
A unified model of CA1/3 pyramidal cells: an investigation into excitability
Upload
u-bordeaux1Category
view
4download
0
Voltage-Clamp Analysis of the Potentiation of the SlowCa21-Activated K1 Current in HippocampalPyramidal Neurons
Michel Borde, Christian Bonansco,David Fernandez de Sevilla, Didier Le Ray,and Washington Buno*
Departamento de Plasticidad Neural, Instituto Cajal,Consejo Superior de Investigaciones Cientıficas,Madrid, Spain
ABSTRACT: Exploring the principles that govern activity-dependentchanges in excitability is an essential step to understand the function ofthe nervous system, because they act as a general postsynaptic controlmechanism that modulates the flow of synaptic signals. We show anactivity-dependent potentiation of the slow Ca21-activated K1 current(sIAHP) which induces sustained decreases in the excitability in CA1pyramidal neurons. We analyzed the sIAHP using the slice technique andvoltage-clamp recordings with sharp or patch-electrodes. Using sharpelectrodes-repeated activation with depolarizing pulses evoked a pro-longed (8-min) potentiation of the amplitude (171%) and duration(208%) of the sIAHP. Using patch electrodes, early after entering thewhole-cell configuration (<20 min), responses were as those reportedabove. However, although the sIAHP remained unchanged, its potentiationwas markedly reduced in later recordings, suggesting that the underlyingmechanisms were rapidly eliminated by intracellular dialysis. Inhibition ofL-type Ca21 current by nifedipine (20 mM) markedly reduced the sIAHP(79%) and its potentiation (55%). Ryanodine (20 mM) that blocks therelease of intracellular Ca21 also reduced sIAHP (29%) and its potentiation(25%). The potentiation of the sIAHP induced a marked and prolonged(>50%; '8 min) decrease in excitability. The results suggest that sIAHP ispotentiated as a result of an increased intracellular Ca21 concentration([Ca21]i) following activation of voltage-gated L-type Ca21 channels,aided by the subsequent release of Ca21 from intracellular stores. Anotherpossibility is that repeated activation increases the Ca21-binding capacityof the channels mediating the sIAHP. This potentiation of the sIAHP couldbe relevant in hippocampal physiology, because the changes in excitabil-ity it causes may regulate the induction threshold of the long-term poten-tiation of synaptic efficacy. Moreover, the potentiation would act as aprotective mechanism by reducing excitability and preventing the accu-mulation of intracellular Ca21 to toxic levels when intense synapticactivation occurs. Hippocampus 2000;10:198–206.© 2000 Wiley-Liss, Inc.
KEY WORDS: hippocampus; voltage-clamp; postsynaptic plasticity;Ca21 homeostasis; Ca21 stores
INTRODUCTION
The electrical activity in neurons is usually shaped andcontrolled by Ca21-activated K1 conductances (e.g., Ad-ams et al., 1982; Madison and Nicoll, 1984; Blatz andMagleby, 1987; Storm, 1990). Two types of Ca21-acti-vated K1 channels with large and small unitary conduc-tance have been described (BK and SK, respectively; e.g.,Marty, 1981; Latorre et al., 1989). BK channels haverapid kinetics, are voltage-sensitive, and are blocked bytetraethylammonium (TEA) and charybdotoxin. Thecurrents mediated through BK channels control actionpotential (AP) repolarization as well as generate AP fastafterhyperpolarization in neurons (e.g., Adams et al.,1982; Storm, 1987) and invertebrate muscle fibers (e.g.,Araque and Buno, 1995). SK channels have slower kinet-ics, are voltage-insensitive, are blocked by apamin, andmediate the current that generates the medium afterhy-perpolarization (mAHP) that follows action-potentialbarrages in many neurons (e.g., Marty, 1981; Sah, 1996;Sah and Clements, 1999). Moreover, a voltage- andapamin-insensitive Ca21-activated K1 current withslower kinetics (sIAHP) mediates the slow AHP (sAHP)(Sah, 1996; Sah and Bekkers, 1996) and controls firingadaptation in CA1 pyramidal neurons (Madison andNicoll, 1984; Lancaster and Adams, 1986; Lancaster andNicoll, 1987).
Repeated activation of CA1 pyramidal neurons by de-polarizing current pulses induces a marked and sustainedreduction in excitability (Borde et al., 1995, 1999). Thisreduction in excitability was probably due to a gradualpotentiation of the slow Ca21-activated K1 current(sIAHP) that mediates the sAHP. This was suggested bythe finding that a potentiated sIAHP paralleled the re-duced synaptic efficacy evoked by repeated activation inCA1 pyramidal cells (Borde et al., 1999).
Using in vitro hippocampal slice preparation and volt-age-clamp recordings, we analyzed the sustained poten-
Grant sponsor: DGES/MEC; Grant number: PB95-0031; Grant sponsor:CEC/CAM; Grant number: 08.5/0038/98.M.B. and C.B. contributed equally to this work.Present addresses: Dr. M. Borde, Lab. Neurofisiologıa, Depto. Fisiologıa,Facultad de Medicina, General Flores 2125, 11800-Montevideo, Uruguay;Dr. C. Bonansco, Facultad de Ciencias, Universidad de Playa Ancha, GranBretana 40, casilla 34-V, Valparaıso, Chile.*Correspondence to: Dr. Washington Buno, Instituto Cajal, Consejo Supe-rior de Investigaciones Cientıficas, 28002-Madrid, Spain.E-mail: [email protected] for publication 22 November 1999
HIPPOCAMPUS 10:198–206 (2000)
© 2000 WILEY-LISS, INC.

tiation of the sIAHP occurring in CA1 pyramidal neurons followingrepeated activation. Inhibition of L-type Ca21 conductance bynifedipine reduced the sIAHP and its potentiation. RestrainingCa21 release from intracellular stores with ryanodine had similarsIAHP and potentiation-blocking effects. Although a robust sIAHP
was recorded with the whole-cell technique, its potentiation byrepeated stimulation was rapidly reduced and eventually elimi-nated.
This activity-dependent potentiation of the sIAHP could be rel-evant in hippocampal physiology because it provides a homeostaticnegative feedback regulation of neuronal excitability that actspostsynaptically, controlling the flow of synaptic information.This control would be under the modulatory influence of the mul-tiple neurotransmitter and second-messenger systems that regulatethe sIAHP (e.g., Benardo and Prince, 1982; Madison and Nicoll,1984; Lancaster and Adams, 1986; Pedarzani and Storm, 1996;Storm, 1990). Moreover, the increased negative feedback providedby the potentiation of the sIAHP may contribute to limit the rise inintracellular Ca21 concentration ([Ca21]i) and stabilize the mem-brane potential following intense synaptic activation. It would thusact as a protective mechanism by reducing excitability and prevent-ing the accumulation of intracellular Ca21 to toxic levels.
METHODS
Full details of the procedures of animal care, surgery, and slicepreparation were described previously (e.g., Garcıa-Munoz et al.,1993; Borde et al., 1995); therefore, it suffices to briefly delineatethem here.
Slice Preparation
Wistar rats (15–25 days old) were anesthetized with ether anddecapitated, and the brain removed and submerged in artificialcerebrospinal fluid (ACSF; see composition below) at 4°C andmaintained at pH 7.4 by bubbling with carbogen (95% O2, 5%CO2). Transverse slices (350 mm) of the hippocampus were cutwith a Vibratome (Pelco 101, St. Louis, MO) and incubated in theACSF (1 h at 20–22°C).
Recording, Stimulation, and Analysis
Slices were transferred to a 2-ml chamber fixed to an invertedmicroscope stage (Nikon Diaphot-TMD, Tokyo, Japan), main-tained at 32–34°C and superfused with carbogen-bubbled ACSF(2 ml/min). Two recording methods were used: 1) intracellularrecordings with sharp, 60–120 MV K1-acetate filled (3-M) mi-cropipettes; 2) patch-recordings in the whole-cell configurationwith 3–6 MV fire-polished pipettes (Hamill et al., 1981) filledwith 150 mM KMeSO4 (Zhang et al., 1994) (both pulled with aSutter Inst., P-87, Novato, USA). Occasionally, micropipetteswere filled with tetrapotassium salt of BAPTA at final concentra-tions of 60–150 mM diluted in K1-acetate (3 M). In some cases,sharp and fire-polished pipettes were Sylgard-coated to decrease
the capacitance of the immersed tip and thus reduce artifacts andimprove the voltage control (VC) in VC experiments (Hamill etal., 1981). Micropipettes were connected to an Axoclamp 2B am-plifier (Axon Instruments, Inc., Foster City, CA), and recordingswere in the bridge or single-electrode voltage-clamp modes (CCand VC, respectively). Micropipettes were placed with a hydraulicmicromanipulator (Narishige, Tokyo, Japan) under direct visual-ization of the CA1 soma layer. The characteristic responses todirect stimulation identified pyramidal neurons. Experimentsstarted after a '5-min stabilization period following penetrationwith sharp electrodes or access to the intracellular compartmentwith patch-electrodes. In the CC mode, 12 successive 200-ms or2-s duration 0.1–0.5-nA depolarizing current pulses repeated atrates of 1 pulse every 5–60 s were used to induce the reduction ofcell excitability and the potentiation of the sIAHP. The reduction ofcell excitability was evaluated in CC mode by calculating the meanrate of firing (i.e., the ratio of the number of APs over time) duringthe last 1,800 ms of the response evoked by 2-s depolarizing cur-rent pulses. The firing rate during the last 1,800 ms corresponds tothe adapted rate (AR). Membrane currents were evoked by 2-s or200-ms depolarizing voltage commands from a 260-mV holdingpotential (Vh) to 0 mV. Currents were obtained before (6–8 min)and after (10 s) inducing the potentiation in CC mode (see above).VC and CC data were high-pass-filtered at 0.3 and 3.0 KHz,respectively, and sampled at rates between 1.0–10.0 KHz. A486/PC computer, a TL-1/DMA interface board, and pCLAMPsoftware (Axon Instruments, Inc.) were used for experimental con-trol, data acquisition, and analysis. Data were also stored on tapewith a PCM-videocassette system (Cybertec, Madrid, Spain) forfurther analysis. The decay time constant (t) of the sIAHP wasestimated from fits (minimum square regression) to a single expo-nential function. All figures are computer printouts (Laserjet 4L or6L, Hewlett Packard, Boise, ID). Values were expressed as themean 6 SD.
Solutions
The standard solution ASCF was as follows (in mM): 124.00NaCl, 2.69 KCl, 1.25 KH2PO4, 2.00 Mg2SO4, 26.00 NaHCO3,2.00 CaCl2, and 10.00 glucose, carbogen-bubbled to stabilized thepH at 7.4. Ca21-free ACSF was prepared by substituting CaCl2with MgCl2. TEA (10 mM), tetrodotoxin (TTX, 1.5 mM),v-conotoxin-GIVA (v-CgTX, 1 mM), v-agatoxin-IVA (100nM), CdCl2 (1 mM), and NiCl2 (10–30 mM) were added to theASCF and superfused. Nifedipine stock solution (1 mM in etha-nol) was dissolved in ASCF to a final concentration of 10–20 mM.Ryanodine stock solution (10 mM in DMSO) was dissolved in theACSF at 5–10 mM. Carbamilcholine chloride (CCh) was admin-istered as a “bolus” (Borde et al., 1995) to an estimated final con-centration of 10 mM, and its effects were recorded after the Vmhad returned to control values (cf. Borde et al., 1995). All chemi-cals were purchased from Sigma-Aldrich (Madrid, Spain) exceptv-CgTX and v-agatoxin-IVA, which were from the Peptide Insti-tute (Osaka, Japan).
_____________________________________________________________________ POTENTIATION OF sIAHP 199
RESULTS
The 53 neurons recorded with sharp electrodes were silent atrest, showed a Vm of 272.1 6 6.2 mV and Rin of 49.1 6 8.6 MV,and fired repetitive APs (78.6 6 5.8 mV, 1.2 6 0.8 ms) whendepolarized above a 255.3 6 7.2 mV threshold (e.g., Fig. 1B).Therefore, the cellular attributes were as described previously (e.g.,Garcıa-Munoz et al., 1993; Borde et al., 1995, 1999). The 33neurons recorded in the whole-cell configuration were also silent atrest and responded in a similar manner to depolarizing pulses (Fig.6B). However, they showed different resting Vm (257.6 6 2.3mV), Rin (147.9 6 12.8 MV), and AP thresholds (245.1 64.0 mV).
sIAHP Was Potentiated by Repeated StimulationWhen Sharp Electrodes Were Used
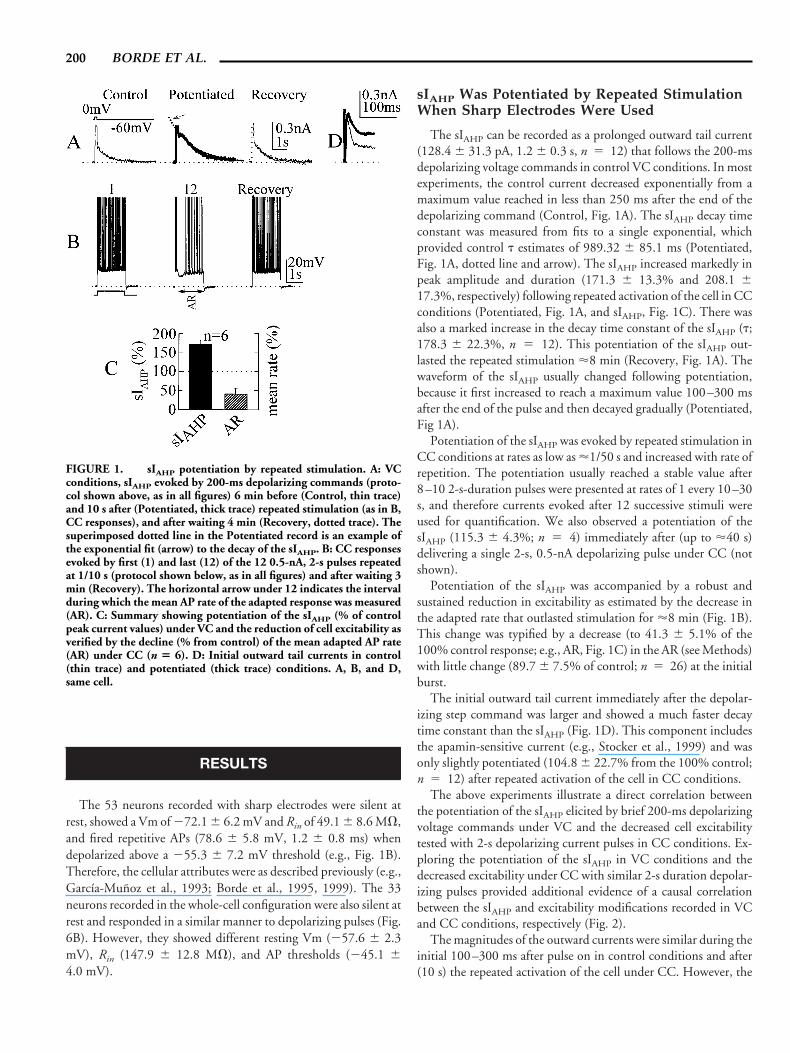
The sIAHP can be recorded as a prolonged outward tail current(128.4 6 31.3 pA, 1.2 6 0.3 s, n 5 12) that follows the 200-msdepolarizing voltage commands in control VC conditions. In mostexperiments, the control current decreased exponentially from amaximum value reached in less than 250 ms after the end of thedepolarizing command (Control, Fig. 1A). The sIAHP decay timeconstant was measured from fits to a single exponential, whichprovided control t estimates of 989.32 6 85.1 ms (Potentiated,Fig. 1A, dotted line and arrow). The sIAHP increased markedly inpeak amplitude and duration (171.3 6 13.3% and 208.1 617.3%, respectively) following repeated activation of the cell in CCconditions (Potentiated, Fig. 1A, and sIAHP, Fig. 1C). There wasalso a marked increase in the decay time constant of the sIAHP (t;178.3 6 22.3%, n 5 12). This potentiation of the sIAHP out-lasted the repeated stimulation '8 min (Recovery, Fig. 1A). Thewaveform of the sIAHP usually changed following potentiation,because it first increased to reach a maximum value 100–300 msafter the end of the pulse and then decayed gradually (Potentiated,Fig 1A).
Potentiation of the sIAHP was evoked by repeated stimulation inCC conditions at rates as low as '1/50 s and increased with rate ofrepetition. The potentiation usually reached a stable value after8–10 2-s-duration pulses were presented at rates of 1 every 10–30s, and therefore currents evoked after 12 successive stimuli wereused for quantification. We also observed a potentiation of thesIAHP (115.3 6 4.3%; n 5 4) immediately after (up to '40 s)delivering a single 2-s, 0.5-nA depolarizing pulse under CC (notshown).
Potentiation of the sIAHP was accompanied by a robust andsustained reduction in excitability as estimated by the decrease inthe adapted rate that outlasted stimulation for '8 min (Fig. 1B).This change was typified by a decrease (to 41.3 6 5.1% of the100% control response; e.g., AR, Fig. 1C) in the AR (see Methods)with little change (89.7 6 7.5% of control; n 5 26) at the initialburst.
The initial outward tail current immediately after the depolar-izing step command was larger and showed a much faster decaytime constant than the sIAHP (Fig. 1D). This component includesthe apamin-sensitive current (e.g., Stocker et al., 1999) and wasonly slightly potentiated (104.8 6 22.7% from the 100% control;n 5 12) after repeated activation of the cell in CC conditions.
The above experiments illustrate a direct correlation betweenthe potentiation of the sIAHP elicited by brief 200-ms depolarizingvoltage commands under VC and the decreased cell excitabilitytested with 2-s depolarizing current pulses in CC conditions. Ex-ploring the potentiation of the sIAHP in VC conditions and thedecreased excitability under CC with similar 2-s duration depolar-izing pulses provided additional evidence of a causal correlationbetween the sIAHP and excitability modifications recorded in VCand CC conditions, respectively (Fig. 2).
The magnitudes of the outward currents were similar during theinitial 100–300 ms after pulse on in control conditions and after(10 s) the repeated activation of the cell under CC. However, the
FIGURE 1. sIAHP potentiation by repeated stimulation. A: VCconditions, sIAHP evoked by 200-ms depolarizing commands (proto-col shown above, as in all figures) 6 min before (Control, thin trace)and 10 s after (Potentiated, thick trace) repeated stimulation (as in B,CC responses), and after waiting 4 min (Recovery, dotted trace). Thesuperimposed dotted line in the Potentiated record is an example ofthe exponential fit (arrow) to the decay of the sIAHP. B: CC responsesevoked by first (1) and last (12) of the 12 0.5-nA, 2-s pulses repeatedat 1/10 s (protocol shown below, as in all figures) and after waiting 3min (Recovery). The horizontal arrow under 12 indicates the intervalduring which the mean AP rate of the adapted response was measured(AR). C: Summary showing potentiation of the sIAHP (% of controlpeak current values) under VC and the reduction of cell excitability asverified by the decline (% from control) of the mean adapted AP rate(AR) under CC (n 5 6). D: Initial outward tail currents in control(thin trace) and potentiated (thick trace) conditions. A, B, and D,same cell.
200 BORDE ET AL.

current evoked after repeated stimulation rose above the controlcurrent to reach a maximum 300–600 ms after pulse on (Poten-tiated, Fig. 2A). The larger peak outward current (143.3 6 8.3%;
n 5 4) evoked after repeated activation is in agreement with apotentiation of the sIAHP. The magnitude and waveform modifi-cations in the outward current matched the reduction in the APrate under CC (Fig. 2A, B). It is noteworthy that the voltagecontrol of the region of AP generation was incomplete, as shown bythe firings in the VC records (arrows, Fig. 2A).
TTX and TEA Did Not Inhibit Potentiation ofthe sIAHP
Adding TEA and TTX to the ACSF did not significantly modifythe activity-dependent potentiation of the sIAHP (149.1 6 14.1%and 161.3 6 18.2% in ACSF and TEA 1 TTX, respectively; n 55), indicating that voltage-gated Na1- and TEA-sensitive K1 con-ductances did not contribute to potentiation of the sIAHP (Fig. 3A,B). The CC responses recorded in TEA 1 TTX solution revealedthat less high-threshold Ca21-mediated spikes were evoked by thesame pulse after repeated stimulation, indicating that the reduc-tion in excitability was also induced by the potentiated sIAHP underTEA 1 TTX (Fig. 3D, E). The first high-threshold Ca21 spikeevoked at pulse-on decreased considerably in duration (to 64 612.8% of the 100% control duration; n 5 4) with repeated stim-ulation (Fig. 3F), due to a reduction in the repolarizing slope; itspeak amplitude of the first Ca21 spike did not change.
When the sIAHP was blocked by superfusion with Ca21-freeCdCl2 (1 mM) or a CCh “bolus” (10 mm), or by intracellularinjection of the fast Ca21-chelator BAPTA, repeated activation
FIGURE 2. Profile of potentiated outward current. A: Currentsevoked by 2-s depolarizing command 6 min before (Control, thintrace) and 10 s after (Potentiated, thick trace) repeated stimulation (asin B). Arrows indicate APs not under the VC. B: CC responses evokedby first (1) and last (12) pulses (12, 0.5 nA, 2 s; 1/20 s). A, B, same cell.
FIGURE 3. Effects of TEA 1 TTX. A: Superimposed currentsevoked before (6 min; thin trace) and after (10 s; thick trace) poten-tiation in standard solution (ACSF). B: Same as A, but after addingTEA (10 mM) and TTX (1.5 mM) (TEA 1 TTX). Potentiation in Aand B were evoked by repeated activation (12, 0.5 nA, 2 s; 1/20 s), asshown in D and E, respectively. C: Summary showing potentiation of
the sIAHP (% of control peak values) in standard solution (ACSF) andTEA 1 TTX (n 5 5). D: First (1) and last (12) responses evoked byrepeated stimulation under CC in ACSF. E: Same as D, but afteradding TEA 1 TTX. F: Superimposed initial Ca21 spikes in E1 (thintrace) and E12 (thick trace) after adding TEA 1 TTX. A, B, and D–F,two cells.
_____________________________________________________________________ POTENTIATION OF sIAHP 201

was less effective in terms of reducing cell excitability. However, asmall (,20%), brief (,1 min) reduction in excitability was stillinduced by repeated activation after the above manipulations (datanot shown; cf. Borde et al., 1995).
Blocking L-Type Ca21 Channels Reduced thesIAHP and Its Potentiation
The effects of repeated activation were tested during superfusionwith nifedipine, v-CgTx, v-agatoxin-IVA, or low concentrationsof Ni21 to determine which of the main types of voltage-gatedCa21 conductances might be responsible for the Ca21 inflow thatactivated sIAHP and induced its potentiation.
Nifedipine (20 mM), a specific L-type channel blocker, mark-edly reduced the sIAHP (78.9 6 8.9% of control values; n 5 6)and its potentiation by repeated stimulation under CC (Fig. 4A–C). The potentiation in ACSF was to 159.1 6 7.3% and wasreduced to 103.2 6 12.4% from controls under nifedipine.
As expected, nifedipine also inhibited the reduction in excitabil-ity as estimated in CC conditions (Fig. 4D, E). Ni21 (10–30 mM;n 5 6), v-CgTx (1 mM; n 5 2), or v-agatoxin-IVA (100 nM;n 5 2), which specifically block T-, N-, and P-type channels,respectively, did not induce significant reductions of the sIAHP orof its potentiation (not shown).
It had been shown by Tanabe et al. (1998) in CA3 neurons inorganotypic cultures and by Marrion and Tavalin (1998) in iso-
lated CA1 pyramidal cells that a significant amount of the Ca21
needed for the activation of the sIAHP flows through L-type Ca21
channels. We extend those observations, showing that L-typeCa21 channel activation is required for full activation of the sIAHP
in CA1 pyramidal neurons in slices. Moreover, we also show thatthe activation of L-type Ca21 channels is a prerequisite for induc-ing the activity-dependent potentiation of the current. Interest-ingly, L-type channels and the channels mediating the sIAHP arestrategically located at the apical dendrites in CA1 pyramidal neu-rons (Magee and Johnston, 1995; Sah and Bekkers, 1996).
sIAHP and Its Potentiation Were Also Reducedby Ryanodine
Another possible source of the Ca21 to activate sIAHP is thatreleased from intracellular stores by an Ca21-activated Ca21-re-lease mechanism (e.g., Tanabe et al., 1998). This intracellularsource of Ca21 may also be important to induce the potentiation ofthe sIAHP. Since the Ca21-release mechanism is blocked by ryan-odine, we tested the effects of the drug on the sIAHP and its poten-tiation (Fig. 5A, B). The sIAHP was inhibited (29.0 6 7.8%; n 54), and its potentiation was reduced from 152.3 6 12.2% in ACSFto 126.9 6 3.5% (n 5 4) after superfusion with 20 mM ryano-dine (Fig. 5C). There was a concomitant reduction of the re-sponses under CC (Fig. 5D, E).
FIGURE 4. Effects of nifedipine. A: Superimposed currentsevoked before (6 min; thin trace) and after (10 s; thick trace) poten-tiation in standard solution (ACSF). B: Same as A, showing inhibitionof sIAHP and of potentiation after adding nifedipine (20 mM)(Nifepine). Potentiations in A and B were evoked by repeated activa-
tion, as shown in D and E, respectively. C: Summary showing inhib-itory effects of nifedipine (n 5 6). D: First (1) and last (12) responsesevoked by repeated stimulation (12, 0.5 nA, 2 s; 1/10 s) under CC instandard solution (ACSF). E: Same as D, but after adding nifedipine(Nifepine). A, B, same cell; D, E, same cell.
202 BORDE ET AL.

The above results suggest that Ca21 released from intracel-lular stores assists in the activation of the sIAHP and its poten-tiation.
sIAHP Persisted, But Its Potentiation RapidlyWaned When Whole-Cell Recordings Were Used
We also tested the effects of repeated activation, using re-cordings in the whole-cell configuration of the patch-clamptechnique. Control responses were essentially identical to thoseevoked using sharp electrodes (Fig. 6). However, repeated acti-vation with depolarizing pulses had different effects if appliedimmediately (,10 –20 min) or later after entering the whole-cell configuration. Repeated stimulation early during record-ings induced sIAHP potentiation that was essentially identical,although somewhat smaller, to that reported above (n 5 26).However, later after the initiation of recordings, there was nopotentiation of the sIAHP (Fig. 6A). The sIAHP increased only to105.1 6 4.1% after repeated stimulation (Fig. 6C; n 5 6).The above results indicate that although the sequence of Ca21
influx through L-type Ca21 channels, the resulting rise in[Ca21]i, and the activation of the sIAHP persist, the mechanismsunderlying potentiation of the sIAHP rapidly fade with time andare finally abolished, probably due to cytoplasmic dialysis. It isnoteworthy that a small decrease in excitability remained underCC conditions (Fig. 6B).
DISCUSSION
The above results show that when sharp electrodes are used,repeated activation of CA1 pyramidal neurons elicited a robustpotentiation of the sIAHP. The potentiated sIAHP acted as an in-creasing negative feedback that caused a marked drop in neurons’excitability (Borde et al., 1995), which induced a generalized re-duction of the efficacy of excitatory synaptic inputs (Borde et al.,1999). The potentiation of the sIAHP persisted for several minutes,could be detected immediately after the first stimulation, and in-creased with the rate of repetition.
Evidence that potentiation of the sIAHP brought about the sus-tained reduction in excitability was provided by the finding thatmanipulations that blocked or reduced the sIAHP also blocked thereduction in excitability. Indeed, superfusion with Ca21-free orCdCl2 ACSF that indirectly blocked the sIAHP by obstructingCa21 inflow (Benardo and Prince, 1982; Lancaster and Adams,1986; Storm, 1987), and BAPTA-loading that chelated intracellu-lar Ca21, thus blocking the sIAHP, prevented the reduction inexcitability (cf. Borde et al., 1995).
Moreover, CCh that directly blocks the apamin-insensitivesIAHP (Storm, 1990; Halliwell and Adams, 1982; Borde et al.,1995, 1999; Sah and Clements, 1999) also prevented potentiation(our results) and the drop in excitability (Borde et al., 1995). Wealso provide evidence indicating that the Ca21 inflow needed to
FIGURE 5. Action of ryanodine. All records as in Figure 4, but ryanodine (10 mM) was addedto ACSF instead of nifedipine. A, B, same cell; D, E, same cell; C, n 5 4.
_____________________________________________________________________ POTENTIATION OF sIAHP 203

increase the [Ca21]i that triggers the sIAHP and its potentiation ismediated by high-voltage-activated L-type Ca21 channels. Indeed,nifedipine, that specifically blocks L-type channels, markedly re-duced the sIAHP and its potentiation, whereas other specific Ca21
channel blockers such as Ni21, v-CgTX, and v-agatoxin-IVA hadno significant effects. It has been shown that Ca21 inflow throughL-type Ca21 channels is needed to activate sIAHP in CA3 and CA1pyramidal neurons (Marrion and Tavalin, 1998; Tanabe et al.,1998; see above). Moreover, there are indications that L-type Ca21
channels and the channels mediating the sIAHP are close by in thedendrite of CA1 pyramidal neurons (e.g., Magee and Johnston,1995; Marrion and Tavalin, 1998; Sah and Bekkers, 1996).
It has also been demonstrated that ryanodine partially inhibitsthe sIAHP in CA3 neurons (Tanabe et al., 1998), thus indicating
that Ca21 released from intracellular stores contributes to the ac-tivation of the sIAHP. We extend this observation by demonstratingthat Ca21 released from intracellular stores assists in the activationof the current also in CA1 pyramidal neurons. We also show thatintracellular stores provide part of the Ca21 needed for the poten-tiation of the sIAHP. Therefore, the most probable sequence ofevents leading to the activation of the current is as follows. Depo-larization induces Ca21-inflow through L-type channels, increas-ing the [Ca21]i, which in turn activates the channels mediating thesIAHP and releases Ca21 by a Ca21-activated Ca21-release mech-anism. The Ca21 released from intracellular stores contributes tothe [Ca21]i elevation, and thus to the activation of the sIAHP.
The results show that the sIAHP was potentiated, whereas theinitial burst changed little in CC conditions. These observationsimply that it was not an increased Ca21 inflow during the initialburst that triggered the potentiation of the sIAHP. Although, adirect measure of the L-type Ca21 current could not be obtained,indirect evidence in favor of the above hypothesis is provided bythe marked reduction of duration of the first high-threshold Ca21
spike evoked by the pulses during repeated stimulation under CC.This finding suggests strongly that the inflow of Ca21 during the“on” response did not increase and was even reduced during re-peated stimulation.
The channels mediating sIAHP are sensitive to low levels of[Ca21]i and are voltage-insensitive (e.g., Lancaster and Adams,1986; Blatz and Magleby, 1987; Wang, 1998). Therefore, al-though it has been shown that the sIAHP lags the intracellular Ca21
signal (Lancaster and Zucker, 1994; Sah and Clements, 1999), thecurrent should show a close temporal correlation with dynamicchanges in the [Ca21]i (e.g., Miyakawa et al., 1992; Regehr andTank, 1992). Nevertheless, the slow kinetics of potassium channelsunderlying the sAHP were recently proposed as an additional fac-tor contributing to the slow time course of the sIAHP (Sah andClements, 1999).
The [Ca21]i is determined by the opposed effects of two pro-cesses: 1) the increased [Ca21]i due both to Ca21 inflow throughvoltage-gated L-type Ca21 channels and Ca21 released from intra-cellular stores; and 2) the intracellular Ca21 buffering capacity.When the sIAHP was potentiated, there was a marked increase inthe decay t of the sIAHP that probably reflected a parallel decreaseof the [Ca21]i decay time constant. It should be emphasized that adecreased efficiency of the Ca21 buffering mechanism would alsojustify the potentiation of the sIAHP in the absence of an increase inCa21 inflow during the initial bursts, as has been shown by mod-eling (Borde et al., 1995). Indeed, the modeled-decreased Ca21
buffering closely simulated the potentiation of the sIAHP and thereduced excitability (Borde et al., 1995). The potentiation pre-dicted by the model fits with the dynamic [Ca21]i changes re-corded using fluorometric techniques in experiments in which theCa21 buffering was modified (e.g., Miller et al., 1996; Shmigol etal., 1995).
In close agreement with our proposal, Sah and Clements (1999)showed that rapid chelation of intracellular Ca21 by release ofcaged BAPTA increased the rate of decay of the sIAHP. Obviously,a decreased chelation would have the opposite effect on sIAHP,
FIGURE 6. sIAHP potentiation is blocked when patch electrodesare used. A–C: As in Figure 1, except that the pulse intensity was 0.1nA, and recovery is not shown. A, B, same cell; C, n 5 6.
204 BORDE ET AL.

therefore paralleling what occurs during potentiation of the cur-rent.
An alternative explanation for the potentiation is that repeatedactivation could also increase the Ca21-binding capacity of thechannels mediating the sIAHP. Moreover, a slowdown of the kinet-ics of the slow K1 channels underlying the sAHP (Sah and Clem-ents, 1999) could be an additional factor contributing to the po-tentiation of the sIAHP. More experimentation, using simultaneousintracellular Ca21 measurement techniques and electrophysiol-ogy, is needed to elucidate the problem. The apamin-sensitiveCa21-actrivated K1 current in CA1 pyramidal neurons has muchfaster kinetics (up to 20-fold) than the sIAHP current that waspotentiated in these experiments. Moreover, it is not blocked byCCh, is inhibited by TEA, and is a only a small component of thenet tail current when brief voltage steps are used (e.g., Sah andClements, 1999; Stocker et al., 1999). Likewise, we show that theinitial outward tail current immediately after the depolarizing step,which includes the apamin-sensitive component, was not signifi-cantly potentiated by repeated activation of the cell. Therefore,although we cannot totally rule out the participation of theapamin-sensitive current, its contribution to potentiation, ifpresent, is small.
The activity-dependent potentiation of the sIAHP was rapidlywashed out when the whole-cell configuration of the patch-clamptechnique was used. However, a robust sIAHP was still evoked.Therefore, the potentiation of the sIAHP is controlled intracellu-larly by a process that can be gradually diminished, probably as aresult of diluting out the factors responsible by the intracellulardialysis that occurs with whole-cell recordings. This process didnot modify the control sIAHP, suggesting that it was not due to awashout of the channels mediating the L-type current, and there-fore could revolve around the intracellular Ca21 buffering machin-ery.
We have shown that Ca21 released from intracellular storescontributed to the activation and potentiation of the sIAHP. There-fore, internal Ca21 amplification by the Ca21-activated Ca21-release mechanism aids the potentiation of the sIAHP. It had beenshown that AP activity could induce Ca21 release from intracellu-lar stores in hippocampal pyramidal cells (Miller et al., 1996; Ja-cobs and Mayer, 1997). Moreover, Ca21 release from intracellularstores contributes to the activation of the sIAHP in CA3 pyramidalcells (Tanabe et al., 1998). Evidence from other systems indicatesthat repeated activation may induce a potentiated slow efflux ofmitochondrial Ca21 (Tang and Zucker, 1997), suggesting thatthis [Ca21]i elevation could assist in the potentiation of the sIAHP.
In conclusion, repeated activation generates a robust and sus-tained potentiation of the sIAHP that intensifies AP frequency ad-aptation by increasing the sAHP. This plastic phenomenon couldbe relevant in hippocampal function, because it causes a markeddecrease in synaptic efficacy (Borde et al., 1999). Indeed, changesin the magnitude of the sAHP in hippocampal pyramidal cells havebeen related to learning and acquisition (e.g., Thompson et al.,1996) and are known to regulate the induction threshold of thelong-term potentiation of synaptic efficacy (Sah and Bekkers,1996). Although the channels mediating sIAHP have not been iden-tified, indirect evidence indicates that they are strategically located
at the proximal apical dendrites in CA1 pyramidal neurons (Mageeand Johnston, 1995; Sah and Bekkers, 1996). This suggests that anincreased shunting action due to potentiation of the sIAHP maybring about a postsynaptic control of dendritic signaling by actingas a variable gain, regulating excitatory synaptic events occurring atmore distal dendritic sites. It may also regulate back-propagatingAPs, which have been shown to modify synaptic efficacy (Markramet al., 1997). Moreover, the potentiation of the sIAHP could play ahomeostatic role by acting as a gradually increasing negative feed-back to reduce activity and unnecessary [Ca21]i elevations duringprolonged excitatory synaptic bombardment. In addition, a varietyof neurotransmitters and second messenger systems can regulatethe sIAHP (e.g., Benardo and Prince, 1982; Madison and Nicoll,1984; Lancaster and Adams, 1986; Storm, 1990; Pedarzani andStorm, 1996). This type of regulation provides synaptic mecha-nisms whereby potentiation of the sIAHP may be controlledthrough the activation of specific inputs to CA1 pyramidal neu-rons.
Acknowledgments
This work was supported by DGES/MEC PB95-0031 andCEC/CAM 08.5/0038/98 grants to W.B. M.B. was a CEC/DGXII, Europe fellow, and C.B. was a “MUTIS”/ICI/MEC,Spain, and AGCI, Chile, fellow. D.F.S. and D.L.R. are FPI/MEC(Spain) and Human Frontier fellows, respectively. Many thanksare due to Dr. J. Storm for his valuable suggestions and to Dr. M.Sefton for correcting the English-language version of the manu-script.
REFERENCES
Adams PR, Constanti A, Brown DA, Clark RB. 1982. Intracellular Ca21
activates a fast voltage-sensitive K1 current in vertebrate sympatheticneurones. Nature 246:746–749.
Araque A, Buno W. 1995. Fast, persistent, Ca21-dependent K1 currentcontrols graded electrical activity in crayfish muscle. Pflugers Arch430:541–551.
Benardo LS, Prince DA. 1982. Ionic mechanisms of cholinergic excitationin mammalian hippocampal pyramidal cells. Brain Res 249:333–344.
Blatz LA, Magleby KL. 1987. Calcium-activated potassium channels.Trends Neurosci 10:463–467.
Borde M, Cazalets JR, Buno W. 1995. Activity-dependent response de-pression in rat hippocampal CA1 pyramidal neurons in vitro. J Neu-rophysiol 74:1714–1729.
Borde M, Bonansco C, Buno W. 1999. The activity-dependent potenti-ation of the slow Ca21-activated K1 current regulates syaptic efficacyin rat CA1 pyramidal neurons. Pflugers Arch 437:261–266.
Garcıa-Munoz A, Barrio LC, Buno W. 1993. Membrane potential oscil-lations in CA1 hippocampal pyramidal neurons in vitro: intrinsicrhythms and fluctuations entrained by sinusoidal injected currents.Exp Brain Res 97:325–333.
Halliwell JV, Adams PR. 1982. Voltage clamp analysis of muscarinicexcitation in hippocampal neurons. Brain Res 250:71–92.
Hamill OP, Marty A, Neher E, Sackmann B, Sigworth FJ. 1981. Im-proved patch-clamp techniques for high-resolution current recordingfrom cells and cell-free membrane patches. Pflugers Arch 391:85–100.
_____________________________________________________________________ POTENTIATION OF sIAHP 205

Jacobs JM, Mayer Y. 1997. Control of action potential-induced Ca21
signaling in the soma of hippocampal neurons by Ca21 release fromintracellular stores. J Neurosci 17:4129–4135.
Lancaster B, Adams PR. 1986. Calcium-dependent current generating theafterhyperpolarization in hippocampal neurons. J Neurophysiol 55:1268–1282.
Lancaster B, Nicoll RA. 1987. Properties of two calcium-activated hyper-polarizations in rat hippocampal neurones. J Physiol (Lond) 389:187–203.
Lancaster B, Zucker RS. 1994. Photolitic manipulation of Ca21 and thetime course of the slow, Ca21-activated potassium current in rat hip-pocampal neurons. J Physiol (Lond) 475:229–239.
Latorre R, Oberhauser A, Labarca P, Alvarez O. 1989. Varieties of calci-um-activated channels. Annu Rev Physiol 51:385–399.
Madison DV, Nicoll RA. 1984. Control of repetitive discharge of rat CA1pyramidal neurones in vitro. J Physiol (Lond) 354:319–331.
Magee JC, Johnston D. 1995. Characterization of single voltage-gatedNa1 and Ca21 channels in apical dendrites of rat CA1 pyramidalneurons. J Physiol (Lond) 487:60–90.
Markram H, Lubke J, Frotscher M, Sackmann B. 1997. Regulation ofsynaptic efficacy by coincidence of postsynaptic APs and EPSPs. Sci-ence 275:213–215.
Marrion NV, Tavalin SJ. 1998. Selective activation of Ca21 activated K1channels by co-localized Ca21 channels in hippocampal neurons. Na-ture 395:900–905.
Marty A. 1981. Ca-dependent potassium channel with large unitary con-ductance in chromaffin cell membranes. Nature 291:497–500.
Miller LD, Petrozzino JJ, Golarai G, Connor JA. 1996. Ca21 release fromintracellular stores induced by afferent stimulation of CA3 pyramidalneurons in hippocampal slices. J Neurophysiol 76:554–562.
Miyakawa H, Ross WN, Jaffe D, Callaway JC, Lasser-Ross N, Lisman JE,Johnston D. 1992. Synaptically activated increases in Ca21 concen-tration in hippocampal CA1 pyramidal cells are primarily due tovoltage-gated Ca21 channels. Neuron 9:1163–1173.
Pedarzani P, Storm JF. 1996. Interaction between alpha- and beta-adrenergic receptor agonists modulating the slow Ca21-activated K1current in hippocampal neurons. Eur J Neurosci 8:2098–2110.
Regehr WG, Tank DW. 1992. Calcium dynamics produced by synapticactivation of CA1 hippocampal pyramidal cells. J Neurosci 12:4202–4223.
Sah P. 1996. Calcium21-activated K1 currents in neurons: types, physi-ological roles and modulation. Trends Neurosci 19:150–154.
Sah P, Bekkers JM. 1996. Apical dendritic location of slow afterhyperpo-larization current in hippocampal pyramidal neurons: implications forthe integration of long-term potentiation. J Neurosci 16:4537–4542.
Sah P, Clements JD. 1999. Photolytic manipulation of [Ca21]i revealsslow kinetics of potassium channels underlying the afterhyperpolariza-tion in hippocampal pyramidal neurons. J Neurosci 19:3657–3664.
Shmigol A, Verkhratsky A, Isenberg G. 1995. Calcium-induced calciumrelease in rat sensory neurons. J Physiol (Lond) 489:627–636.
Stocker M, Krause M, Pedarzani P. 1999. An apamin-sensitive Ca21-activated K1 current in hippocampal pyramidal neurons. Proc NatlAcad Sci USA 96:4662–4667.
Storm JF. 1987. Action potential repolarization and fast afterhyperpolar-ization in rat hippocampal pyramidal cells. J Physiol (Lond) 385:733–759.
Storm JF. 1990. Potassium currents in hippocampal pyramidal cells. ProgBrain Res 83:161–187.
Tanabe M, Gahwiler BH, Gerber U. 1998. L-type Ca21 channels mediatethe slow Ca21-dependent afterhyperpolarization current in rat CA3pyramidal cells in vitro. J Neurophysiol 80:2268–2273.
Tang Y, Zucker RS. 1997. Mitochondrial involvement in post-tetanicpotentiation of synaptic transmission. Neuron 18:483–491.
Thompson LT, Moyer JR Jr, Disterhoft JF. 1996. Transient changes inexcitability of rabbit CA3 neurons with time course appropriate tosupport memory consolidation. J Neurophysiol 76:1836–1849.
Wang XJ. 1998. Calcium coding and adaptive temporal computation incortical pyramidal neurons. J Neurophysiol 79:1549–1566.
Zhang L, Weiner JL, Valiante TA, Velumian AA, Watson PL, JahromiAA, Schertzer S, Pennefather P, Carlen PL. 1994. Whole-cell record-ings of the Ca21-dependent slow after hyperpolarization in hippocam-pal neurons: effects of internally applied anions. Pflugers Arch 426:247–253.
206 BORDE ET AL.
Copyright © 2022 FDOKUMEN



















