Treatment of phenylmercury salts by heterogeneous photocatalysis over TiO2
7
Treatment of phenylmercury salts by heterogeneous photocatalysis over TiO 2 Emmanuel M. de la Fournie `re a,c , Ana G. Leyva b , Eduardo A. Gautier a , Marta I. Litter a,c, * a Unidad de Actividad Quı ´mica, Centro Ato ´ mico Constituyentes, Comisio ´ n Nacional de Energı ´a Ato ´ mica, Av. Gral. Paz 1499, 1650 San Martı ´n, Prov. de Buenos Aires, Argentina b Unidad de Actividad Fı ´sica, Centro Ato ´ mico Constituyentes, Comisio ´ n Nacional de Energı ´a Ato ´ mica, Av. Gral. Paz 1499, 1650 San Martı ´n, Prov. de Buenos Aires, Argentina c Escuela de Posgrado, Universidad de Gral. San Martı ´n, Peatonal Belgrano 3563, 1° Piso, 1650 San Martı ´n, Prov. de Buenos Aires, Argentina Received 16 February 2007; received in revised form 14 May 2007; accepted 16 May 2007 Available online 2 July 2007 Abstract UV/TiO 2 photocatalysis of phenylmercury salts in aqueous solutions has been performed starting from both acetate (C 6 H 5 HgCH 3 CO 2 , PMA) and chloride (C 6 H 5 HgCl, PMC) salts, in the presence or the absence of oxygen at acidic pH. Removal of Hg(II) in solution took place with the simultaneous deposit of dark or pale gray solids on the photocatalyst, identified as metallic Hg (when starting from PMA) or mixtures of Hg(0) and Hg 2 Cl 2 (when starting from PMC). Partial mineralization of the organic part of both compounds has also been achieved. Hg(II) removal and mineralization were enhanced in the absence of oxygen. PMA photo- catalysis followed a saturation kinetics, going from first order at low concentrations to zero order at higher concentrations (>0.5 mM). For PMA, reaction was faster at high pH (11) with formation of mixtures of Hg and HgO. Phenol was detected as a product of the reaction in both cases, PMA and PMC, and no formation of dangerous methyl- or ethylmercury species was observed in the first case. A mechanism for the photocatalytic reaction has been proposed. The fact that calomel was found as a deposit when starting from PMC under nitrogen suggests that the mechanism of Hg(II) transformation proceeds through successive one-electron transfer reactions passing by mercurous forms. Ó 2007 Elsevier Ltd. All rights reserved. Keywords: Heterogeneous photocatalysis; Mercury; Phenylmercury; Titanium dioxide 1. Introduction Heterogeneous photocatalysis over TiO 2 has been widely used for destruction of organic pollutants in waste- waters. Less studied but not less important from an environmental point of view is the photocatalytic transfor- mation of metal ions by oxidation or reduction to species of lower toxicity or easier to separate from the aqueous phase (Serpone et al., 1988; Litter, 1999; Dome `nech et al., 2004; Litter et al., 2004). Mercury(II) is a frequent component of industrial wastewaters and its major use is in agricultural applica- tions, taking part of pesticides, fungicides, herbicides, insecticides and bactericides. It is also used in other indus- tries (e.g., chlorine-alkali, paints, pharmaceuticals, elec- tronics, cosmetics, etc.). This contributes to the large dispersion of the element as a hazardous pollutant. In fact, mercury(II) is remarkably toxic at concentrations higher than 0.005 mg l 1 , and it is included in the list of priority pollutants of the USEPA with a maximum contaminant 0045-6535/$ - see front matter Ó 2007 Elsevier Ltd. All rights reserved. doi:10.1016/j.chemosphere.2007.05.042 * Corresponding author. Address: Unidad de Actividad Quı ´mica, Centro Ato ´ mico Constituyentes, Comisio ´n Nacional de Energı ´a Ato ´ mica, Av. Gral. Paz 1499, 1650 San Martı ´n, Prov. de Buenos Aires, Argentina. Tel./fax: +54 11 6 772 7016. E-mail address: [email protected] (M.I. Litter). www.elsevier.com/locate/chemosphere Chemosphere 69 (2007) 682–688
Transcript of Treatment of phenylmercury salts by heterogeneous photocatalysis over TiO2

www.elsevier.com/locate/chemosphere
Chemosphere 69 (2007) 682–688
Treatment of phenylmercury salts by heterogeneousphotocatalysis over TiO2
Emmanuel M. de la Fourniere a,c, Ana G. Leyva b, Eduardo A. Gautier a,Marta I. Litter a,c,*
a Unidad de Actividad Quımica, Centro Atomico Constituyentes, Comision Nacional de Energıa Atomica, Av. Gral. Paz 1499,
1650 San Martın, Prov. de Buenos Aires, Argentinab Unidad de Actividad Fısica, Centro Atomico Constituyentes, Comision Nacional de Energıa Atomica, Av. Gral. Paz 1499,
1650 San Martın, Prov. de Buenos Aires, Argentinac Escuela de Posgrado, Universidad de Gral. San Martın, Peatonal Belgrano 3563, 1� Piso, 1650 San Martın,
Prov. de Buenos Aires, Argentina
Received 16 February 2007; received in revised form 14 May 2007; accepted 16 May 2007Available online 2 July 2007
Abstract
UV/TiO2 photocatalysis of phenylmercury salts in aqueous solutions has been performed starting from both acetate(C6H5HgCH3CO2, PMA) and chloride (C6H5HgCl, PMC) salts, in the presence or the absence of oxygen at acidic pH. Removal ofHg(II) in solution took place with the simultaneous deposit of dark or pale gray solids on the photocatalyst, identified as metallicHg (when starting from PMA) or mixtures of Hg(0) and Hg2Cl2 (when starting from PMC). Partial mineralization of the organic partof both compounds has also been achieved. Hg(II) removal and mineralization were enhanced in the absence of oxygen. PMA photo-catalysis followed a saturation kinetics, going from first order at low concentrations to zero order at higher concentrations (>0.5 mM).For PMA, reaction was faster at high pH (11) with formation of mixtures of Hg and HgO. Phenol was detected as a product of thereaction in both cases, PMA and PMC, and no formation of dangerous methyl- or ethylmercury species was observed in the first case.A mechanism for the photocatalytic reaction has been proposed. The fact that calomel was found as a deposit when starting from PMCunder nitrogen suggests that the mechanism of Hg(II) transformation proceeds through successive one-electron transfer reactions passingby mercurous forms.� 2007 Elsevier Ltd. All rights reserved.
Keywords: Heterogeneous photocatalysis; Mercury; Phenylmercury; Titanium dioxide
1. Introduction
Heterogeneous photocatalysis over TiO2 has beenwidely used for destruction of organic pollutants in waste-waters. Less studied but not less important from anenvironmental point of view is the photocatalytic transfor-mation of metal ions by oxidation or reduction to species
0045-6535/$ - see front matter � 2007 Elsevier Ltd. All rights reserved.
doi:10.1016/j.chemosphere.2007.05.042
* Corresponding author. Address: Unidad de Actividad Quımica,Centro Atomico Constituyentes, Comision Nacional de Energıa Atomica,Av. Gral. Paz 1499, 1650 San Martın, Prov. de Buenos Aires, Argentina.Tel./fax: +54 11 6 772 7016.
E-mail address: [email protected] (M.I. Litter).
of lower toxicity or easier to separate from the aqueousphase (Serpone et al., 1988; Litter, 1999; Domenechet al., 2004; Litter et al., 2004).
Mercury(II) is a frequent component of industrialwastewaters and its major use is in agricultural applica-tions, taking part of pesticides, fungicides, herbicides,insecticides and bactericides. It is also used in other indus-tries (e.g., chlorine-alkali, paints, pharmaceuticals, elec-tronics, cosmetics, etc.). This contributes to the largedispersion of the element as a hazardous pollutant. In fact,mercury(II) is remarkably toxic at concentrations higherthan 0.005 mg l�1, and it is included in the list of prioritypollutants of the USEPA with a maximum contaminant

E.M. de la Fourniere et al. / Chemosphere 69 (2007) 682–688 683
level of 2 lg l�1 (Salomons et al., 1995; USEPA, 2006). InArgentina, 4 lg l�1 has been established as National LevelQuality Standard in water for human consumption (Sub-secretarıa de Recursos Hıdricos de la Nacion, 2005).
Toxicity of organomercurial compounds such as methyl-or phenylmercuric salts is higher than that of inorganicmercury species. The impact of these organic compoundson the natural environment is of great concern (Nriagu,1979). For example, the massive case of poisoning inJapan, the Minamata Bay incident, was attributed toindustrial discharge of organomercurials, and decliningbird populations in Sweden was blamed on the use of phe-nyl- and methylmercurial pesticides as seed dressings(Baughman et al., 1973). For this reason, our interest isfocused on the study of the treatment of phenylmercuricacetate (PMA) and phenylmercuric chloride (PMC) inaqueous solution. Especially, PMA is a pesticide widelyused in Argentina some decades ago and, although its usehas been forbidden since 1971 (Garcıa et al., 2003), it pos-sibly still remains in soils and water, becoming a noxiouspollutant for living beings.
Removal of mercuric species in aqueous solution is dif-ficult. Common remediation procedures are precipitationwith chemicals, ion exchange, adsorption, and reduction(Botta et al., 2002 and references therein). Heterogeneousphotocatalysis (HP) has been proposed as a convenienttool for mercury reduction because it does not requirethe use of expensive chemicals and only near UV light isneeded; even solar light can be used (Serpone et al., 1988;Litter, 1999; Botta et al., 2002; Domenech et al., 2004;Litter et al., 2004; Wang et al., 2004; Custo et al., 2006).
Photocatalytic studies with inorganic salts of mercury(Hg(NO3)2, HgCl2 and Hg(ClO4)2), carried out previouslyby our group, have shown that HP of mercury speciesdepends on the nature of the starting salt, pH and presenceor absence of oxygen (Botta et al., 2002). Depending on theconditions, different products are formed on the photocat-alyst surface, like Hg(0), HgO or calomel. Presence ofdonors (e.g., EDTA) in the photocatalytic system, acceler-ates Hg(II) reduction due to a synergy between the organiccompound and the metal ion (Custo et al., 2006). However,due to the complicated chemistry of mercury in solutionand in solid phase, and the nature of products that canbe formed, HP processes on this metal ion are still far frombeing completely understood.
Organomercurials are not easily decomposed (Capeloet al., 2000; Gadfeldt et al., 2001). PMA, in aqueous phase,can be bioreduced to the metallic state by some Pseudomo-
nas strains (Mirgain et al., 1989). Short UV-degradation ofphenylmercury compounds has been reported (Baughmanet al., 1973; Cerrillos et al., 1994). Regarding advanced oxi-dation processes, ultrasonic irradiation has been proposedfor conversion of methyl- and phenylmercury to inorganicmercury for the subsequent determination by flow injec-tion-cold vapor atomic absorption spectrometry (Capeloet al., 2000). In this case, the organic moiety of both mer-cury compounds could be decomposed, but reducing spe-
cies in the sonolytic system were not able to reduceremaining Hg(II) to the metallic form; this step had to bedone with a stronger agent like sodium tetrahydroborate.On the other hand, although HP of methylmercury(II)solutions with deposition of mercury on the catalyst wasreported in two previous articles, no further details onmechanism or products were given (Serpone et al., 1987;Prairie et al., 1993). Tennakone et al. (1993) reported thecomplete mineralization of mercurochrome, an organo-mercury compound, by TiO2 photocatalysis, with Hg(0)deposition.
In this paper, photocatalytic studies of transformationof PMA and PMC are presented. The kinetic behavior,nature of the reaction products and influence of oxygenand pH have been analyzed.
2. Experimental
2.1. Materials
TiO2 (Degussa P-25) was provided by Degussa AG Ger-many and used as received. PMA (C6H5HgCH3CO2, Len-nox) and PMC (C6H5HgCl, Fluka) of the highest puritywere used. All other reagents were at least of reagent gradeand used without further purification. Solutions were pre-pared with Milli-Q water (resistivity = 18 MX cm). DilutedNaOH solutions were used in some cases to adjust pHbefore photocatalytic experiments.
2.2. Photocatalytic experiments
Photocatalytic runs were carried out in a recirculatingsystem (1.5 l min�1 flow rate) consisting of an annular reac-tor (415 mm-length, 35 mm-external diameter, 85 ml totalvolume), a peristaltic pump and a thermostatted (298 K)cylindrical reservoir. A black-light tubular UV lamp (FL-BLB, Toshiba Electric, 15 W, 300 < k/nm < 400, 100%maximum emission at 352 nm) was installed inside theannular reactor as the source of illumination. Actinometricmeasurements were performed by the ferrioxalate method(Hatchard and Parker, 1956). The photon flux per unit ofvolume (P0) was 3.1 lEinstein s�1 l�1.
The reaction was conducted with the reactor open to airor under a water-saturated nitrogen stream bubbled in thesuspension at 0.4 l min�1 throughout the experiment. Inall cases, a fresh solution (500 cm3) of the correspondingphenylmercuric salt was used, the catalyst (1 g l�1) was sus-pended in the solution, and the suspension was ultrasoni-cated for 2 min. Prior to irradiation, suspensions werestirred in the dark for 30 min, a time enough to assure sub-strate–surface equilibrium (see Section 3). The extent ofadsorption of the mercury(II) compound onto TiO2 wasdetermined by measuring mercuric concentrations in the fil-trate (see below) before and after stirring in the dark, underthe same conditions (air or nitrogen) used for irradiation.Irradiations were performed under magnetic stirring for240 min. Initial pH of PMA and PMC solutions was

0
0.2
0.4
0.6
0.8
1
0 60 120 180 240Time (min)
[Hg(
II)]/[
Hg(
II)] 0
PMA, airPMA, N2PMC, airPMC, N2
Fig. 1. Time profiles of normalized mercury TiO2-photocatalytic conver-sion starting from phenylmercuric salts. Conditions: [PMA] = [PMC] =0.5 mM, [TiO2] = 1 g l�1, T = 25 �C. Initial pH for PMA: 4.1 and forPMC: 3.95. Near UV-light (350 < k/nm < 400). P0 = 3.1 lEinstein s�1 l�1.
0
0.2
0.4
0.6
0.8
1
0 60 120 180 240Time (min)
TOC
/TO
C0
PMA airPMA N2PMC airPMC N2
Fig. 2. TOC decrease during the photocatalysis of phenylmercuric saltsover TiO2. Conditions of Fig. 1.
684 E.M. de la Fourniere et al. / Chemosphere 69 (2007) 682–688
measured in the suspension after equilibration in the dark,and was left to vary freely during the experiments. Whenthe effect of pH in the photocatalytic reaction was tested,previous adjustment to a prefixed value was made bydiluted NaOH addition. Samples were periodically with-drawn and filtered through 0.22 lm Millipore filters. Atleast, duplicated runs were carried out for each condition,averaging the results; however, several runs were neededin general to assure reproducibility, especially in the pres-ence of oxygen. In all cases, the amount of the mercury saltadsorbed in the dark was discounted to analyze only theeffect of light on the photocatalytic transformation.
Mercury concentration in the filtrate was measured bydetection at 220 nm of the complex formed between mer-cury(II) species (associated or free) in solution and 2-mer-captopropionic acid, by a modification of the methodreported by Parkin (1986), where 6-mercaptopurine wasused to form the complex. A HP 8453 diode-array UV–vis spectrophotometer was employed for all spectrophoto-metric measurements.
Phenol concentration and mercury speciation in the fil-trate was followed by a HPLC method developed by thegroup (de la Fourniere et al., unpublished results). Thechromatographic system consisted of an Alltech 301 HPLCPump, a 100-ll loop, a Thermo C18 column (5 lm,15 cm · 4.6 mm) and a Spectra SYSTEM UV1000 detec-tor. The mobile phase was composed of methanol–acetoni-trile–0.005 M NaH2PO4 (1:4:5) containing 9.14 · 10�2 mM2-mercaptopropionic acid. A carrier flow of 0.8 ml min�1
was employed, and analytical measurements were carriedout at 220 nm. Data acquisition was processed with Konik-rom software.
The deposits on the recovered photocatalyst were ana-lyzed by chemical reactions for identification of differentproducts. A gray deposit, soluble in concentrated nitricacid, indicated metallic Hg (Martı et al., 1994; Tennakoneand Ketipearachchi, 1995). Treatment of the deposit with2.5 M KI, giving a filtrate absorbing at 323 nm, indicatedHgO (Kolthoff et al., 1969; Tennakone et al., 1993). A palegray deposit was an evidence of a mixture of metallic Hgand Hg2Cl2. Samples were also analyzed by X-ray diffrac-tion (XRD) at room temperature, using a Philips PW-3710 diffractometer and CuKa radiation.
The mineralization degree was followed by total organiccarbon (TOC) analysis, using a Shimadzu 5000A TOC ana-lyzer in the non-purgeable organic carbon mode.
3. Results and discussion
Prior to photocatalytic experiments, the degree ofadsorption in the dark and the time needed to reach theadsorption equilibrium were evaluated by stirring suspen-sions of the organomercuric compounds (0.5 mM) inTiO2 (1 g l�1) for 30 min at the original pH (4.1 for PMAand 3.95 for PCM). From the experimental data, it canbe concluded that the phenylmercuric ion presents a con-siderable affinity for TiO2 under all the experimental condi-
tions here essayed, ranging 12–25%. The adsorption degreewas somewhat higher under nitrogen and for PMA. Oncediscounted this initial adsorption, no changes in Hg(II)concentration were registered in the following 240 min ofstirring in the dark, indicating also that there is no trans-formation of the mercuric salt in the absence of light.
Reaction under irradiation in the absence of TiO2 wasnegligible. At the wavelengths used in our experiments,the very well known photolysis of phenylmercuric com-pounds (Baughman et al., 1973) cannot take place.
In Fig. 1, profiles of normalized concentration vs. timeafter UV irradiation over TiO2 (measured as total Hg(II)species in solution, either organic or inorganic) are shown.In all systems, after 4 h of irradiation, a high mercury con-version took place (more than 65% in the less favored case,PMC in air); mercury removal was higher in the absence ofoxygen. PMA was more reactive than PMC, althoughunder N2, at the final time, PMC attained a similar conver-sion. In all cases, pH dropped slightly to more acidic valuesduring the photocatalytic run, varying less than 0.5 after240 min of irradiation. This variation was even lower athigher pH values.
Fig. 2 shows the depletion of TOC for the four condi-tions, which was rather important (more than 40% after240 min irradiation) and almost independent on the anion
0
10000000
20000000
30000000
40000000
50000000
0 5000 10000 15000 20000
1/C0 (M-1)1/
R0
(M-1
s)
0
0.4
0.8
1.2
1.6
0 20 40 60 80C0 x 105 (M)
R0
x 10
7 (M
s-1
)
a
b
100
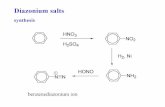
Fig. 4. Kinetic behavior of the photocatalysis of PMA with the reactoropen to air. Photocatalytic runs at different PMA concentrations. Otherconditions: same of Fig. 1, solutions at original pH.
E.M. de la Fourniere et al. / Chemosphere 69 (2007) 682–688 685
of the phenylmercuric salt, suggesting that the aromaticmoiety is the main degraded portion of the molecule.TOC decrease was slightly higher in deaerated conditions,indicating that Hg(II), acting as the oxidant in the system,presents a photocatalytic oxidizing ability higher than O2.
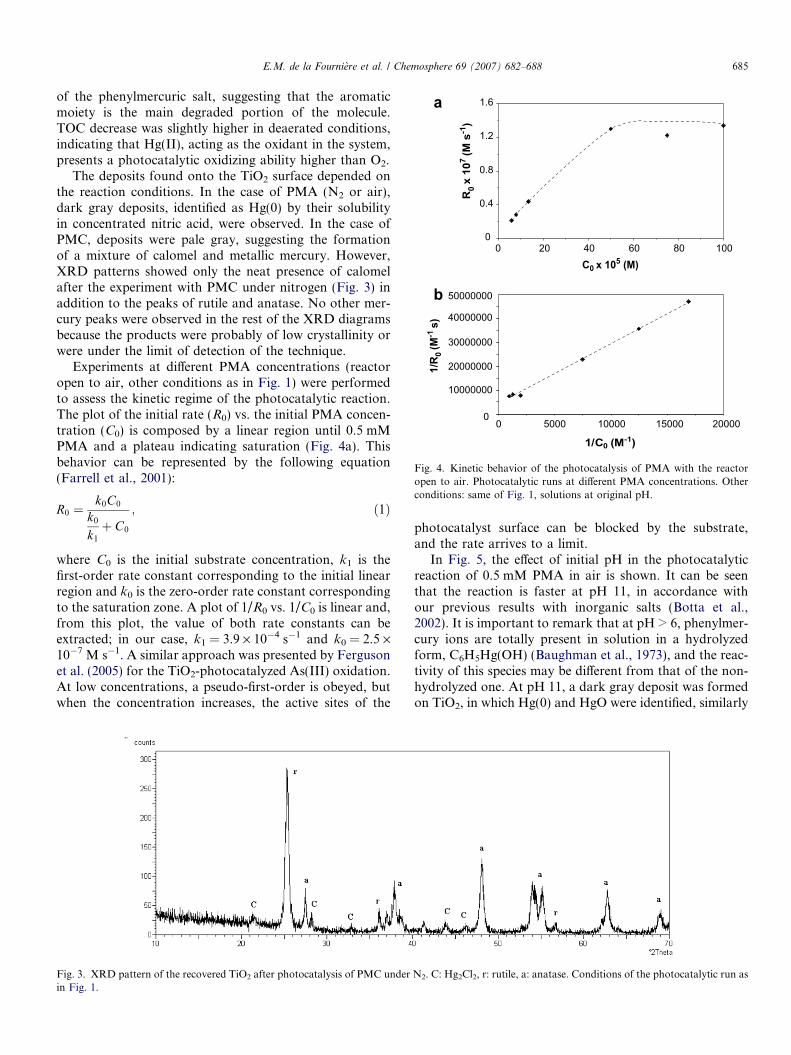
The deposits found onto the TiO2 surface depended onthe reaction conditions. In the case of PMA (N2 or air),dark gray deposits, identified as Hg(0) by their solubilityin concentrated nitric acid, were observed. In the case ofPMC, deposits were pale gray, suggesting the formationof a mixture of calomel and metallic mercury. However,XRD patterns showed only the neat presence of calomelafter the experiment with PMC under nitrogen (Fig. 3) inaddition to the peaks of rutile and anatase. No other mer-cury peaks were observed in the rest of the XRD diagramsbecause the products were probably of low crystallinity orwere under the limit of detection of the technique.
Experiments at different PMA concentrations (reactoropen to air, other conditions as in Fig. 1) were performedto assess the kinetic regime of the photocatalytic reaction.The plot of the initial rate (R0) vs. the initial PMA concen-tration (C0) is composed by a linear region until 0.5 mMPMA and a plateau indicating saturation (Fig. 4a). Thisbehavior can be represented by the following equation(Farrell et al., 2001):
R0 ¼k0C0
k0
k1
þ C0
; ð1Þ
where C0 is the initial substrate concentration, k1 is thefirst-order rate constant corresponding to the initial linearregion and k0 is the zero-order rate constant correspondingto the saturation zone. A plot of 1/R0 vs. 1/C0 is linear and,from this plot, the value of both rate constants can beextracted; in our case, k1 = 3.9 · 10�4 s�1 and k0 = 2.5 ·10�7 M s�1. A similar approach was presented by Fergusonet al. (2005) for the TiO2-photocatalyzed As(III) oxidation.At low concentrations, a pseudo-first-order is obeyed, butwhen the concentration increases, the active sites of the
Fig. 3. XRD pattern of the recovered TiO2 after photocatalysis of PMC underin Fig. 1.
photocatalyst surface can be blocked by the substrate,and the rate arrives to a limit.
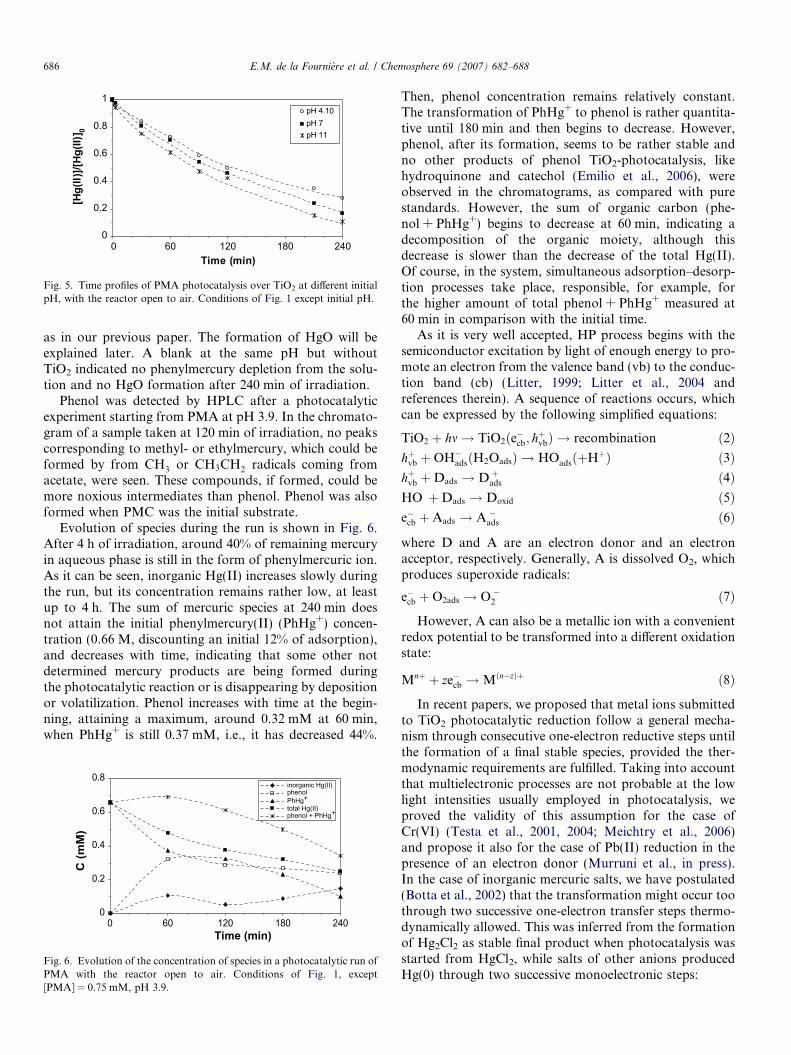
In Fig. 5, the effect of initial pH in the photocatalyticreaction of 0.5 mM PMA in air is shown. It can be seenthat the reaction is faster at pH 11, in accordance withour previous results with inorganic salts (Botta et al.,2002). It is important to remark that at pH > 6, phenylmer-cury ions are totally present in solution in a hydrolyzedform, C6H5Hg(OH) (Baughman et al., 1973), and the reac-tivity of this species may be different from that of the non-hydrolyzed one. At pH 11, a dark gray deposit was formedon TiO2, in which Hg(0) and HgO were identified, similarly
N2. C: Hg2Cl2, r: rutile, a: anatase. Conditions of the photocatalytic run as
0
0.2
0.4
0.6
0.8
1
0 60 120 180 240Time (min)
[Hg(
II)]/[
Hg(II
)] 0
pH 4.10pH 7pH 11
Fig. 5. Time profiles of PMA photocatalysis over TiO2 at different initialpH, with the reactor open to air. Conditions of Fig. 1 except initial pH.
686 E.M. de la Fourniere et al. / Chemosphere 69 (2007) 682–688
as in our previous paper. The formation of HgO will beexplained later. A blank at the same pH but withoutTiO2 indicated no phenylmercury depletion from the solu-tion and no HgO formation after 240 min of irradiation.
Phenol was detected by HPLC after a photocatalyticexperiment starting from PMA at pH 3.9. In the chromato-gram of a sample taken at 120 min of irradiation, no peakscorresponding to methyl- or ethylmercury, which could beformed by from CH�3 or CH3CH�2 radicals coming fromacetate, were seen. These compounds, if formed, could bemore noxious intermediates than phenol. Phenol was alsoformed when PMC was the initial substrate.
Evolution of species during the run is shown in Fig. 6.After 4 h of irradiation, around 40% of remaining mercuryin aqueous phase is still in the form of phenylmercuric ion.As it can be seen, inorganic Hg(II) increases slowly duringthe run, but its concentration remains rather low, at leastup to 4 h. The sum of mercuric species at 240 min doesnot attain the initial phenylmercury(II) (PhHg+) concen-tration (0.66 M, discounting an initial 12% of adsorption),and decreases with time, indicating that some other notdetermined mercury products are being formed duringthe photocatalytic reaction or is disappearing by depositionor volatilization. Phenol increases with time at the begin-ning, attaining a maximum, around 0.32 mM at 60 min,when PhHg+ is still 0.37 mM, i.e., it has decreased 44%.
0
0.2
0.4
0.6
0.8
0 60 120 180 240Time (min)
C (m
M)
inorganic Hg(II)phenolPhHg+total Hg(II)phenol + PhHg+
Fig. 6. Evolution of the concentration of species in a photocatalytic run ofPMA with the reactor open to air. Conditions of Fig. 1, except[PMA] = 0.75 mM, pH 3.9.
Then, phenol concentration remains relatively constant.The transformation of PhHg+ to phenol is rather quantita-tive until 180 min and then begins to decrease. However,phenol, after its formation, seems to be rather stable andno other products of phenol TiO2-photocatalysis, likehydroquinone and catechol (Emilio et al., 2006), wereobserved in the chromatograms, as compared with purestandards. However, the sum of organic carbon (phe-nol + PhHg+) begins to decrease at 60 min, indicating adecomposition of the organic moiety, although thisdecrease is slower than the decrease of the total Hg(II).Of course, in the system, simultaneous adsorption–desorp-tion processes take place, responsible, for example, forthe higher amount of total phenol + PhHg+ measured at60 min in comparison with the initial time.
As it is very well accepted, HP process begins with thesemiconductor excitation by light of enough energy to pro-mote an electron from the valence band (vb) to the conduc-tion band (cb) (Litter, 1999; Litter et al., 2004 andreferences therein). A sequence of reactions occurs, whichcan be expressed by the following simplified equations:
TiO2 þ hm! TiO2ðe�cb; hþvbÞ ! recombination ð2Þ
hþvb þOH�adsðH2OadsÞ ! HO�adsðþHþÞ ð3Þhþvb þDads ! D�þads ð4ÞHO� þDads ! Doxid ð5Þe�cb þAads ! A��ads ð6Þ
where D and A are an electron donor and an electronacceptor, respectively. Generally, A is dissolved O2, whichproduces superoxide radicals:
e�cb þO2ads ! O��2 ð7Þ
However, A can also be a metallic ion with a convenientredox potential to be transformed into a different oxidationstate:
Mnþ þ ze�cb !Mðn�zÞþ ð8Þ
In recent papers, we proposed that metal ions submittedto TiO2 photocatalytic reduction follow a general mecha-nism through consecutive one-electron reductive steps untilthe formation of a final stable species, provided the ther-modynamic requirements are fulfilled. Taking into accountthat multielectronic processes are not probable at the lowlight intensities usually employed in photocatalysis, weproved the validity of this assumption for the case ofCr(VI) (Testa et al., 2001, 2004; Meichtry et al., 2006)and propose it also for the case of Pb(II) reduction in thepresence of an electron donor (Murruni et al., in press).In the case of inorganic mercuric salts, we have postulated(Botta et al., 2002) that the transformation might occur toothrough two successive one-electron transfer steps thermo-dynamically allowed. This was inferred from the formationof Hg2Cl2 as stable final product when photocatalysis wasstarted from HgCl2, while salts of other anions producedHg(0) through two successive monoelectronic steps:

E.M. de la Fourniere et al. / Chemosphere 69 (2007) 682–688 687
Hg2þ þ e�cb ! HgðIÞ ð9ÞHgðIÞ þ e�cb ! Hgð0Þ ð10Þ
As anodic reaction, the oxidation of water by holes, avery slow process, has been proposed for metallic photo-catalytic reductions in the absence of electron donors (Lit-ter, 1999):
H2Oþ hþvb ! HO� þHþ ð11ÞHowever, when in the reaction medium there is a species
more oxidizable than water, mercury reduction is sinergi-cally helped by this species (Wang et al., 2004; Custoet al., 2006). In the case of the phenylmercury salts, we pro-pose that the organic moiety can play the role of the donor,and the following sequence of simplified reactions can bewritten:
C6H5Hgþ(Cl� or CH3COO�) + (HO�) + Hþ
!C6H5OH + Hg2þ(Cl� or CH3COO�) ð12Þfollowed by reactions (9) and (10). The anodic pathwayscan continue through:
C6H5OHþ hþvbðHO�Þ ! � � � ! CO2 þH2O ð13ÞCH3COO� þ hþvbðHO�Þ ! � � � ! CO2 þH2O ð14Þ
It is also possible to propose a simultaneous Hg(II)reduction and oxidation of the organic moiety, taking placeon the initial organomercurial. However, it is not possiblewith our results to distinguish between simultaneous orconsecutive steps.
Global reaction agrees with the observed pH decrease:
C6H5Hgþ+ H2O!C6H5OH + Hg(0) + Hþ ð15ÞThe inhibition of mercury reduction by oxygen is, of
course, explained by the competition between processes(7), (9) and (10), as already described in previous works(Serpone et al., 1988; Litter, 1999; Litter et al., 2004, andreferences therein). However, the fact that a larger TOCdecrease is observed under nitrogen than in air (Fig. 2) isa proof of the synergy between the inorganic and theorganic moiety of the organomercurial compound; it isconcluded that Hg(II) behaves as a better oxidant thanoxygen for mineralization.
Formation of HgO at pH 11 can be explained by reoxi-dation of Hg(0) by holes or HO�, or by direct transforma-tion of Hg(II) at that pH, helped by light and catalyst(Botta et al., 2002). Our experiment was performed in airand, under these conditions, metallic Hg can be also oxi-dized very rapidly (Cotton and Wilkinson, 1998):
Hg(0) + 1/2O2!HgO ð16ÞAs we suggest in our previous paper, this product is alsoexpected when starting from PMC because, if calomel isformed, it will be easily disproportioned at pH 11 to Hgand HgO.
4. Conclusions
Heterogeneous photocatalysis over TiO2 is particularlyconvenient for PMA and PMC treatment because it can
remove at a large extent the content of mercury presentin solution and leads to the mineralization of the organicportion of the compound. Removal of Hg(II) in solutiontook place with deposit of metallic Hg (starting fromPMA) or mixtures of Hg and Hg2Cl2 (starting fromPMC). Hg(II) behaves as a better oxidant than oxygenfor mineralization. Hg(II) removal and mineralization aremore favorable in the absence of oxygen and at pH 11,where HgO is formed together with Hg(0). Phenol wasdetected for both, PMA and PMC, as a product of the pho-tocatalytic reaction, without the risk of the formation ofvery dangerous alkylmercury species in the last case, whichis an advantage for application purposes. However, irradi-ations should be carried out until reaching complete miner-alization, to avoid the risk of formation of noxiousintermediates after the treatment.
Acknowledgements
This work was performed as part of Comision Nacionalde Energıa Atomica P5-PID-36-4 Program, and AgenciaNacional de Promocion Cientıfica y Tecnologica (AN-PCyT) project PICT2003-13-13261. E.F. thanks ANPCyTfor a doctoral fellowship. MIL is a member of CONICET.
References
Baughman, G.L., Gordon, J.A., Lee Wolfe, N., Zepp, R.G., 1973.Chemistry of Organomercurials in Aquatic Systems, EPA-660/3-73-012.
Botta, S.G., Rodrıguez, D.J., Leyva, A.G., Litter, M.I., 2002. Features ofthe transformation of HgII by heterogeneous photocatalysis over TiO2.Catal. Today 76, 247–258.
Capelo, J.L., Lavilla, I., Bendicho, C., 2000. Room temperature sonolysis-based advanced oxidation process for degradation of organomercuri-als: application to determination of inorganic and total mercury inwaters by flow injection-cold vapor atomic absorption spectrometry.Anal. Chem. 72, 4979–4984.
Cerrillos, C., Pradera Adrian, M.A., Navıo, J.A., 1994. UV photolyticdegradation of phenylmercury compounds in water–acetonitrile (1:1)media. J. Photochem. Photobiol. A: Chem. 84, 299–303.
Cotton, F.A., Wilkinson, G., 1998. Quımica Inorganica Avanzada.Limusa, Mexico, D.F., pp. 715–747.
Custo, G., Litter, M.I., Rodrıguez, D., Vazquez, C., 2006. Total reflectionX-ray fluorescence trace mercury determination by trapping complex-ation: application in advanced oxidation technologies. Spectrochim.Acta B 61, 1119–1123.
de la Fourniere, E.M., Litter, M.I., Gautier, E.A., unpublished results.Detection of mercury species in the treatment of phenylmercuric saltsby heterogeneous photocatalysis.
Domenech, X., Jardim, W., Litter, M., 2004. Tecnologıas avanzadas deoxidacion para la eliminacion de contaminantes. In: Blesa, M.A.,Sanchez Cabrero, B. (Eds.), Eliminacion de contaminantes porfotocatalisis heterogenea. Ediciones CIEMAT, Madrid, pp. 7–34.
Emilio, C.A., Litter, M.I., Kunst, M., Bouchard, M., Colbeau-Justin, C.,2006. Phenol photodegradation on platinized-TiO2 photocatalystsrelated to charge-carrier dynamics. Langmuir 22, 3606–3613.
Farrell, J., Wang, J., O’Day, P., Conklin, M., 2001. Electrochemical andspectroscopic study of arsenate removal from water using zero-valentiron media. Environ. Sci. Technol. 35, 2026–2032.
Ferguson, M.A., Hoffmann, M.R., Hering, J.G., 2005. TiO2-photocata-lyzed As(III) oxidation in aqueous suspensions: reaction kinetics andeffects of adsorption. Environ. Sci. Technol. 39, 1880–1886.

688 E.M. de la Fourniere et al. / Chemosphere 69 (2007) 682–688
Gadfeldt, K., Sommar, J., Stromberg, D., Feng, X., 2001. Oxidation ofatomic mercury by hydroxyl radicals and photoinduced decompositionof methylmercury in the aqueous phase. Atmos. Environ. 35, 3039–3047.
Garcıa, S.I., Bovi Mitre, G., Moreno, I., Eiman Grossi, M., Digon, A., deTitto, E., 2003. Regional workshop on intoxications by plaguicidesand harmonization in the collection of the information, Buenos Aires,Health Ministry, Argentina, <www.medioambiente.gov.ar/archivos/web/salud_ambiente/File/tallerplag2003.pdf>.
Hatchard, C.G., Parker, C.A., 1956. A new sensitive chemical actinom-eter. II. Potassium ferrioxalate as a standard chemical actinometer.Proc. Roy. Soc. A 235, 518–536.
Kolthoff, I.M., Sandell, E.B., Meehan-Stanley Bruckenstein, E.J., 1969.Analisis Quımico Cuantitativo, Nigar, Buenos Aires, p. 808.
Litter, M.I., 1999. Heterogeneous photocatalysis: transition metal ions inphotocatalytic systems. Appl. Catal. B: Environ. 23, 89–114.
Litter, M., Domenech, X., Mansilla, H., 2004. Remocion de contaminan-tes metalicos. In: Blesa, M.A., Sanchez Cabrero, B. (Eds.), Eliminacionde contaminantes por fotocatalisis heterogenea. Ediciones CIEMAT,Madrid, pp. 163–187.
Martı, F.B., Conde, F.L., Jimeno, S.A., 1994. Quımica AnalıticaCualitativa, Paraninfo, Madrid, p. 435.
Meichtry, J.M., Brusa, M., Mailhot, G., Grela, M.A., Litter, M.I., 2006.Heterogeneous photocatalysis of Cr(VI) in the presence of citric acidover TiO2 particles: relevance of Cr(V)–citrate complexes. Appl. Catal.B: Environ. 71, 101–107.
Mirgain, I., Werneburg, B., Harf, C., Monteil, H., 1989. Phenylmercuricacetate biodegradation by environmental strains of Pseudomonas
species. Res. Microbiol. 140, 695–707.Murruni, L., Leyva, G., Litter, M.I., in press. Photocatalytic removal of
Pb(II) over TiO2 and Pt–TiO2 powders. Catal. Today.Nriagu, J.O. (Ed.), 1979. The Biogeochemistry of Mercury in the
Environment. Elsevier-North Holland Biomedical, Amsterdam.Parkin, J.E., 1986. Assay of phenylmercuric acetate and nitrate in
pharmaceutical products by high-performance liquid chromatographywith indirect photometric detection. J. Chromatogr. A 370, 210–213.
Prairie, M.R., Stange, B.M., Evans, L.R., 1993. Photocatalysis for thedestruction of organics and the reduction of heavy metals. In: Ollis,D.F., Al-Ekabi, H. (Eds.), Photocatalytic Purification and Treatment
of Water and Air. Elsevier Sci. Publish. B.V., Amsterdam, pp. 353–363.
Salomons, W., Forstner, U., Mader, P. (Eds.), 1995. Heavy Metals,Problems and Solutions. Springer-Verlag, Berlin, Heidelberg, p. 36.
Serpone, N., Ah-You, Y.K., Tran, T.P., Harris, R., Pelizzetti, E., Hidaka,H., 1987. AM1 simulated sunlight photoreduction and elimination ofHg(II) and CH3Hg(II) chloride salts from aqueous suspensions oftitanium dioxide. Sol. Energy 39, 491–498.
Serpone, N., Borgarello, E., Pelizzetti, E., 1988. Photoreduction andphotodegradation of inorganic pollutants: II. Selective reduction andrecovery of Au, Pt, Pd, Rh, Hg, and Pb. In: Schiavello, M. (Ed.),Photocatalysis and Environment. Kluwer Academic Publishers,Dordrecht, pp. 527–565.
Subsecretarıa de Recursos Hıdricos de la Nacion, Diciembre 2005. NivelesGuıa Nacionales de Calidad de Agua Ambiente. Republica Argentina.<http://www.obraspublicas.gov.ar/hidricos/calidad_del_agua_activid-ades.htm>.
Tennakone, K., Thaminimulle, C.T.K., Senadeera, S., Kumarasinghe,A.R., 1993. TiO2-catalysed oxidative photodegradation of mercuro-chrome: an example of an organo-mercury compound. J. Photochem.Photobiol. A 70, 193–195.
Tennakone, K., Ketipearachchi, U.S., 1995. Photocatalytic method forremoval of mercury from contaminated water. Appl. Catal. B 5, 343–349.
Testa, J.J., Grela, M.A., Litter, M.I., 2001. Experimental evidence in favorof an initial one-electron-transfer process in the heterogeneousphotocatalytic reduction of chromium(VI) over TiO2. Langmuir 17,3515–3517.
Testa, J.J., Grela, M.A., Litter, M.I., 2004. Heterogeneous photocatalyticreduction of chromium(VI) over TiO2 particles in the presence ofoxalate: involvement of Cr(V) species. Environ. Sci. Technol. 38, 1589–1594.
US Environmental Protection Agency, 2006. Ground water & drinkingwater. List of drinking water contaminants & MCLs. Nationalprimary drinking water regulations. <http://www.epa.gov/safewater/mcl.html#mcls>.
Wang, X., Pehkonen, S.O., Ray, A.K., 2004. Photocatalytic reduction ofHg(II) on two commercial TiO2 catalysts. Electrochim. Acta 49, 1435–1444.