VISA Is an Adapter Protein Required for Virus-Triggered IFN-β Signaling
14
Molecular Cell, Vol. 19, 727–740, September 16, 2005, Copyright ©2005 by Elsevier Inc. DOI 10.1016/j.molcel.2005.08.014 VISA Is an Adapter Protein Required for Virus-Triggered IFN- Signaling Liang-Guo Xu, 1,4 Yan-Yi Wang, 2,4 Ke-Jun Han, 1 Lian-Yun Li, 2 Zhonghe Zhai, 2 and Hong-Bing Shu 1,2,3, * 1 Department of Immunology National Jewish Medical and Research Center 1400 Jackson Street Denver, Colorado 80206 2 Department of Cell Biology and Genetics College of Life Sciences Peking University Beijing 100871 China 3 College of Life Sciences Wuhan University Wuhan 430072 China Summary Viral infection or stimulation of TLR3 triggers signal- ing cascades, leading to activation of the transcrip- tion factors IRF-3 and NF-B, which collaborate to in- duce transcription of type I interferon (IFN) genes. In this study, we identified a protein termed VISA (for virus-induced signaling adaptor) as a critical compo- nent in the IFN- signaling pathways. VISA recruits IRF-3 to the cytoplasmic viral dsRNA sensor RIG-I. Depletion of VISA inhibits virus-triggered and RIG-I- mediated activation of IRF-3, NF-B, and the IFN- promoter, suggesting that VISA plays a central role in virus-triggered TLR3-independent IFN- signaling. Our data also indicate that VISA interacts with TRIF and TRAF6 and mediates bifurcation of the TLR3-trig- gered NF-B and IRF-3 activation pathways. These findings suggest that VISA is critically involved in both virus-triggered TLR3-independent and TLR3- mediated antiviral IFN signaling. Introduction Viral infection results in induction of type I IFNs, includ- ing IFN-β and IFN-α family cytokines (Durbin et al., 2000; Levy and Garcia-Sastre, 2001; Levy and Marie, 2004). Type I IFNs activate the JAK-STAT signal trans- duction pathways, leading to transcriptional induction of a wide range of genes. The induced downstream gene products, such as IRFs, PKR, IP10, ISG15, and OAS, orchestrate inhibition of viral replication and clearance of virus-infected cells and thus lead to antivi- ral responses (Durbin et al., 2000; Levy and Garcia- Sastre, 2001; Levy and Marie, 2004). Transcriptional activation of the promoters of type I IFN genes requires the coordinate activation of multiple transcription factors and their cooperative assembly into transcriptional enhancer complexes in vivo. For ex- *Correspondence: [email protected] 4 These authors contributed equally to this work. ample, the enhancer of the IFN-β gene contains a κB site recognized by NF-κB, a site for ATF-2/c-Jun, and two IFN-stimulated response elements (ISREs) recog- nized by phosphorylated IRF-3 and/or IRF-7. It has been shown that transcriptional activation of the IFN-β gene requires coordinate and cooperative assembly of an enhanceosome that contains all of these transcrip- tion factors (Maniatis et al., 1998; Wathelet et al., 1998). At least two distinct pathways for the activation of the innate immune responses by viral infection have been proposed (Levy and Garcia-Sastre, 2001; Levy and Marie, 2004; tenOever et al., 2002). First, viruses enter cells by membrane fusion at the plasma mem- brane or through an endocytic process, leading to re- lease of viral nucleocapsids (or ribonucleoproteins) into the cytoplasm. The viral nucleocapsids trigger signal- ing cascades that lead to activation of IRF-3 and NF- κB and subsequent transcription of type I IFNs (Levy and Garcia-Sastre, 2001; tenOever et al., 2002). Sec- ond, viral dsRNA, released upon lysis of infected cells, binds to TLR3 and triggers TRIF-mediated pathways, leading to IRF-3 and NF-κB activation (Alexopoulou et al., 2001; Han et al., 2004; Hoebe et al., 2003; Oshiumi et al., 2003a; Yamamoto et al., 2002, 2003). It has been proposed that TLR3 is important for systemic re- sponses to viral infection but is not required for the ini- tial, cell-autonomous recognition of viral infection that induces the first wave of type I IFN production (Honda et al., 2003; Levy and Marie, 2004; Yoneyama et al., 2004). Until recently, the molecular components involved in virus-triggered TLR3-independent signaling are largely undefined. However, during the past couple of years, major breakthroughs have been achieved. It has been shown that two serine/threonine kinases, TBK1 and IKK, phosphorylate IRF-3 and IRF-7 and are involved in virus-triggered induction of type I IFNs in various cell types (Fitzgerald et al., 2003a; Hemmi et al., 2004; McWhirter et al., 2003; Perry et al., 2004; Sharma et al., 2003). More recently, a CARD module-containing RNA helicase protein, RIG-I, has been identified as a sensor of cytoplasmic viral dsRNA (Levy and Marie, 2004; Yoneyama et al., 2004). Depletion of RIG-I inhibits IFN-β production in response to Newcastle disease virus, suggesting that RIG-I is required for virus-trig- gered signaling. It has been further shown that RIG-I- mediated signaling is independent of TLR3 and TRIF (Yoneyama et al., 2004). The mechanisms of RIG-I- mediated downstream signaling are unknown. The mechanisms of TLR3-mediated signaling have also begun to emerge. It has been shown that the TIR domain-containing adaptor protein TRIF is associated with TLR3 and critically involved in TLR3-mediated ac- tivation of NF-κB and IRF-3 and transcription of the IFN-β gene (Han et al., 2004; Jiang et al., 2004; Oshiumi et al., 2003a; Yamamoto et al., 2002, 2003). TRIF in- teracts with TRAF6, which activates NF-κB through TAK1 and IKK (Han et al., 2004; Jiang et al., 2004; Sato et al., 2003). TRIF is also associated with IRF-3 as well as TBK1 and IKK (Fitzgerald et al., 2003a; Han et al.,
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of VISA Is an Adapter Protein Required for Virus-Triggered IFN-β Signaling

Molecular Cell, Vol. 19, 727–740, September 16, 2005, Copyright ©2005 by Elsevier Inc. DOI 10.1016/j.molcel.2005.08.014
VISA Is an Adapter Protein Requiredfor Virus-Triggered IFN-� Signaling
Liang-Guo Xu,1,4 Yan-Yi Wang,2,4 Ke-Jun Han,1
Lian-Yun Li,2 Zhonghe Zhai,2
and Hong-Bing Shu1,2,3,*1Department of ImmunologyNational Jewish Medical and Research Center1400 Jackson StreetDenver, Colorado 802062Department of Cell Biology and GeneticsCollege of Life SciencesPeking UniversityBeijing 100871China3College of Life SciencesWuhan UniversityWuhan 430072China
Summary
Viral infection or stimulation of TLR3 triggers signal-ing cascades, leading to activation of the transcrip-tion factors IRF-3 and NF-�B, which collaborate to in-duce transcription of type I interferon (IFN) genes. Inthis study, we identified a protein termed VISA (forvirus-induced signaling adaptor) as a critical compo-nent in the IFN-� signaling pathways. VISA recruitsIRF-3 to the cytoplasmic viral dsRNA sensor RIG-I.Depletion of VISA inhibits virus-triggered and RIG-I-mediated activation of IRF-3, NF-�B, and the IFN-�promoter, suggesting that VISA plays a central rolein virus-triggered TLR3-independent IFN-� signaling.Our data also indicate that VISA interacts with TRIFand TRAF6 and mediates bifurcation of the TLR3-trig-gered NF-�B and IRF-3 activation pathways. Thesefindings suggest that VISA is critically involved inboth virus-triggered TLR3-independent and TLR3-mediated antiviral IFN signaling.
Introduction
Viral infection results in induction of type I IFNs, includ-ing IFN-β and IFN-α family cytokines (Durbin et al.,2000; Levy and Garcia-Sastre, 2001; Levy and Marie,2004). Type I IFNs activate the JAK-STAT signal trans-duction pathways, leading to transcriptional inductionof a wide range of genes. The induced downstreamgene products, such as IRFs, PKR, IP10, ISG15, andOAS, orchestrate inhibition of viral replication andclearance of virus-infected cells and thus lead to antivi-ral responses (Durbin et al., 2000; Levy and Garcia-Sastre, 2001; Levy and Marie, 2004).
Transcriptional activation of the promoters of type IIFN genes requires the coordinate activation of multipletranscription factors and their cooperative assemblyinto transcriptional enhancer complexes in vivo. For ex-
*Correspondence: [email protected]
4 These authors contributed equally to this work.ample, the enhancer of the IFN-β gene contains a κBsite recognized by NF-κB, a site for ATF-2/c-Jun, andtwo IFN-stimulated response elements (ISREs) recog-nized by phosphorylated IRF-3 and/or IRF-7. It hasbeen shown that transcriptional activation of the IFN-βgene requires coordinate and cooperative assembly ofan enhanceosome that contains all of these transcrip-tion factors (Maniatis et al., 1998; Wathelet et al., 1998).
At least two distinct pathways for the activation ofthe innate immune responses by viral infection havebeen proposed (Levy and Garcia-Sastre, 2001; Levyand Marie, 2004; tenOever et al., 2002). First, virusesenter cells by membrane fusion at the plasma mem-brane or through an endocytic process, leading to re-lease of viral nucleocapsids (or ribonucleoproteins) intothe cytoplasm. The viral nucleocapsids trigger signal-ing cascades that lead to activation of IRF-3 and NF-κB and subsequent transcription of type I IFNs (Levyand Garcia-Sastre, 2001; tenOever et al., 2002). Sec-ond, viral dsRNA, released upon lysis of infected cells,binds to TLR3 and triggers TRIF-mediated pathways,leading to IRF-3 and NF-κB activation (Alexopoulou etal., 2001; Han et al., 2004; Hoebe et al., 2003; Oshiumiet al., 2003a; Yamamoto et al., 2002, 2003). It has beenproposed that TLR3 is important for systemic re-sponses to viral infection but is not required for the ini-tial, cell-autonomous recognition of viral infection thatinduces the first wave of type I IFN production (Hondaet al., 2003; Levy and Marie, 2004; Yoneyama et al.,2004).
Until recently, the molecular components involved invirus-triggered TLR3-independent signaling are largelyundefined. However, during the past couple of years,major breakthroughs have been achieved. It has beenshown that two serine/threonine kinases, TBK1 andIKK�, phosphorylate IRF-3 and IRF-7 and are involvedin virus-triggered induction of type I IFNs in various celltypes (Fitzgerald et al., 2003a; Hemmi et al., 2004;McWhirter et al., 2003; Perry et al., 2004; Sharma et al.,2003). More recently, a CARD module-containing RNAhelicase protein, RIG-I, has been identified as a sensorof cytoplasmic viral dsRNA (Levy and Marie, 2004;Yoneyama et al., 2004). Depletion of RIG-I inhibitsIFN-β production in response to Newcastle diseasevirus, suggesting that RIG-I is required for virus-trig-gered signaling. It has been further shown that RIG-I-mediated signaling is independent of TLR3 and TRIF(Yoneyama et al., 2004). The mechanisms of RIG-I-mediated downstream signaling are unknown.
The mechanisms of TLR3-mediated signaling havealso begun to emerge. It has been shown that the TIRdomain-containing adaptor protein TRIF is associatedwith TLR3 and critically involved in TLR3-mediated ac-tivation of NF-κB and IRF-3 and transcription of theIFN-β gene (Han et al., 2004; Jiang et al., 2004; Oshiumiet al., 2003a; Yamamoto et al., 2002, 2003). TRIF in-teracts with TRAF6, which activates NF-κB throughTAK1 and IKK (Han et al., 2004; Jiang et al., 2004; Satoet al., 2003). TRIF is also associated with IRF-3 as wellas TBK1 and IKK� (Fitzgerald et al., 2003a; Han et al.,

Molecular Cell728
2004; Oshiumi et al., 2003a; Yamamoto et al., 2002).Several reports have indicated that TBK1 and IKK�phosphorylate IRF-3 and IRF-7 and are critically in-volved in TLR3-mediated production of type I IFNs andcellular antiviral response (Fitzgerald et al., 2003a;Hemmi et al., 2004; McWhirter et al., 2003; Perry et al.,2004; Sharma et al., 2003).
In this study, we identified a protein termed VISA.VISA contains a CARD module and consensus TRAFbinding motifs. VISA recruits IRF-3 to RIG-I and is re-quired for virus-triggered TLR3-independent signaling.VISA also interacts with TRIF and TRAF6 and mediatesbifurcation of the TLR3-mediated NF-κB and IRF-3 acti-vation pathways. Our findings suggest that VISA is anadaptor protein critically involved in virus-triggeredIFN-β signaling.
Results
Sequence and Expression Analysis of VISAIn a large-scale screen for human proteins that activateNF-κB and MAPK signaling pathways, 25 uncharac-terized proteins were identified to activate NF-κB (Mat-suda et al., 2003). The mechanisms of action and bio-logical significance of these proteins were unknown.We made expression plasmids for 12 of these 25 pro-teins and found that only one (designated as clonenumber 031N in the original report) could significantlyactivate NF-κB in reporter assays (data not shown). Wehave designated this protein as VISA.
Alignment of available cDNA and EST sequences inthe GenBank databases indicated that human VISAmRNA has two splice variants. The shorter one is w3.4kb in length, and the longer one contains extra frag-ments at the 3# UTR (data not shown). However, bothtranscripts encode the same 540 amino acid (aa) pro-tein (data not shown). Structural analysis with theEMBL-EBI InterProScan program indicated that VISAcontains a CARD module at its N terminus (Figure 1A).The CARD module of VISA is mostly homologous to theCARDs of RIG-I and MDA5 (Figure 1B), two related RNAhelicase proteins that have been shown to be involvedin virus-triggered IFN signaling (Yoneyama et al., 2004).Other than the CARD module, VISA is not homologousto known proteins. Human VISA shares w45% se-quence identity at the amino acid level with two un-characterized sequences from mouse (Figure 1A) andrat (data not shown), respectively.
Northern blot analysis indicated that human VISAmRNA is expressed in all examined tissues, includingbrain, heart, skeletal muscle, colon, thymus, spleen,kidney, liver, small intestine, placenta, lung, and periph-eral blood leukocytes, as two transcripts of w3.4 andw9.5 kb in sizes (Figure 1C).
We prepared two rabbit polyclonal antibodies againstaa 1–180 and aa 181–360 of human VISA, respectively.Both of these antibodies could detect an endogenousw62 kDa protein in human embryonic kidney 293 andhuman sarcoma SAOS-2, but not in human lung cancerH1299 cells (Figure 1D and data not shown). These anti-bodies could also detect overexpressed HA-taggedVISA, which migrated slightly slower than the endoge-nous VISA because of the additional HA tag (Figure 1D).
Tc
VaTfpits
ttsI2ae2IVIVsiW2
Ilectκv2tTκ
VTBIvRqttv
gvAigk(VS3
hese data suggest that the VISA cDNA we cloned en-odes full-length VISA protein.
ISA Potently Activates NF-�B, ISRE,nd the IFN-� Promotero confirm whether VISA activates NF-κB, we trans-ected 293 cells with a VISA expression plasmid anderformed NF-κB luciferase reporter assays. As shown
n Figure 2A, VISA activated NF-κB more potently thanhe positive control IKKβ in these assays. These datauggest that overexpression of VISA activates NF-κB.We then determined whether VISA activates other
ranscription factors. We found that VISA could po-ently activate ISRE in reporter assays (Figure 2B). Con-istently, overexpression of VISA caused a shift ofRF-3 to a slightly higher molecular weight band (FigureB), which is a hallmark of IRF-3 phosphorylation andctivation (Lin et al., 1999; Servant et al., 2002; Weavert al., 1998; Yoneyama et al., 1998; Zhang and Pagano,002). In vitro kinase assays using recombinant GST-RF-3 as substrate confirmed that overexpression ofISA caused IRF-3 phosphorylation (Figure 2B). Both
RF-3 phosphorylation and ISRE activation caused byISA were as potent as that mediated by IKK�, a kinasehown to be involved in virus-induced type I IFN signal-
ng (Fitzgerald et al., 2003a; Hemmi et al., 2004; Mc-hirter et al., 2003; Perry et al., 2004; Sharma et al.,
003).To further confirm that VISA activates NF-κB and
SRE, we performed gel-shift experiments with 32P-abeled consensus NF-κB and ISRE probes. Thesexperiments indicated that overexpression of VISAaused NF-κB and ISRE activation (Figure 2C). Consis-ent with the observations that VISA activated both NF-B and IRF-3, overexpression of VISA potently acti-ated the promoter of the human IFN-β gene (FigureD). In reporter assays, VISA did not activate CHOP, aranscription factor activated by p38 kinase (Figure 2E).hese data suggest that VISA specially activated NF-B, ISRE, and the IFN-β promoter.
ISA Is Required for Virus-InducedLR3-Independent Signalingecause VISA potently activates NF-κB, ISRE, and the
FN-β promoter, we reasoned that VISA is involved inirus-triggered signaling. To test this, we made sevenNAi expression vectors containing different target se-uences of the human VISA cDNA. Transient transfec-ion and Western blot analysis indicated that these vec-ors could inhibit expression of endogenous VISA toaried levels in 293 cells (Figure 3A).To determine whether VISA plays a role in virus-trig-
ered ISRE activation, we transfected the VISA RNAiectors into 293 cells and performed reporter assays.s shown in Figure 3A, knockdown of VISA expression
nhibited Sendai virus-induced ISRE activation. The de-ree of inhibition was correlated with the efficiency ofnockdown of VISA expression by each RNAi vectorFigure 3A). Consistent with these data, knockdown ofISA expression by RNAi also significantly inhibitedendai virus-induced IRF-3 phosphorylation (FigureB). Taken together, these data suggest that VISA is

VISA Is a Component of Virus-Induced Signaling729
Figure 1. Sequence and Expression Analysis of VISA
(A) Alignment of amino acid sequences of human and mouse VISA. The CARD module, TRAF2 binding motif (T2BM), N-terminal (T6BM1), andC-terminal (T6BM2) TRAF6 binding motifs are boxed. Identical amino acids are shaded and boxed. Similar amino acids are boxed only.(B) Alignment of amino acid sequences of the CARD modules of human VISA, RIG-I, and MDA5.(C) Tissue expression of human VISA mRNA.(D) Expression of human VISA protein in cells. Total lysates from HA-VISA-transfected 293 cells or untransfected 293, H1299, and SAOS-2cells were analyzed by Western blot with a rabbit polyclonal antibody against human VISA-(181–360). Ten times less lysate from HA-VISA-transfected 293 cells was loaded.
required for virus-triggered IRF-3 phosphorylation andISRE activation.
We used the #3 VISA RNAi vector for all the experi-ments described below. Similar results were obtainedwith an independent VISA RNAi vector (#7) (data notshown).
In reporter assays, VISA RNAi also dramatically inhib-
ited Sendai virus-induced activation of NF-κB (Figure3C) and the IFN-β promoter (Figure 3D) in 293 cells.Previously, it has been shown that 293 cells do not ex-press TLR3, and addition of poly(I:C) to the culture me-dium does not activate ISRE in reporter assays (Yone-yama et al., 2004). This observation was also confirmedby us (data not shown). Taken together, these data sug-
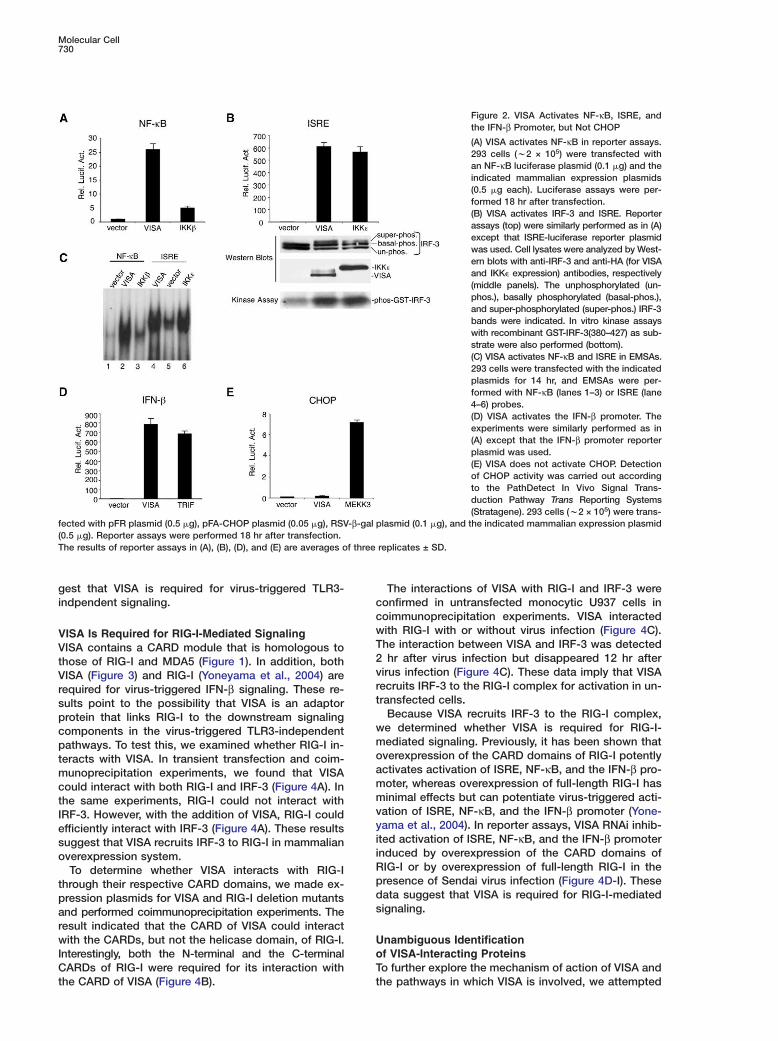
Molecular Cell730
Figure 2. VISA Activates NF-κB, ISRE, andthe IFN-β Promoter, but Not CHOP
(A) VISA activates NF-κB in reporter assays.293 cells (w2 × 105) were transfected withan NF-κB luciferase plasmid (0.1 �g) and theindicated mammalian expression plasmids(0.5 �g each). Luciferase assays were per-formed 18 hr after transfection.(B) VISA activates IRF-3 and ISRE. Reporterassays (top) were similarly performed as in (A)except that ISRE-luciferase reporter plasmidwas used. Cell lysates were analyzed by West-ern blots with anti-IRF-3 and anti-HA (for VISAand IKK� expression) antibodies, respectively(middle panels). The unphosphorylated (un-phos.), basally phosphorylated (basal-phos.),and super-phosphorylated (super-phos.) IRF-3bands were indicated. In vitro kinase assayswith recombinant GST-IRF-3(380–427) as sub-strate were also performed (bottom).(C) VISA activates NF-κB and ISRE in EMSAs.293 cells were transfected with the indicatedplasmids for 14 hr, and EMSAs were per-formed with NF-κB (lanes 1–3) or ISRE (lane4–6) probes.(D) VISA activates the IFN-β promoter. Theexperiments were similarly performed as in(A) except that the IFN-β promoter reporterplasmid was used.(E) VISA does not activate CHOP. Detectionof CHOP activity was carried out accordingto the PathDetect In Vivo Signal Trans-duction Pathway Trans Reporting Systems(Stratagene). 293 cells (w2 × 105) were trans-
fected with pFR plasmid (0.5 �g), pFA-CHOP plasmid (0.05 �g), RSV-β-gal plasmid (0.1 �g), and the indicated mammalian expression plasmid(0.5 �g). Reporter assays were performed 18 hr after transfection.The results of reporter assays in (A), (B), (D), and (E) are averages of three replicates ± SD.
gest that VISA is required for virus-triggered TLR3-indpendent signaling.
VISA Is Required for RIG-I-Mediated SignalingVISA contains a CARD module that is homologous tothose of RIG-I and MDA5 (Figure 1). In addition, bothVISA (Figure 3) and RIG-I (Yoneyama et al., 2004) arerequired for virus-triggered IFN-β signaling. These re-sults point to the possibility that VISA is an adaptorprotein that links RIG-I to the downstream signalingcomponents in the virus-triggered TLR3-independentpathways. To test this, we examined whether RIG-I in-teracts with VISA. In transient transfection and coim-munoprecipitation experiments, we found that VISAcould interact with both RIG-I and IRF-3 (Figure 4A). Inthe same experiments, RIG-I could not interact withIRF-3. However, with the addition of VISA, RIG-I couldefficiently interact with IRF-3 (Figure 4A). These resultssuggest that VISA recruits IRF-3 to RIG-I in mammalianoverexpression system.
To determine whether VISA interacts with RIG-Ithrough their respective CARD domains, we made ex-pression plasmids for VISA and RIG-I deletion mutantsand performed coimmunoprecipitation experiments. Theresult indicated that the CARD of VISA could interactwith the CARDs, but not the helicase domain, of RIG-I.Interestingly, both the N-terminal and the C-terminalCARDs of RIG-I were required for its interaction withthe CARD of VISA (Figure 4B).
ccwT2vrt
wmoammvyiiRpds
UoTt
The interactions of VISA with RIG-I and IRF-3 wereonfirmed in untransfected monocytic U937 cells inoimmunoprecipitation experiments. VISA interactedith RIG-I with or without virus infection (Figure 4C).he interaction between VISA and IRF-3 was detectedhr after virus infection but disappeared 12 hr after
irus infection (Figure 4C). These data imply that VISAecruits IRF-3 to the RIG-I complex for activation in un-ransfected cells.
Because VISA recruits IRF-3 to the RIG-I complex,e determined whether VISA is required for RIG-I-ediated signaling. Previously, it has been shown thatverexpression of the CARD domains of RIG-I potentlyctivates activation of ISRE, NF-κB, and the IFN-β pro-oter, whereas overexpression of full-length RIG-I hasinimal effects but can potentiate virus-triggered acti-
ation of ISRE, NF-κB, and the IFN-β promoter (Yone-ama et al., 2004). In reporter assays, VISA RNAi inhib-ted activation of ISRE, NF-κB, and the IFN-β promoternduced by overexpression of the CARD domains ofIG-I or by overexpression of full-length RIG-I in theresence of Sendai virus infection (Figure 4D-I). Theseata suggest that VISA is required for RIG-I-mediatedignaling.
nambiguous Identificationf VISA-Interacting Proteinso further explore the mechanism of action of VISA andhe pathways in which VISA is involved, we attempted
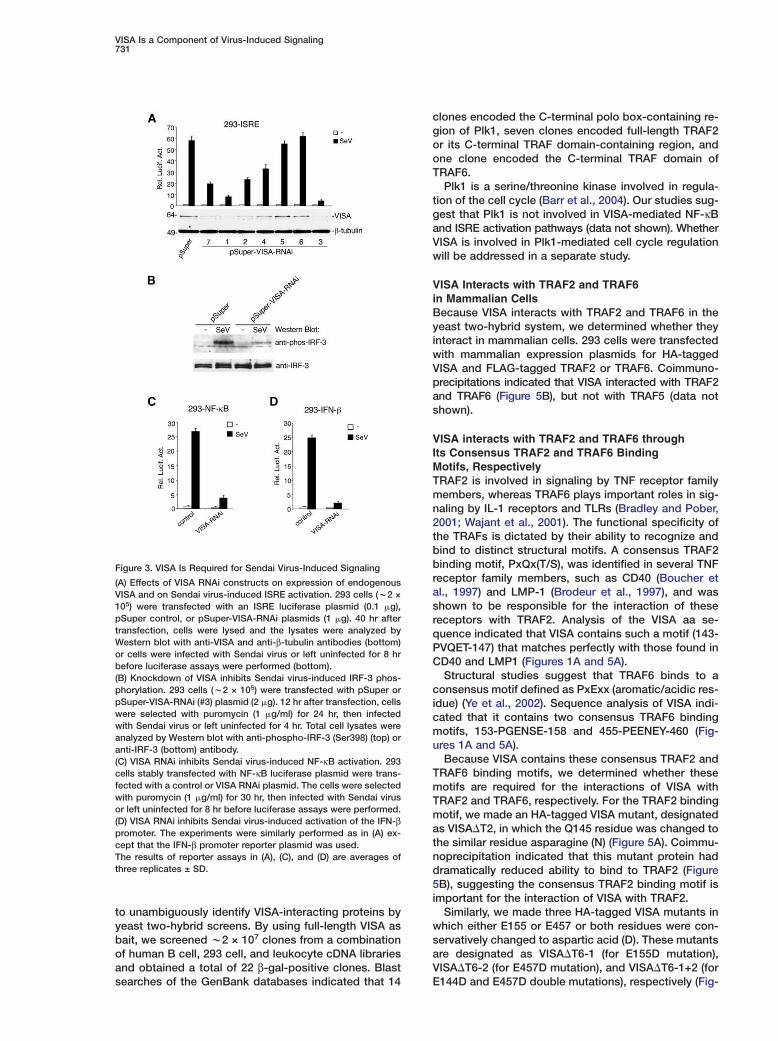
VISA Is a Component of Virus-Induced Signaling731
Figure 3. VISA Is Required for Sendai Virus-Induced Signaling
(A) Effects of VISA RNAi constructs on expression of endogenousVISA and on Sendai virus-induced ISRE activation. 293 cells (w2 ×105) were transfected with an ISRE luciferase plasmid (0.1 �g),pSuper control, or pSuper-VISA-RNAi plasmids (1 �g). 40 hr aftertransfection, cells were lysed and the lysates were analyzed byWestern blot with anti-VISA and anti-β-tubulin antibodies (bottom)or cells were infected with Sendai virus or left uninfected for 8 hrbefore luciferase assays were performed (bottom).(B) Knockdown of VISA inhibits Sendai virus-induced IRF-3 phos-phorylation. 293 cells (w2 × 105) were transfected with pSuper orpSuper-VISA-RNAi (#3) plasmid (2 �g). 12 hr after transfection, cellswere selected with puromycin (1 �g/ml) for 24 hr, then infectedwith Sendai virus or left uninfected for 4 hr. Total cell lysates wereanalyzed by Western blot with anti-phospho-IRF-3 (Ser398) (top) oranti-IRF-3 (bottom) antibody.(C) VISA RNAi inhibits Sendai virus-induced NF-κB activation. 293cells stably transfected with NF-κB luciferase plasmid were trans-fected with a control or VISA RNAi plasmid. The cells were selectedwith puromycin (1 �g/ml) for 30 hr, then infected with Sendai virusor left uninfected for 8 hr before luciferase assays were performed.(D) VISA RNAi inhibits Sendai virus-induced activation of the IFN-βpromoter. The experiments were similarly performed as in (A) ex-cept that the IFN-β promoter reporter plasmid was used.The results of reporter assays in (A), (C), and (D) are averages ofthree replicates ± SD.
to unambiguously identify VISA-interacting proteins byyeast two-hybrid screens. By using full-length VISA asbait, we screened w2 × 107 clones from a combinationof human B cell, 293 cell, and leukocyte cDNA librariesand obtained a total of 22 β-gal-positive clones. Blastsearches of the GenBank databases indicated that 14
clones encoded the C-terminal polo box-containing re-gion of Plk1, seven clones encoded full-length TRAF2or its C-terminal TRAF domain-containing region, andone clone encoded the C-terminal TRAF domain ofTRAF6.
Plk1 is a serine/threonine kinase involved in regula-tion of the cell cycle (Barr et al., 2004). Our studies sug-gest that Plk1 is not involved in VISA-mediated NF-κBand ISRE activation pathways (data not shown). WhetherVISA is involved in Plk1-mediated cell cycle regulationwill be addressed in a separate study.
VISA Interacts with TRAF2 and TRAF6in Mammalian CellsBecause VISA interacts with TRAF2 and TRAF6 in theyeast two-hybrid system, we determined whether theyinteract in mammalian cells. 293 cells were transfectedwith mammalian expression plasmids for HA-taggedVISA and FLAG-tagged TRAF2 or TRAF6. Coimmuno-precipitations indicated that VISA interacted with TRAF2and TRAF6 (Figure 5B), but not with TRAF5 (data notshown).
VISA interacts with TRAF2 and TRAF6 throughIts Consensus TRAF2 and TRAF6 BindingMotifs, RespectivelyTRAF2 is involved in signaling by TNF receptor familymembers, whereas TRAF6 plays important roles in sig-naling by IL-1 receptors and TLRs (Bradley and Pober,2001; Wajant et al., 2001). The functional specificity ofthe TRAFs is dictated by their ability to recognize andbind to distinct structural motifs. A consensus TRAF2binding motif, PxQx(T/S), was identified in several TNFreceptor family members, such as CD40 (Boucher etal., 1997) and LMP-1 (Brodeur et al., 1997), and wasshown to be responsible for the interaction of thesereceptors with TRAF2. Analysis of the VISA aa se-quence indicated that VISA contains such a motif (143-PVQET-147) that matches perfectly with those found inCD40 and LMP1 (Figures 1A and 5A).
Structural studies suggest that TRAF6 binds to aconsensus motif defined as PxExx (aromatic/acidic res-idue) (Ye et al., 2002). Sequence analysis of VISA indi-cated that it contains two consensus TRAF6 bindingmotifs, 153-PGENSE-158 and 455-PEENEY-460 (Fig-ures 1A and 5A).
Because VISA contains these consensus TRAF2 andTRAF6 binding motifs, we determined whether thesemotifs are required for the interactions of VISA withTRAF2 and TRAF6, respectively. For the TRAF2 bindingmotif, we made an HA-tagged VISA mutant, designatedas VISA�T2, in which the Q145 residue was changed tothe similar residue asparagine (N) (Figure 5A). Coimmu-noprecipitation indicated that this mutant protein haddramatically reduced ability to bind to TRAF2 (Figure5B), suggesting the consensus TRAF2 binding motif isimportant for the interaction of VISA with TRAF2.
Similarly, we made three HA-tagged VISA mutants inwhich either E155 or E457 or both residues were con-servatively changed to aspartic acid (D). These mutantsare designated as VISA�T6-1 (for E155D mutation),VISA�T6-2 (for E457D mutation), and VISA�T6-1+2 (forE144D and E457D double mutations), respectively (Fig-
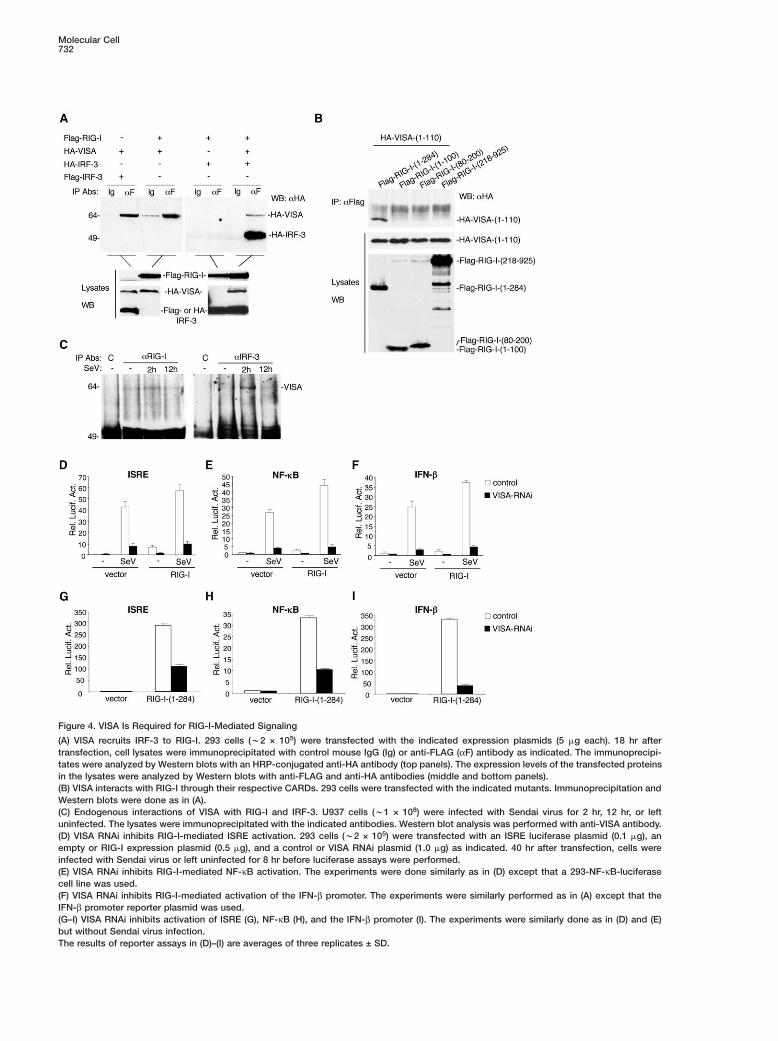
Molecular Cell732
Figure 4. VISA Is Required for RIG-I-Mediated Signaling
(A) VISA recruits IRF-3 to RIG-I. 293 cells (w2 × 105) were transfected with the indicated expression plasmids (5 �g each). 18 hr aftertransfection, cell lysates were immunoprecipitated with control mouse IgG (Ig) or anti-FLAG (αF) antibody as indicated. The immunoprecipi-tates were analyzed by Western blots with an HRP-conjugated anti-HA antibody (top panels). The expression levels of the transfected proteinsin the lysates were analyzed by Western blots with anti-FLAG and anti-HA antibodies (middle and bottom panels).(B) VISA interacts with RIG-I through their respective CARDs. 293 cells were transfected with the indicated mutants. Immunoprecipitation andWestern blots were done as in (A).(C) Endogenous interactions of VISA with RIG-I and IRF-3. U937 cells (w1 × 108) were infected with Sendai virus for 2 hr, 12 hr, or leftuninfected. The lysates were immunoprecipitated with the indicated antibodies. Western blot analysis was performed with anti-VISA antibody.(D) VISA RNAi inhibits RIG-I-mediated ISRE activation. 293 cells (w2 × 105) were transfected with an ISRE luciferase plasmid (0.1 �g), anempty or RIG-I expression plasmid (0.5 �g), and a control or VISA RNAi plasmid (1.0 �g) as indicated. 40 hr after transfection, cells wereinfected with Sendai virus or left uninfected for 8 hr before luciferase assays were performed.(E) VISA RNAi inhibits RIG-I-mediated NF-κB activation. The experiments were done similarly as in (D) except that a 293-NF-κB-luciferasecell line was used.(F) VISA RNAi inhibits RIG-I-mediated activation of the IFN-β promoter. The experiments were similarly performed as in (A) except that theIFN-β promoter reporter plasmid was used.(G–I) VISA RNAi inhibits activation of ISRE (G), NF-κB (H), and the IFN-β promoter (I). The experiments were similarly done as in (D) and (E)but without Sendai virus infection.The results of reporter assays in (D)–(I) are averages of three replicates ± SD.
VISA Is a Component of Virus-Induced Signaling733
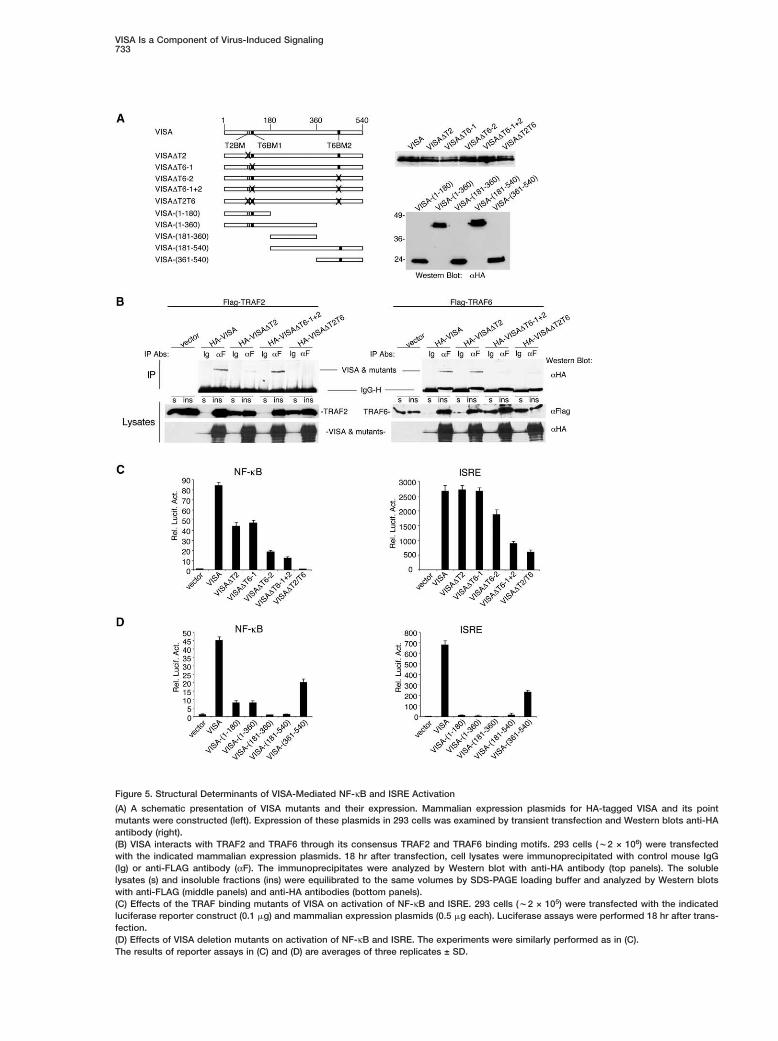
Figure 5. Structural Determinants of VISA-Mediated NF-κB and ISRE Activation
(A) A schematic presentation of VISA mutants and their expression. Mammalian expression plasmids for HA-tagged VISA and its pointmutants were constructed (left). Expression of these plasmids in 293 cells was examined by transient transfection and Western blots anti-HAantibody (right).(B) VISA interacts with TRAF2 and TRAF6 through its consensus TRAF2 and TRAF6 binding motifs. 293 cells (w2 × 106) were transfectedwith the indicated mammalian expression plasmids. 18 hr after transfection, cell lysates were immunoprecipitated with control mouse IgG(Ig) or anti-FLAG antibody (αF). The immunoprecipitates were analyzed by Western blot with anti-HA antibody (top panels). The solublelysates (s) and insoluble fractions (ins) were equilibrated to the same volumes by SDS-PAGE loading buffer and analyzed by Western blotswith anti-FLAG (middle panels) and anti-HA antibodies (bottom panels).(C) Effects of the TRAF binding mutants of VISA on activation of NF-κB and ISRE. 293 cells (w2 × 105) were transfected with the indicatedluciferase reporter construct (0.1 �g) and mammalian expression plasmids (0.5 �g each). Luciferase assays were performed 18 hr after trans-fection.(D) Effects of VISA deletion mutants on activation of NF-κB and ISRE. The experiments were similarly performed as in (C).The results of reporter assays in (C) and (D) are averages of three replicates ± SD.

Molecular Cell734
ure 5A). Coimmunoprecipitation experiments indicatedthat both VISA�T6-1 and VISA�T6-2 could still interactwith TRAF6 (data not shown), whereas VISA�T6-1+2had dramatically reduced ability to bind to TRAF6 (Fig-ure 5B). These data suggest that both consensusTRAF6 binding motifs can independently mediate theinteraction of VISA with TRAF6.
VISA Causes Accumulation of TRAF2and TRAF6 in Insoluble FractionsThe interaction between VISA and TRAF2 or TRAF6was relatively weak in our coimmunoprecipitation ex-periments (Figure 5B). During our experiments, wefound that a major fraction of overexpressed VISA ex-isted in insoluble cellular fractions (data not shown).Overexpression of VISA also caused increased accu-mulation of TRAF2 and TRAF6 in the insoluble fractions(Figure 5B). Mutation of the consensus TRAF2 andTRAF6 binding motifs of VISA abolished its ability tocause accumulation of TRAF2 and TRAF6 in the insolu-ble fractions, respectively (Figure 5B). These data sug-gest that VISA-mediated accumulation of TRAF2 andTRAF6 in insoluble fractions requires its binding toTRAF2 and TRAF6. These observations suggest a func-tional connection between VISA and TRAF2 or TRAF6.In this context, several reports have demonstrated thatactivation of TNF receptor family members caused ac-cumulation of cytoplasmic TRAF2 in insoluble aggre-gates (Arch et al., 2000; Brown et al., 2001; Fotin-Mlec-zek et al., 2002).
Structural Determinants of VISA-MediatedNF-�B and ISRE ActivationTo determine the functional significance of the interac-tion between VISA and TRAF2 or TRAF6, we examinedwhether the TRAF binding motif mutants of VISA couldactivate NF-κB and ISRE in reporter assays. As shownin Figure 5C, VISA�T2T6, a VISA mutant in whichall three TRAF binding motifs are mutated, completelylost its ability to activate NF-κB, whereas VISA�T2,VISA�T6-1, VISA�T6-2, and VISA�T61+2 all had re-duced ability to activate NF-κB. These data suggestthat VISA activates NF-κB through its interactions withTRAF2 and TRAF6, and all three TRAF binding motifscontribute to this ability. Among the TRAF binding mo-tifs of VISA, it seems that the C-terminal TRAF6 bindingmotif is most important for its ability to activate NF-κB,because mutation of this motif had a more dramaticeffect (Figure 5C).
Similarly, we performed ISRE reporter assays withthe VISA mutants. VISA�T2 and VISA�T6-1 did not af-fect the ability of VISA to activate ISRE. VISA�T6-2,VISA�T6-1+2, and VISA�T2/T6 had reduced, but notabolished, ability to activate ISRE activation (Figure5C). These data suggest that VISA-induced ISRE acti-vation is not dependent on TRAF2 and TRAF6. Muta-tion of the C-terminal TRAF6 binding motif reduced theability of VISA to activate ISRE, which can be explainedby the observation that the C terminus of VISA is impor-tant for its ability to activate ISRE (see below).
We also made mammalian expression plasmids for aseries of deletion mutants of VISA, including aa 1–180,aa 1–360, aa 181–360, aa 181–540, and aa 361–540
(cat1bThC
5tim3sau
VTBcTwpialnasVvVn
ViIitaJnκVmaVI
tTeacdaT
Figure 5A). Reporter assays indicated that the mutantsontaining any one of the TRAF binding motifs couldctivate NF-κB but to a lower degree in comparison tohe full-length VISA (Figure 5D). One exception is the aa81–540 mutant, which contains the C-terminal TRAF6inding motif but did not activate NF-κB (Figure 5D).he simplest explanation is that aa 181–360 has an in-ibitory role on NF-κB activation when linked with theterminus of VISA.In reporter assays, the C terminus of VISA (aa 361–
40) was sufficient to activate ISRE, whereas the mu-ants that lack this domain did not significantly activatet (Figure 5D). These data suggest that the CARD do-
ain is not required and the C-terminal domain (aa61–540) is sufficient for activating ISRE. Again, iteems that aa 181–360 had an inhibitory role on ISREctivation when linked with the C terminus of VISA (Fig-re 5D).
ISA Is Involved in TLR3-, but Not TLR4- and TNF-,riggered Signaling Pathwaysecause VISA interacts with TRAF2 and TRAF6, twoomponents that are involved in signaling mediated byNF receptor and TLR family members, we determinedhether knockdown of VISA has any effects on theseathways. In reporter assays, VISA RNAi significantly
nhibited poly(I:C)-induced activation of NF-κB, ISRE,nd the IFN-β promoter in a TLR3-expressing 293 cell
ine (293-TLR3) (Figure 6A). In contrast, VISA RNAi didot inhibit LPS-induced activation of NF-κB and ISRE inn LPS-responsive 293 cell line (293-TLR4/MD2/CD14)tably expressing TLR4, MD2, and CD14 (Figure 6B).ISA RNAi also did not inhibit TNF-induced NF-κB acti-ation in 293 cells (Figure 6C). These data suggest thatISA is specifically involved in TLR3-mediated sig-aling.
ISA Is Associated with Multiple Proteinsn the TLR3-Mediated Signaling Pathwaysn our earlier experiments, we found that VISA couldnteract with TRAF6 in a mammalian expression sys-em. However, TRAF6 is only involved in TLR3-medi-ted activation of NF-κB, but not ISRE (Han et al., 2004;iang et al., 2004). To determine the molecular mecha-isms of the involvement of VISA in TLR3-mediated NF-B and ISRE activation pathways, we examined whetherISA could interact with other components in the TLR3-ediated signaling pathways. In transient transfection
nd coimmunoprecipitation experiments, we found thatISA was associated with TRIF, TAK1, TBK1, IRF-3, and
RF-7, but not with IRF-1 (Figure 7A).We also attempted to detect endogenous interac-
ions between VISA and signaling components of theLR3- and virus-triggered pathways. In 293-TLR3 cells,ndogenous VISA interacted with endogenous TRIFnd TRAF6 and these interactions were not signifi-antly affected by poly(I:C) treatment (Figure 7B). Theseata suggest that VISA is a scaffolding protein that isssociated with multiple signaling components in theLR3-mediated pathways.

VISA Is a Component of Virus-Induced Signaling735
Figure 6. VISA Is Required for TLR3-, but NotTLR4- and TNF-, Induced Signaling
(A) VISA-RNAi inhibits poly(I:C)-induced acti-vation of NF-κB, ISRE, and the IFN-β promoter.293-TLR3 cells (w2 × 105) were transfectedwith the indicated luciferase construct (0.1 �g)and a control or VISA RNAi plasmid (1 �geach). 40 hr after transfection, cells weretreated with poly(I:C) (25 �g/ml) for 8 hr beforeluciferase assays were performed.(B) VISA RNAi does not inhibit LPS-inducedactivation of ISRE and NF-κB. 293-TLR4/MD2/CD14 cells (w2 × 105) were transfectedwith the indicated luciferase reporter con-struct (0.1 �g) and control or VISA RNAiplasmid (1 �g each). 40 hr after transfection,cells were treated with LPS (10 �g/ml) or leftuntreated for 8 hr before luciferase assayswere performed.(C) VISA RNAi did not inhibit TNF-inducedNF-κB activation. 293 cells (w2 × 105) weretransfected with an NF-κB luciferase con-struct (0.1 �g) and a control or VISA RNAiplasmid (1 �g each). 40 hr after transfection,cells were treated with TNF (10 �g/ml) or leftuntreated for 8 hr before luciferase assayswere performed.The results of reporter assays in (A)–(C) areaverages of three replicates ± SD.
ability of VISA to activate NF-κB (Figure 5), suggesting
Figure 7. VISA Interacts with Signaling Com-ponents of the TLR3-Mediated Pathways
(A) VISA is associated with TRIF, TAK1,TBK1, IRF-3, and IRF-7, but not IRF-1. 293cells (w2 × 106) were transfected with the in-dicated plasmids (5 �g each). 18 hr after trans-fection, coimmunoprecipitation and Westernblots were performed with the indicated anti-bodies. The expression levels of the trans-fected proteins in the lysates were compara-ble as indicated by Western blot analysis(data not shown).(B) Endogenous interactions between VISAand TRIF or TRAF6. 293-TLR3 cells (w2 ×107) were treated with poly (I:C) (25 �g/ml) orleft untreated for 10 min. Coimmunoprecipi-tation and Western blots were performedwith the indicated antibodies. Protein A-HRPwas used as the secondary antibody inthese experiments.
ISRE activation (Figure 8H). On the other hand, VISA
A Molecular Order for VISA-Mediated NF-�Band ISRE Activation PathwaysBecause VISA activates both NF-κB and ISRE and isassociated with multiple components in the TLR3-mediated pathways, we attempted to determine a mo-lecular order for the involvement of VISA in these sig-naling pathways. In reporter assays, overexpression ofVISA could activate both NF-κB and ISRE to a similardegree in TRIF−/− and wild-type (wt) mouse embryonicfibroblasts (MEFs) (Figures 8A and 8B). In addition, VI-SA�T2T6 inhibited TRIF-, but not TRAF6-, induced NF-κB activation in a dose-dependent manner (Figure 8E),whereas VISA RNAi inhibited TRIF-induced ISRE acti-vation (Figure 8G). These data suggest that VISA func-tions downstream of TRIF.
Mutation of the TRAF6 binding motifs reduced the
that TRAF6 functions downstream of VISA in NF-κB ac-tivation. To confirm this, we performed NF-κB reporterassays with TRAF6−/− MEFs. As shown in Figure 8C,VISA-mediated NF-κB activation was dramatically de-creased in TRAF6−/− MEFs in comparison to wt controlMEFs, suggesting that TRAF6 is important for VISA-mediated NF-κB activation. Consistently, dominant-negative mutants of TRAF6, TAK1, and IKKβ, but notTRIF, inhibited VISA-mediated NF-κB activation (Figure8F). These data suggest that VISA signals NF-κB acti-vation through a TRAF6-TAK1-IKK-dependent pathway.
In reporter assays, overexpression of VISA activatedISRE in both wt and TRAF6−/− MEFs (Figure 8D), sug-gesting that TRAF6 is not required for VISA-mediatedISRE activation. Dominant-negative mutants of IRF-3,but not those of TRIF and TBK1, inhibited VISA-induced
Molecular Cell736
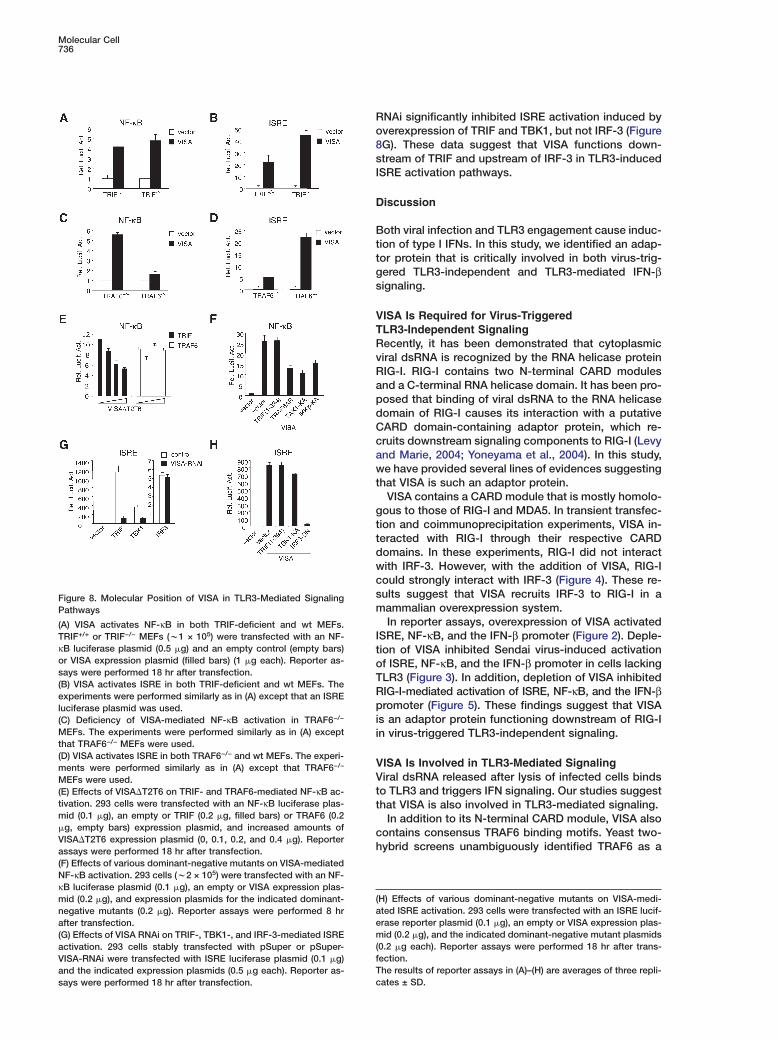
Figure 8. Molecular Position of VISA in TLR3-Mediated SignalingPathways
(A) VISA activates NF-κB in both TRIF-deficient and wt MEFs.TRIF+/+ or TRIF−/− MEFs (w1 × 105) were transfected with an NF-κB luciferase plasmid (0.5 �g) and an empty control (empty bars)or VISA expression plasmid (filled bars) (1 �g each). Reporter as-says were performed 18 hr after transfection.(B) VISA activates ISRE in both TRIF-deficient and wt MEFs. Theexperiments were performed similarly as in (A) except that an ISREluciferase plasmid was used.(C) Deficiency of VISA-mediated NF-κB activation in TRAF6−/−
MEFs. The experiments were performed similarly as in (A) exceptthat TRAF6−/− MEFs were used.(D) VISA activates ISRE in both TRAF6−/− and wt MEFs. The experi-ments were performed similarly as in (A) except that TRAF6−/−
MEFs were used.(E) Effects of VISA�T2T6 on TRIF- and TRAF6-mediated NF-κB ac-tivation. 293 cells were transfected with an NF-κB luciferase plas-mid (0.1 �g), an empty or TRIF (0.2 �g, filled bars) or TRAF6 (0.2�g, empty bars) expression plasmid, and increased amounts ofVISA�T2T6 expression plasmid (0, 0.1, 0.2, and 0.4 �g). Reporterassays were performed 18 hr after transfection.(F) Effects of various dominant-negative mutants on VISA-mediatedNF-κB activation. 293 cells (w2 × 105) were transfected with an NF-κB luciferase plasmid (0.1 �g), an empty or VISA expression plas-mid (0.2 �g), and expression plasmids for the indicated dominant-negative mutants (0.2 �g). Reporter assays were performed 8 hrafter transfection.(G) Effects of VISA RNAi on TRIF-, TBK1-, and IRF-3-mediated ISREactivation. 293 cells stably transfected with pSuper or pSuper-VISA-RNAi were transfected with ISRE luciferase plasmid (0.1 �g)and the indicated expression plasmids (0.5 �g each). Reporter as-says were performed 18 hr after transfection.
Ro8sI
D
Bttgs
VTRvRapdCcawt
gttdwcsm
ItoTRpii
VVtt
ch
(aem(fTc
ybrid screens unambiguously identified TRAF6 as a
H) Effects of various dominant-negative mutants on VISA-medi-ted ISRE activation. 293 cells were transfected with an ISRE lucif-rase reporter plasmid (0.1 �g), an empty or VISA expression plas-id (0.2 �g), and the indicated dominant-negative mutant plasmids
0.2 �g each). Reporter assays were performed 18 hr after trans-ection.he results of reporter assays in (A)–(H) are averages of three repli-ates ± SD.
NAi significantly inhibited ISRE activation induced byverexpression of TRIF and TBK1, but not IRF-3 (FigureG). These data suggest that VISA functions down-tream of TRIF and upstream of IRF-3 in TLR3-inducedSRE activation pathways.
iscussion
oth viral infection and TLR3 engagement cause induc-ion of type I IFNs. In this study, we identified an adap-or protein that is critically involved in both virus-trig-ered TLR3-independent and TLR3-mediated IFN-βignaling.
ISA Is Required for Virus-TriggeredLR3-Independent Signalingecently, it has been demonstrated that cytoplasmiciral dsRNA is recognized by the RNA helicase proteinIG-I. RIG-I contains two N-terminal CARD modulesnd a C-terminal RNA helicase domain. It has been pro-osed that binding of viral dsRNA to the RNA helicaseomain of RIG-I causes its interaction with a putativeARD domain-containing adaptor protein, which re-ruits downstream signaling components to RIG-I (Levynd Marie, 2004; Yoneyama et al., 2004). In this study,e have provided several lines of evidences suggesting
hat VISA is such an adaptor protein.VISA contains a CARD module that is mostly homolo-
ous to those of RIG-I and MDA5. In transient transfec-ion and coimmunoprecipitation experiments, VISA in-eracted with RIG-I through their respective CARDomains. In these experiments, RIG-I did not interactith IRF-3. However, with the addition of VISA, RIG-Iould strongly interact with IRF-3 (Figure 4). These re-ults suggest that VISA recruits IRF-3 to RIG-I in aammalian overexpression system.In reporter assays, overexpression of VISA activated
SRE, NF-κB, and the IFN-β promoter (Figure 2). Deple-ion of VISA inhibited Sendai virus-induced activationf ISRE, NF-κB, and the IFN-β promoter in cells lackingLR3 (Figure 3). In addition, depletion of VISA inhibitedIG-I-mediated activation of ISRE, NF-κB, and the IFN-βromoter (Figure 5). These findings suggest that VISA
s an adaptor protein functioning downstream of RIG-In virus-triggered TLR3-independent signaling.
ISA Is Involved in TLR3-Mediated Signalingiral dsRNA released after lysis of infected cells binds
o TLR3 and triggers IFN signaling. Our studies suggesthat VISA is also involved in TLR3-mediated signaling.
In addition to its N-terminal CARD module, VISA alsoontains consensus TRAF6 binding motifs. Yeast two-

VISA Is a Component of Virus-Induced Signaling737
VISA-interacting protein. This interaction was con-firmed in both a mammalian overexpression systemand untransfected cells (Figures 5 and 7). In addition,VISA also interacts with TRIF, TBK1, and IRF-3, compo-nents in the TLR3-mediated pathways (Figure 7). Thesedata suggest that VISA is a scaffolding protein thathelps to assemble a complex for activation of NF-κBand ISRE in the TLR3-mediated pathways. Knockdownof VISA expression by RNAi inhibited TLR3-mediatedactivation of NF-κB, ISRE, and the IFN-β promoter inpoly(I:C)-responsive 293-TLR3 cells (Figure 6). Theseresults suggest that VISA is involved in TLR3-mediatedsignaling leading to NF-κB and ISRE activation.
Molecular Mechanisms of VISA-MediatedActivation of IRF-3Previously, it has been demonstrated that TBK1 directlyphosphorylates IRF-3 and is critically involved in virus-triggered TLR3-independent IFN signaling pathways(Fitzgerald et al., 2003a; Hemmi et al., 2004; McWhirteret al., 2003; Perry et al., 2004; Sharma et al., 2003).In coimmunoprecipitation experiments, we found thatVISA could interact with both IRF-3 and TBK1 (Figures4 and 7), suggesting that VISA may function as a scaf-folding protein for TBK1 and IRF-3. Upon viral infection,VISA may recruit these proteins to the RIG-I complex inwhich IRF-3 is phosphorylated by TBK1. In the absenceof VISA, IRF-3 may not be efficiently recruited to TBK1for phosphorylation. Consistent with this hypothesis,knockdown of VISA significantly inhibited TBK1-medi-ated activation of ISRE (Figure 8G).
VISA may play a similar scaffolding role in TLR3-mediated signaling pathways. In TRIF-deficient MEFs,VISA activated both NF-κB and ISRE as potently as inwt control cells (Figures 8A and 8B). On the other hand,VISA mutant or RNAi inhibited TRIF-mediated activa-tion of NF-κB and ISRE, respectively (Figures 8E and8G). These results suggest that VISA functions down-stream of TRIF in TLR3-mediated signaling pathways.Previously, it has been shown that TBK1 and IKK� arealso involved in TLR3-mediated IFN signaling by theirassociation with TRIF (Fitzgerald et al., 2003a; Hemmiet al., 2004; McWhirter et al., 2003; Perry et al., 2004;Sharma et al., 2003). Taken together, these observa-tions suggest that VISA recruits IRF-3 to the TRIF com-plex in which IRF-3 is phosphorylated by TBK1 and/or IKK�.
Because the kinase-inactive mutant of TBK1 or IKK�(or their combination) could not significantly inhibitVISA-mediated ISRE activation in reporter assays (Fig-ure 8H and data not shown), this points to the possi-bility that VISA may be capable of activating an addi-tional unidentified IRF-3 and/or IRF-7 kinase(s).
Reporter assays with deletion mutants of VISA indi-cated that the N-terminal CARD domain was not re-quired and the C-terminal domain (aa 360–540) wassufficient for activating ISRE (Figure 5D). One explana-tion for these observations is that the C-terminal do-main of VISA can simultaneously recruit IRF-3 andTBK1/IKK� and is therefore capable of activating ISREwhen overexpressed in cells. Although the CARD do-main of VISA can interact with RIG-I, this mutant maynot be able to recruit IRF-3 and TBK1/IKK� and there-
fore barely activates ISRE when overexpressed (Fig-ure 5D).
Previous studies have demonstrated that both IRF-3and IRF-7 are required for efficient production of type IIFNs (Servant et al., 2002; Zhang and Pagano, 2002).However, their roles are different in these processes. Inthe early phase of viral infection, preexisting IRF-3 isactivated and induces expression of IFN-β and IFN-α4.These early produced IFNs transcriptionally induceIRF-7, and upon viral infection, the induced high-levelIRF-7 is activated and transactivates multiple IFNgenes, leading to robust production of IFNs in responseto viral infection. Recently, gene knockout studies haveconfirmed that IRF-7 plays a critical role in robust pro-duction of type I IFNs induced by viral infection in vari-ous cell types and by engagement of TLR9 in differenti-ated plasmacytoid dendritic cells (Honda et al., 2005a,2005b). Previously, it has been demonstrated that IRF-7is also phosphorylated by TBK1 and IKK� (Han et al.,2004; tenOever et al., 2004). In the present study, wefound that VISA could also interact with IRF-7 (Figure7). These observations point to the possibility that VISAis also involved in signaling that leads to IRF-7 acti-vation.
Molecular Mechanisms of VISA-MediatedActivation of NF-�BOur results suggest that VISA is also involved in virus-triggered and TLR3-mediated activation of NF-κB. Sev-eral observations suggest that TRAF6 is required forVISA-mediated NF-κB, but not ISRE activation. Muta-tion of the TRAF binding motifs abolished the ability ofVISA to activate NF-κB (Figure 5), suggesting thatTRAF6 functions downstream of VISA in NF-κB activa-tion. In TRAF6−/− MEFs, VISA-mediated NF-κB activa-tion was dramatically inhibited in comparison to wtcontrol MEFs (Figure 8C). In addition, dominant-nega-tive mutants of TRAF6, TAK1, and IKKβ inhibited VISA-mediated NF-κB activation (Figure 8F). These resultssuggest that VISA signals NF-κB activation through theTRAF6-TAK1-IKK cascade.
In contrast, VISA still activated ISRE in TRAF6−/−
MEFs, suggesting that TRAF6 is not required for VISA-mediated ISRE activation (Figure 8D). In fact, VISA acti-vates ISRE more potently in TRAF6−/− MEFs than in wtcontrol MEFs (Figure 8D). It is possible that VISA-medi-ated NF-κB and ISRE activation pathways can competewith each other. Deficiency of TRAF6 causes loss ofVISA-mediated NF-κB activation and enhancement ofVISA-mediated ISRE activation.
Taken together, our data suggest that VISA mediatesbifurcation of virus-triggered and TLR3-mediated NF-κB and ISRE activation pathways.
VISA Is Not Involved in TLR4-and TNF-Induced SignalingKnockdown of VISA did not inhibit TLR4-mediated IFN-βsignaling (Figure 6). This is surprising because it wasreported that TLR4 signals IFN-β activation through aTRIF and TRAM-containing complex (Fitzgerald et al.,2003b; Oshiumi et al., 2003b; Yamamoto et al., 2003).It is possible that the TRIF-TRAM complex can signalwithout VISA.

Molecular Cell738
In a mammalian overexpression system, VISA can in-teract with TRAF2 (Figure 5). However, RNAi knock-down experiments suggest that VISA is not involved inTNF-induced NF-κB activation (Figure 6). Whether VISAinteracts with TRAF2 under physiological conditionsand whether VISA plays a role in signaling of uniden-tified pathways will be investigated in a separate study.
Concluding RemarksIn virus-triggered TLR3-independent signaling path-ways, VISA interacts with RIG-I through its respectiveCARD modules and functions as an adaptor to recruitdownstream components, such as IRF-3, to the RIG-Icomplex. In TLR3-mediated signaling pathways, VISAinteracts with TRIF and TRAF6 and mediates bifurca-tion of the NF-κB and ISRE activation pathways. Ourfindings suggest that VISA plays essential roles in anti-viral responses by participating two virus-triggered IFNsignaling pathways.
Experimental Procedures
ReagentsRecombinant human TNF (R&D Systems), poly(I:C) (Invivogen),Sendai virus (Charles River Laboratories), LPS (Sigma), monoclonalantibodies against FLAG (Sigma) and HA (Covance) epitopes, rab-bit polyclonal anti-TRAF6 antibody (Santa Cruz Biotechnology),goat polyclonal anti-TRIF antibody (Imgenex), SAOS-2 and H1299(ATCC), and 293-TLR4/MD2/CD14 cells (Invivogen) were purchasedfrom the indicated manufacturers. Rabbit anti-RIG-I (Tadaatsu Im-aizumi), 293 cells (Dr. Zhaodan Cao), 293-TLR3 cells (Drs. KatherineFitzgerald and Tom Maniatis), and TRAF6−/− and wt MEFs (Drs. TakMak and Wen-Chen Yeh) were provided by the indicated investiga-tors. TRIF-deficient mice were provided by Dr. Bruce Beutler, andTRIF−/− MEFs were prepared from embryos at day 10 of gestation.
ConstructsNF-κB luciferase reporter construct was provided by Dr. GaryJohnson. ISRE and CHOP luciferase reporter constructs were pur-chased from Stratagene. Mammalian expression plasmids forTRAF2, TRAF6, IKKβ, and IKKβ-KA (Drs. David Goeddel and Zhao-dan Cao); TBK1, TBK1-KA, IKK�, and IKK�-KA (Dr. Uli Siebenlist);and IRF-3 (Dr. John Hiscott) were provided by the indicated investi-gators. Mammalian expression plasmids for IRF-7, TRIF, and TRIF-(1–394) (Han et al., 2004) and the IFN-β promoter luciferase reporterplasmid (Han et al., 2004) have been described.
Mammalian expression plasmids for human HA- or FLAG-taggedVISA and RIG-I and their mutants were constructed by standardmolecular biology techniques.
Antibody PreparationHuman VISA-(1–180) and VISA-(181–360) cDNAs were constructedin pET32a vector (Novagen). The recombinant proteins were ex-pressed in E. coli and purified by nickel column chromatography.The recombinant proteins were injected into rabbits to produce an-tisera against VISA-(1–180) and VISA-(181–360), respectively.
Northern Blot HybridizationHuman multiple tissue mRNA blot was purchased from Clontech.The blot was hybridized with P32-dCTP-labeled cDNA probe corre-sponding to human VISA coding sequence. Hybridization was per-formed in the Rapid Hybridization Buffer (Clontech) under high-stringency conditions.
Reporter Gene Assays293 cells and their derivatives (w2 × 105) were seeded in 12-welldishes and transfected the following day by standard calciumphosphate precipitation method. Wt, TRAF6−/−, and TRIF−/− MEFs(w1 × 105) were seeded in 24-well dishes and transfected the fol-lowing day with Lipofectamine 2000 (Invitrogen). Reporter assays
wa
CTa
IToeCITmdTpwn
EETf
RTrtsTA3T
A
WTHgbt
RRAP
R
ARb
Atc
Bt4
B(Cc
Bt
B(sm
ere performed as previous described (Han et al., 2004; Jiang etl., 2004).
oimmunoprecipitation and Western Blot Analysisransient transfection, coimmunoprecipitation, and Western blotnalysis were performed as previous described (Shu et al., 1996).
n Vitro Kinase Assayo determine whether IRF-3 is phosphorylated after overexpressionf VISA and IKK�, 293 cells (w2 × 106) were cotransfected withxpression plasmids for HA-VISA or HA-IKK� (5 �g/each) for 16 hr.ells were lysed, and the lysate was incubated with 25 �l of GST-
RF-3(380–427) bound glutathione Sepharose beads for 1 hr at 4°C.he beads were washed with lysis buffer and then kinase buffer (20M HEPES, [pH 7.4], 10 mM MgCl2, 0.1 mM sodium orthovana-ate, 20 mM β-glycerophosphate, 1 mM DTT, and 50 mM NaCl).he beads were then incubated with 30 �l of kinase buffer in theresence of 1 �l of [32P]-γ-ATP at 30°C for 30 min. Kinase reactionsere resolved by 10% SDS-PAGE. The gel was fixed in 30% metha-ol/10% acetic acid, dried, and exposed to X-ray film.
MSAsMSAs were performed as previously described (Wu et al., 2003).he consensus NF-κB and ISRE oligonucleotides were purchasedrom Santa Cruz Biotechnology.
NAi Constructshe human VISA RNAi constructs were made by using the pSuper.etro vector (OligoEngine) according to protocols recommended byhe manufacturer. The target sequences for the human VISA con-tructs were: #1, 5#-GACAAGACCTATAAGTATA-3#; #2, 5#-GACCTAAAGTATATCTGC-3#; #3, 5#-GTATATCTGCCGCAATTTC-3#; #4, 5#-TGCAGAGAAACAGGAGTC-3#; #5, 5#-TTTCAGCAATTTTTGCAAT-#; #6, 5#-TTTTTGCAATGTGGATGTT-3#; and #7, 5#-GACAAGACCATAAGTATA-3#.
cknowledgments
e thank Laurel Lenz for reading the manuscript and Bruce Beutler,ak Mak, Wen-Chen Yeh, Tadaatsu Imaizumi, David Goeddel, Johniscott, Rongtuan Lin, Gary Johnson, Uli Siebenlist, Katherine Fitz-erald, and Tom Maniatis for reagents. This work was supportedy grants from the National Institutes of Health (R01 AI062739) and
he Chinese High-Technology Program (#2003AA221030).
eceived: July 26, 2005evised: August 8, 2005ccepted: August 15, 2005ublished online: September 8, 2005
eferences
lexopoulou, L., Holt, A.C., Medzhitov, R., and Flavell, R.A. (2001).ecognition of double-stranded RNA and activation of NF-kappaBy Toll-like receptor 3. Nature 413, 732–738.
rch, R.H., Gedrich, R.W., and Thompson, C.B. (2000). Transloca-ion of TRAF proteins regulates apoptotic threshold of cells. Bio-hem. Biophys. Res. Commun. 272, 936–945.
arr, F.A., Sillje, H.H., and Nigg, E.A. (2004). Polo-like kinases andhe orchestration of cell division. Nat. Rev. Mol. Cell Biol. 5, 429–40.
oucher, L.M., Marengere, L.E., Lu, Y., Thukral, S., and Mak, T.W.1997). Binding sites of cytoplasmic effectors TRAF1, 2, and 3 onD30 and other members of the TNF receptor superfamily. Bio-hem. Biophys. Res. Commun. 233, 592–600.
radley, J.R., and Pober, J.S. (2001). Tumor necrosis factor recep-or-associated factors (TRAFs). Oncogene 20, 6482–6491.
rodeur, S.R., Cheng, G., Baltimore, D., and Thorley-Lawson, D.A.1997). Localization of the major NF-kappaB-activating site and theole TRAF3 binding site of LMP-1 defines two distinct signalingotifs. J. Biol. Chem. 272, 19777–19784.

VISA Is a Component of Virus-Induced Signaling739
Brown, K.D., Hostager, B.S., and Bishop, G.A. (2001). Differentialsignaling and tumor necrosis factor receptor-associated factor(TRAF) degradation mediated by CD40 and the Epstein-Barr virusoncoprotein latent membrane protein 1 (LMP1). J. Exp. Med. 193,943–954.
Durbin, J.E., Fernandez-Sesma, A., Lee, C.K., Rao, T.D., Frey, A.B.,Moran, T.M., Vukmanovic, S., Garcia-Sastre, A., and Levy, D.E.(2000). Type I IFN modulates innate and specific antiviral immunity.J. Immunol. 164, 4220–4228.
Fitzgerald, K.A., McWhirter, S.M., Faia, K.L., Rowe, D.C., Latz, E.,Golenbock, D.T., Coyle, A.J., Liao, S.M., and Maniatis, T. (2003a).IKKepsilon and TBK1 are essential components of the IRF3 signal-ing pathway. Nat. Immunol. 4, 491–496.
Fitzgerald, K.A., Rowe, D.C., Barnes, B.J., Caffrey, D.R., Visintin, A.,Latz, E., Monks, B., Pitha, P.M., and Golenbock, D.T. (2003b). LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adaptersTRAM and TRIF. J. Exp. Med. 198, 1043–1055.
Fotin-Mleczek, M., Henkler, F., Samel, D., Reichwein, M., Hausser,A., Parmryd, I., Scheurich, P., Schmid, J.A., and Wajant, H. (2002).Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion ofTRAF2 and IAP proteins and accelerates TNF-R1-dependent acti-vation of caspase-8. J. Cell Sci. 115, 2757–2770.
Han, K.J., Su, X., Xu, L.G., Bin, L.H., Zhang, J., and Shu, H.B. (2004).Mechanisms of the TRIF-induced interferon-stimulated responseelement and NF-kappaB activation and apoptosis pathways. J.Biol. Chem. 279, 15652–15661.
Hemmi, H., Takeuchi, O., Sato, S., Yamamoto, M., Kaisho, T., Sanjo,H., Kawai, T., Hoshino, K., Takeda, K., and Akira, S. (2004). Theroles of two IkappaB kinase-related kinases in lipopolysaccharideand double stranded RNA signaling and viral infection. J. Exp. Med.199, 1641–1650.
Hoebe, K., Du, X., Georgel, P., Janssen, E., Tabeta, K., Kim, S.O.,Goode, J., Lin, P., Mann, N., Mudd, S., et al. (2003). Identification ofLps2 as a key transducer of MyD88-independent TIR signalling.Nature 424, 743–748.
Honda, K., Sakaguchi, S., Nakajima, C., Watanabe, A., Yanai, H.,Matsumoto, M., Ohteki, T., Kaisho, T., Takaoka, A., Akira, S., et al.(2003). Selective contribution of IFN-alpha/beta signaling to thematuration of dendritic cells induced by double-stranded RNA orviral infection. Proc. Natl. Acad. Sci. USA 100, 10872–10877.
Honda, K., Ohba, Y., Yanai, H., Negishi, H., Mizutani, T., Takaoka,A., Taya, C., and Taniguchi, T. (2005a). Spatiotemporal regulation ofMyD88-IRF-7 signalling for robust type-I interferon induction. Na-ture 434, 1035–1040.
Honda, K., Yanai, H., Negishi, H., Asagiri, M., Sato, M., Mizutani, T.,Shimada, N., Ohba, Y., Takaoka, A., Yoshida, N., and Taniguchi, T.(2005b). IRF-7 is the master regulator of type-I interferon-depen-dent immune responses. Nature 434, 772–777.
Jiang, Z., Mak, T.W., Sen, G., and Li, X. (2004). Toll-like receptor3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1receptor domain-containing adapter inducing IFN-beta. Proc. Natl.Acad. Sci. USA 101, 3533–3538.
Levy, D.E., and Garcia-Sastre, A. (2001). The virus battles: IFN in-duction of the antiviral state and mechanisms of viral evasion. Cy-tokine Growth Factor Rev. 12, 143–156.
Levy, D.E., and Marie, I.J. (2004). RIGging an antiviral defense—it’sin the CARDs. Nat. Immunol. 5, 699–701.
Lin, R., Mamane, Y., and Hiscott, J. (1999). Structural and functionalanalysis of interferon regulatory factor 3: localization of the trans-activation and autoinhibitory domains. Mol. Cell. Biol. 19, 2465–2474.
Maniatis, T., Falvo, J.V., Kim, T.H., Kim, T.K., Lin, C.H., Parekh, B.S.,and Wathelet, M.G. (1998). Structure and function of the interferon-beta enhanceosome. Cold Spring Harb. Symp. Quant. Biol. 63,609–620.
Matsuda, A., Suzuki, Y., Honda, G., Muramatsu, S., Matsuzaki, O.,Nagano, Y., Doi, T., Shimotohno, K., Harada, T., Nishida, E., et al.(2003). Large-scale identification and characterization of humangenes that activate NF-kappaB and MAPK signaling pathways. On-cogene 22, 3307–3318.
McWhirter, S.M., Fitzgerald, K.A., Rosains, J., Rowe, D.C., Golen-bock, D.T., and Maniatis, T. (2003). IFN-regulatory factor 3-depen-dent gene expression is defective in Tbk1-deficient mouse embry-onic fibroblasts. Proc. Natl. Acad. Sci. USA 101, 233–238.
Oshiumi, H., Matsumoto, M., Funami, K., Akazawa, T., and Seya, T.(2003a). TICAM-1, an adaptor molecule that participates in Toll-likereceptor 3-mediated interferon-beta induction. Nat. Immunol. 4,161–167.
Oshiumi, H., Sasai, M., Shida, K., Fujita, T., Matsumoto, M., andSeya, T. (2003b). TIR-containing adapter molecule (TICAM)-2, abridging adapter recruiting to toll-like receptor 4 TICAM-1 that in-duces interferon-beta. J. Biol. Chem. 278, 49751–49762.
Perry, A.K., Chow, E.K., Goodnough, J.B., Yeh, W.C., and Cheng, G.(2004). Differential requirement for TANK-binding kinase-1 in type Iinterferon responses to toll-like receptor activation and viral infec-tion. J. Exp. Med. 199, 1651–1658.
Sato, S., Sugiyama, M., Yamamoto, M., Watanabe, Y., Kawai, T.,Takeda, K., and Akira, S. (2003). Toll/IL-1 receptor domain-contain-ing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates twodistinct transcription factors, NF-kappa B and IFN-regulatory fac-tor-3, in the Toll-like receptor signaling. J. Immunol. 171, 4304–4310.
Servant, M.J., Tenoever, B., and Lin, R. (2002). Overlapping anddistinct mechanisms regulating IRF-3 and IRF-7 function. J. In-terferon Cytokine Res. 22, 49–58.
Sharma, S., tenOever, B.R., Grandvaux, N., Zhou, G.P., Lin, R., andHiscott, J. (2003). Triggering the interferon antiviral responsethrough an IKK-related pathway. Science 300, 1148–1151.
Shu, H.B., Takeuchi, M., and Goeddel, D.V. (1996). The tumor necro-sis factor receptor 2 signal transducers TRAF2 and c-IAP1 arecomponents of the tumor necrosis factor receptor 1 signaling com-plex. Proc. Natl. Acad. Sci. USA 93, 13973–13978.
tenOever, B.R., Servant, M.J., Grandvaux, N., Lin, R., and Hiscott,J. (2002). Recognition of the measles virus nucleocapsid as amechanism of IRF-3 activation. J. Virol. 76, 3659–3669.
tenOever, B.R., Sharma, S., Zou, W., Sun, Q., Grandvaux, N., Julku-nen, I., Hemmi, H., Yamamoto, M., Akira, S., Yeh, W.C., et al. (2004).Activation of TBK1 and IKKvarepsilon kinases by vesicular stomati-tis virus infection and the role of viral ribonucleoprotein in the de-velopment of interferon antiviral immunity. J. Virol. 78, 10636–10649.
Wajant, H., Henkler, F., and Scheurich, P. (2001). The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors,kinases and their regulators. Cell. Signal. 13, 389–400.
Wathelet, M.G., Lin, C.H., Parekh, B.S., Ronco, L.V., Howley, P.M.,and Maniatis, T. (1998). Virus infection induces the assembly of co-ordinately activated transcription factors on the IFN-beta enhancerin vivo. Mol. Cell 1, 507–518.
Weaver, B.K., Kumar, K.P., and Reich, N.C. (1998). Interferon regula-tory factor 3 and CREB-binding protein/p300 are subunits ofdouble-stranded RNA-activated transcription factor DRAF1. Mol.Cell. Biol. 18, 1359–1368.
Wu, M., Xu, L.G., Zhai, Z., and Shu, H.B. (2003). SINK is a p65-interacting negative regulator of NF-kappaB-dependent transcrip-tion. J. Biol. Chem. 278, 27072–27079.
Yamamoto, M., Sato, S., Mori, K., Hoshino, K., Takeuchi, O.,Takeda, K., and Akira, S. (2002). Cutting edge: a novel Toll/IL-1 re-ceptor domain-containing adapter that preferentially activates theIFN-beta promoter in the Toll-like receptor signaling. J. Immunol.169, 6668–6672.
Yamamoto, M., Sato, S., Hemmi, H., Hoshino, K., Kaisho, T., Sanjo,H., Takeuchi, O., Sugiyama, M., Okabe, M., Takeda, K., and Akira,S. (2003). Role of adaptor TRIF in the MyD88-independent toll-likereceptor signaling pathway. Science 301, 640–643.
Ye, H., Arron, J.R., Lamothe, B., Cirilli, M., Kobayashi, T., Shevde,N.K., Segal, D., Dzivenu, O.K., Vologodskaia, M., Yim, M., et al.(2002). Distinct molecular mechanism for initiating TRAF6 signal-ling. Nature 418, 443–447.

Molecular Cell740
Yoneyama, M., Suhara, W., Fukuhara, Y., Fukuda, M., Nishida, E.,and Fujita, T. (1998). Direct triggering of the type I interferon systemby virus infection: activation of a transcription factor complex con-taining IRF-3 and CBP/p300. EMBO J. 17, 1087–1095.
Yoneyama, M., Kikuchi, M., Natsukawa, T., Shinobu, N., Imaizumi,T., Miyagishi, M., Taira, K., Akira, S., and Fujita, T. (2004). The RNAhelicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737.
Zhang, L., and Pagano, J.S. (2002). Structure and function ofIRF-7. J. Interferon Cytokine Res. 22, 95–101.
Accession Numbers
The GenBank accession numbers for the human and mouse VISAmRNAs are DQ167126 and DQ167127, respectively.