylated forms of
TRK. Note different expression pattern of human TRKB in human cells (only 145KDa form in Figure 1)
and rodent cells (both 145KDa and 120KDA forms) as previously described.52 (J) Anti-human BDNF
antibody (10µg/ml and 20µg/ml) inhibited growth of leukemic cells in colony-forming assays. Results are
presented as the average percentage of colonies formed in the presence of antibody (100% value
derived from untreated control). Results presented are the mean ± SD of 2 independent experiments.
Figure 5. K252a induces apoptosis of primary AML cells and. Cell viability was analyzed using the
Annexin-V assay. Results are presented as the percentage of living cells in the presence of K252a
(100% value derived from untreated control). (A) Cells were from patient T72 expressing TRKB and
BDNF, both patients T10 and T29 expressing TRKs (TRKA, TRKB and TRKC) and BDNF, and T0 (only
TRKs). Patients T17 and T56 were negative for TRKs and NTs. Results presented for T10 (K 200nM)
and T29 (K 400nM) are the mean ± SD of 2 independent experiments. Note that treatment schedule was
not completely applied to all patients tested due to limited number of cells. The concentrations of K252a
that we used have been well documented for TRK inhibition in the literature.17,47,48 K=K252a. (B) Flow
cytometry diagram of apoptosis of blasts from patient T17 and T29, cultured with K252a for 18h before
analysis.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
A
B
Li et al., 2008Figure 3
CmIL-3
100 102 104100
102
10414%
12%72%
BDNF
100 102 104100
102
10424%
14%59%
w/o cytokines
100 102 104100
102
10470%
21%7%
Annexin
IP
TRKA
0 10 20 30 40 50 600
25
50
75
100no factormIL-3hNGF
Time [hours]
TRKB
0 10 20 30 40 50 600
25
50
75
100no factormIL-3BDNF
Time [hours]
32Dwt
0 10 20 30 40 50 600
25
50
75
100no factormIL-3hNGFBDNF
Time [hours]
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
A
Li et al., 2008Figure 4
B
14% 14%
38%
100 102 104100
102
104
3%
70%4%
100 102 104100
102
104
FN
DB
TRKB
C D
E F
G H
I J
α− pTRKB
α− TRKB
lm/gμ02 bA
Mn001 a252K
detaertnU
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
Top Related
Copyright © 2022 FDOKUMEN

