
A survey on phytochemical and bioactivity of plant extracts from Malaysian forest reserves
Upload
independentCategory
view
1download
0
Triple Hyp?Pro Replacement in Integramide A, a Peptaib Inhibitor ofHIV-1 Integrase: Effect on Conformation and Bioactivity
Marta De Zotti,1 Wim De Borggraeve,2 Bernard Kaptein,3 Quirinus B. Broxterman,3 Sheo B. Singh,4
Peter J. Felock,5 Daria J. Hazuda,5 Fernando Formaggio,1 Claudio Toniolo1
1Department of Chemistry, University of Padova, Institute of Biomolecular Chemistry, CNR, Padova Unit,
35131 Padova, Italy
2Department of Chemistry, Katholieke Universiteit Leuven, 3001 Leuven, Belgium
3DSM, Innovative Synthesis BV, P.O. Box 18, 6160 MD Geleen, The Netherlands
4Merck Research Laboratories, West Point, PA 19486
5Merck Research Laboratories, Rahway, NJ 07065
Received 15 February 2010; revised 31 March 2010; accepted 2 April 2010
Published online 21 August 2010 in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/bip.21461
This article was originally published online as an accepted
preprint. The ‘‘Published Online’’ date corresponds to the
preprint version. You can request a copy of the preprint by
emailing the Biopolymers editorial office at biopolymers@wiley.
com
INTRODUCTION
In the last decade clinical treatments of patients infected
by HIV with inhibitors of inverse transcriptase and HIV
protease resulted in significant improvements in the
reduction of the viral load and AIDS progression. How-
ever, these treatments tend to become ineffective owing
to the high mutation rate of the virus.
The enzyme HIV-1 integrase is a promising target for the
anti-HIV therapy because it is essential for virus replication
Triple Hyp?Pro Replacement in Integramide A, a Peptaib Inhibitor ofHIV-1 Integrase: Effect on Conformation and Bioactivity
Additional Supporting Information may be found in the online version of this article.
Correspondence to: Claudio Toniolo; e-mail: [email protected]
ABSTRACT:
AIDS is produced by HIV-induced infections. HIV
integrase is an important enzyme as it is critical for the
integration of the HIV genome into that of the host cell.
This complex process is exclusively carried out by a viral
enzyme not found in the host cell. Therefore, this protein
represents a safe target for the development of single or
combined anti-HIV therapy. Integramide A is a 16-mer
long, effective peptaib inhibitor of HIV-1 integrase. We
have previously described a versatile synthetic strategy in
solution to afford this natural compound and its
diastereomer at positions 14 and 15. We also found that
both peptides display a significant inhibitory activity.
Here, we present our data on the synthesis in solution,
in-depth conformational analysis, and biological activity
against HIV-integrase of the analogs of the two above
mentioned peptides in which all of the three (2S,4R)-Hyp
residues at positions 2, 9, and 13 are replaced by L-Pro.
This study definitely confirms that the mixed a-/310-
helical conformation of natural integramide A plays a key
role in its mechanism of inhibition. Moreover, our data
provide evidence that the amphipathic character of this
helical structure is not required for the activity of
integramide A against HIV-1 integrase. These
observations will hopefully help us to further clarify the
precise mechanism of inhibition of this interesting peptaib
and to identify shorter peptide sequences active against
HIV-1 integrase. # 2010 Wiley Periodicals, Inc.
Biopolymers (Pept Sci) 96: 49–59, 2011.
Keywords: circular dichroism; infrared absorption; integrase;
nuclear magnetic resonance; peptide helices
VVC 2010 Wiley Periodicals, Inc.
PeptideScience Volume 96 / Number 1 49

and there are no known counterparts in the host cell.
Currently, numerous classes of inhibitors of HIV-1 integrase
have been published.1–7 They also involve some non-Aib
(a-aminoisobutyric acid)/Iva (isovaline) (see Figure 1)
peptides of modest activity.8–16 However, in 2002 a publica-
tion from one of us (S.B.S.)17 has dealt with two naturally
occurring peptide molecules (integramides A and B) which
exhibit interesting inhibitory activities (integramide A inhib-
its the coupled and strand transfer reactions of HIV-1 inte-
grase with IC50 values of 7-10 and 30-34 lM, respectively).
Integramide A is a 16-mer long peptaib compound, i.e. a
linear peptide rich in Aib residues. More specifically, its
primary structure (see Figure 1) is characterized by as many
as nine (56%) Ca-tetrasubstituted a-amino acids (four
Aib and five Iva residues). All chiral amino acids have the
L-configuration, except two D-Iva residues (at positions 1
and 15).17–20 While the C-terminus of integramide A is free,
the N-terminus is blocked by acetylation. In integramide B
the Aib8 residue is replaced by an additional L-Iva.
Recently, we reported the total solution syntheses of inte-
gramide A and its D-Iva14-L-Iva15- diastereomer.19 This
achievement is significant, particularly because of the pres-
ence in these peptides of a large amount (56%) of sterically
demanding, poorly reactive, Ca-methylated a-amino acids.
Furthermore, our in-depth 3D-structural investigation on
integramide A and appropriately chosen short sequences
supported the view that the full-length peptide inhibitor is
folded in a predominantly a-/310-helical structure with
amphipathic features of potential relevance to bioactivity.
In this work, we applied a closely similar synthetic strategy
to prepare two additional analogs of integramide A in which
the three (2S,4R)-Hyp residues at positions 2, 9, and 13 of
each analog are substituted by L-Pro. The results of a detailed
conformational study of these peptides and selected short-
sequences, obtained by the combined use of FTIR absorp-
tion, CD and NMR techniques, will also be presented. Lastly,
our findings on the two analogs, evaluated as inhibitors of
HIV-1 integrase, will be discussed in comparison with those
of their published Hyp-based counterparts.17,18
MATERIALS AND METHODS
Synthesis and Characterization of PeptidesThe strategy of synthesis of the peptides discussed in this work is
reported in Scheme 1. Details of their characterizations (including
analytical and physical properties) are reported in the Supporting
Information. The Z-derivatives of the free amino acids were
prepared as described in Ref. 21. The materials used during the
syntheses were purchased from Acros (Geel, Belgium): free amino
acids, Ac2O, EDC, HOBt, Na-(benzyloxycarbonyloxy) succinimide;
Alphagaz (Milan, Italy): isobutene; Fluka (Buchs, Switzerland):
TFA, 4-(dimethylamino)pyridine, N,N-diisopropyl-N-ethylamine,
N-methylmorpholine; GL Biochem (Shangai, China): HATU; Per-
septive Biosystems (Warrington, UK): HOAt; Riedel-de Haen
(Seelze, Germany): ZnBr2. Melting points were determined using a
Leitz (Wetzlar, Germany) model Laborlux 12 apparatus and are not
corrected. Optical rotations were measured using a Perkin-Elmer
(Norwalk, CT) model 241 polarimeter equipped with a Haake (Karls-
ruhe, Germany) model D thermostat. The solid-state IR absorption
spectra were recorded by use of KBr pellets and a Perkin-Elmer 1720
X spectrophotometer, nitrogen flushed, equipped with a sample-shut-
tle device, at 2 cm�1 nominal resolution, averaging 100 scans. HPLC
measurements were performed using a Gilson (Middleton, WI)
apparatus equipped with a UV detector at 226 nm.
Infrared AbsorptionThe FTIR absorption spectra were recorded by using the Perkin-
Elmer model 1720 X IR spectrometer mentioned above. Cells with
FIGURE 1 Chemical structures of Aib and Iva, and amino acid sequences of integramides A and B.
50 De Zotti et al.
Biopolymers (Peptide Science)

path lengths of 0.1, 1.0, and 10 mm (with CaF2 windows) were
employed. Spectrograde deuterochloroform (99.8% D) was pur-
chased from Aldrich (St. Louis, MO). Solvent (baseline) spectra
were recorded under the same conditions.
Circular DichroismThe circular dichroism (CD) measurements were carried out on a
Jasco (Tokyo, Japan) model J-715 spectropolarimeter at room tem-
perature, using Hellma (Mullheim, Germany) quartz cells with
Suprasil1 windows and an optical path length of 0.1 mm. The sig-
nal to noise ratios were improved by accumulating 16 scans. Data
were processed using the J-700 program for Windows and expressed
in terms of [h]T, the total molar ellipticity (deg 3 cm2 3 dmol�1).
Spectrograde 2,2,2-trifluoroethanol (TFE) and methanol (MeOH)
were Acros (Geel, Belgium) products.
Nuclear Magnetic Resonance Experiments and
Structure CalculationsThe 1D-NMR dimethylsulphoxide (DMSO) titration experiments
were performed at 298 K with a Bruker AVANCE DRX-400 spec-
trometer operating at the frequency of 400 MHz for protons. Pep-
tide concentration in CDCl3 was 13 10�3 M.
The 2D-NMR experiments were performed with a Bruker
AVANCE DMX-600 spectrometer operating at 600 MHz. The NMR
solution of the Ac(acetyl)/OH [L-Pro2,9,13] integramide A analog
was 2 3 10�3 M in TFE-d2. Processing and evaluation of the experi-
mental data were carried out using the programs XWinNMR,
SPARKY, and XEASY (version 1.4).22 The DQF-COSY23 spectra
were acquired in the magnitude mode, while CLEAN-TOCSY24,25
(spin lock period, 70 ms), and NOESY (mixing time, 100 ms) spec-
tra were recorded in the phase-sensitive mode. The -OH signal of
TFE-d2 was suppressed by presaturation during the relaxation delay.
All homonuclear 2D spectra were acquired by collecting 400–
512 experiments of 64–128 scans each, with a relaxation delay of 1 s,
and 2 K data points. The spectral width was 10 ppm in each
dimension.
The assignment of the two methyl groups belonging to the same
Aib residue was obtained by means of 2D 1H-13C correlation spec-
tra. To optimize the digital resolution in the carbon dimension,
HMQC and HMBC26,27 experiments were acquired using selective
excitation by means of Gaussian-shaped pulses with 1% truncation.
The Cb-selective HMQC experiments28 with gradient coherence
selection29 were recorded with 300 t1 increments, of 270 scans and
2K points each. A spectral width of 16 ppm centered at 22 ppm in
F1 was used. HMBC experiments30 with selective excitation in the
CO region were performed using a long-range coupling constant of
7.5 Hz, a spectral width in F1 of 15 ppm centered at 176 ppm,
320 t1 experiments of 400 scans, and 4 K points each. NOESY
experiments were used for sequence-specific assignments. Interpro-
ton distances were obtained by integration of the NOESY spectrum
SCHEME 1 Strategy adopted to prepare the two Na-acetylated, diastereomeric hexadecapeptides
A-B-C-D[L-Pro2,9,13], (-D-Iva14-L-Iva15-) and (-L-Iva14-D-Iva15-). (i): H2/Pd in CH2Cl2; (ii): H2/Pd in
methanol (MeOH); (iii): (Z-Aib)2O in CH2Cl2; (iv): TFA (trifluoroacetic acid) (10 eq.) in CH2Cl2;
(v): EDC [N-ethyl-N0-(3-dimethylaminopropyl)-carbodiimide] and HOAt (7-aza-1-hydroxy-1,2,3-
benzotriazole) (1.3 eq.) in CH2Cl2; pH kept at 8 by adding N-methylmorpholine; (vi): EDC in ace-
tonitrile at reflux for three to five days [via 5(4H)-oxazolone]; (vii): HATU {N-[(dimethylamino)-
1H-1,2,3-triazole(4,5-b)pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate
N-oxide} (1 eq.), HOAt (0.5 eq.) and N,N-diisopropyl-N-ethylamine (2 eq.) in CH2Cl2; (viii) H2/Pd
and AcOH (1 eq.) in MeOH at �108C. (2)Ac2O (5 eq.); (ix): ZnBr2 (10 eq.) in CH2Cl2, four days.
Triple Hyp?Pro Replacement in Integramide A 51
Biopolymers (Peptide Science)

using the SPARKY package. When peaks could not be integrated
because of partial overlap, a distance corresponding to the maxi-
mum limit of detection of the experiment (4.0 A) was assigned to
the corresponding proton pair.
Distance geometry and molecular dynamics (MD) calculations
were carried out using the random simulating annealing (SA) pro-
tocol of the XPLOR-NIH 2.9.6 program.31 For distances involving
equivalent or nonstereo-assigned protons, r�6 averaging was used.
The MD calculations involved a minimization stage of 100 cycles,
followed by SA and refinement stages. The SA consisted of 30 ps of
dynamics at 1500 K (10,000 cycles, in three fs steps) and of 30 ps of
cooling from 1500 to 100 K in 50 K decrements (15,000 cycles, in
two fs steps). The SA procedure, in which the weights of NOE and
nonbonded terms were gradually increased, was followed by
200 cycles of energy minimization. In the SA refinement stage, the
system was cooled from 1000 to 100 K in 50 K decrements (20,000
cycles, in one fs steps). Finally, the calculations were completed with
200 cycles of energy minimization using a NOE force constant of
50 kcal/mol. The generated structures were visualized using the
MOLMOL32 (version 2K.2) program.
Bioactivity MeasurementsThe microtiter plate assay for strand transfer was carried out
with an immobilized 30-bp U5 donor substrate and a 20-bp tar-
get substrate biotinylated at the 30-end of each DNA strand.
Each reaction mixture of 100 ll consisted of 24.06 mM HEPES
(pH 7.8), 25 mM NaCl, 31.25 mM MnCl2, 6.19 mM b-mercap-
toethanol, 62.5 lg/ml bovine serum albumin, and integrase
(50 nM final concentration). The final concentration of DMSO
in all reactions was 10%. Integrase was assembled with immobi-
lized LTR oligonucleotides for 30 min at 378C. For coupled
assays, the strand transfer reaction was initiated by the addition
of 5 ll of the biotinylated target substrate (150 nM final concen-
tration). For the strand-transfer reaction, unbound enzyme was
removed and the reaction and the reaction buffer were
exchanged before addition of the target substrate. Strand-transfer
reaction mixtures were incubated for 30 min at 378C. Strand-
transfer products were detected using alkalinephosphatase-conju-
gated avidin. IC50 values were determined using a series of two-
fold dilutions in duplicate or triplicate starting at 5–200 lM in
100% DMSO. The IC50 was determined using GraphPad Prism 4
software with eight-point titration and curve fit. Representative
data from at least two independent measurements are presented.
The solubility properties of integramide A and its Pro-analogs
investigated in this work in the solvent mixtures used for the
bioactivity measurements are appreciable.
RESULTS AND DISCUSSION
Peptide Synthesis
For the large-scale production of the enantiomerically pure
L-Iva and D-Iva, an economically attractive and generally
applicable chemoenzymatic synthesis developed by DSM was
used.33 The method involves a combination of organic
synthesis for the preparation of the racemic a-amino acid,
followed by optical resolution using a broadly specific
a-amino amidase.
The strategy of our solution-phase synthesis (segment
condensation approach) exploited for the preparation of the
[L-Pro2,9,13] integramide A analog and its diastereomer at
positions 14 and 15 is shown in Scheme 1. Except for the
synthesis and coupling of the amino acid derivative A, it fol-
lows strictly the successful strategy we already reported for
the two original, Hyp-based, peptides.19 Briefly, we first syn-
thesized the Z/OtBu (Z, benzyloxycarbonyl; OtBu, tert-
butoxy) terminally protected segments C, D, and C-D (both
Iva14-Iva15 diastereomers for segments D and C-D). The
step-by-step synthesis of segments C and D was performed
by the EDC/HOAt C-activation method.34 The segment con-
densation of peptide C to D was carried out by activating the
carboxyl function of peptide C (characterized by the nonepi-
merizable -Iva-Aib- sequence at its C-terminus) via 5(4H)-
oxazolone with EDC. The reaction yields were from moder-
ate (63%) to good (90%). Not surprisingly, the lowest yields
were observed for the sterically hindered Iva-Aib (segment
C), Iva-Iva (segment D), and Aib-Pro (segment C-D) cou-
plings. The B + C-D synthetic step was carried out by isolat-
ing and utilizing the intermediate 5(4H)-oxazolone of
segment B (with its C-terminal -Aib-Aib- sequence), pre-
pared in turn by reacting the corresponding peptide free acid
with EDC. The yield for the synthesis of the B-C-D segment
(L-Iva14-D-Iva15 isomer) in anhydrous acetonitrile was 56%,
but in distilled CH2Cl2 (D-Iva14-L-Iva15 isomer) the yield for
this coupling raised to 72%. The A+B-C-D synthetic step
was performed using Z-D-Iva-OH and the HATU34 C-activa-
tion method (yields for the synthesis of the L-Iva14-D-Iva15
and D-Iva14-L-Iva15 isomers: 70% and 62%, respectively). As
mentioned above, this is the only difference between the
present synthetic strategy and that we already published for
integramide A.19 We preferred the use of Z-D-Iva-OH rather
that Ac-D-Iva-OH for coupling because of the remarkably
higher reactivity of the Na-urethane protected derivative.
In the next step, we carried out the synthesis of the two
diastereomeric Ac/OtBu A-B-C-D hexadecapeptide seg-
ments. In our hands, the best method for the combined
Z-deprotection/Na-acetylation reactions (in particular, to
minimize the amounts of 2,5-dioxopiperazine formation
from the D-Iva-L-Pro N-terminal sequences) turned out to be
treatment of the peptides at �108C with H2/Pd/C in MeOH
in the presence of an equimolar amount of AcOH followed
by reaction of the resulting Na-deprotected peptides with an
excess of Ac2O in CH2Cl2 solution at room temperature
overnight. The yields of the final, pure products were 78%
and 63% for the L-Iva14-D-Iva15 and D-Iva14-L-Iva15 peptide
isomers, respectively. Previous attempts, using catalytic
52 De Zotti et al.
Biopolymers (Peptide Science)

hydrogenation either without the addition of AcOH or in the
presence of bulk Ac2O at room temperature, afforded impure
materials in much lower yields. Removal of the C-terminal
(tert-butyl ester) protection was achieved for both diaster-
eomers by reaction with ZnBr235 in CH2Cl2. After three days,
the products were purified by HPLC (40% yield) (see Figure
2). The overall yields for the syntheses of the two Ac/OH 16-
mer peptides (involving 16 coupling steps each) turned out
to be 2.8–3.7%.
FTIR Absorption
The conformations of the two Ac/OH, diastereomeric
[L-Pro2,9,13] 16-mers, as well as those of their Z/OtBu
protected segments C, D, C-D, B-C-D, and A-B-C-D, were
first characterized by FTIR absorption in CDCl3 solution.
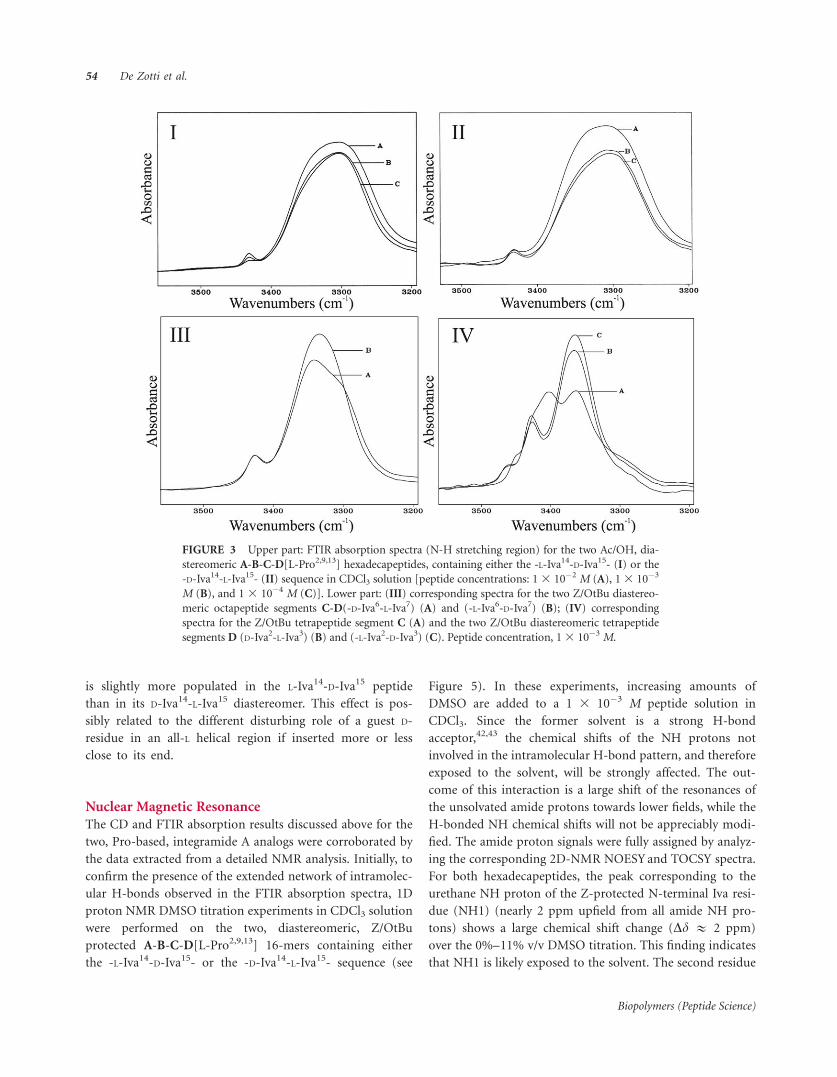
In Figure 3 the conformationally informative N��H
stretching region is reported for the two [L-Pro2,9,13]
integramide analogs. The substitutions of the Pro residues
for Hyp made the peptides soluble in CDCl3. This
property allowed us to record even the spectra of the
Na-acetylated, C-terminally deprotected, final peptides. It
is evident that the variation of peptide concentration
affects the intensity of the very strong band associated
with H-bonded NH groups near 3320 cm�1 only to a
limited extent.36,37 These observations strongly support the
overwhelming occurrence in both peptides of a helical
conformation stabilized by intramolecular H-bonds. Figure
3 also shows the spectra acquired for the two diastereo-
meric, Z/OtBu protected octapeptides C-D, which demon-
strate that they are significantly more folded than their
corresponding tetrapeptides C and D. These latter data are
fully consistent with those reported previously for the
related, Hyp-containing, compounds.19
Circular Dichroism
The conformational preferences of the two Ac/OH, diastereo-
meric [L-Pro2,9,13] 16-mers were investigated using CD in
TFE and 1 3 10�1 M aqueous solution of sodium dodecyl-
sulphate (SDS). As illustrated in Figure 4, both analogs are
largely right-handed, mixed a-/310-helical. This conclusion
was derived from the occurrence of two negative maxima
near 205 and 225 nm and one positive maximum at about
195 nm.38 Not surprisingly, the highest ellipticity and, as a
consequence, the most significant structuration, is seen for
both compounds in the helix-supporting solvent TFE.39 A
preliminary estimate of the helical type adopted was
extracted from the ellipticity ratio R ¼ [Y]225/[Y]205, known
to be <0.35 for a largely prevailing population of 310-helix
and >0.70 for a largely prevailing population of a-helix.40,41
In TFE solution, for both Pro-containing 16-mers the
R value was found to be 0.65. Their a-/310-helix ratio is quite
close to that shown by integramide A (the CD curve of which
is also reported in Figure 4 for comparison) but, overall,
the helix is more developed in the natural compound. In
water solution the shape of the CD patterns of the two Pro-
containing peptides is very similar to that in TFE (with R val-
ues near 0.65), but the ellipticities are about 35% lower. In
aqueous SDS the R values for the two peptides are in the
range 0.8–0.9. Therefore, the conformation in this mem-
brane-mimetic environment seems to be more a-helical forthe [L-Pro2,9,13] analogs with respect to that of integramide
A (R ¼ 0.7). This finding could suggest a somewhat different
organization in the membranes of the highly lipophilic
helices of the Pro-based analogs as compared with that of
the amphipathic integramide A.19 Also, in SDS (more sig-
nificantly than in TFE solution) the a-helix conformation
FIGURE 2 HPLC profiles of the two Ac/OH, diastereomeric hexa-
decapeptides A-B-C-D and [L-Pro2,9,13] -D-Iva14-L-Iva15- (A) and -L-
Iva14-D-Iva15- (B). Conditions: analytical Phenomenex Kromasil C18
column (particle size: 5 lm; pore size: 100 A); gradient system,
90?100%B in 20 min; flow rate, 1 ml/min (eluant A: H2O/acetoni-
trile 9/1 + 0.05% TFA; eluant B: acetonitrile/H2O 9/1 + 0.05% TFA);
room temperature; absorbance detector at 226 nm.
Triple Hyp?Pro Replacement in Integramide A 53
Biopolymers (Peptide Science)
is slightly more populated in the L-Iva14-D-Iva15 peptide
than in its D-Iva14-L-Iva15 diastereomer. This effect is pos-
sibly related to the different disturbing role of a guest D-
residue in an all-L helical region if inserted more or less
close to its end.
Nuclear Magnetic Resonance
The CD and FTIR absorption results discussed above for the
two, Pro-based, integramide A analogs were corroborated by
the data extracted from a detailed NMR analysis. Initially, to
confirm the presence of the extended network of intramolec-
ular H-bonds observed in the FTIR absorption spectra, 1D
proton NMR DMSO titration experiments in CDCl3 solution
were performed on the two, diastereomeric, Z/OtBu
protected A-B-C-D[L-Pro2,9,13] 16-mers containing either
the -L-Iva14-D-Iva15- or the -D-Iva14-L-Iva15- sequence (see
Figure 5). In these experiments, increasing amounts of
DMSO are added to a 1 3 10�3 M peptide solution in
CDCl3. Since the former solvent is a strong H-bond
acceptor,42,43 the chemical shifts of the NH protons not
involved in the intramolecular H-bond pattern, and therefore
exposed to the solvent, will be strongly affected. The out-
come of this interaction is a large shift of the resonances of
the unsolvated amide protons towards lower fields, while the
H-bonded NH chemical shifts will not be appreciably modi-
fied. The amide proton signals were fully assigned by analyz-
ing the corresponding 2D-NMR NOESYand TOCSY spectra.
For both hexadecapeptides, the peak corresponding to the
urethane NH proton of the Z-protected N-terminal Iva resi-
due (NH1) (nearly 2 ppm upfield from all amide NH pro-
tons) shows a large chemical shift change (Dd & 2 ppm)
over the 0%–11% v/v DMSO titration. This finding indicates
that NH1 is likely exposed to the solvent. The second residue
FIGURE 3 Upper part: FTIR absorption spectra (N-H stretching region) for the two Ac/OH, dia-
stereomeric A-B-C-D[L-Pro2,9,13] hexadecapeptides, containing either the -L-Iva14-D-Iva15- (I) or the
-D-Iva14-L-Iva15- (II) sequence in CDCl3 solution [peptide concentrations: 13 10�2 M (A), 13 10�3
M (B), and 1 3 10�4 M (C)]. Lower part: (III) corresponding spectra for the two Z/OtBu diastereo-
meric octapeptide segments C-D(-D-Iva6-L-Iva7) (A) and (-L-Iva6-D-Iva7) (B); (IV) corresponding
spectra for the Z/OtBu tetrapeptide segment C (A) and the two Z/OtBu diastereomeric tetrapeptide
segments D (D-Iva2-L-Iva3) (B) and (-L-Iva2-D-Iva3) (C). Peptide concentration, 13 10�3 M.
54 De Zotti et al.
Biopolymers (Peptide Science)

FIGURE 4 Far-UV CD spectra for the two Ac/OH, diastereomeric A-B-C-D[L-Pro2,9,13] hexade-
capeptides containing either the -L-Iva14-D-Iva15- (LD) or the -D-Iva14-L-Iva15- (DL) sequence in
comparison with those of the natural integramide A (intA) in TFE (A) and in 1 3 10�1 M SDS (B)
solutions. Peptide concentration: 1 3 10�3 M.
FIGURE 5 Plot of NH proton chemical shifts in the 1H-NMR spectra for the two Z/OtBu pro-
tected, diastereomeric A-B-C-D[L-Pro2,9,13] hexadecapeptides containing either the -L-Iva14-D-
Iva15- (A) or the-D-Iva14-L-Iva15-(B) sequence as a function of increasing percentages of DMSO
added to the CDCl3 solution (v/v). Peptide concentration: 13 10�3 M.
Triple Hyp?Pro Replacement in Integramide A 55
Biopolymers (Peptide Science)

(Pro) lacks the NH proton as it is Na-alkylated. The NH3
proton is only slightly deshielded by the interaction with
DMSO, while all other NH proton signals are not affected at
all, so that they are very likely participating in the intramo-
lecularly H-bonded 3D-structure. From these initial data, we
tentatively conclude that the peptides tend to adopt a well
developed, mixed 310-/a-helix conformation in CDCl3.
The two synthetic, diastereomeric [L-Pro2,9,13] integra-
mide A analogs were extensively studied by 2D-NMR spec-
trometry. In Figures S1 and S2 (Supporting Information) we
reported the amide and fingerprint regions, respectively, of
the NOESY spectra acquired in TFE-d2 solution. All NH-NH
sequential connectivities were seen, as well as some NH(i)-
NH(i+2) connectivities, characteristic of the presence of a
helical structure.
The spin systems of the trisubstituted amino acid residues
were assigned according to the procedure described by
Wuthrich,44 using DQF-COSY and TOCSY spectra, while
HMQC and HMBC experiments were exploited for the
assignment of the Ca-tetrasubstituted residues (Aib and Iva).
NOESY spectra were used to obtain the sequential assign-
ments. Although as many as three Pro residues characterize
this sequence, no evidence for a cis configuration at any of
the Xxx-Pro bonds was found. Indeed, the 1D NMR spec-
trum did not exhibit any minor peak and no sequential CaH-
CaH NOESY cross peaks were observed.
The proton chemical shift assignments for the two diaster-
eomeric 16-mers are reported in Tables SIII and SIV (Sup-
porting Information). A summary of the NOE connectivities
found in the NOESY spectrum of the diastereomer contain-
ing the L-Iva14-D-Iva15 sequence (reported in Table SV of the
Supporting Information) clearly highlights the occurrence of
a helical structure, as shown by the presence of a number of
CaH(i)-NH(i+3) connectivities, along with some CaH(i)-
NH(i+2) and CaH(i)-NH(i+4) connectivities. Globally,
the present findings are in favor of the occurrence of a mixed
a-/310-helical conformation44 for these analogues, quite close
to that of native integramide A under the same experimental
conditions.19
The conformational preferences of the L-Iva14-D-Iva15 ana-
log (with the same configurational sequence as that of integra-
mide A) were investigated in more detail by distance geometry
and restrained MD calculations. A set of NOESY spectra was
recorded at 300K using mixing times of 100, 150, 200, 250,
and 300 ms. Analysis of the build-up curve (not shown) indi-
cated that the data to at least up to 250 ms are in the linear
region and could be used for distance determinations. NOE-
volumes were integrated in the 100 ms NOESY spectrum.3JNa coupling constants were measured both in the 1D
spectrum (at 308K) and in the DQF-COSY spectrum to
derive dihedral angle constraints for structural analysis by
restrained MD. Because direct measurement of the coupling
constants in the COSY spectrum is not accurate, a modified
procedure was used.45 For the three proteinogenic amino
acids (Ile, Leu, Gly) present in the sequence, values around 7
Hz were obtained. These values are not in agreement with
those expected (about 4 Hz) for a rigid, mixed 310-/a- helix,but rather they are more indicative for an ensemble of several
structures, including some more extended conformations.
A total of 123 interproton distance restraints were derived
from the 100 ms NOESY spectrum (Table I) and used in the
random SA protocol. Calibration was performed on the
strongest peak in the amide region which was set to 2.4 A.
Distance restraints were then determined and error bounds
of 610% were taken into account. To resolve a few problems
with overlapping signals, some restraints were added from a
NOESY spectrum taken at 288K (100 ms mixing time) using
the same calibration procedure. The interproton distance
restraints were used to generate 150 structures via a random
SA protocol and a further refinement. Structures not con-
taining any violation (bigger than 0.5 A) were retained and
analyzed. The H-bonding pattern present in more than 66%
of the accepted structures was implemented with new
restraints in a subsequent round of dynamics, where another
150 structures were generated. The 22 accepted structures
with a total energy <129 kcal/mol were selected. Their super-
position is illustrated in Figure 6(I). All of these structures
converge to a mixed a-/310-helical conformation with a back-
bone average pairwise root-mean-square deviation of 0.75
(6 0.32) A (Table II). In particular, the N-terminal 1-10 seg-
ment in all accepted structures is closer to the a-helix, while
Table I NOE Constraints, Deviations from Idealized Geometry,
and Mean Energies for the NMR-Based Structure of [L-Pro2,9,13]
Integramide A
Number of NOEs
Total 123
Intraresidue 42
Sequential 50
i,i+n, n¼2, 3, 4 31
Mean rmsda from ideality of accepted structures
Bonds [A] 0.0043
Angles [8] 0.93
Improper [8] 21.41
NOEs [A] 0.09
Mean energies [kcal/mol] of accepted structures
Eoverall 127.4
Ebond 4.5
Eangle 62.0
ENOE 49.6
a Root-mean-square deviation.
56 De Zotti et al.
Biopolymers (Peptide Science)

the C-terminal pentapeptide segment is more biased toward
the 310-helix. The lowest-energy 3D-structure is shown in
Figure 6(II). Taken together, the NMR data and the results of
the MD calculations suggest a helical conformation through-
out the sequence of the integramide A analog, which parallels
the 3D-structural conclusions already published for the natu-
ral peptide.19
Bioactivity Measurements
In our previous paper,18 we evaluated natural integramide A,
its fully synthetic counterpart, and its D-Iva14-L-Iva15 diaster-
eomer as inhibitors of HIV-1 integrase in both coupled and
strand transfer reactions of proviral DNA into host cell DNA.
Natural and synthetic integramide A inhibit coupled and
strand transfer reactions with IC50 of 7-10 and 30-34 3 10�6
M, respectively, while those of the D-Iva14-L-Iva15 diaster-
eomer are 8 and 55 3 10�6 M for the two bioassays, respec-
tively. From these data, it is evident that the stereochemical
FIGURE 6 (I) Superposition of the 22 lowest-energy backbone 3D-structures (energy <129
Kcal/mol) for the Ac/OH A-B-C-D[L-Pro2,9,13] hexadecapeptide containing the -L-Iva14-D-Iva15-
sequence consistent with the NMR-derived distances and dihedral angle restraints. (II) Ribbon rep-
resentation of the lowest-energy 3D-structure obtained for the same peptide. The three L-Pro resi-
dues are labeled.
Table II Average Values [8] of Dihedral Angles /m and wm
and the Relative Standard Deviations Resulting from the
22 Calculated Structures (Energy < 129 kcal/mol) of [L-Pro2,9,13]
Integramide A
Residue /m D/ wm Dw
D-Iva1 — �59.3 61.5
L-Pro2 �71.7 60.3 35.7 60.5
L-Ile3 �57.4 61.2 �45.9 60.2
L-Iva4 �39.6 60.4 �28.4 60.2
L-Leu5 �130.1 60.4 �43.7 60.6
Aib6 �41.0 62.2 �30.5 65.9
Aib7 �65.6 69.0 �32.5 68.5
Aib8 �39.6 68.5 �58.5 63.8
L-Pro9 �68.1 60.7 �45.4 61.4
L-Leu10 �50.7 61.8 �22.9 61.8
L-Iva11 �76.6 60.6 �27.3 62.5
Aib12 �35.8 60.8 �51.2 60.9
L-Pro13 �74.5 60.4 �10.1 61.0
L-Iva14 �60.4 68.0 �34.9 60.2
D-Iva15 �45.8 60.5 �23.0 60.3
Gly16 �149 685 —
Triple Hyp?Pro Replacement in Integramide A 57
Biopolymers (Peptide Science)

inversion in the natural Iva14-Iva15 sequence does not afford
a less active analog.
In this study, we examined the bioactivities of the two dia-
stereomeric [L-Pro2,9,13] integramide A analogs. Our results
show that the triple Pro incorporation leads to IC50 values of
19 and 60 3 10�6 M for the HIV-1 integrase inhibition of the
coupled and strand transfer reactions, respectively, promoted
by the [L-Pro2,9,13] integramide A analog. Moreover, the corre-
sponding IC50 values for the [L-Pro2,9,13] D-Iva14-L-Iva15 dia-
stereomeric analog are 10 and 21 3 10�6 M. However, the
eight-point titration curve for the strand transfer assay of the
latter analog is somewhat flat, making the corresponding IC50
value not completely reliable. In any case, we can safely con-
clude that in general the two Pro-containing integramide A
analogs are only slightly less active than natural (or synthetic)
integramide A itself in inhibiting HIV-1 integrase.
CONCLUSIONSIn our previous study19 on integramides, the Aib-/Iva- based
hexadecapeptide inhibitors of HIV-1 integrase, we concluded
that onset of a stable, mixed a-/310- helical structure with
amphipathic features might be responsible for their bioactiv-
ity. In this work, by using appropriately selected ‘‘synthetic
mutants’’, we decided to investigate in-depth the role of helix
amphipathicity by keeping constant all other structural
parameters.
To this end, for the total syntheses of the two integramide
A analogs we exploited a minor variant of our published, rig-
orously planned, solution-phase, segment-condensation
strategy.19 The two analogs are characterized by an Iva14-
Iva15 diastereomeric sequence, and, more importantly, by a
drastic modification of one of the peptide helical faces via
replacement of the three hydrophilic Hyp residues by Pro.
Our present, detailed FTIR absorption, CD, and NMR
analyses, conducted under the same experimental conditions
as those of the Hyp-based counterparts,19 led us to the con-
clusion that the triple Hyp?Pro replacement does not
induce any significant change in the overall secondary struc-
ture of the integramide peptides. Moreover, the two Pro-
based diastereomers exhibit a comparable, mixed a-/310- hel-ical structure. Quite interestingly, the Pro-containing hexade-
capeptides are almost as active as their Hyp-containing coun-
terparts in inhibiting the two biochemical steps catalyzed by
HIV-1 integrase. We believe that these findings represent a
clear indication that helix conformation, but not its amphi-
pathicity, is a prerequisite for the integramide A molecule to
be bioactive.
In other words, the largely hydrophobic face of the helix
of the peptaib inhibitor seems to be the actual determinant
for the competitive interaction with the hydrophobic surfaces
of the monomer � � �monomer binding site of the C-terminal
domain of the dimeric enzyme.10 Our current efforts tend to
rigorously evaluate the role of peptide main-chain length of
integramide A on its enzyme inhibition properties, particu-
larly in view of the known, remarkable helical stability of
even much shorter sequences of Aib-/Iva- rich peptides.46–49
REFERENCES1. Craigie, R. J Biol Chem 2001, 276, 23213–23216.
2. Singh, S. B.; Pelaez, F.; Hazuda, D. J.; Lingham, R. B. Drugs
Future 2005, 30, 277–299.
3. Dayam, R.; Gundla, R.; Al-Mawsawi, L. Q.; Neamati, N. Med
Res Rev 2008, 28, 118–154.
4. Prikazchikova, T. A.; Sychova, A. M.; Agapkina, Y.; Alexandrov,
D. A.; Gottikh, M. B. Russian Chem Rev 2008, 77, 421–434.
5. Garvey, E. P.; Schwartz, B.; Gartland, M. J.; Lang, S.; Halsey, W.;
Sathe, G.; Carter, H. L., III; Weaver, K. L. Biochemistry 2009,
48, 1644–1653.
6. Beck-Engeser, G. B.; Eilat, D.; Harrer, T.; Jack, H.-M.; Wabl, M.
Proc Nat Acad Sci USA 2009, 106, 20865–20870.
7. Mehellou, Y.; De Clercq, E. J Med Chem 2010, 53, 521–538.
8. Puras Lutzke, R. A.; Eppens, N. A.; Weber, P. A.; Houghten, R.
A.; Plasterk, R. H. A. Proc Natl Acad Sci USA 1995, 92, 11456–
11460.
9. Maroun, R. G.; Krebs, D.; Rashani, M.; Porumb, H.; Auclair, C.;
Troalen, F.; Fermandjian, S. Eur J Biochem 1999, 260, 145–155.
10. de Soultrait, V. R.; Caumont, V.; Parissi, V.; Morellet, N.; Ven-
tura, M.; Lenoir, C.; Litvak, S.; Fournier, M.; Roques, B. J Mol
Biol 2002, 318, 45–48.
11. Zhao, L.; O’Reilly, M. K.; Shultz, M. D.; Chmielewski, J. Bioorg
Med Chem Lett 2003, 13, 1175–1177.
12. Krajewski, K.; Marchand, C.; Long, Y.-Q.; Pommier, Y.; Roller,
P. P. Bioorg Med Chem Lett 2004, 14, 5595–5598.
13. Oz Gleenberg, I.; Avidan, O.; Goldgur, Y.; Herschhorn, A.; Hizi,
A. J Biol Chem 2005, 280, 21987–21996.
14. Cherepanov, P.; Ambrosio, A. L. B.; Rahman, S.; Ellenberger, T.;
Engelman, A. Proc Natl Acad Sci USA 2005, 102, 17308–17313.
15. Li, H.-Y.; Zawahir, Z.; Song, L.-D.; Long, Y.-Q.; Neamati, N.
J Med Chem 2006, 49, 4477–4486.
16. Maes, M.; Levin, A.; Hayouka, Z.; Shalev, D. E.; Loyter, A.; Frie-
dler, A. Bioorg Med Chem 2009, 17, 7635–7642.
17. Singh, S. B.; Hereth, K.; Guan, Z.; Zink, D. L.; Dombrowski, A.
W.; Polishook, J. D.; Silverman, K. C.; Lingham, R. B.; Felock, P.
J.; Hazuda, D. J. Org Lett 2002, 4, 1431–1434.
18. De Zotti, M.; Formaggio, F.; Kaptein, B.; Broxterman, Q. B.;
Felock, P. J.; Hazuda, D. J.; Singh, S. B.; Bruckner, H.; Toniolo,
C. ChemBioChem 2009, 10, 87–90.
19. De Zotti, M.; Damato, F.; Formaggio, F.; Crisma, M.; Schievano,
E.; Mammi, S.; Kaptein, B.; Broxterman, Q. B.; Felock, P. J.;
Hazuda, D. J.; Singh, S. B.; Kirschbaum, J.; Bruckner, H.;
Toniolo, C. Chem Eur J 2010, 16, 316–327.
20. De Zotti, M.; Schievano, E.; Mammi, S.; Kaptein, B.; Broxter-
man, Q. B.; Singh, S. B.; Bruckner, H.; Toniolo, C. Chem Bio-
divers, in press.
21. De Zotti, M. Ph.D. Thesis, University of Padova, 2006.
58 De Zotti et al.
Biopolymers (Peptide Science)

22. XEASY-ETH Automated Spectroscopy 1.4 (version 60801000 of
the XWindow System). Bartels, C.; Xia, T.; Billeter, M.; Guntert,
P.; Wuthrich, K. J Biomol NMR 1995, 6, 1–10.
23. Rance, M.; Sørensen, O. W.; Bodenhausen, G.; Wagner, G.;
Ernst, R. R.; Wuthrich, K. Biochem Biophys Res Commun
1983, 117, 479–485.
24. Bax, A.; Davis, D. G. J Magn Reson 1985, 65, 355–360.
25. Griesinger, C.; Otting, G.; Wuthrich, K.; Ernst, R. R. J Am
Chem Soc 1988, 110, 7870–7872.
26. Bauer, C.; Freeman, R.; Frenkiel, T.; Keeler, J.; Shaka, A. J Magn
Reson 1984, 58, 442–457.
27. Emsley, L.; Bodenhausen, G. J Magn Reson 1989, 82, 211–221.
28. Bax, A.; Subramanian, S. J Magn Reson 1986, 67, 565–569.
29. Bax, A.; Griffey, R. H.; Hawkins, B. L. J Magn Reson 1983, 55,
301–315.
30. Bax, A.; Summers, M. F. J Am Chem Soc 1986, 108, 2093–2094.
31. Schwieters, C. D.; Kuszewski, J. J.; Tjandra, N.; Clore, G. M. J
Magn Reson, 2003, 160, 65–74 (based on X-PLOR 3.851 by
Brunger, A. T.).
32. Koradi, R.; Billeter, M.; Wuthrich, K. J Mol Graph 1996, 14,
51–55.
33. Sonke, T.; Kaptein, B.; Boesten, W. H. J.; Broxterman, Q. B.;
Schoemaker, H. E.; Kamphuis, J.; Formaggio, F.; Toniolo, C.;
Rutjes, F. P. J. T. In Stereoselective Biocatalysis; Patel, R. N., Ed.;
Dekker, New York, NY, 1999; pp 23–58.
34. Carpino, L. A. J Am Chem Soc 1993, 115, 4397–4398.
35. Wu, Y.; Limburg, D. C.; Wilkinson, D. E.; Vaal, M. J.; Hamilton,
G. S. Tetrahedron Lett 2000, 41, 2847–2849.
36. Pysh, E. S.; Toniolo, C. J Am Chem Soc 1977, 99, 6211–6219.
37. Bonora, G. M.; Mapelli, C.; Toniolo, C.; Wilkening, R. R.; Ste-
vens, E. S. Int J Biol Macromol 1984, 6, 179–188.
38. Beychok, S. In Poly-a-Amino Acids: Protein Models for Confor-
mational Studies; Fasman, G. D., Ed.; Dekker, New York, NY,
1967; pp 293–337.
39. Goodman, M.; Verdini, A. S.; Toniolo, C.; Phillips, W. D.; Bovey,
F. A. Proc Natl Acad Sci USA 1969, 64, 444–450.
40. Toniolo, C.; Polese, A.; Formaggio, F.; Crisma, M.; Kamphuis, J.
J Am Chem Soc 1996, 118, 2744–2745.
41. Formaggio, F.; Crisma, M.; Rossi, P.; Scrimin, P.; Kaptein, B.;
Broxterman, Q. B.; Kamphuis, J.; Toniolo, C. Chem Eur J 2000,
6, 4498–4504.
42. Martin, D.; Hauthal, H. G. Dimethyl Sulphoxide; van
Nostrand-Reinhold: Wokingham, UK, 1975.
43. Kopple, K. D.; Ohnishi, M.; Go, A. Biochemistry 1969, 8, 4087–
4095.
44. Wuthrich, K. NMR of Proteins and Nucleic Acids; Wiley: New
York, NY, 1986.
45. Kim, Y. M.; Prestegard, J. H. J Magn Reson 1989, 84, 9–13.
46. Marshall, G. R. In Intra-Science Chemistry Reports; Kharasch,
N., Ed.; Gordon and Breach: New York, NY, 1971; pp 305–326.
47. Karle, I. L.; Balaram, P. Biochemistry 1990, 29, 6747–6756.
48. Toniolo, C.; Crisma, M.; Formaggio, F.; Peggion, C. Biopoly-
mers (Pept Sci) 2001, 60, 396–419.
49. Di Blasio, B.; Pavone, V.; Saviano, M.; Lombardi, A.; Nastri, F.;
Pedone, C.; Benedetti, E.; Crisma, M.; Anzolin, M.; Toniolo, C.
J Am Chem Soc 1992, 114, 6273–6278.
Triple Hyp?Pro Replacement in Integramide A 59
Biopolymers (Peptide Science)
Copyright © 2022 FDOKUMEN












![Problems with a conformation assignment of aryl-substituted resorc[4]arenes](https://static.fdokumen.com/doc/165x107/6324d12685efe380f30661c8/problems-with-a-conformation-assignment-of-aryl-substituted-resorc4arenes.jpg)







