
Understanding''global''systems biology: metabonomics and the continuum of metabolism
11
668 | AUGUST 2003 | VOLUME 2 www.nature.com/reviews/drugdisc PERSPECTIVES even in such simple systems, statistical relation- ships between, for instance, gene expression and protein levels can be weak 4 . We have argued that the multivariate temporal modelling of metabolism and physiological processes following pathophysio- logical stimuli can be important in connect- ing molecular events at the gene and protein level to those occurring at the macrosystem level, including pathological end-points 5–8 . This is likely to be true because changes in the kinetics of specific pathways in cells, tissues and organs are real end-points in their own right. However, there are limitations in the way in which ‘omics’ data types can be mod- elled in higher animals, because of the num- ber and spatial dispersion of the interacting cell types. Furthermore, gene expression and proteomic data might only indicate the poten- tial for pathophysiological effects, because many pathway feedback mechanisms are simply not reflected in protein concentration or gene expression changes. This realization has led to increased efforts by pharmaceutical companies to try to model transcriptomic and proteomic data in relation to metabolic pathway activity, and to map such data onto well-known pathway databases, such as those provided by the Kyoto Encyclopedia of Genes and Genomes (KEGG) 9 . However, the humanbiological ‘system’ is very extensive, and the functional integrity of human physiol- ogy is also dependent on many external fac- tors, even including other genomes from 13. Kennedy, T. Managing the drug discovery/ development interface. Drug Discov. Today 2, 436–444 (1997). 14. van de Waterbeemd, H. & Gifford, E. ADMET in silico modelling: towards prediction paradise? Nature Rev. Drug Discov. 2, 192–204 (2003). 15. Lipinski, C. A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 44, 235–249 (2000). 16. Ajay, A., Walters, W. P. & Murcko, M. A. Can we learn to distinguish between ‘drug-like’ and ‘nondrug-like’ molecules? J. Med. Chem. 41, 3314–3324 (1998). 17. Sadowski, J. & Kubinyi, H. A scoring scheme for discriminating between drugs and nondrugs. J. Med. Chem. 41, 3325–3329 (1998). 18. Teague, S. J., Davis, A. M., Leeson, P. D. & Oprea, T. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. 38, 3743–3748 (1999). 19. Oprea, T. I., Davis, A. M., Teague, S. J. & Leeson, P. D. Is there a difference between leads and drugs? A historical perspective. J. Chem. Inf. Comput. Sci. 41, 1308–1316 (2001). 20. Proudfoot, J. R. Drugs, leads, and drug-likeness: an analysis of some recently launched drugs. Bioorg. Med. Chem. Lett. 12, 1647–1650 (2002). 21. Hann, M. M., Leach, A. R. & Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 41, 856–864 (2001). 22. Carr, R. & Hann, M. The right road to drug discovery? Fragment-based screening casts doubt on the Lipinski route. Modern Drug Discov. April, 45–48 (2002) 23. Lommerse, J. P. M., Price, S. L. & Taylor, R. Hydrogen bonding of carbonyl, ether, and ester oxygen atoms with alkanol hydroxyl groups. J. Comput. Chem. 18, 757–774 (1997). 2. Drews, J. Strategic trends in the drug industry. Drug Discov. Today 8, 411–420 (2003). 3. Hopkins, A. L. & Groom, C. R. The druggable genome. Nature Rev. Drug Discov. 1, 727–730 (2002). 4. Lahana, R. How many leads from HTS? Drug Discov. Today 4, 447–448 (1999). 5. Lipinski, C. A., Lombardo F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and develop- ment settings. Adv. Drug Del. Rev. 23, 3–25 (1997). 6. Ertl, P., Rohde, B. & Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contri- butions and its application to the prediction of drug trans- port properties. J. Med. Chem. 43, 3714–3717 (2000). 7. Cruciani, G., Pastor, M. & Guba, W. VolSurf: a new tool for the pharmacokinetic optimization of lead compounds. Eur. J. Pharm. Sci. 11 (Suppl. 2) S29–S39 (2000). 8. Kelder, J., Grootenhuis, P. D., Bayada, D. M., Delbressine, L. P. & Ploemen, J. P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 16, 1514–1519 (1999). 9. Bergström, C. A. S. et al. Absorption classification of oral drugs based on molecular surface properties. J. Med. Chem. 46, 558–570 (2003). 10. Veber, D. F. et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623 (2002). 11. Wenlock, M. C., Austin, R. P., Barton, P., Davis, A. M. & Leeson, P. D. A comparison of physicochemical property profiles of development and marketed oral drugs. J. Med. Chem. 46, 1250–1256 (2003). 12. Prentis, R. A., Lis, Y. & Walker, S. R. Pharmaceutical innovation by the seven UK-owned pharmaceutical companies (1964–1985). Br. J. Clin. Pharmacol. 25, 387–396 (1988). Understanding ‘Global’ Systems Biology: Metabonomics and the Continuum of Metabolism Jeremy K. Nicholson* and Ian D. Wilson ‡ OPINION To apply genomic knowledge effectively in drug discovery, mechanistic connectivities between genetic variation and disease processes need to be established via systems biology approaches. Humans have hundreds of functionally specialized cell types that interact differentially with environmental factors to influence disease development and to modulate the effects of drugs. Metabonomics can provide a means of modelling these interactions, but the relationships between ‘endogenous’ metabolic processes (coded in the genome and intrinsic to cellular function) and ‘xenobiotic’ (foreign compound) metabolism are poorly understood, especially with respect to environmental factors. We present an overview of ‘global’ mammalian metabolic conversions that should be accounted for in human systems biology models and propose a new probabilistic approach to help understand gene–disease relationships and vexed issues of idiosyncratic drug toxicity. Systems biology — that is, the computational integration of data generated by the suite of genetic, transcriptomic, proteomic and metabonomic platforms to understand func- tion through different levels of biomolecular organization — offers exciting new prospects for determining the causes of human disease and finding possible cures 1 . Certainly the judicious use of ‘omics’ data should give new insights and opportunities for the drug dis- covery and development process and for understanding drug toxicology 2 . From a modelling point of view, considerable advances have been made in correlating gene–protein–metabolite interactions in microorganisms in which linked processes in single cell types can be probed in depth 3 . But 24. Böhm, H.-J., Brode, S., Hesse, U. & Klebe, G. Oxygen and nitrogen in competitive situations: which is the hydrogen-bond acceptor? Chem. Eur. J. 2, 1509–1513 (1996). 25. Kubinyi, H. in Pharmacokinetic Optimization in Drug Research: Biological, Physicochemical, and Computational Strategies (eds Testa, B., van de Waterbeemd, H., Folkers, G. & Guy, R.) 513–524 (Helvetica Chimica Acta and Wiley-VCH, Zurich, 2001). 26. Sneader, W. Drug Prototypes and Their Exploitation. (John Wiley & Sons, Chichester, 1996). 27. Wermuth, C. G. The Practice of Medicinal Chemistry. (Academic, London, 1996). 28. Watson, J. D. The Double Helix: A Personal Account of the Discovery of the Structure of DNA. (Atheneum, New York, 1968). 29. Watson, J. D., with Berry, A. DNA. The Secret of Life. (William Heinemann, London, 2003). 30. Finding the flaws [online] (cited May 2003) <http://www. phy.cam.ac.uk/camphy/dna/ dna13_1.htm> (2003). 31. A working model! [online] (cited May 2003) <http://www. phy.cam.ac.uk/camphy/dna/ dna14_1.htm> (2003). Online links DATABASES LocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/ BCR–ABL FURTHER INFORMATION Cambridge Crystallographic Data Centre: www.ccdc.cam.ac.uk/prods/isostar/ Hugo Kubinyi’s website: http://home.t-online.de/home/kubinyi Access to this interactive links box is free online.
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Understanding''global''systems biology: metabonomics and the continuum of metabolism

668 | AUGUST 2003 | VOLUME 2 www.nature.com/reviews/drugdisc
P E R S P E C T I V E S
even in such simple systems, statistical relation-ships between, for instance, gene expressionand protein levels can be weak4.
We have argued that the multivariatetemporal modelling of metabolism andphysiological processes following pathophysio-logical stimuli can be important in connect-ing molecular events at the gene and proteinlevel to those occurring at the macrosystemlevel, including pathological end-points5–8.This is likely to be true because changes in thekinetics of specific pathways in cells, tissuesand organs are real end-points in their ownright. However, there are limitations in theway in which ‘omics’ data types can be mod-elled in higher animals, because of the num-ber and spatial dispersion of the interactingcell types. Furthermore, gene expression andproteomic data might only indicate the poten-tial for pathophysiological effects, becausemany pathway feedback mechanisms aresimply not reflected in protein concentrationor gene expression changes. This realizationhas led to increased efforts by pharmaceuticalcompanies to try to model transcriptomicand proteomic data in relation to metabolicpathway activity, and to map such data ontowell-known pathway databases, such as thoseprovided by the Kyoto Encyclopedia of Genesand Genomes (KEGG)9. However, thehumanbiological ‘system’ is very extensive,and the functional integrity of human physiol-ogy is also dependent on many external fac-tors, even including other genomes from
13. Kennedy, T. Managing the drug discovery/development interface. Drug Discov. Today 2, 436–444 (1997).
14. van de Waterbeemd, H. & Gifford, E. ADMET in silicomodelling: towards prediction paradise? Nature Rev.Drug Discov. 2, 192–204 (2003).
15. Lipinski, C. A. Drug-like properties and the causes ofpoor solubility and poor permeability. J. Pharmacol.Toxicol. Methods 44, 235–249 (2000).
16. Ajay, A., Walters, W. P. & Murcko, M. A. Can we learn todistinguish between ‘drug-like’ and ‘nondrug-like’molecules? J. Med. Chem. 41, 3314–3324 (1998).
17. Sadowski, J. & Kubinyi, H. A scoring scheme fordiscriminating between drugs and nondrugs. J. Med.Chem. 41, 3325–3329 (1998).
18. Teague, S. J., Davis, A. M., Leeson, P. D. & Oprea, T.The design of leadlike combinatorial libraries. Angew.Chem. Int. Ed. 38, 3743–3748 (1999).
19. Oprea, T. I., Davis, A. M., Teague, S. J. & Leeson, P. D. Isthere a difference between leads and drugs? A historicalperspective. J. Chem. Inf. Comput. Sci. 41, 1308–1316(2001).
20. Proudfoot, J. R. Drugs, leads, and drug-likeness: ananalysis of some recently launched drugs. Bioorg. Med.Chem. Lett. 12, 1647–1650 (2002).
21. Hann, M. M., Leach, A. R. & Harper, G. Molecularcomplexity and its impact on the probability of findingleads for drug discovery. J. Chem. Inf. Comput. Sci. 41,856–864 (2001).
22. Carr, R. & Hann, M. The right road to drug discovery?Fragment-based screening casts doubt on the Lipinskiroute. Modern Drug Discov. April, 45–48 (2002)
23. Lommerse, J. P. M., Price, S. L. & Taylor, R. Hydrogenbonding of carbonyl, ether, and ester oxygen atoms withalkanol hydroxyl groups. J. Comput. Chem. 18,757–774 (1997).
2. Drews, J. Strategic trends in the drug industry. Drug Discov. Today 8, 411–420 (2003).
3. Hopkins, A. L. & Groom, C. R. The druggable genome.Nature Rev. Drug Discov. 1, 727–730 (2002).
4. Lahana, R. How many leads from HTS? Drug Discov.Today 4, 447–448 (1999).
5. Lipinski, C. A., Lombardo F., Dominy, B. W. & Feeney, P. J.Experimental and computational approaches to estimatesolubility and permeability in drug discovery and develop-ment settings. Adv. Drug Del. Rev. 23, 3–25 (1997).
6. Ertl, P., Rohde, B. & Selzer, P. Fast calculation of molecularpolar surface area as a sum of fragment-based contri-butions and its application to the prediction of drug trans-port properties. J. Med. Chem. 43, 3714–3717 (2000).
7. Cruciani, G., Pastor, M. & Guba, W. VolSurf: a new toolfor the pharmacokinetic optimization of lead compounds.Eur. J. Pharm. Sci. 11 (Suppl. 2) S29–S39 (2000).
8. Kelder, J., Grootenhuis, P. D., Bayada, D. M.,Delbressine, L. P. & Ploemen, J. P. Polar molecularsurface as a dominating determinant for oral absorptionand brain penetration of drugs. Pharm. Res. 16,1514–1519 (1999).
9. Bergström, C. A. S. et al. Absorption classification of oraldrugs based on molecular surface properties. J. Med.Chem. 46, 558–570 (2003).
10. Veber, D. F. et al. Molecular properties that influence theoral bioavailability of drug candidates. J. Med. Chem.45, 2615–2623 (2002).
11. Wenlock, M. C., Austin, R. P., Barton, P., Davis, A. M. &Leeson, P. D. A comparison of physicochemical propertyprofiles of development and marketed oral drugs. J. Med. Chem. 46, 1250–1256 (2003).
12. Prentis, R. A., Lis, Y. & Walker, S. R. Pharmaceuticalinnovation by the seven UK-owned pharmaceuticalcompanies (1964–1985). Br. J. Clin. Pharmacol. 25,387–396 (1988).
Understanding ‘Global’ SystemsBiology: Metabonomics and theContinuum of MetabolismJeremy K. Nicholson* and Ian D. Wilson‡
O P I N I O N
To apply genomic knowledge effectively indrug discovery, mechanistic connectivitiesbetween genetic variation and diseaseprocesses need to be established viasystems biology approaches. Humans havehundreds of functionally specialized celltypes that interact differentially withenvironmental factors to influence diseasedevelopment and to modulate the effects ofdrugs. Metabonomics can provide a meansof modelling these interactions, but therelationships between ‘endogenous’metabolic processes (coded in the genomeand intrinsic to cellular function) and‘xenobiotic’ (foreign compound) metabolismare poorly understood, especially withrespect to environmental factors. Wepresent an overview of ‘global’ mammalianmetabolic conversions that should beaccounted for in human systems biologymodels and propose a new probabilistic
approach to help understandgene–disease relationships and vexedissues of idiosyncratic drug toxicity.
Systems biology — that is, the computationalintegration of data generated by the suite ofgenetic, transcriptomic, proteomic andmetabonomic platforms to understand func-tion through different levels of biomolecularorganization — offers exciting new prospectsfor determining the causes of human diseaseand finding possible cures1. Certainly thejudicious use of ‘omics’ data should give newinsights and opportunities for the drug dis-covery and development process and forunderstanding drug toxicology2. From amodelling point of view, considerableadvances have been made in correlatinggene–protein–metabolite interactions inmicroorganisms in which linked processes insingle cell types can be probed in depth3. But
24. Böhm, H.-J., Brode, S., Hesse, U. & Klebe, G. Oxygenand nitrogen in competitive situations: which is thehydrogen-bond acceptor? Chem. Eur. J. 2, 1509–1513(1996).
25. Kubinyi, H. in Pharmacokinetic Optimization in DrugResearch: Biological, Physicochemical, andComputational Strategies (eds Testa, B., van deWaterbeemd, H., Folkers, G. & Guy, R.) 513–524(Helvetica Chimica Acta and Wiley-VCH, Zurich, 2001).
26. Sneader, W. Drug Prototypes and Their Exploitation.(John Wiley & Sons, Chichester, 1996).
27. Wermuth, C. G. The Practice of Medicinal Chemistry.(Academic, London, 1996).
28. Watson, J. D. The Double Helix: A Personal Account ofthe Discovery of the Structure of DNA. (Atheneum, NewYork, 1968).
29. Watson, J. D., with Berry, A. DNA. The Secret of Life.(William Heinemann, London, 2003).
30. Finding the flaws [online] (cited May 2003) <http://www.phy.cam.ac.uk/camphy/dna/ dna13_1.htm> (2003).
31. A working model! [online] (cited May 2003) <http://www.phy.cam.ac.uk/camphy/dna/ dna14_1.htm> (2003).
Online links
DATABASESLocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/BCR–ABL
FURTHER INFORMATIONCambridge Crystallographic Data Centre:www.ccdc.cam.ac.uk/prods/isostar/Hugo Kubinyi’s website:http://home.t-online.de/home/kubinyiAccess to this interactive links box is free online.

P E R S P E C T I V E S
of the host; they might also have significanteffects on metabolism by the host, as gutmicrobes such as Bacteroides thetaiotaomicroneven carry genes that might assist the metabo-lism of other species, including humans23.Dietary composition, in turn, influences theselection of gut microflora and probioticfood products are now widely marketed ‘toimprove gut health’22. Feeding infants bene-ficial bacteria might reduce allergic inflamma-tion, and this has been related to antagonismbetween probiotic bacteria (such as Bifido-bacterium lactis) and some of the inflamma-tory gut microbes25. The general health benefitsand effects of probiotics are yet to be deter-mined, but in a wider context could stillinfluence endogenous metabolism in subtleand important ways that are relevant to drugmetabolism and toxicity.
The role of nutrition in the developmentof human disease at the molecular level iswidely appreciated26, and it is well-known thatcancer and cardiovascular diseases have bothdietary and genetic components27. It has alsobeen shown that selective dietary supplemen-tation can markedly affect the metabolismand toxicity of even commonly used drugs,such as paracetamol (acetaminophen)28.Undoubtedly, all of these nutritional andmicrobial factors also influence the efficacy,metabolism and toxicity of drugs in a varietyof ways in human populations. We now con-sider that only by accounting for all the prin-cipal ‘metabolic axes’ between the diet, theentero-microbiolome and an individual’soverall metabolic status (part genomically,part environmentally conferred) will it bepossible to shed light on the development ofcomplex disease traits and the adverse idio-syncratic drug toxicity reactions that can befatal to both the patient and the drug. Humansystems biology could ultimately turn out tobe more like an ecological problem than oneof molecular biology.
Mammalian metabolic continuaIt must be recognized that intracellular andextracellular metabolites can come fromdiverse sources that cannot be treated equallywith respect to process control exerted by themammalian genome. Hitherto, metaboliteshave been classified simply as being either‘endogenous’ or ‘XENOBIOTIC’, and have beenconsidered from different academic viewpointswith respect to pathway analysis. However,endogenous and xenobiotic metabolites repre-sent ends of a continuum of integrated meta-bolic and non-enzymatic transformations ofmany kinds with numerous intermediate cat-egories that result from multi-site processing(see glossary). Many so-called endogenous
symbiotic organisms. Consideration of theinteractions of the internal constituents andexternal factors (FIG. 1) in mammals indicatesthat simple pathway modelling will nevercapture the information richness needed todescribe a human disease process or a druginteraction, irrespective of the sophisticationof the applied measurement technology. Thisproblem is summarized by two questions:how can we understand disease or drug-induced modulations in transcriptomic, pro-teomic or metabonomic data in relation tostandardized metabolic pathways that have nocompartmental constraints, when mammalianmetabolic control functions are dispersedacross many cell types in topographicallydistinct locations that are in different physio-logical states simultaneously? Also, how doesone interpret the many ‘extra-genomic’sources of metabolites and other influencesnot described in the individual’s genome thatnonetheless have important effects on theintegrated metabolism of the organism andon the disposition, fate and toxicity of drugs?
To answer these questions, we have re-examined the meaning of the term ‘metabo-lism’ as applied to mammals, and propose amore complete classification of the range ofmetabolic processes found in higher animalsand how they might interact probabilisticallyto determine the outcome of a diseaseprocess or drug interaction. This exposessome weaknesses in current bioinformatic
approaches to pathway modelling that willrequire the development of new integrativemetabolic tools for mammalian systems.
Metabolic pathway diagrams have longbeen used as shorthand summaries of cellu-lar biomolecular transformations, and areaccepted as being representative of under-lying cellular order and control. In single-cell systems (especially microbial ones),Metabolic Control Analysis10,11 (MCA) hasbeen appliedto describe the fluxes of metabo-lites through individual pathways or pathwayunits, and these methods can accuratelydescribe kinetic properties of such systems.MCA cannot be applied so easily to mam-mals, because metabolic control is dispersedin many cell types through space and time.Complex interactions occur between the con-trol of ENDOGENOUS METABOLIC pathways and themetabolism of foreign compounds, many ofwhich also induce their own metabolism. Forinstance, human cytochrome P450 enzymesand N-acetyl transferases vary polymorphi-cally in individuals in relation to disease,both within and between human ethnicpopulations12–20. As an example, humanCYP2D6 variations and activities have alsobeen related to possible susceptibility tolung cancer18. Mammals also have well-developed communities of gut microflora(the ‘microbiolome’21, with >500 individualspecies in humans22), which exert a strongcontrolling influence on the immune system
NATURE REVIEWS | DRUG DISCOVERY VOLUME 2 | AUGUST 2003 | 669
Host genome Gene expressionin cells and tissues
Extra-genomic factors
Disease outcome
Dietary factors
Cellular proteincomposition and activity
The gutmicrobiolome
Cellular and tissuemetabolic fluxes
Drugs
Host factors
Figure 1 | Mammalian system–microbial–nutritional–xenobiotic interactions. A summary of somekey factors that limit or augment individual genetic complement and affect the probability of particulardisease outcomes later in life. The moment-to-moment management of cells and tissues is representedby the transcriptomic, proteomic and metabonomic/metabolomic profiles. But these are also influencedby exposure to foreign compounds and interactions with commensal microorganisms, which are in turnmodulated and even selected by the host’s nutrition and drug inputs.
670 | AUGUST 2003 | VOLUME 2 www.nature.com/reviews/drugdisc
P E R S P E C T I V E S
been optimized by evolution. This covers theprincipal intermediary biosynthetic, energeticand catabolic pathways. Defects in these path-ways at the genetic level result in inborn
In truly endogenous processes, the primarydeterminants of the biochemical transforma-tions are under the direct control of enzymesand transporters of the host genome and have
and xenobiotic compounds can be inter-converted by a number of processes medi-ated by extra-genomic elements or by facilechemical reactions.
COOH
H
HO
C
H
HO
O
N
COOHH
C
H
HO
O
NH
SO3H
COOH
OH
HO OH
a Biosynthesis
f Reabsorptioninto blood viahepatic portalsystem
e 7α-dehydroxylationby gut microflora
b Phase II conjugationwith glycine andtaurine
g Phase IIconjugationwith glycineand taurine
i Deconjugationand further reactions
c Secretion into bile
h Secretioninto bile
Cholic acid
Glycocholic acid Taurocholic acid
Glycodeoxyocholic acid Taurodeoxyocholic acid
Deoxycholic acid
d Amino aciddeconjugation bygut microflora andregeneration ofcholic acid
Mammalian liver
Mammalian gut
HO
OH
O
O
COOHHO
OH
OH
Chlorogenic acid (dietary source)
OH
COOH
OH
COOHMammaliangut microflora
3-hydroxycinnamic acid
Absorbedin gut
3-(hydroxyphenyl) propionic acid
Phase II conjugationwith endogenous sulphateand glucuronic acid
Excretedin urine
Conjugatesexcretedin urine
COOH
Mammaliangut microflora
Various dietary aromaticcompounds and aromaticamino acids secreted in bile
Benzoic acid(absorbed in gut)
Conjugateexcretedin urine C
N
COOH
O
H
Hippuric acid
Phase II conjugation withendogenous glycine in livervia acetyl CoA intermediate
+
A
B
C
C
OH
HO OH
O
N
COOHH
H
COOH
OH
HO OHH
C
OH
HO OH
O
NH
SO3HH
H H
H
H
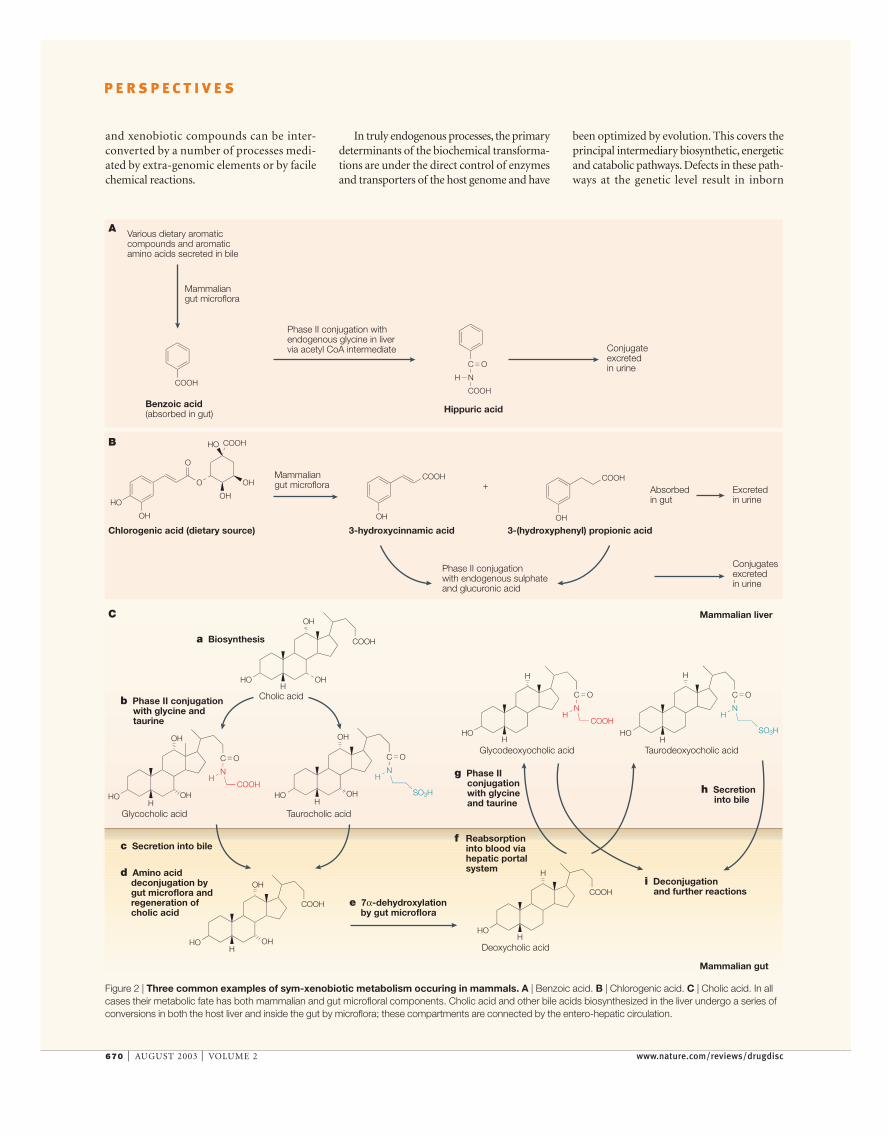
Figure 2 | Three common examples of sym-xenobiotic metabolism occuring in mammals. A | Benzoic acid. B | Chlorogenic acid. C | Cholic acid. In allcases their metabolic fate has both mammalian and gut microfloral components. Cholic acid and other bile acids biosynthesized in the liver undergo a series ofconversions in both the host liver and inside the gut by microflora; these compartments are connected by the entero-hepatic circulation.

P E R S P E C T I V E S
aborigines39; but there might also be influ-ences on drug metabolism exerted by alteredgut microflora or even parasites that have yetto be investigated. In instances of liver diseaseor toxicity, a series of increasingly complexmetabolic host polymorphism–microfloralinteractions become possible, with unpre-dictable biological consequences. Some of themost important interactions are summarizedin FIG. 3. The difficulties in modelling suchinteractions using the bioinformatics toolsavailable at present in terms of pathway analy-sis are obvious, and therefore new approachesto modelling the integrated human metabolicsystem need to be developed.
TRANS-XENOBIOTIC metabolism concernscompounds of extra-genomic or chemical ori-gin that can be converted into true endo-genous metabolites in specific tissue localities,sometimes at high enough concentrations toalter endogenous processes. A well-knownexample is ethanol, which is predominantlyoxidatively converted first into acetaldehydeby liver alcohol dehydrogenase and then toacetate by acetaldehyde dehydrogenase.Acetate is a key metabolic intermediate and isused in a wide range of biosynthetic and ener-getic pathways, including the citric acid cycle.Ethanol is a particularly interesting example ofa compound that fits in the continuum ofmetabolism because it can also result fromfermentation reactions in the gut (mediatedagain by the mircoflora) and so can also beconsidered to be sym-endogenous. Becausemammals have evolved with this source ofethanol, they have also evolved specificenzymes to metabolize, and hence utilize, itsproduct, acetate, as an energy or biosyntheticsource40. Other trans-xenobiotic transforma-tions include the sequential cleavage of fattyacyl chains of phthalate esters and other for-eign compounds with alkyl side chains thatcan by converted into low-molecular-weightfatty acids (including acetate) via ω-, ω-1- andβ-oxidation41,42.
At the end of the continuum of mam-malian metabolism lies the true XENOBIOTIC, thatis, a compound of alien or chemical origin thatcan be metabolized via a wide range ofendogenous oxidation and functional groupmodification systems and/or via conjugationwith endogenous species, but without enteringthe endogenous system. Compound-specific‘detoxification’ mechanisms have not evolvedfor these species, and instead they are subject toa range of generalized, nonspecific conversionsto produce metabolites that have reduced oraltered pharmacological activity or toxicity.Assuch, it is recognized that drug metabolism isof importance with respect to understandingpharmacokinetics, efficacy and for validating
errors of metabolism, many of which are veryrare, but which can give rise to profoundpathologies29. However, it is well known thatnormal endogenous enzymatic processesrequire many diet-derived co-factors, vita-mins, certain amino acids and lipids that arenot in the mammalian biosynthetic blueprint.We term these SYM-ENDOGENOUS compounds,which are essential to endogenous functionand which, in certain cases, are metabolizedfurther. We also identify a SYM-XENOBIOTIC classof compounds, which derive from exoge-nous genomic sources and are non-essential(though little is known about many of them),but nonetheless can compete, interact with andutilize endogenous metabolic resources andhence might influence more important bio-transformations. Hippuric acid (excreted atmillimolar concentrations in urine) is derivedfrom benzoic acid (produced mainly by gutmicrobial degradation of intestinal aromaticcompounds), and is then absorbed and conju-gated with endogenous glycine (FIG. 2). How-ever, benzoate can also be produced from truexenobiotic sources via the cytochrome P450-mediated oxidative metabolism30 of, forexample, toluene — which can come fromindustrial exposure or solvent abuse31 — indi-cating the degree of crossover and interactionof metabolic processes from diversely sourcedsubstrates. Chlorogenic acid (FIG. 2) is of dietaryplant origin, but its metabolic fate involvesboth microfloral and mammalian elements.The observation of high concentrations ofchlorogenic acid metabolites in the urine isassociated with low hippurate and is consistentwith altered different gut microfloral activity,which can in turn be nutritionally selected32,33.
Many bile acids can be regarded as becom-ing sym-endogenous through alien genomeco-metabolism. For example, human bile acidssuch as cheno-deoxycholic acid can be isomer-ized, for instance, to deoxycholic acid or can behydroxylated by the gut microflora34,35; the endproduct is then reabsorbed through theenterohepatic route and can act as an inhibitorof bile acid synthesis. Some bile acids arepotential hepatotoxins and undergo phase IIconjugations with sulphate, glucuronate andamino acids in the liver to produce lower-toxicity metabolites, which are secreted intothe bile34–37. The gut microflora subsequentlydeconjugate, and 7α-dehydroxylate thesecompounds, and can also dehydrate, epimer-ize and oxido-reduce them to produce diversestructures that are returned to the host via themammalian enterohepatic circulation37,38. Theimportance of the establishment of a healthygut microflora and its influence on global hostmetabolism is illustrated in axenic rats (raisedin a microbe-free environment), which haveabnormal gut epithelial structure22 indicatingthe importance of extra-genomic interac-tions on post-natal gut development. Re-introduction of such axenic animals to a‘normal’ enterobacterial background resultsin a sequence of major endogenous and sym-xenobiotic changes in the host that reflect thesuccessive colonization of gut microfloralconstituencies39. Drug metabolic variationbetween human populations can be attrib-uted to genetic polymorphisms in drug-metabolizing enzymes, and it has been shownthat variants of CYP2D6 exist in west Africanpopulations38, as well as variants of CYP2D6,CYP2C19 and CYP2E1 among Australian
NATURE REVIEWS | DRUG DISCOVERY VOLUME 2 | AUGUST 2003 | 671
Gut microflora'The Microbiolome'
Entero-parasite/pathogenpresence and composition
Gut structure
Drugs and drug metabolism
Dietary compositionand nutrient bioavailability
Figure 3 | Possible interactions between diet, gut microfloral composition, parasites and gutstructure and the influence of drugs. There is an interdependence of the biological components(mammal, microbe and pathogen) that can be influenced by the presence of drugs which in turn mayinfluence the metabolism of drugs entering the system at later times.

672 | AUGUST 2003 | VOLUME 2 www.nature.com/reviews/drugdisc
P E R S P E C T I V E S
toxicological studies between species. However,as analytical technology improves, many morelow-level metabolites can be detected and wehave termed this MICROMETABOLISM43.
Chemical and enzymatic transformationNot all cellular transformations of smallmolecules require enzymes. There are manyexamples of these so-called METABONATES,which are well-known in drug degradationstudies, for example, conversion of penicillinsto penicilloic acids via β-lactam ring opening,and these reactions can generate toxicologi-cally or allergenically active species44,45. Animportant example of a metabonate from anendogenous source is acetone, a major ketonebody, which is formed from the spontaneousdecarboxylation of acetoacetic acid that accu-mulates in prolonged fasting or in diabetesmellitus (acetoacetic acid can reach millimolarconcentrations in plasma46). Any compoundof endogenous or exogenous origin that con-tains a carboxylate group can, in principle,undergo enzyme-dependent ester glu-curonidation (via UDP-glucuronosyl trans-ferase) and these glucuronides readily undergofacile internal rearrangement reactions47 thatproduce up to eight positional isomers andanomers48,49. The reactivity of ester glu-curonides is a function of the molecularphysico-chemical properties of the drug50,and subsequent reactivity of the transacylatedglucuronides towards macromolecules mightbe the basis of certain immunological andtoxicological interactions51, although thisremains speculative. Reactive ester glu-curonides can also be formed from endoge-nous metabolites with carboxylate groups,including bile acids. Carbamate formation isanother purely chemical transformation thatoccurs via the reaction of bicarbonate with anamino-containing compound (via a nucleo-philic attack of the amine on the δ-positivecarbon centre52) under slightly alkaline condi-tions. This transforms a zwitterion (for exam-ple, a free amino acid) into a di-anion, therebychanging its physico-chemical interactionswith proteins and other binding sites. Overall,the widespread occurrence of these facile (butconditional) reactions further detract fromthe determinacy of some metabolic processesin complex organisms, as such metabonatescan also influence fluxes through enzyme-controlled pathways in unpredictable ways.
Macro and micrometabolismConcepts such as phase I and phase IImetabolism, and the basic idea of the meta-bolic pathway, have been valuable as theyprovide simplified maps through metabolicspace. However, these reactions need not be
a b
Sourcec
Introduction of sym- ortrans-xenobiotics changesoutcome probability
A polymorphic variationor knockout at this pointchanges outcome probability
Macrometabolites(from high-probabilitytransformation)
Micrometabolite(normally low probability)
Tim
e
Figure 4 | The dynamic Pachinko model of metabolism. a | A typical Pachinko (Japanese pinball)machine, in which the outcome is determined by the probabilistic flow and collisions between the steel ballsand pins. b | Pachinko machines and players in situ. The machines are identical on a short timescale, but theoutcomes are not. c | Pachinko model diagram of xenobiotic metabolism and interactions with endogenous,sym-endogenous, sym-xenobiotic and trans-xenobiotic elements. In this diagram the pins represent keymetabolizing enzymes or transporter molecules, some of which are arranged in sequence to indicateprobable pathways a hole represents an exit point from the system of an excreted metabolite. Somepathways through the machine are more probable than others, dictated by a combination of the chemistryand enzyme-substrate interactions. A mutation or single-nucleotide polymorphism (SNP) variation at anypoint is the equivalent of moving or changing the size of a pin that alters the probabilities of routes through themachine (metabolic fate). The outcome is conditional on the sequence and sites of the previous metabolicevents. Highly probable events lead to macro-metabolites (the most familiar in drug metabolism), the numberand type of these being highly compound specific. Low-probability events lead to the formation ofmicrometabolites. The activity of the system is influenced by the presence numbers and types of othermetabolites flowing through the system, which can be of exogenous origin, for example, sym-xenobiotics —the equivalent of running other types of ball size and shape variation through the Pachinko machine at thesame time as the ‘normal’ balls. This will result in a probabilistic interaction between the species and couldlead to blocking some pathways normally used in drug metabolism. If this results in an increased probabilityof a micrometabolite being formed (increase in concentration), and if the compound is a toxic species, thiscould result in idiosyncratic toxicity of the drug in individuals with unusual combinations of SNPs or multiplecopies of particular genes leading to higher levels of protein/enzyme production and/or endogenous orsym-endogenous pathway activity.
P E R S P E C T I V E S
basis that is determined by molecularphysico-chemical parameters which act atevery recognition, transport and reactionpoint in the system. However, it is not possibleto predict minor metabolites with thisapproach because they can be very numerousand are often produced by a number of con-ditional sequential reactions. Micrometa-bolic diversity in xenobiotic metabolism isexemplified by studies on model compound4-bromoaniline (4BA) which should, in‘classical’ models, have a simple metabolicfate, and, indeed, most of the 4BA dose istransformed to a few major metabolites.However, examination with state-of-the-artanalytical techniques, including directly cou-pled liquid chromatography with inductivelycoupled mass spectrometry and time-of-flightmass spectrometry (LC-ICPMS-TOFMS57)using bromine-detection, reveals >100 com-pound-related 4BA micrometabolites ofremarkably diverse structure showing multi-ple phase I and phase II conversions58. It isprobable that most micrometabolites havelittle biological relevance; however, the possi-bility that this ‘combinatorial’ metabolismwill generate some highly potent com-pounds cannot be ignored. The observedpharmacology or toxicology might be attrib-uted to some readily detectable metabolite,although it actually resides in some unde-tected micrometabolite or micrometabolitemixture. The extent to which these will beobserved depends not only on the fact oftheir production, but also on their stabilityand propensity for further biotransforma-tion. So we suggest that for xenobiotics atleast, regulated metabolic pathways do notreally exist43, and that what we observe is thesum of the conditional probabilities that dif-ferent biotransformations will occur. In thisview, biotransformation will result in theproduction of high-probability (major)metabolites (which might be the sum ofseveral steps), whereas less probable reac-tions will result in minor or micrometabo-lites. The essential logical outcome of thismodel is that if a metabolite is capable ofbeing produced at all, it will be: it is merelythe quantity that is in question. Also, in sucha model of xenobiotic metabolism, it isaxiomatic that it will be impossible to fullycharacterize the metabolic fate of any com-pound. Indeed, a further consequence ofsuch a model is that it is quite likely that even‘un-metabolized’ compounds will undergo awide range of biotransformations, but withsuch a low probability as to be undetectablewith current technology. However, as theirformation is probabilistic, the possibilityremains that these could become significant
ordered and there are many examples ofphase II reactions that are performeddirectly on suitable molecules without theneed for prior phase I metabolism becauseof the presence of a pre-existing functionalgroup that is suitable for conjugation — forexample, sulphation of phenols53. Similarly,phase I metabolites can be excreted in theabsence of phase II metabolism. Further-more, molecules on which phase II metabo-lism has occurred can still undergo furtherphase I and II reactions, and compoundscan go down ‘competing’ pathways withcombinations of phase I or phase II reac-tions, followed by excretion. Sometimes,phase I reactions introduce a functionalgroup to a molecule (which can then beexcreted following conjugation), althoughthe molecule is not conjugated via the newlyintroduced functional group, but to a pre-existing one. This complexity reflects, inpart, the generalized nature of the systemsthat have evolved to eliminate xenobiotics,
and the requirement that such enzyme sys-tems be broad in their substrate specificities.This is in contrast to most of the systemsevolved for endogenous compounds,although some variation must be expectedin endogenous metabolic systems, otherwiseevolution would not be possible! There isalso the potential for a xenobiotic to becomeinvolved with enzyme systems whose func-tion is to deal with endogenous compounds.One potential implication of such complex-ity could be that predictive metabolism isnot practicable, but this is a false conclusion.We have shown that the dominant metabolicfate and urinary excretion (for example,glycine versus glucuronide conjugation forbenzoic acids or sulphate versus glucuronideconjugation for phenols) of simple substi-tuted aromatic compounds can be predictedin vivo using computed molecular parametersand chemometric modelling53–56. This in vivopredictivity is possible precisely because thetotal biological system operates on a statistical
NATURE REVIEWS | DRUG DISCOVERY VOLUME 2 | AUGUST 2003 | 673
1.5
1.0
0.5
0.0
–0.5
–1.0
–1.5
–2.0
–2.5
–3.0
–3.5
M3.t[3]
M3.t[3]M3.t[2]
–5.02.0
1.0
0.0
–1.0–2.0
–3.0–4.0
–4.0–3.0
–2.0–1.0
0.01.0
2.03.0
4.0
Nutrition vectorAge vector
Sexual dimorphism vectorOther physiological or disease vectors
Microfloral compositionMetabolic torque (vectorinteraction)
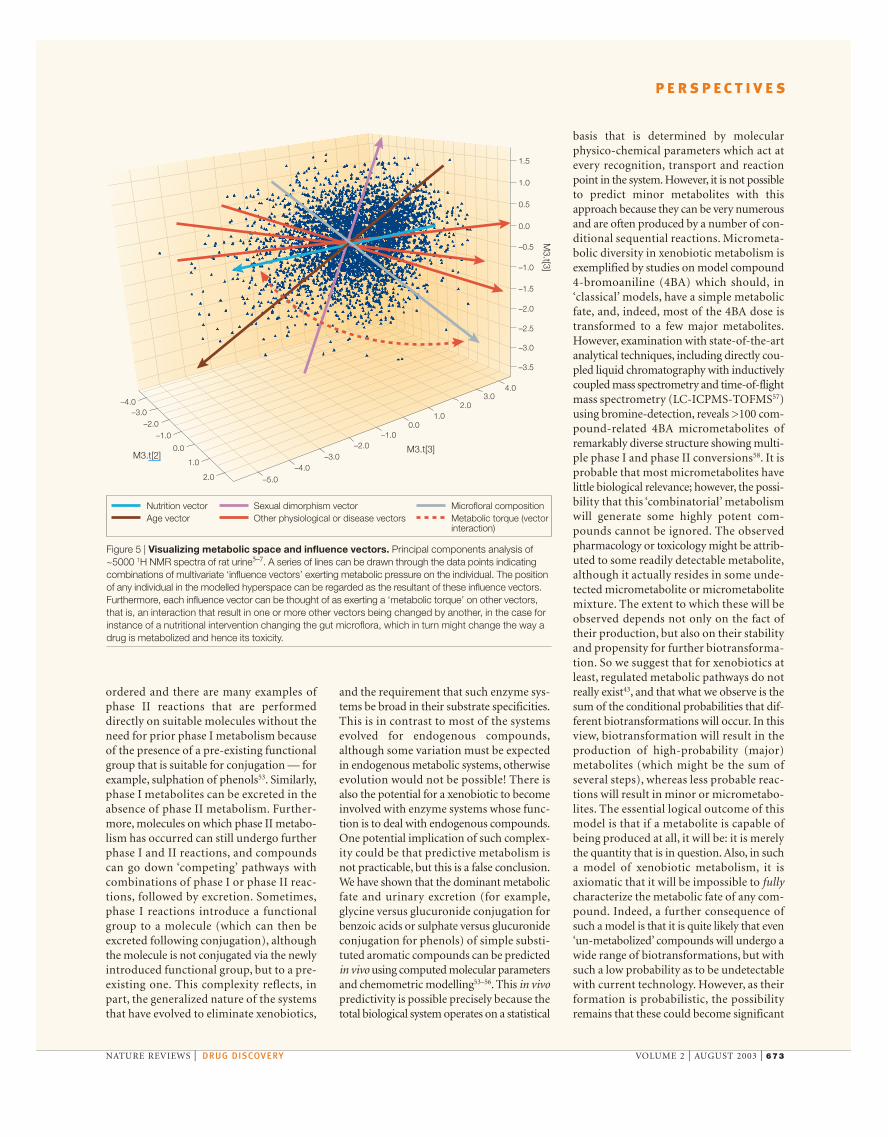
Figure 5 | Visualizing metabolic space and influence vectors. Principal components analysis of~5000 1H NMR spectra of rat urine5–7. A series of lines can be drawn through the data points indicatingcombinations of multivariate ‘influence vectors’ exerting metabolic pressure on the individual. The positionof any individual in the modelled hyperspace can be regarded as the resultant of these influence vectors.Furthermore, each influence vector can be thought of as exerting a ‘metabolic torque’ on other vectors,that is, an interaction that result in one or more other vectors being changed by another, in the case forinstance of a nutritional intervention changing the gut microflora, which in turn might change the way adrug is metabolized and hence its toxicity.

674 | AUGUST 2003 | VOLUME 2 www.nature.com/reviews/drugdisc
P E R S P E C T I V E S
and sized balls (for example, sym-xenobioticor trans-xenobiotic metabolites) will alsoinfluence the path through and hence themetabolic fate of the drug in question. In sucha model it becomes easy to understand howidiosyncratic toxicity could arise in selectedindividuals due to unusual metabolic fates orinteractions of drugs arising because rare, buttoxic, metabolic outcomes become favouredby unusual combinations of genetic andenvironmental factors.
Global systems biologyIf we are to capture the full power of systemsbiology and new genomic tools for drug dis-covery we need to measure and model thewhole system, which includes the environ-mental factors. This is ‘global’ systems biology;simple pathway analysis alone is unlikely tosuffice for explaining many disease processes.Solutions to understanding these metabolicmodelling problems lie in our concepts ofmultivariate analysis and modelling. Currentbioinformatic tools cannot easily incorporatethe rich interactions between genes and dis-ease, between microbes and humans andbetween drugs and metabolism, and henceneed new modelling paradigms. Bioinformatictools have a strong part to play in helping usorganize massive data sets, but by themselvescannot be expected to answer fundamentalquestions on ecological–biological–chemicalinteractions that influence our lives or thatmight affect the way a drug is metabolized oris toxic. There are two potentially usefulapproaches to global metabolic analysis. Thefirst is a mapping strategy in which we try tovisualize an individual occupying a positionin a metabolic (or other ‘-omic’) hyperspace.We can then consider that the position of theindividual is a result of the interactions of aseries of multivariate ‘influence vectors’ thatexert a metabolic pressure on the individual.These pressures could derive from intrinsicgenetic sources or from exogenous factors. Avery simple representation of this is shownin FIG. 5 using a principal components mapthat represents the urinary composition of~5,000 control Sprague–Dawley rats derivedfrom 600 MHz 1H nuclear magnetic reso-nance (NMR) spectra7. A series of hypotheti-cal ‘influence vectors’ are shown superim-posed to indicate possible directions ofmetabolic pressure exerted by several interact-ing macroscopic factors that determine thefinal position in the metabolic hyperspace. Ofcourse, this is a huge simplification as theinfluence vectors can be highly nonlinear, andbecause urine is only one compartment thatcan be analysed. The challenge is to find outthe directions, magnitudes and components
metabolic activation in the liver, transportthrough the blood and further metabolism,in, say, the kidney. In the case of drug metabo-lites, time-displacement of these reactionssignificantly confounds attempts at metabolicmodelling and introduces a series of nonlin-ear convolutions that are hard to predict invivo. Mammalian metabolic transformationscan be considered to be analogous to theoperation of Pachinko machines (Japanesepin-ball, FIG. 4), in which there is a flow ofpinballs (read: drug molecules) through thesystem after introduction (‘dosing’). The des-tination (metabolic fate) of each ball is notdetermined absolutely, but is probabilistic andstrongly influenced by the distribution of pinsand exit holes. The positioning of the pins(enzymes that can transform the com-pounds), and the exact shape and size of theballs (drug properties), determines the routethrough the machine (cell or tissue) andwhich holes in the machine the balls exit(disposition and fate). In the PachinkoMetabolism Model, changing the number ofpins, and their sizes and positions, is analo-gous to changing the presence or activity ofan enzyme; such variation in the presence oractivity of the enzyme could be the result ofgenetic influences of the host and single-nucleotide polymorphisms (SNPs). Putting inother sources of different shaped (eccentric)
under certain circumstances and give rise toproblems of idiosyncratic toxicity in certainindividuals.
Probabilistic (Pachinko) metabolism In cells, all molecules interact with the cellularcomponents in a way that is dependent ontheir molecular physico-chemical propertiesand their probabilistic collisions with poten-tially reactive moieties, the outcomes of whichare determined by the laws of thermodynam-ics and kinetics. However, essential metabolicchemistry has been tuned by evolution toprocess endogenous metabolites in carefullydefined ways under the influence of geneti-cally encoded enzymes and structures, butwith the extra capacity to deal with toxins inthe diet using more generalized strategies.Many drug metabolic reactions occurring in aseries can be considered to be based on condi-tional probabilities (that is, A must go to Bbefore B can go to C) and, therefore, followBayesian statistical rules. For endogenous sys-tems, natural selection has ensured that theprobability of the ‘correct’ reaction occurringat each point in a ‘pathway’ has been opti-mized to maximize functional efficiency, andalthough the degrees of freedom are con-strained they are not completely rigid.Displacement of the ‘sequential’ processes inspace and time occur in the case of initial
Glossary
ENDOGENOUS METABOLISM
Metabolic conversions under direct host cellgenome/proteome control or under mitochondrial control, for example, all major energy-generating pathways and biosynthetic routes.
METABOLOMICS
The measurement of metabolite concentrations andfluxes and secretions in cells and tissues in which thereis a direct connection between the genetic activity(gene expression), protein activity (proteome) and themetabolic activity itself.
METABOLOME
The full set of metabolites within, or that can besecreted by, a given cell type of tissue.
METABONOMICS
The quantitative measurement of the multivariatemetabolic responses of multicellular systems to pathophysiological stimuli or genetic modification5,6.An approach to understanding global metabolic regulation of organism and its commensal and symbiotic partners.
METABONOME
The sum of the cellular metabolomes in a multi-cellular organism and their interaction components plus the products of facile chemical trans-formations and extra-genomically generated metabolites.
METABONATE
A compound that is produced by a facile chemicalrearrangement or reaction within an organism, that can be excreted or further metabolized.
SYM-ENDOGENOUS
Processes or compounds that are essential to host biologi-cal function and which can be metabolized or furtherutilized by host, but for which there is no biosyntheticcapability in the host genome, for example, vitaminsand essential amino acids.
SYM-XENOBIOTIC
Metabolites or processes involving co-metabolism bytwo or more organisms that are commensal or symbi-otic (for example, bile acid metabolism). Not necessarilyessential to the host, but can influence endogenous orother xenobiotic metabolic processes.
TRANS-XENOBIOTIC
Compounds of extra-genomic or chemical origin butwhich are metabolically converted to endogenousspecies or metabolites that can be utilized directly inendogenous processes, for example, ethanol.
XENOBIOTIC
A compound that is foreign to the endogenous processand has no intrinsic biological function but which canhave major effects on endogenous pathway control andcan be extensively metabolized by complexes of hostenzymic systems that have collectively relatively lowsubstrate specificities.

P E R S P E C T I V E S
18. Kivistö, K. T. et al. Analysis of CYP2D6 expression inhuman lung: implications for the association betweenCYP2D6 activity and susceptibility to lung cancer.Pharmacogenetics 7, 295–302 (1997).
19. Griese, E. U., Asante-Poku, S., Ofori-Adjei, D., Mikus, G.& Eichelbaum, M. Analysis of the CYP2D6 genemutations and their consequences for enzyme function ina west African population. Pharmacogenetics 9,715–723 (1999).
20. Griese, E. U. et al. Allele and genotype frequencies ofpolymorphic cytochromes P4502D6, 2C19 and 2E1 inaborigines from Western Australia. Pharmacogenetics11, 69–76 (2001).
21. Gilmore, M. S. & Ferretti, J. J. The thin line between gutcommensal and pathogen. Science 299, 1999–2002(2003).
22. Tannock, G. W. Normal Microflora (Chapman and Hall,1995).
23. Xu, J. et al. A genomic view of the human–Bacteroidesthetaiotaomicron symbiosis. Science 299, 2074–2076(2003).
24. Beale, B. Probiotics: Their tiny worlds are under scrutiny.The Scientist 16, 20–22 (2002).
25. Kirjavainen, P. V. et al. Aberrant composition of gutmicroflora of allergic infants: a target of bifidobacterialtherapy at weaning. Gut 51, 51–55 (2002).
26. Muller, M. & Kersten, S. Nutrigenomics: Goals andStrategies. Nature Gen. 4, 315–322 (2003).
27. Willet, W. C. Balancing life-style and genomics researchfor disease prevention. Science 296, 695–698 (2002).
28. Ghauri, F., McLean, A., Beales, D., Wilson, I. D. &Nicholson, J. K. Induction of 5-oxoprolinuria in the ratfollowing chronic feeding with N-acetyl 4-aminophenol(paracetamol). Biochem. Pharmacol. 46, 953–957(1993).
29. Nyhan, W. L. & Ozand, P. T. Atlas of Metabolic Diseases(Chapman and Hall, 1998).
30. Lof, A. et al. Relationship between uptake and eliminationof toluene and debrisoquine hydroxylation polymorphism.Clin. Pharmacol. Ther. 47, 412–417 (1990).
31. Cok, I., Dagdelen, A. & Gokce, E. Determination ofurinary hippuratic acid and O-cresol levels as biologicalindicators of toluene exposure in shoe-workers and gluresniffers. Biomarkers 8, 119–127 (2003).
32. Phipps, A. N., Stewart, J., Wright, B. & Wilson, I. D. Effectof diet on the urinary excretion of hippuric acid and otherdietary derived aromatics in the rat. Xenobiotica 28,527–537 (1998).
33. Gavaghan, C. L. et al. HPLC-NMR spectroscopic andchemometric studies on metabolic variation in SpragueDawley rats. Anal. Biochem. 291, 245–252 (2001).
34. Murphy, G. M. The Bile Acids Vol. 4 (eds. Katchevsky, D.K. & Nair, P. P.) (Plenum, 1988).
35. Setchell, K. D., Harrison, D. L., Gilbert, J. M. & Mupthy, G. M. Serum unconjugated bile acids: qualitativeand quantitative profiles in ileal resection and bacterialovergrowth. Clin. Chim. Acta 152, 297–306 (1985).
36. Yoneda, M. et al. The biotransformed metabolite profilesin blood after intravenous administration of dehydrocholicacid. Am. J. Gastroenterol. 84, 290–295 (1989).
37. Einarsson, K., Nilsell, K. & Bjorkhem, I. Increasedoxidoreduction of deoxycholic acid incholecystectomised patients. Gut 30, 1275–1278 (1989).
38. Howard, P. J. & Murphy, G. M. Bile physiology: theoryand practise. Curr. Opin. Gastroenterol. 6, 657–667(1990).
39. Nicholls, A. N., Mortishire–Smith, R. & Nicholson, J. K.Metabonomic investigations into the acclimatisation ofaxenic rats to a normal gut microflora. Chem. Res.Toxicol. (in the press).
40. Krebs, H. A. & Perkins, J. R. The physiological role of liveralcohol dehydrogenase.Biochem. J. 118, 635–644(1970).
41. Albro, P. W. & Lavenhar, S. R. Metabolism of di (2-ethyl-hexyl)phthalate. Drug Metab. Rev. 21, 13–34 (1989).
42. Astill, B. D. Metabolism of DEHP: Effects of prefeedingand dose variation, and comparative studies in rodentsand the cynomolgus monkey (CMA Studies). DrugMetab. Rev. 21, 35–53(1989).
43. Wilson, I. D. & Nicholson, J. K. Do metabolic pathwaysfor xenobiotics really exist? Xenobiotica (in the press).
44. Schwartz, M. A. Chemical aspects of penicillin allergy.J. Pharm. Sci. 58, 643–661 (1969).
45. Connor, S. C., Everett, J., Jennings, K. R., Woodnut, G.& Nicholson, J. K. High-resolution 1H NMR spectroscopicstudies of the metabolism and excretion of ampicillin andamoxycillin. J. Pharm. Pharmacol. 46, 128–134 (1994).
46. Nicholson, J. K. et al. Proton NMR studies of serum,plasma and urine from fasting normal, and diabeticsubjects. Biochem. J. 217, 365–375 (1984).
of these ‘vectors’ for each type of disease orphysiological condition under study. This willlead to combinations of biomarkers frommany pathways that are altered in each condi-tion and which probably originate in manydifferent perturbed sites in the body. Theseare useful because they can act both as diag-nostic parameters and also as metrics of effi-cacy for treatments. Mechanistic informationcan be gleaned from a consideration of thepossible pathways (given that our knowledgeof these is far from complete) and the waysthat they are collectively changed. Detailedpathway modelling can only be achievedusing quantitative information derived fromclean kinetic data, which would require sometype of labelling.
Another area for future research (suggestedby Professor John Lindon at Imperial College)is to take a Maximum Entropy Method(MEM)59 approach, which is often used foroptimizing spectral processing60 but whichcan be applied to many other types of prob-lems. An MEM solution would involve usingthe minimum imposed rules or structure toaccount for a given set of metabolic observa-tions, and building a probabilistic system onthe basis of prior knowledge and outcomes byusing a procedure which is the reverse of thatnormally used in pathway analysis. In thistype of model, we would take the observeddata and work forward to find the best modelthat is consistent both with those data (giventhe analytical error) and prior knowledge.This would not necessarily result in anordered sequence of metabolic conversions(pathways), but would relate metabolic para-meters and disease- or toxin-induced move-ments to each other in a probabilistic way.The advantage of this is that it is not necessaryto ascribe every parameter or component to apathway in a particular cell type, but insteadto globally model the important changes for aparticular disease or toxic process. This wouldallow the interventions of lifestyle change ordrug treatment to be evaluated in terms ofbulk metabolic movement of the integratedsystem into ‘beneficial’ or ‘adverse’ hyper-spaces, and therefore enable the creation ofnew metrics of efficacy and/or toxicity on thebasis of probabilities. These models wouldallow the endogenous and xenobiotic compo-nents to be explored together to probe drugtoxicity interactions. If idiosyncratic toxicity isreally idiosyncratic it will not be possible topredict. However, if we assume that it is dueto an ‘unfortunate’ combination of measur-able genetic and environmental factors, thenby use of a broader modelling concept itmight be possible to understand why idiosyn-cratic toxicity has occurred in particular cases
and to find the relationships between gene,environment and metabolism that has led tothis outcome. Although it might not be pos-sible to predict the unpredictable, we mightbe able to predict the likelihood of unpre-dictability. Specifically, we might be able topredict under what circumstances, and withwhich compound classes, unpredictabilitycould cause particular problems of toxicity,or indeed lack of efficacy due to inappropri-ate metabolism. Our ability to do this in thefuture will depend on the construction ofnew modelling paradigms that are not lim-ited by the horizons of metabolic knowledgeof the last century!
*Biological Chemistry, Biomedical SciencesDivision, Faculty of Medicine, Imperial College
London, Sir Alexander Fleming Building,South Kensington, London, SW7 2AZ, UK.
‡Department of Drug Metabolism andPharmacokinetics, AstraZeneca, Mereside,
Alderley Park, Macclesfield, Cheshire SK10 4TG,UK. Correspondence to J. K. N.
e-mail: [email protected]
doi:10.1038/nrd1157
1. Hood, L. & Galas, D. The digital code of DNA. Nature421, 444–448 (2003).
2. Smith, L. L. Key challenges for toxicologists in the 21stCentury. Trend. Pharm. Sci. 22, 281–285 (2001).
3. Raamsdonk, L. M. et al. A functional genomics strategy that uses metabolome data to reveal thephenotype of silent mutations. Nature Biotech. 19,45–50 (2001).
4. Gygi, S. P., Rochon, Y., Franza, B. R. & Aebersold, R.Correlation between protein and mRNA abundance inyeast. Mol. Cell. Biol. 19, 1720–1730 (1999).
5. Nicholson, J. K., Lindon, J. C. & Holmes, E.‘Metabonomics’: understanding the metabolic responsesof living systems to pathophysiological stimuli viamultivariate statistical analysis of biological NMR data.Xenobiotica 29, 1181–1189 (1999).
6. Nicholson, J. K., Connelly, J., Lindon, J. C. & Holmes, E.Metabonomics: a platform for studying drug toxicity andgene function. Nature Rev. Drug Discov. 1, 153–161(2002).
7. Lindon, J. C., Nicholson, J. K., Holmes, E. & Everett, J. R.Metabonomics: Metabolic processes studied by NMRspectroscopy of biofluids. Concepts Magn. Reson. 12,289–320 (2000).
8. Brindle, J. T. et al. Rapid and non-invasive diagnosis ofthe presence of coronary heart disease using 1H NMR-based metabonomics. Nature Med. 8, 1439–1444(2002).
9. KEGG: Kyoto Encyclopaedia of genes and genomes.Release 26. April 2003.
10. Kacser, H. Recent developments beyond metaboliccontrol analysis. Biochem. Soc. Trans. 23, 387–391(1973).
11. Kacser, H. & Burns, J. A. in Rate Control of Biologicalprocesses. Symposium of the Society of ExperimentalBiology 27 (ed. Davies, D. D.) 65–104 (Cambridge Univ.Press, 1973).
12. Meyer, U. A. & Zanger, U. M. Molecular mechanisms ofgenetic polymorphisms of drug metabolism. Annu. Rev.Pharmacol. Toxicol. 37, 269–296 (1997).
13. Srivastava, P. Drug metabolism and individualizedmedicine. Curr. Drug Metab. 4, 33–44 (2003).
14. Ingelman-Sundberg, M. Polymorphism of cytochromeP450 and xenobiotic toxicity. Toxicology 181, 447–452(2002).
15. Eichelbaum, M. & Burk, O. CYP3A genetics in drugmetabolism. Nature Med. 7, 285–288 (2001).
16. Eiselt, R. et al. Identification and functionalcharacterization of eight CYP3A4 protein variants.Pharmacogenetics 11, 447–458 (2001).
17. Scott, R. J. et al. Association of extracolonicmanifestations of familial adenomatous polyposis withacetylation phenotype in a large FAP kindred. Eur. J.Hum. Genet. 5, 43–49 (1997).
NATURE REVIEWS | DRUG DISCOVERY VOLUME 2 | AUGUST 2003 | 675

676 | AUGUST 2003 | VOLUME 2 www.nature.com/reviews/drugdisc
P E R S P E C T I V E S
drug metabolites from HPLC–NMR studies through theuse of quantified maximum entropy processing of NMRspectra. J. Pharm Biomed. Anal. 12, 419–424 (1994).
AcknowledgementsWe thank the BBSRC, EPSRC, The Wellcome Trust and NIH andthe COMET consortium for funding this and related work. We alsothank Professors John Lindon, Paul Elliot and James Scott, FRS,Drs Elaine Holmes, Paul Carmichael, George Tranter, MaryBollard, Hector Keun, Olaf Boeckoenert, Tim Ebbels, Henrik Anttiand Steve Mitchell (Imperial College), Dr Istvan Peltzer, Dr YueurgUtzinger and Professor Burt Singer (Princeton University),Professor Jeremy Everett (Pfizer Global Research, UK) and DrFelicity Nicholson for their helpful comments and discussion onthis work and related subjects.
Online links
DATABASESThe following terms in this article are linked online to:LocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/CYP2C19 | CYP2D6 | CYP2E1
FURTHER INFORMATIONImperial College Division of Biomedical Science:http://www.med.ic.ac.uk/divisions/1/home.aspImperial College Section of Biological Chemistry:http://wwwfom.sk.med.ic.ac.uk/med/about/divisions/bms/biolchem/default.htmlAccess to this interactive links box is free online.
54. Ghauri, F. Y. K. et al. Quantitative structure metabolismrelationships for substituted benzoic acids in the rat usingNMR spectroscopy, computational chemistry and patternrecognition methods. Biochem. Pharmacol. 44,1935–1946(1992).
55. Cupid, B. C., Bedell, C. R., Lindon, J. C., Wilson, I. D. &Nicholson, J. K. Quantitative structure metabolismrelationships for substituted benzoic acids in the rabbit:prediction of urinary excretion of glycine and glucuronideconjugates. Xenobiotica 26, 157–176(1996).
56. Scarfe, G. B., Wilson, I. D., Lindon, J. C., Holmes, E. &Nicholson, J. K. Determination of structure–metabolismrelationships of substituted anilines for prediction of N-acetylation and N-oxanillic acid formation. Xenobiotica32, 267–277(2002).
57. Nicholson, J. K. et al. High-performance liquidchromatography coupled to inductively coupled plasmamass spectrometry (HPLC-ICP-MS) and orthogonalacceleration time-of-flight mass spectrometry (oa-TOF-MS) for the simultaneous analysis and identification of themetabolites of 2-bromo-4-trifluoromethylacetaniline in raturine. Anal. Chem. 73, 1491–1494 (2001).
58. Major, H., Castro-Perez, J., Nicholson, J. K. & Wilson, I. D.Characterisation of putative pentose-containing con-jugates as minor metabolites of 4-bromoaniline presentingthe urine of rats following intraperitoneal administration.Rap. Comm. in Mass Spectrom. 17, 76–80 (2003).
59. Skilling, J. in Maximum Entropy and Bayesian Methods(ed. Paul Fougere) 267–273 (Kluwer, Dordrecht, 1990).
60. Seddon, M. J., Spraul, M., Wilson, I. D., Nicholson, J. K. &Lindon, J. C. Improvement in the characterization of minor
47. Caldwell, J., Hutt, A. J., Marsh, M. V. & Sinclair, K. in DrugMetabolite Isolation and Determination (eds Reid, E. &Leppard, J. P.) 161–179 (Plenum, New York, 1983).
48. Corcoran, O., Mortensen, R. W., Hansen, S. H., Troke, J.& Nicholson, J. K. HPLC/1H NMR spectroscopic studiesof the reactive α-1-O-acyl isomer formed during acylmigration of S-naproxen β-1-O-acyl glucuronide. Chem.Res. Toxicol. 14, 1363–1370 (2001).
49. Mortensen, R. W. et al. S-naproxen-β-1-O-acylglucuronide degradation kinetic studies by stopped-flowHPLC-1H NMR and HPLC-UV. Drug Metab. Dispos. 29,375–380 (2001).
50. Nicholls, A. W. et al. NMR spectroscopic and theoreticalchemistry studies on the internal acyl migration reactionsof the 1-O-acyl-β-D-glucopyranuronate conjugates of 2-,3- and 4-trifluoromethylbenzoic acids. Chem. Res.Toxicol. 9, 1414–1424 (1996).
51. Spahn-Langguth, H. & Benet, L. Z. Acyl glucuronidesrevisited: is the glucuronidation process a toxification aswell as a detoxification mechanism? Drug Metab. Rev.24, 5–48 (1992).
52. Anthony, M., McDowell, P., Holmes, E., Gray, T. &Nicholson, J. K. 1H NMR spectroscopic studies on thereactions of haloalkylamines with bicarbonate ions:Formation of N-carbamates and 2-oxazolidone in cellculture media and blood plasma. Chem. Res. Toxicol. 8,1046–1053 (1995).
53. Holmes, E. et al. Quantitative structure metabolismrelationships and the prediction of phase II conjugationreactions of substituted phenols in the rat. Xenobiotica25,1269–1282(1995).

O N L I N E
Kubinyi
LocusLink:http://www.ncbi.nlm.nih.gov/LocusLink/
BCRhttp://www.ncbi.nlm.nih.gov/LocusLink/LocRpt.cgi?l=613
ABLhttp://www.ncbi.nlm.nih.gov/LocusLink/LocRpt.cgi?l=25
Cambridge Crystallographic Data Centre:www.ccdc.cam.ac.uk/prods/isostar/
Hugo Kubinyi’s website:http://home.t-online.de/home/kubinyi
Biography
Hugo Kubinyi studied chemistry in Vienna, Austria. After his PhD thesisat the Max Planck Institute of Biochemistry in Munich, he continued asa postdoc at the German Cancer Research Centre in Heidelberg. In 1966he joined Knoll AG, later a subsidiary of BASF AG, and in 1985 hemoved to BASF AG. Since 1987, until his retirement in summer 2001, hewas responsible for the Molecular Modelling, X-ray Crystallography andDrug Design group of BASF, since early 1998 also for CombinatorialChemistry in the Life Sciences. He is Professor of PharmaceuticalChemistry at the University of Heidelberg, former Chair of The QSARand Modelling Society, and IUPAC Fellow. His scientific work hasresulted five books on QSAR, 3-D QSAR, and Drug Design (theGerman book Wirkstoffdesign received the 1999 Book Award of the FCI,Association of Chemical Industry) and about 90 publications. He is amember of several Scientific Advisory Boards, co-editor of the Wiley-VCH book series Methods and Principles in Medicinal Chemistry, andmember of the Editorial Boards of several scientific journals.

O N L I N E
Nicholson
LocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/
CYP2C19http://www.ncbi.nlm.nih.gov/LocusLink/LocRpt.cgi?l=1557
CYP2D6http://www.ncbi.nlm.nih.gov/LocusLink/LocRpt.cgi?l=1565
CYP2E1http://www.ncbi.nlm.nih.gov/LocusLink/LocRpt.cgi?l=1571
Professor Jeremy K. Nicholson, BSc PhD C Biol FI Biol FRSA FRC Path CChem FRSC, obtained his BSc from Liverpool University (1977) and hisPhD from London University (1980) working on the application of ana-lytical electron microscopy and energy dispersive X-ray microanalysis ininorganic biochemistry and molecular toxicology. He was appointedLecturer in Chemistry (Birkbeck College, London University, 1981–83)and Lecturer in Experimental Pathology at The London School ofPharmacy (1983–85) returning to Birkbeck as a Senior Lecturer inChemistry, then Reader (1989) and Professor of Biological Chemistry(1992). Since 1998 he has been Professor and Head of BiologicalChemistry at Imperial College, London University and director of theCentre for Biological NMR Spectroscopy. Professor Nicholson is theauthor of more than 400 scientific papers and articles on the develop-ment and application of novel spectroscopic and chemometricapproaches to the investigation of disease processes. This work has beenrecognized by the award of international prizes including: The RoyalSociety of Chemistry (SAC) Silver Medal for Analytical Chemistry(1992); The Royal Society of Chemistry (SAC) Gold Medal for AnalyticalChemistry (1997) for work on NMR spectroscopy of biofluids and thedevelopment of Metabonomics as an investigative tool for studying dis-ease processes; The Chromatographic Society Jubilee Medal (1994) forwork in the development and application of directly coupled chromato-graphic–NMR methods for metabolic analysis; The Pfizer Prize inChemical and Medicinal Technologies (2002) for the development ofNMR-based metabonomics as a toxicological and clinical diagnostic tool.He is a member of the Editorial Advisory Boards of Chemical Researchin Toxicology, The Journal of Proteome Research, Biomarkers and TheJournal of Pharmaceutical and Biomedical Analysis. Professor Nicholsonis a Fellow of the Royal Society of Chemistry, The Royal Society ofMedicine, The Royal College of Pathologists and a Fellow of the Instituteof Biology and is a founder Director and Chief Scientific Officer ofMetabometrix, Ltd, an IC spin-off company specializing in advancedmetabolic profiling and diagnostics for toxicology, human disease, func-tional genomics and phenotyping.
Professor Ian Wilson BSc, MSc, PhD, DSc, CChem, FRSC, EurChem,FChromSoc, FRES is a Principal Scientist in the Department of DrugMetabolism and Pharmacokinetics at the AstraZeneca research site atAlderley Park. He trained as a biochemist at the University ofManchester Institute of Science and Technology, from where he receivedboth BSc (1974) and MSc (1975) degrees. Subsequently, he obtained aPhD (1978) and, more recently, a DSc (1998) from the University ofKeele. He is author or co-author of 300 papers and reviews, and hasreceived a number of awards in separations and analytical science fromthe Royal Society of Chemistry including the SAC Silver Medal forAnalytical Chemistry (1990); the Analytical Separation Methods Medal(1996); and the Analysis and Instrumentation Medal (2002). He has alsorelieved the Jubilee Medal of the Chromatographic Society (1994) andgave the first Desty Memorial lecture for Innovation in SeparationScience (1996). As well as being on the editorial boards of numerousjournals he is an editor of the journals of Planar Chromatography andPharmaceutical and Biomedical Analysis and Editor in Chief of theEncyclopaedia of Separation Science. He is currently a visiting Professorat the Universities of Keele, York, UMIST and Imperial College. Hisresearch is presently centred on the development of hyphenated tech-niques in chromatography and their application to problems in drugmetabolism and metabonomics.




















