
Type I collagen degradation by mouse calvarial osteoblasts stimulated with 1,25-dihydroxyvitamin...
8
Biochimica et Biophysica Acta, 1014(1989) 125-132 125 Elsevier BBAMCR 12582 Type I collagen degradation by mouse calvarial osteoblasts stimulated with 1,25-dihydroxy tamin D-3: evidence for a plasminogen-plasmin-metalloproteinase activation cascade Brian M. Thomson 1, Susan J. Atkinson 1, Anne M. McGarrity 1, Rosalind M. Hembry John J. Reynolds i and Murray C. Meikle 1,2 s Cell Physiolo~ Department, Strangeways Research Laboratory, Worts Causeway, Cambridge and 2 Department of Orthodontics, Eastman Dental Hospital and Institute of Dental Surgery, University of London, London ( U.K.) (Received21 April 1989) 1 9 Key words: Osteoblasts;Collagen degradation; 1,25-Dihydroxyvitamin D-3; Plasminogen activator; Collagenase; Metalloproteinase; Tissueinhibitor of metalloproteinases; (Mouse) To understand the mechanisms regulating esteoid removal by osteoblasts, mouse calvarial osteoblasts were grown on 14C-|abelled type I collagen films and stimulated with 1,25-dihydroxyvitamin D-3 (2.5.10-s M) for 48-72 h. in the presence of $% non-inhibitory rabbit serum this resulted in a 2-3-fold increase in collagen degradation and a dramatic change in osteoblast morphology, when compared with untreated osteoblasts. Collagenolysls was accompanied by increased synthesis and release of latent eollagenase, gelatinase and stromelysin and a concomitant decrease in their specific inhibitor, 2"IMP (tissue inhibitor of metalloproteinases), in serum-free medium, osteohlasts failed to degrade collagen, hut their ability to lyse collagen could be restored by adding plasminogen (5 pg/m|) to the cultures. Plasminogen-dependent collagenolysis was inhibited by human recombinant "lIMP (5 units/ml), demonstrating that plasmin, derived from plasndnogen, activated latent cullagenase and did not itself degrade collagen. Plasminogen activator production was confirmed by culturing osteoblasts on nS|-labeiled fibrin plates. Comparison with urokinase-type and tissue-type plasminogen activator standards suggested that ostcoblast plasminogen activator was predominantly cell-associated and likely to be of the urokinase type. Immunocytochemfistry indicated that osteoblasts also constitu- tively produce plasminogen activator inhibltor-l. These findings provide evidence for the involvement of a plasminogen- plasmin-latent metailoproteinase activation cascade in type I collagen degradation by osteoblasts, and for its regulation by TIMP and plasminogen activator |nhibitor-|. Introduction It has long been known that collagenase production by bone explants cultured with calcium regulating hormones is involved with bone resorption [1-4]. Only recently, however, has the role of collagenase in the bone resorptive process and the controversy surround- ing the cellular source of the enzyme been resolved. Osteoblasts are now established as the cell of orion [5-7]. Since non-resorbing bone surfaces are covered by a layer of nonmineralized osteoid [8-10], and osteo- Abbreviations: 1,25-(OH)2D 3, 1,25-dihydroxyvitamin D-3; TIMP, tissue inhibitor of metalloproteinases; DMEM, Dulbecco'smodifica- tion of Eagle's medium; PBS, phosphate-buffered saline; FCS, fetal calf serum; PAI-1, plasminogen activator inhibitor-1. Correspondence: M.C. Meikle, Strangeways Research Laboratory, Worts Causeway,CambridgeCB1 4RN, U.K. blasts are the target cells for resorptive agents in bone [11-16], it seems likely that osteoblast collagenase might promote bone resorption through osteoid removal (see Ref. 17 for a review). This hypothesis is supported by the observation that isolated osteoclasts in vitro do not resorb bone unless the surface osteoid layer is first removed, either by the action of osteoblasts or by pretreatment with mammalian collagenase [18,19]. To investigate the cellular and molecular basis for osteoid removal we have used model systems in which osteoblasts are cultured on collagea substrates. Re- cently, we reported that mouse calvarial osteoblasts cultured on type I collagen films are stimulated by 1,25-dihydroxyvitamin D-3 (1,25(OH)2D3) to degrade the collagen [20]. Collagenolysis was accompanied by increased secretion of the metalioprcteinases col- lagenase and gelatinase, and by a redaction in their specific inhibitor, TIMP. In this paper we show that osteoblasts do not degrade collagen in the absence of serum and present evidence for the involvement of a 0167-4889/89/$03.50 © 1989 ElsevierSciencePublishersB.V.(BiomedicalDivision)
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Type I collagen degradation by mouse calvarial osteoblasts stimulated with 1,25-dihydroxyvitamin...

Biochimica et Biophysica Acta, 1014 (1989) 125-132 125 Elsevier
BBAMCR 12582
Type I collagen degradation by mouse calvarial osteoblasts stimulated with 1,25-dihydroxy tamin D-3: evidence
for a plasminogen-plasmin-metalloproteinase activation cascade
Brian M. Thomson 1, Susan J. Atkinson 1, Anne M. McGarrity 1, Rosalind M. Hembry John J. Reynolds i and Murray C. Meikle 1,2
s Cell Physiolo~ Department, Strangeways Research Laboratory, Worts Causeway, Cambridge and 2 Department of Orthodontics, Eastman Dental Hospital and Institute of Dental Surgery, University of London, London ( U. K. )
(Received 21 April 1989)
1 9
Key words: Osteoblasts; Collagen degradation; 1,25-Dihydroxyvitamin D-3; Plasminogen activator; Collagenase; Metalloproteinase; Tissue inhibitor of metalloproteinases; (Mouse)
To understand the mechanisms regulating esteoid removal by osteoblasts, mouse calvarial osteoblasts were grown on 14C-|abelled type I collagen films and stimulated with 1,25-dihydroxyvitamin D-3 (2 .5 .10 - s M) for 48-72 h. in the presence of $% non-inhibitory rabbit serum this resulted in a 2-3-fold increase in collagen degradation and a dramatic change in osteoblast morphology, when compared with untreated osteoblasts. Collagenolysls was accompanied by increased synthesis and release of latent eollagenase, gelatinase and stromelysin and a concomitant decrease in their specific inhibitor, 2"IMP (tissue inhibitor of metalloproteinases), in serum-free medium, osteohlasts failed to degrade collagen, hut their ability to lyse collagen could be restored by adding plasminogen (5 p g / m | ) to the cultures. Plasminogen-dependent collagenolysis was inhibited by human recombinant "lIMP (5 uni ts /ml) , demonstrating that plasmin, derived from plasndnogen, activated latent cullagenase and did not itself degrade collagen. Plasminogen activator production was confirmed by culturing osteoblasts on nS|-labeiled fibrin plates. Comparison with urokinase-type and tissue-type plasminogen activator standards suggested that ostcoblast plasminogen activator was predominantly cell-associated and likely to be of the urokinase type. Immunocytochemfistry indicated that osteoblasts also constitu- tively produce plasminogen activator inhibltor-l. These findings provide evidence for the involvement of a plasminogen- plasmin-latent metailoproteinase activation cascade in type I collagen degradation by osteoblasts, and for its regulation by TIMP and plasminogen activator |nhibitor-|.
Introduction
It has long been known that collagenase production by bone explants cultured with calcium regulating hormones is involved with bone resorption [1-4]. Only recently, however, has the role of collagenase in the bone resorptive process and the controversy surround- ing the cellular source of the enzyme been resolved. Osteoblasts are now established as the cell of o r ion [5-7]. Since non-resorbing bone surfaces are covered by a layer of nonmineralized osteoid [8-10], and osteo-
Abbreviations: 1,25-(OH)2D 3, 1,25-dihydroxyvitamin D-3; TIMP, tissue inhibitor of metalloproteinases; DMEM, Dulbecco's modifica- tion of Eagle's medium; PBS, phosphate-buffered saline; FCS, fetal calf serum; PAI-1, plasminogen activator inhibitor-1.
Correspondence: M.C. Meikle, Strangeways Research Laboratory, Worts Causeway, Cambridge CB1 4RN, U.K.
blasts are the target cells for resorptive agents in bone [11-16], it seems likely that osteoblast collagenase might promote bone resorption through osteoid removal (see Ref. 17 for a review). This hypothesis is supported by the observation that isolated osteoclasts in vitro do not resorb bone unless the surface osteoid layer is first removed, either by the action of osteoblasts or by pretreatment with mammalian collagenase [18,19].
To investigate the cellular and molecular basis for osteoid removal we have used model systems in which osteoblasts are cultured on collagea substrates. Re- cently, we reported that mouse calvarial osteoblasts cultured on type I collagen films are stimulated by 1,25-dihydroxyvitamin D-3 (1,25(OH)2D3) to degrade the collagen [20]. Collagenolysis was accompanied by increased secretion of the metalioprcteinases col- lagenase and gelatinase, and by a redaction in their specific inhibitor, TIMP. In this paper we show that osteoblasts do not degrade collagen in the absence of serum and present evidence for the involvement of a
0167-4889/89/$03.50 © 1989 Elsevier Science Publishers B.V. (Biomedical Division)

126
plasminogen-plasmin-latent metalloproteinase activa- tion cascade in the osteoid degradative process.
Materials and Methods
Reagents. Human recombinant TIMP (rTIMP) was supplied by courtesy of Peter Koklitis, Celltech, Slough, Berkshire (spec. act. 4762 units/mg protein as assayed by the collagen fibril assay). 1,25(OH)2D 3 was gener- ously supplied by Dr. Ian Dickson, Department of Medicine, University of Cambridge. Urokinase-type plasminogen activator and tissue-type plasminogen activator standards were obtained from the National Institute for Biological Standards and Control, Hampstead, U.K. The polyclonal rabbit anti-mouse plasminogen activator-1 antibody was generously donated by Professor T.D. Gelehrter, University of Michigan, U.S.A.
Preparation of osteoblasts from neonatal mouse calvariae. Calvariai osteoblasts were prepared and char- acterized as previously described [5]. Briefly, neonatal mouse caivariae (40-50) were dissected free from adher- ent soft tissue, washed in Ca 2+- and Mg2+-free Tyrode's solution (10 min) and sequentially digested with 1 mg/ml trypsin (10 rain), 2 mg/ml dispase (30 rain) and 4 mg/ml collagenase (3 x 30 min). Cells released by the last two collagenase digestions were washed and grown in Dulbeceo's modification of Eagle's medium (DMEM) containing 105 fetal calf serum (FCS; Globepharm, Esher, Surrey) and antibiotics for 5-7 days prior to use. All cultures were maintained at 37°C in a humidified atmosphere of 5~ CO2:955 air.
Preparation of collagen films. Radiolabelled collagen films were prepared as previously described [21]. Aliquots of t4C-acetylated collagen (rat skin type I; 150 ~g in 300 ~tl of 10 mM phosphate buffer (pH 7.4) containing 300 mM NaCl and 0.025 sodium azide) were dispensed into tissue culture wells (Linbro, 16 mm diameter) and dried at 37°C. The collagen was then washed twice with sterile distilled water and once with DMEM.
Preparation of 12~l.labelled fibrin plates. 125I-labelied fibrin plates were prepared by the method of Unkeless et al. [22]. A solution of labelled fibrinogen was pre- pared by diluting unlabelled fibrinogen (Sigma, Poole, Dorset) in water (2.2 mg/ml) and adding sufficient nSl-fibrinogen (Amersham International, Amersham, Buckinghamshire) to give 500000 cpm/ml. The result- ing labelled fibrinogen solution was filter sterilized, dispensed into tissue culture plates (Linbro, 16 nun diameter, 100 ~tl/well) and spread evenly across the bottom of the wells with a plastic pipette. The plates were then air dried at 37°C in a non-humidified incubator. When dry, the fibrinogen was converted to fibrin by adding DMEM (0.5 ml) containing 10~ non- heat-inactivated rabbit serum (SeraLab, Crawley, Sus-
sex) to each well, and incubating the plates for 24 h at 37°C in a humidified incubator. The plates were then washed twice with sterile phosphate-buffered saline (PBS) and stored at - 2 0 °C until required.
Preparation of acid-treated serum and acid-treated plasminogen.depleted serum. To destroy serum inhibitors of neutral proteinases, aliquots (20 ml) of heat-in- activated rabbit serum (Globepharm) were acidified to pH 3.2 with 1 M HCI and incubated for 35 rain at 37°C. The pH was then returned to 7.4 with 1 M NaOH. Plasminogen was subsequently removed from some of the aliquots by lysine Sepharose affinity chro- matography [23,24]. Briefly, a 20 ml lysine Sepharose column was equilibrated with PBS containing 0.02~ NaN 3 and acid-treated rabbit serum (20 ml) was loaded onto the column and circulated for 48 h at room tem- perature (flow rate 10 ml/h). Plasminogen was bound by the column to leave plasminogen-depleted serum (approx. 90% reduction), which was eluted with PBS/0.02~ NaN 3 (35 ml). The eluate was dialyzed against distilled water (2 × 2 1), lyophilized and recon- stituted with water (20 ml). Both acid-treated serum and acid-treated plasminogen-depleted serum were filter sterilized before use.
Culture of osteoblasts on collagen films. Osteoblasts (4.5. 104/well) were settled onto collagen films in 1 ml DMEM plus 10~ FCS, incubated for 16 h at 37 o C and washed with serum-free DMEM. Cells were then cul- tured in either DMEM (1 ml) alone or DMEM (1 ml) supplemented with either plasminogen (5/tg/ml) or 5% non-inhibitory rabbit serum (Burroughs Wellcome, Be- ckenham, Kent). This rabbit serum contained little a 2- macroglobulin or other detectable proteinase inhibitors and was used until supplies were withdrawn from the market: subsequently we have used acid-treated rabbit serum as described above. Either 1,25(OH)2D 3 (final concentration 2.5.10 -s M, added in 10/~1 ethanol), or TIMP (5 U/ml), or 1,25(OH)2D 3 plus TIMP, or 10/ti ethanol alone, were then added to the wells and the cultures maintained at 37°C for 48 h. Corresponding sets of wells without osteoblasts served as cell-free controls. At the end of the culture period, the media were centrifuged (15 rain, 12000 × g) to remove any collagen fibrils and the radioactivity released during collagen degradation quantified by liquid scintillation counting. Residual collagen was digested with bacterial collagenase (50 /tg/ml) and assayed for radioactivity. Collagenolysis was expressed as radioactivity released from the films as a percentage of the total + S.E.
Assays for collagenase, gelatinase, stromelysin and TIMP. Collagenase and TIMP assays were performed as previously described [25]. 1 unit of collagenase de- grades 1 /tg of collagen per min at 35°C. TIMP was assayed by the inhibition of rabbit collagenase; 1 unit is defined as the amount of TIMP which inhibits 2 units of collagenase by 50%. Gelatinase was measured by the

method of Sellers et al. [26]. 1 unit of gelatinase hydro- lyses 1/~g of gelatin per min at 37 o C. Stromelysin was assayed at 37 °C with [14C]casein as substrate, prepared by acetylating casein with [1-14C]acetic anhydride [27], 1 unit of enzyme hydrolyses 1 Fg casein/rain.
Culture of osteoblasts on 1251-fibrin plates. 12Sl-Fibrin plates were thawed, washed with PBS and equilibrated with DMEM. Osteoblasts (4.5- 104/well) were then set- tled onto the wells in DMEM containing 10~ FCS (1 ml) and incubated at 37 o C. After 16 h, the cells were washed with DMEM and cultured in 1 ml of either: (1) DMEM supplemented with 5~ acid-treated rabbit serum; (2) DMEM supplemented with 5~ plasminogen- depleted acid-treated rabbit serum; or (3) DMEM sup- plemented with 5~ plasminogen-depleted acid-treated rabbit serum plus 4.5/Lg/ml plasminogen. Correspond- ing sets of wells without osteoblasts served as cell-free controls. Either 1,25(OH)2D 3 or vehicle was then added and the cultures maintained at 37°C for 24 h. The media were then removed and the radioactivity released into the medium by fibrin degradation quantified by gamma counting. Residual 125I-fibrin was digested with trypsin (100 pg/ml , 16 h) and assayed for radioactivity. Fibrinolysis was expressed as the percentage of the total radioactivity released into the medium + S.E.M.
Quantitation of plasminogen activator by comparison with known plasminogen activator standards. 125I-Fibrin- coated plates were thawed, washed in PBS and equi- librated in DMEM. Osteoblasts (4.104 in DMEM con- taining 10~ FCS; I ml) were then added to the fibrin- coated wells and incubated at 37°C. Similar wells without cells (containing DMEM supplemented with 10~ FCS alone; I ml) served as controls. The wells were washed with serum-free DMEM and then DMEM con- taining 2~ acid-treated rabbit serum and either 1,25(OH)2D 3 (final concentration 2.5.10 -5 M) or vehicle (10 pl ethanol) were added to each well. To prepare a standard curve serial dilutions of urokinase- type or tissue-type plasminogen activator (0 to 0.4 units/ml in DMEM containing 2~ acid-treated rabbit serum) were also added to fibrin-coated wells without osteoblasts. After 24 h at 37 o C, aliquots of the culture medium (200 pl) were removed and assayed for radioac- tivity by gamma counting. Similar aliquots were also added to the 125I-fibrin-coated wells of a 96-well multi- well plate and incubated for a further 24 h alongside serial dilutions of either urokinase-type or tissue-type plasminogen activator and the released 1251 was quanti- fied by gamma counting. Total and soluble plasminogen activator production were then calculated by linear re- gression from the dose-response curves of the plas- minogen activator standards.
Immunolocalization of plasminogen activator inhibitor- 1 (PAl-l). Primary mouse osteoblasts were prepared as above and plated onto glass coverslips in 3 cm tissue culture dishes (Sterilin) at a density of 4.104 cells per
127
dish in 1 ml DMEM with 10~ FCS. The medium was changed on day 1 and the coverslips were used on days 4-7. To block translocation and secretion of secretory proteins, monensin (Sigma, 5 ~M) was added to the culture medium of some coverslips for the final 3 h of the culture period [28]. Additional coverslips were stimulated for 24 h with 1,25(OH)2D 3 prior to fixation. Cells and contact area preparations were obtained as described by PiSll~nen et al. [29]. Briefly, coverslip cul- tures were washed three times with PBS (10 mM NaPO4, 145 mM NaCl, pH 7.4) then fixed in either methanol ( - 2 0 ° C , 20 rain) or paraformaldehyde (4~ w/v in PBS, 20 rain, room temperature). Cells were permeabi- lized with Triton X-100 (0.1~, 5 rain) and washed, then incubated with either rabbit antibody IgG to mouse PAI-1, normal rabbit lgG (50 igg/ml in PBS, 30 min) or PBS alone. After further washing, the coverslips were incubated with fluorescein-conjugated pig IgG anti-rab- bit IgG (Dakopatts a/s, Denmark, 1:200 in PBS, 30 min), then rewashed and mounted onto slides with Citifluor (City University, London). Staining of live cells was done by incubating with either antibody IgG, control lgG or PBS on ice prior to fixation with - 20 ° C methanol. Incubation with the fluorochrome-conjugated second antibody was then carried out at room temper- ature. Cell contact area preparations were made by washing the coverslips three times with PBS at 0 ° C, followed by shaking at 100 rpm in PBS with saponin (Sigma, 0.2%, 0 ° C, 20 min). The coverslips were then intensively pipetted with PBS (at 0 o C) and the material remaining on the coverslips was fixed in 4~ paralormal- dehyde (20 rain, room temperature). Subsequew stain- ing was as described above. The cells were viewed by fluorescence microscopy on a Zeiss Photomicroscope Ill with epifluorescence and standard FITC filters. Photo- graphs were taken on Agfachrome 1000 ASA film up- rated during processing to 2000 ASA.
Osteoblasts were stained for alkaline phosphatase with napthol AS-MX phosphate and fast blue RR (Sigma Diagnostics kit), and counterstained with Mayer's haematoxylin. Photographs were taken on Kodak Pan F film.
Results
Mouse osteoblasts cultured on native type I collagen films in DMEM supplemented with 5% non-inhibitory rabbit serum responded to 1,25(OH)2D 3 with a 2-3-fold increase in collagen degradation over untreated controls (Fig. 1). This was accompanied by enhanced synthesis and secretion of the metalloproteinases collagenase, gelatinase and stromelysin into the culture media, all three enzymes being in the latent form. The results of a typical experiment are shown in Table I. Lysis of the collagen films was visible by phase-contrast microscopy and there was a marked change in osteoblast mor-

128
lOO
75 tt.
g
o O
so
, ,J
o
25
±
I 1 +SUIml TIMP [
i Vehicle 1,2S(OH)2D 3 Vehicle 1.2S(OH)2D 3 Vehicle 1,25(OH)203
5% Non.Inhibitory Serum Iree 5pg/rnl rabbit serum Plasmlnooen
Fig, 1. Degradation of t4C-type I collagen films by mouse osteoblasts stimulated with 1,~(OH)2D 3. Cells were cultured with or without 1.25(OH)2D 3 (2,5.10 -s M) on collagen films for 48 h in either: (1) DMEM supplemented with 55 non-inhibitory rabbit serum; (2) serum-free DMEM; or (3) serum-free DMEM supplemented with plasminogen (5 ttg/ml). The effect of human rTIMP (5 U/no) on collagenolysis in each treatment is also shown. Each bar represents
the mean value+ S.E. of six wells.
phology, the cells adopting a stellate shape as described previously [30,31]. Exogenous human rTIMP reduced basal collagenolysis and abolished 1,25(OH)2D 3- stimulated collagen breakdown, demonstrating that de- gradation was mediated by metalloproteinases (Fig. 1). In contrast, ostcoblasts cultured on collagen films in the
TABLE !
Effect of 1,2$(OH)zD a on rnetalioproteinuse production by mouse osteoblusts cultured on type i collagen films
Mouse ealvarial osteoblasts were obtained by sequential enzymatic digestion [5], cultured on 14C-labelled type I collagen films in DMEM supplemented with 55 non-inhibitory rabbit serum and stimulated with 1,25(OH)2D 3 (2.5.10 -s M) for 72 h as described in the text. Coilagenase and gelatinase activity in culture supernatants were mea- sured by the degradation of native and denatured collagen, respec- tively [25,26[ and strumelysin with casein as substrate [27]. Data are expressed as mean values:t: S.E. of six cultures.
Metalloprot-~m~ ~ 3 s ~ o b l a s t s Osteoblasts + + vehicle 1,25(OH)2D3 (units/no) (units/n0)
Collagenase 0 0.49 + 0.02 Gclatinase 0 1.10 + 0.15 Stmmelysin 0 0.20 + 0.01
absence of serum produced neither basal nor 1,25(OH)2D3-stimulated collagen breakdown, but did do so when plasminogen was added to the cultures (Fig. 1). Plasminogen-dependent collagenolysis was inhibited by human rTIMP, demonstrating that plasmin (from plasminogen) did not degrade the films directly, but activated latent coilagenase. We concluded that matrix degradation by osteoblasts was therefore dependent upon the production of plasminogen activator by the cells.
Although collagenase was present in the culture medium in a latent form in the experiments described above, TIMP-inhibitable collagen degradation in some of the cultures implies that active collagenase was being formed. The amounts of active collagenase were calcu- lated by reference to how much inhibition was brought about by the addition of rTIMP (Table IIA). In serum- free medium (Table IIB) the levels of collagenase and TIMP were at the lower detection limits of both assays as was the case when osteoblasts were cultured in serum-free medium to which plasminogen had been added (Table IIC).
Plasminogen activator production by osteoblasts was confirmed by culturing the cells in multiwell plates coated with t25I-labelled fibrin. Osteoblasts cultured in DMEM supplemented with 5~ acid-treated rabbit serum produced fibrinolysis and this degradation was stimu- lated by 1,25(OH)2D 3 (Fig. 2). DMEM containing plasminogen-depleted acid-treated rabbit serum, how- ever, failed to support fibrinolysis by either unstimu- lated or 1,25(OH)2D3-treated osteoblasts (Fig. 2). Ad- dition of plasminogen to cultures incubated in plas- minogen-depleted acid-treated rabbit serum restored both basal and 1,25(OH)2D3-stimulated fibrinolysis. Comparison of osteoblast-mediated fibrinolysis and secreted plasminogen activator with known concentra- tions of urokinase-type and tissue-type plasminogen activator demonstrated that osteoblast plasminogen activator activity was predominantly cell associated and, therefore, probably of the urokinase-type (Fig. 3). Osteoblast-mediated fibrinolysis was not inhibited by human rTIMP (data not shown).
To obtain additional evidence for the involvement of the plasminogen-plasmin system, mouse osteoblasts (Fig. 4a) were stained immunocytochemically using an antibody to murine PAI-1. Methanol fixation of cells and staining for PAI-1 (Fig. 41)) showed a homogeneous carpet of fluorescence in the cell peripheral area which was interrupted by striae of no staining. There was also intracellular perinuelear staining, suggesting PAI-1 synthesis. Paraformaldehyde fixation at room tempera- ture showed the same staining pattern, but both the striae and the intracellular staining were more clearly defined, the latter locafizing a perinuclear lamella struc- ture-presumably the Golgi apparatus. Paraformal- dehyde fixation at 0°C, as used by P~ll~inen [29],

TABLE lI
Effect of 1,23(OH)2D J on collagenase and TIMP synthesis by mouse osteoblasts cultured on ;¢C.labeiled type I collagen films with different serum additions
Mouse calvarial osteoblasts were obtained as described in Table I and cultured for 48 h on 14C-labelled type I collagen films in either: (a) DMEM supplemented with 5~ non-inhibitory rabbit serum; (B) serum-free DMEM; or (C) serum-free DMEM plus plasminogen (5 /tg/ml). Some cultures were stimulated with 1,25(OH)2 D 3 (2.5.10-s M). Latent collagenase and TIMP activities in culture supernatants were measured by fibril assay [25]° Active collagenase was estimated from the inhibition of collagen degradation produced by the addition of exogenous human rTIMP. Data are expressed as mean values ± S.E. of six cultures.
Treatment Active Latent TIMP collagenase collagenase (units/ml) (units/mi) (units/ml)
(A) Osteoblasts + 5~ non-
inhibitory rabbit serum 0.004 ± 0.002
Osteoblasts + 5~$ non- inhibitory rabbit serum + 1,25(OH) 2 D3 0.028 ± 0.002 *
(e) Osteoblasts in
serum-free DMEM 0 Osteoblasts in
serum-free DMEM + 1,25(OH)2 D 3 0
(O Osteoblasts in
serum-free DMEM + plasminogen 0.029 ± 0.001
Osteoblasts in serum-free DMEM + plasminogen + 1,25(OH) 2 D 3 0.041 ± 0.0 a
0.19±0.02 0 .11±0.~
0.29±0.07 a 0.04±0.05 b
0.01±0.0 0.01±0.01
0.01±0.0 0.02±0.02
0.01 + 0.01 0.02 ± 0.01
0.01 +0.01 0.01 +0.02
a Significantly greater than unstimulated control (P < 0.01). b Significantly less than unstimulated control (P < 0.01).
resulted in substantial loss of cells from the coverslips and a staining pattern similar to that described for cell contact areas below. The monovalent ionophore moaensin has been shown to i~.~erfere with the processing and secretion of proteins from human fibroblasts [30]. When monensin was included in the osteoblast cui'mre medium for 3 h prior to fixation with paraformaldehyde and staining for PAI-1 (Fig. 4d), the perinuclear immunofluorescence became much more prominent and vacuolated, and intracellular cyto- plasmic granules were often visible. The staining pattern at the periphery of the cells was not altered by monen- sin treatment. Cells treated with monensin, but stained with either normal rabbit IgG or PBS in place of the primary antibody, were completely negative (Fig. 4e), as were similar controls for the cells of Figs. 4b and 4c. This intracellular build up of immunofluorescence with
129
c
I
#
25
T
Veh*cle 1,25(OH)2D3 Vehicle
Acid treated rabbit serum
t,25[OH]2D: Veh*cle 1,25(OH)2D 3
Plasminogen- Plasminogen-depleted depleted serum serum + Plasmmogen
Fig. 2. Degradation of 12Sl-fibrin plates by mouse osteoblasts stimu- lated with 1,25(OH)2D 3. Cells were cultured with or without 1,25(OH)2D 3 (2.5.10 -~ M) for 24 h in DMEM supplemented with either: (1) 2% acid-treated rabbit serum; (2) 2~o plasminogen-depleted acid-treated rabbit serum; (3) 2~ plasminogen-depleted acid-treated rabbit serum plus plasminogen (4.5/tg/ml). Each bar represents the
mean value + S.E. of six wells.
2 5 -
20
c~ I 0 15
x ¢E Q.
u3
/ /
/ / / / / / / / / / / / / / / /
NiT o
Vehicle 1.25(OH)2D 3 Vehicle 1,25(OH)2D 3
Total Plasminogen Soluble Plasminogen Activator Activator
Fig. 3. Total and soluble plasminogen activator production by mouse osteoblasts. Fibrin degradation by osteoblasts cultured in 2% acid- treated rabbit serum in the presence or absence of 1,25(OH)eD3 (2.5.10 - s M) was quantified by comparison with known concentra- tions of urokinase-type plasminogen activator and tissue-type plas- minogen activator. Each bar represents the mean value+S.E, of
six wells.
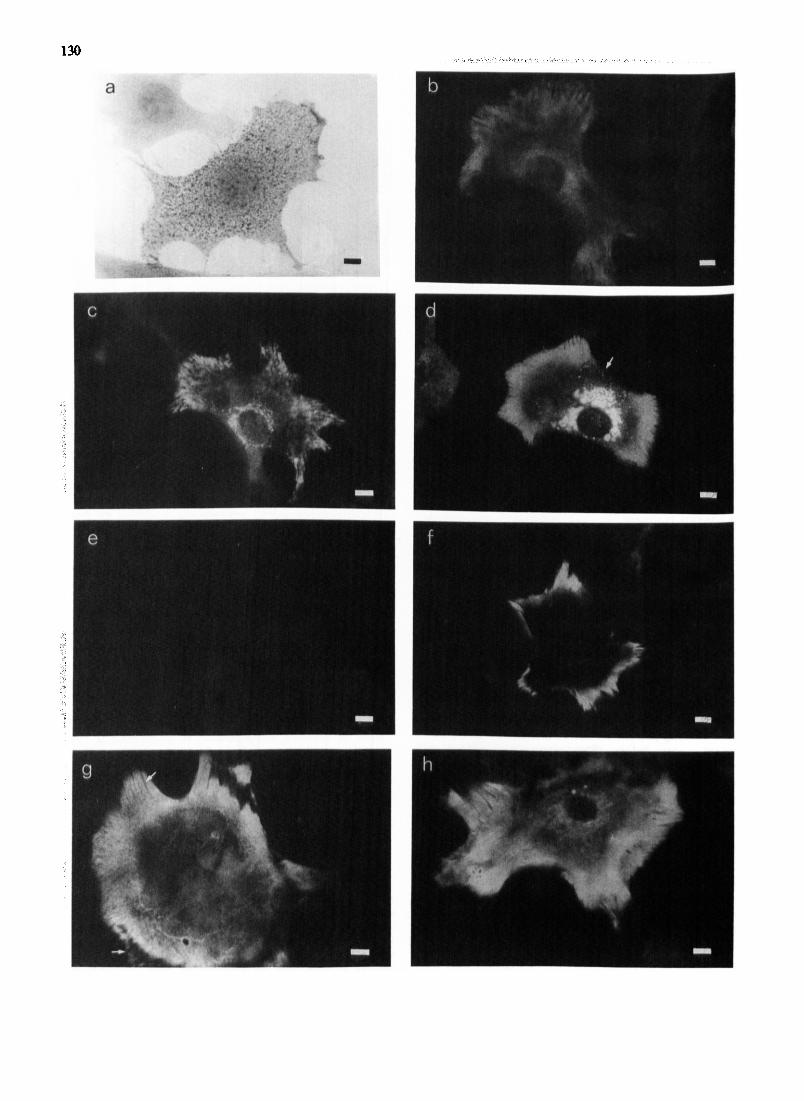

monensin treatment (c.f. Fig. 4c and d) suggests that osteoblasts synthesize PAl-1 constitutively and secre- tion is via the Golgi apparatus. In ceils stained live at 0 ° C with either the antibody to PAI-1, normal rabbit IgG or PBS, immunofluorescence of PAl-1 was re- stricted to the peripheral parts of the cells, whereas the central areas appeared negative (Fig. 4f). The controls were negative. However cell contact preparations, ob- tained by removing the cells with saponin treatment and staining the underlying substratum, showed a homoge- neous carpet of immunofluorescence across the width of the cell, particularly intensive at the periphery, where the striae without staining were also present. The con- trois were negative. These results are similar to those of Piilliinen et al. [29] using HT-1080 sarcoma cells and 2-HES fibroblasts, and show that osteoblast PAI-1 is also located externally on the substratum. The lack of staining under the centre of the cell when stained live at 0 ° C (Fig. 40 suggests that PAI-1 antigen is unavailable to antibody due to close cell-substratum contact. Stimu- lation of osteoblasts with 1,25(OH)2D3 for 24 h prior to fixation and staining did not alter the distribution of the immunofluorescence seen (Fig. 4h).
Discussion
The present experiments were designed to increase our understanding of the mechanisms regulating osteoid removal by osteoblasts. Since 90~ of total bone protein is type I collagen, we cultured mouse calvarial osteob- lasts on type I collagen films as a model. Our results demonstrate that a proteinase cascade involving both serine proteinases and metalloproteinases is required for collagenolysis.
The osteotropic hormone 1,25(OH)2D 3, a potent bone resorptive agent [33], stimulated the synthesis and release of the metalloproteinases gelatinase and stromelysin as well as collagenase. TIMP production was reduced to the lower detection limit of available biochemical assays. Increased metaUoproteinase pro- duction alone was insufficient to initiate collagen de- gradation: activation of the latent collagenase was nec- essary before collagen breakdown could take place and
131
this process was shown to be plasminogen dependent. Plasmin has been implicated as a physiological procol- lagenase activator for some time [34,35] and Gavrilovic et al. [21] have shown in experiments with plasminogen- depleted serum that rabbit articular chondrocytes do not degrade type I collagen films unless plasminogen is added to the cultures. The invasion of human amniotic basement membrane by the tumour cell line B16 has also been shown to be dependent upon activation of procollagenase by plasmin [36]. The role of plasminogen activators is discussed below but it also seems likely that endogenous metalloproteinase activators are in- volved. Recent evidence suggests that the rabbit procoi- lagenase activator purified by r a t e r et al. [37] is identi- cal to stromelysin [38], and stromelysin has been shown to activate procollagenase in vitro in the presence of trypsin [38]. Plasmin could, therefore~ be a physiological substitute for trypsin and the activation cascade in bone may be plasminogen to plasmin and then activation of stromelysin which in turn activates collagenase.
Although 60-70~ of the collagen in films was de- graded by the osteoblasts in our experiments, no active metaUoproteinases could be detected in the culture media using the reconstituted fibril assay. This may have been due to the binding of activated enzyme to collagen and to residual TIMP yielding inactive TIMP- enzyme complexes which cannot be detected by an activity assay. More pertinently, our calculations show that very little active coUagenase is necessary for col- lagen film degradation and that only a small proportion of the latent collagenase secreted needs to be activated (see Table II). These data with osteoblasts are in con- trast to the response of human gingival fibroblasts to recombinant human tumour necrosis factor and inter- leukin-1 in a similar system; collagenase and stromely- sin were detected by activity assays in both active and latent forms [39]. The absence of active enzyme in osteoblast culture supernatants suggests that the attach- ment of osteoblasts to collagen might provide a pro- tected subcellular environment for the sequestration of active enzyme.
Bone resorptive agents including 1,25(OH)2D 3 have previously been shown to stimulate plasminogen activa-
Fig. 4. Immunolocalization of PAI-1 in mouse osteoblasts. (a) Osteoblast stained for alkaline phosphatase showing many blue cytoplasmic granules. (b) Osteoblast fixed in methanol and stained by indirect immunofiuorescence for PAI-I. There is intracellular perinuclear staining and a homogeneous carpet of staining in the cell peripheral area interrupted by striae of no staining. (c) Osteoblast fixed in paraformaldehyde and stained for PAI-1. The pattern of staining is similar to (b) but the striae and perinuclear staining are more sharply defined. (d) Osteoblast cvltured with monensin for 3 h prior to fixation in paraformaldehyde and then stained by indirect immunofluorescence for PAI-I. The perinuclear fluorescence is prominent and vacuolated and fluorescent cytoplasmic granules are also visible (arrow). (e) Osteoblast treated as in Fig. 4d, except that control normal rabbit serum lgG was substituted for the primary antiserum. No fluorescence. (f) Osteoblasts were stained with antibody to PAl-1 at 0 o C before methanol fixation and incubation with the secondary antibody. Only immunofluorescence at the cell periphery is visible. (g) Cell contact area preparations were obtained by removing the cells with saponin treatment and staining the remaining material by indirect immunofluorescence. The substratum has staining across the width of the cell in a homogeneous carpet, and the striae of no staining are still visible around the periphery (arrows). (h) Osteoblasts were stimulated for 24 h with 1,25(OH)2D 3 before fixation with paraformaldehyde and stained with antibody to PAl-1.
The staining pattern is identical to that seen in Fig. 4c. Bars = 20 #m.

132
tor production by osteoblasts [40]. We confirmed this finding and showed that osteoblast plasminogen activa- tor is predominantly cell-associated and therefore prob- ably of the urokinase-type. Although antibodies to localize mouse urokinase-type plasminogen activator immunocytochemically are currently not available, both it and PAl-1 have been immunolocalized in human fibroblasts and sarcoma cells with polyclonal and monoclonal antibodies [29]. Urokinase-type plasmino- gen activator was found to be located at discrete cell- substratum contact sites, and also in areas of cell-cell contact. We found that PAI-1 was distributed as a homogeneous carpet on the substrate under the cells and that mouse osteoblasts constitutively synthesize PAl-1. Recent findings have shown that urokinase-type plasminogen activator is synthesized and released from mammalian cells as a proenzyme with little or no activ- ity, and that cells possess a specific binding site for urokinase-type plasminogen activator and its preform (for a review see Ref. 41). The presence of a urokinase- type plasminogen activator receptor thus allows cells to acquire surface bound plasminogen-activating abifity. The confinement of urokinase-type plasminogen activa- tor to discrete sites may explain how ~ells producing large amounts of PAl-1 can produce plasminogen activator-mediated focal proteolysis [29].
In conclusion, our findings provide evidence for the involvement of a plasminogen-plasmin-latent metal- loproteinase activation cascade in mediating type I col- lagen degradation by osteoblasts, and for its regulation by TIMP and PAl-1. As only a small proportion of the secreted latent collagenase was activated we conclude that these inhibitors are important in controlling the extent of osteoid degradation by osteoblasts.
Acknowledgements
Supported by grants from the Arthritis and Rheuma- tism Council and the Medical Research Council. We thank Christopher Green for preparing the illustration and Eileen Gayfer for secretarial assistance.
References
I Walker, D.G., Lapiere, C.M. and Gross, 5. (1964) Biochem. Bio- phys. Res. Commun. 15, 397-402.
2 Fullmer, H. and Lazarus, G. (1967) Isr. J. Med. Sci. 3, 758-761. 3 Vaes, (3. (1972) Biochem. J. 126, 275-289. 4 Sellers, A., Melkle, M.C. and Reynolds, J.J. (1980) Calcif. Tissue
Int. 31.35-43. $ Heath, J.K., Atkinson, SJ., Meikle, M.C. and Reynolds, J.J. (1984)
Biochim. Biophy.~. Acta 802, 151-154. 6 Sekamoto, M. and Sakamoto, S. (1984) Biomed. Res. 5, 29-38. 70tsuka, K., Sodek, J. and Limeback, H. (1984) Eur. 5. Biochem.
145, 123-129.
8 Raina, V. (1972) J. Clin. PathoL 25, 229-232. 9 Foruasier, V.L. (1980) Metab. Bone Dis. Res. Res. 2S, 103-108.
10 Vandervcil, C.J. (1980) Metab. Bone Dis. Rel. Res. 2S, 109-116. 11 Luben, R.A., Wong, G.L. and Cohn, D.V. (1976) Endocrinology
99, 526-534. 12 Peck, W.A., Burke, J.K., Wilkins, J , Redan, S.B. and Redan, G.A.
(1977) Endocrinology 100, 1357-1364. 13 Silve, C.M., Hradek, G.T., Jones, A.L. and Arnaud, C.D. (1982) 5.
Cell Biol. 94, 379-386. 14 Manolagas, S.C., Haussler, M.R. and Deftos, L.J. (1980) J. Biol.
Chem. 225, 4414-4417. 15 Partridge, N.C., Frampton, R.J., Eisman, J.A., Michelangeli, V.P.,
Elms, E., Bradley, T.R. and Martin, T.J. (1980) FEBS Left. 115, 139-142.
16 Martin, T.5. and Partridge, N.C. (1981) in Hormonal Control of Calcium Metabolism (Cohn, D.V., Talmage, R.V. and Matthews, 5.L., eds.), pp. 147-156, Excerpta Medica, Amsterdam.
17 Sekamoto, S. and Sakamoto, M. (1986) in Bone and Mineral Research, Vol. 4 (Peck, W.A., ed.), pp. 49-102, Elsevier, Amster- dam.
18 Chambers, T.J., Darby, J.A. and Fuller, K. (1985) Cell Tissue K~ 241,671-675.
19 Chambers, T.5. and Fuller, K. (1985) J. Cell Sei. 76, 155-165. 20 Thomson, B.M., Atkinson, S.J., Reynolds, J.J. and Meikle, M.C.
(1987) Biochem. Biophys. Res. Commun. 148, 596-602. 21 Gavrilovic, J., Reynolds, JJ. and Murphy, G. (1985) Cell Biol. int.
Rep. 9, 1097-1107. 22 Unkeless, J.C., Tobia, L., Ossowski, J.P., Rifkin, D.B. and Reich,
E. (1973) J. Exp. Med. 137, 85-111. 23 Deutz, D.G. and Mertz, E.T. (1970) Science 170, 1095-1096. 24 Wu, M.-C., Sehultz~ D.R., Arimura, G.K., Gross, M.A. and Yunis,
A.A. (1975) Exp. Cell Res. 96, 37-46. 25 Sellers, A. and Reynolds, J3. (1977) Biochem. J. 167, 352-360. 26 Sellers, A., Reynolds, 5.J. and Meikle, M.C. (1978) Biochem. J.
171,493-496. 27 Murphy, G., Cawston, T.E., Galloway, W.A., Barnes, M.J., Bun-
ning~ R.A.D., Mercer, E., Reynolds, J.J. and Burgeson, R.E. (1981) Biochem. J. 199, 807-811.
28 Hembry, R.M., Murphy, G. and Reynolds, J.J. (1985) J. Cell Sci. 73, 105-119.
29 Pglllinen, J., Saksela, O., Salonen, E.-M., Andreasen, P., Nielsen, L., Dane, K. and Vaheri, A. (1987) J. Cell Biol. 105, 1085-1096.
30 Miller, S., Watt, A.M. and Arnaud, C.D. (1976) Science 192, 1340-1343.
31 Jones, S.J. and Boyde, A. (1976) Cell Tissue Res. 166, 101-107. 32 Ledger, C.W., Uchida, N. and Tanzer, M.L. (1980) J. Cell Biol. 87,
663-671. 33 Reynolds, J.J., Holick, M.F. and Deluca, H.F. (1974) Calcif.
Tissue Res. 15, 333-339. 34 Eeckhout, Y. and Vaes, G. (1977) Biochem. J. 166, 21-31. 35 Werb, Z., Mainardi, C.L., Vater, C.A. and Harris, E.D. (1977)
New Engl. J. Med. 296, 1017-1023. Mignatti, P., Robbins, E. and Rifldn, D.B. (1986) Cell 47, 487-498. Vater, C.A., Nagase, H. and Harris, E.D. (1983) J. Biol. Chem. 258, 9374-9382. Murphy, G., Cockett, M.I., Stephens, P.E., Smith, B.J. and Docherty, A.J.P. (1987) Biochem. J. 248, 265-268. Meikle, M.C., Atkinson, S.J., Ward, R.V., Murphy, G. and Re- ynolds, J.J. (1989) J. Periodont. Res. 24, 207-213. Hamilton, J.A., Lingelbach, S., Partridge, N.C. and Martin, T.J. (1984) Biochem. Biophys. Res. Commun. 122, 230-236. Blasi, F , Vassalli, J.-D. and Dane, K. (1987) J. Cell Biol. 104, 801-804.
36 37
38
39
40
41












